

Abstract
The use of external controls in genome-wide association study (GWAS) can significantly increase the size and diversity of the control sample, enabling high-resolution ancestry matching and enhancing the power to detect association signals. However, the aggregation of controls from multiple sources is challenging due to batch effects, difficulty in identifying genotyping errors and the use of different genotyping platforms. These obstacles have impeded the use of external controls in GWAS and can lead to spurious results if not carefully addressed. We propose a unified data harmonization pipeline that includes an iterative approach to quality control and imputation, implemented before and after merging cohorts and arrays. We apply this harmonization pipeline to aggregate 27 517 European control samples from 16 collections within dbGaP. We leverage these harmonized controls to conduct a GWAS of Crohn’s disease. We demonstrate a boost in power over using the cohort samples alone, and that our procedure results in summary statistics free of any significant batch effects. This harmonization pipeline for aggregating genotype data from multiple sources can also serve other applications where individual level genotypes, rather than summary statistics, are required.
Introduction
Genome-wide association studies (GWAS) have been successful in identifying genetic loci that confer some risk to disease (1–6). A key factor that determines the ability of GWAS to detect disease-associated variants is sample size. Leveraging external controls that have already been genotyped and shared publicly can increase power for discovery while allowing resources to be focused on collecting and genotyping only cases (7). Integrating external control samples can also supplement existing controls and increase the number of ancestrally matched controls in a study.
Despite these clear advantages, external control resources have not been widely adopted. One reason is the significant administrative and technical barriers to obtaining permission and then acquiring multiple different publicly available data sets (8,9). Furthermore, it is critical to ensure that any allele frequency differences between controls and cases are indeed attributable to the disease or trait being studied, and not due to systematic biases caused by insufficient ancestry matching, technical artifacts or batch effects (10).
The use of external controls typically requires merging genotype data from multiple sources in order to maximize control sample size and provide a large pool of controls from which to match the ancestry of the cases. In this process, many factors such as genotyping error, batch effects, and the use of different genotyping platforms can yield spurious correlations between cases and controls (11). As such, it is crucial to conduct careful quality control (QC) and data harmonization that is targeted towards these different sources of error when merging external controls. Even when genotyping data are derived from a single cohort, its use in GWAS first requires some analyses to ensure the data are of sufficient quality to be employed in association testing. This QC usually involves a series of analytic filters aimed at removing poor quality samples and problematic single nucleotide polymorphisms (SNPs) (12).
To this aim, we have developed a data harmonization pipeline to reliably leverage external genotyped controls, aggregated from multiple sources, to boost power in GWAS without introducing spurious associations. The design of the pipeline addresses two key issues in merging such heterogeneous data—one is errors driven by batch effects, and the other is spurious signals introduced by imputation. Our pipeline iterates through a series of QC filters and imputation to examine samples at the levels of cohort and genotyping platform. We demonstrate the utility of our harmonization pipeline by aggregating 27 517 European control samples from 16 data collections within dbGaP (13,14), and use these as controls in a GWAS of Crohn’s disease. Our work demonstrates the plausibility of harmonizing genotyping data from multiple sources, and enables external controls to be reliably integrated in GWAS.
Several methods were proposed to remove the technical batch effects in using external controls for GWAS. Lee et al. (15) developed iECAT, a method for case-control analysis that corrects batch effects by comparing the odds ratio estimates using internal controls versus using the combination of internal and external controls. Li and Lee (16) further generalized iECAT to allow for covariate adjustment. Hendricks et al. (17) proposed ProxECAT, which does not need internal controls and uses allele counts of non-functional variants as a proxy to adjust the differences between studies. Other related developments include Derkach et al. (18), Hu et al. (19) and Chen and Lin (20). These methods are designed for next generation sequencing data, but we consider in this paper the use of data from SNP arrays. Since the causes of batch effects are different for sequencing-based and array-based genotype data, most of the above methods are not directly applicable. Furthermore, the aforementioned methods assume that there is no batch effect within the external control, so that the focus is only adjusting the batch effects between case and control. However, our aim is to aggregate external controls from multiple sources, where severe batch effects exist within the control sample.
To merge array-based genotype data, a standard QC procedure applies a series of filters on samples and SNPs and then imputes data on a common reference panel. However, this is insufficient to control false positives when we aggregate multiple control samples from different genotype platforms. NSG Network et al. (21) recognized this issue in aggregating 31 collections of ischaemic stroke studies and had to modify the standard QC procedure (see their Supplementary Material). While they aggregated controls from different arrays, all those arrays belong to the Illumni platforms. The 16 collections we aim to aggregate come from HumanHap, Affymetrix and Axiom platforms. Neither a standard QC procedure nor the refined one in NSG Network et al. (21) works satisfactorily. In contrast, our proposed pipeline can harmonize genotype data from completely different platforms.
The application of our data harmonization pipeline is not limited to GWAS, but may be useful for many other analysis methods that require individual-level genotype data rather than summary statistics. One example is the knockoff method for controlling false discovery rates (22,23) and its application to genetic association studies. This method creates surrogate genes (‘knockoff variables’) from individual-level genotypes, handling linkage disequilibrium in a principled manner (24). Another type of analysis utilizes the genetic relationship matrix to capture the pairwise relationship among individuals to compute estimates of heritability, the genetic correlation between traits, and genetic risk scores (25,26). When computing genetic risk scores for highly polygenic traits such as autism spectrum disorder, for which GWAS discoveries have been limited, this approach is more sensitive than traditional polygenic risk score approaches that instead utilize summary statistics (27). Despite the tremendous progress leveraging summary statistics, many valuable avenues of analysis require the aggregation of genotypes, thus a reliable harmonization pipeline is essential to remove batch effects. Although we demonstrate the performance of our pipeline in the context of GWAS, the proposed data harmonization method is also promising for these other applications.
Results
The data harmonization pipeline
The pipeline contains four modules: (i) within-array processing, (ii) imputation, (iii) cross-array comparison and (iv) re-imputation. The within-array processing module aims to group samples by array, cohort and ancestry, so that each group contains homogeneous samples. Within this module, multiple QC filters are applied to samples and variants, within and across cohort. Next, imputation is conducted in each homogeneous group. The post-imputation data are merged within each array, followed by a few standard QC filters. The first two modules resolve issues such as genotyping errors and missing values, however, two issues remain. First, batch effects still exist, which prohibit us from merging data across different arrays. The cross-array comparison module detects batch effects via ‘pseudo-GWAS’, where samples from one array are treated as cases and samples from each other array are treated as controls. Significant variants in this pseudo case–control comparison will be removed. Second, the imputation quality is low for some variants, possibly due to low coverage in the reference panel or high recombination rate. Such low-quality imputation can drive false association signals in GWAS. In the re-imputation module, we aim to detect such spurious signals and remove them, before re-imputing in the surrounding region to recover those QC-failed variants and improve imputation quality. A summary of the data harmonization pipeline is shown in Fig. 1. The description of each module is given in section Materials and Methods.
Figure 1.

A high-level description of the data harmonization pipeline.
Creating a resource of harmonized external controls
We aggregated genotyped data on 27 517 individuals of self-reported European-descent from 16 studies in the dbGaP repository. These cohorts had been genotyped using a plethora of technologies including various Illumina and Affymetrix arrays. A summary of external controls is in Table 1, and a detailed description of 16 cohorts is provided in Supplementary Material, Table S1.
Table 1.
Summary of controls from 16 genotyping studies, grouped by array type. For each array, the number of SNPs after harmonization includes the imputed ones. The number of SNPs in the combined data set is obtained by taking the intersection
| Array | #Cohorts | Before harmonization | After harmonization | ||
|---|---|---|---|---|---|
| #Samples | #SNPs | #Samples | #SNPs | ||
| Affy6 | 3 | 4504 | 907 K | 2936 | 6.18 M |
| Axiom | 1 | 10 000 | 636 K | 9080 | 5.73 M |
| Human300 | 1 | 219 | 300 K | 197 | 6.42 M |
| Human550 | 5 | 5559 | 550 K | 4449 | 6.33 M |
| Human610 | 4 | 4672 | 610 K | 3604 | 6.19 M |
| Human660 | 2 | 2563 | 660 K | 2244 | 6.25 M |
| Combined | 16 | 27 517 | – | 22 510 | 5.52 M |
We applied the harmonization pipeline. Module 1 removed 5007 samples, where 1570 were filtered due to removal of non-European samples (the majority of non-European samples are from the GERA cohort, while other cohorts contain very few non-European individuals), 579 due to high sample missing rate, and 2858 because of family relatedness or abnormal inbreeding coefficients. A total of 22 510 samples were retained. The QC steps in Modules 2–4 only removed variants but not samples. In addition to standard QC filters (missing rate, minor allele frequencies and Hardy–Weinberg Equilibrium) the cross-array pseudo-GWAS removes around 500 variants on average from each genotyping platform. We merged across the different platforms, retaining any SNPs with missing rate < 1%, to obtain the final harmonized dataset of 5 524 462 variants.
Figure 2 shows the projection of the harmonized controls onto the first two PC’s of the 1000 Genomes (1KG) data and onto the first two PCs of its European subset. The left panel shows that the majority of harmonized controls are indeed of European descent, and the right panel shows that they represent a range of European ancestry, including British, Italian, Northern European and Spanish, and are distinct from Finland. The set of harmonized public controls provide a much richer resource of European genotypes as it is approximately 10 times larger than the European subset of 1KG and represents a more continuous sample along the cline of European ancestry. While this particular dataset was developed in the first instance as a European ancestry control resource, due to the availability of larger sample numbers, the same pipeline may be applied to studies of other ancestries to generate harmonized controls for more broad collection of genome-wide association studies. A list of SNPs that were removed in the harmonization pipeline, along with details of the step in which each was filtered out, is available in our GitHub repository (https://github.com/mikkoch/unicorn-qc).
Figure 2.

PC analysis representation of the ancestry distribution of the harmonized public controls. Left: projection of harmonized controls onto the PC space of 1000 Genomes (1KG). Right: projection onto the PC space of 1KG European subset.
Performance of GWAS using the external control resource
To assess the quality and utility of the harmonized public control data set, we conducted a comparison of GWAS using the Crohn’s disease data of the CHOP study (28), obtained from the IBD Genetics Consortium. The case-control analysis was implemented on the software Hail. We computed the first 20 PC’s of the LD-pruned data matrix (i.e. pooling case and control samples and only retaining SNPs with MAF > 0.05, Hardy–Weinberg P-value > 1e-4, and r2 < 0.2). For each SNP, we fit a logistic regression with these 20 PC’s as covariates to calculate the P-value.
The IBD Crohn’s disease study collected 1589 cases and 5950 internal controls. We examined P-values calculated from comparing the IBD cases to (i) the harmonized controls with those obtained from using (ii) the IBD study internal controls, as well as (iii) P-values from the IBD consortium’s meta-analysis, which included 5956 case subjects and 14 927 control subjects (28). The meta-analysis results serve as our best picture of the truth. We applied the same harmonization pipeline to the Crohn’s disease data, but Module 3 (the cross-array comparison) is not required as all of the IBD study data is typed on a single chip (see section Materials and Methods).
We consider two performance characteristics: (a) a boost in power for true signals and (b) the elimination of spurious signals. We compare the association results generated from the harmonized controls and those obtained from using the internal controls in the Manhattan plots of Fig. 3 and see that in general they are highly concordant across the whole genome. The QQ plot in the bottom right panel shows that there is very little evidence of inflation in either version of the GWAS (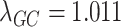 using harmonized controls and
using harmonized controls and 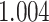 using internal controls). The QQ plot also shows the increase in significance of the most highly associated SNPs when using the harmonized controls compared to the internal controls. The bottom left panels show a zoomed-in view of two regions of genome-wide significant signal in the meta-analysis (on Chromosomes 3 and 5) where, even at this finer scale, the concordance is still very good. Furthermore, the minimum P-value at these loci is lower when using the harmonized controls for GWAS instead of the internal controls. These loci provide examples of regions where there is a boost in power to detect true signals of association when using the harmonized controls.
using internal controls). The QQ plot also shows the increase in significance of the most highly associated SNPs when using the harmonized controls compared to the internal controls. The bottom left panels show a zoomed-in view of two regions of genome-wide significant signal in the meta-analysis (on Chromosomes 3 and 5) where, even at this finer scale, the concordance is still very good. Furthermore, the minimum P-value at these loci is lower when using the harmonized controls for GWAS instead of the internal controls. These loci provide examples of regions where there is a boost in power to detect true signals of association when using the harmonized controls.
Figure 3.

Comparison of GWAS for the Crohn’s disease study using harmonized public controls versus internal controls. Top: whole genome Manhattan plot. Bottom left: zoom-in to regions in individual chromosomes. Bottom right: QQ-plot of P-values (with respect to theoretical null).
Next, with the meta analysis as our best estimate of the truth, we directly compare the P-values obtained in GWAS (i) and (ii), examining separately SNPs that are significant and non-significant in meta-analysis (a P-value < 5e-8 is considered significant). For variants that are not significant in the meta analysis (left panel of Fig. 4), both GWAS determined these SNPs to be non-significantly associated with IBD as well, suggesting that the use of harmonized controls does not result in a higher false positive rate. On the right panel of Fig. 4, looking at variants that are significant in meta-analysis, the majority of these sites are more significant in the GWAS of harmonized controls than in the GWAS of the internal controls. In particular, there are a number of variants that are deemed significant using the harmonized controls but missed using the internal controls. These results shown in Figs 3 and 4 suggest that the use of harmonized controls yields no apparent/strong p-value inflation at null SNPs and boosts power at signal SNPs.
Figure 4.

Comparison of P-values from using harmonized controls with those from using internal controls. Left: SNPs with P-values 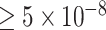 in meta-analysis (for a better visualization, we plot the absolute Z-scores). Right: SNPs with P-values
in meta-analysis (for a better visualization, we plot the absolute Z-scores). Right: SNPs with P-values 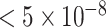 in meta-analysis (the dashed lines correspond to
in meta-analysis (the dashed lines correspond to 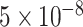 ).
).
We next show examples of the ability of the harmonization pipeline to detect and correct for spurious associations that are driven by poor quality imputation, that otherwise would not be filtered in a standard GWAS pipeline. Fig. 5 shows the Manhattan plots of the association results of analyses (i) and (iii) for two 20 Mb regions of Chromosome 1 (top row) and Chromosome 2 (bottom row). The left two plots show the results from GWAS (i) both before and after applying the re-imputation module, against the background of those from the meta analysis (GWAS (iii)) shown in gray. The red points correspond to genotyped variants with EmpRsq < 0.1. A large fraction of these red variants indeed generate spurious signals in the surrounding region in the first round of imputation: the P-values based on aggregated controls (before re-imputation) are small, but the meta-analysis suggests that they are not true signals. Comparing the lower and upper halves of the Manhattan plots, we can see that removal and re-imputation of these red SNPs removes the spurious peaks around those points (these peaks were the consequence of poor imputation driven by the red variants). In the right two plots, we compare P-values calculated from the harmonized controls (GWAS (i)) with P-values from internal controls (GWAS (ii)) and observe that they are highly concordant. These results suggest that the re-imputation module is effective in removing spurious signals that would otherwise appear when using the harmonized controls.
Figure 5.

Examples of spurious signals being removed in the harmonization pipeline (top: Chromosome 1; bottom: Chromosome 2). The red dots are typed variants that drive poor quality imputation, flagged by their EmpRsq values, which co-localize with batch effects. These spurious signals are corrected by re-imputation.
Finally, we compare our approach with a more conventional QC procedure. The standard QC pipeline first filters out problematic samples and SNPs, next imputes data, and then removes the SNPs with low imputation quality. We mimic the standard procedure by applying modules 1–2 of our harmonization pipeline, followed by removing the SNPs with low EmpRsq (using the same threshold as in the Filtering step of Module 4). This procedure is more refined than the standard procedure (because our Module 1 is more than merely applying QC filters on samples and SNPs), hence, its performance serves as an upper estimate of the performance of the standard QC procedure. The results are shown in Fig. 6. The aforementioned ‘standard’ procedure yields a much more severe P-value inflation, where 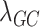 is 1.320 for this procedure and 1.011 for our procedure. It suggests that our proposed pipeline significantly improves the standard QC pipeline in terms of controlling false positives.
is 1.320 for this procedure and 1.011 for our procedure. It suggests that our proposed pipeline significantly improves the standard QC pipeline in terms of controlling false positives.
Figure 6.

Comparison of GWAS results using our harmonization pipeline versus using a standard QC pipeline. Left: QQ-plot of P-values. Right: zoom-in of the left panel.
Discussion
We have shown that it is possible to aggregate disparate genotyping data sets, even those assayed using different genotyping arrays, through a harmonization pipeline that involves iterative QC and imputation steps to control for batch effects and technical artifacts. This approach is valuable in constructing a large harmonized data set of external controls for use in GWAS, and we have shown it can deliver more powerful association tests while being robust to spurious signals driven by batch effects or insufficient ancestry matching.
The strength of our pipeline comes from the thorough and agglomerative approach to QC, that first operates within an array type for a single ancestry, and then across different arrays. The identification of problematic SNPs (through the EmpRsq metric) that are driving poor quality imputation, and the re-imputation after removing these sites is a key insight that allows the aggregation across different array types. Since multiple genotyping arrays have been used in human genetic studies, and as new arrays are developed over time, this step is essential to bring together different datasets from various sources.
While our approach permits existing external controls to be integrated into GWAS, the extent to which these resources can be useful depends upon whether the ancestry spanned by the control set sufficiently covers that of the cases to which it is being compared. That is, for a case collection from a population that is underrepresented in publicly available controls, there will be a paucity of control samples of matched ancestry. If the harmonized control set does not sufficiently capture the ancestry space spanned by the cases, then the projection of cases on to the control-generated PC space (this can be used as a diagnostic plot) will be biased (29), giving an inaccurate depiction of their ancestry relative to the axes spanned by the control set. This mismatching of controls to cases can lead to spurious associations and subsequent false findings, and highlights the unmet need for the inclusion of more ancestrally diverse samples in public genetic resources.
We have not examined the application of this method to admixed samples, which pose a challenge as their ancestry is heterogeneous across the genome. This means that clustering individuals by PCA will group those that share similar proportions of ancestry genome-wide, and will not necessarily match samples by their ancestral origins at any specific genomic site. We propose the extension of this method to admixed populations as a future research direction.
Although we have demonstrated that it is possible to leverage multiple different sources of genotyping data for their use in a unified GWAS, the extensive QC pipeline that we developed speaks to the many challenges and potential pitfalls involved in this process. We advocate for broad consent and data use agreements that enable public sharing of individual-level data to enable direct GWAS for health-related research. As public data sets grow increasingly larger, through biobanking efforts for example, there will be less need to harmonize between different sources of control samples. Until then, the careful aggregation of multiple smaller resources can be valuable in enabling well powered GWAS at no additional cost.
Materials and Methods
Description of the data harmonization pipeline
We give a high-level description of each module of the pipeline. The details can be found in Supplementary material.
Module 1: Within-array processing consists of the following main steps:
Cohort-level QC. Remove samples with high missingness rate and abnormal inbreeding coefficients, as well as variants with a high missingness rate.
Ancestry matching. Each sample is assigned to one of the five ancestries, East Asian, South Asian, African, American and European. Here, we focus on European ancestry, where samples are further split into three sub-groups—major European, Finnish and Ashkenazi Jewish. The ancestry assignment is determined by a pre-trained classifier, where the training data is 1000 Genomes data (with self-reported ancestry labels) and the classification method is a standard random forest algorithm (30) on leading PC’s.
Merging. Based on the first two steps, a stratum has been formed for each array-cohort-ancestry combination. For each stratum, we further remove variants with low minor allele frequencies and small Hardy–Weinberg Equilibrium P-values. We then merge samples of the same genotyping array and ancestry group.
Array-level QC. To remove batch effects, two rounds of pseudo-GWAS are performed iteratively within each array-ancestry stratum. In the first round, all the samples in this stratum are compared with the 1000 Genomes samples belonging to the same ancestry group. In the second round, the samples from one cohort are compared with those from each other cohort in the same array-ancestry stratum. In these pseudo-GWAS comparisons, the first 20 principal components (PCs) are included as covariates to account for population structure. Significant variants identified in either round are removed.
Module 2: Imputation. Module 1 produces a data stratum per array per ancestry. These strata are imputed separately. The motivation is that each stratum contains relatively homogeneous samples, which can improve the imputation quality compared with imputing all strata together. We use the Michigan Imputation server with 1000 Genomes data as the reference panel.
Module 3: Cross-array comparison. While Module 1 (through the array-level QC) is aimed at accounting for batch effects due to genotyping across independent studies, we include a second module to target batch effects that arise due to imputation. We expect that the quality of imputation at different regions of the genome will vary for different arrays, owing to the design of their particular backbone and to technical characteristics. This will induce considerable batch effects due to the large number of variants that are imputed in Module 2 (about 46.8 M per stratum in our experiment). This module aims to remove such batch effects by cross-array pseudo-GWAS.
Post-imputation QC. Remove variants with low minor allele frequencies, or small Hardy–Weinberg Equilibrium P-values, or low imputation info scores.
Cross-array pseudo-GWAS. We first merge samples genotyped on the same array type and take the intersection of the SNP sets on these samples. Next, a pseudo-GWAS is performed for each array, where samples on this array are treated as cases and those on each other array are used as controls. Since ancestry groups have been merged, we include 20 leading PC’s as covariates in the pseudo case–control comparison, to account for cross-ancestry heterogeneity. Significant variants are removed.
Module 4: Re-imputation. The last module deals with spurious association signals introduced by poor quality imputation, first removing the poorly typed variants that drive the imputation and then re-imputing the surrounding region.
Filtering. There are multiple metrics to assess imputation quality (31–34). We use EmpRsq, which is the squared correlation between the leave-one-out imputed dosages and the observed (i.e. typed) genotypes. Any typed variant with EmpRsq below the minimum threshold is removed.
Re-imputation. With those poorly typed variants already removed, we re-do the whole imputation procedure, where we use the same strata as in Module 1, conduct imputation in a similar way as in Module 2, and then apply similar post-imputation steps as in Module 3.
The strength of this pipeline comes from the division of QC procedures into steps that are aimed at capturing genotyping batch effects and those that are designed to identify bad sites arising from poor imputation. The order of modules in the pipeline makes this possible, and the cross-array pseudo-GWAS ensures that the allele frequencies are consistent between chips.
The re-imputation module plays a key role in removing poorly genotyped SNPs that pass earlier QC filters but show evidence of driving low-quality inference at nearby imputed sites. The first, rather strict, filter aims to ensure that the retained (typed) variants are of high quality. We then conduct a second imputation, so that the poorly imputed variants in the previous round are corrected. Subsequent QC of the re-imputed data indeed removes much less variants, confirming the benefit of the re-imputation module.
We have constructed our data harmonization pipeline as a series of modules containing multiple filters, the thresholds and parameters of which may be adjusted by the user. We have selected their values to be effective in our applications. The code to execute the pipeline is shared publicly in our GitHub repository (https://github.com/mikkoch/unicorn-qc).
Using the harmonized controls for GWAS
When our proposed pipeline is used to harmonize external control data, a similar process should be applied to the case data. We consider a common scenario where all the case samples come from one study. The four modules in the harmonization pipeline need minor modifications. In module 1, we conduct cohort-level QC using the same filters and ancestry matching using the same classifier. We then merge data to create the array-ancestry strata. The step of array-level QC is modified as follows: The previous harmonization pipeline produces a ‘blacklist’ consisting of the variants removed when aggregating public controls; we remove this ‘blacklist’ on the case data. Module 2 is the same as before. In Module 3, we implement post-imputation QC using the same filters but skip cross-array pseudo-GWAS. Module 4 is optional: Since we already removed variants in the ‘blacklist’ (this list includes those variants removed in the re-imputation of controls), the benefit of re-imputing the case data is relatively marginal, and we often skip this module.
In this process, we deal with the batch effects between case and control by (i) using the same QC filters on the case and control, (ii) imputing them to the same reference panel and (iii) removing the ‘blacklist’ of SNPs. To see the role of (iii), we consider a common scenario where the external controls include some samples genotyped on the same array as the case data. The SNPs that suffer from batch effects between case and control will be blacklisted in the cross-array comparison module of harmonizing external controls and thus removed from the case data.
Given the harmonized control and processed case, we can conduct GWAS via standard statistical methods. Note that although the above processing of case and control includes ancestry matching, it is only for QC purpose. We still need careful ancestry stratification in GWAS, for which several methods are available (7,35).
To get reliable GWAS results, we need some minimum requirements on data. First, the ancestry space of the external controls should properly ‘cover’ the ancestry of the case data. This can be checked by projecting both case samples and harmonized control samples onto the PC space of 1000 Genomes (7). Second, the external controls should include at least some samples genotyped on the same array as the case data. As we have explained, this ensures that the SNPs causing batch effects between case and control will be included in the ‘blacklist.’ If these requirements are violated, we should either expand the external controls (and re-apply the harmonization pipeline) or use a more stringent QC procedure. For example, suppose the cases come from an array not covered by the external controls. If an internal control is available, we can conduct a pseudo-GWAS between the internal control and the harmonized control to further identify those SNPs suffering batch effects between case and control.
Acknowledgements
We thank the people at Broad Institute of MIT and Harvard and for their help and support.
Conflict of Interest Statement. Benjamin M. Neale is a member of the scientific advisory board at Deep Genomics and RBNC Therapeutics, a member of the scientific advisory committee at Milken and a consultant for Camp4 Therapeutics, Merck and Biogen.
Contributor Information
Danfeng Chen, Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, 08544 NJ, USA.
Katherine Tashman, Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital, Boston, 02114 MA, USA; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA.
Duncan S Palmer, Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital, Boston, 02114 MA, USA; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA.
Benjamin Neale, Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital, Boston, 02114 MA, USA; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA; Program in Medical and Population Genetics, Broad Institute of Harvard and MIT, Cambridge, 02142 MA, USA.
Kathryn Roeder, Department of Statistics, Carnegie Mellon University, Pittsburgh, 15213 PA, USA.
Alex Bloemendal, Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital, Boston, 02114 MA, USA; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA.
Claire Churchhouse, Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital, Boston, 02114 MA, USA; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA; Program in Medical and Population Genetics, Broad Institute of Harvard and MIT, Cambridge, 02142 MA, USA.
Zheng Tracy Ke, Department of Statistics, Harvard University, Cambridge, 02138 MA, USA.
Author Contributions
D.C., A.B., C.C., K.T., D.P. designed the data harmonization pipeline. D.C. implemented the pipeline, performed all analyses and produced the figures. K.T. conducted the imputation. A.B., K.R. and B.N. supervised the project. Z.T.K. and C.C. wrote the manuscript and several authors provided valuable edits.
Funding
National Institute of Mental Health (Grants R37MH057881); National Institute of Health (Grants R01 MH101244, R37 MH107649); National Science Foundation (Grants DMS-1925845 and DMS-1943902).
References
- 1. Ripke, S., Neale, B.M., Corvin, A., Walters, J.T.R., Farh, K.-H., Holmans, P.A., Lee, P., Bulik-Sullivan, B., Collier, D.A., Huang, H.et al. (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stahl, E.A., Breen, G., Forstner, A.J., McQuillin, A., Ripke, S., Trubetskoy, V., Mattheisen, M., Wang, Y., Coleman, J.R.I., Gaspar, H.A.et al. (2019) Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet., 51, 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu, J.Z., Van Sommeren, S., Huang, H., Ng, S.C., Alberts, R., Takahashi, A., Ripke, S., Lee, J.C., Jostins, L., Shah, T.et al. (2015) Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet., 47, 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xue, A., Wu, Y., Zhu, Z., Zhang, F., Kemper, K.E., Zheng, Z., Yengo, L., Lloyd-Jones, L.R., Sidorenko, J., Wu, Y.et al. (2018) Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun., 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nelson, C.P., Goel, A., Butterworth, A.S., Kanoni, S., Webb, T.R., Marouli, E., Zeng, L., Ntalla, I., Lai, F.Y., Hopewell, J.C.et al. (2017) Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet., 49, 1385. [DOI] [PubMed] [Google Scholar]
- 6. Chang, D., Nalls, M.A., Hallgrímsdóttir, I.B., Hunkapiller, J., Van Der Brug, M., Cai, F., Kerchner, G.A., Ayalon, G., Bingol, B., Sheng, M.et al. (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet., 49, 1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bodea, C.A., Neale, B.M., Ripke, S., Barclay, M., Peyrin-Biroulet, L., Chamaillard, M., Colombel, J.-F., Cottone, M., Croft, A., DIncà, R.et al. (2016) A method to exploit the structure of genetic ancestry space to enhance case-control studies. Am. J. Hum. Genet., 98, 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaye, J., Boddington, P., deVries, J., Hawkins, N. and Melham, K. (2010) Ethical implications of the use of whole genome methods in medical research. Eur. J. Hum. Genet., 18, 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Im, H.K., Gamazon, E.R., Nicolae, D.L. and Cox, N.J. (2012) On sharing quantitative trait GWAS results in an era of multiple-omics data and the limits of genomic privacy. Am. J. Hum. Genet., 90, 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mitchell, B.D., Fornage, M., McArdle, P.F., Cheng, Y.-C., Pulit, S., Wong, Q., Dave, T., Williams, S.R., Corriveau, R., Gwinn, K.et al. (2014) Using previously genotyped controls in genome-wide association studies (GWAS): application to the stroke genetics Network (SiGN). Front. Genet., 5, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leek, J.T., Scharpf, R.B., Bravo, H.C., Simcha, D., Langmead, B., Johnson, W.E., Geman, D., Baggerly, K. and Irizarry, R.A. (2010) Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet., 11, 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Laurie, C.C., Doheny, K.F., Mirel, D.B., Pugh, E.W., Bierut, L.J., Bhangale, T., Boehm, F., Caporaso, N.E., Cornelis, M.C., Edenberg, H.J.et al. (2010) Quality control and quality assurance in genotypic data for genome-wide association studies. Genet. Epidemiol., 34, 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mailman, M.D., Feolo, M., Jin, Y., Kimura, M., Tryka, K., Bagoutdinov, R., Hao, L., Kiang, A., Paschall, J., Phan, L.et al. (2007) The NCBI dbGaP database of genotypes and phenotypes. Nat. Genet., 39, 1181–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koike, A., Nishida, N., Inoue, I., Tsuji, S. and Tokunaga, K. (2009) Genome-wide association database developed in the Japanese integrated database project. J. Hum. Genet., 54, 543–546. [DOI] [PubMed] [Google Scholar]
- 15. Lee, S., Kim, S. and Fuchsberger, C. (2017) Improving power for rare-variant tests by integrating external controls. Genet. Epidemiol., 41, 610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li, Y. and Lee, S. (2021) Novel score test to increase power in association test by integrating external controls. Genet. Epidemiol., 45, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hendricks, A.E., Billups, S.C., Pike, H.N.C., Farooqi, I.S., Zeggini, E., Santorico, S.A., Barroso, I. and Dupuis, J. (2018) ProxECAT: proxy external controls association test. A new case-control gene region association test using allele frequencies from public controls. PLoS Genet., 14, e1007591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Derkach, A., Chiang, T., Gong, J., Addis, L., Dobbins, S., Tomlinson, I., Houlston, R., Pal, D.K. and Strug, L.J. (2014) Association analysis using next-generation sequence data from publicly available control groups: the robust variance score statistic. Bioinformatics, 30, 2179–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu, Y.-J., Liao, P., Johnston, H.R., Allen, A.S. and Satten, G.A. (2016) Testing rare-variant association without calling genotypes allows for systematic differences in sequencing between cases and controls. PLoS Genet., 12, e1006040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen, S. and Lin, X. (2020) (2020) analysis in case–control sequencing association studies with different sequencing depths. Analysis in case–control sequencing association studies with different sequencing depths. Biostatistics, 21, 577–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Network, N.S.G., Pulit, S.L., McArdle, P.F., Wong, Q., Malik, R., Gwinn, K., Achterberg, S., Algra, A., Amouyel, P., Anderson, C.D.et al. (2016) Loci associated with ischaemic stroke and its subtypes (SiGN): a genome-wide association study. Lancet Neurol., 15, 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barber, R.F. and Candès, E.J. (2015) Controlling the false discovery rate via knockoffs. Ann. Stat., 43, 2055–2085. [Google Scholar]
- 23. Candès, E., Fan, Y., Janson, L. and Lv, J. (2018) Panning for gold: model-X knockoffs for high dimensional controlled variable selection. J. R. Stat. Soc. Series B Stat. Methodol., 80, 551–577. [Google Scholar]
- 24. Sesia, M., Katsevich, E., Bates, S., Candès, E. and Sabatti, C. (2020) Multi-resolution localization of causal variants across the genome. Nat. Commun., 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Los Campos, G., Gianola, D. and Allison, D.B. (2010) Predicting genetic predisposition in humans: the promise of whole-genome markers. Nat. Rev. Genet., 11, 880–886. [DOI] [PubMed] [Google Scholar]
- 26. Yang, J., Lee, S.H., Goddard, M.E. and Visscher, P.M. (2011) GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet., 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klei, L., McClain, L.L., Mahjani, B., Panayidou, K., De Rubeis, S., Gramat, A.-C.S., Karlsson, G., Lu, Y., Melhem, N., Xu, X.et al. (2020) How rare and common risk variation jointly affect liability for autism spectrum disorder. medRxiv (31 October 2020. 10.1101/2020.10.27.20220095Arxiv medrxiv;2020.10.27.20220095v1, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jostins, L., Ripke, S., Weersma, R.K., Duerr, R.H., McGovern, D.P., Hui, K.Y., Lee, J.C., Schumm, L.P., Sharma, Y., Anderson, C.A.et al. (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature, 491, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martin, A.R., Gignoux, C.R., Walters, R.K., Wojcik, G.L., Neale, B.M., Gravel, S., Daly, M.J., Bustamante, C.D. and Kenny, E.E. (2017) Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet., 100, 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Breiman, L. (2001) Random forests. Mach. Learn., 45, 5–32. [Google Scholar]
- 31. Lin, P., Hartz, S.M., Zhang, Z., Saccone, S.F., Wang, J., Tischfield, J.A., Edenberg, H.J., Kramer, J.R., Goate, A.M., Bierut, L.J.et al. (2010) A new statistic to evaluate imputation reliability. PLoS One, 5, e9697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hancock, D.B., Levy, J.L., Gaddis, N.C., Bierut, L.J., Saccone, N.L., Page, G.P. and Johnson, E.O. (2012) Assessment of genotype imputation performance using 1000 genomes in African American studies. PLoS One, 7, e50610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramnarine, S., Zhang, J., Chen, L.-S., Culverhouse, R., Duan, W., Hancock, D.B., Hartz, S.M., Johnson, E.O., Olfson, E., Schwantes-An, T.-H.et al. (2015) When does choice of accuracy measure alter imputation accuracy assessments? PLoS One, 10, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Das, S., Forer, L., Schönherr, S., Sidore, C., Locke, A.E., Kwong, A., Vrieze, S.I., Chew, E.Y., Levy, S., McGue, M.et al. (2016) Next-generation genotype imputation service and methods. Nat. Genet., 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Price, A.L., Patterson, N.J., Plenge, R.M., Weinblatt, M.E., Shadick, N.A. and Reich, D. (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet., 38, 904–909. [DOI] [PubMed] [Google Scholar]