

Abstract
Flowers are produced by floral meristems, groups of stem cells that give rise to floral organs. In grasses, including the major cereal crops, flowers (florets) are contained in spikelets, which contain one to many florets, depending on the species. Importantly, not all grass florets are developmentally equivalent, and one or more florets are often sterile or abort in each spikelet. Members of the Andropogoneae tribe, including maize (Zea mays), produce spikelets with two florets; the upper and lower florets are usually dimorphic, and the lower floret is greatly reduced compared to the upper floret. In maize ears, early development appears identical in both florets but the lower floret ultimately aborts. To gain insight into the functional differences between florets with different fates, we used laser capture microdissection coupled with RNA-sequencing to globally examine gene expression in upper and lower floral meristems in maize. Differentially expressed genes were involved in hormone regulation, cell wall, sugar, and energy homeostasis. Furthermore, cell wall modifications and sugar accumulation differed between the upper and lower florets. Finally, we identified a boundary domain between upper and lower florets, which we hypothesize is important for floral meristem activity. We propose a model in which growth is suppressed in the lower floret by limiting sugar availability and upregulating genes involved in growth repression. This growth repression module may also regulate floret fertility in other grasses and potentially be modulated to engineer more productive cereal crops.
Floret-specific differences in cell wall composition and sugar accumulation likely contribute to growth suppression in the lower floret of maize spikelets.
Introduction
Flowers are essential for plant reproduction and also form fruits and seeds, which are consumed as food. Flowers are produced by floral meristems (FMs), undifferentiated groups of stem cells that generate floral organs (Bartlett and Thompson, 2014). Grass flowers (florets) are contained in spikelets, which contain two bracts (glumes) and one to many florets depending on the species. Like other grass flowers, maize (Zea mays) florets are highly derived structures. Within two enclosing organs, the lemma and palea, maize flowers contain two lodicules (homologous to petals), three stamens, and three carpels, two of which fuse to form the silk (Figure 1).
Figure 1.
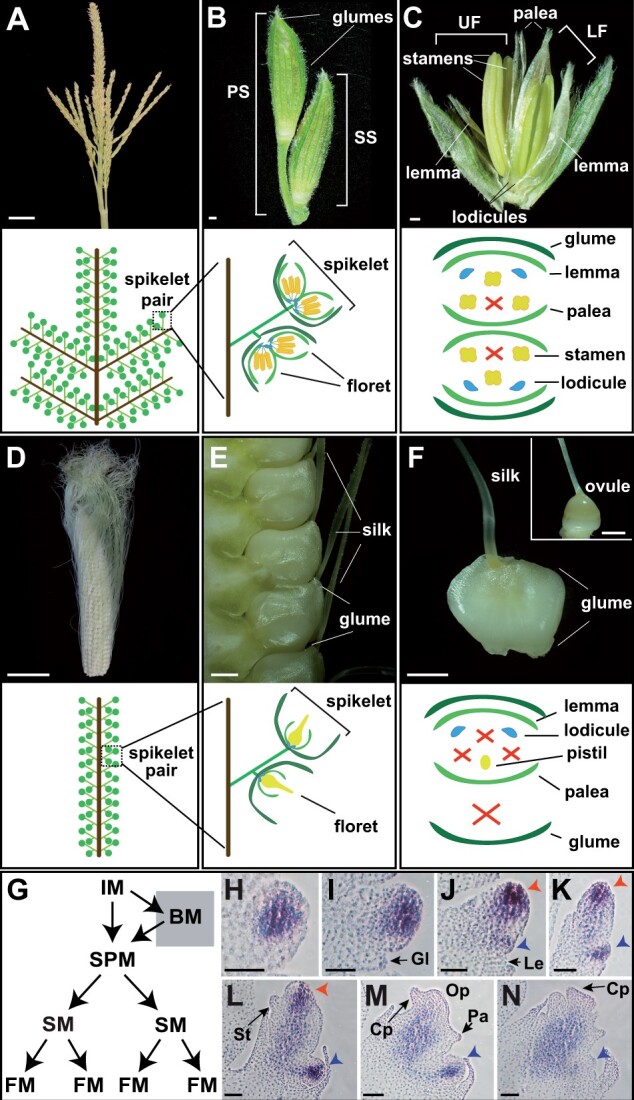
Normal maize floral development. A, Mature tassel, the male inflorescence. B, Pair of tassel spikelets. C, Dissected tassel spikelet, exposing two male florets. D, Mature ear, the female inflorescence. E, Mature ear spikelets. F, Dissected ear spikelet, containing a single female floret. Inset is a mature ovule with glumes and other floral organs removed. G, Diagram depicting meristems in the inflorescence. H–N, RNA in situ hybridization of the meristem marker, kn1, in developing ear spikelets; Red and blue arrowheads indicate upper and lower FM, respectively. PS, pedicellate spikelet; SS, sessile spikelet; UF, upper floret; LF, lower floret; IM, inflorescence meristem; BM, branch meristem; SPM, spikelet pair meristem; SM, spikelet meristem; Gl, glume; Op, ovule primordia; Cp, carpel primordia; Lo, lodicule; St, stamen; Le, lemma; Pa, palea. Scale bars: (A and D) = 5 cm, (B, C, E, and F) = 500 μm, (H–N) = 50 μm.
Maize produces two inflorescences, the tassel and ear, which produce male and female flowers, respectively (Cheng et al., 1983). Unlike Arabidopsis (Arabidopsis thaliana), in which the inflorescence meristem directly initiates FMs on its flanks, grass inflorescence meristems produce a series of higher order meristems before initiating FM. Upon the transition to flowering, the shoot apical meristem (SAM; tassel) or an axillary meristem (ear) transitions to an indeterminate inflorescence meristem; the inflorescence meristem initiates ordered rows of spikelet pair meristems, which in turn give rise to two spikelet meristems (Figure 1G;Thompson and Hake, 2009; Whipple, 2017). The spikelet meristem first initiates the proximal/lower FM (LFM) in the axil of a lemma on the abaxial side of the spikelet (Figure 1, H–L). The origin of the distal/upper FM (UFM) is less clear; one model proposes that the UFM is also initiated as an axillary meristem by the spikelet meristem, whereas the second model proposes the spikelet is itself converted to the UFM (Irish, 1997; Chuck et al., 1998).
Both the tassel and ear initiate bisexual flowers and early floral development is very similar in upper and lowers florets (Irish and Nelson, 1989). Carpels abort via programmed cell death in the tassel and stamens arrest shortly after anther formation in the ear (Cheng et al., 1983). In the ear, lower floret abortion is initiated by programmed cell death similar to the carpel abortion program in the tassel (Cheng et al., 1983). Thus, mature ear spikelets contain a single female floret, whereas mature tassel spikelets contain two male florets (Figure 1, A–F).
Spikelets containing sterile or aborted florets are common in the grasses, including cereal crops. In some species (e.g. maize and barley; Hordeum vulgare), floret abortion/sterility is genetically preprogrammed and invariable between individuals whereas in other species (e.g. wheat; Triticum aestivum), the number of aborted florets in a spikelet is variable and influenced by the environment. A few regulators of floral abortion have been identified (i.e. jasmonic acid [JA] in maize, six-rowed spike/vrs genes in barley, and Grain Number Increase 1/GNI1 in wheat); however, the importance of floral abortion is still unknown and we know very little about the processes downstream of these high-level regulators (Sakuma and Schnurbusch, 2020). To gain insight into the functional differences between florets with different developmental fates, we used laser capture microdissection (LCM) coupled with RNA-sequencing (RNA-seq) to globally survey gene expression in UFM and LFM of maize ears.
Results
Upper and lower FMs have distinct gene expression profiles
Gene expression is dynamic during floral development; to ensure we isolated upper and lower FM at similar developmental stages, we isolated FM from ear primordia after initiation of lemma, but before stamen primordia (Figure 2, A–F). Because LFM development is delayed relative to the UFM (Cheng et al., 1983), spikelets for LFM dissections were older than those used for UFM dissections. Principal component analysis (PCA) and Pearson’s correlation analysis confirmed UFM and LFM biological samples clustered together and had high reproducibility (Supplemental Figure S1). Approximately 700 genes were differentially expressed between UFM and LFM (238 UFM-enriched, 456 LFM-enriched; fold change ≥2 and q < 0.05; Figure 2G;Supplemental File S1). Importantly, our data included three UFM-enriched genes with known RNA expression patterns (Supplemental Figure S2 and Supplemental File S2). zmm8/GRMZM2G102161 and zmm14/GRMZM2G099522 encode MADS-box transcription factors (TFs) that are broadly expressed in the meristem and floral organs of the upper floret, but not detected in the lower floret (Cacharron et al., 1999; Du et al., 2021). barren stalk1 (ba1)/GRMZM2G397518 encodes a basic helix–loop–helix (bHLH) protein required for axillary meristem initiation and is expressed in a diffuse pattern in UFM and in a group of cells at the UFM/LFM boundary, but not detected in LFM (Gallavotti et al., 2004).
Figure 2.

Maize upper and lower FMs are enriched for genes belonging to distinct functional groups. Scanning electron micrographs (SEMs) of spikelets at developmental stages of UFM (A) and LFM (D) dissections. Representative images of FM before (B and E) and after (C and F) LCM. False coloring indicates UFM (red), LFM (blue), and lemma primordia (green). G, Venn diagram depicting DEGs (q < 0.05 and fold change ≥2) in UFM (red) and LFM (blue). H, Distribution of DEGs in MapMan-annotated functional groups. Note difference in scale of the X-axis. Asterisk indicates P < 0.05 in Wilcoxon rank sum test. Genes unassigned to a functional group (99 UFM-enriched and 227 LFM-enriched) are not shown. I and J, GO-enrichment maps for UFM and LFM DEGs. Nodes (circles) indicate significantly enriched GO terms. Node size is proportional to number of DEGs in each node; node color indicates statistical significance. Edges (lines) link similar GO terms. Edge thickness is proportional to the number of DEGs shared between GO terms. Node clusters were manually labeled based on corresponding GO terms in each cluster. Scale bars = 50 μm.
To gain insight into the biological function of differentially expressed genes (DEGs), we predicted gene function using MapMan (Schwacke et al., 2019), gene ontology (GO) enrichment analysis (Gene Ontology Consortium, 2015), and CornCyc, which predicts metabolic pathways (Schläpfer et al., 2017; Figure 2, H–J; Supplemental File S2). To facilitate the interpretation of hierarchical GO enrichment groups, we used the Cytoscape plug-in, Enrichment Map, to construct functional GO networks (Merico et al., 2010). In general, the UFM was enriched for genes in functional groups associated with growth and primary metabolism, including RNA synthesis and processing, protein synthesis, vesicle trafficking, nucleotide metabolism, and sugar response and transport (Figure 2, H and I; Supplemental File S2). In contrast, the LFM was enriched for genes in functional groups associated with secondary metabolism and dormancy, including phytohormones, protein degradation, amino acid catabolism, and cell wall-related genes (Figure 2, H–J; Supplemental File S2).
We further examined select functional groups to gain insight into the functional patterns of DEGs (Supplemental Figures S2–S4 and Supplemental File S2). DEGs in the RNA biosynthesis group contains several classes of TFs with well-known roles in plant growth and development, including APETALA2/ETHYLENE-RESPONSIVE FACTOR (AP2/ERF), myeloblastosis (MYB), homeobox, bHLH, TEOSINTE BRANCHED1/CYCLOIDEA/PROLIFERATING (TCP) CELL NUCLEAR ANTIGEN FACTOR, and WRKY TFs (Supplemental Figure S2). DEGs also included TFs with known functions in maize floral development, including ba1/GRMZM2G397518 (Gallavotti et al., 2004), GRF-interacting factor1 (gif1)/GRMZM2G180246 (Zhang et al., 2018), zmm8/GRMZM2G102161, and zmm14/GRMZM2G099522 (Du et al., 2021) in the UFM and branched silkless1 (bd1)/GRMZM2G307119 (Chuck et al., 2002), gnarley1 (gn1)/GRMZM2G452178 (Foster et al., 1999a, 1999b), teosinte branched1 (tb1)/AC233950.1_FG002 (Hubbard et al., 2002), Wavy auricle in blade1 (Wab1)/branched angle defective1 (bad1)/GRMZM2G110242 (Hay and Hake, 2004; Bai et al., 2012; Lewis et al., 2014), and zfl2/GRMZM2G180190 (Bomblies et al., 2003) in the LFM.
DEGs in the phytohormone group function in metabolism and signaling of multiple hormones, including cytokinin, auxin, gibberellin (GA), and JA; Figure 2, H–J; Supplemental Figure S3 and Supplemental File S2). Of the four cytokinin-related genes in our DEGs set, two cytokinin biosynthesis genes (czog1/GRMZM2G168474, GRMZM2G008726) were UFM-enriched and two A-type ARR negative regulators of cytokinin signaling (crr2/GRMZM2G392101, GRMZM2G179827) were LFM-enriched, suggesting that cytokinin signaling may be higher in UFM relative to LFM. In contrast, auxin-, GA-, and JA-related genes were predominantly enriched in LFM. JA is required for lower floret abortion in the ear (DeLong et al., 1993; Acosta et al., 2009; Lunde et al., 2019; Wang et al., 2020) and three JA biosynthesis genes were LFM-enriched (lox9/GRMZM2G017616; tasselseed1 (ts1)/GRMZM2G104843; GRMZM2G168404). Seven auxin-related DEGs were LFM-enriched and functioned in auxin synthesis (tar2/GRMZM2G066345), transport (pin3/GRMZM2G149184; GRMZM2G085236; GRMZM2G037386), and signaling (aas8/GRMZM2G053338; iaa37/GRMZM2G359924; bif4/GRMZM5G864847). GA-related DEGs were also LFM-enriched and function in the GA synthesis (ga20ox1/AC203966.5_FG005), inactivation (ga2ox3/GRMZM2G022679; ga2ox9/GRMZM2G152354), and signaling (gras46/GRMZM2G001426; GRMZM2G040278; GRMZM2G440543). The LFM was also enriched for three genes encoding Gibberellic Acid Stimulated Arabidopsis cysteine-rich polypeptides (gsl1/GRMZM2G062527; GRMZM2G077845; GRMZM2G150688), which in Arabidopsis are induced by GA and have broad functions in defense and development (Roxrud et al., 2007; Zhong et al., 2015). These gene expression profiles suggest that hormone accumulation and signaling differs between UFM and LFM of maize ears, with high cytokinin in the upper floret and high auxin, GA and JA in the lower floret.
We investigated the spatial expression of DEGs by RNA in situ hybridization in developing spikelets and determined specific expression patterns for 10 genes. AC217050.4_FG006 (encodes a 14-3-3 protein, log2FC = 1.124), AP2/EREBP transcription factor 26 (ereb26)/GRMZM2G317160 (log2FC = −1.162), and chromatin complex subunit A 101 (chr101)/GRMZM2G177165 (log2FC = −1.251) were broadly expressed in both upper and lower FM (Figure 3, A–C); AC217050.4_FG006 and chr101/GRMZM2G177165 were also present in stamen and carpel primordia (Figure 3, A and C; Supplemental Figure S5). As previously shown, gif1/GRMZM2G180246 (log2FC = 1.063) was expressed in a ring around developing UFM and at the base of palea in upper florets (Zhang et al., 2018), and showed a similar expression pattern in lower florets (Figure 3D). GRMZM2G101682 (grass-specific gene of unknown function, log2FC = −1.009) was also expressed in both UFM and LFM (Figure 3E), with strong expression restricted to the outermost cell layer. We also observed expression in the outer cell layer of spikelet meristem, stamen and carpel primordia (Figure 3E;Supplemental Figure S5). In developing shoots, GRMZM2G101682 is localized to the L1 layer of boundary regions between initiating organs and the preligular band of developing leaves, but is not expressed in the meristem itself (Johnston et al., 2014). Histone H1-like/GRMZM2G069911 (log2FC = −1.233) and GRMZM2G180870 (XYLOGLUCAN ENDOTRANSGLUCOSYLASE/HYDROLASE 9 (XTH9) homolog, log2FC = −1.059) were expressed in punctate patterns characteristic of genes involved in cell division (Figure 3, F and G; Supplemental Figure S6; Asai et al., 2002; Kim et al., 2007; Ikeda-Kawakatsu et al., 2009). In the SAM, stem cells at the tip of the meristem have lower cell division rates compared to cells in axillary primordia (Satterlee et al. 2020). If this pattern of cell division also occurs in FM, enrichment of cell division genes in the LFM could reflect the axillary meristem of LFM, whereas the UFM is likely converted from the spikelet meristem. Alternatively, because UFM were larger than LFM at the time of dissection, we likely captured a higher proportion of “tip stem cells” (with lower cell division rates) in UFM relative to LFM samples. Finally, three genes were localized to a unique boundary region between the upper and lower florets. GRMZM2G114552 (log2FC = 2.616) encodes a Bowman–Birk-type trypsin inhibitor (BBTI) and was expressed in a discrete domain on the abaxial side of UFM but not detectable in LFM (Figure 3H). BBTI was also expressed at the boundaries of initiating spikelet pair and spikelet meristems, and at the base of palea in the upper floret (Supplemental Figure S7). A pectate lyase homolog, GRMZM2G131912 (log2FC = −1.281), and arginine decarboxylase1 (adc1)/GRMZM2G396553 (log2FC = −1.216) were present in discrete domains on the adaxial side of the LFM at the boundary with the upper floret (Figure 3, I and J; Supplemental Figure S7). BBTI, adc1, and pectate lyase were also expressed in this boundary region in tassel florets (Supplemental Figure S7), indicating that this boundary expression is not unique to the ear.
Figure 3.

UFM and LFM DEGs have distinct RNA expression patterns. A, AC217050.4_FG006, 14-3-3 protein. B, GRMZM2G317160/ereb26. C, GRMZM2G177165/chr101. D, GRMZM2G180246/gif1. E, GRMZM2G101682, unknown function. F, GRMZM2G069911, histone H1-like. G, GRMZM2G180870, AtXTH9 (XYLOGLUCAN ENDOTRANSGLUCOSYLASE/HYDROLASE 9) homolog. H, GRMZM2G114552/BBTI, Bowman–Birk-type (proteinase/bran trypsin) inhibitor. I, GRMZM2G131912, pectate lyase homolog. J, GRMZM2G396553/adc1. Developmental stages at which UFM (top) and LFM (bottom) were dissected are shown for each gene. St, Stamen; Cp, carpel primordia. Scale bars = 50 μm.
Pectin modification is dynamic during spikelet development and differs between upper and lower florets
GO and MapMan functional analyses indicated that DEGs belonged to multiple functional groups (Figure 2), several of which made sense based on their well-established roles in development (i.e. transcription, development, morphogenesis, hormones); however, other functional groups were more surprising. We were particularly intrigued by enrichment of cell wall-related genes in LFM and sugar-related genes in UFM, and thus further investigated these functional groups.
Our DEG set included 20 MapMan-annotated cell wall-related genes, 18 of which were enriched in LFM (Figure 4). Indeed, RNA in situ hybridization confirmed that GRMZM2G131912 (pectate lyase homolog), is expressed in the lower floret, adjacent to the UFM/LFM boundary (Figure 3I). Cell wall-related DEGs were involved in synthesis or modification of all major cell wall components, including cellulose (one gene), lignin (two genes), hemicellulose (four genes), and pectin (five genes), as well as arabinogalactan proteins (six genes) and expansins (two genes; Figure 4A;Supplemental File S2). Most of these genes are involved in synthesis and modification of the primary cell wall, which is synthesized and continuously deposited around dividing or expanding cells, including meristems (Cavalier et al., 2008; Keegstra, 2010; Sampathkumar et al., 2019).
Figure 4.

The UFM and LFM have distinct cell wall compositions. A, Expression profiles for DEG in the MapMan Cell Wall functional group. Left part indicates log2(fold change) for each gene. UFM-enriched genes are plotted on the left (red) and LFM-enriched genes on the right (blue). Middle shows an expression heatmap. Red asterisk indicates gene with known RNA in situ hybridization patterns. Ruthenium red staining of acidic pectin in SM (B), early (B′), and late (B″) ear florets. C–G, LM19 immunostaining of low methylesterified HG in developing spikelets. H, Negative control lacking primary antibody to show background autofluorescence using the same laser settings as (C–G). White boxes indicate zoomed in areas shown in (C′–H′). I–M, LM20 immunostaining of high methylesterified HG in developing spikelets. N, Negative control lacking primary antibody to show background autofluorescence using the same laser settings as (I–M). White boxes indicated zoomed in areas corresponding to UFM (I′–N′) and LFM (I″–N″). Weak, diffuse cytoplasmic signal in (C–N) is background autofluorescence, which varies due to incomplete quenching. Micrographs were false colored using ImageJ (Orange Blue icb look-up table) to visualize signal intensity. SM, spikelet meristem; Gl, glume; Cp, carpel primordia; Le, lemma. Scale bars = 50 μm.
Differential expression of cell wall-related genes suggested that UFM and LFM have different cell wall compositions and/or modifications. Therefore, we stained the major cell wall components in developing spikelets, including cellulose (calcofluor white/fluorescent brightener 28), lignin (phloroglucinol–HCl), and pectin (ruthenium red). To confirm our staining protocols accurately reflected cell wall composition, we first stained vasculature tissue, where cell wall composition is well-characterized (Supplemental Figure S8; Chen et al., 2006; Verhertbruggen et al., 2009; Pesquet et al., 2013; Hu et al., 2017; Torode et al., 2018). Lignin is predominantly found in secondary cell walls, which only form after cells have stopped expansion (Zhong et al., 2019). As expected for meristematic tissue, lignin staining was weak or undetectable in inflorescence primordia (Supplemental Figure S8), with no indication of floret-specific accumulation. Cellulose accumulated at the periphery of all cells and appeared similar in both florets (Supplemental Figure S8). We visualized pectin using ruthenium red, which preferentially stains acidic pectin (Ruzin, 1999), and observed striking differences in pectin distribution between UFM and LFM. Ruthenium red strongly stained the L1 layer of spikelet meristems and glume primordia; staining persisted in the L1 of UFM, however, was much weaker or absent in LFM (Figure 4B).
To examine pectin composition in more detail, we used two monoclonal antibodies, LM19 and LM20, which recognize the low methylesterified (acidic) and high methylesterified forms of homogalacturonan (HG), the most abundant pectic polysaccharide (Verhertbruggen et al., 2009). We observed dynamic LM19 staining during floral development, suggesting that HG methylesterification is developmentally regulated (Figure 4, C–H). LM19 (low methylesterification) strongly stained the L1 layer of spikelet meristems and glume primordia, similar to ruthenium red staining (Figure 4C). LM19 also stained cells at the base of glumes, and strongly stained incipient and initiating floral organ primordia (Figure 4, D–G). We observed weak staining at the base of the LFM, and occasionally observed a couple of brightly staining cells that appeared to correspond to palea primordia (Figure 4G).
LM20 (high methylesterified HG) showed a dramatically different staining pattern than LM19 (Figure 4, I–N). LM20 weakly stained the periphery of most or all cells in developing spikelets and intensely stained the apical surface of the L1 layer of spikelet meristems and young UFM (Figure 4, I–K). LM20 staining persisted throughout UFM development, but its localization became more punctate and primarily accumulated at cell junctions (Figure 4, K–M). In LFM, LM20 also stained cell peripheries, often forming puncta at cell junctions (Figure 4, J–M). Although variable, we never observed strong LM20 staining in LFM as we did in UFM.
We next asked if differential pectin accumulation also occurred in spikelets where the lower floret does not abort. Specifically, we examined tassel spikelets, which produce two staminate florets, and ramosa3; grassy tillers1 (ra3; gt1) ear spikelets, in which the lower floret does not abort and produce two pistillate florets (Klein et al., 2021). Pectin preferentially accumulated in the upper floret of tassel and ra3; gt1 ear spikelets, similar to what we observed in normal ear spikelets (Supplemental Figure S9). In the upper floret of tassel spikelets, however, LM19 also strongly stained aborting carpels (Figure 5, A–C). Indeed, demethylesterified pectin was generally associated with carpel abortion; LM19 strongly stained aborting carpels in the lower florets of both tassels and ears (Figure 5, D–I). Lower florets of ra3; gt1 double mutants, however, lacked intense LM19 staining characteristic of aborting carpels and resembled initiating and elongating carpels in upper florets (Figure 5, J–L). Together, these results indicate that pectin is dynamically regulated during floral development and pectin modification differs between upper and lower FM. Both methylesterified and demethylesterified pectin preferentially accumulate in the upper floret, even in spikelets where the lower floret does not abort. Demethylesterified pectin strongly accumulates in incipient and initiating organ primordia of UFM, indicating pectin may also play a role in organ initiation in maize. Finally, demethylesterified pectin is strongly associated with aborting carpels in the lower floret of ears and both florets in tassels.
Figure 5.
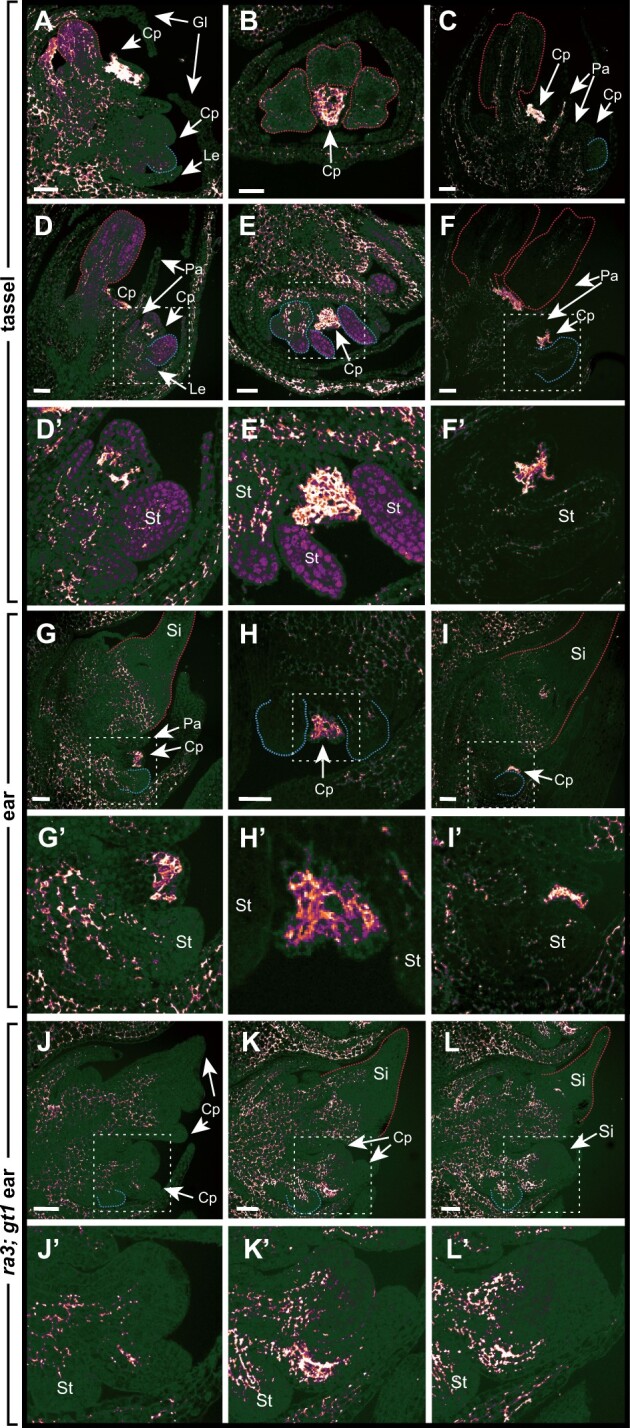
Pectin demethylesterification is associated with aborting carpels. LM19 immunostaining of low methylesterified HG in aborting carpels in the upper (A–C) and lower (D–F) florets of tassel spikelets, and lower florets of ear spikelets (G–I). In ra3;gt1 mutant ear spikelets (J–L), where the lower floret does not abort, LM19 stains initiating carpel primordia, but lacks the intense staining associated with aborting carpels. White boxes indicate zoomed in areas corresponding to regions shown in (D′–L′). Red and blue dashed lines outline stamen primordia in upper and lower florets, respectively. Silks in the upper floret are also outlined in red in G, I, K, and L. LM19 staining use the same laser settings as Figure 4, C–G. Cp, carpel primordia; Gl, glume; Le, lemma; Pa, palea; Si, silk; St, stamen primordia. Scale bars = 50 μm.
Sugar-related genes and starch are differentially regulated between upper and lower FMs
The ability to coordinate energy and carbon availability with plant growth is critical. Hexose sugars generated through photosynthesis in source tissues are converted to sucrose for transport to sink tissues and starch for storage. Carbohydrates are required not only to provide chemical energy required for plant growth, but also to generate nucleotides and construct cell walls around newly divided and expanding cells (Sampathkumar et al., 2019). GO and MapMan analysis indicated UFM were enriched for genes involved in carbohydrate/sugar transport and response (Figure 2, H and I; Supplemental File S2), while the LFM was enriched for multiple members of the SNF1-related protein kinase 1 (SnRK1) signaling pathway (Figure 6, A and B).
Figure 6.

Sugar metabolism likely differs in UFM and LFM. A, Summary of predicted SnRK1 signaling network genes expressed in FM samples. B, Expression profiles for sugar-related DEG; layout is the same as cell wall-related genes in Figure 4. C–G, Starch accumulates at the boundary between the UFM and LFM in developing spikelets. Red and blue arrowheads indicate UFM and LFM respectively. SM, spikelet meristem; Gl, glume; Cp, carpel primordia; Op, ovule primordia; St, stamen. Scale bars = 50 μm.
The SnRK1 pathway is a key regulator of plant growth and energy homeostasis (Baena-González et al., 2007) and is regulated both by trehalose-6-phosphate (T6P) levels (Baena-González and Lunn, 2020) and interactions with FCS-like zinc (FLZ) finger proteins (Nietzsche et al., 2014; Jamsheer et al., 2018b, 2019). Trehalose is present in trace amounts in plants and its primary role is likely in sugar sensing and signaling, rather than chemical energy storage (Wingler, 2002; Figueroa and Lunn, 2016). The trehalose precursor, T6P, is synthesized from uridine diphosphate-glucose (UDP)-glucose and glucose-6-P by T6P synthase (TPS) and thought to be the active signaling molecule; T6P is converted to trehalose by trehalose-6-phosphate phosphatases (TPPs; Cabib and Leloir, 1958). Increased T6P is associated with high sucrose and T6P sensing may be a key mechanism by which plants monitor energy status (Figueroa and Lunn, 2016). The maize genome contains 13 TPP genes, of which ramosa3 (ra3)/tpp10/GRMZM2G014729 is the most extensively studied and is required for spikelet pair and spikelet meristem determinacy in the inflorescence and carpel abortion in florets (Satoh-Nagasawa et al., 2006). Interestingly, ra3 promotes meristem determinacy at least in part independent of its enzymatic activity and likely functions in transcriptional regulation (Claeys et al., 2019; Demesa-Arevalo et al., 2021). Both ra3 and tpp3/GRMZM2G117564 were LFM-enriched, suggesting that sugar signaling could be critical in the LFM. Ra3 functions redundantly with the HD-ZIP TF, grassy tillers1 (gt1) to repress carpel growth in tassel florets. Particularly relevant to this work, the lower floret fails to abort in ra3; gt1 double mutants, demonstrating that our approach identified genes with functional differences in the upper and lower florets (Klein et al. 2021).
The plant-specific FLZ gene family is defined by the presence of a approximately 50 amino acid FLZ domain, which interacts with SnRK1 (Jamsheer and Laxmi, 2014; Jamsheer et al., 2018b). While the function of many FLZ genes is unknown, they have been implicated in abscisic acid, sugar, and energy response in Arabidopsis (Jamsheer and Laxmi, 2015; Jamsheer et al., 2018a) and are thought to act as adapters between SnRK1 and other proteins (Tsai and Gazzarrini, 2014; Jamsheer et al., 2018b, 2019). Based on MapMan annotations, the maize genome contains 39 FLZ genes in the “multiprocess regulation” functional group, 28 of which were expressed in our FM samples. Strikingly, nearly one-third (8/28) of the FM-expressed FLZ genes were differentially expressed between UFM and LFM, all of which were LFM-enriched (P < 4.7×10−7 in a two-sided Fisher’s exact test; Figure 6;Supplemental File S2). All core components of the SnRK1 signaling pathway were expressed in our FM samples, but only FLZ genes were differentially expressed between UFM and LFM (Figure 6A).
Determining sugar accumulation and distribution in situ is challenging due to a lack of dyes or other mechanisms to detect specific sugars. Most sugar analysis requires grinding tissue and measuring the overall sugar levels, which precludes the cellular-level resolution required to detect spatial differences in sugar accumulation. Starch, the major storage carbohydrate in plants, however, can be easily visualized by iodine staining (Zhang et al., 2019). In the inflorescence, starch accumulates at the base of developing spikelet meristem, but not in the spikelet meristem itself (Figure 6C). After LFM initiation, starch begins to accumulate at the base of UFM, near the boundary with LFM (Figure 6, D and E). Starch accumulation intensifies and becomes more defined at the boundary between UFM and LFM in older spikelets (Figure 6, E–G). Strikingly, we did not observe detectable starch in LFM at any stage of spikelet development (Figure 6, C–G). We observed similar starch accumulation in tassels (Supplemental Figure S8, N–R) and in ra3; gt1 ears (Supplemental Figure S8, S–W), in which both upper and lower florets fully develop, indicating starch distribution is carefully regulated in developing spikelets of both male and female inflorescences and sugars accumulate differently in UFM and LFM independent of lower floret abortion.
Discussion
Upper and lower FMs are not developmentally equivalent
FMs are typically regarded as functionally equivalent, regardless of where they are initiated on the plant. In Arabidopsis, for example, all FM form as axillary meristems on the flanks on an inflorescence meristem, produce identical flowers, and appear to have the same developmental potential (Liu et al., 2009). In maize, spikelet meristems are often depicted as initiating two equivalent FM (Figure 1G). However, this depiction is misleading; upper and lower FM are likely functionally divergent from the time of initiation. The Andropogoneae tribe, which includes maize along with the key crops sorghum (Sorghum bicolor) and sugarcane (Saccharum officinarum), produce paired spikelets with two florets per spikelet. Upper and lower florets in the Andropogoneae are typically dimorphic; the upper floret is often hermaphroditic whereas the lower floret is usually reduced or sterile (Le Roux and Kellogg, 1999). Ear spikelets, in which the lower floret aborts, may be more representative of the Andropogoneae and its findings more relevant to other species in the tribe. Floral abortion and sterility are common in the cereals and the mechanisms that regulate lower floret growth in maize may also apply to other cereal crops.
We sought to understand the functional differences of the upper and lower florets by globally surveying gene expression in the UFM and LFM of maize ears. Both UFM and LFM expressed a broad set of genes, including genes previously implicated in floral development and/or meristem function. Approximately 3.5% of genes are differentially expressed between UFM and LFM (Figure 2G;Supplemental File S1), which is consistent with previous molecular and genetic analyses. At least two maize mutants differentially affect the upper and lower florets. In bearded-ear (bde) mutants, UFM are indeterminate whereas LFM initiate additional FMs and lose FM fate (Thompson et al., 2009). In restorer of fertility2 (rf2) mutants, stamens arrest in lower, but not upper florets (Liu et al., 2001). Microarray analysis indicates that ∼9% of genes are differentially expressed in equivalently staged anthers from the upper and lower florets (Skibbe et al., 2008); thus, floret-specific gene expression persists even in differentiated floral organs. These data support the model that UFM and LFM use distinct gene regulatory networks and have divergent developmental fates from the very earliest stages of development.
The divergent developmental fates of UFM and LFM may be due in part to their distinct ontogenies. The LFM is clearly an axillary meristem and associated with the formation of auxin maxima and with the novel expression of shoot and FM markers, such as knotted1 (kn1) and bde, respectively (Figure 1, H–N; Jackson et al., 1994; Gallavotti et al., 2008; Thompson et al., 2009). In contrast, the formation of the UFM is not associated with an auxin maximum, which supports the model that the UFM is not an axillary meristem but rather that the spikelet meristem is converted to the UFM (Gallavotti et al., 2008). We found the LFM was enriched for auxin-related genes (Figure 2, H and J; Supplemental Figure S3 and Supplemental File S2), which could reflect the axillary meristem identity of LFM, but not UFM.
Transcriptional regulatory networks also differ between upper and lower florets. One of the largest groups of DE genes identified in our analysis were transcriptional regulatory proteins, which included TF classes with key functions in plant development (i.e. TCP, WRKY, homeobox, AP2/ERF). These experiments were motivated in part by the bde mutant phenotype, which as previously mentioned, promotes FM determinacy in the upper floret and FM fate in the lower floret. bde encodes a MADS-box TF and we hypothesized that these floret-specific phenotypes were caused by disruption of distinct BDE-containing complexes in the UFM and LFM, resulting in the misregulation of different target genes (Thompson et al., 2009). Surprisingly, zmm8 and zmm14 were the only two DE MADS-box genes identified in our samples (Supplemental Figure S2 and Supplemental File S2), both of which were previously shown to be strongly UFM-enriched and have been hypothesized to act as upper floret selector genes (Cacharron et al., 1999). Recent analysis of zmm8;zmm14 double mutants, however, indicate that zmm8/zmm14 promote FM meristem determinacy in both florets and do not have floret-specific functions (Du et al., 2021). Although zmm8/zmm14 is highly UFM-enriched in our data, they are also expressed in LFM (Supplemental File S1). Combined with the fact that we did not identify clear candidates for lower floret selector genes, these data suggest that upper versus lower floret selector genes may not exist. We favor the hypothesis that the developmental history and anatomy of upper versus lower florets lead to physiological differences between the florets (i.e. energy availability or hormone status), which can cause floret-specific mutant phenotypes and ultimately determine UFM versus LFM fate.
Genes associated with growth repression are enriched in the lower FM
Plants must be able to alter growth and development in response to both internal and external cues, including energy status. Sugar, mainly in the form of glucose and fructose, is produced by photosynthesis in source tissues and transported as sucrose to sink tissues, such as developing seeds. Once localized to sink tissues, sucrose can be converted to glucose and used for chemical energy, as a structural component of cells (e.g. cell walls) or stored for later use. Sugar is also an important signaling molecule and functions in diverse processes. The lower floret is enriched for genes involved in growth repression, and our data suggests low sugar availability in the lower floret may contribute to this growth repression. Indeed, feeding sucrose through the flag leaf in wheat decreases floral abortion, suggesting that sugar signaling and homeostasis can regulate floral abortion in cereal crops (Ghiglione et al., 2008).
The conserved SnRK1 protein kinase (homologous to yeast Snf1 and animal AMPK1) is a key mechanism by which plants sense nutrient availability and maintain energy homeostasis. SnRK1 stimulates pathways that inhibit growth and increase catabolism in response to energy starvation (Baena-González et al., 2007). SnRK1 senses energy status primarily through repression by T6P, which is a proxy for carbon availability (Smeekens, 2015; Figueroa and Lunn, 2016). Our data suggest a model in which low sugar availability in the lower floret suppresses growth via the SnRK1 signaling pathway (Figure 7). The LFM is enriched for RNAs encoding two TPP enzymes (ra3/tpp10 and tpp3; Figure 6B), consistent RA3 localization at the UFM/LFM boundary (Satoh-Nagasawa et al., 2006; Klein et al., 2021). The UFM is enriched for tpp12, consistent with data showing that ra3 and tpp12 expression is inversely correlated (Claeys et al., 2019). In other developmental contexts, the RA3 (and other TPPs) primarily function in transcriptional regulation and not direct modulation of T6P levels (Claeys et al. 2019; Demesa-Arevalo et al. 2021). Regardless, RA3/TPPs likely regulate or respond to sugar levels, suggesting that sugar levels are carefully regulated in the spikelet.
Figure 7.
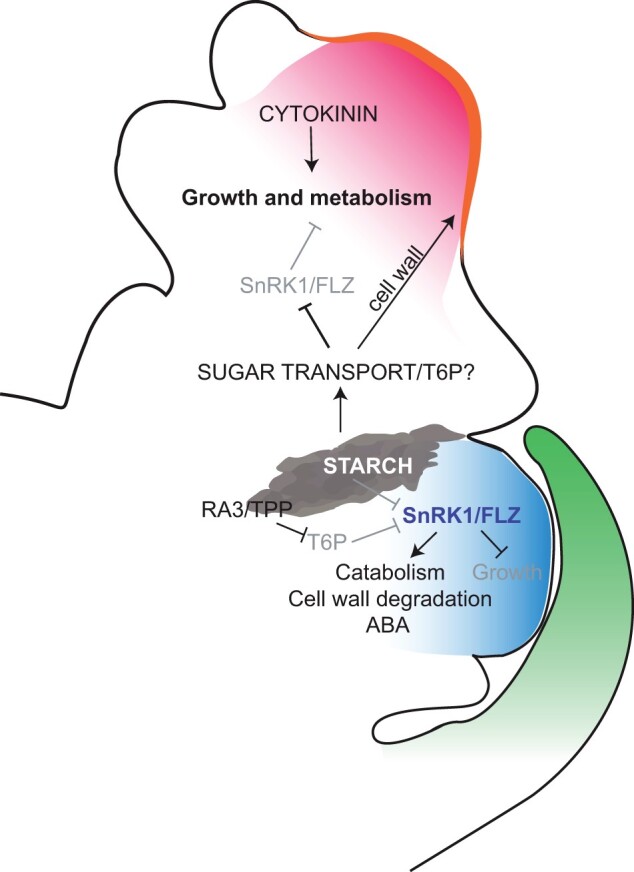
Model for growth regulation in developing florets. In UFM (red), high cytokinin and sugar availability promote growth and metabolism, including synthesis of new cell walls (orange). In LFM (blue), low sugar availability activates the SnRK1/FLZ pathway to repress growth and increase catabolism. Starch accumulates at the UFM/LFM boundary and may supply sugar to the UFM and/or be important for boundary formation.
Both the UFM and LFM express RNAs corresponding to all core components of the SnRK1 signaling pathway, most of which are not differentially expressed (Figure 6A). The LFM, however, showed enrichment of eight FLZ genes (Figure 6, A and B), which likely act as adaptors for SnRK1, and FLZ RNA levels respond to sugar, hormones and abiotic stress (Jamsheer and Laxmi, 2015; Jamsheer et al., 2018a). In addition to SnRK1 subunits, FLZ interacts with developmental regulators, including homologs of LFM-enriched genes (TCP, homeobox TFs, GAI, DELLA; Nietzsche et al., 2016; Jamsheer et al., 2019). Thus, LFM-enriched FLZ genes may direct SnRK1 to specific targets that function in floral development. The LFM also showed enrichment of genes involved in protein degradation and amino acid catabolism (Figure 2, H and I; Supplemental Figure S4), consistent with active SnRK1 in LFM. High T6P levels have been correlated with increased expression of genes involved in primary metabolism (Oszvald et al., 2018), and indeed, the UFM was enriched for genes involved in RNA processing and protein biosynthesis (Figure 2H;Supplemental File S2).
T6P also promotes starch accumulation, the major storage carbohydrate in plants (Figueroa and Lunn, 2016). Starch gradually accumulates during spikelet development (Figure 6, C–G), presumably increasing the strength of the inflorescence as a sink tissue. Starch does not accumulate throughout the spikelet, but rather accumulates in a defined region at the boundary between the upper and lower floret and appears to be excluded from the lower floret. In animals, the SnRK1 homolog, AMPK1, binds to and is negatively regulated by glycogen (Janzen et al., 2018), which is analogous to starch in plants, raising the intriguing possibility that SnRK1 directly binds to and is regulated by starch. Starch accumulation is similar in spikelets where the lower floret does not abort (Supplemental Figure S8, N–W), suggesting that low starch does not directly signal floral abortion, but may be important for general growth repression of the lower floret.
In addition to its function in sugar signaling and homeostasis within a tissue, T6P can affect sugar utilization and distribution at the whole plant level and is a key regulator of the source/sink balance (Figueroa and Lunn, 2016). The interaction between source and sink tissues affects timing of senescence and many DEGs in our data set have been implicated in senescence. For example, the UFM-enriched senescence-inducible chloroplast stay-green protein, GRMZM2G091837, is associated with delayed senescence (Li et al., 2020), whereas the LFM-enriched Malate synthase 1 (Mas1/GRMZM2G102183) and Arabidopsis FLZ genes are induced in senescing tissues (Yandeau-Nelson et al., 2011; Jamsheer and Laxmi, 2015; Jamsheer et al., 2018a). Furthermore, we noted an overlap between genes that regulate the natural variation of senescence in maize (Sekhon et al. 2019) and our DEGs, including glutathione S-transferases (GSTs; five LFM-enriched, two UFM-enriched, three senescence-associated; Supplemental Figure S4); indeed gst41/GRZM2G097989 overlapped both lists. The cell wall has also emerged as an important contributor to senescence, which can act as a secondary sink and may affect the source/sink balance (Sekhon et al., 2012, 2019). The Arabidopsis senescence-inducible promoter, pSAG12, is sufficient to drive transcription in the lower floret and indeed pSAG12-induced expression of the cytokinin biosynthesis gene, isopentenyl transferase1 (ipt1) in the lower floret inhibits floret abortion (Young et al., 2004). While not directly implicated in senescence, we also noted the LFM was enriched for two Rapid Alkalinization Factor (RALF)/RALF-like (RALFL) peptides, which are associated with repressed growth (Blackburn et al., 2020). We propose the LFM executes a senescence-like program to repress growth in the lower floret (Figure 7).
Plant growth requires the synthesis of new cell walls as cells divide and modification of existing cell walls to allow for cell expansion. In eudicots, pectin composition and modification are developmentally regulated and have been implicated in multiple aspects of plant growth and development (Saffer, 2018). Pectins are complex galacturonic acid-rich polysaccharides, of which HG is the most abundant (Harholt et al., 2010). HG is deposited in the cell wall in a highly methylesterified form and can be demethylesterified by pectin methylesterases; in Arabidopsis, demethylesterification of HG regulates primordia initiation and phyllotaxy (Peaucelle et al., 2008, 2011). Grass cell walls contain substantially less pectin than eudicot cells walls (∼5% in grasses versus 20%–35% in eudicots) (Vogel, 2008) and what role, if any, pectin modification plays in primordia initiation and phyllotaxy in grasses is unclear. Our data show that demethylesterified pectin is clearly associated with floral organ primordia initiation in the UFM (Figure 4, C–H), strongly suggesting that pectin’s role in organ initiation is conserved in grasses. Furthermore, we show that demethylesterified pectin strongly accumulates in aborting carpels (Figure 5). While it is unclear if this pectin demethylesterification is a trigger for or the result of carpel abortion, these data clearly show that pectin is dynamically modified during maize floral development and may be critical for floret fertility.
Does the upper/lower FM boundary affect FM activity?
Boundary regions between meristems and initiating primordia are essential to separate groups of cells with different developmental fates and can also affect the activity of adjacent cell populations (Wang et al., 2016; Richardson and Hake 2018). In grass inflorescences, meristem determinacy is controlled by groups of genes expressed in regions adjacent to the meristem that form boundaries with organ primordia. For example, the ramosa regulatory module functions at the base of spikelet pair meristem and is required to restrict its determinacy (Eveland et al., 2014). Similarly, bd1 and indeterminate spikelet1 (ids1) transcription factors are expressed at the base of the spikelet meristems and limit determinacy (Chuck et al., 1998, 2002). In barley compositum1 (com1) mutants, determinate spikelets on the main rachis are transformed into indeterminate branches due to defective boundary formation. The ra3 ortholog, along with other sugar and cell wall-related genes are misexpressed in com1 mutants, suggesting that sugar signaling and cell wall changes are critical for boundary formation (Poursarebani et al., 2020). Thus, boundary regions adjacent to meristems may function as signaling centers that regulate meristem activity (Whipple, 2017), although the mechanism is unclear.
Our data suggest that a similar boundary program may function at the UFM/LFM boundary. First, RA3 is localized to the boundary (Satoh-Nagasawa et al., 2006; Klein et al., 2021) and overlaps with starch accumulation (Figure 6, C–G). We also identified the com1 ortholog, Wab1/bad1, as a LFM-enriched gene in our samples (Supplemental Figure S2 and Supplemental File S2). Because of the small size of LFM, we likely isolated boundary genes in our LFM samples that were excluded from UFM samples in which we were able to isolate the “tips” of the meristems. Our data also suggest cell walls are differentially regulated in UFM and LFM (Figure 4). Indeed, pectate lyase was localized to a discrete domain at the UFM/LFM boundary (Figure 3I;Supplemental Figure S7) and boundary regions are often characterized by stiffer cell walls (Richardson and Hake, 2018). Arginine decarboxylase1 (adc1), a key enzyme required for synthesis of the polyamines, is also expressed at the UFM/LFM boundary (Figure 3J;Supplemental Figure S7). Polyamines have diverse functions in plants ranging from stress responses to growth and development, including flower bud formation (Chen et al., 2018). ba1 is also expressed in the domain at the UFM/LFM boundary and later below the palea (Gallavotti et al., 2004). Finally, BBTI is expressed in the upper floret at the UFM/LFM boundary and at the base of the palea (Figure 3H;Supplemental Figure S7). The palea expression of BBTI and ba1 is particularly intriguing; in barley, com1 is also expressed in palea and com1 mutants have enlarged palea cells with thinner cell walls (Poursarebani et al., 2020). Thus, palea may also have important boundary functions or, alternatively, boundary regulatory modules may be redeployed in palea development.
Understanding the genetic and physiological processes that regulate floret abortion and sterility is a necessary first step to engineer maize and other cereal crops with increased floret fertility. Our data suggest that upper versus lower FM fate in maize is not determined by master regulatory genes, but rather by differences in core physiological processes that coordinate sugar availability, energy homeostasis, and plant growth. This foundational work provides important insights into the downstream processes that likely regulate floret abortion and provides a rich set of candidate genes to potentially increase floret fertility in cereal crops and enhance yield.
Materials and methods
Laser capture microdissection, RNA isolation, and amplification
Ear primorida (1–2 cm) were dissected from greenhouse grown (16-h light at 27°C, 8-h dark at 21°C) maize (Z. mays) B73 plants and immediately fixed and embedded for LCM as previously described (Takacs et al., 2012). Longitudinal sections (8 μm) were made using a Reichert-Jung (Leica) 2030 rotary microtome and mounted on Zeiss Membrane Slide (1.0 polyethylene naphthalate); LCM was performed using the Zeiss PALM MicroBeam System. A minimum 350,000 μm2 tissue was dissected for each of six replicates (three UFM and three LFM; 1–2 ear primordia used per replicate). Because LFM are developmentally delayed relative to UFM, LFM samples were dissected from later stage spikelets than UFM samples. Total RNA was extracted using Arcturus PicoPure RNA Isolation kit (Applied Biosystems) and DNase treated using the Qiagen RNase-free DNase set. RNA was amplified (Epicenter TargetAmp 2-Round aRNA Amplification Kit 2.0, Epicentre Biotechnologies), DNase-treated (RapidOut DNA Removal kit, ThermoFisher Scientific) and purified (RNeasy MinElute Cleanup Kit, Qiagen). Quality and size of aRNA was assessed using Bioanalyzer 2100 (Agilent Technologies).
RNA-seq and data analysis
Library construction (TruSeq Stranded mRNA Sample Prep LS kit) and RNA-seq was performed on an Illumina HiSeq 2500 system by Genomic Sciences Laboratory at North Carolina State University. Raw data were trimmed and low-quality reads were filtered out using trim_galore. Reads were mapped to the maize genome (V3) using Tophat2 (v2.1.0; Kim et al., 2013) with parameters: –library-type fr-secondstrand –b2-very-sensitive -i 20 and quantified using the htseq-count package with default parameters except: –stranded = yes (Anders et al., 2015). Count tables were analyzed using DESeq2 in the R environment for differential expression analysis (Love et al., 2014). Genes with a minimum read count of 10 in at least two biological replicates, fold change ≥2 and adjusted P <0.05, were considered differentially expressed. PCA was performed using DESeq2 (Love et al., 2014) in the R environment and correlation analysis was performed using R package “psych” pairs.panels function.
GO was analyzed using g:Profiler (Raudvere et al., 2019) with default options except statistical settings: Benjamini–Hochberg false discovery rate (FDR) and all known genes. Gene enrichment maps were generated using Cytoscape (version 3.7.1) plug-in, Enrichment Map (Merico et al., 2010), with default options except FDR q-value cutoff = 0.05, connectivity = second degree sparse, and size of functional category = 1–5,000. Functional groups were predicted using MapMan 3.6.0RC1 (X4 annotation), with the maize v3 mapping file (retrieved from Mercator4 Fasta validator with the protein option; Schwacke et al., 2019). CornCyc 9.0 was used to predict metabolic pathways (Schläpfer et al., 2017). Gene ID description analysis was analyzed with g:Profiler g:Convert functional tab (Raudvere et al., 2019).
RNA in situ hybridization and histochemistry
Inflorescence primordia (1–2 cm) for RNA in situ hybridization, lignin/cellulose staining, and immunohistochemistry, were fixed and embedded as described in Thompson et al. (2009) and sectioned (10 µm) using a Microm HM315 Microtome. Inflorescence primordia for ruthenium red and starch staining were directly frozen with optimal cutting temperature embedding medium on dry ice and sectioned (60–80 µm) with a Microm HM550 Cryostat Microtome at −20°C.
RNA in situ hybridization was performed as described previously (Jackson, 1991), with the following modifications. Pronase digestion was performed for 25 min at 37°C; incubated in blocking solution (Sigma Roche) for 1 h at room temperature before incubation with anti-DIG antibody (1:4,000–5,000 in blocking solution). After antibody incubation, slides were washed with Buffer A without Triton X-100. Slides were imaged using an Olympus BX-41 compound light microscope and processed using Adobe Photoshop. Probes were generated as described in Bortiri et al. (2006), using primer sequences listed in Supplemental Table S1.
For pectin, lignin, and cellulose staining, cryosections and rehydrated sections were stained as previously described (Gunawardena et al., 2007; Pradhan Mitra and Loqué, 2014) except staining was performed at room temperature in the dark. Phloroglucinol–HCl-stained samples were immediately imaged with Olympus BX-41 compound light microscope. Calcofluor white/fluorescent brightener 28-stained samples were visualized with an Olympus IX2-DSU Confocal Compound Light Microscope using eDAPI or emDAPI filters. Lugol’s Iodine Solution (Electron Microscopy Sciences) was used to stain starch, washed with 90% isopropanol or ethanol and mounted with histoclear. Maize stem tissue was processed in parallel for all stains.
Immunofluorescence labeling was modified from (Xue et al., 2013). Briefly, rehydrated sections were blocked using 1× PBS with 5% BSA (w/v) for 30 min at room temperature and stained with primary antibodies, LM19 and LM20 (Kerafast, diluted 1:10) overnight at 4°C. After washing in 1× PBS (three washes, 5 min each) sections were incubated for 2 h at room temperature with goat anti-Rat IgG (H + L) Alexa Fluor 488 secondary antibody (ThermoFisher Scientific, diluted 1:200). Antibodies were diluted in 1× PBS with 5% BSA (w/v) and incubated in the dark. Sections were washed three times (5 min each) with 1× PBS, incubated with 0.02% Toluidine Blue O (1×PBS; w/v) for 5 min to quench autofluorescence, and rinsed twice with 1× PBS prior to mounting with antifade medium (Hinnant et al., 2017). Slides were stored at 4°C in the dark prior to imaging with a Zeiss LSM700 laser scanning microscope (488-nm laser, Z-stacks with 1-μm optical sections). Negative controls lacking primary antibody were processed in parallel. Images were processed in Adobe Photoshop and false colored using the Blue Orange icb look up table in ImageJ (Schneider et al., 2012).
Accession numbers
RNA sequencing data were deposited into NCBI Sequence Read Archive under accession number PRJNA717335.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Cluster and reproducibility analysis of LFM and UFM biological replicates.
Supplemental Figure S2. Summary of DEGs in the MapMan RNA biosynthesis functional group.
Supplemental Figure S3. Summary of DEGs in the MapMan phytohormone functional group.
Supplemental Figure S4. Summary of DEGs in the MapMan protein modification (A) and degradation (B) functional groups.
Supplemental Figure S5. RNA in situ hybridization of AC217050.4_FG006, chr101/GRMZM2G177165, and GRMZM2G101682 in developing inflorescences.
Supplemental Figure S6. RNA in situ hybridization of histone H1-like/GRMZM2G069911 and XTH9 homolog/GRMZM2G180870 in developing inflorescences.
Supplemental Figure S7. RNA in situ hybridization of BBTI, adc1, and pectate lyase in developing inflorescences.
Supplemental Figure S8. Histochemical staining in maize stems and spikelets.
Supplemental Figure S9. The UFM and LFM have distinct cell wall composition regardless of LFM fate.
Supplemental Table S1. Primers used in this study.
Supplemental File S1. Summary of mapping and differential analysis.
Supplemental File S2. Summary of GO, MapMan, and CornCyc analysis.
Supplementary Material
Acknowledgments
We are thankful to Julie Marik, Cathy Herring, and Jim Holland for greenhouse and field support, Cindy Kukoly for technical assistance with LCM, Ross Sozzani and Angel de Balaguer for help with RNA-seq and early analysis, Katherine Novitzky for technical discussion with RNA in situ hybridization, and Allison Beachum, Tom Fink, and Elizabeth Ables for help with microscopy. Critical friend Madelaine Bartlett provided invaluable discussions and comments as well as ra3; gt1 seeds. We also thank Clint Whipple, Patrick Horn, and two anonymous reviewers for helpful comments on the manuscript.
Funding
This work was supported by grants from the North Carolina Biotechnology Center (#2011-BRG-1213) and the National Science Foundation (IOS-1148971).
Conflict of interest statement. None declared.
K.N., Q.D, H.Y., and B.T designed the research; K.N., Q.D., and H.Y. performed the experiments and analyzed the data with B.T. H.Y. and B.T. wrote the manuscript.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plphys/pages/general-instructions) is: Beth Thompson (thompsonb@ecu.edu).
References
- Acosta IF, Laparra H, Romero SP, Schmelz E, Hamberg M, Mottinger JP, Moreno MA, Dellaporta SL (2009) tasselseed1 is a lipoxygenase affecting jasmonic acid signaling in sex determination of maize. Science 323:262–265 [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai K, Satoh N, Sasaki H, Satoh H, Nagato Y (2002) A rice heterochronic mutant, mori1, is defective in the juvenile-adult phase change. Development 129:265–273 [DOI] [PubMed] [Google Scholar]
- Baena-González E, Lunn JE (2020) SnRK1 and trehalose 6-phosphate – two ancient pathways converge to regulate plant metabolism and growth. Curr Opin Plant Biol 55:52–59 [DOI] [PubMed] [Google Scholar]
- Baena-González E, Rolland F, Thevelein JM, Sheen J (2007) A central integrator of transcription networks in plant stress and energy signalling. Nature 448:938–942 [DOI] [PubMed] [Google Scholar]
- Bai F, Reinheimer R, Durantini D, Kellogg EA, Schmidt RJ (2012) TCP transcription factor, BRANCH ANGLE DEFECTIVE 1 (BAD1), is required for normal tassel branch angle formation in maize. Proc Natl Acad Sci USA 109:12225–12230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett ME, Thompson B (2014) Meristem identity and phyllotaxis in inflorescence development. Front Plant Sci 5:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn MR, Haruta M, Moura DS (2020) Twenty years of progress in physiological and biochemical investigation of RALF peptides. Plant Physiol 182:1657–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomblies K, Wang R-L, Ambrose BA, Schmidt RJ, Meeley RB, Doebley J (2003) Duplicate FLORICAULA/LEAFY homologs zfl1 and zfl2 control inflorescence architecture and flower patterning in maize. Development 130:2385–2395 [DOI] [PubMed] [Google Scholar]
- Bortiri E, Chuck G, Vollbrecht E, Rocheford T, Martienssen R, Hake S (2006) ramosa2 encodes a LATERAL ORGAN BOUNDARY domain protein that determines the fate of stem cells in branch meristems of maize. Plant Cell 18:574–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabib E, Leloir LF (1958) The biosynthesis of trehalose phosphate. J Biol Chem 231:259–275 [PubMed] [Google Scholar]
- Cacharron J, Saedler H, Theissen G (1999) Expression of MADS box genes ZMM8 and ZMM14 during inflorescence development of Zea mays discriminates between the upper and the lower floret of each spikelet. Dev Genes Evol 209:411–420 [DOI] [PubMed] [Google Scholar]
- Cavalier DM, Lerouxel O, Neumetzler L, Yamauchi K, Reinecke A, Freshour G, Zabotina OA, Hahn MG, Burgert I, Pauly M, et al. (2008) Disrupting two Arabidopsis thaliana Xylosyltransferase genes results in plants deficient in xyloglucan, a major primary cell wall component. Plant Cell 20:1519–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Shao Q, Yin L, Younis A, Zheng B (2018) Polyamine function in plants: metabolism, regulation on development, and roles in abiotic stress responses. Front Plant Sci 9:1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PC, Greyson RI, Walden DB (1983) Organ initiation and the development of unisexual flowers in the tassel and ear of Zea mays. Am J Bot 70:450–462 [Google Scholar]
- Chen K-M, Wang F, Wang Y-H, Chen T, Hu Y-X, Lin J-X (2006) Anatomical and chemical characteristics of foliar vascular bundles in four reed ecotypes adapted to different habitats. Flora 201:555–569 [Google Scholar]
- Chuck G, Meeley RB, Hake S (1998) The control of maize spikelet meristem fate by the APETALA2-like gene indeterminate spikelet1. Genes Dev 12:1145–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuck G, Muszynski M, Kellogg E, Hake S, Schmidt RJ (2002) The control of spikelet meristem identity by the branched silkless1 gene in maize. Science 298:1238–1241 [DOI] [PubMed] [Google Scholar]
- Claeys H, Vi SL, Xu X, Satoh-Nagasawa N, Eveland AL, Goldshmidt A, Feil R, Beggs GA, Sakai H, Brennan RG, et al. (2019) Control of meristem determinacy by trehalose 6-phosphate phosphatases is uncoupled from enzymatic activity. Nat Plants 5:352–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong A, Calderon-Urrea A, Dellaporta SL (1993) Sex determination gene TASSELSEED2 of maize encodes a short-chain alcohol dehydrogenase required for stage-specific floral organ abortion. Cell 74:757–768 [DOI] [PubMed] [Google Scholar]
- Demesa-Arevalo E, Abraham-Juarez MJ, Xu X, Bartlett M, Jackson D (2021) Maize RAMOSA3 accumulates in nuclear condensates enriched in RNA POLYMERASE II isoforms during the establishment of axillary meristem determinacy. bioRxiv: 10.1101/2021.04.06.438639
- Du Y, Lunde C, Li Y, Jackson D, Hake S, Zhang Z (2021) Gene duplication at the Fascicled ear1 locus controls the fate of inflorescence meristem cells in maize. Proc Natl Acad Sci USA 118: e2019218118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eveland AL, Goldshmidt A, Pautler M, Morohashi K, Liseron-Monfils C, Lewis MW, Kumari S, Hiraga S, Yang F, Unger-Wallace E, et al. (2014) Regulatory modules controlling maize inflorescence architecture. Genome Res 24:431–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa CM, Lunn JE (2016) A tale of two sugars: trehalose 6-phosphate and sucrose. Plant Physiol 172:7–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster T, Veit B, Hake S (1999a) Mosaic analysis of the dominant mutant, Gnarley1-R, reveals distinct lateral and transverse signaling pathways during maize leaf development. Development 126:305–313 [DOI] [PubMed] [Google Scholar]
- Foster T, Yamaguchi J, Wong BC, Veit B, Hake S (1999b) Gnarley1 is a dominant mutation in the knox4 homeobox gene affecting cell shape and identity. Plant Cell 11:1239–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallavotti A, Yang Y, Schmidt RJ, Jackson D (2008) The relationship between auxin transport and maize branching. Plant Physiol 147:1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallavotti A, Zhao Q, Kyozuka J, Meeley RB, Ritter MK, Doebley JF, Pè ME, Schmidt RJ (2004) The role of barren stalk1 in the architecture of maize. Nature 432:630–635 [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium (2015) Gene ontology consortium: going forward. Nucleic Acids Res 43:D1049–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiglione HO, Gonzalez FG, Serrago R, Maldonado SB, Chilcott C, Curá JA, Miralles DJ, Zhu T, Casal JJ (2008) Autophagy regulated by day length determines the number of fertile florets in wheat. Plant J 55:1010–1024 [DOI] [PubMed] [Google Scholar]
- Gunawardena AHLAN, Arunika H L A, Greenwood JS, Dengler NG (2007) Cell wall degradation and modification during programmed cell death in lace plant, Aponogeton madagascariensis (Aponogetonaceae). Am J Bot 94:1116–1128 [DOI] [PubMed] [Google Scholar]
- Harholt J, Suttangkakul A, Scheller HV (2010) Biosynthesis of pectin. Plant Physiol 153:384–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay A, Hake S (2004) The dominant mutant wavy auricle in blade1 disrupts patterning in a lateral domain of the maize leaf. Plant Physiol 135:300–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnant TD, Alvarez AA, Ables ET (2017) Temporal remodeling of the cell cycle accompanies differentiation in the Drosophila germline. Dev Biol 429:118–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard L, McSteen P, Doebley J, Hake S (2002) Expression patterns and mutant phenotype of teosinte branched1 correlate with growth suppression in maize and teosinte. Genetics 162:1927–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Xu Y, Yu C, He K, Tang Q, Jia C, He G, Wang X, Kong Y, Zhou G (2017) Transcriptome analysis of genes involved in secondary cell wall biosynthesis in developing internodes of Miscanthus lutarioriparius. Sci Rep 7:9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda-Kawakatsu K, Yasuno N, Oikawa T, Iida S, Nagato Y, Maekawa M, Kyozuka J (2009) Expression level of ABERRANT PANICLE ORGANIZATION1 determines rice inflorescence form through control of cell proliferation in the meristem. Plant Physiol 150:736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish EE (1997) Experimental analysis of tassel development in the maize mutant tassel seed 6. Plant Physiol 114:817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish EE, Nelson T (1989) Sex determination in monoecious and dioecious plants. Plant Cell 1:737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson D (1991) In-situ hybridisation in plants. In DJ Bowles, SJ Gurr and P McPherson, eds, Molecular Plant Pathology: A Practical Approach. Oxford University Press, Oxford pp 163–174
- Jackson D, Veit B, Hake S (1994) Expression of maize KNOTTED1 related homeobox genes in the shoot apical meristem predicts patterns of morphogenesis in the vegetative shoot. Development 120:405–413 [Google Scholar]
- Jamsheer KM, Sharma M, Singh D, Mannully CT, Jindal S, Shukla BN, Laxmi A (2018a) FCS-like zinc finger 6 and 10 repress SnRK1 signalling in Arabidopsis. Plant J 94:232–245 [DOI] [PubMed] [Google Scholar]
- Jamsheer KM, Shukla BN, Jindal S, Gopan N, Mannully CT, Laxmi A (2018b) The FCS-like zinc finger scaffold of the kinase SnRK1 is formed by the coordinated actions of the FLZ domain and intrinsically disordered regions. J Biol Chem 293:13134–13150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzen NR, Whitfield J, Hoffman NJ (2018) Interactive roles for AMPK and glycogen from cellular energy sensing to exercise metabolism. Int J Mol Sci 19:3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston R, Wang M, Sun Q, Sylvester AW, Hake S, Scanlon MJ (2014) Transcriptomic analyses indicate that maize ligule development recapitulates gene expression patterns that occur during lateral organ initiation. Plant Cell 26:4718–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegstra K (2010) Plant cell walls. Plant Physiol 154:483–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JC, Laparra H, Calderón-Urrea A, Mottinger JP, Moreno MA, Dellaporta SL (2007) Cell cycle arrest of stamen initials in maize sex determination. Genetics 177:2547–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein H, Gallagher J, Demesa-Arevalo E, Abraham-Juárez MJ, Heeney M, Feil R, Lunn JE, Xiao Y, Chuck G, Whipple C, et al. (2021) Recruitment of an ancient branching program to suppress carpel development in maize flowers. bioRxiv. 10.1101/2021.09.03.458935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamsheer KM, Jindal S, Laxmi A (2019) Evolution of TOR–SnRK dynamics in green plants and its integration with phytohormone signaling networks. J Exp Bot 70:2239–2259 [DOI] [PubMed] [Google Scholar]
- Jamsheer KM, Laxmi A (2014) DUF581 is plant specific FCS-like zinc finger involved in protein-protein interaction. PLoS One 9:e99074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamsheer KM, Laxmi A (2015) Expression of Arabidopsis FCS-Like Zinc finger genes is differentially regulated by sugars, cellular energy level, and abiotic stress. Front Plant Sci 6:746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roux LG, Kellogg EA (1999) Floral development and the formation of unisexual spikelets in the Andropogoneae (Poaceae). Am J Bot 86:354–366 [PubMed] [Google Scholar]
- Lewis MW, Bolduc N, Hake K, Htike Y, Hay A, Candela H, Hake S (2014) Gene regulatory interactions at lateral organ boundaries in maize. Development 141:4590–4597 [DOI] [PubMed] [Google Scholar]
- Liu C, Thong Z, Yu H (2009) Coming into bloom: the specification of floral meristems. Development 136:3379–3391 [DOI] [PubMed] [Google Scholar]
- Liu F, Cui X, Horner HT, Weiner H, Schnable PS (2001) Mitochondrial aldehyde dehydrogenase activity is required for male fertility in maize. Plant Cell 13:1063–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Tang J, Srivastava R, Bassham DC, Howell SH (2020) The transcription factor bZIP60 links the unfolded protein response to the heat stress response in maize. Plant Cell 32:3559–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde C, Kimberlin A, Leiboff S, Koo AJ, Hake S (2019) Tasselseed5 overexpresses a wound-inducible enzyme, ZmCYP94B1, that affects jasmonate catabolism, sex determination, and plant architecture in maize. Commun Biol 2:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, Bader GD (2010) Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5:e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nietzsche M, Landgraf R, Tohge T, Börnke F (2016) A protein–protein interaction network linking the energy-sensor kinase SnRK1 to multiple signaling pathways in Arabidopsis thaliana. Curr Plant Biol 5:36–44 [Google Scholar]
- Nietzsche M, Schießl I, Börnke F (2014) The complex becomes more complex: protein-protein interactions of SnRK1 with DUF581 family proteins provide a framework for cell- and stimulus type-specific SnRK1 signaling in plants. Front Plant Sci 5:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oszvald M, Primavesi LF, Griffiths CA, Cohn J, Basu SS, Nuccio ML, Paul MJ (2018) Trehalose 6-phosphate regulates photosynthesis and assimilate partitioning in reproductive tissue. Plant Physiol 176:2623–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peaucelle A, Braybrook SA, Le Guillou L, Bron E, Kuhlemeier C, Höfte H (2011) Pectin-induced changes in cell wall mechanics underlie organ initiation in Arabidopsis. Curr Biol 21:1720–1726 [DOI] [PubMed] [Google Scholar]
- Peaucelle A, Louvet R, Johansen JN, Höfte H, Laufs P, Pelloux J, Mouille G (2008) Arabidopsis phyllotaxis is controlled by the methyl-esterification status of cell-wall pectins. Curr Biol 18:1943–1948 [DOI] [PubMed] [Google Scholar]
- Pesquet E, Zhang B, Gorzsás A, Puhakainen T, Serk H, Escamez S, Barbier O, Gerber L, Courtois-Moreau C, Alatalo E, et al. (2013) Non-cell-autonomous postmortem lignification of tracheary elements in Zinnia elegans. Plant Cell 25:1314–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poursarebani N, Trautewig C, Melzer M, Nussbaumer T, Lundqvist U, Rutten T, Schmutzer T, Brandt R, Himmelbach A, Altschmied L, et al. (2020) COMPOSITUM 1 contributes to the architectural simplification of barley inflorescence via meristem identity signals. Nat Commun 11:5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan Mitra P, Loqué D (2014) Histochemical staining of Arabidopsis thaliana secondary cell wall elements. J Vis Exp 87: 51381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J (2019) g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 47:W191–W198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson AE, Hake S (2018) Drawing a line: grasses and boundaries. Plants 8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roxrud I, Lid SE, Fletcher JC, Schmidt EDL, Opsahl-Sorteberg H-G (2007) GASA4, one of the 14-member Arabidopsis GASA family of small polypeptides, regulates flowering and seed development. Plant Cell Physiol 48:471–483 [DOI] [PubMed] [Google Scholar]
- Ruzin SE (1999) Plant Microtechnique and Microscopy. Oxford University Press, Oxford [Google Scholar]
- Saffer AM (2018) Expanding roles for pectins in plant development. J Integr Plant Biol 60:910–923 [DOI] [PubMed] [Google Scholar]
- Sakuma S, Schnurbusch T (2020) Of floral fortune: tinkering with the grain yield potential of cereal crops. New Phytol 225:1873–1882 [DOI] [PubMed] [Google Scholar]
- Sampathkumar A, Peaucelle A, Fujita M, Schuster C, Persson S, Wasteneys GO, Meyerowitz EM (2019) Primary wall cellulose synthase regulates shoot apical meristem mechanics and growth. Development 146:dev179036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh-Nagasawa N, Nagasawa N, Malcomber S, Sakai H, Jackson D (2006) A trehalose metabolic enzyme controls inflorescence architecture in maize. Nature 441:227–230 [DOI] [PubMed] [Google Scholar]
- Satterlee JW, Strable J, Scanlon MJ (2020) Plant stem-cell organization and differentiation at single-cell resolution. Proc Natl Acad Sci USA 117:33689–33699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schläpfer P, Zhang P, Wang C, Kim T, Banf M, Chae L, Dreher K, Chavali AK, Nilo-Poyanco R, Bernard T, et al. (2017) Genome-wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiol 173:2041–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwacke R, Ponce-Soto GY, Krause K, Bolger AM, Arsova B, Hallab A, Gruden K, Stitt M, Bolger ME, Usadel B (2019) MapMan4: a refined protein classification and annotation framework applicable to multi-omics data analysis. Mol Plant 12:879–892 [DOI] [PubMed] [Google Scholar]
- Sekhon RS, Childs KL, Santoro N, Foster CE, Buell CR, de Leon N, Kaeppler SM (2012) Transcriptional and metabolic analysis of senescence induced by preventing pollination in maize. Plant Physiol 159:1730–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhon RS, Saski C, Kumar R, Flinn BS, Luo F, Beissinger TM, Ackerman AJ, Breitzman MW, Bridges WC, de Leon N, et al. (2019) Integrated genome-scale analysis identifies novel genes and networks underlying senescence in maize. Plant Cell 31:1968–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibbe DS, Wang X, Borsuk LA, Ashlock DA, Nettleton D, Schnable PS (2008) Floret-specific differences in gene expression and support for the hypothesis that tapetal degeneration of Zea mays L. occurs via programmed cell death. J Genet Genomics 35:603–616 [DOI] [PubMed] [Google Scholar]
- Smeekens S (2015) From leaf to kernel: trehalose-6-phosphate signaling moves carbon in the field. Plant Physiol 169:912–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs EM, Li J, Du C, Ponnala L, Janick-Buckner D, Yu J, Muehlbauer GJ, Schnable PS, Timmermans MCP, Sun Q, et al. (2012) Ontogeny of the maize shoot apical meristem. Plant Cell 24:3219–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BE, Bartling L, Whipple C, Hall DH, Sakai H, Schmidt R, Hake S (2009) bearded-ear encodes a MADS box transcription factor critical for maize floral development. Plant Cell 21:2578–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BE, Hake S (2009) Translational biology: from Arabidopsis flowers to grass inflorescence architecture. Plant Physiol 149:38–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torode TA, O’Neill R, Marcus SE, Cornuault V, Pose S, Lauder RP, Kračun SK, Rydahl MG, Andersen MCF, Willats WGT, et al. (2018) Branched pectic galactan in phloem-sieve-element cell walls: implications for cell mechanics. Plant Physiol 176:1547–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai AY-L, Gazzarrini S (2014) Trehalose-6-phosphate and SnRK1 kinases in plant development and signaling: the emerging picture. Front Plant Sci 5:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhertbruggen Y, Marcus SE, Haeger A, Ordaz-Ortiz JJ, Knox JP (2009) An extended set of monoclonal antibodies to pectic homogalacturonan. Carbohydr Res 344:1858–1862 [DOI] [PubMed] [Google Scholar]
- Vogel J (2008) Unique aspects of the grass cell wall. Curr Opin Plant Biol 11:301–307 [DOI] [PubMed] [Google Scholar]
- Wang F, Yuan Z, Zhao Z, Li C, Zhang X, Liang H, Liu Y, Xu Q, Liu H (2020) Tasselseed5 encodes a cytochrome C oxidase that functions in sex determination by affecting jasmonate catabolism in maize. J Integr Plant Biol 62:247–255 [DOI] [PubMed] [Google Scholar]
- Wang Q, Hasson A, Rossmann S, Theres K (2016) Divide et impera: boundaries shape the plant body and initiate new meristems. New Phytol 209:485–498 [DOI] [PubMed] [Google Scholar]
- Whipple CJ (2017) Grass inflorescence architecture and evolution: the origin of novel signaling centers. New Phytol 216:367–372 [DOI] [PubMed] [Google Scholar]
- Wingler A (2002) The function of trehalose biosynthesis in plants. Phytochemistry 60:437–440 [DOI] [PubMed] [Google Scholar]
- Xue J, Bosch M, Paul Knox J (2013) Heterogeneity and glycan masking of cell wall microstructures in the stems of miscanthus x giganteus, and its parents M. sinensis and M. sacchariflorus. PLoS One 8:e82114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandeau-Nelson MD, Laurens L, Shi Z, Xia H, Smith AM, Guiltinan MJ (2011) Starch-branching enzyme IIa is required for proper diurnal cycling of starch in leaves of maize. Plant Physiol 156:479–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young TE, Geisler-Lee J, Gallie DR (2004) Senescence-induced expression of cytokinin reverses pistil abortion during maize flower development. Plant J 38:910–922 [DOI] [PubMed] [Google Scholar]
- Zhang D, Sun W, Singh R, Zheng Y, Cao Z, Li M, Lunde C, Hake S, Zhang Z (2018) GRF-interacting factor1 regulates shoot architecture and meristem determinacy in maize. Plant Cell 30:360–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, von Mogel KJH, Lor VS, Hirsch CN, De Vries B, Kaeppler HF, Tracy WF, Kaeppler SM (2019) Maize sugary enhancer1 (se1) is a gene affecting endosperm starch metabolism. Proc Natl Acad Sci USA 116:20776–20785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C, Xu H, Ye S, Wang S, Li L, Zhang S, Wang X (2015) Gibberellic acid-stimulated Arabidopsis6 serves as an integrator of gibberellin, abscisic acid, and glucose signaling during seed germination in Arabidopsis. Plant Physiol 169:2288–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong R, Cui D, Ye Z (2019) Secondary cell wall biosynthesis. New Phytol 221:1703–1723 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.