

Abstract
Activity of dorsal raphe neurons is controlled by noradrenaline afferents. In this brain region, noradrenaline activates Gαq-coupled α1-adrenergic receptors (α1-AR), causing action potential (AP) firing and serotonin release. In vitro, electrical stimulation elicits vesicular noradrenaline release and subsequent activation of α1-AR to produce an EPSC (α1-AR-EPSC). The duration of the α1-AR-EPSC (∼27 s) is much longer than that of most other synaptic currents, but the factors that govern the spatiotemporal dynamics of α1-AR are poorly understood. Using an acute brain slice preparation from adult male and female mice and electrophysiological recordings from dorsal raphe neurons, we found that the time course of the α1-AR-EPSC was slow, but highly consistent within individual serotonin neurons. The amount of noradrenaline released influenced the amplitude of the α1-AR-EPSC without altering the time constant of decay suggesting that once released, extracellular noradrenaline was cleared efficiently. Reuptake of noradrenaline via noradrenaline transporters was a primary means of terminating the α1-AR-EPSC, with little evidence for extrasynaptic diffusion of noradrenaline unless transporter-dependent reuptake was impaired. Taken together, the results demonstrate that despite slow intrinsic signaling kinetics, noradrenaline-dependent synaptic transmission in the dorsal raphe is temporally and spatially controlled and noradrenaline transporters are critical regulators of serotonin neuron excitability. Given the functionally distinct types of neurons intermingled in the dorsal raphe nucleus and the unique roles of these neural circuits in physiological responses, transporters may preserve independence of each synapse to encode a long-lasting but discrete signal.
SIGNIFICANCE STATEMENT The dorsal raphe nucleus is the predominant source of serotonin in the brain and is controlled by another monoamine, noradrenaline. In this brain region, noradrenaline activates G-protein-coupled α1-adrenergic receptors (α1-AR) causing action potential (AP) firing and serotonin release. Despite high interest in pharmacotherapies to enhance serotonin signaling, the factors that govern noradrenaline α1-AR signaling have received little attention. Here, we show using mouse brain slices that the time course of α1-AR signaling is slow, persisting for tens of seconds. Despite slow intrinsic signaling kinetics, noradrenaline-dependent synaptic transmission in the dorsal raphe is controlled temporally and spatially by efficient noradrenaline transporter-dependent clearance of extracellular noradrenaline. Thus, noradrenaline transporters are critical regulators of serotonin neuron excitability.
Keywords: dorsal raphe nucleus, GPCR, noradrenaline, serotonin, synaptic transmission, transporter
Introduction
The dorsal raphe nucleus is the central source of serotonin-containing neurons in the brain. It also contains a diverse array of non-serotonin neurons, including GABA, glutamate, dopamine, and peptide neurons (Huang et al., 2019). Neural activity of each population influences affective and social behaviors, reward processing, and energy balance, including feeding, arousal, and thermogenesis. In vivo studies using rodent models have shown that dorsal raphe neurons are controlled by noradrenaline afferents. In this brain region, noradrenaline activates Gαq-coupled α1-adrenergic receptors (α1-AR), causing membrane depolarization, action potential (AP) firing, and serotonin release (Baraban and Aghajanian, 1980a; Pudovkina et al., 2003). In the absence of noradrenaline, dorsal raphe serotonin neurons are either silent or fire slowly and erratically (Svensson et al., 1975; Baraban et al., 1978; Baraban and Aghajanian, 1980a,b). These studies indicate that α1-AR activation provides the necessary subthreshold depolarization to initiate tonic firing of APs. But the α1-AR are not typically maximally activated. In vivo, local application of noradrenaline increases the firing rate in the majority of dorsal raphe neurons (Couch, 1970; Baraban and Aghajanian, 1980a; Judge and Gartside, 2006) and the amounts of noradrenaline and serotonin in the dorsal raphe are positively correlated (Agren et al., 1986). Thus, α1-AR signaling is a critical bidirectional modulator of the excitability of dorsal raphe neurons and the release of serotonin. Despite the prevalence of pharmacotherapies aimed at enhancing serotonin signaling (e.g., selective serotonin reuptake inhibitors; SSRIs) and an established role of α1-AR signaling in serotonin neuron excitability, the factors that govern α1-AR signaling have received little attention.
The synaptic connections between noradrenaline terminals and dorsal raphe neurons can be studied in detail ex vivo. In rodent dorsal raphe brain slices, electrical stimulation evokes AP-dependent, release of noradrenaline-filled vesicles (Yoshimura et al., 1985; Gantz et al., 2020). Subsequently, activation of α1-AR produces an EPSC (α1-AR-EPSC) via coupling to ionotropic δ 1 glutamate receptor (GluD1R) channels (Gantz et al., 2020). The duration of the α1-AR-EPSC in the dorsal raphe (∼27 s) is much longer than that of most other synaptic currents (typically 3 ms to 1 s, Gantz et al., 2020), but it is not known how much of the duration is because of the lifetime of extracellular noradrenaline versus the downstream second messenger signaling cascade.
Here, we show that the kinetics of the α1-AR-EPSC are variable across neurons but highly consistent with each stimulation within an individual neuron. The α1-AR-EPSC was present in serotonin neurons, but not non-serotonin neurons, indicating selectivity in noradrenaline signaling. The amount of noradrenaline released influenced the amplitude of the α1-AR-EPSC without altering the time constant of decay, suggesting that once released, extracellular noradrenaline was cleared efficiently. Reuptake of noradrenaline via noradrenaline transporters and intrinsic second messenger signaling kinetics were the primary means of terminating the α1-AR-EPSC, with little evidence for extrasynaptic diffusion of noradrenaline unless transporter-dependent reuptake was impaired. Taken together, the results demonstrate that despite slow intrinsic signaling kinetics, noradrenaline-dependent synaptic transmission in the dorsal raphe is temporally and spatially controlled.
Materials and Methods
Animals
All studies were conducted in accordance with and approval by the University of Iowa Institutional Animal Care and Use Committee or the National Institute on Drug Abuse Animal Care and Use Committee. Male and female wild-type C57BL/6J (more than two months old) mice were used. Mice were group-housed on a 12/12 h light/dark cycle.
Brain slice preparation and electrophysiological recordings
Mice were deeply anesthetized with isoflurane and killed by decapitation. Brains were removed and placed in warmed modified Krebs' buffer containing the following: 126 mm NaCl, 2.5 mm KCl, 1.2 mm MgCl2, 1.2 mm CaCl2, 1.2 mm NaH2PO4, 21.5 mm NaHCO3, and 11 mm D-glucose with 5 μm MK-801 to reduce excitotoxicity and increase slice viability, bubbled with 95/5% O2/CO2. In the same solution, coronal dorsal raphe slices (240 µm) were obtained using a vibrating microtome (Leica VT1000S, Leica Biosystems) and incubated at 28°C for >30 min before recording.
Electrophysiological recordings were made at 35°C with Multiclamp 700B amplifiers (Molecular Devices), Digidata 1440A or 1550B A/D converter (Molecular Devices), and Clampex 10.4 or 11.1 software (Molecular Devices) with borosilicate glass electrodes wrapped with Parafilm to reduce pipette capacitance. Pipette resistances were 3.5–4.2 MΩ when filled with an internal solution containing 104.56 mm K-methyl sulfate, 3.73 mm KCl, 5.30 mm NaCl, 4.06 mm MgCl2, 4.06 mm CaCl2, 7.07 mm HEPES (K), 3.25 mm BAPTA(K4) generating predicted-free [Ca2+]i of ∼180 nm (WebMaxC Standard), 0.26 GTP (sodium salt), 4.87 ATP (sodium salt), 4.59 creatine phosphate (sodium salt), pH 7.27–7.32 with KOH, mOsm ∼270, for whole-cell patch-clamp recordings. Series resistance was monitored throughout the experiment. Reported voltages are corrected for a liquid junction potential of −8 mV between the internal solution and external solution. All drugs were applied by bath application except noradrenaline, which was also iontophoretically applied (−200-nA backing current, 200-nA ejection current, 200 ms) onto the soma of the recorded neuron. Synaptic currents were evoked on 60- or 90-s intervals by applying brief pulses (0.5 ms, 60 Hz) of electrical stimulation to the brain slice via a borosilicate glass monopolar stimulating electrode (World Precision Instruments) placed within 200 µm of the recorded neuron in the presence of NMDA (MK-801), AMPA/kainate (DNQX or NBQX, 3 μm), GABAA (picrotoxin, 100 μm), and 5-HT1a (WAY-100635, 300 nm) receptor blockers to isolate the α1-AR-EPSC. Noradrenaline was applied in the presence of idazoxan (1 μm). To evoke the maximal amplitude α1-AR-EPSCs, stimulation intensity was gradually increased until the amplitude did not increase further.
In situ hybridization
To examine the coexpression of Adra1a, Adra1b, Grid1, or Grid2 mRNA and either (SERT) or (vGAT) mRNA puncta in cell bodies in the dorsal raphe, mRNA was identified with RNAscope Fluorescent Multiplex Assay, a highly sensitive fluorescent in situ hybridization method (ACD Bio, Newark, CA). Commercially available probes were used for Adra1a (#408611), Adra1b (#413561), Slc6a4 (SERT; #315851), Slc32a1 (vGAT; #319191), Grid1 (#552311), and Grid2 (#512701). In brief, mice were anesthetized with isoflurane before decapitation. Brains were flash frozen in chilled 2-methyl-butane for 20 s, sectioned with a cryostat (16 µm) onto SuperFrost+ slides and stored in −80°C. Tissue was fixed in formalin, dehydrated with EtOH, and treated with a protease solution. Signals were amplified, washed, and stained with a DAPI solution for 20–30 s. Finished slides were cover-slipped and stored at 4°C. Z-stack images were collected using an Olympus laser scanning confocal microscope (40× objective) within the next few days. Counts were made using Fiji software. Neurons were counted as positive for RNA expression if the fluorescent signal was greater than the mean ± 2 SDs of the background signal obtained from an extracellular area in the same brain slice.
Immunohistochemistry and confocal microscopy
To determine tryptophan hydroxylase (TpH) expression in neurons, 0.1% of Neurobiotin (biotin ethylenediamine, hydrobromide; Biotium; Kita and Armstrong, 1991) was mixed into the internal solution and delivered via the glass patch-pipette. After recording, brain slices were fixed for 1 h at room temperature using 4% paraformaldehyde in PBS. Slices were washed three times (15 min) with PBS, permeabilized and blocked in 0.5% Triton X-100 and 10% donkey serum in PBS for 5 h. Afterwards, slices were incubated in rabbit anti-TpH antibody (1:1000, MilliporeSigma) overnight at room temperature, washed in PBS, and incubated in Alexa Fluor 488 donkey anti-rabbit secondary antibody (1:1000, Invitrogen) for 2 h at room temperature. Slices were then washed in PBS and incubated in Cy5-conjugated streptavidin (1:1000, Invitrogen) for 2 h at room temperature. Slices were again washed in PBS before being mounted with DAPI Fluoromount-G (Electron Microscopy Sciences). Fluorescent images were collected with a Zeiss 510 confocal laser scanning microscope with 10× and 40× objectives and processed using Fiji software.
Materials
MK-801, DNQX/NBQX, nisoxetine, noradrenaline, picrotoxin, and WAY-100635 were from Tocris. All other materials were from Sigma-Aldrich.
Experimental design and statistical analysis
Data were analyzed using Clampfit 11.1. To determine the rate of activation and decay, recordings were post hoc filtered (Gaussian low pass filter, 60 Hz) and the rising and falling phases were fit with a single exponential. Data are presented as representative traces, or in scatter plots where each point is an individual cell, and bar graphs with mean ± SEM. Representative traces of synaptic currents are an average of two to five consecutive currents and are presented baselined, where the whole-cell current immediately before stimulation was set to 0 pA. Unless otherwise noted, n = number of cells as biological, not technical, replicates. Data sets with n > 30 were tested for normality with a Shapiro–Wilk test. When possible (within-group comparisons), significant differences were determined for two group comparisons by paired t tests, Wilcoxon matched-pairs signed rank test, and in more than two group comparisons by nonparametric repeated-measures (RM) ANOVA (Friedman test). Significant mean differences in between-group comparisons were determined for two group comparisons by Mann–Whitney tests. ANOVAs were followed, when p < 0.05 by Dunn's multiple comparisons post hoc test. A difference of p < 0.05 was considered significant. Exact values are reported unless p < 0.0001 or p > 0.999. Statistical analysis was performed using GraphPad Prism 8 (GraphPad Software).
Results
Whole-cell voltage-clamp recordings were made from dorsal raphe neurons in acute brain slices from wild-type mice at 35°C in the presence of NMDAR, AMPAR, KainateR, GABA-AR, and 5-HT1AR antagonists. A train of 5 electrical stimuli (60 Hz, delivered to the brain slice at 90-s intervals via a monopolar stimulating electrode placed within 200 µm of the recorded neuron), produced an α1-AR-dependent EPSC (α1-AR-EPSC; Fig. 1A) as previously described (Gantz et al., 2020). Figure 1A illustrates marked variability with respect to amplitude, time-to-peak, duration, charge transfer, and kinetics of the α1-AR-EPSC across neurons (n = 51). The amplitude ranged from −9.0 to −79.5 pA (median: −24.6 pA; Fig. 1B). There was a delay between the end of the stimulus and the onset of the α1-AR-EPSC (243 ± 11 ms; Fig. 1C). By comparison, this is >3× and 1.5× slower than the onset of “slow” synaptic transmission mediated by dopamine D2 receptor-mediated and α2-adrenergic receptor-mediated activation of GIRK channels, respectively (Ford et al., 2009; Courtney and Ford, 2014). The α1-AR-EPSC peaked 1.9 s after the end of the stimulus (range of 0.6–8.4 s), with a time constant of activation (τ-activation) of 417 ms (Fig. 1D,E). The α1-AR-EPSC lasted 4.4–72.3 s (median: 22 s), with a time constant of decay (τ-decay) of 8.3 s (Fig. 1F,G). The total charge transfer was −30 to −936 pA*s (median: −251 pA*s; Fig. 1H). Despite the marked variability of the α1-AR-EPSC across neurons, these parameters were highly consistent within individual neurons when α1-AR-EPSCs were evoked at 90-s intervals during a single recording (Fig. 1A,I). When the α1-AR-EPSCs in brain slices from male or female mice were compared, there were no significant differences except that the τ-activation and time-to-peak were significantly faster in brain slices from female mice (τ-activation: p = 0.006; time-to-peak: p = 0.02, n = 22 and 28, Mann–Whitney tests; Table 1).
Figure 1.
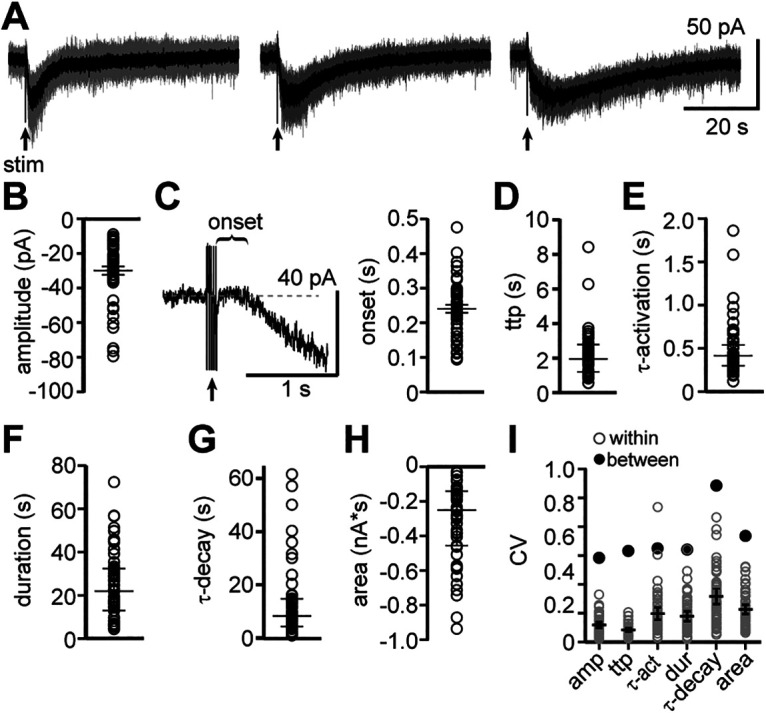
Basic properties of the α1-AR-dependent EPSC are variable across neurons but highly consistent within a single neuron. A, Example traces of electrically evoked (arrow) α1-AR-EPSCs from three neurons (gray: five successive sweeps; black: averaged trace). B, Plot of the maximal amplitude of α1-AR-EPSCs. C, Left, Example trace of the delay between the end of the stimulus and the initiation of the α1-AR-EPSC (onset). Right, Plot of the onset of the α1-AR-EPSCs. D, Plot of the time-to-peak (ttp) of α1-AR-EPSCs. E, Plot of activation kinetics (τ-activation) of α1-AR-EPSCs. F, Plot of the duration of α1-AR-EPSCs. G, Plot of decay kinetics (τ-decay) of α1-AR-EPSCs. H, Plot of charge transfer (area, nA*s) of α1-AR-EPSCs. I, Plot of coefficient of variation (CV) for each parameter in B–G (gray: CV for each sweep within a single neuron; black: CV for grouped data) demonstrating marked variability between neurons, but low variability within an individual neuron. Line and error bars in B, D–I represent median and quartiles, line and error bars in C represent mean ± SEM; n = 51.
Table 1.
Characteristics of the α1-AR-EPSC in dorsal raphe neurons from male and female mice
| Sex | Male | Female |
|---|---|---|
| Amplitude (pA) | −27.6 (−20.5 to −39.4) | −24.3 (−15.9 to −33.3) |
| Onset (ms) | 264.7 ± 16.3 | 225.2 ± 15.4 |
| Time-to-peak (s) | 2.4 (1.9–3.0) | 1.3 (1.2–2.5) |
| Duration (s) | 20.8 (15.7–32.4) | 21.9 (6.7–33.9) |
| Area (pA*s) | −268.0 (−169.0 to −579.3) | −165.9 (−122.9 to −395.3) |
| τ-Activation (ms) | 468.9 (389.0–717.1) | 341.4 (269.5–494.3) |
| τ-Decay (s) | 8.6 (5.9–17.3) | 7.9 (2.4–13.9) |
Median (quartiles), except onset which is mean ± SEM.
N = 22 and 28–29 for males and females, respectively.
Bold type designates statistical significance.
Serotonin and GABA neurons contain mRNA for α1-AR and GluD channels
The dorsal raphe contains a heterogenous population of serotonin and non-serotonin (GABA and dopamine) neurons (Fu et al., 2010; Huang et al., 2019). The majority of noradrenaline terminals in the dorsal raphe form synaptic specializations onto serotonin neuron dendrites (Baraban and Aghajanian, 1981), but α1-AR-dependent excitation of non-serotonin neurons has not been ruled out. Indeed, bath application of an α1-AR agonist, phenylephrine, depolarizes serotonin and non-serotonin neurons in rat brain slices (Kirby et al., 2003). Therefore, to determine whether heterogeneity in the α1-AR-EPSC was explained by recording from both serotonin and GABA neurons, we first assayed for the presence of mRNA for α1-AR (Adra1a and/or Adra1b) in both populations of neurons using in situ hybridization with serotonin transporter (Slc6a4, SERT) and vesicular GABA transporter (Slc32a1, vGAT) mRNA as markers for serotonin and GABA neurons respectively (Fig. 2A,B). The data show that nearly all SERT+ (97%) and vGAT+ (85%) neurons contained mRNA for α1-AR (Fig. 2A–C). The majority of SERT+ (65%) and vGAT+ (55%) neurons contained mRNA for two α1-AR isoforms, α1A-AR and α1B-AR (Fig. 2C). Neurons that contained both isoforms had a higher density of total α1-AR mRNA when compared with neurons that contained only one isoform (one-way ANOVA: SERT+: F(2,1979) = 141.5, p < 0.0001, Tukey's post hoc test, p < 0.0001; vGAT+: F(2,1317) = 148.2, p < 0.0001, Tukey's post hoc test, p < 0.0001; Fig. 2D,E). There was a distinction between SERT+ and vGAT+ neurons in the relative abundance of the two isoforms. Predominately, SERT+ neurons with only one isoform contained α1B-AR whereas vGAT+ neurons contained α1A-AR (Fig. 2C). Further, the abundance of α1B-AR mRNA was greater than α1A-AR in SERT+ neurons (Tukey's post hoc test, p < 0.0001; Fig. 2D,F), whereas the abundance of α1A-AR mRNA was greater than α1B-AR in vGAT+ neurons (Tukey's post hoc test, p = 0.03; Fig. 2E,F). These results indicate that both serotonin and GABA neurons may express α1-AR, with a bias in isoform predominance of α1B-AR in serotonin neurons and α1A-AR in GABA neurons (two-way ANOVA interaction, F(1,5488) = 1238.0, p < 0.0001; Fig. 2F).
Figure 2.
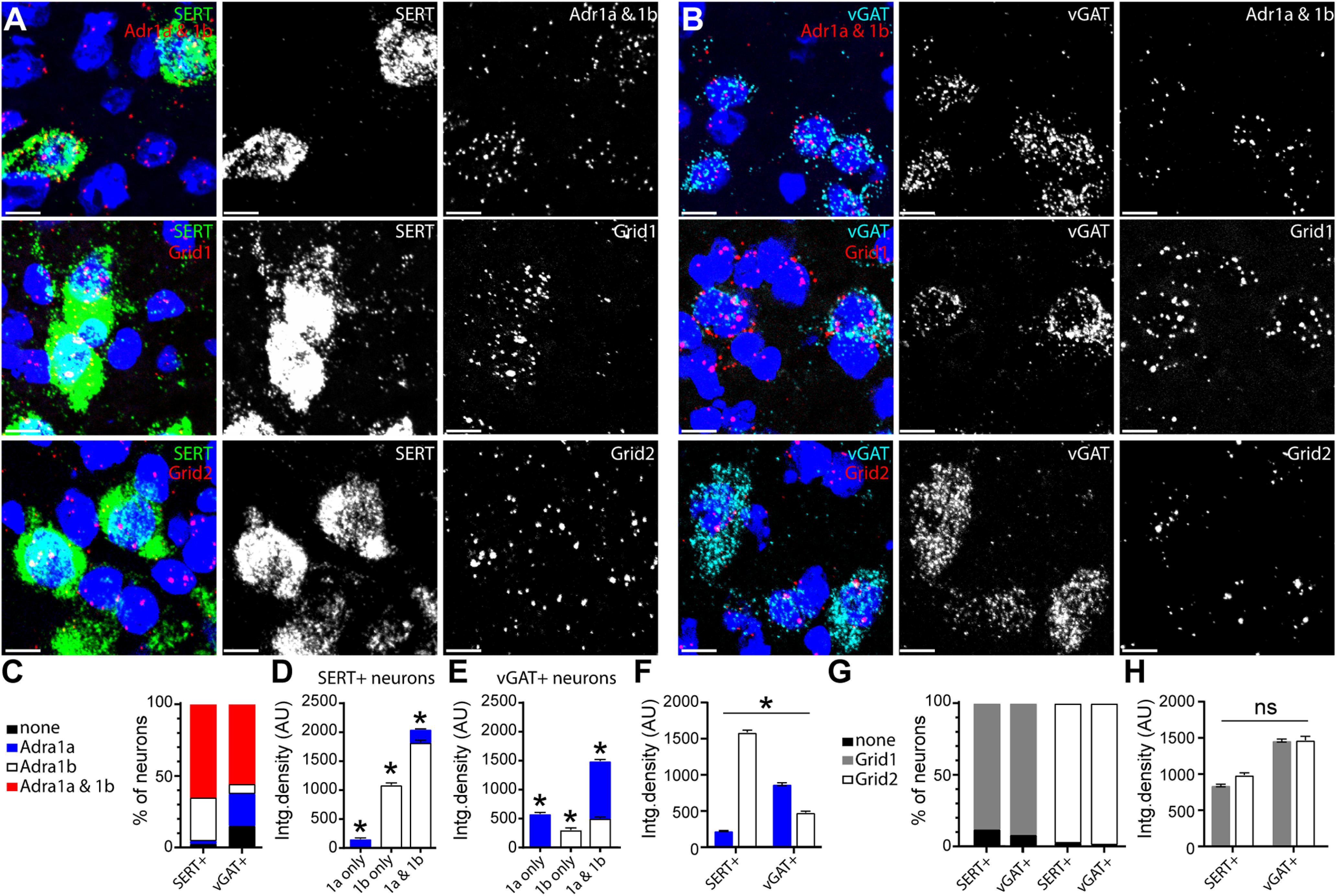
Dorsal raphe serotonin and GABA neurons have mRNA for α1-AR and GluD channels. A, B, Representative maximum intensity projection images of fluorescent in situ hybridization (RNAscope) labeling Slc6a (left, SERT, green), Slc32a (right, vGAT, cyan), Adra1a and Adra1b (top, red), Grid1 (middle, red), and Grid2 (bottom, red) RNA transcript in serotonin neurons (A) and GABA neurons (B). Scale bars: 10 µm. C, Percentage of serotonin (SERT+) and GABA (vGAT+) neurons that expressed only Adra1a, only Adra1b, both isoforms (1a and 1b), or did not express Adra1 mRNA above background values (none). D, Plot of the integrated density of Adra1 mRNA in SERT+ neurons that expressed Adra1a-only (blue, n = 41), Adra1b-only (white, n = 616), or expressed mRNA for both isoforms (1a and 1b, n = 1325). Serotonin neurons that expressed 1a and 1b had higher total density of mRNA and serotonin neurons that expressed Adra1b-only had a higher density that Adra1a-only (p < 0.0001 for all comparisons). E, Plot of the integrated density of Adra1 mRNA in vGAT+ neurons that expressed Adra1a-only (blue, n = 348), Adra1b-only (white, n = 107), or expressed mRNA for both isoforms (1a and 1b, n = 865). GABA neurons that expressed 1a and 1b had higher total density of mRNA (p < 0.0001) and GABA neurons that expressed Adra1a only had a higher density that Adra1b only (p = 0.03). F, Plot of the integrated density of Adra1 and Adra1b mRNA in SERT+ and vGAT+ neurons showing a predominance of Adra1b in serotonin neurons and Adra1a in GABA neurons (two-way ANOVA, interaction, p < 0.0001). G, Percentage of serotonin (SERT+) and GABA (vGAT+) neurons that expressed Grid1 or Grid2 mRNA or did not express either above background values (none). H, Plot of the integrated density of Grid1 and Grid2 mRNA in serotonin (n = 1284 and 396) and vGAT+ (n = 2055 and 202) neurons showing higher values of Grid1 and Grid2 in GABA neurons compared with serotonin neurons (two-way ANOVA, main effect of cell type, p < 0.0001) but no bias in Grid1 or Grid2 predominance between the cell types (two-way ANOVA, interaction, p = 0.07). Line and error bars represent mean ± SEM; * denotes statistical significance, and ns denotes not statistically significant.
Next, we evaluated the presence of mRNA for the ion channel established to carry the ionic current (Gantz et al., 2020), the ionotropic δ 1 glutamate receptor channel (Grid1), as well as the related isoform δ 2 (Grid2) since both are found in the dorsal raphe (Hepp et al., 2015; Nakamoto et al., 2020). The data show that nearly all SERT+ and vGAT+ neurons contained comparable levels of mRNA for Grid1 and Grid2 (Fig. 2A,B,G,H). On average, the density of Grid1 and Grid2 mRNA was greater in vGAT+ neurons when compared with SERT+ neurons (two-way ANOVA, main effect of cell type, F(1,3933) = 210.2, p < 0.0001; Fig. 2H). There was no bias in isoform predominance of Grid1 and Grid2 mRNA that distinguished SERT+ from vGAT+ neurons (two-way ANOVA interaction, F(1,3933) = 3.3, p = 0.07; Fig. 2H). Taken together, these data indicate that both serotonin and GABA neurons may express the receptor and channel necessary to produce an α1-AR synaptic current.
Serotonin neurons have an α1-AR-dependent EPSC
Next, we recorded from dorsal raphe neurons with an internal solution containing 0.1% Neurobiotin. After recording, the brain slices were immunostained for TpH, an enzyme required for the synthesis of serotonin (Fig. 3A). When selecting neurons, efforts were made to label neurons in the ventral-midline, dorsal-midline, and dorsal-lateral subregions of the dorsal raphe. Collectively, 44/63 neurons immunostained for TpH (TpH+), while 19/63 were TpH–. Of the TpH– neurons, 11/19 were found in the midline regions, while 8/19 were in the dorsal-lateral subregions. Consistent with previous reports in rats (Vandermaelen and Aghajanian, 1983; Li et al., 2001; Kirby et al., 2003; Marinelli et al., 2004; Calizo et al., 2011) and mice (Gocho et al., 2013), many of the electrophysiological membrane properties of TpH+ and TpH– neurons were similar (Table 2), but there were differences in AP firing. When APs were evoked with somatic current injection (20 pA increments, 1.5 s), on average, TpH– neurons fired at faster rates (p = 0.001 with Mann–Whitney test; Fig. 4A–C) and had significantly narrower APs (p = 0.003 with Mann–Whitney test; Fig. 4D,E) with larger afterhyperpolarizations (p = 0.009 with Mann–Whitney test; Fig. 4D,F) than APs evoked in TpH+ neurons. Although there was some overlap between the two groups of neurons. However, when the brain slice was stimulated to evoke an α1-AR-EPSC, the α1-AR-EPSC was recorded consistently in TpH+ neurons (37/39), but not TpH– neurons (0/19; Fig. 3B,C). On average, the amplitude of the α1-AR-EPSC in Neurobiotin-filled TpH+ neurons (median −18.5 pA) was comparable to those recorded with standard internal solution in Figure 1B (p = 0.09 with Mann–Whitney test), making it unlikely that Neurobiotin interfered with detection of the α1-AR-EPSC. To determine whether non-serotonin neurons respond to exogenous α1-AR agonists, as reported in rat brain slices (Kirby et al., 2003), phenylephrine (3 μm) and noradrenaline (10 μm) were applied. In TpH+ neurons, both phenylephrine and noradrenaline produced a noisy inward current (n = 15–16; Fig. 3D,E), of comparable amplitude. In contrast, neither agonist produced an inward current in TpH– neurons (n = 8; Fig. 3D,E). These data indicate that despite the presence of mRNA for the receptor and channel, GABA neurons are not depolarized by α1-AR activation. Thus, heterogeneity in the α1-AR-EPSC was not because of recording from serotonin and non-serotonin neurons but stems from another source from variability among the population of serotonin neurons.
Figure 3.

The α1-AR-EPSC is readily recorded in serotonin, but not non-serotonin neurons in the dorsal raphe. A, Representative maximum intensity projection confocal images of Neurobiotin-filled dorsal raphe neurons immunostained for streptavidin (SA; red) and TpH (marker of serotonin neurons, green) showing identification of TpH+ and TpH– neurons. Scale bars: 50 µm (left) and 20 µm (right). B, Example traces of the α1-AR-EPSC recorded in a TpH+ neuron but not a TpH– neuron in response to the same electrical stimulation paradigm. C, Plot of the amplitude of the α1-AR-EPSC in Neurobiotin-filled TpH+ neurons. There was no inward current to electrical stimulation in TpH– neurons (p < 0.0001). Numbers in parentheses indicate the number of neurons with an inward current to electrical stimulation over the total number of neurons assayed. D, Example traces of the inward currents induced by application of the α1-AR agonists phenylephrine (Phe) and noradrenaline (NA) in a TpH+ neuron, but not a TpH– neuron. E, Plot of the amplitude of the inward current induced by Phe and NA in Neurobiotin-filled TpH+ neurons. There were no inward currents to Phe or NA in TpH– neurons (p = 0.0008 for each agonist). Line and error bars represent mean ± SEM; * denotes statistical significance.
Table 2.
Serotonin and non-serotonin neurons in the dorsal raphe exhibit many similar electrophysiological membrane properties
| Immunostained | TpH+ | TpH– |
|---|---|---|
| Vr (mV) | −81.0 ± 2.2 | −75.2 ± 3.3 |
| Rm (mΩ) | 397.1 ± 30.9 | 628.5 ± 95.8 |
| tau (ms) | 22.5 ± 1.2 | 27.4 ± 2.9 |
| AP threshold (mV) | −41.1 ± 0.8 | −44.8 ± 2.3 |
| slope (mV/ms) | 0.7 ± 0.4 | 1.8 ± 1.6 |
Properties measured were resting membrane potential (Vr), membrane resistance (Rm) determined by a voltage step from −65 to −70 mV, and the time constant of the membrane (tau) determined by fitting the hyperpolarization produced by negative current injection (−40 pA) with a single exponential. APs were evoked with somatic current injection (150 pA) to determine AP threshold, and the interspike slope (measured in the middle 60% of the interspike interval). Mean ± SEM.
N = 27–44 and 10–19 for TpH+ and TpH– neurons, respectively.
Figure 4.
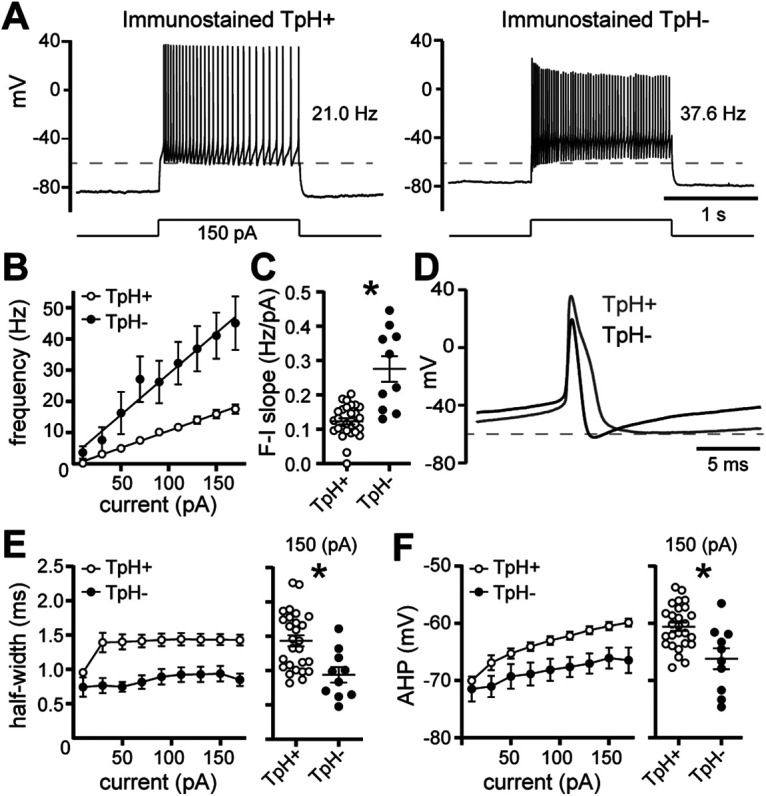
Serotonin neurons fire broad APs at slower rates than neighboring non-serotonin neurons in the dorsal raphe. A, Representative traces of whole-cell current-clamp recordings of APs evoked by current injection (150 pA, 1.5 s) in a serotonin neuron (left, TpH+) and non-serotonin neuron (TpH–, right). Dashed line is at −60 mV. B, Plot of the frequency of AP firing versus injected current in TpH+ (white) and TpH– (black) neurons. Linear fit from 10 to 170 pA indicates mean F-I slope. C, F-I slopes for each neuron, determined by linear fit from the first increment of current injection to evoke APs to 170 pA, demonstrating the steeper F-I slope in TpH– neurons when compared with TpH+ neurons (p = 0.001), but there was some overlap. D, Representative AP waveforms (average of the second through fifth AP) recorded in a TpH+ (gray) and TpH– (black) neuron. E, Left, Plot of the half-width of the AP versus injected current in TpH+ (white) and TpH– (black) neurons. Right, AP half-widths for each neuron evoked by 150-pA current injection, demonstrating that on average, TpH+ neurons fired broader APs than TpH– neurons (p = 0.003). F, left, Plot of the afterhyperpolarization of the second AP (AHP) versus injected current in TpH+ (white) and TpH– (black) neurons. Right, AHPs for each neuron evoked by 150-pA current injection, demonstrating that on average, TpH+ neurons had more depolarized AHPs than TpH– neurons (p = 0.009); n = 28 and 10 for all comparisons. Line and error bars represent mean ± SEM; * denotes statistical significance.
The presence of extracellular noradrenaline is controlled
At some synapses, “spill-over” of neurotransmitter outside of the synaptic cleft produces variable and prolonged synaptic currents from activation of extrasynaptic receptors (Szabadics et al., 2007; Balakrishnan et al., 2009), particularly with repetitive stimulation producing neurotransmitter release at a rate that exceeds the clearance from the extracellular space (Scanziani, 2000). Spill-over of noradrenaline occurs at somatodendritic synapses in the locus coeruleus (Courtney and Ford, 2014). In this brain region, larger amplitude α2-adrenergic receptor-dependent synaptic currents are correlated with slower decay rates (Courtney and Ford, 2014). Therefore, initially we examined the relationship between the maximal amplitude and the time course of the α1-AR-EPSC across serotonin neurons. Figure 5A illustrates α1-AR-EPSCs recorded in three neurons that had similar amplitudes but different decay rates. The grouped data revealed no correlation in the amplitude of the α1-AR-EPSC and τ-activation (p = 0.999, r = −0.000, Spearman correlation; Fig. 5B), nor a correlation in amplitude and τ-decay (p = 0.24, r = −0.19, Spearman correlation; Fig. 5C). Instead, there was a positive correlation between τ-activation and τ-decay (p = 0.005, r = 0.42, Spearman correlation; Fig. 5D). These data indicate that the time course of the α1-AR-EPSC was not determined by variability in the amount of noradrenaline released within the population of serotonin neurons. Therefore, we next tested how increasing the amount of neurotransmitter released alters the synaptic response within the same neuron.
Figure 5.
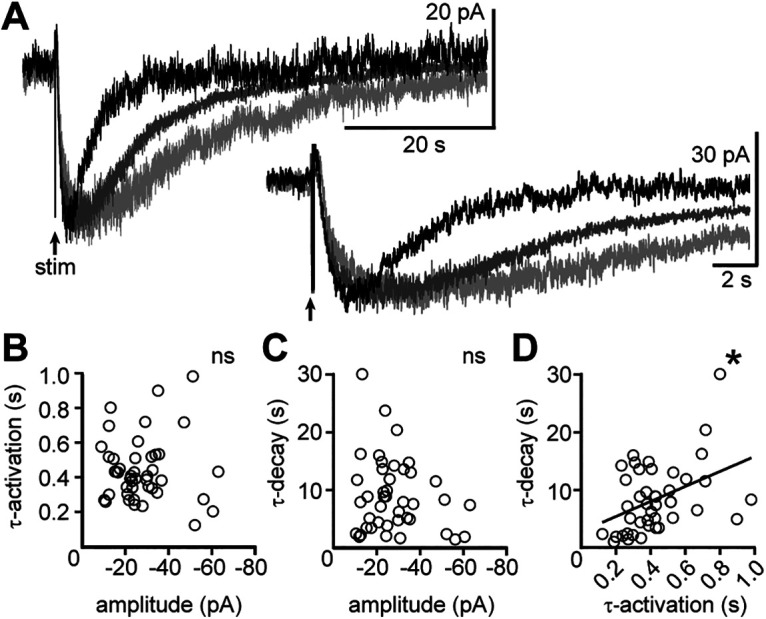
The amplitude of the α1-AR-EPSC does not determine the time course of signaling. A, Example traces of the α1-AR-EPSC in three different neurons demonstrating similar amplitudes but different decay kinetics. B, There was no correlation between amplitude of the α1-AR-EPSC and the activation kinetics (τ-activation, p = 0.999, n = 44 pairs). C, There was no correlation between the amplitude of the α1-AR-EPSC and the decay kinetics (τ-decay, p = 0.19, n = 42 pairs). D, There was a significant correlation between the activation and decay kinetics of the α1-AR-EPSC (p = 0.005, n = 43 pairs). Line represents best-fit line by linear regression for visual aid; * denotes statistical significance, and ns denotes not statistically significant.
Noradrenaline release was varied by changing the intensity of stimulation in the same neurons. First, a maximal amplitude α1-AR-EPSC was evoked, then the stimulation intensity was lowered; reducing the α1-AR-EPSC amplitude by 43.5±1.6% and decreasing the total duration (p = 0.002 with Wilcoxon test for both comparisons, n = 10; Fig. 6A,B). Despite the decrease in amplitude, the time-to-peak and τ-decay did not change (time-to-peak: p = 0.13; τ-decay: p = 0.92, Wilcoxon tests, n = 10; Fig. 6C,D). As a further test, noradrenaline release was varied by changing the number of electrical stimuli delivered to the brain slice at maximal intensity, using either a single stimulus, or a train of 3, 5, or 10 stimuli (60 Hz) in the same neurons. In 9/15 neurons, a single stimulus was sufficient to evoke an α1-AR-EPSC (−4.6 ± 0.3 pA; Fig. 6E–I) that could be detected over baseline noise. On average, increasing the number of stimuli produced α1-AR-EPSCs of larger amplitude, longer duration, and thus greater total charge transfer (mixed model RM ANOVAs, amplitude: F(1.9,20.0) = 50.1, p < 0.0001; duration: F(2.3,24.0) = 12.4, p = 0.0001; charge: F(1.1,11.6) = 20.2, p = 0.0007, n = 9–15; Fig. 6E–H). However, increasing the number of stimuli did not change the time-to-peak nor τ-decay [mixed model RM ANOVAs, time-to-peak: F(0.4,4.7) = 1.4, p = 0.23 (Fig. 6I); τ-decay: F(1.8,21.9) = 0.9, p = 0.41 (Fig. 6J)]. When scaled to the peak amplitude, the α1-AR-EPSCs evoked by 3, 5, or 10 stimuli were similar (Fig. 6K). When the number of electrical stimuli was varied using half-maximal intensity, similar results were obtained. A single stimulus was sufficient to evoke an α1-AR-EPSC in 5/10 neurons. Increasing the number of stimuli produced α1-AR-EPSCs of larger amplitude, longer duration, and thus greater total charge transfer (mixed model RM ANOVAs, amplitude: F(1.2,8.6) = 37.2, p = 0.0002; duration: F(1.9,14.1) = 19.4, p = 0.0001; charge: F(1.1,8.0) = 20.6, p = 0.0017, n = 5–10), but did not change the time-to-peak nor τ-decay (mixed model RM ANOVAs, time-to-peak: F(1.1,8.3) = 0.2, p = 0.72; τ-decay: F(1.9,17.2) = 2.4, p = 0.12). Thus, the amount of noradrenaline released influences the amplitude of the α1-AR synaptic response without affecting the rates of current activation and decay. These data suggest extracellular noradrenaline is likely cleared from the synaptic cleft before the peak of the α1-AR-EPSC (∼2 s from stimulation), otherwise the rates of α1-AR-EPSC activation and decay would be increased by repetitive stimuli.
Figure 6.

Increasing the amount of noradrenaline released increases the amplitude of the α1-AR-EPSC without changing the kinetics of signaling. A, By adjusting the stimulation intensity, maximal and half-maximal α1-AR-EPSCs were evoked (arrow) shown in representative traces (left, inset below shows the same trace on an expanded timescale) and grouped data (right, p = 0.002). B, Increasing the stimulation intensity increased the duration of the α1-AR-EPSC (p = 0.002). C, Increasing the stimulation intensity did not change the time-to-peak of the α1-AR-EPSC (ttp, p = 0.13), nor (D) the decay rate of the α1-AR-EPSC (τ-decay, p = 0.92). E, Example traces of electrically evoked (arrow) α1-AR-EPSCs from a single neuron using a single or train of electrical stimuli delivered at 60 Hz (1, 3, 5, or 10 stims). F–H, Increasing the number of electrical stimuli increased the (F) amplitude (p < 0.0001), (G) area (p = 0.0007), and (H) duration of the α1-AR-EPSC (p = 0.001). I, Increasing the number of electrical stimuli did not change the time-to-peak of the α1-AR-EPSC (ttp, p = 0.23). J, Increasing the number of electrical stimuli did not change the decay kinetics of the α1-AR-EPSC (τ-decay, p = 0.41). The α1-AR-EPSCs evoked by a single stimulus were too small for accurate exponential fits and thus excluded from τ-decay analysis. K, Example traces of α1-AR-EPSCs evoked by 3, 5, or 10 stimuli in the same neuron, scaled to the peak and superposed. F–J, Mixed model RM ANOVA for all comparisons, n = 9–15. Line and error bars represent mean ± SEM; * denotes statistical significance, and ns denotes not statistically significant.
Noradrenaline transporters limit the duration of the α1-AR synaptic current
The presence of extracellular neurotransmitter is often limited by efficient transporter-dependent reuptake. Indeed, the nonselective monoamine transporter blocker, cocaine, prolongs the α1-AR-dependent synaptic response without increasing the amplitude (Oleskevich and Williams, 1995). In the present study, blocking reuptake with a noradrenaline transporter inhibitor, nisoxetine (1 μm) increased the amplitude of the maximal α1-AR-EPSC by ∼35% (evoked by a train of five stimuli at 60 Hz, p = 0.04 with Wilcoxon test, n = 17; Fig. 7A–C), but the increase was transient (Fig. 7C) and accompanied by a small, tonic inward current (−7.3 ± 2.9 pA, p = 0.01 with Wilcoxon test, n = 23; Fig. 7D). Nisoxetine produced a more substantial and persistent change in the activation and decay rates. Blocking reuptake delayed the time-to-peak by 4.0 ± 0.7 s (p < 0.0001 with Wilcoxon test, n = 17; Fig. 7A,E,G) and prolonged the τ-decay by 12.3 ± 3.1 s (p = 0.0002 with Wilcoxon test; Fig. 7A,F). Similar results were obtained when half-maximal α1-AR-EPSCs (49.6 ± 8.3% of max, n = 9) were evoked. Nisoxetine increased the amplitude of the half-maximal α1-AR-EPSC (p = 0.009 with Wilcoxon test) to a similar extent as the maximal α1-AR-EPSC (37%; Fig. 7C). There was also a significant increase in the time-to-peak (3.7 ± 0.7 s, p = 0.004 with Wilcoxon test, n = 9) and the τ-decay of the half-maximal α1-AR-EPSC (32.1 ± 12.8 s, p = 0.004 with Wilcoxon test, n = 9). There was a disproportionate increase in the time-to-peak of α1-AR-EPSC, such that α1-AR-EPSCs with faster activation rates were more affected by reuptake blockade than those with slower activation rates (Spearman correlations, maximal: r = −0.70, p = 0.002, n = 17, half-maximal: r = −0.73, p = 0.03, n = 9; Fig. 7G). Taken together, the results indicate that reuptake of noradrenaline via transporters limits the duration of the α1-AR-EPSC and the efficiency of reuptake may be a factor that contributes to the heterogeneity in the time course of signaling.
Figure 7.
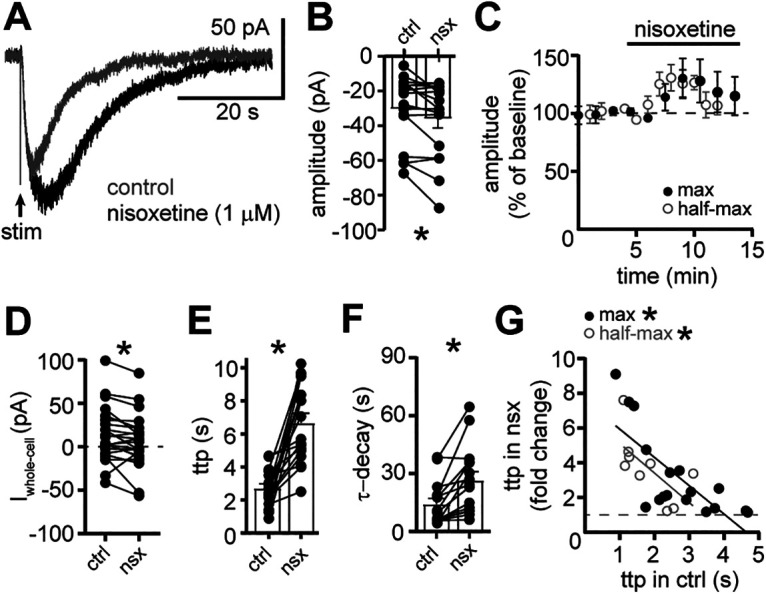
Clearance of noradrenaline by transporter-dependent reuptake limits the duration of the α1-AR-EPSC. A, Example traces of the α1-AR-EPSC in control conditions (gray) and nisoxetine (1 μm, black). B, Plot of the amplitude of the α1-AR-EPSC in control conditions (ctrl) and nisoxetine (nsx; p = 0.04, n = 17). C, Plot of the normalized amplitude of the maximal and half-maximal α1-AR-EPSCs versus time demonstrating the transient increase by nisoxetine. D, Plot of the whole-cell current in control conditions (ctrl) and nisoxetine (nsx; p = 0.01, n = 23). E, Plot of the time-to-peak (ttp) of the α1-AR-EPSC in control conditions (ctrl) and nisoxetine (nsx; p < 0.0001, n = 17). F, Plot of the τ-decay of the α1-AR-EPSC in control conditions (ctrl) and nisoxetine (nsx; p = 0.0002, n = 15). G, There were significant correlations between time-to-peak of the α1-AR-EPSC in control conditions (ctrl) and the increase in the time-to-peak of the maximal (black) and half-maximal (gray) α1-AR-EPSC in nisoxetine (nsx), shown as a fold-change (maximal: r = −0.70, p = 0.002, n = 17 pairs; half-maximal: r = −0.73, p = 0.03, n = 9, Spearman correlations). Solid lines represent best-fit lines by linear regression. Dashed line demarks no change in time-to-peak. Line and error bars represent mean ± SEM; * denotes statistical significance.
To determine whether extracellular noradrenaline was also cleared via passive diffusion, we tested the effect of extracellular dextran (5%, Molecular Weight: 35–45 kDa), which by macromolecular crowding slows diffusion of neurotransmitter in brain slices as a function of distance (Ford et al., 2010; Courtney and Ford, 2016). To determine how impairing diffusion impacts the time course of the α1-AR-dependent current, noradrenaline was focally applied ∼5 µm from the soma via a high-resistance pipette and iontophoresis (200 ms). Iontophoretically applied noradrenaline (delivered to the brain slice at 90-s intervals 90 s) reliably produced an inward current (INA, −30.1 ± 4.6 pA; Fig. 8A,B). Relative to the α1-AR-EPSC (243 ± 11 ms; Fig. 1C), the onset of INA was markedly slower (444.9 ± 22.3 ms, p < 0.0001; Fig. 8A,C). Restricting diffusion with extracellular dextran reduced the amplitude of INA (p = 0.02; Fig. 8A,B,E) and delayed the onset by 254 ± 55.6 ms (p = 0.02; Fig. 8A,C). Dextran had no effect on τ-decay of INA (p = 0.94, Fig. 8D; Wilcoxon tests, n = 7 for each comparison), consistent with τ-decay determined primarily by transporter-dependent clearance and the rate of intrinsic second messenger signaling. In contrast to INA, dextran had no effect on the amplitude of the α1-AR-EPSC (p = 0.41, n = 22; Fig. 8E,F), nor the onset (p = 0.22, n = 13; Fig. 8G). These data indicate that diffusion over extended distance is not required for afferent-derived noradrenaline to activate postsynaptic α1-AR. Further, passive diffusion is not a primary mechanism by which extracellular noradrenaline is cleared. Unlike reuptake blockade, dextran also had no effect on τ-decay of the α1-AR-EPSC (p = 0.77, n = 22, Wilcoxon tests; Fig. 8H). However, when reuptake was impaired by nisoxetine, then dextran reduced the amplitude of the α1-AR-EPSC (p = 0.02 with Wilcoxon test, n = 8; Fig. 8I). There was no change in the onset (p = 0.88 with Wilcoxon test, n = 6; Fig. 8J). Together, these data indicate that under conditions of reuptake blockade, afferent-derived noradrenaline can diffuse over extended distance to prolong the time course of the α1-AR-EPSC. Interestingly, τ-decay remained significantly prolonged in nisoxetine and dextran (p = 0.38 with Wilcoxon test, n = 7; Fig. 8K). It may be that dextran incompletely prevented extended diffusion, which was observed for INA, or nisoxetine also prolonged the lifetime of noradrenaline in the synaptic cleft. We also examined the role of enzymatic degradation of noradrenaline by catechol-O-methyltransferase (COMT). Acute inhibition of COMT with OR-486 (1 μm) increased the amplitude of the α1-AR-EPSC by 44.9 ± 18.5% (p = 0.049) without changing τ-activation (p = 0.92), time-to-peak (p = 0.91), or τ-decay (p = 0.77, Wilcoxon tests, n = 10 for each comparison, data not illustrated). Thus, enzymatic degradation of noradrenaline does not influence the time course of the α1-AR-EPSC. Taken together, the results indicate that the activation of α1-AR is limited primarily by noradrenaline transporter-dependent clearance of noradrenaline with little role of extrasynaptic diffusion unless the primary mode of clearance is impaired.
Figure 8.

Slowing diffusion of noradrenaline does not affect the α1-AR-EPSC unless clearance via transporters is impaired. A, Example traces of the inward current produced by focal iontophoretic application of noradrenaline (ionto, 200 ms) in control conditions (gray) and in 5% dextran (black). Inset below shows the same traces on an expanded timescale. B, Plot of the amplitude of the noradrenaline-induced inward current (INA) in control conditions (ctrl) and in dextran (dex, p = 0.02, n = 7). C, Plot of the onset of INA in control conditions and dextran (p = 0.02, n = 7). D, Plot of the τ-decay of INA in control conditions and dextran (p = 0.94, n = 7). E, α1-AR current was produced every 90s by iontophoretic application of noradrenaline (ionto, INA) or electrical stimulation (stim, α1-AR-EPSC). Dextran reduced the amplitude of INA but not the α1-AR-EPSC. F, Dextran had no effect on the amplitude of the α1-AR-EPSC shown in representative traces of the in control conditions (gray) and in 5% dextran (black; left) and grouped data (right, p = 0.41, n = 22). G, Plot of the onset of the α1-AR-EPSC in control conditions and dextran (p = 0.22, n = 13). H, Plot of the τ-decay of the α1-AR-EPSC in control conditions and dextran (p = 0.77, n = 22). I, left, Example traces of the α1-AR-EPSC in control conditions (dark gray), nisoxetine (1 μm, light gray), and nisoxetine with dextran (black). Inset below shows the same traces on an expanded timescale. Right, Plot of the amplitude of the α1-AR-EPSC in nisoxetine (nsx) and nisoxetine with dextran (+dex; p = 0.02, n = 8). J, Plot of the onset of the α1-AR-EPSC in nisoxetine (nsx) and nisoxetine with dextran (+dex; p = 0.88, n = 6). K, Plot of the τ-decay of the α1-AR-EPSC in nisoxetine (nsx) then nisoxetine with dextran (+dex; p = 0.60, n = 7). Line and error bars represent mean ± SEM; * denotes statistical significance, and ns denotes not statistically significant.
Discussion
Spatiotemporal control of noradrenaline in the dorsal raphe
At many synapses, spill-over of neurotransmitter outside of the synaptic cleft extends the action of neurotransmitter in time and space (Isaacson et al., 1993; Otis et al., 1996; Min et al., 1998; Carter and Regehr, 2000; Scanziani, 2000; Beenhakker and Huguenard, 2010). Spill-over can lead to pooling of neurotransmitter from neighboring presynaptic release sites (Otis et al., 1996; Balakrishnan et al., 2009), and activation extrasynaptic receptors (Isaacson et al., 1993; Scanziani, 2000; Szapiro and Barbour, 2009). In some instances, receptors are only located at extrasynaptic sites and spill-over is required for their activation and the generation of the postsynaptic response (Otis and Mody, 1992; Beenhakker and Huguenard, 2010). When spill-over is required, neurotransmitter release must often be evoked by trains of stimuli, such that extracellular neurotransmitter overwhelms the clearance mechanisms. In contrast, in the majority of recordings here, a single stimulus was sufficient to evoke the α1-AR-EPSC. Moreover, impairing diffusion of noradrenaline from the release site with dextran had no effect on the α1-AR-EPSC. These results are consistent with ultrastructural evidence for direct innervation of noradrenaline axon terminals onto serotonin neuron dendrites (Baraban and Aghajanian, 1981), suggesting that spill-over of noradrenaline to extrasynaptic sites is not required for the generation of the α1-AR-EPSC.
More commonly, spill-over of neurotransmitter activates both synaptic and extrasynaptic receptors. Spill-over not only prolongs synaptic currents mediated by ionotropic receptors, but also prolongs synaptic currents mediated by G-protein-coupled receptors despite their slow signaling kinetics (Isaacson et al., 1993; Scanziani, 2000; Beenhakker and Huguenard, 2010; Courtney and Ford, 2016). In the locus coeruleus, spill-over of noradrenaline shapes the time course of α2-adrenergic receptor-dependent synaptic currents with larger amplitude currents, generated by increasing noradrenaline release, having slower decay rates (Courtney and Ford, 2014). In contrast, increasing noradrenaline release did not prolong the decay rate of the α1-AR-EPSC, suggesting limited spill-over in the dorsal raphe under basal conditions. Since increasing the amount of noradrenaline released did not affect the kinetics of the α1-AR-EPSC, the presence of extracellular noradrenaline is likely shorter in duration than the time-to-peak (<2 s) of the α1-AR-EPSC, indicating that ∼18 s (∼22-s total duration minus ∼2-s time-to-peak; Fig. 1) of the α1-AR-EPSC is caused by second messenger signaling and modulation of GluD1R channels.
Noradrenaline transporters limit spill-over of noradrenaline
Given the α1-AR-EPSC is much slower than other G-protein-coupled receptor-dependent synaptic currents, it is possible that the second messenger signal cascade would obscure spatiotemporal spread of noradrenaline signaling. Since spill-over of neurotransmitter is often limited by transporter-dependent uptake (Isaacson et al., 1993; Otis et al., 1996; Min et al., 1998; Carter and Regehr, 2000; Scanziani, 2000; Szabadics et al., 2007; Beenhakker and Huguenard, 2010; Courtney and Ford, 2014, 2016), we examined the effect of blocking reuptake with a noradrenaline transporter inhibitor, nisoxetine. Consistent with a previous study using rat brain slices (Oleskevich and Williams, 1995), blocking reuptake prolonged the α1-AR-EPSC substantially. Thus, despite the slow signaling kinetics, the time course of the α1-AR-EPSC is affected by extending the presence of extracellular noradrenaline in time and space, and reuptake of noradrenaline by transporters is the primary mechanism by which extracellular noradrenaline is cleared from the cleft. There was marked heterogeneity in the time course of the α1-AR-EPSC across neurons that was not explained by membrane properties nor the amount of noradrenaline released. Instead, activation and decay rates were positively correlated within the same neuron. Moreover, α1-AR-EPSCs with faster activation rates under control conditions were more affected by reuptake blockade, suggestive of greater transporter activity at these synapses. Therefore, heterogeneity in α1-AR-EPSC kinetics may arise from varied density of noradrenaline transporters across subregions of the dorsal raphe, as observed in human brain tissue (Ordway et al., 1997). However, the possibility that the depth of the neuron in the brain slice contributes to heterogeneity cannot be excluded.
The efficiency of transporter-dependent reuptake also limited the spatial spread of noradrenaline. When reuptake was impaired, restricting diffusion of noradrenaline with dextran reduced the amplitude of the α1-AR-EPSC, indicating that under conditions of reuptake blockade, noradrenaline diffuses out of the synaptic cleft and activates extrasynaptic receptors. As observed under conditions of spill-over (Isaacson et al., 1993; Min et al., 1998), additional effects of impairing clearance or diffusion of noradrenaline on the α1-AR-EPSC via activation of presynaptic inhibitory autoreceptors (Frankhuijzen et al., 1988; Garratt et al., 1991) cannot be ruled out. Taken together, the data indicate that noradrenaline signaling may occur at discrete synapses without pooling of presynaptic release sites and spill-over of noradrenaline to extrasynaptic sites. This form of synaptic transmission resembles dopamine D2 receptor-dependent synaptic transmission in the ventral midbrain, where release sites and postsynaptic receptors are closely apposed (Ford et al., 2010; Gantz et al., 2013). Despite dramatic differences in the time course of the D2R and α1-AR-dependent synaptic currents, it is likely noradrenaline signaling in the dorsal raphe is similarly localized, with transporters maintaining synapse specificity by limiting spill-over.
Utility of independent synapses in a heterogenous brain region
The dorsal raphe nucleus contains a remarkably diverse array of serotonin and non-serotonin neurons, including GABA, glutamate, dopamine, and peptide neurons (Huang et al., 2019). Each class of neurons influences distinct behaviors, largely through nonredundant or reciprocal changes in firing rates in response to rewarding, aversive, or salient stimuli (Liu et al., 2014; Cohen et al., 2015; Li et al., 2016; Matthews et al., 2016; Cho et al., 2017, 2021; Nectow et al., 2017; Seo et al., 2019; Gazea et al., 2021; Yu et al., 2021). Differences in noradrenaline α1-AR-dependent signaling across the types of dorsal raphe neurons have received little attention, whether at the level of individual synapses or neural circuits and behavior. In the present study, we found that nearly all serotonin neurons expressed the necessary machinery for α1-AR-dependent synaptic transmission and had an α1-AR-EPSC. In addition, the GABA neurons also contained mRNA for α1-AR and GluD1R channels, which carry the ionic current after α1-AR activation (Gantz et al., 2020). However, using conventional brain slice stimulation, α1-AR-EPSCs were not recorded in nonserotonin neurons. These results are consistent with a prior in vivo study that concluded dorsal raphe GABA neurons exert tonic inhibitory influence on neighboring serotonin neurons but are not directly targeted by noradrenergic afferents (Baraban and Aghajanian, 1980b).
Within the population of serotonin neurons, there are subtypes distinguished by anatomic and molecular properties, genetic sublineages, or gene expression (Jensen et al., 2008; Calizo et al., 2011; Kiyasova et al., 2011; Spaethling et al., 2014; Fernandez et al., 2016; Huang et al., 2019). Accumulating evidence demonstrates these subtypes of serotonin neurons are also functionally distinct, influencing separate physiological responses (Brust et al., 2014; Okaty et al., 2015; Niederkofler et al., 2016; Ren et al., 2018) or in some cases, exerting opposing effects in the same behavior (e.g., anxiolytic vs anxiogenic; for review, see Hale et al., 2012). Whether the heterogeneous rates of noradrenaline signaling among the population of serotonin neurons segregate by the different subtypes remains unknown. But given the functionally distinct types of serotonin neurons intermingled in the dorsal raphe, limiting cross talk across neurons with efficient clearance of noradrenaline may preserve independence of each synapse to encode a discrete signal.
Health implications and concluding remarks
Dysregulation of the noradrenaline system has been linked to sleep disturbances, irritability, hyperaggression, and posttraumatic stress disorder. Imbalances in the serotonin system have been linked to complex emotional disorders including anxiety, bipolar, impulsivity, and depression. Disorders caused by imbalances in noradrenaline and serotonin signaling are frequently comorbid. This is likely because of a significant but not fully understood interaction between the serotonin and noradrenaline systems.
We have previously shown in brain slices that noradrenaline release and subsequent synaptic activation of α1-AR depolarize serotonin neurons and produces AP firing for ∼10 s (Gantz et al., 2020). Here, we show that the delays intrinsic to the second messenger cascade likely constitute the largest portion of the time course of noradrenaline-dependent excitation. Despite the slow kinetics of signaling, reuptake via noradrenaline transporters limited the temporal and spatial spread of extracellular noradrenaline, such that inhibition of noradrenaline transporters substantially prolonged the α1-AR-EPSC and allowed for diffusion to extrasynaptic sites. Thus, noradrenaline transporters are critical regulators of serotonin neuron excitability. Noradrenaline transporter inhibition is a leading pharmacotherapy, namely, by anti-depressants or psychostimulants. The results of this study suggest that the behavioral effects of noradrenaline transporter inhibition may arise, at least in part, via modulation of the dorsal raphe neural circuits and serotonin release. Furthermore in mouse models, noradrenaline release in the dorsal raphe is required for the acute anti-depressant behavioral responses to SSRIs (Cryan et al., 2004; O'Leary et al., 2007). Thus, altered expression of noradrenaline transporters may contribute to the etiology of neurologic and neuropsychiatric disease classically linked to perturbances in serotonin signaling.
Footnotes
This work was supported by a startup award from the University of Iowa Carver College of Medicine (S.C.G.). A portion of this work was supported by the Intramural Research Program at the National Institute on Drug Abuse (H.S.H. and S.C.G.).
The authors declare no competing financial interests.
References
- Agren H, Koulu M, Saavedra JM, Potter WZ, Linnoila M (1986) Circadian covariation of norepinephrine and serotonin in the locus coeruleus and dorsal raphe nucleus in the rat. Brain Res 397:353–358. 10.1016/0006-8993(86)90638-4 [DOI] [PubMed] [Google Scholar]
- Balakrishnan V, Kuo SP, Roberts PD, Trussell LO (2009) Slow glycinergic transmission mediated by transmitter pooling. Nat Neurosci 12:286–294. 10.1038/nn.2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban JM, Aghajanian GK (1980a) Suppression of firing activity of 5-HT neurons in the dorsal raphe by alpha-adrenoceptor antagonists. Neuropharmacology 19:355–363. 10.1016/0028-3908(80)90187-2 [DOI] [PubMed] [Google Scholar]
- Baraban JM, Aghajanian GK (1980b) Suppression of serotonergic neuronal firing by alpha-adrenoceptor antagonists: evidence against GABA mediation. Eur J Pharmacol 66:287–294. 10.1016/0014-2999(80)90461-6 [DOI] [PubMed] [Google Scholar]
- Baraban JM, Aghajanian GK (1981) Noradrenergic innervation of serotonergic neurons in the dorsal raphe: demonstration by electron microscopic autoradiography. Brain Res 204:1–11. 10.1016/0006-8993(81)90646-6 [DOI] [PubMed] [Google Scholar]
- Baraban JM, Wang RY, Aghajanian G (1978) Reserpine suppression of dorsal raphe neuronal firing: mediation by adrenergic system. Eur J Pharmacol 52:27–36. 10.1016/0014-2999(78)90018-3 [DOI] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR (2010) Astrocytes as gatekeepers of GABAB receptor function. J Neurosci 30:15262–15276. 10.1523/JNEUROSCI.3243-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust RD, Corcoran AE, Richerson GB, Nattie E, Dymecki SM (2014) Functional and developmental identification of a molecular subtype of brain serotonergic neuron specialized to regulate breathing dynamics. Cell Rep 9:2152–2165. 10.1016/j.celrep.2014.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calizo LH, Akanwa A, Ma X, Pan Y-Z, Lemos JC, Craige C, Heemstra LA, Beck SG (2011) Raphe serotonin neurons are not homogenous: electrophysiological, morphological and neurochemical evidence. Neuropharmacology 61:524–543. 10.1016/j.neuropharm.2011.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG (2000) Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci 20:4423–4434. 10.1523/JNEUROSCI.20-12-04423.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JR, Treweek JB, Robinson JE, Xiao C, Bremner LR, Greenbaum A, Gradinaru V (2017) Dorsal raphe dopamine neurons modulate arousal and promote wakefulness by salient stimuli. Neuron 94:1205–1219.e8. 10.1016/j.neuron.2017.05.020 [DOI] [PubMed] [Google Scholar]
- Cho JR, Chen X, Kahan A, Robinson JE, Wagenaar DA, Gradinaru V (2021) Dorsal raphe dopamine neurons signal motivational salience dependent on internal state, expectation, and behavioral context. J Neurosci 41:2645–2655. 10.1523/JNEUROSCI.2690-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JY, Amoroso MW, Uchida N (2015) Serotonergic neurons signal reward and punishment on multiple timescales. Elife 4:e06346. 10.7554/eLife.06346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch JR (1970) Responses of neurons in the raphe nuclei to serotonin, norepinephrine and acetylcholine and their correlation with an excitatory synaptic input. Brain Res 19:137–150. 10.1016/0006-8993(70)90243-X [DOI] [PubMed] [Google Scholar]
- Courtney NA, Ford CP (2014) The timing of dopamine- and noradrenaline-mediated transmission reflects underlying differences in the extent of spillover and pooling. J Neurosci 34:7645–7656. 10.1523/JNEUROSCI.0166-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney NA, Ford CP (2016) Mechanisms of 5-HT1A receptor-mediated transmission in dorsal raphe serotonin neurons. J Physiol 594:953–965. 10.1113/JP271716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan JF, O'Leary OF, Jin S-H, Friedland JC, Ouyang M, Hirsch BR, Page ME, Dalvi A, Thomas SA, Lucki I (2004) Norepinephrine-deficient mice lack responses to antidepressant drugs, including selective serotonin reuptake inhibitors. Proc Natl Acad Sci USA 101:8186–8191. 10.1073/pnas.0401080101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez SP, Cauli B, Cabezas C, Muzerelle A, Poncer J-C, Gaspar P (2016) Multiscale single-cell analysis reveals unique phenotypes of raphe 5-HT neurons projecting to the forebrain. Brain Struct Funct 221:4007–4025. 10.1007/s00429-015-1142-4 [DOI] [PubMed] [Google Scholar]
- Ford CP, Phillips PEM, Williams JT (2009) The time course of dopamine transmission in the ventral tegmental area. J Neurosci 29:13344–13352. 10.1523/JNEUROSCI.3546-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Gantz SC, Phillips PEM, Williams JT (2010) Control of extracellular dopamine at dendrite and axon terminals. J Neurosci 30:6975–6983. 10.1523/JNEUROSCI.1020-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankhuijzen AL, Wardeh G, Hogenboom F, Mulder AH (1988) Alpha 2-adrenoceptor mediated inhibition of the release of radiolabelled 5-hydroxytryptamine and noradrenaline from slices of the dorsal region of the rat brain. Naunyn Schmiedebergs Arch Pharmacol 337:255–260. 10.1007/BF00168835 [DOI] [PubMed] [Google Scholar]
- Fu W, Le Maître E, Fabre V, Bernard J-F, David Xu Z-Q, Hökfelt T (2010) Chemical neuroanatomy of the dorsal raphe nucleus and adjacent structures of the mouse brain. J Comp Neurol 518:3464–3494. 10.1002/cne.22407 [DOI] [PubMed] [Google Scholar]
- Gantz SC, Bunzow JR, Williams JT (2013) Spontaneous inhibitory synaptic currents mediated by a G protein-coupled receptor. Neuron 78:807–812. 10.1016/j.neuron.2013.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz SC, Moussawi K, Hake HS (2020) Delta glutamate receptor conductance drives excitation of mouse dorsal raphe neurons. Elife 9:e56054. 10.7554/eLife.56054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garratt JC, Crespi F, Mason R, Marsden CA (1991) Effects of idazoxan on dorsal raphe 5-hydroxytryptamine neuronal function. Eur J Pharmacol 193:87–93. 10.1016/0014-2999(91)90204-4 [DOI] [PubMed] [Google Scholar]
- Gazea M, Furdan S, Sere P, Oesch L, Molnár B, Giovanni GD, Fenno LE, Ramakrishnan C, Mattis J, Deisseroth K, Dymecki SM, Adamantidis AR, Lőrincz ML (2021) Reciprocal lateral hypothalamic and raphé GABAergic projections promote wakefulness. J Neurosci 41:4840–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocho Y, Sakai A, Yanagawa Y, Suzuki H, Saitow F (2013) Electrophysiological and pharmacological properties of GABAergic cells in the dorsal raphe nucleus. J Physiol Sci 63:147–154. 10.1007/s12576-012-0250-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale MW, Shekhar A, Lowry CA (2012) Stress-related serotonergic systems: implications for symptomatology of anxiety and affective disorders. Cell Mol Neurobiol 32:695–708. 10.1007/s10571-012-9827-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepp R, Hay YA, Aguado C, Luján R, Dauphinot L, Potier MC, Nomura S, Poirel O, Mestikawy El S, Lambolez B, Tricoire L (2015) Glutamate receptors of the delta family are widely expressed in the adult brain. Brain Struct Funct 220:2797–2815. 10.1007/s00429-014-0827-4 [DOI] [PubMed] [Google Scholar]
- Huang KW, Ochandarena NE, Philson AC, Hyun M, Birnbaum JE, Cicconet M, Sabatini BL (2019) Molecular and anatomical organization of the dorsal raphe nucleus. Elife 8:e46464. 10.7554/eLife.46464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Solís JM, Nicoll RA (1993) Local and diffuse synaptic actions of GABA in the hippocampus. Neuron 10:165–175. 10.1016/0896-6273(93)90308-e [DOI] [PubMed] [Google Scholar]
- Jensen P, Farago AF, Awatramani RB, Scott MM, Deneris ES, Dymecki SM (2008) Redefining the serotonergic system by genetic lineage. Nat Neurosci 11:417–419. 10.1038/nn2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge SJ, Gartside SE (2006) Firing of 5-HT neurones in the dorsal and median raphe nucleus in vitro shows differential alpha1-adrenoceptor and 5-HT1A receptor modulation. Neurochem Int 48:100–107. 10.1016/j.neuint.2005.09.003 [DOI] [PubMed] [Google Scholar]
- Kirby LG, Pernar L, Valentino RJ, Beck SG (2003) Distinguishing characteristics of serotonin and non-serotonin-containing cells in the dorsal raphe nucleus: electrophysiological and immunohistochemical studies. Neuroscience 116:669–683. 10.1016/S0306-4522(02)00584-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H, Armstrong W (1991) A biotin-containing compound (N-(2-aminoethyl)biotinamide for intracellular labeling and neuronal tracing studies: comparison with biocytin. J Neurosci Meth 37:141–150. 10.1016/0165-0270(91)90124-I [DOI] [PubMed] [Google Scholar]
- Kiyasova V, Fernandez SP, Laine J, Stankovski L, Muzerelle A, Doly S, Gaspar P (2011) A genetically defined morphologically and functionally unique subset of 5-HT neurons in the mouse raphe nuclei. J Neurosci 31:2756–2768. 10.1523/JNEUROSCI.4080-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YQ, Li H, Kaneko T, Mizuno N (2001) Morphological features and electrophysiological properties of serotonergic and non-serotonergic projection neurons in the dorsal raphe nucleus. An intracellular recording and labeling study in rat brain slices. Brain Res 900:110–118. 10.1016/s0006-8993(01)02272-7 [DOI] [PubMed] [Google Scholar]
- Li Y, Zhong W, Wang D, Feng Q, Liu Z, Zhou J, Jia C, Hu F, Zeng J, Guo Q, Fu L, Luo M (2016) Serotonin neurons in the dorsal raphe nucleus encode reward signals. Nat Comms 7:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhou J, Li Y, Hu F, Lu Y, Ma M, Feng Q, Zhang JE, Wang D, Zeng J, Bao J, Kim JY, Chen ZF, Mestikawy El S, Luo M (2014) Dorsal raphe neurons signal reward through 5-HT and glutamate. Neuron 81:1360–1374. 10.1016/j.neuron.2014.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Schnell SA, Hack SP, Christie MJ, Wessendorf MW, Vaughan CW (2004) Serotonergic and nonserotonergic dorsal raphe neurons are pharmacologically and electrophysiologically heterogeneous. J Neurophysiol 92:3532–3537. 10.1152/jn.00437.2004 [DOI] [PubMed] [Google Scholar]
- Matthews GA, Nieh EH, Vander Weele CM, Halbert SA, Pradhan RV, Yosafat AS, Glober GF, Izadmehr EM, Thomas RE, Lacy GD, Wildes CP, Ungless MA, Tye KM (2016) Dorsal raphe dopamine neurons represent the experience of social isolation. Cell 164:617–631. 10.1016/j.cell.2015.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM (1998) Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron 21:561–570. 10.1016/s0896-6273(00)80566-8 [DOI] [PubMed] [Google Scholar]
- Nakamoto C, Konno K, Miyazaki T, Nakatsukasa E, Natsume R, Abe M, Kawamura M, Fukazawa Y, Shigemoto R, Yamasaki M, Sakimura K, Watanabe M (2020) Expression mapping, quantification, and complex formation of GluD1 and GluD2 glutamate receptors in adult mouse brain. J Comp Neurol 528:1003–1027. 10.1002/cne.24792 [DOI] [PubMed] [Google Scholar]
- Nectow AR, Schneeberger M, Zhang H, Field BC, Renier N, Azevedo E, Patel B, Liang Y, Mitra S, Tessier-Lavigne M, Han MH, Friedman JM (2017) Identification of a brainstem circuit controlling feeding. Cell 170:429–442.e11. 10.1016/j.cell.2017.06.045 [DOI] [PubMed] [Google Scholar]
- Niederkofler V, Asher TE, Okaty BW, Rood BD, Narayan A, Hwa LS, Beck SG, Miczek KA, Dymecki SM (2016) Identification of serotonergic neuronal modules that affect aggressive behavior. Cell Rep 17:1934–1949. 10.1016/j.celrep.2016.10.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okaty BW, Freret ME, Rood BD, Brust RD, Hennessy ML, deBairos D, Kim JC, Cook MN, Dymecki SM (2015) Multi-scale molecular deconstruction of the serotonin neuron system. Neuron 88:774–791. 10.1016/j.neuron.2015.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary OF, Bechtholt AJ, Crowley JJ, Valentino RJ, Lucki I (2007) The role of noradrenergic tone in the dorsal raphe nucleus of the mouse in the acute behavioral effects of antidepressant drugs. Eur Neuropsychopharmacol 17:215–226. 10.1016/j.euroneuro.2006.06.012 [DOI] [PubMed] [Google Scholar]
- Oleskevich S, Williams JT (1995) Cocaine prolongs norepinephrine synaptic potentials in rat dorsal raphe. J Neurophysiol 73:687–692. 10.1152/jn.1995.73.2.687 [DOI] [PubMed] [Google Scholar]
- Ordway GA, Stockmeier CA, Cason GW, Klimek V (1997) Pharmacology and distribution of norepinephrine transporters in the human locus coeruleus and raphe nuclei. J Neurosci 17:1710–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Mody I (1992) Differential activation of GABAA and GABAB receptors by spontaneously released transmitter. J Neurophysiol 67:227–235. 10.1152/jn.1992.67.1.227 [DOI] [PubMed] [Google Scholar]
- Otis TS, Wu YC, Trussell LO (1996) Delayed clearance of transmitter and the role of glutamate transporters at synapses with multiple release sites. J Neurosci 16:1634–1644. 10.1523/JNEUROSCI.16-05-01634.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pudovkina OL, Cremers TIFH, Westerink BHC (2003) Regulation of the release of serotonin in the dorsal raphe nucleus by alpha1 and alpha2 adrenoceptors. Synapse 50:77–82. 10.1002/syn.10245 [DOI] [PubMed] [Google Scholar]
- Ren J, Friedmann D, Xiong J, Liu CD, Ferguson BR, Weerakkody T, DeLoach KE, Ran C, Pun A, Sun Y, Weissbourd B, Neve RL, Huguenard J, Horowitz MA, Luo L (2018) Anatomically defined and functionally distinct dorsal raphe serotonin sub-systems. Cell 175:472–487.e20. 10.1016/j.cell.2018.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M (2000) GABA spillover activates postsynaptic GABA(B) receptors to control rhythmic hippocampal activity. Neuron 25:673–681. 10.1016/s0896-6273(00)81069-7 [DOI] [PubMed] [Google Scholar]
- Seo C, Guru A, Jin M, Ito B, Sleezer BJ, Ho YY, Wang E, Boada C, Krupa NA, Kullakanda DS, Shen CX, Warden MR (2019) Intense threat switches dorsal raphe serotonin neurons to a paradoxical operational mode. Science 363:538–542. 10.1126/science.aau8722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaethling JM, Piel D, Dueck H, Buckley PT, Morris JF, Fisher SA, Lee J, Sul J-Y, Kim J, Bartfai T, Beck SG, Eberwine JH (2014) Serotonergic neuron regulation informed by in vivo single-cell transcriptomics. FASEB J 28:771–780. 10.1096/fj.13-240267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson TH, Bunney BS, Aghajanian GK (1975) Inhibition of both noradrenergic and serotonergic neurons in brain by the alpha-adrenergic agonist clonidine. Brain Res 92:291–306. 10.1016/0006-8993(75)90276-0 [DOI] [PubMed] [Google Scholar]
- Szabadics J, Tamás G, Soltesz I (2007) Different transmitter transients underlie presynaptic cell type specificity of GABAA,slow and GABAA,fast. Proc Natl Acad Sci USA 104:14831–14836. 10.1073/pnas.0707204104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szapiro G, Barbour B (2009) Parasynaptic signalling by fast neurotransmitters: the cerebellar cortex. Neuroscience 162:644–655. 10.1016/j.neuroscience.2009.03.077 [DOI] [PubMed] [Google Scholar]
- Vandermaelen CP, Aghajanian GK (1983) Electrophysiological and pharmacological characterization of serotonergic dorsal raphe neurons recorded extracellularly and intracellularly in rat brain slices. Brain Res 289:109–119. 10.1016/0006-8993(83)90011-2 [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Higashi H, Nishi S (1985) Noradrenaline mediates slow excitatory synaptic potentials in rat dorsal raphe neurons in vitro. Neurosci Lett 61:305–310. 10.1016/0304-3940(85)90481-1 [DOI] [PubMed] [Google Scholar]
- Yu W, Pati D, Pina MM, Schmidt KT, Boyt KM, Hunker AC, Zweifel LS, McElligott ZA, Kash TL (2021) Periaqueductal gray/dorsal raphe dopamine neurons contribute to sex differences in pain-related behaviors. Neuron 109:1365–1380.e5. 10.1016/j.neuron.2021.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]