

Abstract
The lack of knowledge about extreme conservation in genomes remains a major gap in our understanding of the evolution of gene regulation. Here, we reveal an unexpected role of extremely conserved 5’UTRs in non-canonical translational regulation that is linked to the emergence of essential developmental features in vertebrate species. Endogenous deletion of conserved elements within these 5’UTRs decreased gene expression, and extremely conserved 5’UTRs possess cis-regulatory elements that promote cell-type specific regulation of translation. We further developed in-cell mutate-and-map (icM2), a novel methodology that maps RNA structure inside cells. Using icM2, we determined that an extremely conserved 5’UTR encodes multiple alternative structures and that each single nucleotide within the conserved element maintains the balance of alternative structures important to control the dynamic range of protein expression. These results explain how extreme sequence conservation can lead to RNA-level biological functions encoded in the untranslated regions of vertebrate genomes.
Keywords: conserved non-coding sequences, translation control, mRNA structure, vertebrate genome evolution, developmental gene regulation
Introduction
One of the most fascinating findings from comparative analysis of vertebrate genomes is the existence of extreme sequence conservation in non-coding regions, at levels often greater than coding regions with perfectly invariant polypeptides1–11. These regions are undergoing strong purifying selection in humans and are not merely mutational coldspots12. However, the fundamental problem initially raised a decade ago still remains unsolved: why does such extreme conservation arise during evolution, and what are the functional roles for such sequences in the genome?
To date, efforts to understand the phenomenon of extreme conservation have heavily focused on intergenic sequences, suggesting possible roles of these elements as transcriptional enhancers13–15. However, early in vivo knockout studies paradoxically yielded viable mice lacking grossly deleterious phenotypes, raising uncertainties about the relevance and contribution of highly conserved elements to organismal development16,17. Only more recently have mice with loss of single or pairwise deletions of ultraconserved enhancer elements been shown to produce more subtle developmental phenotypes due to their impact on the transcription of neighboring genes18,19.
However, beyond its significance in transcriptional regulation, the biological meaning of extreme conservation in post-transcriptional regulation remains largely unknown. While few examples - such as the functional roles for ultraconserved regions transcribed as long non-coding RNAs or alternatively spliced poison cassette exons - have been described20–24, RNA-level mechanisms for extreme conservation have not been explored widely. The observation of extreme sequence conservation across extended stretches of 5’ untranslated regions (UTRs) suggests the presence of specialized translational cis-regulatory elements. In a paradigmatic example, the Hoxa9 5’UTR contains a ~650nt extremely conserved region that mediates non-canonical translation initiation through a structured IRES-like RNA element25. Knockout of an ~150bp functional element within this conserved region in mice results in diminished spatio-temporal Hoxa9 protein expression and a pronounced axial skeleton phenotype leading to a homeotic transformation, demonstrating how 5’UTR RNA sequences important for specialized translational regulation in the developing embryo can undergo extraordinary negative selection. We were thus inspired to ask if there could be a broader, systematic trend for extreme conservation to reveal currently unknown translational regulatory sequences, and conversely, if such regulatory sequences could help to explain the functional basis of extreme non-coding conservation in mRNAs.
Results
Hyperconserved 5’UTRs in vertebrate genomes
To address the function of extreme non-coding conservation for mRNA 5’UTRs, we used the conservation pattern of the aforementioned Hoxa9 5’UTR as our archetype in selecting a set of other 5’UTRs in the genome. The length of the extremely conserved stretch in Hoxa9 5’UTR is ~650nt; the size of the functional element within the conserved stretch is around 350nt25. We used PhastCons with a log odds score (LOD) minimum of 500, which marked such large blocks of extremely conserved sequences throughout the genome that are 100nt long, on average26. Using mouse RefSeq gene annotations, we intersected mouse 5’UTRs with the LOD≥500 PhastCons elements (representing the top 8.25% of all PhastCons elements identified in the genome), requiring at least ≥250nt overlap. This resulted in a set of 589 5’UTRs for 499 genes (Fig. 1a, Supplementary Table 1). The median nucleotide identity between mouse and human genomes in the conserved regions in the selected 5’UTRs is 92.3% (80% identity at 5th percentile). The average total length and the average number of nucleotides overlapping PhastCons elements for these 589 5’UTRs are 674nt and 389nt, respectively, and they tend to be found more frequently closer to the start codon than to the 5’ end (Extended Data Figs. 1a,b,c). For the remainder of the text, we will refer to these 589 5’UTRs as hyperconserved 5’UTRs (h5UTR) and the LOD≥500 PhastCons elements within the h5UTRs as 5’UTR hyperconserved elements (HCE).
Figure 1 |. Hyperconserved 5’UTRs in vertebrate genomes.

a, Schematic illustrating selection of hyperconserved vertebrate 5’UTRs. We begin with 60-way multiple species alignment of vertebrate genomes, its per-nucleotide PhastCons probabilities, and conserved element prediction tracks. High scoring (≥LOD 500) PhastCons elements are overlapped with RefSeq annotated mouse 5’UTRs. We define those with overlap ≥250nt to be hyperconserved (also see Supplementary Table 1).
b, Distributions of cross-tissue transcriptome-proteome correlations (GTEX consortium data across 32 human tissues) for all genes, genes with h5UTRs, or genes with size-matched non-conserved 5’UTRs. Indicated p-values are from two-sided Wilcoxon rank sum tests for cross-tissue correlation values between h5UTR genes and all genes or between h5UTR genes and size-matched non-conserved controls.
c, Scatter plot illustrating the term enrichment strategy and criteria. X-axis and y-axis plots expected and the observed number of genes for each term. Blue dashed line indicates the minimum observed/expected ratio cutoff of 3. Green line indicates expected and observed counts where two-tailed Fisher’s test p-value (pf) is estimated to have FDR=0.05. Neighbor-weighted test p-value (pfw) ≤0.05 is further used as an additional cutoff. The final set of enriched terms passing filter is colored by pf and sized by pfw.
d, Visualization of representative gene ontology terms significantly enriched for the h5UTRs according to criteria in Fig. 1c. A number of genes mapping to each term are also displayed (also see Supplementary Table 2).
We next asked if h5UTRs are more likely to be discordant in their mRNA:protein expression levels, which would suggest post-transcriptional regulation. Using the GTex consortium transcriptomics and proteomics dataset, we determined if genes with h5UTRs have a different distribution of per-gene cross-tissue correlations in mRNA versus protein levels compared to genes with similarly sized, non-conserved (defined as no overlap with LOD≥500 PhastCons elements) 5’UTRs27,28. For 181 h5UTR genes, both RNA and protein expression was detectable in at least 10 tissues and the h5UTRs were annotated in both human and mouse RefSeq databases. Compared to all genes or to size-matched non-conserved controls, we observe significantly lower (Wilcoxon rank-sum test p=0.0013, p=0.0017 respectively) cross-tissue correlations (Pearson) for h5UTR genes (Fig. 1b). We also compared cross-tissue correlations of h5UTR genes with RNA variance-matched non-conserved controls to eliminate a model in which h5UTRs impact the correlations only through a different dynamic range of variation in RNA expression. The correlations were still lower for the h5UTR group (p=0.03) (Extended Data Fig. 1d). Alternative 5’UTRs are also more frequently annotated for genes with h5UTRs than all genes or non-conserved controls (Extended Data Fig. 1e). In summary, protein levels of h5UTR genes, as a group, are more difficult to predict with RNA levels alone than those of non-conserved 5’UTR genes, suggesting that extreme sequence conservation in the 5’UTR may be due to tissue-specific post-transcriptional control.
To describe the potential biological functions of genes with h5UTRs, we surveyed gene ontology (GO) terms enriched in the h5UTR gene set. To ensure the specificity of the enrichment, we also analyzed a length-matched set of non-conserved 5’UTR genes, which did not yield any enriched term (Extended Data Fig. 1f). h5UTR GO terms highlighted genes critical for vertebrate embryonic developmental processes (Figs. 1c,d, Supplementary Table 2). For example, h5UTR genes are involved in morphogenesis of major tissues and organs, especially the nervous system. Genes that are part of signaling pathways involving the molecules Wnt, retinoic acid, GABA, Fgf, activin, BMP, Pdgf, Notch, Vegf, Hedgehog or Semaphorins are also abundantly present. We also note the genes involved in epigenetics such as chromatin remodeling and histone acetylation. Additionally, when we intersected known disease-associated variants with h5UTRs, we identified 5 potentially interesting associations that suggest that these regions may also play a functional role in disease (Supplementary Table 3)29. Overall, these annotation enrichments suggest that h5UTRs may play an important role in the post-transcriptional control of core embryonic developmental regulators.
Hyperconserved 5’UTRs impact translation efficiency
To experimentally address whether the h5UTRs could impact the translational efficiency of mRNAs, we chose five candidates - Chrdl1, Gdf5, Dlx1, Sema3a, and Zfx - that function in contexts where spatiotemporal expression patterns are important for embryonic development. Chrdl1 is a BMP antagonist with numerous functional roles in cell differentiation and synapse plasticity, implicated in multiple neurological disorders30–35. Gdf5 is a TGF beta family protein with roles in skeletal and nervous system development36–39. Dlx1 is a homeobox transcription factor that has critical roles in craniofacial patterning, as well as in the differentiation and survival of neurons in the brain40,41. Sema3a is a semaphorin family protein that is secreted and functions as a guidance cue for axons and vasculatures42–46. Zfx is a X-linked transcription factor protein that regulates self-renewal of embryonic and hematopoietic stem cells47.
To examine the contribution of h5UTRs, we introduced deletions into the 5’UTRs of Chrdl1, Gdf5, Dlx1, Sema3a, and Zfx using pairs of CRISPR/Cas9 sgRNAs targeting segments ranging between 50 to 200nt within the HCEs (Supplementary Figs. 2–6, Supplementary Table 4) in mouse embryonic stem cells (mESC), mESCs treated with retinoic acid to promote differentiation, or NIH3T3 cells, reflecting cell types and conditions where these transcripts are expressed (Supplementary Fig. 1a). Polysome profiling allows quantification of translational efficiency independent from effects on transcript levels (Fig. 2a). The mRNAs that are more highly translated are expected to be present in heavier polysomes as they are bound by more ribosomes. As expected, global translation levels displayed no difference between the wild-types and CRISPR/Cas9-mediated HCE knockout cells (Supplementary Fig. 1b–f). We however observe that for all five candidates tested, the deletion mutants exhibited a shift in the distribution of the targeted mRNA species from the heavier polysomes into the lighter polysomes, indicating a decrease in translation efficiency (Fig. 2b–f). These findings suggest that h5UTRs may frequently harbor uncharacterized, additional cis-enhancers of translation initiation.
Figure 2 |. Hyperconserved 5’UTRs impact translation efficiency.

a, Schematic of experimental design for testing the impact of hyperconserved 5’UTRs on translation of coding genes. Shift in the distribution of the mRNAs across sucrose gradient fractions towards the right (heavier polysomes) indicates more average ribosome loading and higher translation efficiency, while shift towards the left indicates lower translation efficiency.
b-f, Polysome profiles of wild-type versus hyperconserved element (HCE) knockout cells. Distribution of mRNAs across sucrose gradient fractions are plotted. Y-axis (the line) plots the mean percent mRNA for each fraction. Error bars indicate standard error. Asterisk indicates two-sided t-test p≤0.05 for each fraction between the knockout and the wild-type, n=4. Indicated p-value is calculated by Fisher’s method across all fractions.
Non-canonical translation enhancer in hyperconserved 5’UTRs
There has been growing evidence for the importance of less understood, alternative mechanisms of initiation independent of the cap-eIF4E interaction that have the potential for enhancing transcript-specific regulation of gene expression25,48–53. For example, it has been estimated that 5–10% of cellular mRNAs may undergo cap-independent translation54,55. The HCE of Hoxa9 contains a functional RNA element previously shown to direct translation initiation in a cap-independent manner that is required for proper embryonic development25. Therefore, we asked whether other h5UTRs can similarly activate non-canonical translation initiation.
To test this hypothesis, we performed a large-scale reporter assay to measure the levels of non-canonical translation initiation from the h5UTRs. We synthesized and cloned a library of 253 full-length h5UTRs into a bicistronic reporter construct, containing two reporter genes, Renilla and Firefly luciferase that are transcribed as one mRNA. The first cistron, Renilla luciferase, is positioned immediately downstream of the promoter and is translated by cap-dependent translation. The second cistron, Firefly luciferase, can only be efficiently translated if the intercistronic inserted sequence enhances non-canonical translation initiation. To perform the reporter assays, we initially selected the mouse 10T1/2 cell line, a mesodermal cell line, as a pilot cell type and further expanded our analysis to include other murine cell types – mESCs, neural stem cells (NSC), embryoid bodies, neurons, and primary cultures of limb bud mesenchyme – to better represent a repertoire of lineages and differentiation trajectories in the developing embryo and capture instances of cell type-specific translation control.
We noticed two groups of reporter activities distributed in a bimodal distribution for each of the cell types (Extended Data Fig. 2a). Within the higher group, we found the three positive controls we included in the reporter assays which all promote cap-independent translation - hepatitis C virus (HCV) internal ribosome entry site (IRES), encephalomyocarditis virus (EMCV) IRES, and the Hoxa9 h5UTR. The “empty” negative control reporter activity is found in the lower group near its median. Thus, the lower component appears to represent the background noise level present in our reporter assays. Using mixture modeling of the bimodal distribution, we estimated the false discovery rate (FDR) for each tested h5UTR as the probability that the reporter activity of the tested h5’UTR could have come from the lower noise group. Using the maximum reporter activity across all six assayed cell types, we estimated that the proportion of the tested h5UTRs with non-canonical initiation activity is 33%. At 10% FDR, we are able to identify 90 h5UTRs with high non-canonical translation activity in at least one cell type (Fig. 3a, Supplementary Table 5). Of the 90 5’UTRs with non-canonical translation activity, two are previously known to the literature56,57. The five genes (Chrdl1, Gdf5, Dlx1, Sema3a, Zfx) for which we demonstrated evidence of translational enhancers (Fig. 2B–F) fall in this class of 5’UTRs promoting non-canonical translation initiation. We observe that for 36 5’UTRs, their reporter activities are significantly variable across different primary cell types that we tested (Fig. 3b). To additionally examine whether cell-type specific non-canonical translation may be relevant in an endogenous context, we compared the polysome profiles of h5UTRs that show increased bicistronic reporter activity in NSCs relative to mESCs. Of the 9 h5UTRs analyzed, 5 (Gpbp1, Ppp2r5e, Pten, Trpm7, Senp3) showed shifts that indicated significant increase in translation in NSCs over mESCs, despite lower global translation in NSCs compared to mESCs (Extended Data Figs. 2b–l). These results suggest that non-canonical translation initiation mediated by h5UTRs could be controlled in a highly regulatable fashion across different cell types to differentially control post-transcriptional gene expression.
Figure 3 |. Non-canonical translation enhancer in hyperconserved 5’UTRs.

a, Measurement of non-canonical translation initiation activity from 253 hyperconserved 5’UTRs by bicistronic reporter assay. Each dot is a 5’UTR, where x-axis is the maximum luciferase reporter ratio across six different cell types and y-axis is the rank of the reporter ratio from low to high. The skewing is reflective of the bimodal distribution of the activities (see also Extended Data Fig. 2a), and color of the dot indicates estimated proportion of false positives based on mixture modeling of two Gaussian distributions. Dashed line indicates the reporter ratio above which 10% of the hits are expected to be false positives. Genes labeled in red: HCV and EMCV are positive control viral IRES; others are select h5UTRs with annotated biological functions in embryonic development.
b, Heatmap of non-canonical translation initiation activity for 36 significantly varying h5UTRs across five indicated cell types (F-test, FDR≤0.05). N=4 for C10T1/2, mESC and EB; N=6 for NSCs, neurons, limb mesenchyme culture. The color shows row z-scaled mean log2 reporter activities. The 5’UTRs are ordered by clustering similar reporter activity patterns across cell types.
c, Violin plot of bicistronic reporter activities from hyperconserved and non-conserved 5’UTRs in 10T1/2 cells. p indicates two-sided Wilcoxon rank sum test p-value. Box hinges: 25% quantile, median, 75% quantile, respectively from left to right. Whiskers: lower or upper hinge ±1.5*IQR.
d, The effect of various truncations of the h5UTRs on non-canonical initiation and total translation efficiency. Also see Extended Data Fig. 3b. Left: positions of truncations. Dashed lines indicate truncations and bars indicate the remaining sequences. Purple horizontal lines within bars indicate uORFs. Middle: non-canonical initiation efficiency. Right: total translation efficiency. X-axis indicates the geometric mean of luciferase reporter ratios relative to the wild-type. Error bars indicate geometric standard error. Dashed line marks the reporter ratio for the wild-type 5’UTR. Asterisk indicates two-sided t-test p≤0.05 for each truncation mutant versus the full-length wild-type. The numbers to the left of the bars indicate exact n and p-values for each comparison versus the full-length.
e, Comparison of translational activities between the full-length h5UTR versus the only first 300nt of the h5UTR. 38 different pairs are tested. X-axis indicates the mean log2 luciferase reporter ratios of each truncation relative to its full-length wild-type. Error bars indicate standard error of the log2 luciferase activity ratios. Bars colored in red indicate significantly reduced translation in the shorter, truncated 300nt fragment; black indicates significant increase (two-sided t-test, p≤0.05, paired n=3, marked by asterisk). The numbers to the left of the bars indicate exact p-values for each comparison versus the full-length.
We determined that the higher reporter ratios are not due to cryptic transcriptional and splicing effects, since the ratios of the mRNA levels of the two luciferase genes measured by qPCR in transfected cells are not skewed or correlated with ratios of the two luciferase reporter activities (Extended Data Fig. 3a). In addition, we selected 23 non-conserved mouse 5’UTR sequences and tested their activities in 10T1/2 cells. The distribution of non-conserved 5’UTRs are unimodal near the lower noise component of the mixture observed for the conserved set (Wilcoxon rank sum test p=0.015, Fig. 3c). This result further indicates that the extreme conservation in the 5’UTRs enriches for non-canonical translation initiation, suggesting their predictive value in identifying such elements genome-wide.
It has been often argued that cap-independent translation typically makes only minimal contribution to overall translation efficiency, except under conditions during which cap-dependent translation is globally reduced58–60. To understand the contribution of the non-canonical translation activation by the h5UTRs, we performed two different reporter assays with a series of truncated h5UTRs. The first reporter assay was the bicistronic reporter assay as described above. In the second reporter assay, the endogenous capped monocistronic Renilla luciferase was employed to measure total translational levels for truncated h5UTRs where cap-dependent translation is active. For 9 out of 11 h5UTR truncations in the two reporters transfected to 10T1/2 cells, we observed that at least one truncation significantly reduced non-canonical initiation as well as the total translational levels (Fig. 3d, Extended Data Fig. 3b). The trend for truncations to frequently reduce total translation activity is notable, since cap-dependent translation initiation typically increases in efficiency when the 5’UTR is shortened. We asked if this is more generally true in a larger set of 38 h5UTRs, by comparing the total translation directed by the full-length versus only the first 300 nucleotides of the h5UTR without a large proportion of the HCE in each h5UTR. 20 out of 38 decreases significantly in the shorter truncated 5’UTR relative to the full-length, while only 5 significantly increases (Fig. 3e). In contrast, truncating long, non-conserved 5’UTRs do not show the same trend for decreased translation (Extended Data Figs. 3c,d). Furthemore, there is no correlation between the change in the density of upstream AUGs and change in reporter activities (Extended Data Fig. 3e). Taken together, non-canonical translation enhancer elements in h5UTRs widely impact total translation efficiency in physiological cellular conditions, suggesting that h5UTR genes may be translated via more specialized initiation mechanisms that utilize evolutionarily constrained, sequence-specific cis-regulatory features.
Cellular remodeling of hyperconserved 5’UTR RNA structures
Higher order structures are inherent features of RNA molecules that underpin their biochemical function. The majority of previous co-variation based predictions of RNA structures in vertebrates occur in “moderately” conserved regions of the genome and miss the HCEs61–64. This is because covariation analysis requires sufficient conservation for alignment but also sufficient variation for statistical power65,66. Since extreme conservation limits the extent to which covariation signals can be informative, addressing this question currently requires additional experimental data.
We postulated that specific regions of mRNA that display localized sensitivity in their structures to active cellular remodeling by RNA helicases could potentially lead us to functionally relevant structures within h5UTRs that guide translation initiation. To obtain a high coverage accessibility data for a large number of h5UTRs, we initially performed a highly multiplexed amplicon sequencing adaptation of dimethyl sulfate (DMS) mutational profiling67–69. DMS profiling was performed in mESCs under the conditions of no treatment or depletion of ATP to eliminate helicase activity (Fig. 4a,b). We successfully profiled 161 tiling amplicons of size 250nt across 69 endogenously expressed h5UTRs. We identified 140 11nt windows over 20 h5UTRs that were significantly different (FDR≤0.05) between ATP depletion and no treatment (Fig. 4c, Supplementary Table 6). One known source of RNA structure remodeling in the cell is ribosome unwinding of mRNAs during translation, and thus the presence of upstream open reading frames (uORF) may lead to differential accessibilities upon ATP depletion70. We tested whether differential accessibility windows (FDR≤0.05) are overrepresented in upstream AUGs or potential uORFs but did not observe significant enrichment for either case, arguing against this possibility (Extended Data Figs. 4a,b). Together, these results suggest the frequent presence of secondary structures under active energy-dependent cellular remodeling within h5UTRs.
Figure 4 |. Cellular remodeling hyperconserved 5’UTR RNA structures.
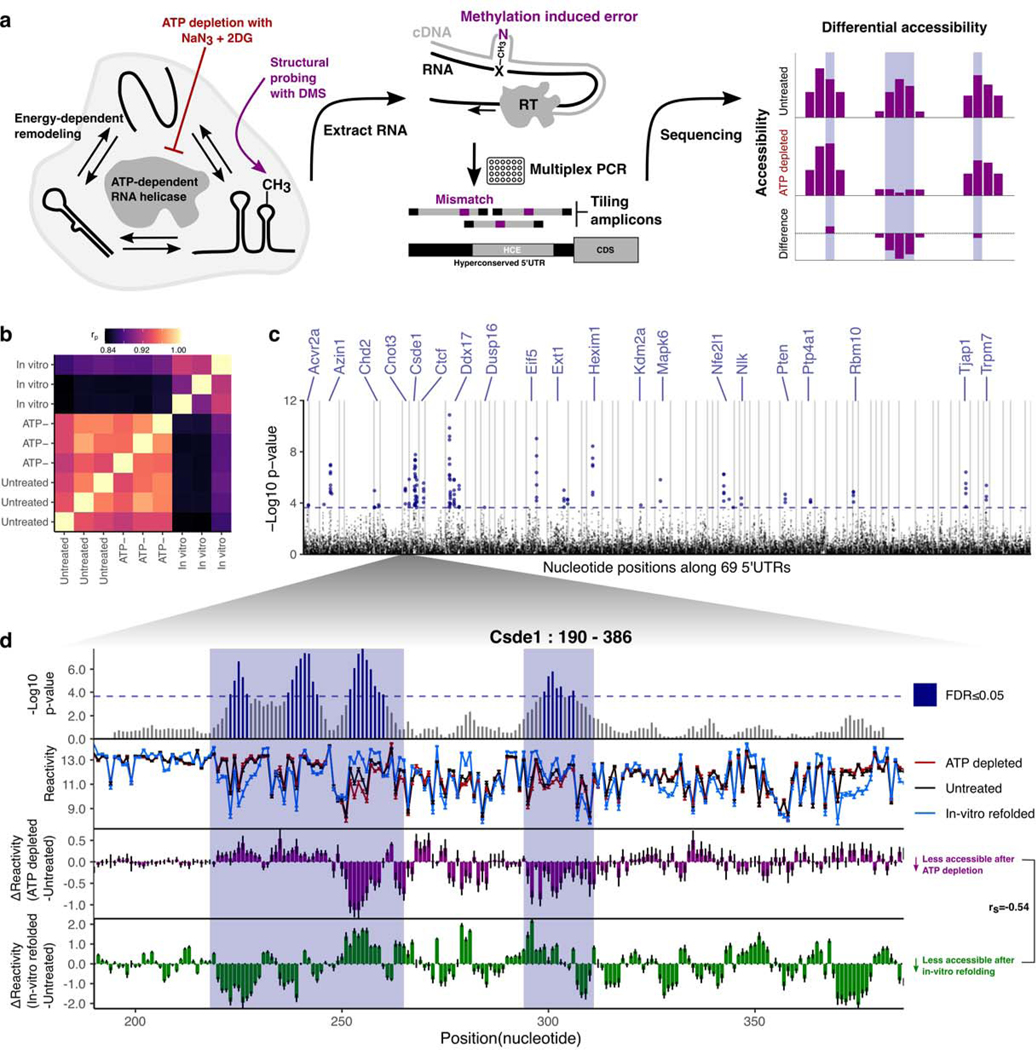
a, Schematic of identifying RNA structures under cellular remodeling in h5UTRs. Multiplexed, targeted DMS chemical probing of 69 h5UTRs inside cells from their endogenous mRNAs is performed following ATP depletion treatment to stop RNA helicase activity.
b, Heatmap of correlation (Pearson’s) matrix across replicate samples for untreated, ATP depleted, and in vitro refolded samples (three each). The correlation values are calculated from a vector of normalized accessibility values for all nucleotides passing per-amplicon reproducibility cutoff.
c, Manhattan plot of differential accessibility tests in 11nt overlapping windows across the 5’UTRs. Y-axis indicates -log10 KS-test p-value for each window along 69 5’UTRs in x-axis. Dashed line indicates the p-value cutoff at which permutation FDR is at 5%.
d, Zoomed-in view of differential accessibilities along the Csde1 5’UTR in from positions 190 to 386. Top plot shows -log10 p-value for each window. Highlighted boxes mark significantly different windows, above the dashed line indicating 5% FDR. Middle plot shows differential accessibility on the y-axis, where greater than zero indicates increased accessibility upon ATP depletion and less than zero indicates decreased accessibility. Bottom plot shows differential accessibility for in cell versus in vitro refolded RNA. Error bars in each plot show standard error, n=3.
For the 20 significant h5UTRs with ATP-dependent differential accessibility, we found strong enrichment of mammalian phenotype ontology terms that indicate essential early developmental gene function: 16 out of the 20 were annotated for either embryonic or neonatal lethality (Supplementary Table 7). We also found known associations with human genetic diseases for 6 out of the 20 (Supplementary Table 8). This suggests that the extremely conserved, structured RNA elements could be impacting post-transcriptional regulation of key developmental genes.
Among the most striking patterns of ATP-dependent differential accessibility observed is in the 5’UTR of Csde1, also known as upstream of N-ras (Unr). Csde1 encodes a RNA binding protein (RBP) that regulates translation and stability of its target mRNAs. It is known to impact cell cycle, stem cell differentiation, apoptosis, and dosage compensation71–77. Csde1 is implicated in a variety of human diseases including Diamond-Blackfan anemia, autism spectrum disorders, and cancers78–81. We identified an approximate 150bp stretch from positions 215 to 365 encompassing an HCE that shows large scale accessibility changes upon ATP depletion (Fig. 4d). Notably, the accessibility changes observed in Csde1 h5UTR following ATP depletion are different from the changes observed between in cell and in vitro refolded RNA - there is even a slight negative correlation (rs=−0.54). Such discordance is also observed in a number of other h5UTRs with ATP-dependent differential accessibility (Extended Data Fig. 4c). Thus, active remodeling by RNA helicases can be important for the formation of cellular structures distinct from those formed under in vitro conditions.
Csde1 5’UTR encodes alternative functional RNA structures
As a paradigm to investigate cellular RNA structure and its remodeling in HCEs, we sought to further characterize the helicase-sensitive structures in Csde1 h5UTR. In particular, we developed in-cell mutate-and-map (icM2), a powerful new methodology that enables the application of the M2 strategy, wherein systematic mutagenesis of RNA is coupled with chemical mapping to generate accessibility profiles for every mutated nucleotide, inside the native cellular context (Fig. 5a,b,c)82. In icM2, the target sequence of interest is mutagenized using error-prone PCR, cloned as a pool into an expression plasmid and transfected into cells. Following the treatment of transfected cells with DMS, total RNAs are extracted and subjected to read-through reverse transcription, where modified nucleotides are misincorporated as mutations on the cDNA that are amplified and sequenced. Correlated mutations in sequencing reads are then quantified, and the resultant covariation matrix is analyzed for signature perturbation patterns. icM2 is particularly suited for analysis of h5UTRs, as it directly addresses what RNA structural changes occur if each of the extremely conserved nucleotides are mutated during evolution.
Figure 5 |. icM2 reveals structured elements in the hyperconserved Csde1 5’UTR.

a, Schematic of localized perturbation patterns that may be observed in M2 data. Here, the mutant does not disrupt the overall structure and “releases” its base pairing partner. This results in an increase of chemical accessibility signal at the interacting nucleotide. Systematic profiling of accessibilities by M2 results in an array of such mutant accessibility data into an approximate contact map.
b, Schematic of global rearrangement patterns that may be observed in M2 data. Here, multiple conformations of the RNA molecule are present together in an ensemble at non-negligible relative proportions. Mutations can shift this balance, such that one structural state is favored over the other. In this case, M2 reveals large-scale accessibility perturbations across a longer stretch of the RNA molecule. Multiple mutations often impact the relative proportions in similar ways, which manifests as correlated arrays accessibility changes in M2 data matrix.
c, Schematic of the icM2 method. Mutagenesis library of the target RNA of interest is first generated using error-prone PCR followed by cloning into an expression vector. The cells are transfected with the library and treated with DMS. Total RNAs are extracted. Read-through reverse transcription encodes DMS-modified nucleotides as mutations on the cDNA, which are read out by high-throughput sequencing. Correlated mutations in sequencing reads are then quantified and the resultant covariation matrix is analyzed for signature perturbation patterns.
d, Heatmap of icM2 accessibility matrix for Csde1 5’UTR from position 190 to 386. For each row, the chemical mapping profile of a single-nucleotide variant of the RNA is plotted across the columns, where the colors indicate z-scaled accessibility change values from the wild-type RNA. 1D data from each mutant are vertically stacked to display a 2D matrix. White boxes mark the two regions (A: positions 334–363 and B: positions 215–315) that display strong perturbation signals that reveal their structures.
e, A structure model (structure W) of region A. Bases colored in red indicate mutations with accessibility changes observed in icM2 data that are consistent with the model.
f, Scatter plot showing correlations of per-nucleotide accessibilities between each mutant versus the “wild-type” (wild-type accessibilities are not directly measured, but mean accessibilities of 10 lowest variable mutants are used as a close approximation) on the y-axis and nucleotide positions along the x-axis. p indicates two-sided Wilcoxon rank sum test p-value for the difference in distributions of correlations between region B versus other nucleotides.
g, Multiple species alignment for Csde1 5’UTR from position 125 to 548. For each row, the sequence alignment of a species is plotted across the columns, where the colors indicate match/substitution/insertion/deletion at each nucleotide. The alignment positions are relative to the mouse sequence. The top row is the mouse alignment, colored separately from other rows as a reference to indicate the identity of the bases in each position in the multiple species alignment.
We applied icM2 in three windows tiling across Csde1 5’UTR in mESCs. We observed strong perturbation signals in the 215–365 positions along the 5’UTR, where we had originally observed large differential accessibilities in response to elimination of RNA helicase unwinding activities (Fig. 5d). The visualization of the icM2 accessibility matrix immediately highlighted two subregions. The first region is around positions 334–363 (region A), where short range localized perturbations indicated the presence of a small stem loop motif. Here, the data corresponded well to the expected accessibility changes for the lowest free energy structure (structure W) predicted for the region (Fig. 5e). The second region is around positions 215–315 (region B), where correlated global perturbations across a long stretch of about 100 nucleotides indicated the presence of multiple conformations. Remarkably, these correlated global perturbations occur for almost every mutation across the 100nt stretch, revealing the strong sensitivity of the ensemble state to the precise sequence identity of each base. This is highlighted by the correlations of per-nucleotide accessibilities between each mutant versus the “wild-type” (Fig. 5f, Wilcoxon rank-sum test p=0.0015 for mutants in region B versus other mutants). Therefore, at least two conformational states exist whose relative proportions inside of the cell are affected by a mutation in almost any of the extremely conserved nucleotides. In addition, we observe the strongest conservation signal of the Csde1 5’UTR in region B, where amongst placental mammals there is a near-perfect sequence identity (Fig. 5g). These results suggest a structural explanation for why such extreme conservation levels may be required. Furthermore, examining the conservation levels and ATP-dependent accessibility profiles across all other h5UTRs reveals that the average per-nucleotide conservation levels in significantly differential accessibility regions (FDR≤0.05) display exceedingly high conservation levels compared to the rest of the RNA (Extended Data Figs. 5a,b). Thus, encoding of actively remodeled cellular RNA structures may be a broadly occurring phenomenon associated with the extreme conservation levels in h5UTRs.
We next asked what candidate structures might comprise and explain the observed alternative states of the ensemble in region B. We used the average accessibility change profiles for the two clusters as two separate constraints for RNA folding (Fig. 6a). Constraining by cluster 1 average accessibility profile revealed a well defined conformation (structure X) disrupted by cluster 1 mutants in the helices and stabilized by cluster 2 mutants in the loops (Fig. 6b). Constraining by cluster 2 profile resulted in a higher entropy fold, which was nevertheless readily visualizable by two representative medoid conformations (structures Y and Z, Figs. 6c,d). To estimate the relative mixing ratios of these structures, we chose to apply RNA ensemble extraction from footprinting insights technique (REEFFIT)83. For the wild-type sequence, REEFFIT yielded proportions of 67±9%:10±4%:23±9% for the representative structures X, Y, and Z, respectively (Figs. 6b,c,d). It also predicted how these proportions are expected to change across the individual mutants, adding quantitative estimates to our initially qualitative observations of alternative structural states. For example, cluster 1 mutants disrupt structure X to favor Y and Z, changing the relative proportions of X:Y:Z to 30%:13%:57% on average, while cluster 2 mutants act in the opposite direction, shifting it to 91%:6%:3% (Fig. 6e). We further discovered that the accessibility change profiles of the two clusters of mutants are closely correlated with helicase-dependent accessibility change (Fig. 6). This observation suggests that elimination of RNA helicase unwinding activity decreases the proportion of structure X in the cell and does so to increase the fraction of the alternative structures Y and Z. Notably, structure X has multiple long stems (positions 232–282) - i.e. the helicase activity promotes a low free energy structure and potentially may act as chaperones84. It is formally possible for other direct contacts on the exact methylation sites of the nucleotides, such as a direct RBP interaction on the base-pairing face, to produce localized “footprints” on the accessibility profiles; however this would not drastically impact our model. Taken together, we propose three candidate conformations to account for our icM2 signal observed in region B of Csde1 5’UTR and hypothesize that the cell is actively expending energy to maintain the precise relative balance of these conformations in the cellular structural ensemble.
Figure 6 |. Csde1 5’UTR tunes translation efficiency by encoding multiple alternative structures that are actively maintained by RNA helicases.

a, Multidimensional scaling (MDS) plot showing dimensionality reduction (K=2) of icM2 data matrix (positions 190–386). Dots indicate each single nucleotide variant of the RNA, where the colors/annotations mark the mutants grouped into two clusters, determined heuristically by visual inspection of the positions on the plot.
b-d, Structure models (X, Y, Z) for the alternative conformations in region B. Bases colored in red or blue indicate mutations with similar patterns of accessibility changes grouped into two clusters as shown in A). Yellow outline indicates mutants that are also tested for function in luciferase reporter assay (shown in Fig. 6g). Percentages indicate relative proportions of the three structures estimated by REEFFIT.
e, REEFFIT estimates of the mixing proportions (relative to the maximal amount of change that can be observed by the single mutations) for structures X,Y, Z upon introduction of single nucleotide mutations. Y-axis indicates the stacked bars indicating proportions, along the variants from the two clusters in X-axis.
f, Comparison of average accessibility changes for each of the two clusters with accessibility changes observed upon ATP depletion at region B. Top plot shows the reactivity differences upon ATP depletion, where greater than zero indicates increased accessibility upon ATP depletion and vice versa. Middle and bottom shows the cluster average accessibility change z-scores. Spearman correlation between cluster 1 and ATP depletion is 0.83 and −0.8 between cluster 2 and ATP depletion.
g, The effect of shifting the relative balance of the alternative conformations at region B on translation. Y-axis shows changes in luciferase reporter activities compared to the wild-type sequence when the single variants affecting mixing proportions (shown in Fig. 6e) are introduced into the full-length Csde1 5’UTR upstream of the luciferase reporter. Plotted are the mean; error bars indicate standard error. The bars and labels along x-axis are colored according to whether they are wild-type, cluster 1 mutants, or cluster 2 mutants. Dashed line indicates the wild-type luciferase reporter level. Asterisk indicates a significant difference between each mutant and wild-type (two-sided t-test p≤0.05, n=3).
In our initial one-dimensional DMS profiling analysis of h5UTRs in mESCs, we had observed that the accessibility profiles of many RNAs refolded in vitro were discordant from those of RNAs in cells (Figs. 4d, Extended Data Figs. 5c). To further expand on these differences and actually compare in-cell vs in-vitro RNA structures, we also performed in vitro M2 on Csde1 h5UTR. We observed a strikingly different accessibility matrix (Extended Data Figs. 6a,b). These results highlight the importance of resolving flexible conformations that can occur uniquely under cellular conditions.
Lastly, we asked whether such a shift in the balance of structural conformations has a functional consequence on the translation of the downstream gene. We performed luciferase reporter assays with mutant Csde1 5’UTRs carrying a number of substitutions from each of the two clusters that are predicted to change the relative proportions. We selected four different nucleotide positions from cluster 1 and three from cluster 2, hypothesizing similar patterns of expression level changes may be observed among each cluster. We observed that all cluster 1 mutants decreased Firefly luciferase activities by 15–20% compared to the wild-type 5’UTR (Fig. 6g). In contrast, cluster 2 mutants increased the reporter activities by 5–15%. When the three individual single mutations from cluster 2 are combined, the effect size is increased to about 50%. The dynamic range of final protein levels can thus be tuned according to the relative proportions of the multiple conformations along the RNA structural landscape of Csde1 5’UTR. Together, these results suggest that the exact proportions and properties of the RNA structural ensemble is a critical functional requirement under negative selection in hyperconserved vertebrate 5’UTRs.
Discussion
Extreme sequence conservation has long been observed in non-coding regions of vertebrate genomes, yet our current functional knowledge of these elements falls short in explaining why and how such conservation levels exist. Here, we uncover a functional role for hyperconserved 5’UTRs in regulation of translation and report their unexpected enrichment in non-canonical initiation sites particularly within those transcripts critical for development in vertebrate species. We speculate that there may potentially be many different types of unknown non-canonical mechanisms that are adopted by these 5’UTRs and that further investigations may identify new classes of RNA elements that accommodate more specialized mechanisms of translational control. The activities of h5UTRs may vary across cells and tissues, which may result in differential translatability of these mRNAs.
A crucial component of decoding cis-regulatory features at the level of RNAs is the determination of their higher order structures beyond the primary sequence. To this end, we developed a new technique, icM2, to examine the RNA structural ensemble within cells. We found that cells precisely tune protein expression levels by remodeling the hyperconserved Csde1 5’UTR to maintain the relative proportions of multiple functional conformations. While icM2 revealed a highly dense array of mutations that disrupt such actively enforced balance of dynamic structures in the Csde1 5’UTR across a ~100nt long stretch, the same mutations are negatively selected against in nature across vertebrate species. This suggests that selective pressures for translational regulation can lead to extreme sequence constraints when an ensemble of multiple functional conformations must be encoded over a single stable structure to ensure a dynamic range of translational outputs. The observation that regions of h5UTRs under helicase-dependent structural remodeling in general display the highest conservation levels further suggests that a similar phenomenon could extend more broadly to other h5UTRs and may at least in part explain extreme conservation in 5’UTRs at the level of RNA.
Flexible structural states can potentially endow multiple functional states in regulatory elements that respond to environmental or cellular cues. Most current genome-wide efforts to identify functional structures have focused on single stable conformations. Our results underscore the necessity of the ensemble perspective of RNA structure in understanding the cellular activities of regulatory RNAs and the potential utility of extreme conservation in detecting such dynamically structured elements in the untranslated regions of mRNAs. We envision that hyperconserved 5’UTRs will aid the discovery of functional RNA structures in vertebrate genomes and advance our broader understanding of post-transcriptional gene regulation in development, disease, and evolution.
Online methods
Data sources
See Supplementary Notes for the publicly available data used in this study.
h5UTR definition
60-way vertebrate PhastCons elements were downloaded from UCSC mouse genome database, and LOD≥500 elements were subsetted. For each mouse RefSeq transcript record, the total number of 5’UTR, CDS, and 3’UTR nucleotides overlapping LOD≥500 elements are calculated (Supplementary Table 1). 5’UTRs with ≥250nt overlap are labeled hyperconserved. See Supplementary Notes for the full description.
Transcriptome-proteome correlations
Cross-tissue TMT mass spectrometry data and matching RNA-seq data were obtained from GTEx quantitative proteomics analysis of 32 human tissues from 14 individuals27. Pearson’s correlation coefficient is calculated between per-tissue medians of RNA expression and per-tissue medians of protein expression. See Supplementary Notes for the full description of data processing steps.
Term enrichment analysis
GO term enrichment analysis is performed using topGO (version 2.38.1)85. GO term-gene mappings are obtained from Bioconductor annotation package org.Mm.eg.db. Mammalian phenotype ontology term enrichment analysis is performed using MouseMine86. See Supplementary Notes for the full description.
CRISPR knockouts
sgRNAs were designed using CRISPOR87. sgRNA sequences were synthesized as ssDNA oligos and were cloned into the BbsI-digested expression plasmid bearing both sgRNA scaffold backbone (BB) and Cas9 nuclease, pX330-U6-Chimeric_BB-CBh-hSpCas9. For ESCs, ~0.5×106 cells were plated onto a single 6-well plate (See Supplementary Table 9 for sgRNA sequences and genotyping primers). After 4 hours, 1.25ug each of the plasmids carrying sgRNA pairs were transfected using 2.5uL P3000 reagent and 12uL Lipofectamine 3000 (ThermoScientific, L3000001). 12 hours after transfection, media was changed to puromycin (ThermoScientific, A1113803) containing media at 1ug/mL. After 24 hours of puromycin selection, cells were washed with PBS, trypsinized, and plated at 1000 cells/10 cm plate. 10 days later, single colonies were picked and replica plated to 2×96 well plates. One plate was used for genotyping. For 3T3 cells, the transfection and selection were performed using the same methods, but the cells were plated at limiting dilution of 0.5 cells/well into 96 well plate for expansion and split for genotyping. Cells in the genotyping plate were lysed by removing the media, adding 100uL 50mM NaOH per well, and heating at 95°C for 10min. After cooling to room temperature, 500uL 500mM Tris-HCl pH 8.0 was added to neutralize and 1:100 dilution was taken for genotyping PCR. Genotyping PCR reaction is as follows: 1× MyTaq HS Red Mix (BIO-25047), 300nM forward primer, 300nM reverse primer, 1uL 1:100 diluted crude lysate in 10uL total reaction volume. Cycling conditions are: 95°C 3min initial denaturation, followed by 30 cycles of 95°C 15s, 68°C 15s, 72°C 30s. Clones with expected shorter amplicons were further expanded. DNA from expanded clones was isolated with Wizard Genomic DNA Purification kit (Promega, A1120). Genotyping PCR reaction from expanded clones are as follows: 0.02U/uL Kapa HiFi HotStart polymerase (Roche, KR0369), 1× Kapa HiFi HotStart buffer, 300uM dNTP each, 300nM forward primer, 300nM reverse primer, 10ng gDNA in 20uL reaction. Cycling conditions are: 95°C 3min initial denaturation, followed by 30 cycles of 98°C 20s, 68°C 15s, 72°C 30s. The amplicons were Sanger sequenced at Quintara Biosciences.
Cell culture
See Supplementary Notes for the description of cell culture conditions.
Mouse husbandry
All animal work was reviewed and approved by the Stanford Administrative Panel on Laboratory Animal Care (APLAC). The Stanford APLAC is accredited by the American Association for the Accreditation of Laboratory Animal Care (AAALAC). All mice used in the study were housed at the Research Animal Facility (RAF) and at the SIM-1 Barrier Facility at Stanford University. All mice used for experiments were between 2 and 6 months old. All animal studies were performed in accordance with Stanford University Animal Care and Use guidelines.
Polysome profiling
Cells were harvested 2min after replacing media with cycloheximide (MilliporeSigma, C7698–1G) containing media at 100ug/mL. ~10×106 cells were resuspended in 400uL of following lysis buffer on ice for 30min, vortexing every 10min: 25mM Tris-HCl pH 7.5, 150mM NaCl, 15mM MgCl2, 1mM DTT, 8% glycerol, 1% Triton X-100, 100ug/mL cycloheximide, 0.2U/uL Superase-In RNase inhibitor (ThermoFisher Scientific, AM2694), 1× Halt protease inhibitor cocktail (ThermoFisher Scientific, 78430), 0.02U/uL TURBO DNase (ThermoFisher Scientific, AM2238). Nuclei were removed by two step centrifuging, first at 1300g for 5min and second at 10000g for 5min, taking the supernatants from each. 25%−50% sucrose gradient was prepared in 13.2mL ultracentrifuge tubes (Beckman Coulter, 331372) using Biocomp Gradient Master with the following recipe: 25 or 50% sucrose (w/v), 25mM Tris-HCl pH 7.5, 150mM NaCl, 15mM MgCl2, 1mM DTT, 100ug/mL cycloheximide. The lysate was layered onto the sucrose gradient and ultracentrifuged on Beckman Coulter SW-41Ti rotor at 40000rpm for 150min at 4°C. The gradient was density fractionated using Brandel BR-188 into 16×750uL fractions. 50pg in vitro transcribed spike-in luciferase RNA was added to each fraction. 700uL of each fraction was mixed with 100uL 10% SDS, 200uL 1.5M sodium acetate, and 900uL acid phenol-chloroform, pH 4.5 (ThermoFisher Scientific, AM9720), heated at 65°C for 5min, and centrifuged at 20000g for 15min at 4°C for phase separation. 600uL aqueous phase was mixed with 600uL 100% ethanol and RNA was purified on silica columns (Zymo, R1013). For each fraction, up to 5ug RNA was DNase treated at 37°C for 30min using 0.2U/uL TURBO DNase with 1U/uL Superase-In in 30uL and purified again on silica column. 100ng RNA was reverse transcribed using iScript reverse transcriptase (Biorad, 1708890) in 10uL reactions. qPCR was performed using Ssoadvanced Universal SYBR Green Supermix (Biorad, 1725270) with 2uL of 1:4 diluted reverse transcription reaction and primer pairs targeting the HCE knockout h5UTR genes or the spike-in (See Supplementary Table 9 for primer sequences).
Ct values were normalized to Ct values of spike-in luciferase and plotted as proportions across the 16 fractions. T-statistic and p-value is calculated for difference in means between the two genotypes for each fraction. Each replicate comprises an independent culture (per genotype), sucrose gradient fractionation and qPCR quantification. Fisher combined p-value is calculated for no difference across all fractions.
Reporter constructs for luciferase assays
Bicistronic reporter gateway plasmid, pRF_gwy, is constructed from pRF vector which has SV40 promoter and two reporter genes, Renilla luciferase and Firefly luciferase, with multiple cloning sites in between them88. Gateway cassette A (ThermoFisher Scientific, 11828029) was inserted in between Renilla and Firefly luciferases, replacing the cloning sites using two EcoRI sites.
RNA normalizing reporter gateway plasmid, pRF_D1, is constructed from pRF vector by replacing the cloning sites with HCV IRES and inserting gateway cassette A in between AvrII and EcoRV sites upstream of Renilla luciferase. Downstream of the Renilla luciferase is the Firefly luciferase and the HCV IRES between them such that the HCV IRES-translated downstream Firefly luciferase normalizes for differences in RNA levels to enable measurement of translation efficiency.
Full-length h5UTRs were synthesized and cloned into pENTR1A (ThermoFisher Scientific, A10462) by SGI (sequences in Supplementary Table 1). Truncation variants are either synthesized or cloned by PCR from synthesized full-length sequences into pENTR1A vectors. Gateway LR Clonase II (ThermoFisher Scientific, 11791020) is used to recombine the full-length or truncation variants into either pRF_gwy or pRF_D1 vectors.
Mutant Csde1 5’UTRs were cloned by Gibson assembly reaction (NEB, E2621S) using mutation containing ssDNA templates with homology arms and two upstream/downstream fragments (sequences in Supplementary Table 9). Full-length wild-type and mutant Csde1 5’UTRs were inserted pGL3 (Promega, E1751) plasmid in between EcoRI and NcoI sites upstream of Firefly luciferase gene. In vitro transcription template was amplified with T7 promoter sequence containing primer and in vitro transcribed using T7 RNA polymerase (NEB, E2040S). The IVT RNAs were capped using Vaccinia virus capping enzyme (Cellscript, C-SCCE0625) and polyA tailed using polyA polymerase (Cellscript, C-PAP5104H).
Reporter transfection for luciferase assays
For DNA transfections, 200ng plasmid DNA is transfected to cells plated on 96-well plates. For 10T1/2 cells, mESCs, NSCs, limb mesenchyme culture, and embryoid bodies, 0.5uL Lipofectamine 2000 (ThermoFisher Scientific, 11668030) is used per one well. For neurons, 0.2uL Viafect (Promega, E4981) is used per well. Cells were incubated for 4 hrs with transfection reagent, DNA in OptiMEM media (ThermoFisher Scientific, 31985062). Cells were then washed with PBS, and the media was changed back to regular growth media.
For RNA transfection, 200ng Firefly luciferase RNA and 10ng Renilla luciferase RNA is transfected to cells plated on 96-well plates. 0.5uL Lipofectamine 2000 is used per one well.
Luciferase assays
For DNA transfections, cells were lysed using Passive Lysis Buffer (Promega, E1941) for 30min at room temperature, 48 hours after transfection. For RNA transfections, cells were lysed 6 hours after transfection. Firefly and Renilla luciferase values were read using Dual-Glo Luciferase Assay System (Promega, E2920) for >96 samples or Dual-Luciferase Reporter Assay System (Promega, E1910) for fewer samples, on Promega GloMax-Multi plate reader. In all experiments, log ratios of the two luciferase activities are taken for each well.
For bicistronic reporter assays across multiple cell types, the data are quantile normalized across all replicate samples. normalmixEM function from R package mixtools (version 1.2.0) is used for mixture modeling of maximum replicate-average values. False discovery estimate at a cutoff is calculated as the proportion of the mixture distribution above the chosen cutoff that comes from the lower component. For identification of h5UTRs with significant differential activity across cell types, F-statistic is calculated, and Benjamini-Hochberg procedure is used with their p-values to estimate the FDR. Each replicate comprises an independent reporter transfection, lysate collection and luciferase activity quantification.The luciferase activity data across cell types was clustered using pairwise Euclidean distance metric between h5UTRs and average linkage hierarchical clustering. For other reporter assays, all statistics are calculated from log ratios of the luciferase activities (two-sided T-test, Welch). Each replicate comprises an independent reporter transfection, lysate collection and luciferase activity quantification. Mean and error bars when plotted in linear scale are back transformed from the mean and standard errors of the log scale values.
In-cell DMS probing following ATP depletion and multiplexed mutational profiling
100 h5UTRs were chosen for amplicon sequencing based on coverage profiles from ENCODE E14 mESC RNA-seq data. See Supplementary Notes for the description of amplicon sequencing primer design and pooling. For ATP depletion, 10×106 mESCs were incubated for 10min in ATP depletion media: DMEM without glucose (ThermoFisher Scientific, A1443001), 10mM 2-Deoxy-D-glucose (MilliporeSigma, 25972), 10mM sodium azide (MilliporeSigma, 71289). The cells were washed, trypsinized and harvested using PBS, trypsin, and finally resuspended in 3.5mL mESC media all containing 10mM 2DG and 10mM NaN3. 1mL 1M bicine (Millipore Sigma, B3876), titrated to pH 8.5 at 25°C, is added to resuspended cells (250mM final bicine concentration). 500uL 16% dimethyl sulfate (MilliporeSigma, D186309) in ethanol is added (1.6% final concentration). Cells are mixed and incubated for 6min at 37°C. 2.5mL ice-cold 30% BME (MilliporeSigma, M3148) in ethanol is added to quench the reaction. This DMS modification protocol is adapted from protocol and data reported in69. Following centrifuge to remove the supernatant, the cells are lysed in Trizol (ThermoFisher Scientific, 15596026). For the untreated condition without ATP depletion treatment, all procedures are the same except for initial 10min incubation in ATP depletion media and inclusion of 2DG and NaN3 in all media. Three independent samples were collected for each condition. Total RNA is phase extracted with chloroform and aqueous phase is purified on silica columns (Zymo, R1013). 10–20ug RNA is DNase treated at 37°C for 30min using 0.2U/uL TURBO DNase (ThermoFisher Scientific, AM2238) with 1U/uL Superase-In (ThermoFisher Scientific, AM2696) in 60uL and purified again on silica column.
1ug RNA is mixed with 1uL 1uM 96× primer pool (96×1uM amplicon reverse primers for total of 1uM oligos) and denatured in 6.25uL total volume (with H2O) at 65°C for 2min, then chilled to 4°C. Reverse transcription reaction conditions are as follows: 20mM Tris-HCl pH7.5, 75mM KCl, 10mM MgCl2, 5mM DTT, 500nM TGIRT (InGex, TGIRT50), 1U/uL Superase-In; 19uL total reaction volume. The RT reaction is preincubated for 25°C for 30min, then initiated with addition of 1uL 12.5mM dNTP each. After incubation at 60°C for 1hour, 1uL 2.5M NaOH is added and the reaction is heated at 95°C for 3min. 1uL 2.5M HCl and 1uL 500mM Tris-HCl pH 7.5 is added to neutralize. 29uL SPRIselect beads (Beckman Coulter, B23318) is used for purification of cDNA; elution volume is 6uL. 4×96 pooled reactions for a total of 384 targets are performed for each sample. To each set of 96 pool cDNA, multiplex PCR is performed with 5uL cDNA, 0.2mM dNTP, 2uM 96× forward primer pool (96×2uM each primer for total of 2uM oligos), 2uM reverse primer pool, 1× SYBR Green I (ThermoFisher Scientific, S7563), Q5 Hot Start DNA polymerase (NEB, M0493S), 1× Q5 Hot Start Reaction Buffer, 1× Q5 Hot Start High GC Enhancer, in total 30uL reaction volume. Cycling conditions are: 98°C 30s initial denaturation, followed by 15–25 cycles (terminated before plateau) of 98°C 10s, 56°C 40s, 76°C 10s. PCR is run for 15–25 cycles. Each 96 pool multiplex PCR reaction is then used in a master mix of second PCR with 96 individual primer pair reactions: 0.2mM dNTP, 500nM forward primer, 500nM reverse primer, 1× SYBR Green I, Q5 Hot Start DNA polymerase, 1× Q5 Hot Start Reaction Buffer, 1× Q5 Hot Start High GC Enhancer, in total 6uL reaction volume. Cycling conditions are: 98°C 30s initial denaturation, followed by 20 cycles of 98°C 10s, 62°C 10s, 76°C 10s. 384 individual reactions are pooled and purified on silica columns (NEB, T1030S). Amplicon pool is end-prepared, Illumina adaptor sequences are ligated, adaptor-ligated DNA is size selected with SPRIselect beads for 370bp, and 3 cycle barcoding PCR is performed (NEB, E7645S). 2×150bp paired end sequencing data is generated on Illumina HiSeq 4000 at Novogene.
1D accessibility data analysis
Briefly, per-nucleotide statistical significance of differential mutation rates are calculated using voom-limma (version 3.42.2) method from TMM-normalized mutation count matrix89–91. For per-window accessibility pattern differences, we calculate Anderson-Darling statistic in sliding 11nt windows between per-nucleotide T-statistic values of the window versus the whole amplicon. False discovery rates are estimated by Benjamini-Hochberg procedure. See Supplementary Notes for the full description.
In-cell mutate-and-map
Error-prone PCR was performed as follows: 0.05U/uL Mutazyme II (Agilent, 200550), 1× Mutazyme II buffer, 1× SYBR, 200uM dNTP each, 300nM forward primer, 300nM reverse primer, 100pg Csde1 5’UTR fragment; 95°C 2min initial denaturation, 10 cycles of 95°C 30s, 63°C 20s, 72°C 1min. See Supplementary Table 9 for the primers. 100pg input amount was determined by initially varying the input amounts to determine the amount at which the PCR is in exponential phase (50% of signal plateau). These parameters result in a mutation rate of approximately 1 per 200nt (Fig. S5C). The error-prone PCR amplicon has homology arms for Gibson assembly into pcDNA5/FRT (ThermoFisher Scientific, V601020) plasmid with EGFP. The final construct has a flanking primer region for cDNA amplification upstream of the mutagenized 5’UTR and EGFP open reading frame downstream. NEB 10-beta E. coli cells (NEB, C3020K) are transformed by electroporation and plated over a total of 2000cm2 area to give ~10000 colonies. The plate is scraped and the mutagenesis library plasmid pool is purified.
mESCs are grown, dissociated, and resuspended at 5×105 cells/mL. 3ug mutagenesis library plasmid pool DNA and 7.5uL Lipofectamine 2000 in 100uL OptiMEM (ThermoFisher Scientific, 31985062) are mixed with 2mL cells (1×106 cells) in mESC media. The cells are incubated for 10min in suspension, centrifuged, washed with mESC media, and plated into one well in a 6-well plate. 24 hours after transfection, the cells are washed, trypsinized and resuspended in 3.5mL mESC media. 1mL 1M bicine (Millipore Sigma, B3876), titrated to pH 8.5 at 25°C, is added to resuspended cells (250mM final bicine concentration). 500uL 16% dimethyl sulfate (MilliporeSigma, D186309) in ethanol is added (1.6% final concentration). Cells are mixed and incubated for 6min at 37°C. 2.5mL ice-cold 30% BME (MilliporeSigma, M3148) in ethanol is added to quench the reaction. Following centrifuge to remove the supernatant, the cells are lysed in Trizol (ThermoFisher Scientific, 15596026). Total RNA is phase extracted with chloroform and aqueous phase is purified on silica columns (Zymo, R1013). 10–20ug RNA is DNase treated at 37°C for 30min using 0.2U/uL TURBO DNase (ThermoFisher Scientific, AM2238) with 1U/uL Superase-In (ThermoFisher Scientific, AM2696) in 60uL and purified again on silica column.
1ug RNA is mixed with 1uL 1uM RT primer targeting the downstream EGFP and denatured in 6.25uL total volume (with H2O) at 65°C for 2min, then chilled to 4°C. Reverse transcription reaction conditions are as follows: 20mM Tris-HCl pH7.5, 75mM KCl, 10mM MgCl2, 5mM DTT, 500nM TGIRT (InGex, TGIRT50), 1U/uL Superase-In, 19uL total reaction volume. The RT reaction is preincubated for 25°C for 30min, then initiated with addition of 1uL 12.5mM dNTP each. After incubation at 60°C for 1hour, 1uL 2.5M NaOH is added and the reaction is heated at 95°C for 3min. 1uL 2.5M HCl and 1uL 500mM Tris-HCl pH 7.5 is added to neutralize. 29uL SPRIselect beads (Beckman Coulter, B23318) is used for purification of cDNA; elution volume is 7uL. PCR reaction is performed as follows: 0.2mM dNTP, 300nM forward primer, 300nM reverse primer, 1× SYBR Green I (ThermoFisher Scientific, S7563), Q5 Hot Start DNA polymerase (NEB, M0493S), 1× Q5 Hot Start Reaction Buffer, 1× Q5 Hot Start High GC Enhancer, 2uL cDNA, in total 20uL reaction volume. Cycling conditions are: 98°C 30s initial denaturation, 20 cycles of 98°C 10s, 64°C 10s, 72°C 30s; 20 cycles with full-length amplicon primers. The reaction is diluted 100 fold and used as template in second round PCR with same conditions but using primers for 3×250nt shorter tiling amplicons for 10 cycles. See Supplementary Table 9 for the RT and PCR primers. The reactions are purified on silica columns and pooled. The pooled DNA is end-prepared, Illumina adaptor sequences are ligated, adaptor-ligated DNA is size selected with SPRIselect beads for 370bp, and 3 cycle barcoding PCR is performed (NEB, E7645S). 2×150bp paired end sequencing data is generated on Illumina HiSeq 4000 at Novogene.
2D accessibility data analysis and structure models
Briefly, the normalized co-variation matrix is clustered using multidimensional scaling, N=2. Cluster average accessibility z-scores are used to constrain partition function calculation in Vienna RNA (version 2.4.14)92. 250 structures are sampled for each cluster and used as input suboptimals to REEFFIT (version 0.6.3)83. For visualization of the landscape, we used pairwise distance metrics, structure clustering, and medoid assignment produced by REEFFIT, and sum of weights for structures belonging to each of the 3 clusters represented by a medoid structure is presented in the main figure. Bootstrapping is used to estimate population fraction errors. See Supplementary Notes for the full description.
Data availability
Raw sequencing data (related to Figs. 4, 5 and 6) are deposited to GEO with accession code GSE155656. Sources for publicly available data are described in methods.
Code availability
All softwares used to analyze the study data are listed in the methods section and in the Nature Research Reporting Summary and are publicly available. All codes used to analyze icM2 data are available through a Github repository: github.com/barnalab/icm2p.
Extended Data
Extended Data Fig. 1. Hyperconserved 5’UTRs in vertebrate genomes.

a, Left: heatmap of the positions of LOD≥500 PhastCons elements in each h5UTR. Middle: heatmap of the relative positions (calculated in 100 bins across the h5UTRs) of the elements. Right: plot of average element overlap across the 100 bins to illustrate the positional preference.
b, Histogram of the length of h5UTRs. Average length is 674nt.
c, Histogram of the number of nucleotides overlap between LOD≥500 PhastCons elements and h5UTRs. Average overlap is 389nt.
d, Distributions of cross-tissue transcriptome-proteome correlations for all genes, genes with h5UTRs, or genes with variance-matched non-conserved 5’UTRs. Indicated p-values are from two-sided Wilcoxon rank sum tests for cross-tissue correlation values between h5UTR genes and all genes or between h5UTR genes and variance-matched non-conserved controls.
e, Distributions of the number of annotated alternative 5’UTRs for all genes, genes with h5UTRs, or genes with size-matched non-conserved 5’UTRs. Indicated p-values are from two-sided Wilcoxon rank sum tests for the number of alternative 5’UTRs between h5UTR genes and all genes or between h5UTR genes and size-matched non-conserved controls.
f, Scatter plot illustrating the lack of significant term enrichments for a size-matched set of non-conserved 5’ UTRs. X-axis and y-axis plots expected and the observed number of genes for each term. Blue dashed line indicates the minimum observed/expected ratio cutoff of 3. Green line indicates expected and observed counts where Fisher’s test p-value (p.) is estimated to have FDR=0.05. Neighbor-weighted test p-value (p.) ≤0.05 is further used as an additional cutoff. The final set of enriched terms passing filter is colored by pf and sized by p.
Extended Data Fig. 2. Non-canonical translation activation by hyperconserved 5’UTRs across cell types.

a, Density plots of non-canonical translation initiation activities from h5UTRs by bicistronic reporter assay. X-axis is the luciferase reporter activity ratios. Jittered dots mark individual reporter ratios for each h5UTR in each cell type.
b, Summarized plot of ribosome load (sum of % mRNA times the ribosome number for each fraction) differential ratio between NSCs and ESCs calculated from polysome profiles for each gene shown in Extended Data Figs. 2c–l. Red indicates significant increase in NSCs and black indicates significant decrease (two-sided t-test p≤0.05, n=3, marked by asterisk).
c-l, Endogenous polysome profiles of NSCs versus ESCs for genes with h5UTRs that show high non-canonical translation reporter activities in NSCs compared to ESCs. Distribution of mRNAs across sucrose gradient fractions are plotted. Y-axis plots the mean percent mRNA. Error bars indicate standard error. Asterisk indicates two-sided t-test p≤0.05 for each fraction between the two cell types. n=3 for each cell type. Indicated p-value (pf) is calculated by Fisher’s method across all fractions. Note that Extended Data Fig. 2c shows the profile of 18S rRNA, which indicates lower global translation in NSCs compared to ESCs.
Extended Data Fig. 3. Non-canonical activation by hyperconserved 5’UTRs significantly contributes to translation.

a, Scatter plot of luciferase activity versus RNA level ratios (mean from n=3) observed for the bicistronic reporters of 90 h5UTRs measured in 10T1/2 cells. Dashed line marks the 10% FDR used in Fig. 3a. Spearman correlation indicated on top left.
b, The effect of various truncations of the h5UTRs on non-canonical initiation and total translation efficiency (also see Fig. 3d). Left: positions of truncations. Dashed lines indicate truncations. Purple horizontal lines indicate uORFs; yellow and red lines indicate in-frame and out-of-frame uAUGs, respectively. Middle: non-canonical initiation efficiency. Right: total translation efficiency. X-axis indicates the mean of luciferase reporter ratios relative to the wild-type. Error bars indicate standard error. Dashed line marks the wild-type 5’UTR activity. Asterisk indicates two-sided t-test p≤0.05 for each truncation versus the full-length. The numbers to the left of the bars indicate n and p-values.
c, Comparison of translational activities between the full-length long, non-conserved 5’UTRs versus the only first 300nt truncation. 11 different pairs are tested. X-axis indicates the mean log2 luciferase reporter ratios of each truncation relative to its full-length wild-type. Error bars indicate standard error. Bars colored in red indicate significantly reduced translation in the shorter, truncated 300nt fragment; black indicates significant increase (two-sided t-test, paired n=3, p≤0.05, marked by asterisk). The numbers to the left of the bars indicate p-values.
d, Violin plot of full-length/truncated reporter activity ratios (log2) from hyperconserved and non-conserved 5’UTRs. p indicates two-sided Wilcoxon rank sum test p-value. Box hinges: 25% quantile, median, 75% quantile, respectively from left to right. Whiskers: lower or upper hinge ±1.5*IQR.
e, Scatter plot of change in translation efficiency between full-length and truncated h5UTRs shown in Fig. 3e versus change in uAUG density (change in number of AUGs / change in length between each pair of full-length and truncated h5UTRs). r indicates pearson’s correlation coefficient and p indicates two-tailed p-value.
Extended Data Fig. 4. Cellular remodeling hyperconserved 5’UTR RNA structures.

a, Stacked bar plots showing proportions of significant (FDR≤0.05) or not significant windows that overlap uAUG in black versus that do not overlap uAUG in red. OR indicates odds ratio for overlaps uAUG / does not overlap uAUG, and p indicates Fisher’s test p-value (one-sided, Ha=odds ratio>0).
b, Stacked bar plots showing proportions of significant (FDR≤0.05) or not significant windows that overlap uORF in black versus that do not overlap uORF in red. OR indicates odds ratio for overlaps uORF / does not overlap uORF, and p indicates Fisher’s test p-value (one-sided, Ha=odds ratio>0).
c, Zoomed-in view of differential accessibilities along h5UTRs with one or more significantly different windows under ATP depletion. Top plot shows -log10 p-value for each window. Highlighted boxes mark significantly different windows, above the dashed line indicating 5% FDR. Middle plot shows differential accessibility on the y-axis, where greater than zero indicates increased accessibility upon ATP depletion and less than zero indicates decreased accessibility. Bottom plot shows differential accessibility for in vitro refolded RNA. Error bars in each plot show standard error, n=3. The three profiled regions shown on the left side exhibit discordant profiles between accessibility changes observed in cells following ATP depletion and accessibility changes observed for in cell versus in vitro refolded RNA. The other three on the right side exhibit concordant profiles.
Extended Data Fig. 5. icM2 reveals structured elements in the hyperconserved Csde1 5’UTR.

a, Boxplot of average PhastCons scores in significant windows of ATP-dependent remodeling versus all windows shown in Fig. 4c. p indicates two-sided Wilcoxon rank sum test p-value.
b, Same as Extended Data Figure 5a, but showing the distribution of average PhyloP scores.
Extended Data Fig. 6. In-vitro M2 analysis of Csde1 5’UTR.

a, Heatmap of in-vitro M2 accessibility matrix for Csde1 5’UTR from position 190 to 386. For each row, the chemical mapping profile of a single-nucleotide variant of the RNA is plotted across the columns, where the colors indicate z-scaled accessibility change values from the wild-type RNA. 1D data from each mutant are vertically stacked to display a 2D matrix. White boxes mark perturbation signals that support the model shown in Extended Data Fig. 6b; color bars at the bottom indicate the nucleotide positions of the stems that match the same color in the model.
b, The model for the in-vitro structure of Csde1 5’UTR from position 190 to 386. Also see Extended Data Fig. 6a
Supplementary Material
Acknowledgements
We would like to thank the members of the Barna lab for constructive criticism of the manuscript. This work was supported by New York Stem Cell Foundation grant NYSCF-R-I36 (M.B.), NIH grant 1R01HD086634 (M.B.), Alfred P. Sloan Research Fellowship (M.B.), Pew Scholars Award (M.B.), Mallinckrodt Foundation Award (M.B.), Benchmark Stanford Graduate Fellowship (G.W.B.), Walter and Idun Berry Foundation (E.S.C.). M.B. is a New York Stem Cell Robertson Investigator.
Footnotes
Competing interests
The authors declare no competing interests.
References (main text)
- 1.Dermitzakis ET, Reymond A. & Antonarakis SE Conserved non-genic sequences - an unexpected feature of mammalian genomes. Nat. Rev. Genet 6, 151–157 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Harmston N, Baresic A. & Lenhard B. The mystery of extreme non-coding conservation. Philos. Trans. R. Soc. Lond. B. Biol. Sci 368, 20130021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halligan DL et al. Positive and negative selection in murine ultraconserved noncoding elements. Mol. Biol. Evol 28, 2651–2660 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Bejerano G. et al. Ultraconserved elements in the human genome. Science 304, 1321–1325 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Dimitrieva S. & Bucher P. Genomic context analysis reveals dense interaction network between vertebrate ultraconserved non-coding elements. Bioinformatics 28, i395–i401 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boffelli D, Nobrega MA & Rubin EM Comparative genomics at the vertebrate extremes. Nat. Rev. Genet 5, 456–465 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Lindblad-Toh K. et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438, 803–819 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Sandelin A. et al. Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics 5, 99 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de la Calle-Mustienes E. et al. A functional survey of the enhancer activity of conserved non-coding sequences from vertebrate Iroquois cluster gene deserts. Genome Res. 15, 1061–1072 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakuraba Y. et al. Identification and characterization of new long conserved noncoding sequences in vertebrates. Mamm. Genome 19, 703–712 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Dermitzakis ET et al. Comparison of human chromosome 21 conserved nongenic sequences (CNGs) with the mouse and dog genomes shows that their selective constraint is independent of their genic environment. Genome Res. 14, 852–859 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katzman S. et al. Human genome ultraconserved elements are ultraselected. Science 317, 915 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Pennacchio LA et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Visel A. et al. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat. Genet 40, 158–160 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visel A. et al. A high-resolution enhancer atlas of the developing telencephalon. Cell 152, 895–908 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahituv N. et al. Deletion of ultraconserved elements yields viable mice. PLoS Biol. 5, e234 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McLean C. & Bejerano G. Dispensability of mammalian DNA. Genome Res. 18, 1743–1751 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickel DE et al. Ultraconserved enhancers are required for normal development. Cell 172, 491–499.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osterwalder M. et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lareau LF, Inada M, Green RE, Wengrod JC & Brenner SE Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446, 926–929 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Ni JZ et al. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 21, 708–718 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas JD et al. RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat. Genet 52, 84–94 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calin GA et al. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 12, 215–229 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Liz J. et al. Regulation of pri-miRNA processing by a long noncoding RNA transcribed from an ultraconserved region. Mol. Cell 55, 138–147 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Xue S. et al. RNA regulons in Hox 5’ UTRs confer ribosome specificity to gene regulation. Nature 517, 33–38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siepel A. et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang L. et al. A quantitative proteome map of the human body. Cell 183, 269–283.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aguet F. et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. BioRxiv (2019). doi: 10.1101/787903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steri M, Idda ML, Whalen MB & Orrù V. Genetic variants in mRNA untranslated regions. Wiley Interdiscip. Rev. RNA 9, e1474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blanco-Suarez E, Liu T-F, Kopelevich A. & Allen NJ Astrocyte-Secreted Chordin-like 1 Drives Synapse Maturation and Limits Plasticity by Increasing Synaptic GluA2 AMPA Receptors. Neuron 100, 1116–1132.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakuta H. et al. Ventroptin: a BMP-4 antagonist expressed in a double-gradient pattern in the retina. Science 293, 111–115 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Webb TR et al. X-linked megalocornea caused by mutations in CHRDL1 identifies an essential role for ventroptin in anterior segment development. Am. J. Hum. Genet 90, 247–259 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gandal MJ et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu T. et al. Chordin-Like 1 Improves Osteogenesis of Bone Marrow Mesenchymal Stem Cells Through Enhancing BMP4-SMAD Pathway. Front Endocrinol (Lausanne) 10, 360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pei Y-F et al. Hypermethylation of the CHRDL1 promoter induces proliferation and metastasis by activating Akt and Erk in gastric cancer. Oncotarget 8, 23155–23166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osório C. et al. Growth differentiation factor 5 is a key physiological regulator of dendrite growth during development. Development 140, 4751–4762 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Keeffe GW et al. Region-specific role of growth differentiation factor-5 in the establishment of sympathetic innervation. Neural Dev. 11, 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H, Li J, Xu D, Zhang Q. & Cui T. Growth differentiation factor 5 improves neurogenesis and functional recovery in adult mouse hippocampus following traumatic brain injury. Front. Neurol 9, 592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buxton P, Edwards C, Archer CW & Francis-West P. Growth/differentiation factor-5 (GDF-5) and skeletal development. J. Bone Joint Surg. Am 83-A Suppl 1, S23–30 (2001). [PubMed] [Google Scholar]
- 40.Panganiban G. & Rubenstein JLR Developmental functions of the Distal-less/Dlx homeobox genes. Development 129, 4371–4386 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Depew MJ, Simpson CA, Morasso M. & Rubenstein JLR Reassessing the Dlx code: the genetic regulation of branchial arch skeletal pattern and development. J. Anat 207, 501–561 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polleux F, Morrow T. & Ghosh A. Semaphorin 3A is a chemoattractant for cortical apical dendrites. Nature 404, 567–573 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Serini G. et al. Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature 424, 391–397 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Shelly M. et al. Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron 71, 433–446 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polleux F, Giger RJ, Ginty DD, Kolodkin AL & Ghosh A. Patterning of cortical efferent projections by semaphorin-neuropilin interactions. Science 282, 1904–1906 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Good PF et al. A role for semaphorin 3A signaling in the degeneration of hippocampal neurons during Alzheimer’s disease. J. Neurochem 91, 716–736 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Galan-Caridad JM et al. Zfx controls the self-renewal of embryonic and hematopoietic stem cells. Cell 129, 345–357 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee ASY, Kranzusch PJ & Cate JHD eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 522, 111–114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilbert WV, Zhou K, Butler TK & Doudna JA Cap-independent translation is required for starvation-induced differentiation in yeast. Science 317, 1224–1227 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Martin F. et al. Cap-assisted internal initiation of translation of histone H4. Mol. Cell 41, 197–209 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Legnini I. et al. Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol. Cell 66, 22–37.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pamudurti NR et al. Translation of CircRNAs. Mol. Cell 66, 9–21.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leppek K. et al. Gene- and Species-Specific Hox mRNA Translation by Ribosome Expansion Segments. Mol. Cell 80, 980–995.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hershey JWB, Sonenberg N. & Mathews MB Principles of translational control: an overview. Cold Spring Harb. Perspect. Biol 4, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weingarten-Gabbay S. et al. Comparative genetics. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 351, (2016). [DOI] [PubMed] [Google Scholar]
- 56.Xiao Z-S, Simpson LG & Quarles LD IRES-dependent translational control of Cbfa1/Runx2 expression. J. Cell. Biochem 88, 493–505 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Jang GM et al. Structurally distinct elements mediate internal ribosome entry within the 5’-noncoding region of a voltage-gated potassium channel mRNA. J. Biol. Chem 279, 47419–47430 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Holcik M. & Sonenberg N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol 6, 318–327 (2005). [DOI] [PubMed] [Google Scholar]
- 59.El-Naggar AM & Sorensen PH Translational control of aberrant stress responses as a hallmark of cancer. J. Pathol 244, 650–666 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Spriggs KA, Bushell M. & Willis AE Translational regulation of gene expression during conditions of cell stress. Mol. Cell 40, 228–237 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Washietl S, Hofacker IL, Lukasser M, Hüttenhofer A. & Stadler PF Mapping of conserved RNA secondary structures predicts thousands of functional noncoding RNAs in the human genome. Nat. Biotechnol 23, 1383–1390 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Torarinsson E. et al. Comparative genomics beyond sequence-based alignments: RNA structures in the ENCODE regions. Genome Res. 18, 242–251 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parker BJ et al. New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res. 21, 1929–1943 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith MA, Gesell T, Stadler PF & Mattick JS Widespread purifying selection on RNA structure in mammals. Nucleic Acids Res. 41, 8220–8236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eddy SR Computational analysis of conserved RNA secondary structure in transcriptomes and genomes. Annu. Rev. Biophys 43, 433–456 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rivas E, Clements J. & Eddy SR Estimating the power of sequence covariation for detecting conserved RNA structure. Bioinformatics 36, 3072–3076 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Homan PJ et al. Single-molecule correlated chemical probing of RNA. Proc Natl Acad Sci USA 111, 13858–13863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zubradt M. et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 14, 75–82 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mustoe AM, Lama NN, Irving PS, Olson SW & Weeks KM RNA base-pairing complexity in living cells visualized by correlated chemical probing. Proc Natl Acad Sci USA 116, 24574–24582 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beaudoin J-D et al. Analyses of mRNA structure dynamics identify embryonic gene regulatory programs. Nat. Struct. Mol. Biol 25, 677–686 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patalano S, Mihailovich M, Belacortu Y, Paricio N. & Gebauer F. Dual sex-specific functions of Drosophila Upstream of N-ras in the control of X chromosome dosage compensation. Development 136, 689–698 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Elatmani H. et al. The RNA-binding protein Unr prevents mouse embryonic stem cells differentiation toward the primitive endoderm lineage. Stem Cells 29, 1504–1516 (2011). [DOI] [PubMed] [Google Scholar]
- 73.Mitchell SA, Brown EC, Coldwell MJ, Jackson RJ & Willis AE Protein factor requirements of the Apaf-1 internal ribosome entry segment: roles of polypyrimidine tract binding protein and upstream of N-ras. Mol. Cell. Biol 21, 3364–3374 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schepens B. et al. A role for hnRNP C1/C2 and Unr in internal initiation of translation during mitosis. EMBO J. 26, 158–169 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guo A-X, Cui J-J, Wang L-Y & Yin J-Y The role of CSDE1 in translational reprogramming and human diseases. Cell Commun. Signal 18, 14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moore KS et al. Csde1 binds transcripts involved in protein homeostasis and controls their expression in an erythroid cell line. Sci. Rep 8, 2628 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wurth L. et al. UNR/CSDE1 Drives a Post-transcriptional Program to Promote Melanoma Invasion and Metastasis. Cancer Cell 30, 694–707 (2016). [DOI] [PubMed] [Google Scholar]
- 78.Horos R. et al. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood 119, 262–272 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Guo H. et al. Disruptive variants of CSDE1 associate with autism and interfere with neuronal development and synaptic transmission. Sci. Adv 5, eaax2166 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Saltel F. et al. Unr defines a novel class of nucleoplasmic reticulum involved in mRNA translation. J. Cell Sci 130, 1796–1808 (2017). [DOI] [PubMed] [Google Scholar]
- 81.Sanders SJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kladwang W, VanLang CC, Cordero P. & Das R. A two-dimensional mutate-and-map strategy for non-coding RNA structure. Nat. Chem 3, 954–962 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cordero P. & Das R. Rich RNA Structure Landscapes Revealed by Mutate-and-Map Analysis. PLoS Comput. Biol 11, e1004473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bhaskaran H. & Russell R. Kinetic redistribution of native and misfolded RNAs by a DEAD-box chaperone. Nature 449, 1014–1018 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
References (online methods)
- 85.Alexa A, Rahnenführer J. & Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Motenko H, Neuhauser SB, O’Keefe M. & Richardson JE MouseMine: a new data warehouse for MGI. Mamm. Genome 26, 325–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Concordet J-P & Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yoon A. et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 312, 902–906 (2006). [DOI] [PubMed] [Google Scholar]
- 89.Robinson MD & Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Law CW, Chen Y, Shi W. & Smyth GK voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lorenz R. et al. ViennaRNA Package 2.0. Algorithms Mol. Biol 6, 26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data (related to Figs. 4, 5 and 6) are deposited to GEO with accession code GSE155656. Sources for publicly available data are described in methods.