

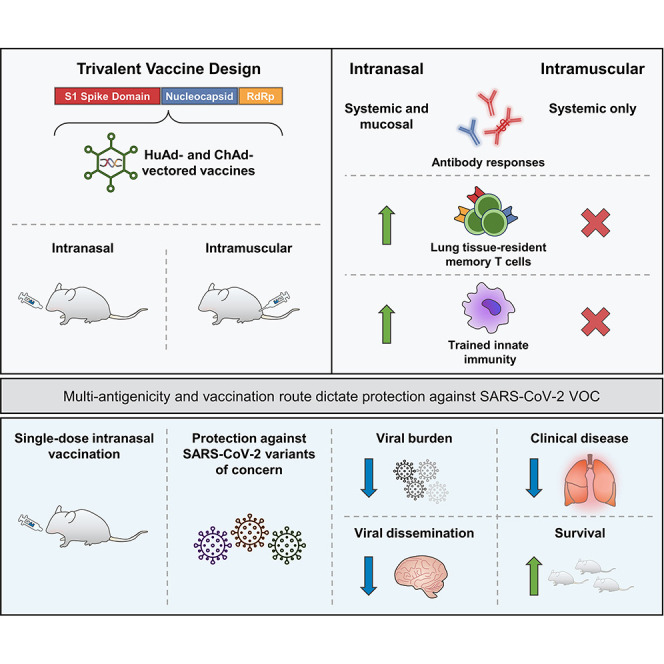
Abstract
The emerging SARS-CoV-2 variants of concern (VOCs) threaten the effectiveness of current COVID-19 vaccines administered intramuscularly and designed to only target the spike protein. There is a pressing need to develop next-generation vaccine strategies for broader and long-lasting protection. Using adenoviral vectors (Ad) of human and chimpanzee origin, we evaluated Ad-vectored trivalent COVID-19 vaccines expressing spike-1, nucleocapsid, and RdRp antigens in murine models. We show that single-dose intranasal immunization, particularly with chimpanzee Ad-vectored vaccine, is superior to intramuscular immunization in induction of the tripartite protective immunity consisting of local and systemic antibody responses, mucosal tissue-resident memory T cells and mucosal trained innate immunity. We further show that intranasal immunization provides protection against both the ancestral SARS-CoV-2 and two VOC, B.1.1.7 and B.1.351. Our findings indicate that respiratory mucosal delivery of Ad-vectored multivalent vaccine represents an effective next-generation COVID-19 vaccine strategy to induce all-around mucosal immunity against current and future VOC.
Keywords: SARS-CoV-2, COVID-19, variants of concern, next-generation vaccines, intramuscular immunization, respiratory mucosal immunization, multi-valent vaccine, adenoviral vector, human adenoviral vector, chimpanzee adenoviral vector, respiratory mucosal immunity, humoral immunity, T cell immunity, trained innate immunity, animal models
Graphical abstract
Respiratory mucosal immunization with a next-generation adenoviral-vectored trivalent COVID-19 vaccine expressing spike, nucleocapsid, and RdRp antigens, induces all-around protective mucosal immunity against SARS-CoV-2 via induction of systemic and local antibodies, lung-tissue-resident memory T cells, and trained alveolar macrophages.
Introduction
Since its outbreak in Wuhan China in 2019, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has globally infected 359 M people and claimed 5.6 M lives and counting. Besides general mitigation/infection control measures, the only effective way to control the pandemic of coronavirus disease 2019 (COVID-19) is to establish herd immunity through vaccination (Fontanet and Cauchemez, 2020; Jeyanathan et al., 2020). Thus, based on a pandemic vaccine paradigm (Lurie et al., 2020), there have been at least 100 vaccines tested in clinical trials and another 180 under preclinical development. These efforts have led a growing number of first-generation COVID-19 vaccines to receive emergency use authorization in various countries. Notably, several authorized vaccines are based on mRNA and adenoviral platforms to express the spike protein of the ancestral SARS-CoV-2 and elicit neutralizing antibody responses following 1–2 intramuscular injections (Jeyanathan et al., 2020).
The global rollout of COVID-19 vaccines has played a critical role in reducing viral transmission, hospitalizations, and deaths. However, since September 2020 there have been five SARS-CoV-2 variants of concern (VOCs) emerged which are B.1.1.7 (UK, Alpha), B.1.351 (South Africa, Beta), P.1 (Brazil, Gamma), B.1.617.2 (India, Delta), and B.1.1.529 (South Africa, Omicron) (Andreano and Rappuoli, 2021; Gupta, 2021). While they all have multiple mutations in the spike protein, B.1.351, P.1, and B.1.1.529 harbor multiple mutations within the receptor-binding domain (RBD) that reduce their neutralization by antibodies present in convalescent or vaccine-induced sera (Chen et al., 2021; Garcia-Beltran et al., 2021; Geers et al., 2021; Hoffmann et al., 2021b; Planas et al., 2021; Wang et al., 2021; Wilhelm et al., 2021). Some B.1.617 sub-lineages also carry E484Q and L452R mutations that reduce antibody binding (Starr et al., 2021). Of importance, several first-generation vaccines including ChAdOx1 nCoV-19 (Astra Zeneca/Oxford) (Madhi et al., 2021), Ad26.COV2.S (J&J) (Sadoff et al., 2021), NVX-CoV2373 (Novavax) (Shinde et al., 2021), and BNT162b2 (Pfizer-BioNTech) (Abu-Raddad et al., 2021) have demonstrated reduced effectiveness in protecting from mild to moderate COVID-19 caused by B.1.351. Likewise, sera from those immunized with mRNA-1273 (Moderna) show reduced neutralization of B.1.351 (Shen et al., 2021). Thus, the emerging VOCs capable of escaping the immunity by first-generation vaccines constantly threaten to impede or disrupt the establishment and sustainability of vaccine-induced herd immunity (Aschwanden, 2021; Harvey et al., 2021).
To meet the challenges from VOC and limited durability of first-generation vaccine-induced immunity, there is an urgent need to develop next-generation COVID-19 vaccine strategies (Callaway and Ledford, 2021; Gupta, 2021; Jeyanathan et al., 2020). Although updating the spike antigen to specific VOC represents one such strategy (Callaway and Ledford, 2021; Gupta, 2021), it is cumbersome and expensive and requires selection of specific VOC sequence(s), which may result in inherently inaccurate prediction of antigenic drift, akin to current seasonal influenza vaccines. An alternative strategy is to develop recombinant viral-vectored multivalent vaccines amenable to respiratory mucosal immunization (Jeyanathan et al., 2020). Besides the spike antigen, such vaccines express additional conserved SARS-CoV-2 antigens to broaden T cell immunity. Since antigenic changes in conserved, internal viral proteins that are the primary focus of T cell responses are rare/improbable in SARS-CoV-2 viruses including VOCs (Alter et al., 2021; Geers et al., 2021), such multivalent vaccines do not require frequent updating while they can boost the spike-specific immunity induced by first-generation vaccines. Thus, these vaccines are expected to be effective against both ancestral and variants of SARS-CoV-2. Furthermore, adenoviral vectors (Ad) delivered via the respiratory tract confer protection via eliciting mucosal-tissue-resident trained innate and adaptive immunity at the site of viral entry (Jeyanathan et al., 2020; Teijaro and Farber, 2021; Xing et al., 2020; Yao et al., 2018). Unfortunately, to-date, there has been a paucity of next-generation COVID-19 vaccine strategies capable of robust and durable protection against the ancestral strain and variants of SARS-CoV-2.
Herein, we have developed and evaluated a next-generation COVID-19 vaccine strategy in murine models. Our vaccine is built upon adenoviral vectors of human (Tri:HuAd) or chimpanzee (Tri:ChAd) origin, expressing three SARS-CoV-2 antigens (spike protein 1, full-length nucleocapsid protein, and truncated polymerase), and is suitable for respiratory mucosal delivery. We show that single-dose intranasal, but not intramuscular, immunization, particularly with the Tri:ChAd vaccine, induces all-around respiratory mucosal immunity against both ancestral SARS-CoV-2 and B.1.1.7 and B.1.351 VOC. Our study thus indicates that respiratory mucosal delivery of multivalent viral-vectored COVID-19 vaccine is an effective next-generation COVID-19 vaccine strategy.
Results
Construction and characterization of HuAd- and ChAd-vectored trivalent COVID-19 vaccines
Currently approved recombinant first-generation COVID-19 vaccines only encode the spike (S) protein from the Wuhan-Hu-1 ancestral SARS-CoV-2 and were designed primarily to induce neutralizing antibodies following intramuscular injections, a strategy inadequate to combat VOC (Aschwanden, 2021; Harvey et al., 2021). To develop next-generation adenoviral-vectored COVID-19 vaccines, we utilized human serotype 5 (Tri:HuAd) and chimpanzee serotype 68 (Tri:ChAd) adenoviral vectors. Our vaccines were constructed to include the full-length S1 domain of spike which contains the NTD and RBD and numerous T cell epitopes (Geers et al., 2021; Tarke et al., 2021). The S1 was fused to the vesicular stomatitis virus G protein (VSVG) transmembrane/trimerization domain (Figure 1 A) to anchor it to the membrane and facilitate its trimerization and exosomal targeting for enhanced antibody responses (Bliss et al., 2020; Kuate et al., 2007). To broaden T cell immunity against additional viral antigens, the full-length nucleocapsid (N) and truncated nsp12 (RNA-dependent RNA polymerase [RdRp]) proteins were included in vaccine design as a single polyprotein downstream of a porcine teschovirus 2A sequence (P2A) (Figure 1A). N is the most abundant viral protein rich in T cell epitopes in humans including convalescent COVID-19 patients (Altmann and Boyton, 2020; Dai and Gao, 2021; Peng et al., 2020), and the genetic vaccines expressing N were shown to induce protective immunity in preclinical COVID-19 models (Class et al., 2021; Hajnik et al., 2021; Matchett et al., 2021). A region of RdRp was selected for maximal high-affinity T cell epitopes based on bioinformatic analysis. T cells specific for N and RdRp are also cross-reactive with other bat-derived coronaviruses (Alter et al., 2021; Altmann and Boyton, 2020; Geers et al., 2021; Tarke et al., 2021).
Figure 1.

Single-dose intranasal immunization leads to superior anti-spike protein humoral responses over intramuscular immunization
(A) Transgene cassette diagram.
(B) Western blot analysis of expression of S1-VSVG and N/RdRp protein from whole-cell lysates from A549 cells untransduced or transduced with Tri:HuAd or Tri:ChAd. GAPDH was used as a loading control for each condition.
(C) Experimental schema.
(D) Serum anti-spike IgG reciprocal endpoint antibody titers at 2 (red) and 4 (blue) weeks post-immunization.
(E) Serum anti-RBD IgG reciprocal endpoint antibody titers at 2 (red) and 4 (blue) weeks post-immunization.
(F) Reciprocal endpoint titer ratios of anti-spike IgG2a:IgG1 at 2 (red) and 4 (blue) weeks post-immunization.
(G) Bar graph depicting serum neutralizing antibody responses 4 weeks post-immunization, measured by percent (%) inhibition with a surrogate virus neutralization test (sVNT). Green bar (+) indicates assay positive control. Gray bar (−) indicates assay negative control.
(H) BAL anti-spike IgG reciprocal endpoint antibody titers at 4 weeks post-immunization.
(I) Experimental schema.
(J) Serum anti-spike (red) or anti-RBD (blue) IgG reciprocal endpoint antibody titers at 8 weeks post-immunization.
(K) Bar graph depicting serum neutralizing antibody responses 8 weeks post-immunization, measured by percent (%) inhibition with a surrogate virus neutralization test (sVNT). Green bar (+) indicates assay positive control. Gray bar (−) indicates assay negative control.
(L) Serum neutralizing antibody responses at 8 weeks post-immunization, measured by percent (%) neutralization utilizing a live SARS-CoV-2 microneutralization (MNT) assay.
(M) BAL anti-spike IgG (left) or IgA (right) reciprocal endpoint antibody titers at 8 weeks post-immunization.
(N) Bar graphs depicting the absolute number of class-switched IgG1+ RBD-specific B cells in the spleen (left) or lung (right) at 8 weeks post-immunization.
Data presented in (D–H and J–N) represent mean ± SEM. Statistical analysis for (D, E, H, and J) were Kruskal-Wallis tests with Dunn’s multiple comparisons test. Statistical analysis for (G, K, and N) were two-tailed t tests. Data are from 2 pooled independent experiments, n = 3–12 mice/group. ns, not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
See also Figures S1, S2, and S3.
Prior to viral rescue, the transgene cassette was verified to be in-frame by Sanger sequencing, with translation initiating at the TpA signal sequence. Following virus rescue, amplification, and purification, A549 cells were transduced with Tri:HuAd or Tri:ChAd and transgene expression at the protein level was verified by western blot for S1-VSVG and Nucleocapsid:RdRp fusion proteins of expected sizes (Figure 1B).
In anticipation of their further clinical evaluation, we assessed the safety of Tri:HuAd and Tri:ChAd during the acute phase following intramuscular (i.m.) or intranasal (i.n.) delivery in mice (Figure S1 A). There was little change in body weight following single-dose i.m. or i.n. vaccination (Figure S1B). Regardless of immunization route or vaccine vector, there was no significant elevations in the lung or bronchoalveolar lavage fluid (BAL) in either neutrophils (Figure S1C) or pro-inflammatory cytokines (Figure S1D), relative to naive controls. Furthermore, there was no indications of hepatic or renal toxicity based on alkaline phosphatase/alanine aminotransferase and creatinine, respectively (Figure S1E). These data support an overall satisfactory safety profile of Tri:HuAd and Tri:ChAd COVID-19 vaccines.
Figure S1.

Acute safety assessment of intramuscularly or intranasally administered Tri:HuAd and Tri:ChAd COVID-19 vaccines, related to Figure 1
(A) Experimental schema.
(B) Changes in body weight over 3 days post-vaccination.
(C) Absolute number of neutrophils in the lung at 3 days post-immunization.
(D) Cytokine levels in bronchoalveolar lavage fluids at 3 days post-immunization.
(E) Serum levels of biomarkers for hepatotoxicity and nephrotoxicity at 3 days post-immunization.
Data presented in (B, C, and E) represent mean ± SEM from n = 3–4 mice/group.
Single-dose intranasal immunization leads to superior anti-spike protein humoral responses compared with intramuscular immunization
Given the close relationship between spike-specific humoral responses and protective immunity (Huang et al., 2020; Khoury et al., 2021; Krammer, 2021), we first examined the kinetics of spike-specific antibody responses following i.m. or i.n. immunization with a single dose of Tri:HuAd or Tri:ChAd vaccine. Serum and BAL were collected from BALB/c mice 2 and 4 weeks post-immunization (Figure 1C). IgG responses against spike and RBD were quantified by enzyme-linked immunosorbent assay (ELISA). While the control sera from i.m. or i.n. empty viral vector (HuAd:EV or ChAd:EV)-treated animals had little anti-spike/RBD IgG responses (Figures S2 A and S2B), the magnitude of spike- and RBD-specific IgG responses for Tri:HuAd vaccine were higher in serum following i.n. immunization and sustained for at least 4 weeks (Figures 1D and 1E). Likewise, i.n. Tri:ChAd vaccine also induced significantly greater spike- and RBD-specific IgG responses in serum than i.m. delivery at 4 weeks (Figures 1D and 1E). Both i.m. and i.n. Tri:HuAd and Tri:ChAd vaccines also induced N-specific IgG responses in serum (Figure S2C).
Figure S2.

Characterization of immune responses following intramuscularly or intranasally administered HuAd- and ChAd-empty vectors, related to Figures 1, 2, and 3
(A) Experimental schema.
(B) Serum anti-spike (red) or anti-RBD (blue) IgG reciprocal endpoint antibody titers at 4 weeks post-immunization.
(C) Serum anti-nucleocapsid IgG responses based on optical density versus reciprocal serum dilutions following i.m. (red) or i.n. (blue) immunization with either Tri:HuAd or Tri:ChAd (top) or empty vector equivalents (bottom).
(D) Bar graphs depicting absolute number of S1 (left), nucleocapsid (middle), or RdRp (right) specific IFN-γ+ CD8+ (top) or IFN-γ+ CD4+ (bottom) T cells in the BAL at 2 (red) and 4 (blue) weeks post-immunization following ex vivo stimulation with overlapping peptide pools.
(E) Flow cytometric dot plots of CD44+ CD8+ T cells for CD69 and CD103 from the lung (left) or BAL (right) at 4 weeks post-immunization.
Data presented in (B–E) represent mean ± SEM. Data are representative of 1–2 independent experiments, n = 3–9 mice/group.
Since vaccine-associated enhanced respiratory disease (VAERD) is potentially associated with Th2-biased immune responses to certain viral infection and has also been experimentally observed post-inactivated SARS-CoV-1 vaccination (Bournazos et al., 2020; Jeyanathan et al., 2020), we determined the ratio of S-specific IgG2a/IgG1 antibodies as a surrogate of the Th1/Th2 immune response. Regardless of vaccine route or vector, no Th2-skewing of antibody responses was seen at either timepoint (Figure 1F).
We next assessed the neutralizing capacity of serum antibodies 4 weeks post-immunization by a surrogate virus neutralization test (sVNT) (Tan et al., 2020). Whereas immunization route had no significant effect on the neutralizing potential of serum antibodies in Tri:HuAd-vaccinated animals (i.m. 6.1% ± 0.2% versus i.n. 11.92% ± 2.7%), i.n. Tri:ChAd generated antibody responses with markedly enhanced neutralizing potential (87.70% ± 2.3%) over that by i.m. route or by Tri:HuAd immunization (Figure 1G).
To assess humoral responses at the respiratory mucosa, BAL fluids collected 4 weeks post-immunization with either trivalent vaccine were assessed for S-specific IgG. As expected, we were only able to reliably detect S-specific antibodies in the airway following i.n., but not i.m., immunization (Figure 1H). Of note, airway S-specific IgG responses following Tri:ChAd immunization almost doubled that by Tri:HuAd.
We next assessed the durability of antibody responses at 8 weeks post-vaccination (Figure 1I). Overall, compared with 4 weeks data (Figures 1D and 1E), serum S- and RBD-specific IgG responses largely sustained following i.m. immunization and remained significantly higher following i.n. immunization with either vaccine (Figure 1J). Once again, the serum neutralization profile determined by sVNT at 8 weeks (Figure 1K) was similar to that at 4 weeks (Figure 1G), showing i.n. Tri:ChAd to induce the highest titers of neutralizing antibodies. Given the robust neutralizing capacity exhibited by serum from i.n. Tri:ChAd mice, we next tested it in a microneutralization (MNT) assay with live SARS-CoV-2. Congruent with the sVNT results, i.m. immunization with either vaccine afforded minimal neutralization against live SARS-CoV-2 (Figure 1L). In contrast, while i.n. immunization with either vaccine increased their respective neutralization capacities, i.n. Tri:ChAd elicited superior neutralization capacity over Tri:HuAd counterpart (Figure 1L). Compared with 4 weeks BAL data (Figure 1H), anti-S IgG from the BAL fluid was somewhat increased at 8 weeks following i.n. immunization with higher levels induced by Tri:ChAd vaccine while i.m. immunization with either vaccine failed to induce anti-S IgG in the airway (Figure 1M). Moreover, significant amounts of anti-S IgA were detected only in the BAL of i.n. Tri:ChAd animals (Figure 1M).
To examine the relationship of vaccine vector and immunization route to detectable antigen-experienced memory B cells in systemic lymphoid and local lung tissues, we tetramerized biotinylated RBD conjugated to a fluorochrome and probed for RBD-specific B cells by FACS (Hartley et al., 2020; Rodda et al., 2021). A decoy tetramer was included during staining to gate out vector-specific B cells (Figure S3 A). While all immunizations induced a detectable population of RBD-specific B cells in the spleen, i.n. Tri:ChAd induced significantly higher levels than i.n. Tri:HuAd (Figure 1N). In addition, only i.n. Tri:ChAd vaccine induced detectable RBD-specific B cells in the lung tissue (Figure 1N).
Figure S3.

Flow cytometric gating strategies, related to Figures 1 and 3
(A) Gating strategy in this study used to distinguish antigen-specific, class-switched B cells.
(B) Gating strategy in this study used to distinguish bona fide pulmonary tissue-resident memory CD8+ (top) or CD4+ (bottom) T cells.
(C) Gating strategy in this study used to distinguish neutrophils, alveolar macrophages (AMs), and interstitial macrophages (IMs) from other major pulmonary myeloid cell populations.
Examples shown are representative from BALB/c mice i.n. vaccinated with Tri:ChAd at 4 weeks post-immunization.
The above data indicate that single-dose intranasal immunization, particularly with Tri:ChAd vaccine, induces superior functional humoral responses both systemically and locally in the lung over the intramuscular route.
Single-dose intranasal immunization induces superior airway T cell responses over intramuscular immunization
We next examined T cell responses with a focus on those within the airways. Besides antibodies, airway T cells play pivotal roles in immunity against coronaviruses (Jeyanathan et al., 2020; Zhao et al., 2016). BALB/c mice were immunized intramuscularly or intranasally with a single dose of either trivalent COVID-19 vaccine. Antigen-specific T cells in mononuclear cells from the BAL harvested 2 and 4 weeks post-immunization were analyzed by FACS for intracellular IFN-γ, TNF-α, IL-2, and Granzyme B expression upon ex vivo stimulation with 15-mer peptide pools for S1 (132 peptides), N (82 peptides), and RdRp (12 peptides). As expected, both CD8+ and CD4+ T cells from 4 weeks i.m. or i.n. empty vector (HuAd:EV or ChAd:EV)-treated animals did not respond to any of these peptide pools (Figure S2D). In agreement with our previous work (Jeyanathan et al., 2017; Lai et al., 2015; Santosuosso et al., 2005), i.m. immunization failed to induce antigen-specific CD8+ T cells in the airways, irrespective of vaccine vector (Figures 2A, 2D, and 2G). In contrast, i.n. immunization induced a significant number of IFNγ+CD8+ T cells specific for S1, N, or RdRp in the airways. Of interest, S1-specific T cells were dominant relative to those for N or RdRp. Compared with Tri:HuAd, i.n. Tri:ChAd vaccine induced greater airway CD8+ T cell responses to the three antigens, particularly at 2 weeks (Figures 2A, 2D, and 2G). I.n., but not i.m., immunization also induced a similar profile of antigen-specific CD4+ T cell responses in the airways, but to a lesser degree than the CD8+ T cell response (Figure S4 A). Of importance, such CD4+ T cells were predominantly of Th1 phenotype based on their IFN-γ versus IL-4 production capacity and ratios, upon spike-specific or polyclonal (αCD3/αCD28) stimulation (Figure S4B), in keeping with a Th1-skewed S-specific IgG2a antibody response (Figure 1F).
Figure 2.

Single-dose intranasal immunization induces superior airway T cell responses over intramuscular immunization
(A) Left, bar graphs depicting absolute number of S1-specific IFNγ+ CD8+ T cells in the BAL at 2 (red) and 4 (blue) weeks post-immunization. Right, representative flow cytometric dotplots of IFN-γ+ CD8+ T cells in the BAL following ex vivo stimulation with overlapping peptide pools for S1.
(B) Bar graphs depicting multifunctional CD8+ T cell responses in the BAL as measured by production of IFN-γ, TNF-α, and/or IL-2 at 4 weeks post-immunization, following ex vivo stimulation with overlapping peptide pools for S1.
(C) Stacked bar graph depicting the frequency of cytotoxic CD8+ T cells in the BAL as measured by Granzyme B production at 4 weeks post-immunization, following ex vivo stimulation with DMSO (red) or overlapping peptide pools for S1 (blue).
(D–F) is the same as (A–C) but following stimulation with overlapping peptide pools for nucleocapsid.
(G–I) is the same as (A–C) but following stimulation with overlapping peptide pools for RdRp.
Data presented in (A–I) represent mean ± SEM. Statistical analysis were Mann-Whitney tests. Data are pooled from 2 independent experiments, n = 3–6 mice/group. ∗p < 0.05; ∗∗p < 0.01.
See also Figures S2 and S4.
Figure S4.

Comparison of antigen-specific CD4 and CD8 T cells in BAL and spleen following single-dose immunization with Tri:HuAd or Tri:ChAd vaccine, related to Figure 2
(A) Bar graphs depicting absolute number of S1 (left), nucleocapsid (middle), or RdRp (right) specific IFN-γ+ CD4+ T cells in the BAL at 2 (red) and 4 (blue) weeks post-immunization following ex vivo stimulation with overlapping peptide pools.
(B) Left, Schema of vaccination regimen. BALB/c mice were intranasally (i.n.) vaccinated with a single dose of either Tri:HuAd or Tri:ChAd. Animals were sacrificed at 3 weeks post-immunization for immunological analysis. Bar graphs depicting frequency of IFN-γ+ CD4+ T cells (middle left), or IL4+ CD4+ T cells (middle right) following ex vivo stimulation with S1 overlapping peptide pools (red), or anti-CD3/CD28 (blue). Right, Ratio of IFNγ:IL-4 producing CD4+ T cells, based on data from middle panels.
(C) Bar graphs depicting absolute number of S1 (left), nucleocapsid (middle), or RdRp (right) specific IFN-γ+ CD8+ (top) or IFN-γ+ CD4+ (bottom) T cells in the spleen at 2 (red) and 4 (blue) weeks post-immunization following ex vivo stimulation with overlapping peptide pools.
Data presented in (A–C) represent mean ± SEM. Data are representative of 1 independent experiment, n = 3–4 mice/group.
We further assessed the multifunctionality of CD8+ T cells with a focus on 4 weeks time point post-i.n. immunization when T cells entered the contraction/memory phase. The majority of S1-specific CD8+ T cells induced by either Tri:HuAd or Tri:ChAd vaccine were multifunctional, co-expressing IFN-γ and TNF-α whereas N- and RdRp-specific T cells were mostly monofunctional (Figures 2B, 2E, and 2H). Using Granzyme B as an indicator of cytotoxic capability, we found that while i.n. immunization with both vaccines generated cytotoxic CD8+ T cells against all vaccine-encoded antigens, Tri:ChAd induced significantly greater frequencies of these T cells in the airways than Tri:HuAd (Figures 2C, 2F, and 2I).
We also assessed systemic antigen-specific T cells in the spleen at 4 weeks post-immunization. In keeping with our previous findings (Santosuosso et al., 2005), i.m., but not i.n., immunization with either vaccine induced robust systemic CD8+ T cell responses to S1, N, and RdRp (Figure S4C, top panels). Once again, S1-specific T cell responses were dominant compared with those to N or RdRp. Antigen-specific CD4+ T cells were also induced in the spleen, but again to a lesser degree than CD8+ T cells (Figure S4C, bottom panels).
The above data indicate that single-dose intranasal, but not intramuscular, immunization, particularly with Tri:ChAd vaccine, is able to induce multifunctional CD8+ T cells with cytotoxic potential within the respiratory tract.
Single-dose intranasal, but not intramuscular, immunization induces multifunctional respiratory mucosal tissue-resident memory T cell responses
Compelling evidence indicates a critical role of mucosal tissue-resident memory T cells (TRM) in host defense (Szabo et al., 2019) and i.n., but not i.m., vaccination can induce such T cells (Jeyanathan et al., 2018, 2020; Teijaro and Farber, 2021). To investigate whether Ad-vectored trivalent COVID-19 vaccine could induce respiratory mucosal TRM, we first established t-SNE maps based on pooled CD3+/CD8+/CD4− mononuclear cells from lung tissues of all animals 8 weeks post-i.m. or i.n. delivery of Tri:HuAd or Tri:ChAd vaccine (Figure 3 A, top panel). Upon overlaying these cells concatenated from i.m. and i.n. animals, two unique CD8+ T cell clusters (orange-yellow color) were identified to be associated only with i.n. immunization (Figure 3A, bottom panel). We next generated heatmaps to overlay expression intensities for the surface markers CD44, CD69, CD103, and CD49a associated with mucosal TRM (Szabo et al., 2019). This analysis shows the two unique clusters of T cells of CD8+ TRM phenotype induced by i.n. immunization (Figures 3B and 3C). Upon quantification according to vaccine routes and vectors and our gating strategy (Figure S3B), we found that while i.n., but not i.m., immunization with either Tri:HuAd or Tri:ChAd induced significant numbers of CD8+ TRM, Tri:ChAd vaccine induced markedly more CD8+ TRM in the lung (Figures 3D, S5 A, and S5B).
Figure 3.

Single-dose intranasal, but not intramuscular, immunization induces multifunctional respiratory mucosal tissue-resident memory T cells
(A) Top: t-SNE maps were generated from concatenating CD3+ CD8+ CD4− gated lung mononuclear cells from 12 individual animals (3 per group of route/vaccine). Analysis was performed utilizing default FlowJo V.10 software settings. Bottom: overlay of populations arising after intramuscular (i.m., green) or intranasal (i.n., yellow) onto t-SNE maps.
(B) Heatmap projections of CD44, CD69, CD103, or CD49a on t-SNE maps. Hashed circles indicate bona fide tissue-resident memory CD8+ T cells.
(C) Overlap of populations arising after i.m. or i.n. Tri:HuAd (red) or i.m. or i.n. Tri:ChAd (blue). Hashed circles indicate two unique clusters of CD8+ T cells induced following i.n. immunization.
(D) Bar graph depicting absolute number of tissue-resident memory CD8+ T cells in the lung at 8 weeks post-immunization.
(E) Left: flow cytometric dot plots of CD44+ CD8+ T cells for CD69 and CD103 from the BAL at 8 weeks post-immunization. Right: histogram depicting expression of CD49a on CD69/CD103 double-positive CD44+ CD8+ T cells.
(F) Bar graphs depicting absolute number of S1, nucleocapsid, or RdRp-specific IFN-γ+ CD8+ T cells in the BAL at 8 weeks post-immunization, following ex vivo stimulation with overlapping peptide pools for S1, nucleocapsid, or RdRp.
(G) Sunburst plots depicting functionality (IFN-γ, TNF-α, and/or IL-2) of CD8+ T cells at 8 weeks post-immunization, following ex vivo stimulation with either S1, nucleocapsid, or RdRp peptide pools.
Data presented in (D–F) represent mean ± SEM. Data are representative of 2 independent experiments, n = 3–6 mice/group.
See also Figures S2, S3, and S5.
Figure S5.

Single-dose intranasal immunization induces respiratory mucosal tissue-resident memory T cells, related to Figure 3
(A) Experimental schema.
(B) Left: frequency of CD44+ CD8+ T cells in the lung. Right: flow cytometric dot plots of CD44+ CD8+ T cells for CD69 and CD103 from the lung at 8 weeks post-immunization.
(C) Flow cytometric dot plots of CD44+ CD4+ T cells for CD69 and CD11a from the BAL at 8 weeks post-immunization.
(D) Experimental schema.
(E) Left: frequency of CD44+ CD8+ T cells in the lung. Right: flow cytometric dot plots of CD44+ CD8+ T cells for CD69 and CD103 from the lung at 4 weeks post-immunization.
(F) Flow cytometric dot plots of CD44+ CD4+ T cells for CD69 and CD103 from the BAL at 4 weeks post-immunization. Histograms depicting expression of CD49a on CD69/CD103 double-positive CD44+ CD8+ T cells are shown.
Data presented in (B, C, E, and F) represent mean ± SEM. Data are representative of 1 independent experiment, n = 3 mice/group.
We next examined CD8+ TRM within the airways (BAL) 8 weeks post-immunization. Given the lack of airway T cells following i.m. immunization (Figures 2A, 2D, and 2G), we focused our analysis on BAL cells from i.n.-immunized animals. We found that regardless of vaccine vector, approximately 50% of antigen-experienced CD44+CD8+ T cells in the BAL co-expressed TRM surface markers CD69, CD103, and CD49a (Figure 3E). Similar observations were made with antigen-experienced CD69+CD11a+CD4+ TRM in the airways except that they were present in smaller frequencies compared with CD8+ TRM (Figure S5C). Potent induction of TRM in the lung by i.n. immunization, particularly with Tri:ChAd, was also observed at 4 weeks (Figures S5D–S5F). In contrast, i.n. delivery of an empty vector (HuAd:EV or ChAd:EV) had a minimal effect on TRM induction in the lung or airways (Figure S2E).
We further determined the multi-functionality of long-term memory CD8+ T cells in the airways 8 weeks post-i.n. immunization. Compared with the 4-week time point (Figures 2A, 2D, and 2G), airway antigen-specific IFN-γ+CD8+ T cells further contracted, irrespective of vaccine vector, with the majority being specific for S1 (Figure 3F). However, multi-cytokine expression analysis reveals that the memory CD8+ T cells induced by i.n. Tri:ChAd were more functional than those by Tri:HuAd based on co-expression of IFN-γ, TNF-α, and/or IL-2 (Figure 3G). Of note, S1-specific memory T cells showed a greater breadth of multifunctionality than those specific to N or RdRp.
These data indicate that intranasal, but not intramuscular, COVID-19 immunization is able to induce durable multifunctional respiratory mucosal TRM responses. Furthermore, Tri:ChAd vaccine is more potent than Tri:HuAd platform.
Single-dose intranasal, but not intramuscular, immunization induces trained airway macrophages
Since alveolar macrophages (AMs) as the main respiratory mucosal-resident innate immune cells have been shown to interact with SARS-CoV-2 (Grant et al., 2021) and other RNA viruses (Kumagai et al., 2007; Schneider et al., 2014), they likely play a critical role in early innate immune control of SARS-CoV-2. Aside from producing robust adaptive immunity within the airways, we have shown that Ad-vectored i.n., but not i.m., TB immunization induces long-lasting airway-resident memory AM and trained innate immunity (TII) (D’Agostino et al., 2020; Xing et al., 2020; Yao et al., 2018). Such trained AM are phenotypically defined by their high surface expression of MHC II (Yao et al., 2018). To investigate whether Ad-vectored COVID-19 vaccines induces trained AM, mice were immunized intramuscularly or intranasally with Tri:HuAd or Tri:ChAd vaccine, and the immune phenotype of airway (BAL) macrophages was analyzed 8 weeks post-immunization (Figure 4 A). To better enable our flow cytometric characterization of CD45+CD11b+CD11c+ airway macrophages, t-SNE maps were first generated with the BAL cells pooled from all animals to visualize the overall differences between both vaccine vectors and routes (Figures 4B and 4C). AM were distinguishable as a large cluster co-expressing high levels of AM surface markers CD11c and Siglec-F (Figure 4B, yellow color). Of importance, while high MHC II expression was seen within the large AM cluster, another clear cluster branched off the main AM population, co-expressing high levels of MHC II and CD11b (Figure 4B, top right panels). A small neutrophil cluster was discernable by co-expression of CD11b and Ly6G (Figure 4B).
Figure 4.

Single-dose intranasal, but not intramuscular, immunization induces trained airway macrophages
(A) Experimental schema.
(B) Left, t-SNE maps were generated from concatenating CD45+ CD11b+/CD11c+ gated BAL mononuclear cells from 12 individual animals (3 per group of route/vaccine condition). Analysis was performed utilizing default FlowJo V.10 software settings. Right, heatmap projections of CD11c, CD11b, MHC II, Siglec-F, Ly6G, or Ly6C on t-SNE maps. Hashed circle indicates an MHC-II-high population.
(C) Overlap of populations arising after i.m. or i.n. Tri:HuAd (red) or i.m. or i.n. Tri:ChAd (blue). Hashed circles indicate a unique MHC-II-high population induced following i.n. immunization.
(D) MFI of MHC II expression on AM (left) and IM (right) in BAL at 8 weeks post-immunization.
(E) Experimental schema.
(F) Left, t-SNE maps were generated from concatenating CD45+ CD11b+/CD11c+ gated BAL mononuclear cells from 12 individual animals (3 per group of route/vaccine condition). Analysis was performed utilizing default FlowJo V.10 software settings. Right, heatmap projections of CD11c, CD11b, MHC II, Siglec-F, Ly6G, or Ly6C on t-SNE maps. Hashed circle indicates an MHC-II-high population.
(G) Overlap of populations arising after i.n. Tri:HuAd (red) or Tri:ChAd (blue) or empty vector equivalent controls. Hashed circles indicate a unique MHC-II-high population induced following i.n. immunization with either Tri:HuAd or Tri:ChAd.
(H) MFI of MHC II expression on AM (left) and IM (right) in BAL at 4 weeks post-immunization.
Data presented in (D and H) represent mean ± SEM. Data are representative of 2 independent experiments, n = 3–6 mice/group. Statistical analysis for (D and H) were one-way ANOVA with Tukey’s multiple comparisons test. ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Alveolar macrophage (AM), interstitial macrophage (IM), and median fluorescence intensity (MFI).
We next specifically determined the extent to which COVID-19 vaccine vectors and routes induced trained MHC IIhigh airway macrophages. Whereas i.m. Tri:HuAd or Tri:ChAd vaccine resulted in hardly any MHC IIhigh AM (Figure 4C, left 2 panels), i.n. immunization, notably with Tri:ChAd, generated several distinct populations expressing high levels of MHC II or CD11b separate from the large AM cluster (Figure 4C, right 2 panels). This is phenotypically consistent with an influx of interstitial macrophages (IMs) and trained AM observed within the airway following i.n. vaccination with a HuAd-vectored TB vaccine (Yao et al., 2018). Thus, using a comprehensive gating strategy, we compared MHC II expression in both AM and IM in the airways. While in keeping with our previous findings with a TB vaccine (D’Agostino et al., 2020; Yao et al., 2018), i.m. Tri:HuAd or Tri:ChAd COVID-19 vaccine was unable to induce trained AM or IM (Figure 4D), i.n. immunization with either vaccine induced markedly increased MHC IIhigh AM and IM (Figure 4D). Furthermore, in line with t-SNE analysis (Figure 4C), i.n. Tri:ChAd induced significantly further increased MHC II median fluorescence intensity (MFI) on both AM and IM over that by Tri:HuAd (Figure 4D).
Furthermore, we also examined whether i.n. immunization induced trained airway macrophages even earlier at 4 weeks with the empty vector (HuAd:EV or ChAd:EV) included as control (Figure 4E). Visualization of t-SNE plots of CD45+CD11C+CD11b+ BAL cells showed the absence of MHC IIhigh macrophages in the airways of HuAd:EV or ChAd:EV animals, compared with i.n. Tri:HuAd- or Tri:ChAd animals (Figures 4F and 4G), consistent with MHC II MFI analysis profiles on both AM and IM (Figure 4H). In keeping with 8 weeks of data (Figure 4D), i.n. Tri:ChAd vaccine induced significantly further increased MHC II MFI on airway macrophages over that by i.n. Tri:HuAd (Figure 4H). The above data together suggest that single-dose respiratory mucosal immunization with Ad-vectored COVID-19 vaccines induces trained airway macrophages, besides its potent effects on inducing memory B and T cells in the lung.
Intranasal, but not intramuscular, immunization provides potent B- and T-cell-dependent protection from SARS-CoV-2 infection
Having demonstrated the robust immunogenicity by intranasal immunization with trivalent COVID-19 vaccines, we next assessed the protection by both vaccine vectors against a mouse-adapted virus (SARS-CoV-2 MA10) (Leist et al., 2020). Wild-type BALB/c mice were i.m.- or i.n.-immunized with single-dose Tri:HuAd or Tri:ChAd and challenged 4 weeks later with a lethal dose of 1 × 105 PFU SARS-CoV-2 MA10 (Figure 5 A). Animals were monitored for weight loss and mortality. All unvaccinated animals succumbed, reaching humane endpoint by 5 days post-infection (Figure 5B). Likewise, i.m. immunization with either vaccine failed to protect, with 80% animals reaching humane endpoint by 4–5 days (Figure 5B; numbers of surviving animals in each group indicated in brackets after legends). In contrast, i.n. Tri:HuAd-immunized animals showed slight (∼5%), transient weight loss but rapidly rebounded to pre-infection weights over the course of experimentation (Figure 5B). Notably, i.n. Tri:ChAd-immunized animals were fully protected, showing no weight loss throughout (Figure 5B). To further evaluate protection, a cohort of mice was sacrificed 2 days post-infection for determination of lung viral burden in the acute phase of infection. In accordance with weight loss, i.m. immunization with either vaccine only modestly reduced viral loads (Figure 5C). However, in keeping with differentially improved clinical outcomes (Figure 5B), i.n. immunization provided vaccine-vector-dependent reductions in viral burden, with Tri:ChAd-immunized animals having the most significantly reduced viral burden (>3 log) compared with >1 log reduction with Tri:HuAd (Figure 5C). The markedly enhanced protection by i.n. immunization, particularly with Tri:ChAd, correlated with markedly reduced lung pathology at 14 days post-infection relative to the severe pathology seen in unvaccinated animals (kept alive via fluid supplementation) (Figures S6 A, S6B, and S6C). As expected, i.n. delivery of the empty vector (HuAd:EV and ChAd:EV) failed to offer any significant protection from clinical outcomes or lung viral burden (Figures S6D, S6E, and S6F). These data show a correlation of the relative magnitude of vaccine-induced mucosal B and T cell immunity and TII as described earlier with protective efficacy against SARS-CoV-2 infection, and support the protective superiority of i.n. Tri:ChAd over Tri:HuAd vaccine.
Figure 5.

Intranasal, but not intramuscular, immunization provides potent B- and T-cell-dependent protection from SARS-CoV-2 infection
(A) Experimental schema.
(B) Changes in body weight over 2 weeks post-SARS-CoV-2 infection.
(C) Viral burden (Log10TCID50) in the lung at 2 days post-SARS-CoV-2 MA10 infection.
(D) Experimental schema.
(E) Changes in body weight of unvaccinated BALB/c, T cell depleted BALB/c, or Jh−/− mice over 2 weeks post-SARS-CoV-2 infection.
(F) Changes in body weight of i.n. Tri:ChAd-vaccinated BALB/c, T-cell-depleted BALB/c, or Jh−/− mice for 2 weeks post-SARS-CoV-2 infection.
(G) Viral burden (Log10TCID50) in the lung of unvaccinated animals at 4 days post-infection.
(H) Viral burden (Log10TCID50) in the lung of Tri:ChAd vaccinated animals at 4 days post-infection.
(I) Experimental schemas.
(J) Changes in body weight of over 2 weeks post-SARS-CoV-2 infection. Black circles indicate unvaccinated mice, blue circles indicate Tri:ChAd-vaccinated mice, red circles indicate Tri:ChAd-vaccinated mice with continuous T cell depletion, purple circles indicate Tri:ChAd-vaccinated mice with T cell depletion prior to infection.
(K) Heatmap representing cumulative acute clinical observations: ruffled fur, lethargy/depression, and erratic/labored respiration.
(L) Gross pathological changes from the lungs of vaccinated mice at 4 (left) and 14 (right) days post-infection. Hashed circles encompass areas of visible lung damage.
(M) Viral burden (Log10TCID50) in the lung of animals at 4 days post-infection.
Data presented in (B, C, E–H, J, and M) represent mean ± SEM. Data are representative of 1–2 independent experiments, n = 5 mice/group. Statistical analysis for (C, G, H, and M) were one-way ANOVA with Tukey’s multiple comparisons test. ns, not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
See also Figures S6 and S7.
Figure S6.

Assessment of intranasal administration of empty vector HuAd:EV and ChAd:EV following SARS-CoV-2 infection, related to Figure 5
(A–C) Representative histopathological images of lungs 14 days post-SARS-CoV-2 MA10 infection in animals intranasally (i.n.) vaccinated with either Tri:HuAd (B) or Tri:ChAd (C), in comparison with unvaccinated controls (A), as per Figure 5A.
(D) Experimental schema.
(E) Changes in body weight over 2 weeks post-SARS-CoV-2 infection.
(F) Viral burden (Log10TCID50) in the lung at 4 days post-SARS-CoV-2 MA10 infection.
Data presented in (E and F) represent mean ± SEM. Statistical analysis for (F) were one-way ANOVA with Tukey’s multiple comparisons test. Data in (E) are representative of 1 independent experiment, n = 5 mice/group. Data in (F) is pooled from 2 independent experiments, n = 10 mice per group. ns, not significant.
We next investigated the relative contribution of B and T cell immunity to i.n. vaccine-induced protection, focusing on i.n. Tri:ChAd vaccine strategy. To this end, wild-type BALB/c mice (wild type), BALB/c mice deficient in J segments (B cell KO) of the immunoglobulin heavy-chain locus (lacking mature B cells or circulating IgG), and BALB/c mice depleted of both CD4+ and CD8+ T cells (T cell dep.) were left unvaccinated or intranasally vaccinated with Tri:ChAd. Mice were then infected with SARS-CoV-2 MA10 at 4 weeks post-immunization (Figure 5D). T cell depletion was carried out from day 25 post-immunization when B cells/antibody responses were fully developed and prior to viral challenge by 2–3 repeated intraperitoneal injections of a T-cell-depleting antibody cocktail (Yao et al., 2018). Unvaccinated wild-type mice rapidly succumbed to infection, reaching humane endpoint by 4 days (Figure 5E). Unvaccinated mice lacking either T cells or B cells showed similar weight loss kinetics, with 80% T cell dep. and 20% B cell KO mice reaching humane endpoint at the same time as unvaccinated wild-type controls (Figure 5E). In contrast, i.n. Tri:ChAd vaccine protected wild-type, T cell dep., and B cell KO animals equally well as they did not show any weight losses throughout (Figure 5F). These data indicate that the superior immunogenicity of i.n. Tri:ChAd vaccine is capable of compensating for the lack of either T or B cells to retain protection against clinical disease.
Besides clinical outcomes, we examined the role of B and T cells in control of viral burden in the lung 4 days post-infection (Figure 5D). Compared with wild-type hosts, lack of T or B cells in unvaccinated animals had little effects on high lung viral burden (Figure 5G). In contrast, i.n. Tri:ChAd vaccination of wild-type control animals led to complete viral clearance at 4 days (Figure 5H). However, lack of either T or B cells in i.n. Tri:ChAd-vaccinated animals resulted in significantly elevated lung viral titers (2–4 log) (Figure 5H), despite no changes in morbidity/mortality (Figure 5F). In comparison, similar to i.n. Tri:ChAd vaccine, i.n. Tri:HuAd also well protected wild-type and B cell KO mice but vaccinated mice lacking T cells experienced a transient moderate weight loss with 20% of them succumbing to infection (Figures S7 A and S7B). However, while similar to the 2-day data (Figure 5C), i.n. Tri:HuAd vaccine at 4 days only moderately reduced viral loads in the lung of wild-type animals by ∼2 logs, lack of either T or B cells in these animals also resulted in increased lung viral burden (∼1 log) (Figure S7C).
Figure S7.

Assessment of B- and T-cell-dependent protection from SARS-CoV-2 infection following intranasal immunization with Tri:HuAd, and characterization of immunogenicity of intranasal immunization with Tri:ChAd vaccine in wild-type C57BL/6 or K18-hACE2 mice, related to Figures 5 and 6
(A) Experimental schema.
(B) Changes in body weight of i.n. Tri:HuAd-vaccinated BALB/c, T-cell-depleted BALB/c, or Jh−/− mice for 2 weeks post-SARS-CoV-2 infection.
(C) Viral burden (Log10TCID50) in the lung of Tri:HuAd vaccinated animals at 4 days post-infection.
(D) Experimental schema.
(E) Serum neutralizing antibody responses at 4 weeks post-immunization in C57BL/6 mice, measured by percent (%) neutralization utilizing a live SARS-CoV-2 microneutralization (MNT) assay.
(F) Absolute number of antigen-specific IFNγ+ CD8+ T cells in the airway at 4 weeks post-immunization in C57BL/6 mice, following ex vivo stimulation with overlapping peptide pools for S1, nucleocapsid, or RdRp.
(G) Flow cytometric dot plots showing frequencies of spike-specific IFN-γ+ CD8+ T cells in lung mononuclear cells at 4 weeks post-immunization in C57BL/6 mice, upon stimulation with either ancestral or variant SARS-CoV-2 spike protein at 4 weeks post-immunization.
(H) MFI of MHC II expression on AM (left) and IM (right) in BAL at 4 weeks post-immunization in C57BL/6 mice.
(I) Left, serum neutralizing antibody responses at 4 weeks post-immunization of K18-hACE2 mice, measured by percent (%) neutralization utilizing a live SARS-CoV-2 ancestral (red), B.1.1.7 (blue), or B.1.351 (purple) microneutralization (MNT) assay. Right, MNT50 values.
Data presented in (B, C, and E–I) represent mean ± SEM. Statistical analysis for (C) was one-way ANOVA with Tukey’s multiple comparisons test. Statistical analysis for (H) was two-tailed t tests. Data are representative of 1 independent experiment, n = 3–5 mice/group. ∗p < 0.05.
To further understand the mechanisms of i.n. Tri:ChAd-induced protection in either T-cell-depleted or B-cell-deficient hosts, we determined the protection in i.n. Tri:ChAd-vaccinated B cell KO mice depleted of T cells. Since we have reported that T cell help is required for vaccine-mediated airway macrophage priming and TII only at early (3–5 days), but not later, times post-i.n. Ad-vectored vaccination (Yao et al., 2018), this model also provided the opportunity to address the protective role of TII in the absence of both T and B cell immunity. Thus, a continuous T cell depletion protocol initiated shortly after i.n. Tri:ChAd vaccination was employed in B cell KO mice to render deficiencies in TII, and T and B cells (Figure 5I, top schema). Additionally, a prior T cell depletion protocol initiated right before infection and from day 25 post-i.n. vaccination on was employed in B cell KO mice to render TII induction in airway macrophages but lack of both T and B cells throughout SARS-CoV-2 infection (Figure 5I, bottom schema). Unvaccinated wild-type BALB/c mice were used as naive controls. Consistent with the earlier data (Figures 5E and 5F), unvaccinated BALB/c mice experienced significant morbidity as measured by weight loss, whereas both i.n.-vaccinated B cell KO animals (Jh−/− Tri:ChAd) and B cell KO animals depleted of T cells but with intact TII (Jh−/− Tri:ChAd/prior T cell depletion) appeared healthy in clinical outcomes (Figures 5J and 5K, blue and purple circle, respectively). In contrast, the i.n.-vaccinated B cell KO hosts lacking both T cells and TII (Jh−/− Tri:ChAd/continuous T cell depletion) suffered not only an early body weight loss but also the most severe clinical signs (Figures 5J and 5K, red circle), consistent with their heightened gross pathology in the lungs (Figure 5L). Despite their normal or near-normal clinical outcomes, B cell KO Jh−/− Tri:ChAd animals had significant, though reduced, viral burden (though a bit variable compared with B cell KO data in Figure 5H). Those lacking both B and T cells (Jh−/− Tri:ChAd/prior T cell depletion) and those lacking B and T cells and TII (Jh−/− Tri:ChAd/continuous T cell depletion) all had high viral burden in the lung (Figure 5M).
The above data indicate that intranasal immunization provides superior protection against a mouse-adapted SARS-CoV-2 over intramuscular route, Tri:ChAd vaccine is more potent than Tri:HuAd, both humoral and T cell immunity are required for optimal protection by intranasal immunization, and vaccine-induced TII contributes to protection primarily by improving clinical outcomes while it plays a minor role in control of viral burden.
Single-dose intranasal immunization with ChAd-vectored trivalent COVID-19 vaccine protects against lethal infection by SARS-CoV-2 variants of concern
We next assessed vaccine-induced protection against wild-type ancestral and VOC strains of SARS-CoV-2 in highly susceptible K18-hACE2 mouse model (Khoury et al., 2020). Since our data indicate the superiority of i.n. Tri:ChAd vaccine in both immunogenicity and protection, we focused on evaluating the protection by i.n. Tri:ChAd immunization in K18-hACE2 models.
Since the bulk of our immunogenicity data were from BALB/c mice, we began by assessing vaccine immunogenicity in C57BL/6 K18-hACE2 mice at 4 weeks post-i.n. Tri:ChAd immunization (Figure S7D). Similar to the neutralization titers in BALB/c hosts (Figure 1L), sera from i.n. Tri:ChAd-immunized C57BL/6 animals showed robust SARS-CoV-2 neutralization (Figure S7E). Similarly, airway antigen-specific CD8+ T cell responses in C57BL/6 mice were also comparable (Figure S7F) with those in BALB/c mice (Figures 2A, 2D, and 2G), and these cells responded to ancestral and B.1.351 variant spike antigens equally well (Figure S7G). Also, consistent with our findings in BALB/c mice (Figure 4D), there was markedly increased MHC II expression on airway macrophages (AM & IM) only following i.n. Tri:ChAd immunization (Figure S7H). These data suggest that i.n. Tri:ChAd vaccine is similarly immunogenic in C57BL/6 and BALB/c hosts and that T cell antigens are well conserved among ancestral and VOC strains of SARS-CoV-2.
We then evaluated the protection of i.n. Tri:ChAd-vaccinated K18-hACE2 mice post-challenge with an ancestral SARS-CoV-2 virus. As controls, some mice were left unvaccinated (naive) or inoculated with an empty ChAd vector (ChAd:EV). Mice were infected 4 weeks later with 1 × 105 PFU of SARS-CoV-2/SB3 (Figure 6 A), an early pandemic strain isolated in Toronto, Canada (Banerjee et al., 2020). Both naive and ChAd:EV animals quickly succumbed, reaching the clinical endpoint based on weight loss and/or neurological symptoms between 5 and 7 days (Figures 6B and 6C). In contrast, i.n. Tri:ChAd vaccine largely prevented weight loss (Figure 6B) and mortality (Figure 6C), with 10/11 mice surviving infection. In agreement with the clinical outcomes, while both naive and ChAd:EV animals had high lung viral burden, i.n. Tri:ChAd vaccine provided sterilizing immunity in the lung (Figure 6D). These data indicate that a single i.n. dose of Tri:ChAd vaccine is sufficient to protect K18-hACE2 mice from lethal challenge with ancestral SARS-CoV-2.
Figure 6.

Intranasal Tri:ChAd immunization protects against lethal challenge with SARS-CoV-2 variants of concern
(A) Experimental schema.
(B) Changes in body weight over 2 weeks post-ancestral SARS-CoV-2 infection.
(C) Survival of mice post-ancestral SARS-CoV-2 SB3 infection.
(D) Viral burden (Log10TCID50) in the lung at 4 days post-infection.
(E) Changes in body weight over 2 weeks post-SARS-CoV-2 B.1.1.7 infection.
(F) Survival of mice post-SARS-CoV-2 B.1.1.7 infection.
(G) Changes in body weight over 2 weeks post-SARS-CoV-2 B.1.351 infection.
(H) Survival of mice post-SARS-CoV-2 B.1.351 infection.
(I) Viral burden (Log10TCID50) in the lung at 4 days post-B.1.1.7 or B.1.351 infection.
(J) Viral burden (Log10TCID50) in the brain at 4 days post-B.1.1.7 or B.1.351 infection.
Data presented in (B, D, E, G, I, and J) represent mean ± SEM. Statistical analysis for (D, I, and J) were one-way ANOVA with Tukey’s multiple comparisons test. Data in (B and C) are pooled from 2 independent experiments, n = 5–11 mice/group. Data in (D–J) are representative of 1 experiment, n = 5 mice/group. ns, not significant; ∗p < 0.05; ∗∗∗∗p < 0.0001.
See also Figure S7.
Having observed superb protective efficacy of i.n. Tri:ChAd vaccine against infection by both mouse-adapted (Figure 5) and wild-type ancestral (Figures 6B, 6C, and 6D) strains of SARS-CoV-2, we evaluated its protection in K18-hACE2 model against two VOC, B.1.1.7 and B.1.351, with the latter shown to evade the immunity by first-generation vaccines (Harvey et al., 2021). Indeed, while the immune serum from i.n. Tri:ChAd-vaccinated K18-hACE2 animals neutralized both ancestral and B.1.1.7 viruses equally well, it had much reduced capacity (MNT50: 70.70%) to neutralize B.1.351 variant (Figure S7I). Thus, i.n. Tri:ChAd-vaccinated K18-hACE2 mice and control groups were challenged with a lethal dose (1 × 105 PFU) of B.1.1.7 or B.1.351 variant as described in Figure 6A. Similar to what was observed with ancestral SARS-CoV-2 (Figures 6B and 6C), both unvaccinated naive and ChAd:EV animals showed quick weight losses (Figure 6E), reaching the clinical endpoint between 5 and 6 days post-B.1.1.7 infection (Figure 6F). In contrast, i.n. Tri:ChAd vaccine completely protected the animals from morbidity/mortality (Figures 6E and 6F). Similarly, upon B.1.351 variant infection, while 80% of unvaccinated naive and all of ChAd:EV animals succumbed by 7–8 days, the vast majority of i.n. Tri:ChAd animals were well protected (Figures 6G and 6H). We next assessed viral burden in the lung at 4 days post-infection (Figure 6A). Since SARS-CoV-2 infection of K18-hACE2 mice also causes viral dissemination to the brain (Zheng et al., 2021), the brain was included in the assay. Unvaccinated naive mice had similarly high B.1.1.7 or B.1.351 viral burden in the lung (Figure 6I) and brain (Figure 6J). Of interest, while ChAd:EV mice had similarly high viral titers in the lung as in naive mice (Figure 6I), they showed moderately reduced viral burden in the brain (Figure 6J). Remarkably, i.n. Tri:ChAd-immunized mice developed sterilizing immunity against both B.1.1.7 and B.1.351 variants in the lung (Figure 6I) and brain (Figure 6J).
The above data indicate that single-dose respiratory mucosal immunization with trivalent ChAd-vectored COVID-19 vaccine induces robust mucosal immunity against lethal infection by not only ancestral SARS-CoV-2 but also immune-evasive VOC.
Comparison of protective efficacy of ChAd-vectored trivalent vaccine with its bi-valent and mono-valent counterparts
We next examined the protective role of the N and RdRp antigens included in Tri:ChAd vaccine design. To this end, we developed a bi-valent and a mono-valent ChAd vaccine only expressing the N/RdRp (Bi:ChAd) and the S1 (Mono:ChAd), respectively (Figure 7 A). We first examined the protection by i.n. Bi:ChAd vaccine and compared it with Tri:ChAd in BALB/c mouse model (Figure 7B). In contrast to severe clinical illness in unvaccinated animals, i.n. Bi:ChAd protected mice as well as i.n. Tri:ChAd against infection (Figure 7C). On the other hand, while i.n. Tri:ChAd led to no detectable viruses, i.n. Bi:ChAd markedly reduced lung viral titers by 3 logs relative to unvaccinated controls (Figure 7D).
Figure 7.

Comparison of protective efficacy of ChAd-vectored trivalent vaccine with its bi-valent and mono-valent counterparts
(A) Transgene cassette diagrams for bi-valent (Bi:ChAd, left) and mono-valent (Mono:ChAd, right) ChAd vaccines.
(B) Experimental schema.
(C) Changes in body weight over 2 weeks post-SARS-CoV-2 infection.
(D) Viral burden (Log10TCID50) in the lung at 4 days post-SARS-CoV-2 MA10 infection.
(E) Experimental schema.
(F) Changes in body weight over 2 weeks post-SARS-CoV-2 B.1.351 infection (open circles: 1 surviving animal).
(G) Viral burden (Log10TCID50) in the lung at 4 days post-B.1.351 infection.
(H) Viral burden (Log10TCID50) in the brain at 4 days post-B.1.351 infection.
(I) Experimental schema.
(J) Changes in body weight over 2 weeks post-SARS-CoV-2 B.1.351 infection.
(K) Gross pathological changes from the lungs of vaccinated mice at 4 days post-infection. Hashed circles encompass areas of visible lung damage.
(L) Viral burden (Log10TCID50) in the lung at 4 days post-B.1.351 infection.
Data presented in (C, D, F–H, J, and L) represent mean ± SEM. Statistical analysis for (D, G, H, and L) were one-way ANOVA with Tukey’s multiple comparisons test. Data in (C, D, F, G, and H) are pooled from 2 independent experiments, n = 3–11 mice/group. Data in (J–L) are representative of 1 experiment, n = 5 mice/group. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
We further assessed the role of N and RdRp by evaluating i.n. Bi:ChAd-mediated protection in the K18-hACE2 model of B.1.351 challenge (Figure 7E). While unvaccinated animals experienced severe disease, i.n. Bi:ChAd delayed body weight losses and i.n. Tri:ChAd offered the best protection (Figure 7F), in keeping with earlier data (Figures 6G and 6H). Unvaccinated K18-hACE2 mice had high B.1.351 viral burden whereas i.n. Tri:ChAd led to sterilizing immunity in the lung (Figure 7G) and brain (Figure 7H). In comparison, i.n. Bi:ChAd markedly reduced viral burden by 2 and 3 logs in the lung and brain, respectively (Figures 7G and 7H).
Using a different approach to examine the role of N/RdRp, we tested i.n. S1-expressing Mono:ChAd vaccine in B.1.351 K18-hACE2 model and compared it with Bi:ChAd and Tri:ChAd vaccines (Figure 7I). Mono:ChAd was similarly S1-immunogenic as Tri:ChAd. Over the 4-day period post-B.1.351 infection, unvaccinated and i.n. Mono:ChAd animals showed comparable body weight losses (Figure 7J). In contrast, both i.n. Bi:ChAd and Tri:ChAd animals remained clinically stable (Figure 7J). Indeed, extensive gross pathology was seen in the lungs of Mono:ChAd animals whereas the Bi:ChAd lungs appeared nearly free of gross pathology as did the Tri:ChAd animals (Figure 7K). Furthermore, unvaccinated animals had high viral loads in the lung whereas i.n. Tri:ChAd significantly reduced viral loads by 3.5 logs (Figure 7L). In comparison, both i.n. Bi:ChAd and Mono:ChAd vaccines only moderately reduced viral load.
The above data indicate the protective superiority of ChAd-vectored vaccine when expressing the S1, N, and RdRp antigens over its bi-valent and mono-valent counterparts. Inclusion of N/RdRp antigens in our trivalent vaccine design offers additional protection via neutralizing antibody-independent T cell and TII.
Discussion
The effective global control of COVID-19 via immunization with first-generation vaccines has been threatened by the VOC and waning vaccine-induced antibody immunity (Goldberg et al., 2021; Harvey et al., 2021; Krause et al., 2021). This situation calls for the development of not only next-generation/“universal” vaccines but also diversified vaccine strategies. In response, we have developed Ad-vectored next-generation trivalent COVID-19 vaccines expressing the original S1 antigen and highly conserved T cell antigens N and RdRp for respiratory mucosal route of delivery. We show that respiratory mucosal immunization is superior to intramuscular immunization at inducing neutralizing antibodies, mucosal tissue-resident memory T cells and trained airway macrophages. We further show that the choice of Ad vector also is of importance, with chimpanzee-derived Ad68 platform (Tri:ChAd) outperforming the human Ad5 counterpart (Tri:HuAd) and that inclusion of both the spike and conserved internal T cell antigens in vaccine design is required for optimal protection against both ancestral SARS-CoV-2 and VOC. We also reveal that both B and T cells and TII are required for robust respiratory mucosal immunity.
To our knowledge, our study is the first to have developed a multivalent next-generation vaccine strategy against both ancestral SARS-CoV-2 and emerging VOC in animal models. Although a recent murine study shows the ability of a first-generation S-encoding mRNA vaccine (CVnCoV) to protect from B.1.351 infection (Hoffmann et al., 2021a), this vaccine has led to disappointing efficacy results from clinical trials. It is widely accepted that the next-generation vaccine strategies ought to take into consideration both vaccine multivalency and route of delivery (Jeyanathan et al., 2020; Teijaro and Farber, 2021). While almost all first-generation genetic COVID-19 vaccines were designed for intramuscular delivery and to express only the S protein, there is a growing interest in studying their utility for respiratory mucosal delivery in preclinical models (Bricker et al., 2021; Cao et al., 2021; Hassan et al., 2020, 2021a; Ku et al., 2021). However, most of these studies did not compare the intranasal with the intramuscular route of immunization, nor did they test their protection against VOC. The studies by scientists at Washington University in St. Louis (WUSTL) are the only ones to compare the intranasal delivery with intramuscular immunization (Bricker et al., 2021; Hassan et al., 2020) and this group also extended their study to show the ability of intranasal ChAd-S immunization to protect against a virus displaying B.1.351 spike protein (Hassan et al., 2021b). By comparison, with the goal of developing next-generation COVID-19 vaccines, we have bioengineered two Ad-vectored trivalent vaccines (Tri:HuAd & Tri:ChAd) and extensively compared their immunogenicity and protection against ancestral and variant strains of SARS-CoV-2 following single-dose intramuscular or intranasal immunization. Our findings indicate the respiratory mucosal delivery of a trivalent ChAd-vectored vaccine to be the most effective next-generation COVID-19 vaccine strategy. Our study thus supports its further clinical development.
The superiority of i.n. COVID-19 immunization at inducing both protective humoral and mucosal T cell immunity over the i.m. route observed in our study is well aligned with the established paradigm associated with other vaccines (Belyakov and Ahlers, 2009; Jeyanathan et al., 2018; Neutra and Kozlowski, 2006). It has also been observed in animal models of COVID-19 using a ChAd-vectored first-generation vaccine (Bricker et al., 2021; Hassan et al., 2020). The high degree of tissue compartmentalization of immunity dictated by the route of immunization is not only limited to animal models. Inhaled aerosol MVA TB vaccine induced respiratory mucosal immunity In humans whereas intradermal injection of the same vaccine failed to do so (Satti et al., 2014). We have also recently reported that inhaled aerosol, but not intramuscular delivery, of HuAd-vectored TB vaccine induces respiratory mucosal immunity in humans (Jeyanathan et al., 2022). Given that all of the currently approved viral-vectored COVID-19 vaccines including ChAdOx1nCov-19 (Astra Zeneca/Oxford), Ad26.COV2-S (J&J), Gam-COVID-Vac (Gamaleya), and Ad5-nCoV (CanSino) are intramuscularly administered, they are unlikely to induce protective respiratory mucosal immunity (Jeyanathan et al., 2020). Although aerosol delivery of Ad5-nCoV was tested in humans, it is unclear whether it induced respiratory mucosal immunity (Wu et al., 2021). We have also recently shown that SARS-CoV-2-specific IgA enriched at mucosal surfaces like the lung can induce neutrophils to undergo NETosis capable of trapping and killing virus, thereby limiting spread (Stacey et al., 2021). Thus, the well-recognized limitations of i.m. vaccine delivery, along with our current findings and those from others (Bricker et al., 2021; Hassan et al., 2020), should bolster the global effort in developing respiratory mucosal-delivered next-generation COVID-19 vaccines. In this regard, there are at least two clinical trials testing inhaled aerosol ChAdOx1nCov-19 (Singh et al., 2021) or intranasally delivered ChAd-SARS-CoV-2-S (clinical trial NCT04751682).
Our study has also provided the evidence that both B and T cells are required for optimal protection. We find that the clinical outcomes/illness do not always corroborate with viral burden. Indeed, while i.n. Tri:ChAd vaccine protected wild-type and B-cell- and T-cell-deficient mice in terms of clinical outcomes, lack of B or T cells led to partially impaired viral clearance in the lung. Importantly, we observed that while lack of both B and T cells at the time of infection completely abolished the control of viral infection, the animals remained reasonably protected from clinical illness and lung pathology due to the presence of vaccine-induced TII. Thus, the optimal protection in both clinical disease and viral clearance is accomplished when vaccines effectively elicited functional antibodies, T cells and TII.
Globally, while waning vaccine-induced antibody immunity may account for recent increases in break-through infections by VOC (Wilhelm et al., 2021), the first-generation vaccines have thus far largely protected against severe disease (Scott et al., 2021), supporting the critical role of T cell immunity in controlling established infection in human lungs. In our study, besides using a T cell depletion approach, the role of vaccine-induced T cell immunity is supported further by our observations from both BALB/c and K18-hACE2 models immunized i.n. with a bi-valent ChAd-vectored vaccine (Bi:ChAd) that only expresses T cell antigens N/RdRp. Intranasal Bi:ChAd vaccine protected against clinical illness as well as Tri:ChAd vaccine in BALB/c hosts and offered partial protection against viral load in the lung. Since K18-hACE2 animals are much more susceptible than BALB/c animals to SARS-CoV-2, due in part to early viral dissemination to the brain, i.n. Bi:ChAd vaccination was less protective against clinical illness but still significantly reduced viral burden both in the lung and brain. Furthermore, when Bi:ChAd and S1-expressing Mono:ChAd vaccines were compared in our K18-hACE2/B.1.351 model, i.n. Bi:ChAd vaccine better protected against lung pathology than Mono:ChAd vaccine, whereas both vaccines reduced viral loads in the lungs. These findings together support the relevance of broadening the breadth of T cell immunity in COVID-19 vaccine design.
Delivery of Ad-vectored vaccines via the respiratory mucosal route to humans helps to bypass pre-existing anti-vector immunity which is more prevalent in the circulation than in the lung. This is particularly relevant to the use of human Ad5 and Ad26 vectors (Jeyanathan et al., 2020). Indeed, we have seen little presence of pre-existing anti-Ad5 antibodies in human airways, contrast to the peripheral blood (Jeyanathan et al., 2022). Nonetheless, ChAd-vectored vaccines have an advantage over Ad5 and Ad26 vectors in that humans have little pre-existing immunity against ChAd viruses. We previously found that not only intranasal ChAd-vectored vaccine was not impacted by anti-Ad5 immunity in murine lungs but it also triggered T cell responses to additional antigenic epitopes (Jeyanathan et al., 2015). Besides these potential advantages, our current study provides strong evidence that ChAd-vectored trivalent COVID-19 vaccine is also much more potent than its HuAd-vectored counterpart. Furthermore, since intramuscular injection of ChAdOx1nCov-19 (Astra Zeneca/Oxford) and Ad26.COV2-S (J&J) has been associated with rare cases of vaccine-induced thrombotic thrombocytopenia (VITT) which is likely related to the activation of platelets and endothelium by accidental intravenous introduction of adenovirus (Nicolai et al., 2021; Tsilingiris et al., 2021), respiratory mucosal delivery of Ad-vectored vaccines may avert this adverse outcome.
In summary, we have developed a next-generation COVID-19 vaccine strategy which is unique in both vaccine design and route of delivery. We show the superiority of a single-dose intranasal immunization with trivalent ChAd-vectored vaccine in inducing the tripartite respiratory mucosal immunity against both ancestral and variant strains of SARS-CoV-2.
Limitations of the study
We show respiratory mucosal Ad-vectored immunization to induce the tripartite mucosal immunity which includes TII. Although our evidence supports the protective role of TII against COVID-19, its potency may be underestimated given the limitations of murine models. Unlike infected humans, mice infected with relatively large viral inoculums do not have a pre-symptomatic period where TII may play a critical role (Jeyanathan et al., 2020). Likewise, while we show strong evidence that intranasal ChAd-vectored vaccine is more potent than HuAd-vectored counterpart, whether the same holds true in humans remains to be investigated. We have recently begun a phase 1 clinical trial (clinicaltrials.gov: NCT05094609) to compare these two COVID-19 vaccines following inhaled aerosol delivery to mRNA-vaccinated humans.
Although our experimental evidence supports the superiority of intranasal immunization over the intramuscular route in inducing respiratory mucosal immunity, murine lungs differ from human lungs in that the latter are not “naive” and are therefore potentially amenable to the translocation of circulating antibodies following intramuscular COVID-19 immunization. This may explain vaccine-mediated protection in humans at least within the first 6–8 months. In addition, our data showing the immunodominance of S1 over N and RdRp antigens should be interpreted with caution. This is particularly relevant to T cell responses. It is known that the immunodominance of T cell epitopes in mice is dictated by the mouse-strain-dependent homogeneous MHC haplotype, a scenario different from genetically heterogeneous human populations. Thus, we expect a stronger and more diverse T cell response to N and RdRp antigens in humans following immunization with our trivalent vaccines. Likewise, although we have observed a predominantly CD8+ T cell response to our COVID-19 vaccine in murine models, a better balanced CD4+ and CD8+ T cell response is expected in humans. Indeed, while we observed a predominantly CD8+ T cell response to our HuAd-vectored TB vaccine in mice, a robust CD4+ T cell response was seen in human lungs following inhaled aerosol delivery of the same vaccine (Jeyanathan et al., 2022).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified Rat Anti-Mouse CD16/CD32 (clone 2.4G2) | BD BioSciences | Cat# 553141, RRID:AB_394656 |
| Anti-mouse CD45 APC-Cy7 (clone 30-F11) | BD Biosciences | Cat# 557659, RRID:AB_396774 |
| Anti-mouse CD11b PE-Cy7 (clone M1/70) | BD Biosciences | Cat# 552850, RRID:AB_394491 |
| Anti-mouse CD11c APC (clone HL3) | BD Biosciences | Cat# 550261, RRID:AB_398460 |
| Anti-mouse MHC II AF700 (clone M5/114.15.2) | Thermo Fisher Scientific | Cat# 56-5321-82, RRID:AB_494009 |
| Anti-mouse CD3 V450 (clone 17A2) | BD Biosciences | Cat# 561389, RRID:AB_10679120 |
| Anti-mouse CD45R (B220)-V450 (clone RA3-6B2) | BioLegend | Cat# 103204 RRID:AB_312989 |
| Anti-mouse Ly6C Biotin (clone HK1.4) | BioLegend | Cat# 128004 RRID:AB_1236553 |
| Streptavidin Qdot800 | Thermo Fisher Scientific | Cat# Q10171MP |
| Anti-mouse CD24 BV650 (clone M1/69) | BD Biosciences | Cat# 563545 RRID:AB_2738271 |
| Anti-mouse CD64 PE (clone X54-5/7.1) | BioLegend | Cat# 139304, RRID:AB_10612740 |
| Anti-mouse Ly6G BV605 (clone 1A8) | BD Biosciences | Cat# 563005 RRID:AB_2737946 |
| Anti-mouse Siglec-F PE-CF594 (clone E50-2440) | BD Biosciences | Cat# 562757, RRID:AB_2687994 |
| Anti-mouse CD4 APC-Cy7 (clone GK1.5) | BD Biosciences | Cat# 552051 RRID:AB_394331 |
| Anti-mouse CD8 PE-Cy7 (clone 53-6.7) | BD Biosciences | Cat# 552877 RRID:AB_394506 |
| Anti-mouse IFNy APC (clone XMG1.2) | Thermo Fisher Scientific | Cat# 17-7311-82 RRID:AB_469504 |
| Anti-mouse TNFa FITC (clone MP6-XT22) | BD Biosciences | Cat# 554418 RRID:AB_395379 |
| Anti-mouse IL2 BV605 (clone JES6-5H4) | BioLegend | Cat#503829 RRID:AB_11204084 |
| Anti-mouse Granzyme B PE (clone NGZB) | Thermo Fisher Scientific | Cat# 12-8898-82 RRID:AB_10870787 |
| Anti-mouse CD44 PE (clone IM7) | BD BioSciences | Cat# 553134 RRID:AB_394649 |
| Anti-mouse CD69 BV605 (clone H1.2F3) | BD BioSciences | Cat# 563290 RRID:AB_2738120 |
| Anti-mouse CD103-Biotin (clone M290) | BD BioSciences | Cat# 557493 RRID:AB_396730 |
| Anti-mouse CD11a FITC (clone 2D7) | BD BioSciences | Cat# 553120 RRID:AB_10892820 |
| Anti-mouse IgD BV711 (clone 11-26c.2a) | BioLegend | Cat# 405731 RRID:AB_2563342 |
| Anti-IgG1-BV421 (clone RMG1-1) | BioLegend | Cat# 406616 RRID:AB_2562234 |
| Goat anti-mouse IgG, human ads-BIOT | SouthernBiotech | Cat# 1030-08 RRID:AB_2794296 |
| Goat anti-mouse IgG1-BIOT | SouthernBiotech | Cat# 1071-08 RRID:AB_2794427 |
| Goat anti-mouse IgG2a-BIOT | SouthernBiotech | Cat# 1081-08 RRID:AB_2794495 |
| Streptavidin-Alkaline phosphatase | SouthernBiotech | Cat# 7105-04 |
| Purified anti-mouse CD4 (clone GK1.5) | Dr. Zhou Xing McMaster University, Hamilton, Canada |
(Yao et al., 2018) |
| Purified anti-mouse CD8 (clone 2.43) | Dr. Zhou Xing McMaster University, Hamilton, Canada |
(Yao et al., 2018) |
| IRDye® 800CW donkey anti-rabbit IgG secondary antibody | LI-COR Biosciences | Cat# 926-33213 RRID:AB_621848 |
| IRDye® 680RD donkey anti-mouse IgG secondary antibody | LI-COR Biosciences | Cat# 926_68072 RRID:AB_10953628 |
| VSV-G rabbit polyclonal antibody | Invitrogen | Cat# PA1-30235 RRID:AB_1961361 |
| GAPDH monoclonal antibody | Sigma | Cat# G8795 |
| Bacterial and Virus Strains | ||
| Tri:ChAd | This paper | N/A |
| ChAd:EV | This paper | N/A |
| Tri:HuAd | This paper | N/A |
| HuAd:EV | This paper | N/A |
| SARS-CoV-2 MA10 | Dr. Ralph Baric | (Leist et al., 2020) |
| SARS-CoV-2 strain SB3 | Dr.Banerjee Dr.Mossman Dr.Mubareka |
(Banerjee et al., 2020) |
| SARS-CoV-2 strain hCoV-19/England/204820464/2020 (B.1.1.7) | BEI Resources | NR-54000 |
| SARS-CoV-2 strain hCoV-19/South Africa/KRISP-K005325/2020 (B.1.351) | BEI Resources | NR-54009 |
| Biological samples | ||
| FBS | ThermoFisher Scientific | Cat# 16140071 |
| Penicillin-Streptomycin | ThermoFisher Scientific | Cat# 15140122 |
| L-Glutamine | ThermoFisher Scientific | Cat# A2916801 |
| BSA | Sigma-Aldrich | Cat# 10735086001 |
| RPMI 1640 | ThermoFisher Scientific | Cat# 11875093 |
| DMEM | ThermoFisher Scientific | Cat# 11965118 |
| MEM Non-Essential Amino Acids Solution | ThermoFisher Scientific | Cat# 11140050 |
| Sodium pyruvate | ThermoFisher Scientific | Cat# 11360070 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase type I | ThermoFisher Scientific | Cat# 17100-017 |
| ACK lysing buffer | ThermoFisher Scientific | Cat# A10492-01 |
| BSA | Sigma-Aldrich | Cat# 10735086001 |
| GolgiPlug | BD Biosciences | Cat# BDB555029 |
| Aqua Staining Kit | ThermoFisher Scientific | Cat# L34957 |
| Cytofix/Cytoperm | BD Biosciences | Cat# 554714 |
| HEPES | ThermoFisher Scientific | Cat# 15630080 |
| Turks blood dilution | RICCA Chemicals | Cat# 8850-16 |
| S1 peptide pool | PEPSCAN | N/A |
| Nucleocapsid peptide pool | PEPSCAN | N/A |
| RdRp peptide pool | PEPSCAN | N/A |
| Ancestral RBD | Dr. Florian Krammer Icahn School of Medicine, New York, United States |
N/A |
| Ancestral SARS-CoV-2 Spike | Dr. Florian Krammer Icahn School of Medicine, New York, United States |
N/A |
| B.1.351 SARS-CoV-2 Spike | Dr. Matthew S. Miller McMaster University, Hamilton, Canada |
N/A |
| RIPA | VWR | Cat# 76236-534 |
| Protease Inhibitor Cocktail | ThermoFisher Scientific | Cat# 78492 |
| Cell TiterGlo2.0 | Promega | Cat#G9241 |
| PnPP One component substrate | Southern Biotech | Cat#OB042101 |
| Critical Commercial Assays | ||
| Aqua dead cell staining kit | ThermoFisher Scientific | Cat# L34957 |
| BCA protein quantification kit | ThermoFisher Scientific | Cat# 23225 |
| SARS-CoV-2 Surrogate Virus Neutralization Test (sVNT) kit | GenScript | L00847 |
| ExpiFectamine 293 transfection kit | Invitrogen | Cat# A14524 |
| EZ-Link Sulfo-NHS-LC-Biotin No Weigh Format | ThermoFisher Scientific | Cat# A39257 |
| Streptavidin (SA)-PE | Agilent | Cat# PJRS25 |
| Alexa Fluor 647 protein labeling kit | ThermoFisher Scientific | Cat# A20173 |
| Amicon Ultra spin columns | Millipore | Cat# Z648043 |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | Cat# CRL-1573 |
| Expi293F cells | Invitrogen | Cat# A14527 |
| Vero E6 | ATCC | Cat# CRL-1586 |
| A549 | ATCC | Cat# CCL-185 |
| Experimental Models: Organisms/Strains | ||
| BALB/cJ | Charles River | Cat# CRL:547RRID:IMSR_CRL:547 |
| C57BL/6J | Charles River | Cat# CRL:027RRID:IMSR_CRL:027 |
| C.Cg-Igh-Jtm1Dhu | Taconic Biosciences | Cat# 1147-F |
| B6.Cg-Tg(K18-ACE2)2Prlmn/J | The Jackson Laboratory | Cat# JAX:034860RRID: ISMR_JAX:034860 |
| Software and Algorithms | ||
| GraphPad Prism, version 9 | https://www.graphpad.com/scientific-software/prism/ | SCR_002798 |
| FlowJo, version 10 | https://www.flowjo.com | SCR_008520 |
| SnapGene v5.3 | https://www.snapgene.com/ | SCR_015052 |
| BioTek Synergy Gen5 software | https://www.biotek.com/products/software-robotics-software/gen5-microplate-reader-and-imager-software/ | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Zhou Xing (xingz@mcmaster.ca).
Materials availability
All requests for resources and reagents should be directed to and will be fulfilled by the lead contact or correspondence authors. All reagents including antibodies, viruses and vaccines may be made available on request after completion of a Materials Transfer Agreement.
Experimental model and subject details
Mice
Age-matched 6–8-wk-old wild-type female or male BALB/c, C57BL/6J, or B6.Cg-Tg (K18-ACE2) 2Prlmn/J mice were purchased from either Charles River Laboratories (Saint Constant, QC, Canada) or The Jackson Laboratory (Bar Harbor, ME, United States). B cell-deficient mice C.Cg-Igh-J tm1Dhu of BALB/c background were purchased from Taconic Biosciences (Germantown, NY, United States). Animals were housed in either a specific pathogen-free level B or a Containment Level 3 Facility at McMaster University, Hamilton, ON, Canada. All experiments were performed in accordance with institutional guidelines from the Animal Research and Ethics Board.
Method details
Vaccine construction
The transgene cassette was constructed through a series of overlapping polymerase chain reactions (PCR) wherein transgene expression is under control of the murine CMV (mCMV) promoter and protein translation is initiated with the human tissue plasminogen (tPA) signal sequence (Figure 1A). The first overlapping PCR product contained the tPA signal sequence upstream of the S1 sequence of the Wuhan-Hu-1 Isolate of SARS-CoV-2 (GenBank: MN908947.3) fused to the vesicular stomatitis virus G protein transmembrane (VSVG TM) domain to facilitate trimerization and exosome targeting. This PCR product was cloned in pCY1 plasmid which contains the mCMV promoter. The second overlapping PCR product contained the porcine teschovirus-1 2A (P2A) skip sequence upstream of the full-length nucleocapsid sequence from the same SARS-CoV-2 isolate fused to a highly conserved region of nsp12 (RNA-dependent RNA polymerase (RdRp)). The sequence of RdRp was chosen based on conserved sequence homology to bat coronaviruses and further refined to include several predicted high affinity human CD8 T cell epitopes on HLA 0101, 0201, and 0301. The second overlapping PCR product was cloned downstream of the VSVG TM domain in pCY1 to generate the complete expression cassette. The same transgene cassette was cloned in the shuttle plasmids used during co-transfection to rescue the trivalent, replication-defective human serotype 5 adenoviral-vectored (Tri:HuAd) and chimpanzee serotype 68 adenoviral-vectored (Tri:ChAd) COVID-19 vaccines.
Tri:HuAd was packaged and rescued in HEK293 cells through a two-plasmid co-transfection system as previously described (Wang et al., 2004). Tri:ChAd was also constructed and rescued in HEK293 cells via direct subcloning or similarly through a two-plasmid co-transfection system. Briefly, the transgene cassette was PCR amplified to incorporate restriction enzyme sites and cloned in a shuttle vector containing a unique FspI cut site. The shuttle vector was then linearized with FspI and used for co-transfection with an SrfI-linearized plasmid containing the E1/E3-deficient ChAd68 genomic backbone. Both trivalent vaccines were further amplified in HEK293 cells and subsequently purified by cesium chloride density gradient ultracentrifugation.
Both Mono- and Bi-valent ChAd vaccines (Mono:ChAd & Bi:ChAd) were constructed, purified and characterized as described above except that they were bio-engineered to express only the S1/VSVG TM (Mono:ChAd) or N/RdRp (Bi:ChAd).
Cell lines and SARS-CoV-2 viruses
Vero E6 (CRL-1586, American Type Culture Collection (ATCC), Manassas, VA, United States) were cultured at 37°C in Dulbecco’s Modified Eagle medium (DMEM) supplemented with 10 % fetal bovine serum (FBS), 1 % HEPES pH7.3, 1 mM sodium pyruvate, 1% L-Glutamine and 100 U/mL of penicillin–streptomycin. SARS-CoV-2 strain SB3-TYAGNC was provided by Dr. Arinjay Banerjee, Dr. Karen Mossman, Dr. Samira Mubareka, and Dr. Rob Kozak and isolated as described previously (Banerjee et al., 2020). SARS-CoV-2 strain MA10 was generously provided by Dr. Ralph Baric (Leist et al., 2020). SARS-CoV-2 strain hCoV-19/England/204820464/2020 (B.1.1.7 variant, NR-54000, Public Health England) and strain hCoV-19/South Africa/KRISP-K005325/2020 (B.1.351 variant, NR-54009, African Health Research Institute) were both obtained from BEI Resources (Manassas, VA, United States).
Immunization and infection
Animals were anesthetized with isoflurane and vaccinated intramuscularly or intranasally with 5x107 PFU of a recombinant human adenovirus (Ad) serotype 5 SARS-CoV-2 vaccine (Tri:HuAd) or 1x107 PFU of a recombinant chimpanzee adenovirus serotype 68 SARS-CoV-2 vaccine (Tri:ChAd). In a subset of experiments where mono-, bi-, and trivalent ChAd-vectored vaccines were compared for protective efficacy in K18-hACE2 mouse model, an intranasal dose of 1x106 PFU was used. Wherever indicated, an empty Ad vector (the same adenoviral backbone lacking the vaccine transgene) was included as a control. Intranasal vaccinations were performed with a final volume of 25μL diluted in PBS. Intramuscular vaccinations were performed with a final volume of 100μL administered in equal volumes into the quadricep muscle of each hind leg. Infections were carried out with 1x105 PFU SARS-CoV-2 administered intranasally in a final volume of 40μL diluted in PBS. Mice were monitored for clinical signs and weight loss daily, with 80% of initial weight considered humane endpoint, in accordance with institutional guidelines.
In vivo T cell depletion
T cell depletion was carried out utilizing previously published and validated protocols (Yao et al., 2018). 200μg of anti-CD4 (clone GK1.5) and anti-CD8 (clone 2.43) depleting, or an IgG isotype control antibodies (MilliporeSigma, Etobicoke, ON, Canada) were intraperitoneally administered as a single bolus either 3- or 25-days post-vaccination. A second 100μg dose was administered 2-days following the first dose, and repeated every 7-days to maintain depletion, as per experimental requirements.
Bronchoalveolar lavage, lung, and spleen mononuclear cell isolation
Mice were euthanized by exsanguination. Cells from bronchoalveolar lavage (BAL), lung tissue, and spleen, were isolated as previously described (D’Agostino et al., 2020; Jeyanathan et al., 2015; Yao et al., 2018). Briefly, BAL was performed by instillation with 250 μL, followed by 200 μL of PBS. This fraction was utilized for downstream soluble factor analysis. Further instillation of 3x 300 μL of PBS was performed for BAL cell retrieval. Lungs were minced into small pieces and digested with collagenase type 1 (ThermoFisher Scientific Waltham, MA, United States) at 37°C in an agitating incubator. A single cell suspension was obtained by crushing the digested tissue through 100 μm basket filter (BD Biosciences, San Jose, CA, United States), with red blood cells being lysed with Ammonium-Chloride-Potassium (ACK) buffer for 2 minutes. Spleens were ground between frosted glass microscope slides prior to passing through a 100 μm basket filter. Isolated cells were resuspended in complete RPMI 1640 (10 % FBS, 1% L-glutamine, 100 U/mL penicillin/streptomycin, 1% HEPES pH 7.3, 1% MEM non-essential amino acids (Gibco, Gaithersburg, MD, United States), 1% sodium-pyruvate (Gibco, Gaithersburg, MD, United States). Cell numbers were quantified in Turk’s Blood Dilution Fluid (RICCA Chemical, Arlington, TX, United States) and counted under a microscope. Where required, cells were counted automatically by a Sceptre 3.0 Cell Counter and Software Pro (Millipore Sigma, Etobicoke, ON, Canada).
Transgene protein analysis by Western blot
A549 cells (CCL-185, ATCC, Manassas, VA, United States) were cultured at 37°C in DMEM supplemented with 10% FBS, 1 % HEPES pH 7.3, 1% L-glutamine and 100 U/mL of penicillin–streptomycin. Cells were seeded in 24-well plates (7.5x104 cells/well) 24 hours prior to infection. Infections were carried out with Tri:HuAd (MOI 100) and Tri:ChAd (MOI 50) diluted in PBS (with Mg2+ and Ca2+). 18 hours post-infection, cells were lysed using RIPA buffer (VWR, Mississauga, ON, Canada) containing a protease inhibitor cocktail (ThermoFisher Scientific Waltham, MA, United States). Lysates were quantified using a BCA kit (ThermoFisher Scientific Waltham, MA, United States) and 60 μg of each lysate was boiled at 98°C with 1X sample buffer (6.35% v/v 1M Tris, pH 6.8, 46.5% v/v 10X SDS, 20% v/v glycerol, and 5% v/v β-mercaptoethanol; MilliporeSigma, Etobicoke, ON, Canada) for 10 minutes. The samples were run on a 4-12% SDS-PAGE gel (ThermoFisher Scientific Waltham, MA, United States) for 1.5 hours at 100 V and transferred to nitrocellulose membrane (VWR, Mississauga, ON, Canada) using wet-transfer at 125 mA for 1.5 hours. The membrane was incubated with primary antibodies (1:1000 Anti-VSV-G (ThermoFisher Scientific Waltham, MA, United States), 1:2000 Anti-NP (ThermoFisher Scientific Waltham, MA, United States) and 1:5000 GAPDH (MilliporeSigma, Etobicoke, ON, Canada) diluted in 5% skim milk, followed by anti-mouse and anti-rabbit IRDye secondary antibodies (LI-COR, Lincoln, NE, United States) diluted in 5 % skim milk. The membrane was developed using an Odyssey CLx (LI-COR, Lincoln, NE, United States).
Recombinant antigen production
Plasmids encoding mammalian cell codon optimized sequences for the receptor binding domain (RBD) and full-length spike of SARS-CoV-2 was generously gifted from the lab of Dr. Florian Krammer (Amanat et al., 2020)(Icahn School of Medicine, NY, United States). Proteins were produced in Expi293F cells (ThermoFisher Scientific Waltham, MA, United States) according to the manufacturers’ instructions and purified as previously described (Stadlbauer et al., 2021). Briefly, when culture viability reached 40%, supernatants were collected and spun at 500 x g for 5 minutes. The supernatant was then incubated by shaking overnight at 4°C with 1 mL of Ni-NTA agarose (Qiagen, Germantown MD, United States) per 25 mL of transfected cell supernatant. The following day 10 ml polypropylene gravity flow columns (Qiagen, Germantown, MD, United States) were used to elute the protein. Recombinant RBD was concentrated in a 10 kDa Amicon centrifugal units (Millipore Sigma, Etobicoke, ON, Canada), and recombinant Spike was concentrated in a 50kDa Amicon centrifugal unit (Millipore Sigma, Etobicoke, ON, Canada) prior to being resuspended in phosphate buffered saline (PBS).
RBD tetramer construction
The recombinant RBD B cell tetramer was produced through biotinylation and tetramerization as previously described (Taylor et al., 2012). A decoy tetramer was created as previously described (Taylor et al., 2012), to gate out non-RBD binding B cells. The decoy tetramer was constructed through conjugating streptavidin-PE to Alexa Fluor 647 (Thermo Fisher Scientific) for 1 hour at room temperature. Excess Alexa Fluor 647 was removed through washing and centrifugation with 100 kDa Amicon spin columns (MilliporeSigma, Etobicoke, ON, Canada). The solution was then incubated with an irrelevant biotinylated protein at sixfold molar excess for 30 minutes at room temperature. The concentration of the resulting decoy tetramer was calculated by the absorbance of PE at 565 nM and diluted to 1 μM.
Enzyme Linked Immunosorbent Assay (ELISAs) for antibody measurement
96-well NUNC- MaxiSorp™ plates (Thermo Scientific, Waltham, MA, United States) were coated overnight at 4°C with SARS-CoV-2 RBD, or full-length spike, diluted to 2 μg/mL in bicarbonate-carbonate coating buffer (pH 9.4). Plates were blocked by shaking for 1 hour at 37°C with reagent diluent (0.5% bovine serum albumin (BSA), 0.02 % sodium azide, in 1X Tris-Tween buffer). Samples were serially diluted from 1:10 (serum), or 1:4 (BAL) starting dilution. BAL samples were first concentrated through Pierce™ Protein Concentrators with a 50 kDa molecular weight cut-off (MWCO) (ThermoFisher Scientific Waltham, MA, United States) according to the manufacturer’s instructions, with volumes normalized prior to concentration. Samples were arranged such that one row contained only antigen and secondary antibodies and served as the plate blank. Following a 1 hour incubation with shaking at 37°C, plates were washed three times with 1X Tris-Tween wash buffer. After washing, goat anti-mouse-biotin antibodies (Southern Biotech, Birmingham, AL, United States) IgA (1:2000), IgG (1:5000), IgG1 (1:5000), IgG2a (1:5000)) were diluted in reagent diluent and added to all wells. Plates were again incubated for 1 hour, with shaking, at 37 °C, followed by three washes with 1X Tris-Tween buffer. A streptavidin-alkaline phosphatase secondary antibody (1:2000, Southern Biotech, Birmingham, AL, United States) was added to all wells for 1 hr with shaking at 37°C. Plates were subsequently washed three times prior to addition of pNPP one component microwell substrate solution (Southern Biotech, Birmingham, AL, United States) to each well. Plates were developed for 10 minutes and the reaction was quenched with an equal volume 3N sodium hydroxide. The optical density (O.D.) at 405 nm was read on a SpectramaxI3 (Molecular Devices, San Jose, CA, United States). Endpoint titers were defined by the lowest dilution at which the O.D. was three standard deviations above the mean of the blank wells.
SARS-CoV-2 neutralization assays
Microneutralization assays were performed as described previously (Huynh et al., 2021). Briefly, Vero E6 cells (CRL-1586, ATCC, Manassas, VA, United States) were seeded at a density of 2.5x104 cells per well in opaque 96-well flat-bottom plates ((Millipore Sigma, Etobicoke, ON, Canada) in complete DMEM (supplemented with 10% FBS, 1% L-glutamine, 100 U/mL penicillin-streptomycin). 24 hours later, serum was inactivated by incubating at 56°C for 30 minutes, then diluted 1:10 in low serum DMEM (supplemented with 2% FBS, 1% L-glutamine, 100 U/mL penicillin-streptomycin), followed by a 1:2 dilution series in 96 well U-bottom plates resulting in a final volume of 55 μL diluted serum per well. An equal volume of SARS-CoV-2 consisting of 330 PFU per well was then added to the diluted serum. The serum-virus mixture was then incubated at 37°C for 1 hour. The Vero E6 culture media was then replaced with 100 μL of the serum-virus mixture and was incubated at 37°C for 72 hours. The plates were read by removing 50 μL of culture supernatant and adding 50μL of CellTiter-Glo 2.0 Reagent (Promega, Madison, WI, United States) to each well. The plates were then shaken at 282 cpm at 3 mm diameter for 2 minutes, incubated for 5 minutes at room temperature, then luminescence was read using a BioTek Synergy H1 microplate reader with gain of 135 and integration time of 1 second.
In certain experiments, serum neutralizing antibodies were assessed utilizing a surrogate SARS-CoV-2 virus neutralization test (sVNT). sVNT assays were performed utilizing the cPass Neutralization Antibody Detection kit (GenScript, Piscataway, NJ, United States), according to manufacturer’s instructions.
Cytokine and serum chemistry
Evaluation of serum and BAL cytokines were performed by Eve Technologies (Alberta, Canada) through a mouse cytokine array/chemokine array 44-plex (MD44). Serum chemistry was performed by Antech Diagnostics (Ontario, Canada) through a chemistry panel (BioChem 5 panel).
Flow cytometry
Cell immunostaining and flow cytometry were performed as previously described (D’Agostino et al., 2020; Jeyanathan et al., 2015; Yao et al., 2018). Briefly, tissue isolated mononuclear cells were plated in U-bottom, 96-well plates at a concentration of 2x107 cells/mL in PBS. Following staining with The LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (ThermoFisher Scientific Waltham, MA, United States) at room temperature for 30 min, cells were washed and blocked with anti-CD16/CD32 (clone 2.4G2) in 0.5 % BSA-PBS for 15 min on ice and then stained with fluorochrome-labeled mAbs for 30 min on ice. Fluorochrome-labeled mAbs used for staining cells were anti-CD45–APC-Cy7 (clone 30-F11), anti-CD11b–PE-Cy7 (clone M1/70), anti-CD11c–APC (clone HL3), anti–MHC class II (MHC II)–Alexa Fluor 700 (clone M5/114.15.2; eBioscience, ThermoFisher Scientific Waltham, MA, United States), anti-CD3-V450 (clone 17A2), anti-CD45R (B220)-V450 (clone RA3- 6B2), anti–Ly-6C–Biotin (clone HK1.4; BioLegend, San Diego, CA, United States), Streptavidin Qdot 800 (ThermoFisher Scientific Waltham, MA, United States), anti-CD24–BV650 (clone M1/69), anti-CD64–PE (clone 354-5/7.1; BioLegend, San Diego, CA, United States), anti–Ly-6G–BV605 (clone 1A8), anti–Siglec-F–PE-CF594 (clone E50-2440), anti-CD4 APC-Cy7 (clone GK1.5), anti-CD8 PE-Cy7 (clone 53-6.7), anti-IFNγ APC (clone XMG1.2), anti-TNFα FITC (clone MP6-XT22), anti-IL2 BV605 (clone JES6-5H4), anti-Granzyme B PE (clone NGZB; ThermoFisher Scientific Waltham, MA, United States), anti-CD44 PE (clone IM7), anti-CD69 BV605 (clone H1.2F3), anti-CD103-Biotin (clone M290), anti-CD11a FITC (clone 2D7), anti-IgD BV711 (clone 11-26c.2a; BioLegend San Diego, CA, United States), and anti-IgG1-BV421 (clone RMG1-1; BioLegend San Diego, CA, United States). Stained cells were fixed and permeabilized with BD Cytofix/Cytoperm before incubation in BD Perm/Wash buffer (BD Biosciences, San Jose, CA, United States). All mAbs and reagents were purchased from BD Biosciences unless otherwise indicated. Stained cells were processed according to BD Biosciences instructions for flow cytometry and run on a BD LSR II flow cytometer. Data were analyzed using FlowJo software (version 10.1; Tree Star, Ashland, OR, United States).
SARS-CoV-2 viral burden determination in tissues
Lung and brains were homogenized using a Bead Mill 24 homogenizer (ThermoFisher Scientific Waltham, MA, United States). Homogenates were clarified by centrifugation at 300 x g and frozen at -80°C. Homogenates were then thawed, and serially diluted 1:10 in serum-free DMEM supplemented with 1% HEPES pH 7.3, 1 mM sodium pyruvate, 1% L-Glutamine and 100 U/mL of penicillin–streptomycin. 100 μL of viral inoculum was plated on Vero E6 cells in 96-well plates (4 x 104 cells per well) for 1hr at 37°C. 5% CO2, at which point the inoculum was replaced with low-serum DMEM supplemented with 2% fetal bovine serum (FBS), 1% HEPES pH 7.3, 1 mM sodium pyruvate, 1% L-Glutamine and 100 U/mL of penicillin–streptomycin. Wells were assessed for cytopathic effect at 5-days post-infection using an EVOS M5000 microscope (ThermoFisher Scientific Waltham, MA, United States).
Peptide library construction and stimulation
Peptide libraries consisting of 10 amino acid, 15mer synthetic overlapping peptides for vaccine encoded antigens S1, nucleocapsid, and RdRp were synthesized by Pepscan (Lelystad, The Netherlands). Peptides were reconstituted in DMSO according to manufacturer’s instructions to a final concentration of 40 μg/μL. Antigen peptide pools were generated with each pool containing 0.2 μg/μL of each peptide. Unless otherwise stated, peptide stimulations were carried for each vaccine antigen individually with their respective peptide pools, utilizing 2 μg of each peptide/mL of culture media.
Quantification and statistical analysis
Asterisks in the figures indicate the level of statistical significance (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001) as determined using either Mann-Whitney test, two-tailed unpaired Student t test, Kruskal-Wallis test with Dunn’s multiple comparisons test, or one-way ANOVA with a Tukey multiple comparisons test, as defined in figure captions. Tests were performed using GraphPad Prism software (Version 9, Graphpad Software, La Jolla, CA, United States). Data are expressed as mean ± SEM unless otherwise stated.
Acknowledgments
The work was supported by the Canadian Institutes of Health Research (CIHR) COVID-19 Rapid Research Project (Z.X. et al.), CIHR Foundation Program (Z.X.), the Innovative Research Program of National Sanitarium Association of Canada (Z.X.), CIHR New Investigator Award and Ontario Early Research Award, and Canada Research Chair (Tier 2) in Viral Pandemics (M.S.M.), CIHR Canada Graduate Scholarship Doctoral Award and Physicians’ Services Incorporated Research Trainee Fellowship (A.Z.), Ontario Graduate Scholarship (M.R.D.), Ontario Graduate Scholarship and Canadian Society for Virology United Supermarket Studentship (H.D.S.), and Canadian Foundation for Innovation. Authors are thankful to Turnstone Biologics Inc. for provision of ChAd68 vector, to Dr. Ralph Baric for provision of mouse-adapted SARS-CoV-2, and to Dr. Carolina Ilkow for provision of subcloning materials.
Author contributions
S.A., M.R.D., Y.W., K.L.M., M.J., F.S., B.D.L., M.S.M., and Z.X. conceived and designed the study. S.A., M.R.D., A. Zhang, H.D.S., A.M., A.K., R.S., J.B., G.Y., X.L., F.W., J.C.A., A. Zganiacz, S.F.G., S.T., and A.G. performed experiments. J.F.E.K., A.P., and M.J. constructed RBD tetramers. S.A., M.R.D., A.Z., H.D.S., A.M., F.W., and J.C.A. analyzed data. U.S., N.K., M.F.C.M., S.A., and B.D.L. constructed and purified vaccines. S.A., M.R.D., M.S.M., and Z.X. wrote the paper.
Declaration of interests
B.D.L., F.S., and Z.X. are inventors on a US provisional patent application no. 63/222723, entitled “Novel COVID vaccine and method for delivery.” All other authors declare no competing interests.
Published: March 3, 2022
Data and code availability
All data supporting the findings of this study are available within the paper and are available from the lead contact or correspondence authors upon request. This paper does not report original code.
References
- Abu-Raddad L.J., Chemaitelly H., Butt A.A. Effectiveness of the BNT162b2 COVID-19 vaccine against the B.1.1.7 and B.1.351 variants. N. Engl. J. Med. 2021;385:187–189. doi: 10.1056/NEJMc2104974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter G., Yu J., Liu J., Chandrashekar A., Borducchi E.N., Tostanoski L.H., McMahan K., Jacob-Dolan C., Martinez D.R., Chang A., et al. Immunogenicity of Ad26.COV2.S vaccine against SARS-CoV-2 variants in humans. Nature. 2021;596:268–272. doi: 10.1038/s41586-021-03681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann D.M., Boyton R.J. SARS-CoV-2 T cell immunity: specificity, function, durability, and role in protection. Sci. Immunol. 2020;5:eabd6160. doi: 10.1126/sciimmunol.abd6160. [DOI] [PubMed] [Google Scholar]
- Amanat F., Stadlbauer D., Strohmeier S., Nguyen T.H.O., Chromikova V., McMahon M., Jiang K., Arunkumar G.A., Jurczyszak D., Polanco J., et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020;26:1033–1036. doi: 10.1038/s41591-020-0913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreano E., Rappuoli R. SARS-CoV-2 escaped natural immunity, raising questions about vaccines and therapies. Nat. Med. 2021;27:759–761. doi: 10.1038/s41591-021-01347-0. [DOI] [PubMed] [Google Scholar]
- Aschwanden C. Five reasons why COVID herd immunity is probably impossible. Nature. 2021;591:520–522. doi: 10.1038/d41586-021-00728-2. [DOI] [PubMed] [Google Scholar]
- Banerjee A., Nasir J.A., Budylowski P., Yip L., Aftanas P., Christie N., Ghalami A., Baid K., Raphenya A.R., Hirota J.A., et al. Isolation, sequence, infectivity, and replication kinetics of severe acute respiratory syndrome coronavirus 2. Emerg. Infect. Dis. 2020;26:2054–2063. doi: 10.3201/eid2609.201495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyakov I.M., Ahlers J.D. What role does the route of immunization play in the generation of protective immunity against mucosal pathogens? J. Immunol. 2009;183:6883–6892. doi: 10.4049/jimmunol.0901466. [DOI] [PubMed] [Google Scholar]
- Bliss C.M., Parsons A.J., Nachbagauer R., Hamilton J.R., Cappuccini F., Ulaszewska M., Webber J.P., Clayton A., Hill A.V.S., Coughlan L. Targeting antigen to the surface of EVs improves the in vivo immunogenicity of human and non-human adenoviral vaccines in mice. Mol. Ther. Methods Clin. Dev. 2020;16:108–125. doi: 10.1016/j.omtm.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournazos S., Gupta A., Ravetch J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020;20:633–643. doi: 10.1038/s41577-020-00410-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker T.L., Darling T.L., Hassan A.O., Harastani H.H., Soung A., Jiang X., Dai Y.-N., Zhao H., Adams L.J., Holtzman M.J., et al. A single intranasal or intramuscular immunization with chimpanzee adenovirus vectored SARS-CoV-2 vaccine protects against pneumonia in hamsters. Cell Rep. 2021;36:109400. doi: 10.1016/j.celrep.2021.109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E., Ledford H. How to redesign COVID vaccines so they protect against variants. Nature. 2021;590:15–16. doi: 10.1038/d41586-021-00241-6. [DOI] [PubMed] [Google Scholar]
- Cao H., Mai J.M., Zhou Z., Li Z., Duan R., Watt J., Chen Z., Bandara R.A., Li M., Ahn S.K., et al. Intranasal HD-Ad vaccine protects the upper and lower respiratory tracts of hACE2 mice against SARS-CoV-2. Cell Biosci. 2021;11:202. doi: 10.1186/s13578-021-00723-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.E., Zhang X., Case J.B., Winkler E.S., Liu Y., VanBlargan L.A., Liu J., Errico J.M., Xie X., Suryadevara N., et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021;27:717–726. doi: 10.1038/s41591-021-01294-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Class J., Dangi T., Richner J.M., Penaloza-MacMaster P., Richner J. A SARS CoV-2 nucleocapsid vaccine protects against distal viral dissemination. Preprint at bioRxiv. 2021 2021.04.26.440920. [Google Scholar]
- D’Agostino M.R., Lai R., Afkhami S., Khera A., Yao Y., Vaseghi-Shanjani M., Zganiacz A., Jeyanathan M., Xing Z. Airway macrophages mediate mucosal vaccine-induced trained innate immunity against Mycobacterium tuberculosis in early stages of infection. J. Immunol. 2020;205:2750–2762. doi: 10.4049/jimmunol.2000532. [DOI] [PubMed] [Google Scholar]
- Dai L., Gao G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021;21:73–82. doi: 10.1038/s41577-020-00480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanet A., Cauchemez S. COVID-19 herd immunity: where are we? Nat. Rev. Immunol. 2020;20:583–584. doi: 10.1038/s41577-020-00451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Beltran W.F., Lam E.C., St. Denis K., Nitido A.D., Garcia Z.H., Hauser B.M., Feldman J., Pavlovic M.N., Gregory D.J., Poznansky M.C., et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell. 2021;184:2372–2383.e9. doi: 10.1016/j.cell.2021.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geers D., Shamier M.C., Bogers S., den Hartog G., Gommers L., Nieuwkoop N.N., Schmitz K.S., Rijsbergen L.C., van Osch J.A.T., Dijkhuizen E., et al. SARS-CoV-2 variants of concern partially escape humoral but not T-cell responses in COVID-19 convalescent donors and vaccinees. Sci. Immunol. 2021;6:eabj1750. doi: 10.1126/sciimmunol.abj1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg Y., Mandel M., Bar-On Y.M., Bodenheimer O., Freedman L., Haas E.J., Milo R., Alroy-Preis S., Ash N., Huppert A. Waning immunity after the BNT162b2 vaccine in Israel. N. Engl. J. Med. 2021;385:e85. doi: 10.1056/NEJMoa2114228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant R.A., Morales-Nebreda L., Markov N.S., Swaminathan S., Querrey M., Guzman E.R., Abbott D.A., Donnelly H.K., Donayre A., Goldberg I.A., et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature. 2021;590:635–641. doi: 10.1038/s41586-020-03148-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.K. Will SARS-CoV-2 variants of concern affect the promise of vaccines? Nat. Rev. Immunol. 2021;21:340–341. doi: 10.1038/s41577-021-00556-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnik R.L., Plante J.A., Liang Y., Alameh M.-G., Tang J., Zhong C., Adam A., Scharton D., Rafael G.H., Liu Y., et al. Combinatorial mRNA vaccination enhances protection against SARS-CoV-2 delta variant. Preprint at bioRxiv. 2021 2021.12.08.471664. [Google Scholar]
- Hartley G.E., Edwards E.S.J., Aui P.M., Varese N., Stojanovic S., McMahon J., Peleg A.Y., Boo I., Drummer H.E., Hogarth P.M., et al. Rapid generation of durable B cell memory to SARS-CoV-2 spike and nucleocapsid proteins in COVID-19 and convalescence. Sci. Immunol. 2020;5:eabf8891. doi: 10.1126/sciimmunol.abf8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., Harrison E.M., Ludden C., Reeve R., Rambaut A., et al. COVID-19 Genomics UK (COG-UK) Consortium SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021;19:409–424. doi: 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A.O., Feldmann F., Zhao H., Curiel D.T., Okumura A., Tang-Huau T.-L., Case J.B., Meade-White K., Callison J., Chen R.E., et al. A single intranasal dose of chimpanzee adenovirus-vectored vaccine protects against SARS-CoV-2 infection in rhesus macaques. Cell Rep. Med. 2021;2:100230. doi: 10.1016/j.xcrm.2021.100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A.O., Kafai N.M., Dmitriev I.P., Fox J.M., Smith B.K., Harvey I.B., Chen R.E., Winkler E.S., Wessel A.W., Case J.B., et al. A single-dose intranasal ChAd vaccine protects upper and lower respiratory tracts against SARS-CoV-2. Cell. 2020;183:169–184.e13. doi: 10.1016/j.cell.2020.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A.O., Shrihari S., Gorman M.J., Ying B., Yaun D., Raju S., Chen R.E., Dmitriev I.P., Kashentseva E., Adams L.J., et al. An intranasal vaccine durably protects against SARS-CoV-2 variants in mice. Cell Reports. 2021;36:109452. doi: 10.1016/j.celrep.2021.109452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann D., Corleis B., Rauch S., Roth N., Mühe J., Halwe N.J., Ulrich L., Fricke C., Schön J., Kraft A., et al. CVnCoV and CV2CoV protect human ACE2 transgenic mice from ancestral B BavPat1 and emerging B.1.351 SARS-CoV-2. Nat. Commun. 2021;12:4048. doi: 10.1038/s41467-021-24339-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Arora P., Groß R., Seidel A., Hörnich B.F., Hahn A.S., Krüger N., Graichen L., Hofmann-Winkler H., Kempf A., et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell. 2021;184:2384–2393.e12. doi: 10.1016/j.cell.2021.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A.T., Garcia-Carreras B., Hitchings M.D.T., Yang B., Katzelnick L.C., Rattigan S.M., Borgert B.A., Moreno C.A., Solomon B.D., Trimmer-Smith L., et al. A systematic review of antibody mediated immunity to coronaviruses: kinetics, correlates of protection, and association with severity. Nat. Commun. 2020;11:4704. doi: 10.1038/s41467-020-18450-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh A., Arnold D.M., Smith J.W., Moore J.C., Zhang A., Chagla Z., Harvey B.J., Stacey H.D., Ang J.C., Clare R., et al. Characteristics of anti-SARS-CoV-2 antibodies in recovered COVID-19 subjects. Viruses. 2021;13:697. doi: 10.3390/v13040697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyanathan M., Afkhami S., Khera A., Mandur T., Damjanovic D., Yao Y., Lai R., Haddadi S., Dvorkin-Gheva A., Jordana M., et al. CXCR3 signaling is required for restricted homing of parenteral tuberculosis vaccine-induced T cells to both the lung parenchyma and airway. J. Immunol. 2017;199:2555–2569. doi: 10.4049/jimmunol.1700382. [DOI] [PubMed] [Google Scholar]
- Jeyanathan M., Afkhami S., Smaill F., Miller M.S., Lichty B.D., Xing Z. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 2020;20:615–632. doi: 10.1038/s41577-020-00434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyanathan M., Fritz D.K., Afkhami S., Aguirre E., Howie K.J., Zganiacz A., Dvorkin-Gheva A., Thompson M.R., Silver R.F., Cusack R.P., et al. Aerosol delivery, but not intramuscular injection, of adenovirus-vectored tuberculosis vaccine induces respiratory-mucosal immunity in humans. JCI Insight. 2022;7:e155655. doi: 10.1172/jci.insight.155655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyanathan M., Thanthrige-Don N., Afkhami S., Lai R., Damjanovic D., Zganiacz A., Feng X., Yao X.-D., Rosenthal K.L., Medina M.-F., et al. Novel chimpanzee adenovirus-vectored respiratory mucosal tuberculosis vaccine: overcoming local anti-human adenovirus immunity for potent TB protection. Mucosal Immunol. 2015;8:1373–1387. doi: 10.1038/mi.2015.29. [DOI] [PubMed] [Google Scholar]
- Jeyanathan M., Yao Y., Afkhami S., Smaill F., Xing Z. New tuberculosis vaccine strategies: taking aim at un-natural immunity. Trends Immunol. 2018;39:419–433. doi: 10.1016/j.it.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Khoury D.S., Cromer D., Reynaldi A., Schlub T.E., Wheatley A.K., Juno J.A., Subbarao K., Kent S.J., Triccas J.A., Davenport M.P. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 2021;27:1205–1211. doi: 10.1038/s41591-021-01377-8. [DOI] [PubMed] [Google Scholar]
- Khoury D.S., Wheatley A.K., Ramuta M.D., Reynaldi A., Cromer D., Subbarao K., O’Connor D.H., Kent S.J., Davenport M.P. Measuring immunity to SARS-CoV-2 infection: comparing assays and animal models. Nat. Rev. Immunol. 2020;20:727–738. doi: 10.1038/s41577-020-00471-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer F. Correlates of protection from SARS-CoV-2 infection. Lancet. 2021;397:1421–1423. doi: 10.1016/S0140-6736(21)00782-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause P.R., Fleming T.R., Longini I.M., Peto R., Briand S., Heymann D.L., Beral V., Snape M.D., Rees H., Ropero A.-M., et al. SARS-CoV-2 variants and vaccines. N. Engl. J. Med. 2021;385:179–186. doi: 10.1056/NEJMsr2105280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku M.-W., Bourgine M., Authié P., Lopez J., Nemirov K., Moncoq F., Noirat A., Vesin B., Nevo F., Blanc C., et al. Intranasal vaccination with a lentiviral vector protects against SARS-CoV-2 in preclinical animal models. Cell Host Microbe. 2021;29:236–249.e6. doi: 10.1016/j.chom.2020.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuate S., Cinatl J., Doerr H.W., Überla K. Exosomal vaccines containing the S protein of the SARS coronavirus induce high levels of neutralizing antibodies. Virology. 2007;362:26–37. doi: 10.1016/j.virol.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai Y., Takeuchi O., Kato H., Kumar H., Matsui K., Morii E., Aozasa K., Kawai T., Akira S. Alveolar macrophages are the primary interferon-α producer in pulmonary infection with RNA viruses. Immunity. 2007;27:240–252. doi: 10.1016/j.immuni.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Lai R., Afkhami S., Haddadi S., Jeyanathan M., Xing Z. Mucosal immunity and novel tuberculosis vaccine strategies: route of immunisation-determined T-cell homing to restricted lung mucosal compartments. Eur. Respir. Rev. 2015;24:356–360. doi: 10.1183/16000617.00002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist S.R., Dinnon K.H., Schäfer A., Tse L.V., Okuda K., Hou Y.J., West A., Edwards C.E., Sanders W., Fritch E.J., et al. A mouse-adapted SARS-CoV-2 induces acute lung injury and mortality in standard laboratory mice. Cell. 2020;183:1070–1085.e12. doi: 10.1016/j.cell.2020.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurie N., Saville M., Hatchett R., Halton J. Developing Covid-19 vaccines at pandemic speed. N. Engl. J. Med. 2020;382:1969–1973. doi: 10.1056/NEJMp2005630. [DOI] [PubMed] [Google Scholar]
- Madhi S.A., Baillie V., Cutland C.L., Voysey M., Koen A.L., Fairlie L., Padayachee S.D., Dheda K., Barnabas S.L., Bhorat Q.E., et al. Efficacy of the ChAdOx1 nCoV-19 COVID-19 vaccine against the B.1.351 variant. N. Engl. J. Med. 2021;384:1885–1898. doi: 10.1056/NEJMoa2102214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matchett W.E., Joag V., Stolley J.M., Shepherd F.K., Quarnstrom C.F., Mickelson C.K., Wijeyesinghe S., Soerens A.G., Becker S., Thiede J.M., et al. Cutting edge: nucleocapsid vaccine elicits spike-independent SARS-CoV-2 protective immunity. J. Immunol. 2021;207:376–379. doi: 10.4049/jimmunol.2100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutra M.R., Kozlowski P.A. Mucosal vaccines: the promise and the challenge. Nat. Rev. Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- Nicolai L., Leunig A., Pekayvaz K., Anjum A., Riedlinger E., Eivers L., Hoffknecht M.-L., Rossaro D., Escaig R., Kaiser R., et al. Thrombocytopenia and splenic platelet directed immune responses after intravenous ChAdOx1 nCov-19 administration. Preprint at bioRxiv. 2021 2021.06.29.450356. [Google Scholar]
- Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas D., Bruel T., Grzelak L., Guivel-Benhassine F., Staropoli I., Porrot F., Planchais C., Buchrieser J., Rajah M.M., Bishop E., et al. Sensitivity of infectious SARS-CoV-2 B.1.1.7 and B.1.351 variants to neutralizing antibodies. Nat. Med. 2021;27:917–924. doi: 10.1038/s41591-021-01318-5. [DOI] [PubMed] [Google Scholar]
- Rodda L.B., Netland J., Shehata L., Pruner K.B., Morawski P.A., Thouvenel C.D., Takehara K.K., Eggenberger J., Hemann E.A., Waterman H.R., et al. Functional SARS-CoV-2-specific immune memory persists after mild COVID-19. Cell. 2021;184:169–183.e17. doi: 10.1016/j.cell.2020.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoff J., Gray G., Vandebosch A., Cárdenas V., Shukarev G., Grinsztejn B., Goepfert P.A., Truyers C., Fennema H., Spiessens B., et al. Safety and efficacy of single-dose Ad26.COV2.S vaccine against Covid-19. N. Engl. J. Med. 2021;384:2187–2201. doi: 10.1056/NEJMoa2101544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santosuosso M., Zhang X., McCormick S., Wang J., Hitt M., Xing Z. Mechanisms of mucosal and parenteral tuberculosis vaccinations: adenoviral-based mucosal immunization preferentially elicits sustained accumulation of immune protective CD4 and CD8 T cells within the airway lumen. J. Immunol. 2005;174:7986–7994. doi: 10.4049/jimmunol.174.12.7986. [DOI] [PubMed] [Google Scholar]
- Satti I., Meyer J., Harris S.A., Manjaly Thomas Z.-R., Griffiths K., Antrobus R.D., Rowland R., Ramon R.L., Smith M., Sheehan S., et al. Safety and immunogenicity of a candidate tuberculosis vaccine MVA85A delivered by aerosol in BCG-vaccinated healthy adults: a phase 1, double-blind, randomised controlled trial. Lancet Infect. Dis. 2014;14:939–946. doi: 10.1016/S1473-3099(14)70845-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C., Nobs S.P., Heer A.K., Kurrer M., Klinke G., van Rooijen N., Vogel J., Kopf M. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. 2014;10:e1004053. doi: 10.1371/journal.ppat.1004053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott J., Richterman A., Cevik M. COVID-19 vaccination: evidence of waning immunity is overstated. BMJ. 2021;374:n2320. doi: 10.1136/bmj.n2320. [DOI] [PubMed] [Google Scholar]
- Shen X., Tang H., Pajon R., Smith G., Glenn G.M., Shi W., Korber B., Montefiori D.C. Neutralization of SARS-CoV-2 variants B.1.429 and B.1.351. N. Engl. J. Med. 2021;384:2352–2354. doi: 10.1056/NEJMc2103740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde V., Bhikha S., Hoosain Z., Archary M., Bhorat Q., Fairlie L., Lalloo U., Masilela M.S.L., Moodley D., Hanley S., et al. Efficacy of NVX-CoV2373 COVID-19 vaccine against the B.1.351 Variant. N. Engl. J. Med. 2021;384:1899–1909. doi: 10.1056/NEJMoa2103055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Kang A., Luo X., Jeyanathan M., Gillgrass A., Afkhami S., Xing Z. COVID-19: current knowledge in clinical features, immunological responses, and vaccine development. FASEB J. 2021;35:e21409. doi: 10.1096/fj.202002662R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey H.D., Golubeva D., Posca A., Ang J.C., Novakowski K.E., Zahoor M.A., Kaushic C., Cairns E., Bowdish D.M.E., Mullarkey C.E., et al. IgA potentiates NETosis in response to viral infection. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2101497118. e2101497118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadlbauer D., Tan J., Jiang K., Hernandez M.M., Fabre S., Amanat F., Teo C., Arunkumar G.A., McMahon M., Capuano C., et al. Repeated cross-sectional sero-monitoring of SARS-CoV-2 in New York City. Nature. 2021;590:146–150. doi: 10.1038/s41586-020-2912-6. [DOI] [PubMed] [Google Scholar]
- Starr T.N., Greaney A.J., Dingens A.S., Bloom J.D. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep. Med. 2021;2:100255. doi: 10.1016/j.xcrm.2021.100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo P.A., Miron M., Farber D.L. Location, location, location: tissue resident memory T cells in mice and humans. Sci. Immunol. 2019;4:eaas9673. doi: 10.1126/sciimmunol.aas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C.W., Chia W.N., Qin X., Liu P., Chen M.I.-C., Tiu C., Hu Z., Chen V.C.-W., Young B.E., Sia W.R., et al. A SARS-CoV-2 surrogate virus neutralization test based on antibody-mediated blockage of ACE2-spike protein-protein interaction. Nat. Biotechnol. 2020;38:1073–1078. doi: 10.1038/s41587-020-0631-z. [DOI] [PubMed] [Google Scholar]
- Tarke A., Sidney J., Methot N., Zhang Y., Dan J.M., Goodwin B., Rubiro P., Sutherland A., da Silva Antunes R., Frazier A., et al. Negligible impact of SARS-CoV-2 variants on CD4+ and CD8+ T cell reactivity in COVID-19 exposed donors and vaccinees. Preprint at bioRxiv. 2021 2021.02.27.433180. [Google Scholar]
- Taylor J.J., Martinez R.J., Titcombe P.J., Barsness L.O., Thomas S.R., Zhang N., Katzman S.D., Jenkins M.K., Mueller D.L. Deletion and anergy of polyclonal B cells specific for ubiquitous membrane-bound self-antigen. J. Exp. Med. 2012;209:2065–2077. doi: 10.1084/jem.20112272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijaro J.R., Farber D.L. COVID-19 vaccines: modes of immune activation and future challenges. Nat. Rev. Immunol. 2021;21:195–197. doi: 10.1038/s41577-021-00526-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsilingiris D., Vallianou N.G., Karampela I., Dalamaga M. Vaccine induced thrombotic thrombocytopenia: the shady chapter of a success story. Metabol. Open. 2021;11:100101. doi: 10.1016/j.metop.2021.100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Thorson L., Stokes R.W., Santosuosso M., Huygen K., Zganiacz A., Hitt M., Xing Z. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J. Immunol. 2004;173:6357–6365. doi: 10.4049/jimmunol.173.10.6357. [DOI] [PubMed] [Google Scholar]
- Wang P., Nair M.S., Liu L., Iketani S., Luo Y., Guo Y., Wang M., Yu J., Zhang B., Kwong P.D., et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature. 2021;593:130–135. doi: 10.1038/s41586-021-03398-2. [DOI] [PubMed] [Google Scholar]
- Wilhelm A., Widera M., Grikscheit K., Toptan T., Schenk B., Pallas C., Metzler M., Kohmer N., Hoehl S., Helfritz F.A., et al. Reduced neutralization of SARS-CoV-2 Omicron Variant by Vaccine Sera and monoclonal antibodies. Preprint at medRxiv. 2021 2021.12.07.21267432. [Google Scholar]
- Wu S., Huang J., Zhang Z., Wu J., Zhang J., Hu H., Zhu T., Zhang J., Luo L., Fan P., et al. Safety, tolerability, and immunogenicity of an aerosolised adenovirus type-5 vector-based COVID-19 vaccine (Ad5-nCoV) in adults: preliminary report of an open-label and randomised phase 1 clinical trial. Lancet Infect. Dis. 2021;21:1654–1664. doi: 10.1016/S1473-3099(21)00396-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Z., Afkhami S., Bavananthasivam J., Fritz D.K., D’Agostino M.R., Vaseghi-Shanjani M., Yao Y., Jeyanathan M. Innate immune memory of tissue-resident macrophages and trained innate immunity: re-vamping vaccine concept and strategies. J. Leukoc. Biol. 2020;108:825–834. doi: 10.1002/JLB.4MR0220-446R. [DOI] [PubMed] [Google Scholar]
- Yao Y., Jeyanathan M., Haddadi S., Barra N.G., Vaseghi-Shanjani M., Damjanovic D., Lai R., Afkhami S., Chen Y., Dvorkin-Gheva A., et al. Induction of autonomous memory alveolar macrophages requires T cell help and is critical to trained immunity. Cell. 2018;175:1634–1650.e17. doi: 10.1016/j.cell.2018.09.042. [DOI] [PubMed] [Google Scholar]
- Zhao J., Zhao J., Mangalam A.K., Channappanavar R., Fett C., Meyerholz D.K., Agnihothram S., Baric R.S., David C.S., Perlman S. Airway memory CD4 + T cells mediate protective immunity against emerging respiratory coronaviruses. Immunity. 2016;44:1379–1391. doi: 10.1016/j.immuni.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J., Wong L.R., Li K., Verma A.K., Ortiz M.E., Wohlford-Lenane C., Leidinger M.R., Knudson C.M., Meyerholz D.K., McCray P.B., et al. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature. 2021;589:603–607. doi: 10.1038/s41586-020-2943-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data supporting the findings of this study are available within the paper and are available from the lead contact or correspondence authors upon request. This paper does not report original code.