

Abstract
Calcium signaling via mitochondrial calcium uniporter (MCU) complex coordinates mitochondrial bioenergetics with cellular energy demands. Emerging studies show that the stability and activity of the pore-forming subunit of the complex, MCU, is dependent on the mitochondrial phospholipid, cardiolipin (CL), but how this impacts calcium-dependent mitochondrial bioenergetics in CL-deficiency disorder like Barth syndrome (BTHS) is not known. Here we utilized multiple models of BTHS including yeast, mouse muscle cell line, as well as BTHS patient cells and cardiac tissue to show that CL is required for the abundance and stability of the MCU-complex regulatory subunit MICU1. Interestingly, the reduction in MICU1 abundance in BTHS mitochondria is independent of MCU. Unlike MCU and MICU1/MICU2, other subunit and associated factor of the uniporter complex, EMRE and MCUR1, respectively, are not affected in BTHS models. Consistent with the decrease in MICU1 levels, we show that the kinetics of MICU1-dependent mitochondrial calcium uptake is perturbed and acute stimulation of mitochondrial calcium signaling in BTHS myoblasts fails to activate pyruvate dehydrogenase, which in turn impairs the generation of reducing equivalents and blunts mitochondrial bioenergetics. Taken together, our findings suggest that defects in mitochondrial calcium signaling could contribute to cardiac and skeletal muscle pathologies observed in BTHS patients.
Introduction
Mitochondria respond to increased cellular energy demands through calcium (Ca2+) uptake via the mitochondrial calcium uniporter (MCU) complex (1,2). Elevation in mitochondrial Ca2+ stimulates the activities of mitochondrial matrix dehydrogenases resulting in increased NADH generation, which fuels an increase in mitochondrial respiration and ATP production. Ca2+ uptake through MCU, the pore-forming subunit, is regulated by a number of accessory factors, including the mitochondrial calcium uptake proteins 1, 2 and 3 (MICU1, MICU2, MICU3), essential MCU regulator (EMRE), dominant-negative MCU β-subunit (MCUb) and MCU Regulator 1 (MCUR1) (3–9). Structural studies have shown that MCU, EMRE, MICU1 and MICU2 together form a holocomplex, which is often referred to as the uniporter complex or uniplex (10,11).
MICU1 is the principal regulator of the MCU channel that monitors cytosolic Ca2+ [Ca2+]i transients and allows its entry into the mitochondria only under conditions of high [Ca2+]i (12). MICU1 binds to Ca2+ and adopts a structural conformation conducive for Ca2+ inflow through the MCU pore (10,13). In addition, MICU1 governs the spatial distribution of MCU in the inner mitochondrial membrane, which is required for the activation of the uniporter complex (14). As a result, genetic ablation of MICU1 causes dysregulation of uniporter complex function, both by allowing mitochondrial Ca2+ accumulation at low [Ca2+]i and by abolishing cooperative activation of the complex at high [Ca2+]i (15). The importance of MICU1 in mitochondrial Ca2+ signaling is further highlighted by the fact that loss-of-function mutations in MICU1 result in brain and skeletal muscle defects and a progressive and debilitating movement disorder in humans (16,17). Till date, no patients have been identified with a mutation in any of the other subunits of the uniporter complex.
Utilizing a heterologous expression system consisting of yeast mutants of mitochondrial phospholipid biosynthesis, we recently reported a specific requirement of cardiolipin (CL) in the stability and activity of MCU (18). CL is a signature phospholipid of the mitochondria, and its deficiency results in Barth syndrome (BTHS), an X-linked cardio- and skeletal-muscle myopathy that is caused by mutations in the CL remodeling enzyme TAFAZZIN (previously referred to as TAZ) (19,20). Consistent with our findings in the yeast surrogate system, we showed that BTHS patient-derived cells and cardiac tissues also exhibit a reduction in MCU (18). However, currently we do not know if CL-deficiency in BTHS also impacts other subunits of the uniporter complex and whether mitochondrial Ca2+ signaling-dependent downstream bioenergetic processes are disrupted in BTHS disease models. In this study, we utilized multiple BTHS models, including patient-derived cells, cardiac tissue, a murine skeletal muscle cell model of BTHS, and CL-deficient yeast to uncover an essential requirement of CL for MICU1 stability and MICU1-dependent mitochondrial Ca2+ signaling. Importantly, we found that the disruption of MCU and MICU1 blunts mitochondrial bioenergetic pathways in response to an extra-mitochondrial Ca2+ signal in BTHS cells.
Results
MICU1 abundance and stability are reduced in a murine myoblast model of BTHS
We recently showed that the abundance and stability of the pore-forming subunit, MCU, is reduced in multiple cellular models and patient samples of BTHS (18). However, it is not known if CL-requirement is specific to MCU or is it also required for the abundance, assembly, or activity of the other regulatory components of the uniporter complex. A previous study had shown that MICU1, the principal regulatory subunit of the uniporter complex, binds CL in vitro (21). To uncover the role of CL in MICU1 function in vivo, we determined MICU1 abundance in a C2C12 Taz-KO murine myoblast cell line model of BTHS by SDS-PAGE/immunoblotting and observed a ~50% reduction in the steady-state levels of MICU1 in C2C12 Taz-KO myoblasts as compared to the wild type (WT) cells (Fig. 1A and B). Consistent with a decrease in the monomeric protein, the abundance of MICU1 containing uniporter complexes was also moderately reduced in Taz-KO cells, as observed by Blue Native Polyacrylamide gel electrophoresis/immunoblotting (BN-PAGE) (Fig. 1C). These findings suggest that a reduction in CL levels and/or an accumulation of its precursor monolysocardiolipin (MLCL) results in the reduced abundance of MICU1 in Taz-KO cells.
Figure 1.
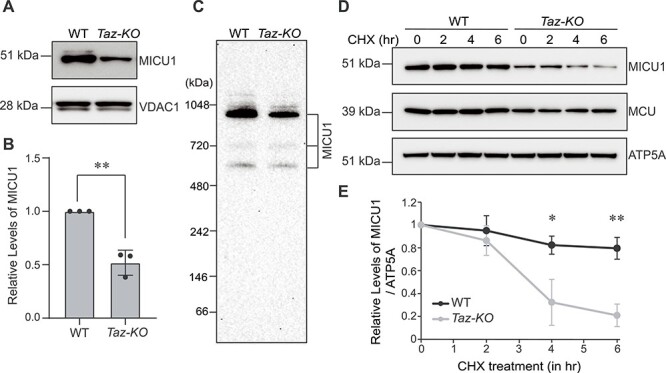
MICU1 abundance and stability are reduced in C2C12 Taz-KO cells. (A) SDS-PAGE immunoblot analysis of MICU1 in mitochondria isolated from C2C12 WT and Taz-KO myoblasts. VDAC1 is used as a loading control. (B) Quantification of relative MICU1 levels from (A) by densitometry using ImageJ software. Data shown as mean ± SD. n = 3. **P < 0.005. (C) BN-PAGE immunoblot analysis of MICU1 in digitonin-solubilized mitochondria isolated from C2C12 WT and Taz-KO myoblasts. Blot is representative of three independent experiments. (D) SDS-PAGE immunoblot analysis of MICU1 and MCU in C2C12 WT and Taz-KO myoblasts treated with 20 μg/ml cycloheximide (CHX) for the indicated time-points. ATP5A is used as loading control. (E) Quantification of relative MICU1 levels from (D) by densitometry using ImageJ software. Data are represented as mean ± SD. n = 3. *P < 0.05, **P < 0.005.
MICU1 associates with the inner mitochondrial membrane peripherally but its interactions with other integral membrane components of the uniporter complex, MCU and EMRE (6,22), and its ability to bind CL in vitro (21) suggests that loss of CL may prevent membrane anchoring of MICU1 in vivo, which in turn would trigger its degradation. To test whether the stability of MICU1 protein in vivo is dependent on CL, we performed a cycloheximide chase assay in C2C12 WT and Taz-KO cells. Inhibition of nascent protein synthesis by cycloheximide allows measurement of the turnover rate of a protein in vivo. Using this assay, we observed a more rapid rate of turnover of MICU1 in CL-depleted Taz-KO cells as compared to WT cells (Fig. 1D and E).
We had previously shown that MCU turnover is increased in C2C12 Taz-KO cells at a much longer timescale of around 72 h (18). Therefore, following cycloheximide treatment, we measured the levels of MCU at the same time-points at which MICU1 was degraded. Consistent with our previous study (18), we found that the basal levels of MCU were reduced in Taz-KO cells when compared to the WT cells; however, the abundance of MCU remained unchanged within the 6 h timeframe in WT as well as in Taz-KO cells (Fig. 1D). This result suggests that reduced MICU1 stability upon loss of CL is independent of MCU.
Uniporter components do not exhibit a generic requirement for CL
CL requirement for the abundance and stability of both MCU and MICU1 led us to ask if there is a generic requirement of CL for uniporter subunits. To answer this, we utilized yeast, Saccharomyces cerevisiae, which provides a ‘clean’ background to test the effect of CL on the uniporter complex subunits, as it does not contain any homologs of the core components of the uniporter machinery (23). Upon heterologous expression of the human MICU1 protein alone, without the co-expression of MCU and EMRE, we observed a ~50% reduction in the abundance of MICU1 in CL-deficient crd1Δ yeast mitochondria compared to WT (Fig. 2A and B). These data clearly established that the effect of CL on MICU1 abundance is independent of MCU and because crd1Δ cells do not accumulate MLCL, the reduced abundance of MICU1 could be attributed to CL deficiency. In contrast to MICU1, the levels of human MCUR1 protein remained unaltered when expressed in WT and crd1Δ cells (Fig. 2C and D), suggesting that the requirement of CL is specific to MICU1.
Figure 2.
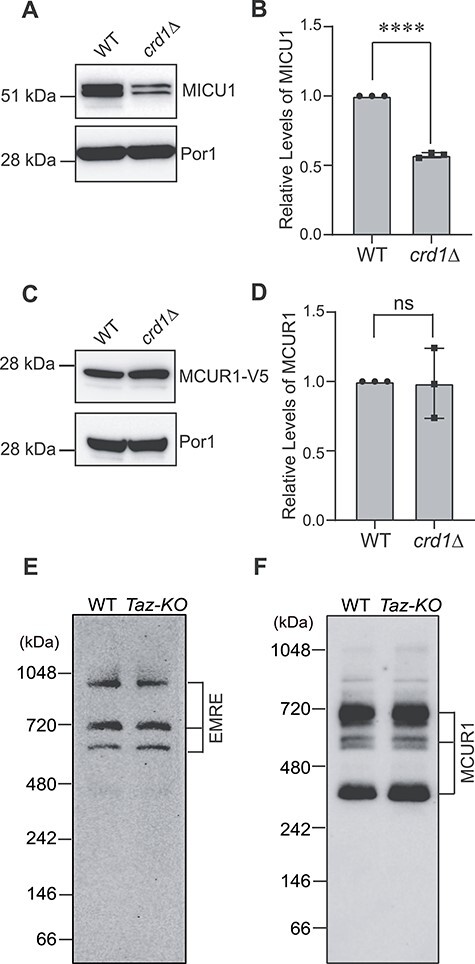
Loss of CL does not cause a generic decline in uniporter subunits. (A) SDS-PAGE immunoblot analysis of MICU1 in mitochondria isolated from WT and crd1Δ yeast expressing human MICU1. Por1 is used as a loading control. (B) Quantification of relative MICU1 levels from (A) using ImageJ software. Data shown as mean ± SD. n = 3. ****P < 0.0001. (C) SDS-PAGE immunoblot of MCUR1-V5 in mitochondria isolated from WT and crd1Δ yeast expressing human MCUR1 tagged with V5. Por1 is used as a loading control. (D) Quantification of relative MCUR1 levels from (C) using ImageJ software. Data shown as mean ± SD. n = 3. ns, not significant. (E) BN-PAGE immunoblot of EMRE in mitochondria isolated from C2C12 WT and Taz-KO cells. Blot is representative of three independent experiments. (F) BN-PAGE immunoblot of MCUR1 in mitochondria isolated from C2C12 WT and Taz-KO cells. Blot is representative of three independent experiments WT, wild type.
Next, we measured the abundance of endogenous EMRE and MCUR1 in C2C12 WT and Taz-KO cells to test whether CL-depletion also affects these integral membrane subunits of the uniporter complex. BN-PAGE/immunoblot analysis of EMRE in C2C12 WT and Taz-KO mitochondria revealed that the abundance of EMRE containing oligomeric uniporter complexes were unaltered under CL deficiency (Fig. 2E). Similarly, the levels of MCUR1 containing uniporter complexes also remained unchanged in CL-deficient cells (Fig. 2F). Taken together, these data reveal that the uniporter subunits do not have a generic requirement of CL.
MICU1 abundance is reduced in BTHS patient-derived cells and cardiac tissue
To directly test if CL deficiency impacts MICU1 abundance in BTHS patient-derived samples we used a BTHS patient B-lymphocyte cell line, which have reduced levels of CL (18). Immunoblot analysis showed a marked reduction (~70%) in the levels of MICU1 in the patient B-lymphocyte cells as compared to the control (Fig. 3A and B). Consistent with our cellular models, we found a reduction in MICU1 abundance in four out of five cardiac tissue samples obtained from five different BTHS patients (Fig. 3C and D). The differences in the levels of MICU1 in patient cardiac tissues could be due to differences in the age, the genetic background, or the type of mutation in these BTHS patients. Additionally, we found that the abundance of MICU2, a paralog of MICU1 (2,7), was also reduced to ~50% in BTHS patient B-lymphocytes when compared to the control cells (Fig. 3E and F).
Figure 3.
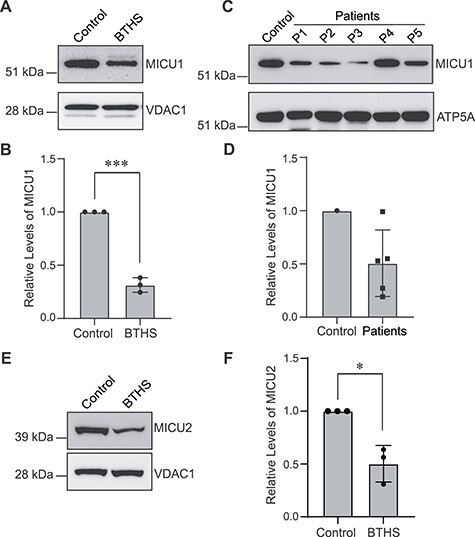
MICU1 is reduced in BTHS patient-derived B-lymphocytes and cardiac tissue. (A) SDS-PAGE immunoblot analysis of MICU1 in mitochondria isolated from control and BTHS patient B-lymphocytes. VDAC1 is used as a loading control. (B) Quantification of relative MICU1 levels using ImageJ software. Data shown as mean ± SD. n = 3. ***P < 0.0005. (C) SDS-PAGE immunoblot analysis of MICU1 in cardiac tissue lysates obtained from control and five BTHS patients (P1–P5). ATP5A is used as a loading control. (D) Quantification of immunoblots from (C) using ImageJ software. Data shown as mean ± SD. n = 5. (E) SDS-PAGE immunoblot analysis of MICU2 in mitochondria isolated from control and BTHS B-lymphocytes. VDAC1 is used as a loading control. (F) Quantification of relative MICU2 levels using ImageJ software. Data shown as mean ± SD. n = 3. *P < 0.05.
MCU-complex-dependent mitochondrial Ca2+ uptake is disrupted in C2C12 Taz-KO myoblasts
A decrease in MICU1 abundance predicts a reduction in MICU1-dependent Ca2+ uptake kinetics in C2C12 Taz-KO myoblasts. MICU1 has been previously shown to perform a ‘gatekeeper’ role in mitochondrial Ca2+ uptake, such that under low [Ca2+]i it blocks Ca2+ entry into the mitochondria and under high [Ca2+]i it opens the MCU pore to facilitate rapid entry of Ca2+ into the mitochondria (12,15). Therefore, we measured mitochondrial Ca2+ uptake with increasing concentrations of exogenously added Ca2+. We found that compared to WT mitochondria, at low concentrations of exogenous Ca2+, BTHS mitochondria showed an increased tendency to uptake Ca2+ (Fig. 4A–C and E) however, at higher concentrations we observed a significant reduction in mitochondrial Ca2+ uptake (Fig. 4D and E). These results suggest a disruption in MICU1-mediated control of Ca2+ uptake, which is likely due to reduced mitochondrial CL in Taz-KO myoblasts.
Figure 4.
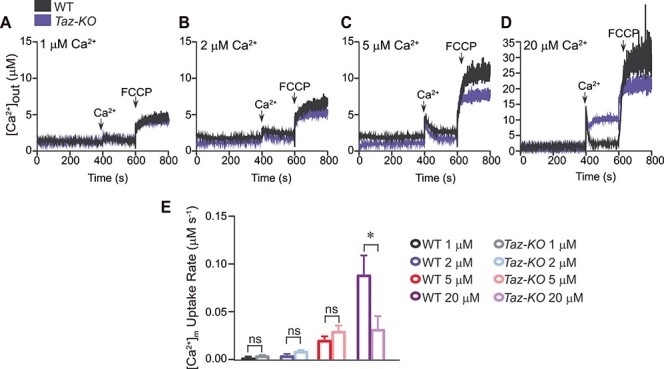
MCU-mediated mitochondrial Ca2+ uptake is reduced in C2C12 Taz-KO cells. (A–D) Permeabilized C2C12 WT and Taz-KO cells were pulsed with different [Ca2+] as indicated. Representative traces show bath [Ca2+] (μM). (E) Rate of [Ca2+]m as function of MCU channel activity. Data are represented as mean ± SEM. n = 3. *P < 0.05, ns, not significant. [Ca2+]m, mitochondrial Ca2+.
Ca2+-stimulated mitochondrial bioenergetics is disrupted in CL-deficient C2C12 cells
Mitochondrial Ca2+ regulates the activities of mitochondrial matrix dehydrogenases including pyruvate dehydrogenase (PDH) (2). Mitochondrial matrix [Ca2+] regulates PDH activity by stimulating PDH phosphatase, which dephosphorylates the E1-α subunit of the PDH complex, thereby activating the PDH enzyme. Previous studies have shown that genetic ablation of MCU in cardiac and skeletal muscle mitochondria results in increased levels of the phosphorylated form of PDH, which leads to a decrease in overall PDH activity (24,25). Thus, we asked whether decreased mitochondrial Ca2+ uptake in Taz-KO cells results in the accumulation of phosphorylated PDH in the mitochondria. To test this, we first validated our assay conditions and the antibodies against the phosphorylated form of PDH in C2C12 WT cells by using a λ-phosphatase enzyme (Fig. 5A).
Figure 5.
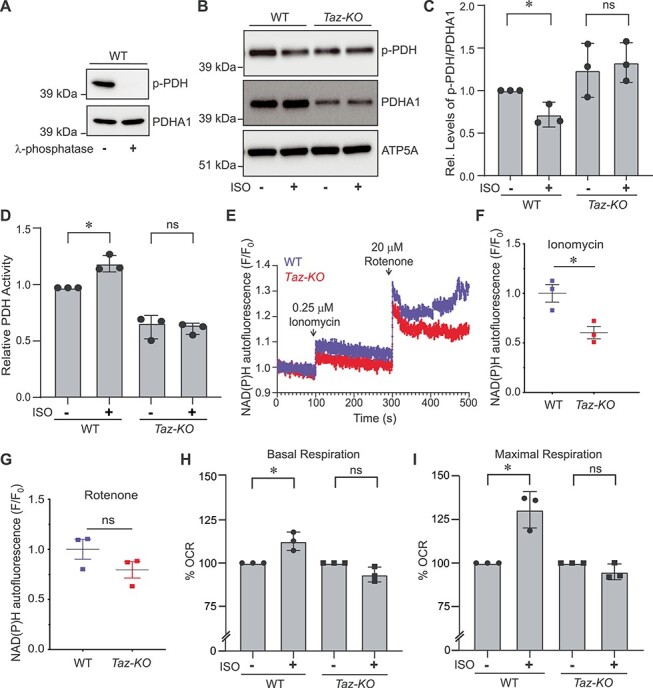
Ca2+-stimulated mitochondrial bioenergetics is impaired in Taz-KO cells. (A) SDS-PAGE immunoblot analysis of phosphorylated PDH (p-PDH) and total PDH (PDHA1) in C2C12 WT mitochondrial lysate treated with or without λ-phosphatase for 10 min. (B) SDS-PAGE immunoblot analysis of p-PDH and total PDHA1 in mitochondria isolated from C2C12 WT and Taz-KO myoblasts treated either with DMSO or 25 μM ISO for 40 min. ATP5A is used to show equal loading. (C) Quantification of relative p-PDH levels from (B) normalized to total PDHA1 by densitometry using ImageJ software. Data shown as mean ± SD. n = 3. *P < 0.05. ns, not significant. (D) Relative PDH activity in mitochondria isolated from WT and Taz-KO myoblasts treated either with DMSO or 25 μM ISO for 40 min. Data shown as mean ± SD. n = 3. *P < 0.05. ns, not significant. (E) NAD(P)H fluorescence changes in C2C12 WT and Taz-KO cells after sequential additions of ionomycin (0.25 μM) and rotenone (20 μM). (F) Quantification of ΔNAD(P)H fluorescence after ionomycin addition. (G) Quantification of ΔNAD(P)H fluorescence after rotenone addition. Data shown as mean ± SEM. n = 3. *P < 0.05. ns, not significant. (H and I) Basal (H) and maximal (I) oxygen consumption rate (OCR) in intact C2C12 WT and Taz-KO cells treated with either DMSO or 25 μM ISO. Data represented as mean ± SD (n = 15, three biological replicates each with five technical replicates per experiment). *P < 0.05, ns, not significant. CCCP and antimycin A were used to measure maximum respiratory capacity and mitochondria-specific respiration, respectively. OCR data are reported as % OCR normalized to total protein content.
Next, we treated C2C12 WT and Taz-KO cells with isoprenaline hydrochloride (ISO), a β-adrenergic agonist, known to elevate intracellular [Ca2+]i followed by an increase in mitochondrial Ca2+ uptake (24,25). Upon a 40 min treatment with ISO, WT cells showed an expected decrease in the levels of PDH phosphorylation (Fig. 5B and C). However, the Taz-KO cells were refractory to the mitochondrial Ca2+ uptake-mediated decrease in PDH phosphorylation status (Fig. 5B and C). Additionally, we found that the overall abundance of the PDH subunit (PDHA1) was reduced and the relative levels of phosphorylated PDH was higher in Taz-KO cells as compared to the WT cells (Fig. 5B and C). We further validated this observation by directly measuring PDH activity in WT and Taz-KO cells. Treatment with ISO significantly elevated PDH activity in WT cells but not in Taz-KO cells (Fig. 5D), suggesting that a decrease in the mitochondrial Ca2+-mediated dephosphorylation of PDH translates to blunted PDH activity in BTHS cells upon a β-adrenergic signal. Moreover, consistent with the reduction in total PDHA1 levels in Taz-KO cells, the basal PDH activity was also reduced by ~40% in Taz-KO cells when compared to WT (Fig. 5D). Mitochondrial Ca2+ uptake enhances the activities of matrix dehydrogenases, which results in increased mitochondrial NADH production (24,26). Therefore, we measured NADH production in C2C12 WT and Taz-KO cells upon ionomycin stimulation. As expected ionomycin treatment resulted in increased production of NADH in WT cells as compared to Taz-KO cells (Fig. 5E and F). Inhibiting NADH dehydrogenase by rotenone increased NADH signal in both WT and Taz-KO cells to a similar extent (Fig. 5G). These data indicate that the mitochondrial NADH production is controlled by MCU-mediated Ca2+ dynamics.
Elevation in mitochondrial Ca2+ has previously been shown to enhance mitochondrial respiration (24,25). Therefore, we asked if a reduction in the activation of PDH enzyme and a concomitant decrease in NADH production diminish mitochondrial respiration. To test this idea, we measured oxygen consumption rates (OCR) in intact C2C12 WT and Taz-KO cells after stimulating them with ISO to increase [Ca2+]i and subsequent mitochondrial Ca2+ uptake. WT cells showed a modest but significant increase in both basal and CCCP-driven maximal respiration following ISO stimulation (Fig. 5H and I). However, CL-deficient Taz-KO cells were unable to stimulate either basal or maximal mitochondrial respiration following treatment with ISO (Fig. 5H and I). These data suggest that mitochondrial Ca2+-stimulated elevation in respiration following an acute stress signal is blunted in BTHS Taz-KO cells. Collectively, these results show that optimal levels of CL are essential for stimulating Ca2+-driven mitochondrial bioenergetic functions in BTHS cells.
Discussion
Mitochondrial dysfunction caused by a decline in the levels and composition of CL is a hallmark of BTHS, an inherited X-linked genetic disorder characterized by debilitating cardiomyopathy and skeletal muscle myopathy (27,28). Although cellular and mitochondrial functions of CL have been extensively studied (29,30), we still do not know the full repertoire of CL-dependent mitochondrial functions that underlie BTHS disease pathogenesis (20). CL depletion leads to a decrease in mitochondrial energy generation and an increase in oxidative stress, both of which are regulated by mitochondrial Ca2+ signaling (2,31). However, the biochemical link between CL depletion, mitochondrial Ca2+ signaling and mitochondrial bioenergetics has not been determined in BTHS models. Here, we uncover the link between these processes by demonstrating a disruption in mitochondrial Ca2+ import machinery and a suppression of Ca2+-stimulated mitochondrial bioenergetics in CL-depleted BTHS cells, a finding that could explain some of the clinical symptoms present in BTHS patients.
Our recent study demonstrating an essential requirement of CL for the stability and activity of MCU identified the mitochondrial membrane phospholipid composition as an important physiological determinant of uniporter function in vivo (18). This functional study was later corroborated by a structural study, which mapped physical interactions between CL and MCU (11) as well as an in vitro proteoliposome-based study that demonstrated a requirement of CL for optimal MCU activity (32). CL has previously been shown to closely interact and stabilize multiple integral inner mitochondrial membrane proteins (33). This is true for MCU (18) but in this study, we found that the loss of CL in C2C12 Taz-KO cells does not affect the abundance of EMRE or MCUR1, two of the other integral membrane proteins that interact with MCU (Fig. 2E and F). These observations suggest that the requirement of CL for the integral inner mitochondrial membrane proteins is not universal. Interestingly, we found that CL is required for the stability of MICU1, a peripheral membrane protein, and its stability is independent of MCU because MICU1 turnover occurred with much faster kinetics compared to that of MCU (Fig. 1D and E). We validated this assertion by showing that MICU1 abundance is reduced in CL-deficient crd1Δ yeast mitochondria, which lack MCU and EMRE (Fig. 2A and B). Notably, a previous study had shown that purified MICU1 could bind CL in vitro (21). This in vitro study, together with our in vivo findings, jointly suggests that CL directly tethers MICU1 to the inner mitochondrial membrane and that this tethering is independent of MCU-MICU1 interactions.
Ca2+ signaling mediated up-regulation of mitochondrial bioenergetics is primarily manifested by the enhanced activities of matrix dehydrogenases, such as PDH. Mitochondrial Ca2+ serves as a key regulator of PDH by stimulating the activity of the Ca2+-dependent PDH-phosphatase, which dephosphorylates PDH, and thereby activates it. Interestingly, a recent study has shown an increase in the levels of phosphorylated PDH in BTHS murine myoblasts, however the biochemical mechanism underlying the change in PDH phosphorylation status was not determined (34). Our results showing an ISO-based decrease in the levels of phosphorylated PDH in C2C12 WT cells, but not in Taz-KO cells (Fig. 5B and C), suggests that the CL-mediated control of the PDH phosphorylation status and activity (Fig. 5D) is likely through diminished mitochondrial Ca2+ signaling.
A previous study comprising of 15 BTHS patients demonstrated diminished oxygen uptake and utilization in skeletal and cardiac muscles, when compared to age-matched controls while going from rest to peak exercise (35). This finding was recapitulated in a BTHS mouse model that showed significantly reduced mitochondrial OCR during increased workload with no changes at resting conditions (36). During aerobic exercise, β-adrenergic signaling mediates excitation-contraction coupling to stimulate mitochondrial bioenergetics in skeletal muscles through intracellular Ca2+ signaling (37–39). Our finding that BTHS myoblasts are unable to stimulate mitochondrial oxygen consumption in response to a treatment with ISO, a β-adrenergic agonist (Fig. 5H and I), suggests that reduced mitochondrial Ca2+ signaling could be responsible for the exercise intolerance defect often observed in BTHS patients.
Taken together, our data support a model that under conditions of acute energetic needs, the reduced abundance of MCU and MICU1 in CL-depleted BTHS cells leads to decreased mitochondrial Ca2+ uptake, which in turn prevents Ca2+-dependent stimulation of PDH dephosphorylation leading to diminished PDH activity that results in reduced NADH production and mitochondrial respiration (Fig. 6). These defects in Ca2+-stimulated mitochondrial bioenergetics could help explain some of the pathophysiology of BTHS including cardiac arrhythmia and proximal muscle myopathy that are commonly observed in these patients. Additional evidence for the role of defective mitochondrial Ca2+ signaling in BTHS pathology comes from human genetic studies with MICU1 patients, who share overlapping clinical features with those seen in BTHS patients (16,17,27). In summary, our results highlight mitochondrial Ca2+ signaling as an important signaling pathway that could contribute to BTHS disease pathology.
Figure 6.
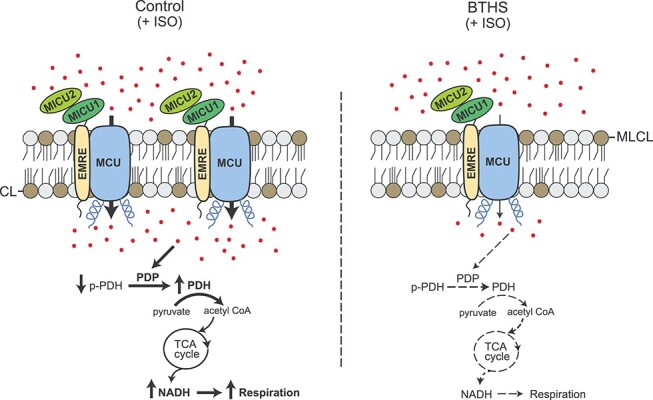
A model depicting perturbed mitochondrial calcium signaling in BTHS cells. Depletion of CL and an accumulation of its precursor monolysocardiolipin (MLCL) in BTHS mitochondria result in a reduced abundance of MCU, MICU1 and MICU2. Addition of β-adrenergic agonist (ISO) fails to stimulate uptake of Ca2+ (shown as red dots) in BTHS mitochondria to the same extent as in the control mitochondria. This prevents Ca2+-dependent dephosphorylation of pyruvate dehydrogenase leading to reduced PDH activity. This in turn decreases tricarboxylic acid (TCA) cycle output in the form of NADH resulting in reduced mitochondrial respiration in BTHS mitochondria. CL, cardiolipin; MLCL, monolysocardiolipin; ISO, isoprenaline hydrochloride; PDP, pyruvate dehydrogenase phosphatase; PDH, pyruvate dehydrogenase; p-PDH, phosphorylated PDH.
Materials and Methods
Mammalian cell culture and BTHS heart tissue samples
The control (ND11500) and BTHS patient (GM22194) B-lymphocytes were obtained from the Coriell Institute for Medical Research. B-lymphocytes were cultured in high glucose Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 15% fetal bovine serum (Sigma). Murine C2C12 WT and Taz-KO myoblasts were generously provided by Dr Miriam L. Greenberg and were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. All cell lines were incubated in 5% CO2 at 37°C. Deidentified BTHS patient heart samples were obtained from Barth syndrome Registry and DNA Bank. Whole cell protein was extracted in lysis buffer (BP-115, Boston BioProducts) supplemented with protease inhibitor cocktail (cOmplete Mini EDTA-free; Roche Diagnostics) and the protein concentrations were determined by BCA assay (Pierce™ BCA Protein Assay).
Yeast strains and growth conditions
Yeast Saccharomyces cerevisiae crd1Δ mutant used in this study was purchased from Open Biosystems and the isogenic WT BY4741 strain was obtained from Dr Miriam L. Greenberg. The authenticity of crd1Δ was confirmed by PCR, replica plating on dropout plates, and phospholipid analyses. Yeast cells were precultured in YPD (1% yeast extract, 2% peptone and 2% dextrose). The final cultures were grown in synthetic complete (SC) medium, which contained 0.17% yeast nitrogen base without amino acids, 0.5% ammonium sulfate, 0.2% dropout mix containing amino acids and 2% galactose (SC-galactose). Human MICU1 was cloned in pRS313-TEF yeast expression vector and human MCUR1 was cloned in pRS41N vector, as described previously (40). Cells expressing MICU1 and V5-tagged MCUR1 were grown in SC-galactose histidine drop-out media and SC-galactose media supplemented with ClonNAT antibiotic, respectively, to resist plasmid curing. Growth in liquid media was measured spectrophotometrically at 600 nm. Final cultures were started at an optical density of 0.1 and were grown to late logarithmic phase at 30°C.
Mitochondria isolation
Mitochondria from mammalian cells were isolated using the Mitochondria Isolation Kit from Abcam (110 170; Abcam) and stored at −80°C until further use. Protein concentration was determined by BCA assay (Pierce™ BCA Protein Assay). Isolation of crude mitochondria from yeast was performed as previously described (41). Briefly, yeast cells grown to late logarithmic phase were pelleted and resuspended in DTT buffer (0.1 M Tris–HCl, pH 9.4, 10 mM DTT) for 20 min at 30°C. The cells in DTT buffer were pelleted by centrifugation at 3000 × g for 5 min and were resuspended in spheroplasting buffer (1.2 M sorbitol, 20 mM potassium phosphate, pH 7.4) and treated with 3 mg zymolyase (US Biological Life Sciences) per gram of cell pellet for 45 min at 30°C. Spheroplasts were homogenized in homogenization buffer (0.6 M sorbitol, 10 mM Tris–HCl, pH 7.4, 1 mM EDTA, 1 mM PMSF [Phenylmethanesulfonyl fluoride], 0.2% [w/v] BSA (fatty acid-free, Sigma–Aldrich) with 15 strokes using a glass teflon homogenizer. After subsequent centrifugation steps, the mitochondrial pellet was centrifuged at 12000 × g for 15 min. Crude mitochondrial fractions were resuspended in SEM buffer (250 mM sucrose, 1 mM EDTA, 10 mM MOPS-KOH, pH 7.2) and stored at −80°C.
SDS-PAGE, BN-PAGE and immunoblotting
Denatured and native proteins were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Blue Native PAGE (BN-PAGE), respectively. For SDS-PAGE, mitochondrial lysate (20 μg) was separated on NuPAGE 4%–12% Bis-Tris gels (Thermo Fisher Scientific, Carlsbad, CA, USA) and transferred onto polyvinylidene fluoride (PVDF) membranes using a Trans-Blot SD semi-dry transfer cell (Bio-Rad, Hercules, CA, USA). For BN-PAGE, mitochondria were first solubilized in buffer containing 1% digitonin (Thermo Fisher Scientific, Carlsbad, CA, USA) at 9 g/g of mammalian mitochondrial protein, followed by incubation for 15 min at 4°C and centrifugation at 20 000 × g for 30 min. Clear supernatant containing native protein complexes was collected and 50X G-250 sample additive was added. Twenty micrograms of protein was loaded on 3%–12% native PAGE Bis-Tris gel (Thermo Fisher Scientific, Carlsbad, CA, USA) and transferred onto PVDF membranes using a wet transfer method. After transfer, the membranes were probed with the following primary antibodies against: MICU1, 1:2000 (12524S; Cell Signaling); MCU, 1:2500 (14997S; Cell Signaling); MICU2, 1:1000 (ab101465; Abcam); VDAC1, 1:2500 (ab14734; Abcam); ATP5A, 1:2500 (ab14748; Abcam); and V5-tag, 1:5000 (R960–25; Invitrogen). For PDH phosphorylation measurements, the following primary antibodies were used: p-PDH (S293) (ab177461; Abcam) and PDHA1 (168 379; Abcam). Antimouse or antirabbit secondary antibodies (1:5000) were incubated for 1 h at room temperature, and membranes were developed using Clarity™ Western ECL (Bio-Rad Laboratories).
Protein turnover measurement
C2C12 cells were grown in DMEM high glucose media supplemented with 10% FBS for ~14 h. Cells were washed with PBS and treated with 20 μg/ml cycloheximide for 0, 2, 4 and 6 h, respectively. At the indicated time points, cells were washed with PBS again and lysed using RIPA lysis buffer supplemented with 1× protease inhibitor cocktail (Roche). Total protein content of the lysate was quantified by BCA assay and 20 μg of protein was separated and detected by SDS PAGE/immunoblotting.
Mitochondrial Ca2+ uptake measurement in permeabilized cells
WT and Taz-KO C2C12 cells were washed with Ca2+ free PBS, pH 7.4. An equal number of cells (~4 × 106 cells) were resuspended and permeabilized with 40 μg/ml digitonin in 1.5 ml of intracellular medium (ICM) composed of 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM HEPES-Tris, pH 7.2. The experiments were performed in the presence of 5 mM succinate and 1 mM Mg-ATP at 37°C with constant stirring. For measuring mitochondrial Ca2+ uptake, the permeabilized cells were suspended in a medium containing 1.0 μM Fura-2FF. Fluorescence was monitored in a multiwavelength excitation, dual-wavelength emission fluorimeter (DeltaRAM, Photon Technology International). Extramitochondrial Ca2+ was recorded at an excitation ratio (340 nm/380 nm) and emission at 510 nm of Fura-2FF fluorescence. For individual experiments, Ca2+ pulses of 1 μM, 2 μM, 5 μM and 20 μM were added at 400 s, and the changes in extramitochondrial Ca2+ fluorescence were monitored. Mitochondrial Ca2+ uptake rate was derived from the decay of bath [Ca2+] after Ca2+ pulses.
NAD(P)H measurement in mammalian cells
C2C12 WT and Taz-KO cells (~5 × 106 cells) were suspended in Hanks’ balanced salt solution (Sigma). Autofluorescence of NAD(P)H was monitored at 350/460 nm (excitation/emission) using a multiwavelength excitation, dual-wavelength emission fluorimeter (DeltaRAM, Photon Technology International). These experiments were conducted at 37°C. C2C12 cells were stimulated with 0.25 μM ionomycin at 100 s after baseline recording followed by 20 μM rotenone at 300 s.
PDH phosphorylation measurement in mammalian cells
C2C12 cells (~2 × 106) were seeded in 150 mm tissue-culture treated dishes and grown in DMEM media supplemented with 10% FBS at 37°C, 5% CO2 for ~14 h. At this point, cells were treated with 25 μM ISO or DMSO (vehicle) for 40 min and harvested. Mitochondria were isolated using the Mitochondria Isolation Kit from Abcam (110 170; Abcam). Proteins were extracted from the mitochondria using RIPA lysis buffer supplemented with 1× protease inhibitor cocktail (Roche) and 1× PhosSTOP phosphatase inhibitor (Roche) and the protein content was quantified by BCA Assay. Lysates (20 μg) were separated by SDS-PAGE, transferred onto PVDF membranes, and immunoblotted by the indicated primary antibodies.
PDH activity measurement in mammalian cells
PDH activity was measured using PDH Activity Assay Kit (Sigma–Aldrich, MAK183) according to manufacturers’ instructions. Briefly, C2C12 cells were grown in 10 cm dish to ~90% confluency in DMEM media supplemented with 10% FBS at 37°C, 5% CO2. Cells were treated with 25 μM ISO or DMSO (vehicle) for 40 min and the cells were harvested followed by two washing with phosphate buffered saline. Cells were then homogenized in 200 μl of ice-cold PDH assay buffer using pellet pestle followed by centrifugation at 10 000 × g for 5 min and the supernatant was collected. Next, in clear 96-well plate, supernatant corresponding to 150 μg protein in 50 μl of PDH assay buffer at room temperature was mixed with 50 μl of the reaction mix, and incubated at 37°C for 2 min. The absorbance was then measured at 450 nm every 2 min at 37°C using Synergy™ (BioTek) microplate reader till the most active sample was near the end of the linear range of the standard curve. The PDH activity in all groups was calculated and represented as relative PDH activity compared to WT levels.
OCR measurement in intact mammalian cells
OCR measurements were carried out as previously described (42) with minor modifications. Briefly, C2C12 WT and Taz-KO myoblasts were cultured in high glucose DMEM growth media supplemented with 10% FBS (Sigma) and 1 mM sodium pyruvate (Life Technologies). The cells were then seeded in XF24-well cell culture microplates (Agilent Technologies) at 10 000 cells/well in 250 μl of DMEM growth media containing 10 mM galactose as sugar source and incubated at 37°C, 5% CO2 for ~14 h. Before the OCR was measured, 475 μl of prewarmed growth medium was added to each well and cells were incubated at 37°C for 30 min. OCR measurements were carried out in intact cells using Seahorse XF24 Extracellular Flux Analyzer (Agilent Technologies). ISO (Sigma) was injected through port A at a final concentration of 25 μM. Mix, wait and measure cycles were set to 2, 3 and 2 min, respectively. For the mitochondrial stress test, oligomycin, carbonyl cyanide 3-cholorophenylhydrazone (CCCP) and antimycin A were sequentially injected through ports B, C and D to achieve final concentrations of 1 μM, 10 μM and 1 μM, respectively. Immediately after the assay, the cell culture microplates were washed twice with 500 μl PBS and 50 μl RIPA lysis buffer (25 mM Tris. HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS supplemented with 1× protease inhibitor cocktail) was added to each well and incubated on ice for 15 min. Protein concentration in each well was measured by BCA assay (Thermo Scientific) and the OCR values were normalized to the total protein content.
Statistical analyses
Statistical analyses were performed in GraphPad Prism software using two-tailed unpaired Student’s t test on data obtained from three independent experiments that were performed on different days with different biological replicates.
Acknowledgements
We thank Dr Miriam L. Greenberg for generously sharing BY4741 WT yeast strain and C2C12 WT and Taz-KO cell lines and Dr Vamsi K. Mootha for kindly providing EMRE antibody. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest statement. None declared.
Contributor Information
Sagnika Ghosh, Department of Biochemistry and Biophysics, Texas A&M University, College Station, TX 77843, USA.
Mohammad Zulkifli, Department of Biochemistry and Biophysics, Texas A&M University, College Station, TX 77843, USA.
Alaumy Joshi, Department of Biochemistry and Biophysics, Texas A&M University, College Station, TX 77843, USA.
Manigandan Venkatesan, Department of Medicine, Cardiology Division, Center for Precision Medicine, University of Texas Health Science Center at San Antonio, San Antonio, TX 78229, USA.
Allen Cristel, Department of Medicine, Cardiology Division, Center for Precision Medicine, University of Texas Health Science Center at San Antonio, San Antonio, TX 78229, USA.
Neelanjan Vishnu, Department of Medicine, Cardiology Division, Center for Precision Medicine, University of Texas Health Science Center at San Antonio, San Antonio, TX 78229, USA.
Muniswamy Madesh, Department of Medicine, Cardiology Division, Center for Precision Medicine, University of Texas Health Science Center at San Antonio, San Antonio, TX 78229, USA.
Vishal M Gohil, Department of Biochemistry and Biophysics, Texas A&M University, College Station, TX 77843, USA.
Funding
Barth Syndrome Foundation ‘Idea’ Grant ( to V.M.G.), the Welch Foundation Grant (A-1810 to V.M.G.) and the National Institutes of Health grants (R01GM111672 to V.M.G and R01GM135760 to M.M.).
References
- 1. Clapham, D.E. (2007) Calcium signaling. Cell, 131, 1047–1058. [DOI] [PubMed] [Google Scholar]
- 2. Kamer, K.J. and Mootha, V.K. (2015) The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol., 16, 545–553. [DOI] [PubMed] [Google Scholar]
- 3. Baughman, J.M., Perocchi, F., Girgis, H.S., Plovanich, M., Belcher-Timme, C.A., Sancak, Y., Bao, X.R., Strittmatter, L., Goldberger, O., Bogorad, R.L.et al. (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature, 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Stefani, D., Raffaello, A., Teardo, E., Szabò, I. and Rizzuto, R. (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature, 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Perocchi, F., Gohil, V.M., Girgis, H.S., Bao, X.R., McCombs, J.E., Palmer, A.E. and Mootha, V.K. (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature, 467, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sancak, Y., Markhard, A.L., Kitami, T., Kovács-Bogdán, E., Kamer, K.J., Udeshi, N.D., Carr, S.A., Chaudhuri, D., Clapham, D.E., Li, A.A.et al. (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science, 342, 1379–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Plovanich, M., Bogorad, R.L., Sancak, Y., Kamer, K.J., Strittmatter, L., Li, A.A., Girgis, H.S., Kuchimanchi, S., De Groot, J., Speciner, L.et al. (2013) MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One, 8, e55785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Raffaello, A., De Stefani, D., Sabbadin, D., Teardo, E., Merli, G., Picard, A., Checchetto, V., Moro, S., Szabò, I. and Rizzuto, R. (2013) The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J., 32, 2362–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tomar, D., Dong, Z., Shanmughapriya, S., Koch, D.A., Thomas, T., Hoffman, N.E., Timbalia, S.A., Goldman, S.J., Breves, S.L., Corbally, D.P.et al. (2016) MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep., 15, 1673–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fan, M., Zhang, J., Tsai, C.W., Orlando, B.J., Rodriguez, M., Xu, Y., Liao, M., Tsai, M.F. and Feng, L. (2020) Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature, 582, 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhuo, W., Zhou, H., Guo, R., Yi, J., Zhang, L., Yu, L., Sui, Y., Zeng, W., Wang, P. and Yang, M. (2021) Structure of intact human MCU supercomplex with the auxiliary MICU subunits. Protein Cell, 12, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mallilankaraman, K., Doonan, P., Cardenas, C., Chandramoorthy, H.C., Muller, M., Miller, R., Hoffman, N.E., Gandhirajan, R.K., Molgo, J., Birnbaum, M.J.et al. (2012) MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell, 151, 630–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang, Y., Han, Y., She, J., Nguyen, N.X., Mootha, V.K., Bai, X.C. and Jiang, Y. (2020) Structural insights into the Ca2+-dependent gating of the human mitochondrial calcium uniporter. elife, 9, e60513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gottschalk, B., Klec, C., Leitinger, G., Bernhart, E., Rost, R., Bischof, H., Madreiter-Sokolowski, C.T., Radulović, S., Eroglu, E., Sattler, W.et al. (2019) MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex. Nat. Commun., 10, 3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Csordás, G., Golenár, T., Seifert, E.L., Kamer, K.J., Sancak, Y., Perocchi, F., Moffat, C., Weaver, D., Perez, S.F., Bogorad, R.et al. (2013) MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab., 17, 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Logan, C.V., Szabadkai, G., Sharpe, J.A., Parry, D.A., Torelli, S., Childs, A.M., Kriek, M., Phadke, R., Johnson, C.A., Roberts, N.Y.et al. (2014) Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet., 46, 188–193. [DOI] [PubMed] [Google Scholar]
- 17. Lewis-Smith, D., Kamer, K.J., Griffin, H., Childs, A.M., Pysden, K., Titov, D., Duff, J., Pyle, A., Taylor, R.W., Yu-Wai-Man, P.et al. (2016) Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol. Genet., 2, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ghosh, S., Basu Ball, W., Madaris, T.R., Srikantan, S., Madesh, M., Mootha, V.K. and Gohil, V.M. (2020) An essential role for cardiolipin in the stability and function of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. U.S.A., 117, 16383–16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bione, S., D’Adamo, P., Maestrini, E., Gedeon, A.K., Bolhuis, P.A. and Toniolo, D. (1996) A novel X-linked gene, G4.5. Is responsible for Barth syndrome. Nat. Genet., 12, 385–389. [DOI] [PubMed] [Google Scholar]
- 20. Ghosh, S., Iadarola, D.M., Basu Ball, W. and Gohil, V.M. (2019) Mitochondrial dysfunctions in Barth syndrome. IUBMB Life, 71, 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kamer, K.J., Grabarek, Z. and Mootha, V.K. (2017) High-affinity cooperative Ca2+ binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep., 18, 1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Phillips, C.B., Tsai, C.W. and Tsai, M.F. (2019) The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex. elife, 8, e41112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bick, A.G., Calvo, S.E. and Mootha, V.K. (2012) Evolutionary diversity of the mitochondrial calcium uniporter. Science, 336, 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luongo, T.S., Lambert, J.P., Yuan, A., Zhang, X., Gross, P., Song, J., Shanmughapriya, S., Gao, E., Jain, M., Houser, S.R.et al. (2015) The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep., 12, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pan, X., Liu, J., Nguyen, T., Liu, C., Sun, J., Teng, Y., Fergusson, M.M., Rovira, I.I., Allen, M., Springer, D.A.et al. (2013) The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol., 15, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shanmughapriya, S., Rajan, S., Hoffman, N.E., Zhang, X., Guo, S., Kolesar, J.E., Hines, K.J., Ragheb, J., Jog, N.R., Caricchio, R.et al. (2015) Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci. Signal., 8, ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clarke, S.L.N., Bowron, A., Gonzalez, I.L., Groves, S.J., Newbury-Ecob, R., Clayton, N., Martin, R.P., Tsai-Goodman, B., Garratt, V., Ashworth, M.et al. (2013) Barth syndrome. Orphanet J. Rare Dis., 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gaspard, G.J. and McMaster, C.R. (2015) Cardiolipin metabolism and its causal role in the etiology of the inherited cardiomyopathy Barth syndrome. Chem. Phys. Lipids, 193, 1–10. [DOI] [PubMed] [Google Scholar]
- 29. Joshi, A.S., Zhou, J., Gohil, V.M., Chen, S. and Greenberg, M.L. (2009) Cellular functions of cardiolipin in yeast. Biochim. Biophys. Acta, 1793, 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ren, M., Phoon, C.K.L. and Schlame, M. (2014) Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res., 55, 1–16. [DOI] [PubMed] [Google Scholar]
- 31. Finkel, T., Menazza, S., Holmström, K.M., Parks, R.J., Liu, J., Sun, J., Liu, J., Pan, X. and Murphy, E. (2015) The ins and outs of mitochondrial calcium. Circ. Res., 116, 1810–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang, C., Baradaran, R. and Long, S.B. (2020) Structure and reconstitution of an MCU-EMRE mitochondrial Ca2+ uniporter complex. J. Mol. Biol., 432, 5632–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Musatov, A. and Sedlák, E. (2017) Role of cardiolipin in stability of integral membrane proteins. Biochimie, 142, 102–111. [DOI] [PubMed] [Google Scholar]
- 34. Li, Y., Lou, W., Raja, V., Denis, S., Yu, W., Schmidtke, M.W., Reynolds, C.A., Schlame, M., Houtkooper, R.H. and Greenberg, M.L. (2019) Cardiolipin-induced activation of pyruvate dehydrogenase links mitochondrial lipid biosynthesis to TCA cycle function. J. Biol. Chem., 294, 11568–11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spencer, C.T., Byrne, B.J., Bryant, R.M., Margossian, R., Maisenbacher, M., Breitenger, P., Benni, P.B., Redfearn, S., Marcus, E. and Cade, W.T. (2011) Impaired cardiac reserve and severely diminished skeletal muscle O2 utilization mediate exercise intolerance in Barth syndrome. Am. J. Physiol. Heart Circ. Physiol., 301, H2122–H2129. [DOI] [PubMed] [Google Scholar]
- 36. Powers, C., Huang, Y., Strauss, A. and Khuchua, Z. (2013) Diminished exercise capacity and mitochondrial bc1 complex deficiency in tafazzin-knockdown mice. Front. Physiol., 4, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Voltarelli, V.A., Coronado, M., Fernandes, L.G., Campos, J.C., Jannig, P.R., Ferreira, J.C.B., Fajardo, G., Brum, P.C. and Bernstein, D. (2021) β2-adrenergic signaling modulates mitochondrial function and morphology in skeletal muscle in response to aerobic exercise. Cell, 10, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cairns, S.P. and Borrani, F. (2015) β-Adrenergic modulation of skeletal muscle contraction: key role of excitation-contraction coupling. J. Physiol., 593, 4713–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blackwood, S.J. and Katz, A. (2019) Isoproterenol enhances force production in mouse glycolytic and oxidative muscle via separate mechanisms. Pflugers Arch., 471, 1305–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zulkifli, M., Neff, J.K., Timbalia, S.A., Garza, N.M., Chen, Y., Watrous, J.D., Murgia, M., Trivedi, P.P., Anderson, S.K., Tomar, D.et al. (2020) Yeast homologs of human MCUR1 regulate mitochondrial proline metabolism. Nat. Commun., 11, 4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meisinger, C., Pfanner, N. and Truscott, K.N. (2006) Isolation of yeast mitochondria. Methods Mol. Biol., 313, 33–39. [DOI] [PubMed] [Google Scholar]
- 42. Gohil, V.M., Sheth, S.A., Nilsson, R., Wojtovich, A.P., Lee, J.H., Perocchi, F., Chen, W., Clish, C.B., Ayata, C., Brookes, P.S. and Mootha, V.K. (2010) Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat. Biotechnol., 28, 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]