

Abstract
This review describes the clinical and pathological features of oxalate nephropathy (ON), defined as a syndrome of decreased renal function associated with deposition of calcium oxalate crystals in kidney tubules. We review the different causes of hyperoxaluria, including primary hyperoxaluria, enteric hyperoxaluria and ingestion-related hyperoxaluria. Recent case series of biopsy-proven ON are reviewed in detail, as well as the implications of these series. The possibility of antibiotic use predisposing to ON is discussed. Therapies for hyperoxaluria and ON are reviewed with an emphasis on newer treatments available and in development. Promising research avenues to explore in this area are discussed.
Keywords: acute kidney injury, oxalate nephropathy, pathology, primary hyperoxaluria, secondary hyperoxaluria
INTRODUCTION
Oxalate nephropathy (ON) is defined by an acute and/or chronic decrease in kidney function associated with the deposition of calcium oxalate crystals, in kidney tubules. There is also typically acute tubular injury and an associated acute/chronic interstitial nephritis or fibrosis [1–5]. The term ON is used in this review in preference to nephrocalcinosis as the latter term also (and according to some authors, exclusively) [6] includes calcium phosphate deposition. Furthermore, nephrocalcinosis is also often used as a term to describe calcification of the renal parenchyma as seen on radiological imaging [7], while ON implies a pathological diagnosis. See Figure 1 for representative light microscopic images including that of intratubular oxalate crystals as seen under polarized light. Calcium oxalate crystals are birefringent under polarized light, unlike calcium phosphate crystals. ON can often cause progressive kidney dysfunction and there is a significant risk of end-stage kidney disease (ESKD) [1–5]. This review will focus on the causes of ON, which in most cases is due to hyperoxaluria (defined by a 24-h urine oxalate of >40–45 mg/day) [8]. We will review the four largest series of ON that have been published recently and discuss the implications of these series. Then we will discuss the latest treatments being developed for hyperoxaluria and ON and outline areas in need of further attention. Issues of kidney transplant will be touched on briefly, but are not emphasized in this review.
FIGURE 1:

Renal oxalosis: (A) Massive deposition of oxalate crystals is noted in tubules with associated advanced chronic tubulointerstitial disease with atrophy and dropout of tubules and prominent interstitial fibrosis and nonspecific inflammation (H&E, bright field, 40X). (B) Same area visualized under polarized light reveals numerous intratubular crystals (H&E, polarized light, 40X). (C) Intratubular oxalate crystals are often transparent or reveal yellow or gray color, with needle or other shapes of crystals (H&E, bright field, 600X). (D) Same area visualized under polarized light reveals colorful crystals (H&E, polarized light, 600X)
PREVALENCE
The overall prevalence of ON has not been clear as a significant amount of the literature has been based on case reports, but two recent reviews have addressed this. Buysschaert et al. [2] found that ON made up 1% of native kidney biopsies in a Belgian series from 2010 to 2018. Only cases with progressive kidney dysfunction were included. Cases were excluded if there were other kidney diseases aside from non-specific nephrosclerosis or diabetic nephropathy. Our group, in a recently published study, which included other concomitant diagnoses, found that the prevalence of any oxalate deposition was as high as 4.07% of biopsies in a New York City metro area population [3]. Oxalate deposition in this study was considered the primary or contributing cause of kidney disease progression in 88% of the cases, with an estimated prevalence of 3.6%.
CAUSES OF ON
Primary hyperoxaluria
This topic has been reviewed in greater detail elsewhere by Cochat and Rumsby [9], but this review will emphasize a number of salient points. Primary hyperoxaluria (PH) is a group of autosomal recessive disorders causing primarily hepatic overproduction of oxalate, due to accumulation of the oxalate precursor glyoxylate. This leads to calcium oxalate nephrolithiasis and multisystem deposits of calcium oxalate, including in the kidneys, and accounts for 1–2% of pediatric ESKD. While the median age of onset is 5.5 years, it can sometimes present in adulthood with kidney stones or kidney failure, and should be considered in cases of hyperoxaluria and ON without another obvious cause. PH Type 1 (PH1), which accounts for ∼80% of PH cases, is also the most severe subtype. It is due to a deficiency of hepatic alanine glyoxylate aminotransferase (AGT), which normally catalyzes the metabolism of glyoxylate to glycine. A definitive diagnosis of PH requires genetic testing, such as for a mutation in AGXT, which encodes AGT, in PH1. See Figure 2 for a simplified summary of hepatic pathways of oxalate metabolism. It is notable that the urinary oxalate excretion tends to be higher in PH (>88 mg/day) as opposed to 44–70 mg/day in enteric hyperoxaluria [8]. However, there is enough overlap that a definitive distinction between PH and enteric hyperoxaluria cannot generally be made based on urinary oxalate excretion alone. Systemic oxalosis, due to very high plasma oxalate levels, is common in PH as renal failure progresses, but this is seen only very rarely in other forms of hyperoxaluria [10]. In patients with renal insufficiency, very high plasma oxalate levels might be useful in distinguishing PH from other forms of kidney disease, including secondary hyperoxaluria [11].
FIGURE 2:

Simplified hepatic pathways of glyoxylate metabolism
Enteric hyperoxaluria
Enteric hyperoxaluria (EH) is defined by hyperoxaluria occurring in the setting of fat malabsorption or steatorrhea [10, 12]. Normally, calcium binds oxalate in the bowel to form insoluble calcium oxalate that is excreted in the feces. In a state of fat malabsorption, calcium is bound by free fatty acids and becomes unavailable for oxalate binding. There is then increased soluble oxalate available to be absorbed by the bowel. Free fatty acids and bile salts may also directly increase colonic permeability to oxalate [13]. An intact colon appears likely to be important for oxalate absorption, and hyperoxaluria in EH has generally not been observed in patients where the colon is not utilized such as in patients with ileostomies after colectomy [14, 15]. Why this is so is not certain, and oxalate absorption also occurs in the proximal gut as evidenced by an increase in urinary oxalate in response to an oral oxalate load in patients with ileostomies in one study [16], though these particular patients did not have baseline EH. Solute-linked carrier 26 (SCL26) anion exchangers, which are a family of transporters that mediate transcellular oxalate transport, are differentially expressed along the gut [8], though it is felt that most oxalate absorption likely occurs paracellularly [17]. The risk of calcium oxalate precipitation is likely worsened by volume depletion from diarrhea as well as bicarbonate loss, which can lead to metabolic acidosis and hypocitraturia.
Both nephrolithiasis and ON were frequent complications of one of the first surgical treatments for obesity, jejunoileal bypass. This procedure involved bypassing much of the small bowel, causing significant malabsorption. This procedure was largely abandoned by 1979 due to high morbidity, including the development of renal failure in as many as 35% of patients [18, 19]. More recently, Roux-en-Y gastric bypass, which replaced jejunoileal bypass as the procedure of choice for malabsorptive bariatric surgery, has been recognized as a cause of ON [4]. So too have other malabsorptive states such as chronic pancreatitis [5], and after bowel resections for inflammatory bowel disease (IBD) [20]. Orlistat, a weight loss agent that also causes fat malabsorption, has similarly been recognized to cause hyperoxaluria and ON [21]. It is important to note that while IBD is often listed as a cause of ON, reports of ON in IBD have virtually all been in patients who have had previous bowel resections, and with presumed malabsorption. Similarly, while malabsorptive bariatric surgery, such as jejunoileal bypass and Roux-en-Y surgery, is strongly associated with ON, this has not been seen with restrictive weight loss surgeries such as sleeve gastrectomy, where the small bowel is not bypassed.
Ingestions
Ingestions include the direct consumption of oxalate in foods with high oxalate content, and also the ingestion of oxalate precursors. A classic and dramatic cause of ingestion-related ON is ethylene glycol (EG). EG is the active ingredient in antifreeze, but is also present in a number of solvents, paints and other industrial and commercial products [22]. Ingested EG is metabolized in the liver to oxalic acid (Figure 3) and causes acute ON with acute tubular injury and oxalate crystal deposition in tubules. There are frequently numerous urine calcium oxalate crystals present in the urine that can be a sign of the diagnosis. See Figure 4 for an image of monohydrate and dihydrate calcium oxalate crystals in urine.
FIGURE 3:
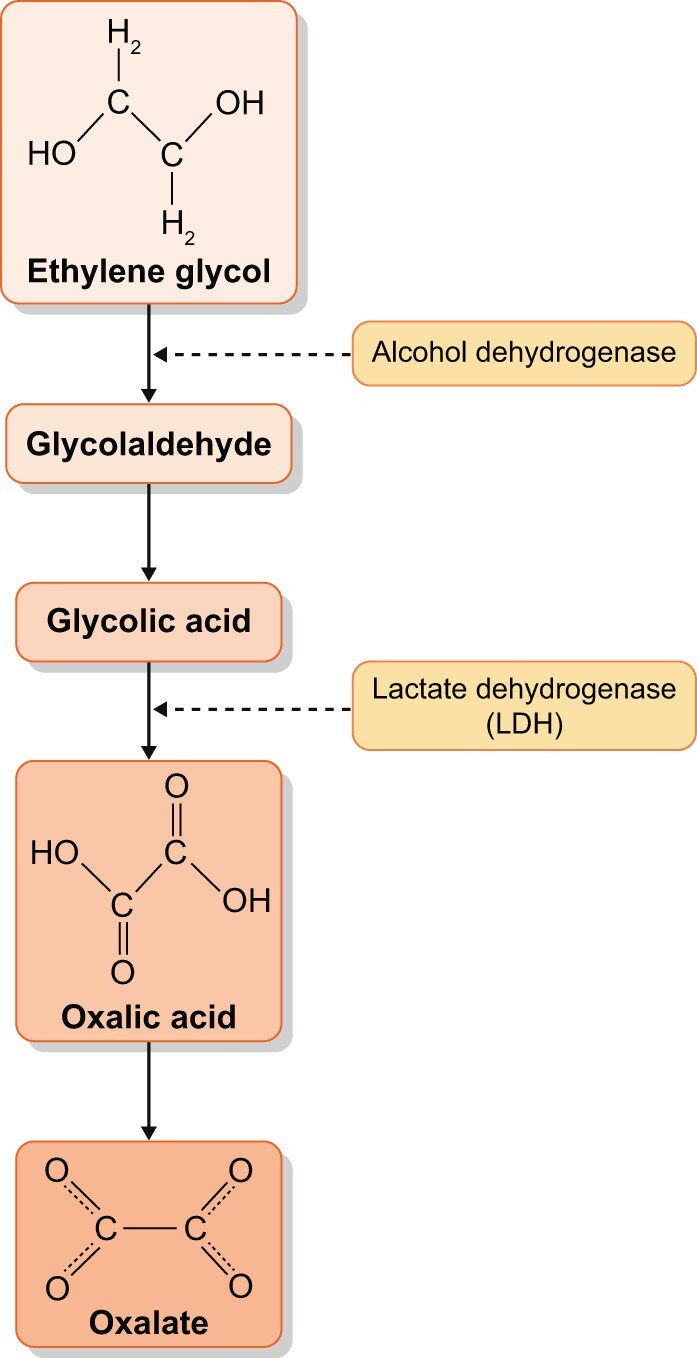
Metabolism of ethylene glycol to oxalate
FIGURE 4:
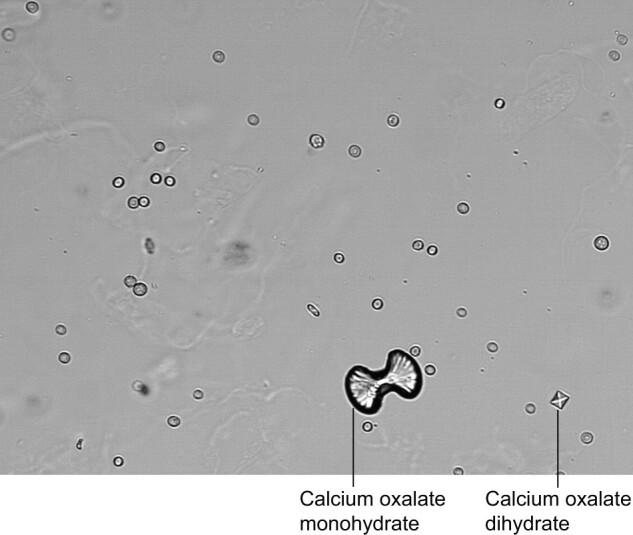
Calcium Oxalate Monohydrate and Calcium Oxalate Dihydrate crystals in spun urine Reproduced according to License CC BY-SA 3.0. Photo by Doruk Salanci
There have also been multiple case reports of ON associated with high dietary oxalate intake from a number of sources, as well as due to ingestions of the oxalate precursor vitamin C (ascorbic acid) [23–55]. Figure 5 shows the non-enzymatic metabolism of vitamin C to oxalate. These reports have generally involved excessive intake of high oxalate foods or megadoses of vitamin C, though some cases have been reported involving more moderate amounts, especially with chronic intake (Table 1). There have been reports of ON from juicing of vegetables, such as spinach smoothies in one report [55] and an assortment of high oxalate vegetables (together with vitamin C) in another [47]. It has been speculated that juiced oxalate foods may be more effectively absorbed in the intestine via the paracellular pathway via solvent drag and because of dilution of calcium by water [56]. Recently, the most frequent case reports have seemed to involve star fruit (carambola) [28–32] and vitamin C supplements [23–27, 35, 46, 50–54, 57], while other ingestions are reported less commonly. The reason for this is not clear. Vitamin C is likely a more bioavailable source of oxalate than food. This is because oxalate in food is complexed with calcium (and to a lesser degree magnesium), limiting absorption. Furthermore, vitamin C is taken by a large segment of the population, sometimes at very high doses. It has recently also used at high intravenous doses as a treatment for sepsis, including in patients with COVID-19 infection [23, 52]. It should also be noted that there is a significant amount of vitamin C in common beverages such as apple juice or orange juice (∼800 mg/L).
FIGURE 5:
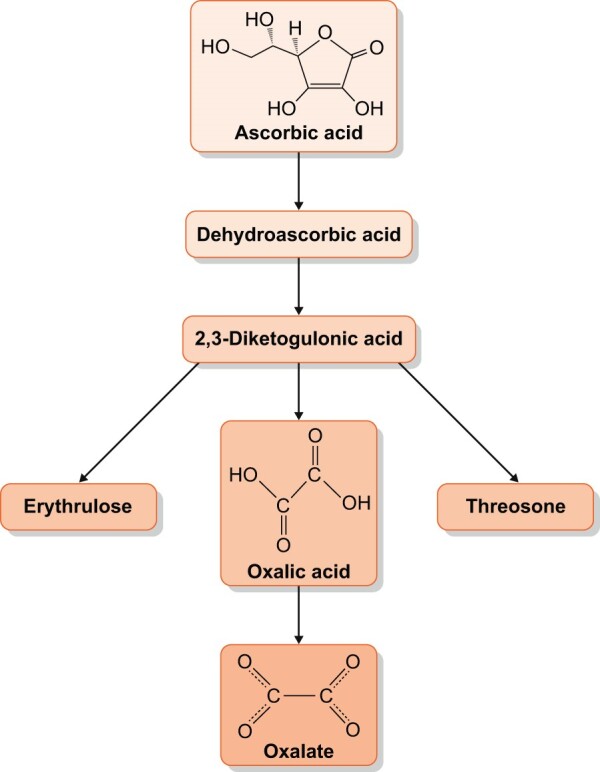
Metabolism of ascorbic acid (vitamin C) to oxalate
Table 1.
Reported ingestions causing ON
| Substance ingested | Quantity of typical reported ingestion causing biopsy confirmed ON | Notes |
|---|---|---|
| Star fruit (Averrhoa carambola) | 200–3000 mL of pure juice | One case reported ingestion of only 200 mL as remedy for diabetes. One case only reported chronic intake of 5–6 fruit over 1 month and then four fruit over 4 days [29] |
| 6–12 fruit in 1 setting [28–32] | ||
| Vitamin C [23–27, 46, 49–54, 57] | ||
| Oral | 2–6.5 g daily | One case reported ingestion as low as 480–960 mg vitamin C daily for 4 months [26] |
| IV | 4–5 g daily | Two cases reported in COVID+ patients receiving 50 mg/kg 4×/day vitamin C for sepsis [23] |
| Irumban puli (Averrhoa bilimbi) | 150–400 mL juice/day [43] | Irumban puli (A. bilimbi) is a local fruit in South India which has relatively high oxalic acid content and is drunk as a beverage |
| Oxalate content of the fruit was 25.1 mg/100 g of the fruit [42] | ||
| Peanuts | 100–243 g peanuts daily for 2–3 months [39–40] | – |
| Cashews | 1 kg of cashews/week for 4 months [38] | – |
| Almonds and almond-containing marzipan | 150–200 g of almonds and 50–100 g of almond-containing marzipan daily [41] | – |
| Rhubarb | 500 g fresh weight/day for >4 weeks [44] | – |
| Chaga mushroom powder | 4–5 teaspoons/day of Chaga mushroom powder for 6 months [48] | 11.2 g of oxalate in 100 g of the powder; it was used as a remedy for liver cancer [48] |
| Black iced tea | Sixteen 8 oz glasses daily [45] | Daily consumption of oxalate >1500 mg in one case report [45] |
| Juicing | Celery, carrots, parsley beets with greens and spinach taken with Vitamin C in one [47] and two cups spinach/day in the other [55] | The oxalate content was estimated at ∼1300 mg/day in each report |
| Nafronyl oxalate | 7 g over 2 days [36] | 19 mg oxalate/100 mg Nafril capsule |
| Was given to patient for toothache and otalgia | ||
| Used to treat peripheral and cerebrovascular disease [36] |
Majority of reports are isolated cases other than for vitamin C, star fruit and Irumban puli. IV, intravenous.
Star fruit may be a frequent culprit, even though it is estimated to contain less oxalate than spinach or rhubarb by weight, possibly because it can be consumed as a concentrated juice or can be eaten easily in large quantities. It has been suggested that star fruit may contain a neurotoxin as well, and there have been reports of acute neurological symptoms from star fruit ingestion in patients already being dialyzed that responded to intensified dialysis [58]. It is possible that star fruit ingestion might come to medical attention more frequently because of these associated symptoms.
See Table 2 for a list of high oxalate foods. It must be noted, as can be seen in the table, that there is a broad range of oxalate content for each item, as amounts vary based on how it is measured, the way the food is prepared, the particular cultivar (genetic variation within a species) of food, and even the season. The oxalate content can also vary widely between the leaf (usually highest) and the stem and root [59–63].
Table 2.
Foods with high oxalate content and estimated amounts
| Substance | Oxalate content in mg/100 g |
|---|---|
| Purslane | 910–1679 |
| Spinach varieties | 320–1260 |
| Garden orach | 300–1500 |
| Rhubarb | 260–1235 |
| Sorrel | 270–730 |
| Cocoa | 170–623 |
| Beet leaves | 121–920 |
| Beet root | 76–675 |
| Almonds | 431–490 |
| Cashews | 231–262 |
| Hazelnuts | 167–223 |
| Peanuts | 96–705 |
| Carambola/star fruit | 80–730 |
| Buckwheat | 269–271 |
| Soy | 179–187 |
| Coffee | 50–150 |
| Black tea (100 mL brewed)a | 48–92 |
Furthermore, the toxicity may depend more on the bioavailability of the item, which depends on the calcium content as well, as mentioned above. It is notable that in many of the case reports involving acute ingestions and acute kidney injury, patients frequently had renal improvement or recovery, suggesting that this may have a better prognosis than the other forms of ON.
SERIES
Most published cases of ON have been in the form of individual case reports, but there have been four published series of patients with biopsy-proven ON, comprising more than a few cases, published relatively recently. Table 3 summarizes the key clinical data for these series. The first series was published in 2008 in a series of 11 patients who had Roux-en-Y gastric bypass [4]. Most of the patients had underlying diabetes and all had hypertension. All patients presented with acute renal failure (ARF), often superimposed on chronic kidney disease (CKD), with a mean serum creatinine at presentation of 5.0 mg/dL. The mean and median times from surgery to ARF were 33 and 12 months, respectively. Only three patients had urine or serum oxalate levels done and they were reported as ‘elevated’. Two patients had crystalluria on urinalysis. No imaging was reported. Biopsies were notable for abundant tubular calcium oxalate deposits in intraluminal and intracellular areas, and also focally in the interstitium. The oxalate crystalline deposits were accompanied by diffuse tubular injury as well as tubular atrophy and interstitial fibrosis (IF). Areas of IF also had interstitial inflammation without tubulitis. Eight of the 11 patients progressed to ESKD and the remaining three had stable to improved renal function at follow-up ranging from 2.5 to 22 months.
Table 3.
Key clinical data in four largest case series
| Clinical data | Nasr et al. [4] (2018) (n = 11) | Cartery et al. [5] (2011) (n = 12) | Buysschaert et al. [2] (2020) (n = 21) | Yang et al. [3] (2020) (n = 25) |
|---|---|---|---|---|
| Age, years (range) | 61.3 (45–79) | 67 (41–91) | 61 ± 20 | 63.6 ± 9.1 |
| Gender, male | 5 (45) | 9 (75) | 14 (67) | 13 (52) |
| White race | 8 (72.7) | 21 (100) | ||
| Diabetes | 9 (81.8) | 9 (75) | 12 (57) | 16 (64) |
| Hypertension | 11 (100) | 8 (66.7) | 16 (76) | 19 (76) |
| Baseline CKD | 7 (58.3) | 13 (62) | ||
| Urinary stones | 3 (25) | 3 (14) | 1 (4) | |
| RAAS inhibitor use | 3 (27.3) | 8 (66.7) | 8 (38) | |
| Diuretic use | 3 (27.3) | 5 (41.6) | 9 (43) | |
| Baseline creatinine, mg/dL (range) | 1.5 (0.9–2.5) | 1.1 (0.79–2.02) | ||
| GFR baseline, mL/min/1.73 m2 (range) | 57 (36–89) | 36 ± 7 | ||
| Serum creatinine at time of presentation, mg/dL (range) | 5.0 (2.4–9.2) | 6.6 (3.3–9.6) | 8.0 ± 4.5 | 6.3 ± 3.2 |
| EH | 11 (100) | 12 (100) | 10 (48) | 10 (40) |
| Ingestion related | – | 1 (8.3) | 2 (10) | 4 (16) |
| Recent antibiotic use | – | 4 (33.3) | 3 (14) | 13 (52) |
| Uncertain cause | – | 3 (14) | 11 (44) | |
| Presence of hypocalcemia | – | 9 (75) | 6 (24) | |
| Microscopic hematuria | 3 (27.3) | 3 (25) | 5 (24) | |
| Leukocyturia | 6 (54.5) | 10 (83.3) | 5 (24) | |
| Urine protein (range) | 24 h, 1.4 g/day (0.37–6.00) | 0.34 g/day (0.05–1.01) | 1.4 g/g ± 2.0 | 52.04 mg/g ± 71.38 |
| Diabetic glomerulopathy | 7 (63.6) | 3 (25) | 6 (28.6) | 8 (29.6) |
| Acute tubular injury | 11 (100) | 12 (100) | 21 (100) | 17 (63) |
| Acute/chronic tubulointerstitial nephritis | 11 (100) | 9 (75) | (Acute) 18 (85.7) |
(Acute) 9 (33.3) (Chronic) 8 (32) |
Data are reported in mean ± SD or n (%) unless otherwise noted. Urine protein is random protein/creatinine ratio unless specified.
The second series, published in 2011, was a series of 12 patients with chronic pancreatitis [5]. Hypertension was present in 67%, diabetes in 75% and one patient had a renal transplant due to diabetic nephropathy. Many of the patients had a recent history of diarrhea, diuretic use and/or use of angiotensin-converting enzyme inhibitors or angiotensin receptor blockers. One-third of patients had been given antibiotics shortly before presentation and one had been given a course of intravenous vitamin C. On presentation with ON, mean serum creatinine was 587 µmol/L or 6.6 mg/dL. Renal imaging was unremarkable except renal atrophy in three cases and non-obstructive nephrolithiasis in three. In the 11 patients it was measured, 24-h urine oxalate was elevated and ranged from 52 to 92 mg/day. No patient had crystalluria on urinalysis. Renal biopsies showed acute tubular injury in all cases with interstitial inflammation and edema. All cases had large calcium oxalate crystals in tubular lumens, epithelial cells and interstitium. After a mean of 7 months of follow-up, one-quarter of patients required long-term dialysis, and the rest had a mean serum creatinine of 212 µmol/L (2.4 mg/dL) and eGFR of 25 mL/min/1.73 m2.
The latest two series were published in 2020. Unlike the previously mentioned series, which focused on specific populations at risk for ON, these surveyed biopsy databases for ON cases. Buysschaert et al. [2] identified 22 cases of ON from a Belgian database, comprising 1% of native biopsies. Twenty-one of the cases had adequate clinical data. Fifty-seven percent were diabetic and 76% were hypertensive. Sixty-two percent had underlying CKD with a mean eGFR of 36 mL/min/1.73 m2 prior to development of ON. The majority took a renin–angiotensin–aldosterone system inhibitor and/or a diuretic. All patients presented with acute kidney injury (with or without underlying CKD), with a mean serum creatinine of 8.0 ± 4.5. Urine oxalate/creatinine was reported as 86 ± 58 mg in 16 cases. Three patients had a nephrolithiasis history but renal ultrasounds showed normal-sized kidneys without mention of nephrocalcinosis or stones. Pathologically, aside from oxalate deposition in all cases, most cases had mild-to-moderate acute interstitial nephritis and mild-to-moderate acute tubular necrosis.
The predominant cause of ON in this series was EH, present in 71.5% of cases. This was mostly due to chronic pancreatitis and gastric bypass. There were two cases attributed to orlistat use, one to small bowel resection in a patient with Crohn’s disease, and lenalidomide-induced bile acid malabsorption in one patient with multiple myeloma. Ten of 15 patients with EH presented with clinical steatorrhea at diagnosis of ON. Toxic ingestion made up 13.6% and included two cases of EG intoxication and one of massive vitamin C intake. The other three (13.6%) cases did not have a clear cause for ON despite a work-up for malabsorption as well as genetic causes.
All patients were treated with intravenous fluids and calcium supplements as well as low fat and low oxalate diets. Pancreatic enzymes were started or increased as needed and orlistat and vitamin C were withdrawn. During a mean follow-up of 29 ± 67 months, 52% progressed to ESKD. Serum creatinine and interstitial fibrosis/tubular atrophy score were most predictive of progression. Of those who did not progress, there was still significant CKD and the mean eGFR at follow-up was 32 ± 19 mL/min/1.73 m2. Only one patient, who had a history of gastric bypass, had complete renal recovery.
Finally, 25 patients with oxalate deposits were identified from a New York City metro area database, for whom this was a major finding consistent with ON in 22 [3]. Seventy-six percent were hypertensive and 64% had diabetes. Mean serum creatinine at presentation was 6.31 ± 3.23 mg/dL. In the five patients for which it was measured, 24-h urine oxalate was elevated. No imaging was documented. On biopsy, aside from oxalate deposition, acute tubular injury was present in 64%, chronic interstitial nephritis in 32% and acute interstitial nephritis in 12%.
Forty percent of patients were felt to have a primarily enteric cause of hyperoxaluria. Three additional patients were ingesting large amounts of oxalate, including one who drank 10 cups a day of black tea, another who took large amounts of bitter melon tea, and a third who consumed large amounts of nuts and kale smoothies. One patient ingested antifreeze. One patient was diagnosed with likely PH. Only five cases in the series had 24-h urine oxalate available and it was elevated in all. In as many as 44% of cases the cause of the oxalate deposition was not clear.
Five patients were treated with some form of oral calcium to lower oxalate absorption and four received sevelamer. One patient with possible PH received pyridoxine. This study had short follow-up of only 3 months but the risk of ESKD was 24%. Serum creatinine at diagnosis was the primary risk factor for progression, but this study was unique in looking at oxalate density determined by percentiles of crystal density ranging between <0.4/mm2 tissue (lowest) and >1.5/mm2 (highest) as a risk factor. This was found to be of borderline significance.
In summary, these four series suggest that ON is likely not rare, especially among those with risk factors such as bowel disease. Most cases have other underlying risk factors for worsening kidney disease such as older age, diabetes and/or hypertension, and underlying CKD. Aside from oxalate deposits, tubular injury is the most common pathologic finding. Renal imaging was generally unremarkable when done, without evidence of nephrocalcinosis, though some cases had renal stones present. This appears to distinguish these causes from PH. In the Rare Kidney Stone Consortium registry, kidney stones were found in 61% and nephrocalcinosis in 37% on imaging (primarily ultrasound) of PH patients [7]. The prognosis of ON was poor in these series, with a high percentage of progression to ESKD or persistent advanced CKD.
It is also notable that in the two series of patients who were not selected for a particular underlying condition, the cause of ON could not always be determined. In fact, in the latter study up to 44% did not have a clear etiology. It is possible that PH may be underdiagnosed, or that there are other secondary causes that are not easily identified. This brings up the issue of antibiotic use.
ANTIBIOTIC USE
As mentioned above, a significant percentage of cases in the previous two series did not have a clear cause of ON. It has been suggested that antibiotic use, especially antibiotics that deplete intestinal Oxalobacter formigenes, which metabolizes oxalate, could lead to hyperoxaluria [64]. Studies have shown that depletion of gut Oxalobacter was associated with increased urinary oxalate, especially in kidney stone-forming patients [65, 66]. A recent study by Tasian et al. [66], showed that antibiotic exposure was associated with a risk of kidney stones ≥3 months after exposure (though the type of stones was not known in this study). This appeared to be independent of an effect on Oxalobacter as this was observed with antibiotics that Oxalobacter was resistant to in many cases. The effect may be related to some other alteration in the intestinal microbiome, though this study did not have information on either the microbiome or on urine chemistries in order to document what urinary changes were present. In our study, 13 of the patients (52%) had been exposed to antibiotics within a month of the biopsy with ON. This certainly does not prove causation, but further research is needed on this possible association.
DIABETES
It is notable that ∼35% of patients in these studies had documented diabetic glomerulopathy on biopsy. To some extent, this could be because the population was older (mean age 60 years), and diabetes is more common with aging. Diabetes has also been associated with increased oxalate excretion, perhaps via an increase in oxalate precursors such as glyoxylate and glyoxal that has been observed in diabetes [67]. Furthermore, diabetes is associated with dysfunction of the gastrointestinal tract, including gastroparesis and diabetes-related enteropathy [68], which would make these patients prone to volume depletion and an increase in urine supersaturation of calcium oxalate. Not all patients with hyperoxaluria develop ON and a concomitant insult such as volume depletion is likely one key factor in precipitating it.
TREATMENT
The main intervention used in most patients in case series and case reports is, as expected, intravenous or oral hydration, which would serve to lower the concentration of urinary oxalate. Oral citrate has been used to inhibit crystallization in both primary and secondary hyperoxaluria, though outcome data are limited [9, 10, 69]. The treatment of PH depends on the mutation, and pyridoxine may be useful for some with Type 1 mutation as well as liver transplantation. In November 2020, the US Food and Drug Administration approved lumasiran as the first drug treatment for PH1. Lumasiran is an RNA interference (RNAi) agent that degrades the messenger RNA for the hepatic enzyme glycolate oxidase. By preventing the conversion of glycolate to glyoxylate this agent decreases the amount of glyoxylate available to be converted to oxalate. The Illuminate-A study [70] showed that in 39 PH1 patients randomized to lumasiran versus placebo, urinary oxalate excretion decreased by 65.4% in the lumasiran group. At 6 months, 84% had 24-h urinary oxalate within 1.5 times normal versus none in the placebo group. There was no difference in eGFR in this short-term study.
The anti-epileptic agent stiripentol has been found to inhibit the lactate dehydrogenase (LDH) isoenzyme 5 that converts glyoxylate to oxalate. A recent study [71] found that stiripental protected rat kidneys from oxalate deposition in models of dietary-induced ON and EG poisoning. Children who took this medicine as an anti-epileptic were found to have lower urine oxalate compared with controls. One patient with PH1 was given stiripental with significant decrease in urine oxalate. An RNAi of LDH was developed (Nedosiran) and was used in a single patient with ESKD with significant decrease in blood oxalate levels [72]. A study of this agent in patients with both PH1 and PH2 is ongoing (NCT03847909).
For acute ingestions, it is crucial to identify the source of oxalate or precursor and remove it from the patient’s diet. EG ingestion is treated with ethanol or fomepizole to competitively inhibit the metabolism of EG by alcohol dehydrogenase (Figure 3). For EH, or if the cause of the ON is not clear, a number of treatments have been utilized. Non-specific treatments include lowering oxalate intake as well as increasing oral calcium intake via diet or calcium supplements as a means of binding oxalate in the bowel. Studies have shown these interventions to have a variable effect on urinary oxalate [10, 12]. It is important to maintain a high fluid intake to avoid an increase in calcium oxalate supersaturation with an increase in calcium intake. Lowering fat intake has been used as well. Sevelamer hydrochloride, a phosphate binder that also binds fatty acids, was used in a single small trial of patients with EH and caused a non-significant decrease in urinary oxalate [73]. Cholestyramine, a bile acid binder, has been studied with the understanding that it may decrease bile acid effect on colon permeability and also may directly bind oxalate. There have been conflicting results with this in EH, with some studies showing decreased, increased or no difference in oxalate absorption [8, 9]. Manipulating the microbiome with probiotics or specifically with oral O. formigenes has been studied. Two randomized controlled trials of oral O. formigenes in PH patients did not lower urine oxalate [74, 75]. Other probiotic agents have also thus far not proven very effective in EH [64, 76].
Despite the lack of benefit so far in studies of probiotic and Oxalobacter trials, pilot studies of an orally administered, non-absorbed, oxalate-degrading enzyme have shown promise for EH. Reloxaliase (formerly known as ALLN-177) is a recombinant oxalate decarboxylase. It lowered urine oxalate in two patients with CKD Stage 3b and plasma oxalate in seven patients with CKD Stage 5 (in both groups by ∼30%), in a recent open-label, single-arm trial [77]. A randomized trial of reloxaliase in EH is now recruiting and is estimated to be completed in November 2023 (NCT03847090).
It is of course important to identify specific causes of EH. For example, chronic pancreatitis is treated with pancreatic enzymes. Use of orlistat, which is available over the counter under the trade name Alli, can be an easily reversible cause of EH. It is more difficult to treat EH related to bariatric surgery. Jejunoileal bypass has been reversed successfully in number of cases of hyperoxaluria [78], and there has been a recent report of reversal of Roux-en-Y for ON with resolution of hyperoxaluria and stabilization of renal dysfunction [79].
Finally, it has become clear that there is downstream inflammation in hyperoxaluric patients that perpetuates chronic interstitial damage and progressive kidney disease. Pro-inflammatory molecules are released from tubular cells damaged by crystals that recruit immune cells to the renal interstitium and activate local macrophages and dendritic cells [80]. Animal models of ON have suggested that suppressing mediators of inflammation including cytokines like tumor necrosis factor, as well as components of cytokine activators in macrophages and dendritic cells called inflammasomes, can be beneficial in limiting progressive damage. One inflammasome component called NLRP3 appears to be crucial and agents are available that can be used in humans and are under investigation for a number of different illnesses [80, 81]. See Table 4 for summary of possible treatments for oxalate nephropathy.
Table 4.
Treatments discussed in this review
| Clinical data | Mechanism | Notes | Current trials |
|---|---|---|---|
| PH | |||
| High fluid intake | Lowers urinary calcium oxalate supersaturation | Prompt initiation of high fluid intake with urinary alkalinization may slow progression [82] | – |
| Pyridoxine | Increase function of AGT | Useful in some PH1 [9] | – |
| Citrate | Inhibit calcium oxalate crystallization | May stabilize or improve renal function in some cases [69] | – |
| Liver transplant | Restore oxalate metabolism primarily in PH1 [9] | PH2 may not necessarily respond and no data in PH3 [9] | – |
| Lumasiran | RNAi of glycolate oxidase enzyme [70] | FDA approved—no long-term data on outcomes. | Single-arm study in advanced kidney disease ongoing—NCT04152200 |
| Nedosiran | RNAi of LDH enzyme [72] | Trial ongoing in PH1 and PH2 | NCT03847909 |
| Secondary hyperoxalurias | – | ||
| High fluid intake | Lowers urinary calcium oxalate supersaturation [10] | – | |
| EH | – | ||
| Increased calcium and low fat intake | Use calcium to bind oxalate in gut | Generally can lower urine oxalate in short term studies [10, 12] | – |
| Lower oxalate intake | decrease gut oxalate | Variable results [10, 12] | – |
| Citrate | Inhibit calcium oxalate crystallization | Only data is in stone patients with low urine citrate [10, 12] | – |
| Sevelamer | Fatty acid binding | Non-significant decrease in urine oxalate in single trial [76] | – |
| Cholestyramine | Decrease bile acids | Conflicting results [10, 12] | – |
| Microbiome manipulation | Increase oxalate degradation in gut | Have generally not been effective [64] | – |
| Reversal of bariatric surgery | Reverse malabsorption | Single case report with Roux-en-Y [79] | |
| Reloxilase (ALLN-177) | Recombinant oxalate decarboxylase | Limited data—clinical trial ongoing [77] | NCT03847090 |
| Cytokine/inflammasome inhibition | Block downstream inflammation leading to fibrosis |
Animal studies only so far [80, 81] Potentially also could be useful in PH and ingestions |
– |
| Ingestions | – | ||
| Identify and remove offending agent from diet | – | ||
| EG | – | ||
| Ethanol | Competitively inhibits metabolism with alcohol dehydrogenase | Reduces formation of toxic metabolites [22] | – |
| Fomepizole | Competitively inhibits metabolism with alcohol dehydrogenase | Reduces formation of toxic metabolites [22] | – |
FDA, Food and Drug Administration.
TRANSPLANT
There is a risk of both de novo and recurrent ON in renal transplants. One should consider the possibility of ON in any patient with unexplained graft dysfunction, especially if there is a risk factor for EH or a history of ON [1, 9, 10].
SUMMARY AND AREAS FOR FURTHER INVESTIGATION
In conclusion, ON is a serious condition that may be less uncommon than previously recognized. There is a high risk of progression to renal failure. It is crucial to have a high index of suspicion in those with unexplained renal dysfunction as it is likely that early identification and treatment may afford a better outcome, as renal function at presentation appears to be a strong marker for prognosis.
Much remains to be better defined in this area. Why some patients with hyperoxaluria develop only ON, while others only nephrolithiasis and yet others both is not known. Whether one can use plasma oxalate levels (in patients with renal dysfunction) to non-invasively distinguish between PH, EH and non-hyperoxaluric kidney disease was the subject of a recent study [11] and needs to be further evaluated. The possible impact of antibiotic use as a cause or contributor to ON, via its effect on the intestinal biome, requires further study. The role of the SCL26 oxalate transporter family (present in both the gut and the kidney), in oxalate physiology as well as in the pathophysiology of hyperoxaluria needs further elucidation. These transporters may also potentially be targets in managing hyperoxaluric states. Therapies are being developed for both PH and EH, including the use of RNAi to target affected enzymes in PH, and recombinant oxalate decarboxylase in EH. The role of inflammation in ON, including by targeting the NLRP3-associated inflammasome, as well as inflammatory cytokines, is under investigation. This is an actively evolving field.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare.
Contributor Information
Jordan L Rosenstock, Division of Nephrology, Lenox Hill Hospital, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell, New York, NY, USA.
Tatyana M J Joab, Division of Nephrology, Lenox Hill Hospital, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell, New York, NY, USA.
Maria V DeVita, Division of Nephrology, Lenox Hill Hospital, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell, New York, NY, USA.
Yihe Yang, Department of Pathology, North Shore University Hospital and Long Island Jewish Medical Center, Donald and Barbara Zucker School of Medicine at Hostra/Northwell, New York, USA.
Purva D Sharma, Division of Kidney Diseases and Hypertension, North Shore University Hospital and Long Island Jewish Medical Center, Donald and Barbara Zucker School of Medicine at Hostra/Northwell, New York, NY, USA.
Vanesa Bijol, Department of Pathology, North Shore University Hospital and Long Island Jewish Medical Center, Donald and Barbara Zucker School of Medicine at Hostra/Northwell, New York, USA.
REFERENCES
- 1. Lumlertgul N, Siribamrungwong M, Jaber BL et al. Secondary oxalate nephropathy: a systematic review. Kidney Int Rep 2018; 3: 1363–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buysschaert B, Aydin S, Morelle J et al. Etiologies, clinical features, and outcome of oxalate nephropathy. Kidney Int Rep 2020; 5: 1503–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang Y, Sharma PD, Nair V et al. Kidney oxalate crystal deposition in adult patients: a relatively common finding. Clin Nephrol 2020; 93: 243–250 [DOI] [PubMed] [Google Scholar]
- 4. Nasr SH, D’Agati VD, Said SM et al. Oxalate nephropathy complicating Roux-en-Y gastric bypass: An underrecognized cause of irreversible renal failure. Clin J Am Soc Nephrol 2008; 3: 1676–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cartery C, Faguer S, Karras A et al. Oxalate nephropathy associated with chronic pancreatitis. Clin J Am Soc Nephrol 2011; 6: 1895–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Markowitz GS, Nasr SH, Klein P et al. Renal failure due to acute nephrocalcinosis following oral sodium phosphate bowel cleansing. Hum Pathol 2004; 35: 675–684 [DOI] [PubMed] [Google Scholar]
- 7. Tang X, Bergstralh EJ, Mehta RA et al. Nephrocalcinosis is a risk factor for kidney failure in primary hyperoxaluria. Kidney Int 2015; 87: 623–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Robijn S, Hoppe B, Vervaet BA et al. Hyperoxaluria: a gut-kidney axis. Kidney Int 2011; 80: 1146–1158 [DOI] [PubMed] [Google Scholar]
- 9. Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med 2013; 369: 649–658 [DOI] [PubMed] [Google Scholar]
- 10. Nazzal L, Puri S, Goldfarb DS. Enteric hyperoxaluria: an important cause of end-stage kidney disease. Nephrol Dial Transplant 2016; 31: 375–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perinpam M, Enders F, Mara K et al. Plasma oxalate in relation to eGFR in patients with primary hyperoxaluria, enteric hyperoxaluria and urinary stone disease. Clin Biochem 2017; 50: 1014–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Asplin JR. The management of patients with enteric hyperoxaluria. Urolithiasis 2016; 44: 33–43 [DOI] [PubMed] [Google Scholar]
- 13. Dobbins JW, Binder HJ. Effect of bile salts and fatty acids on the colonic absorption of oxalate. Gastroenterology 1976; 70: 1096–1100 [PubMed] [Google Scholar]
- 14. Hylander E, Jarnum S, Jensen HJ et al. Enteric hyperoxaluria: dependence on small intestinal resection, colectomy, and steatorrhoea in chronic inflammatory bowel disease. Scand J Gastroenterol 1978; 13: 577–588 [DOI] [PubMed] [Google Scholar]
- 15. Dobbins JW, Binder HJ. Importance of the colon in enteric hyperoxaluria. N Engl J Med 1977; 296: 298–301 [DOI] [PubMed] [Google Scholar]
- 16. Earnest DL, Johnson G, Williams HE et al. Hyperoxaluria in patients with ileal resection: an abnormality in dietary oxalate absorption. Gastroenterology 1974; 66: 1114–1122 [PubMed] [Google Scholar]
- 17. Knauf F, Ko N, Jiang Z et al. Net intestinal transport of oxalate reflects passive absorption and SLC26A6-mediated secretion. J Am Soc Nephrol 2011; 22: 2247–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Requarth JA, Burchard KW, Colacchio TA et al. Long-term morbidity following jejunoileal bypass. Arch Surg 1995; 130: 318–325 [DOI] [PubMed] [Google Scholar]
- 19. Mole DR, Tomson CRV, Mortensen N et al. Renal complications of jejuno-ileal bypass for obesity. QJM 2001; 94: 69–77 [DOI] [PubMed] [Google Scholar]
- 20. Mandell I, Krauss E, Millan JC. Oxalate-induced acute renal failure in Crohn’s disease. Am J Med 1980; 69: 628–632 [DOI] [PubMed] [Google Scholar]
- 21. Singh A, Sarkar SR, Gaber LW et al. Acute oxalate nephropathy associated with orlistat, a gastrointestinal lipase inhibitor. Am J Kidney Dis 2007; 49: 153–157 [DOI] [PubMed] [Google Scholar]
- 22. Kruse JA. Methanol and ethylene glycol intoxication. Crit Care Clin 2012; 28: 661–711 [DOI] [PubMed] [Google Scholar]
- 23. Fontana F, Cazzato S, Giovanella S et al. Oxalate nephropathy caused by excessive vitamin C administration in 2 patients with COVID-19. Kidney Int Rep 2020; 5: 1815–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mashour S, Turner JF, Merrell R. Acute renal failure, oxalosis, and vitamin C supplementation: a case report and review of the literature. Chest 2000; 118: 561–563 [DOI] [PubMed] [Google Scholar]
- 25. Gurm H, Sheta M, Nivera N et al. Vitamin C-induced oxalate nephropathy: a case report. J Community Hosp Intern Med Perspect 2012; 2: 1–3. 10.3402/jchimp.v2i2.17718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lamarche J, Nair R, Peguero A et al. Vitamin C-induced oxalate nephropathy. Int J Nephrol 2011; 2011: 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lawton JM, Conway LT, Crosson JT et al. Acute oxalate nephropathy after massive ascorbic acid administration. Arch Intern Med 1985; 145: 950–951 [PubMed] [Google Scholar]
- 28. Niticharoenpong K, Chalermsanyakorn P, Panvichian R et al. Acute deterioration of renal function induced by star fruit ingestion in a patient with chronic kidney disease. J Nephrol 2006; 19: 682–686 [PubMed] [Google Scholar]
- 29. Abeysekera RA, Wijetunge S, Nanayakkara N et al. Star fruit toxicity: a cause of both acute kidney injury and chronic kidney disease: a report of two cases. BMC Res Notes 2015; 8: 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barman AK, Goel R, Sharma M et al. Acute kidney injury associated with ingestion of star fruit: Acute oxalate nephropathy. Indian J Nephrol 2016; 26: 446–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen CL, Fang HC, Chou KJ et al. Acute oxalate nephropathy after ingestion of star fruit. Am J Kidney Dis 2001; 37: 418–422 [DOI] [PubMed] [Google Scholar]
- 32. Neto MM, Silva GEB, Costa RS et al. Star fruit: Simultaneous neurotoxic and nephrotoxic effects in people with previously normal renal function. NDT Plus 2009; 2: 485–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamamoto R, Morita S, Aoki H et al. Acute renal failure and metabolic acidosis due to oxalic acid intoxication: a case report. Tokai J Exp Clin Med 2011; 36: 116–119 [PubMed] [Google Scholar]
- 34. Dassanayake U, Gnanathasan CA. Acute renal failure following oxalic acid poisoning: a case report. J Occup Med Toxicol 2012; 7: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McHugh GJ, Graber ML, Freebairn RC. Fatal vitamin C-associated acute renal failure. Anaesth Intensive Care 2008; 36: 585–588 [DOI] [PubMed] [Google Scholar]
- 36. Kim MJ, Lee JS, Kim SW. Acute kidney injury associated with nafronyl oxalate overdose. Clin Exp Nephrol 2013; 17: 437–438 [DOI] [PubMed] [Google Scholar]
- 37. Konta T, Yamaoka M, Tanida H et al. Acute renal failure due to oxalate ingestion. Intern Med 1998; 37: 437–438 [DOI] [PubMed] [Google Scholar]
- 38. Bernardino M, Parmar MS. Oxalate nephropathy from cashew nut intake. CMAJ 2017; 189: E405–E408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Park H, Eom M, Won Yang J et al. Peanut-induced acute oxalate nephropathy with acute kidney injury. Kidney Res Clin Pract 2014; 33: 109–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sasaki M, Murakami M, Matsuo K et al. Oxalate nephropathy with a granulomatous lesion due to excessive intake of peanuts. Clin Exp Nephrol 2008; 12: 305–308 [DOI] [PubMed] [Google Scholar]
- 41. Haaskjold YL, Drotningsvik A, Leh S et al. Renal failure due to excessive intake of almonds in the absence of oxalobacter formigenes. Am J Med 2015; 128: e29–e30 [DOI] [PubMed] [Google Scholar]
- 42. Bakul G, Unni VN, Seethaleksmy NV et al. Acute oxalate nephropathy due to “Averrhoa bilimbi” fruit juice ingestion. Indian J Nephrol 2013; 23: 297–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nair S, George J, Kumar S et al. Acute oxalate nephropathy following ingestion of Averrhoa bilimbi juice. Case Rep Nephrol 2014; 2014: 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Albersmeyer M, Hilge R, Schröttle A et al. Acute kidney injury after ingestion of rhubarb: secondary oxalate nephropathy in a patient with type 1 diabetes. BMC Nephrol 2012; 13: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Syed F, Mena-Gutierrez A, Ghaffar U. A case of iced-tea nephropathy. N Engl J Med 2015; 372: 1377–1378 [DOI] [PubMed] [Google Scholar]
- 46. Nasr SH, Kashtanova Y, Levchuk V et al. Secondary oxalosis due to excess vitamin C intake. Kidney Int 2006; 70: 1672. [DOI] [PubMed] [Google Scholar]
- 47. Getting JE, Gregoire JR, Phul A et al. Oxalate nephropathy due to “juicing”: case report and review. Am J Med 2013; 126: 768–772 [DOI] [PubMed] [Google Scholar]
- 48. Kikuchi Y, Seta K, Ogawa Y et al. Chaga mushroom-induced oxalate nephropathy. Clin Nephrol 2014; 81: 440–444 [DOI] [PubMed] [Google Scholar]
- 49. Neild GH, Poulin LD, Riopel J et al. Acute oxalate nephropathy induced by oral high-dose vitamin C alternative treatment. Clin Kidney J 2014; 7: 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sunkara V, Pelkowski TD, Dreyfus D et al. Acute kidney disease due to excessive vitamin C ingestion and remote Roux-en-y gastric bypass surgery superimposed on CKD. Am J Kidney Dis 2015; 66: 721–724 [DOI] [PubMed] [Google Scholar]
- 51. Yaich S, Chaabouni Y, Charfeddine K et al. Secondary oxalosis due to excess vitamin C intake: a cause of graft loss in a renal transplant recipient. Saudi J Kidney Dis Transpl 2014; 25: 113–116 [DOI] [PubMed] [Google Scholar]
- 52. Colliou E, Mari A, Delas A et al. Oxalate nephropathy following vitamin C intake within intensive care unit. Clin Nephrol 2017; 88: 354–358 [DOI] [PubMed] [Google Scholar]
- 53. Buehner M, Pamplin J, Studer L et al. Oxalate nephropathy after continuous infusion of high-dose vitamin C as an adjunct to burn resuscitation. J Burn Care Res 2016; 37: 446–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cossey LN, Rahim F, Larsen CP. Oxalate nephropathy and intravenous vitamin C. Am J Kidney Dis 2013; 61: 1032–1035 [DOI] [PubMed] [Google Scholar]
- 55. Makkapati S, D’Agati VD, Balsam L. “Green smoothie cleanse” causing acute oxalate nephropathy. Am J Kidney Dis 2018; 71: 281–286 [DOI] [PubMed] [Google Scholar]
- 56. Lien YHH. Juicing is not all juicy. Am J Med 2013; 126: 755–756 [DOI] [PubMed] [Google Scholar]
- 57. Neild GH, Poulin LD, Riopel J et al. Acute oxalate nephropathy induced by oral high-dose vitamin C alternative treatment. Clin Kidney J 2014; 7: 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nieto MM, da Costa JAC, Garcia-Cairasco N, Netto JC, Nakagawa B, Dantas M. Intoxication by star fruit in 32 uraemic patients : treatment and outcome. Nephrol Dial Transplant 2003; 18: 120–125 [DOI] [PubMed] [Google Scholar]
- 59. Noonan SC, Savage GP. Oxalate content of foods and its effect on humans. Asia Pac J Clin Nutr 1999; 8: 64–74 [PubMed] [Google Scholar]
- 60. Massey LK. Food oxalate: factors affecting measurement, biological variation, and bioavailability. J Am Diet Assoc 2007; 107: 1191–1194 [DOI] [PubMed] [Google Scholar]
- 61. Tsai J, Huang J, Wu TT et al. Comparison of oxalate content in foods and beverages in Taiwan. J Taiwan Urol Assoc 2005; 16: 93–99 [Google Scholar]
- 62. Chai W, Liebman M. Oxalate content of legumes, nuts, and grain-based flours. J Food Compos Anal 2005; 18: 723–729 [Google Scholar]
- 63. Attalla K, De S, Monga M. Oxalate content of food: a tangled web. Urology 2014; 84: 555–560 [DOI] [PubMed] [Google Scholar]
- 64. Liu M, Nazzal L. Enteric hyperoxaluria: role of microbiota and antibiotics. Curr Opin Nephrol Hypertens 2019; 28: 352–359 [DOI] [PubMed] [Google Scholar]
- 65. Kumar R, Ghoshal UC, Singh G et al. Infrequency of colonization with Oxalobacter formigenes in inflammatory bowel disease: possible role in renal stone formation. J Gastroenterol Hepatol 2004; 19: 1403–1409 [DOI] [PubMed] [Google Scholar]
- 66. Tasian GE, Jemielita T, Goldfarb DS et al. Oral antibiotic exposure and kidney stone disease. J Am Soc Nephrol 2018; 29: 1731–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Efe O, Verma A, Waikar SS. Urinary oxalate as a potential mediator of kidney disease in diabetes mellitus and obesity. Curr Opin Nephrol Hypertens 2019; 28: 316–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Krishnan B, Babu S, Walker J et al. Gastrointestinal complications of diabetes mellitus. World J Diabetes 2013; 4: 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Leumann E, Hoppe B, Neuhaus T et al. Efficacy of oral citrate administration in primary hyperoxaluria. Nephrol Dial Transpl 1995; 10: 14–16 [DOI] [PubMed] [Google Scholar]
- 70. Garrelfs SF, Frishberg Y, Hulton SA et al. Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N Engl J Med 2021; 384: 1216–1226 [DOI] [PubMed] [Google Scholar]
- 71. Le Dudal M, Huguet L, Perez J et al. Stiripentol protects against calcium oxalate nephrolithiasis and ethylene glycol poisoning. J Clin Invest 2019; 129: 2571–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shee K. Nedosiran dramatically reduces serum oxalate in dialysis-dependent primary hyperoxaluria. Urology 2021. (in press) [DOI] [PubMed] [Google Scholar]
- 73. Lieske JC, Regnier C, Dillon JJ. Use of sevelamer hydrochloride as an oxalate binder. J Urol 2008; 179: 1407–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Milliner D, Hoppe B, Groothoff J. A randomised phase II/III study to evaluate the efficacy and safety of orally administered Oxalobacter formigenes to treat primary hyperoxaluria. Urolithiasis 2018; 46: 313–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hoppe B, Groothoff JW, Hulton SA et al. Efficacy and safety of Oxalobacter formigenes to reduce urinary oxalate in primary hyperoxaluria. Nephrol Dial Transplant 2011; 26: 3609–3615 [DOI] [PubMed] [Google Scholar]
- 76. Lieske JC, Tremaine WJ, De Simone C et al. Diet, but not oral probiotics, effectively reduces urinary oxalate excretion and calcium oxalate supersaturation. Kidney Int 2010; 78: 1178–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pfau A, Grujic D, Keddis MT et al. Pilot study of reloxaliase in patients with severe enteric hyperoxaluria and hyperoxalemia. Nephrol Dial Transplant 2021; 36: 945–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dhar NB, Grundfest S, Jones JS et al. Jejunoileal bypass reversal: effect on renal function, metabolic parameters and stone formation. J Urol 2005; 174: 1844–1846 [DOI] [PubMed] [Google Scholar]
- 79. Agrawal V, Wilfong JB, Rich CE et al. Reversal of gastric bypass resolves hyperoxaluria and improves oxalate nephropathy secondary to Roux-en-Y gastric bypass. Case Rep Nephrol Dial 2016; 6: 114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Martin-Higueras C, Ludwig-Portugall I, Hoppe B et al. Targeting kidney inflammation as a new therapy for primary hyperoxaluria? Nephrol Dial Transplant 2019; 34: 908–914 [DOI] [PubMed] [Google Scholar]
- 81. El-Sharkawy L, Brough D, Freeman S. Inhibiting the NLRP3 inflammasome. Multidiscip Digit Publ Inst 2020; 25: 5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fargue S, Harambat J, Gagnadoux MF et al. Effect of conservative treatment on the renal outcome of children with primary hyperoxaluria type 1. Kidney Int 2009; 76: 767–773 [DOI] [PubMed] [Google Scholar]