

Abstract
The nuclear pore complex (NPC) is a multi-protein complex that regulates the trafficking of macromolecules between the nucleus and cytoplasm. Genetic variants in components of the NPC have been shown to cause a range of neurological disorders, including intellectual disability and microcephaly. Translocated promoter region, nuclear basket protein (TPR) is a critical scaffolding element of the nuclear facing interior of the NPC. Here, we present two siblings with biallelic variants in TPR who present with a phenotype of microcephaly, ataxia and severe intellectual disability. The variants result in a premature truncation variant, and a splice variant leading to a 12-amino acid deletion respectively. Functional analyses in patient fibroblasts demonstrate significantly reduced TPR levels, and decreased TPR-containing NPC density. A compensatory increase in total NPC levels was observed, and decreased global RNA intensity in the nucleus. The discovery of variants that partly disable TPR function provide valuable insight into this essential protein in human disease, and our findings suggest that TPR variants are the cause of the siblings’ neurological disorder.
Introduction
Nuclear pore complexes (NPCs) are nuclear envelope channels that regulate the trafficking of macromolecules between the nucleus and cytoplasm within a cell. The NPCs are assembled from a number of proteins that form a complex basket of proteins that allow passive diffusion of macromolecules up to ~40 kDa through a channel of ~90–100 Å width (1), whilst larger cargo requires active translocation facilitated by soluble transport receptors that interact with the NPC. NPCs are critical not only for the transport of macromolecules between the nuclear compartment and the cytoplasm, but also the transport of these macromolecules is central to multiple other cellular functions which include mRNA processing, chromatin remodelling, transcription and protein modifications.
The NPC is assembled from over 30 different highly conserved proteins. Multiple copies of these subunits arrange into a basket-shaped macromolecule channel totalling upward of ~500–1000 individual proteins, and an overall a size of ~110 MDa (1,2). The total number of NPCs per cell can vary from several hundred to tens of thousands, and density is not directly linked to the available nuclear surface area (3). Most NPC proteins, (nucleoporins denoted Nup, followed by a number reflecting their molecular weight) form a series of concentric rings within the nuclear envelope. Some proteins of the NPC protrude into the cytoplasm, forming ‘cytoplasmic filaments’, whilst on the nuclear face the nuclear basket structure is formed, with both these structures providing binding sites for cargo. The nuclear basket provides anchors for mRNA export and interacts directly with transcriptional regulatory machinery (4). This includes the highly conserved transcription elongation and RNA export (TREX-2) complex that associates stably with the NPC, and this association requires TPR and NUP153 (5,6,7). This directly couples transcriptional regulation, mRNA processing and export factors (8,9). The nuclear basket is the least understood component of the NPC, and in humans is assembled from only three proteins; Nup153, Nup50 and TPR. To date, there are no known disease associations with any of the nuclear basket proteins.
Here we report on biallelic variants in TPR identified by whole genome sequencing in a family with two siblings with ataxia, microcephaly and severe intellectual disability. We provide evidence that the variants affect TPR splicing, reduce steady-state TPR levels, abnormal nuclear pore composition and density, and altered global RNA distribution in patient fibroblasts.
Results
Case descriptions
Individual 1
Individual 1 is a female currently aged 21 years. She is the first child, born to healthy non-consanguineous Maltese parents. She was delivered at 41 weeks and 3 days gestation after an uncomplicated pregnancy with a birthweight of 3040 g (10th centile) and her occipitofrontal circumference (OFC) was 34 cm (third centile). From approximately eight months of age, her OFC trajectory dropped to below the 1st centile. Her height and weight have remained around the 60th centile.
In infancy, a small ventricular septal defect was detected, which closed spontaneously. No other congenital anomalies were noted.
During the neonatal period, she developed mild jaundice and was a slow feeder who coughed with bottle feeds. She had ongoing difficulties with feeding incoordination, and swallowing was hampered by tongue thrusting and marked food aversion. She was noted to be mildly hypotonic in the neonatal period. She experienced episodes of stiffening during the first year of life, which became more frequent, and developed into jerky movements of her head, arms and legs. She had global developmental delay. She rolled at six months, sat unsupported at 12 months, crawled at 17 months and walked unsupported by 25 months.
She has severe intellectual disability, which has been difficult to formally assess, but as a young adult, she remains dependent on carers for tasks of daily living. She has a range of vocalizations and a few words and can understand simple commands. She can express pleasure and distress and shows an interest in people and her surroundings. She has not demonstrated any developmental regression. Her hearing is normal, and she has structurally normal eyes with a pseudo-strabismus and normal vision.
She has had dry eczematous skin. She developed pubarche at age nine years, and menarche at age 15 years with subsequent oligomenorrhea. She has otherwise been healthy and has not had seizures.
On examination, she has deep-set eyes, a prominent and broad nasal bridge, a bulbous nasal tip, featureless philtrum and a thin upper vermilion border. Some facial features were familial. She has long, thin fingers with fifth finger clinodactyly, hyperextensible elbows and knees, and tight interphalangeal joints, but no contractures. She has short great toes and flat feet. She has a head nod and intention tremor, with a propensity to hand flapping behaviours. She has increased tone in her upper limbs bilaterally, and normal tone in her lower limbs. Her deep tendon reflexes are normal. She has a jerky, wide-based gait.
Extensive investigations include a brain MRI brain at age one year, which showed appearances suggestive of a leukodystrophy. However, subsequent MRI brain scans at ages two years and eight years showed non-progressive white matter changes with delayed myelination but did show features of a leukodystrophy. Electroencephalograms were normal. Nerve conduction studies were normal. A renal ultrasound was normal. Testing of white cell lysosomal enzymes, very long chain fatty acids, serum transferrin isoforms, urinary amino acids, plasma amino and organic acids, plasma acylcarnitine profile, paired cerebrospinal fluid and blood glucose and urinary guanidino-acetic acid were all normal. Creatine kinase levels were mildly raised at 284 IU/L (normal range 40–240 U/L). Serum lactate and pyruvate levels were mildly raised at 2.7 mmol/L (NR 1.0–1.8 mmol/L) and 0.16 mmol/L (0.03–0.10 mmol/L), respectively. Cytomegalovirus polymerase chain reaction testing on her Guthrie card was negative. Blood folate level was low at age 10 years, presumed to be dietary related. Maternal phenylketonuria testing was negative.
Genetic testing included a 46XX G-banded karyotype, and normal Illumina HumanCytoSNP-12 v2.1 microarray, chromosome breakage studies, Fragile X testing, and methylation studies for Angelman and Prader–Willi syndromes.
Individual 2
Individual 2 is a male currently aged 16 years, the younger brother of individual 1 (Fig. 1). He was naturally conceived after three intervening early-gestation miscarriages, leading to the use of aspirin and clexane during the pregnancy. The pregnancy was complicated by bleeding at 11 weeks of gestation, but no other major concerns. Foetal movements were normal and there were no teratogenic exposures. His mother underwent a planned induction of labour at 38 weeks of gestation.
Figure 1.
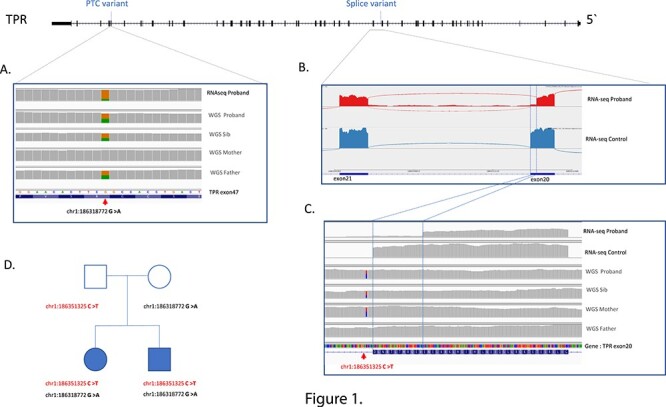
Schematic of TPR indicating location of PTC and splice variants. (A) IGV browser showing the premature termination codon (PTC) in Proband RNAseq data, indicating the reduced level of transcript derived from the allele containing the PTC (16% PTC, 84% WT). It is presumed the remaining RNA containing the PTC will undergo NMD. The WGS sequencing of the proband, sibling, mother and father, showing the variant in all samples, but not the mother (coverage over variants equal or great than 30 reads). (B) Sashimi plot of TPR gene transcript between exon 20 and exon 21 (NM_003292.2), with the proband top and control below, illustrating the premature splicing of the probands TPR exon 20, which removes 12 amino acids from TPR protein with the remaining protein still in frame. There is also a small amount of intron read-through. (C) IGV genome browser view focused on the premature splicing in the probands RNAseq (top) compared to an RNAseq control sample (bottom). The causative variant is located 5 bp into the intron as illustrated in the proband, sibling, and the mother’s WGS. (D) Family pedigree with affected individuals shaded. All coordinates are in hg38 human genome. WGS: whole genome sequencing. RNA-seq: RNA sequencing from fibroblasts.
His birth weight was 2850 g (10th centile), length 49.3 cm (50th centile), OFC 33 cm (10th centile). From approximately five months of age, his OFC trajectory dropped to below the 1st centile. His height and weight have remained around the 25th and 5th centiles, respectively. He developed mild neonatal jaundice, and he had mild feeding difficulties during the newborn period, which required thickened formula feeds. He was not hypotonic during the newborn period. He rolled from front-to-back at four months, was able to sit with support at 15 months, and walked unaided at 27 months. He did not have any stiffening episodes like those observed in his sister.
He also has severe intellectual disability, with assistance required for all tasks of daily living. He is non-verbal, and mainly babbles with vowel sounds. He can understand simple commands such as ‘clapping hands’, knows his name and recognizes his family. He makes limited eye contact and shows variable interest in people. He has not demonstrated any developmental regression. His hearing is normal. He has structurally normal eyes, with a pseudo-strabismus, and normal vision. He has generalized eczema, and multiple allergies to eggs and nuts. He has had occasional ear infections, but otherwise has kept good health, and has not had seizures.
On examination, he shares a number of features with his sister, including deep-set eyes, and a broad nasal tip with a low-lying columella. He has thin, long fingers. He has generalized increased tone, with normal deep tendon reflexes. He has a wide-based ataxic gait. He has a propensity to hand twirling.
An MRI and MRS brain scan showed prominent optic sheaths with relative thinning of the optic nerves, but an otherwise normal brain structure. Urinary and serum metabolic investigations similar to his sister were undertaken, without any informative result. Serum lactate and pyruvate were mildly raised at 2.0 and 0.16 mmol/L, respectively.
Genetic testing includes a 46XY G-banded karyotype, Illumina HumanCytoSNP-12 v2.1 microarray showing a 6 Mb long contiguous region of homozygosity on chromosome 10, and multiplex ligation-dependent probe amplification subtelomeric deletion studies were normal.
Sequencing and in silico analysis
The two affected siblings and their unaffected parents underwent whole genome sequencing and RNA-seq analysis by the Genomics Platform at the Broad Institute of Harvard and MIT (Broad Institute, Cambridge, MA, USA). Analysis of genomic SNP and indels through seqR identified compound heterozygous variants in the gene encoding translocated promoter region (TPR).
The first TPR variant (Hg38 chr1:186318772 G > A, NM_003292.2: c.6625C > T, NP_003283.2: p.Arg2209Ter) identified in heterozygous state in both affected siblings and their father, causes a premature stop codon in exon 47 in the TPR gene, which is predicted to induce nonsense-mediated decay (NMD). This variant is absent in all gnomAD databases as well as TopMed and 1000 genomes, and is predicted to be disease causing by Mutation Taster (10). RNAseq analysis of Individual 1 confirmed that premature stop codon induced nonsense mediated decay by the reduced number of reads mapping over the variant contained the stop-gain variant (16% of total reads contained stop variant instead of expected 50%: 13/80 reads; Fig. 1A).
A second TPR variant was found in heterozygous state in both affected children and their mother, variant (Hg38 chr1:186351325 C > T, NM_003292.2: c.2610 + 5G > A), which is located 5 bp into the start of intron 20 of TPR. This variant was also absent from all population databases and was predicted to affect splicing (SpliceAI (0.86)). Analysis of the RNAseq data, illustrated that the splice variant caused an in-frame 12 amino acid deletion from the TPR transcript (Fig. 1B, 1C). Forty-four (85%) split reads confirmed the splicing defect, with only 8 (15%) reads split reads supporting the canonical splicing.
Biallelic TPR variants lead to decreased TPR protein levels and decreased TPR pore densities in the nuclear envelope
To assess the consequences of the TPR variants on TPR steady-state levels, we studied skin fibroblasts from affected individual I. The variants were confirmed in patient fibroblasts (Supplementary Material, Fig. S1). We used a polyclonal antibody that recognizes the N-terminal region of TPR to perform immunoblotting on these fibroblasts (Fig. 2A). There was a significant decrease (P < 0.05) in TPR levels in patient fibroblasts compared to the paediatric control fibroblast lines (Fig. 2B).
Figure 2.
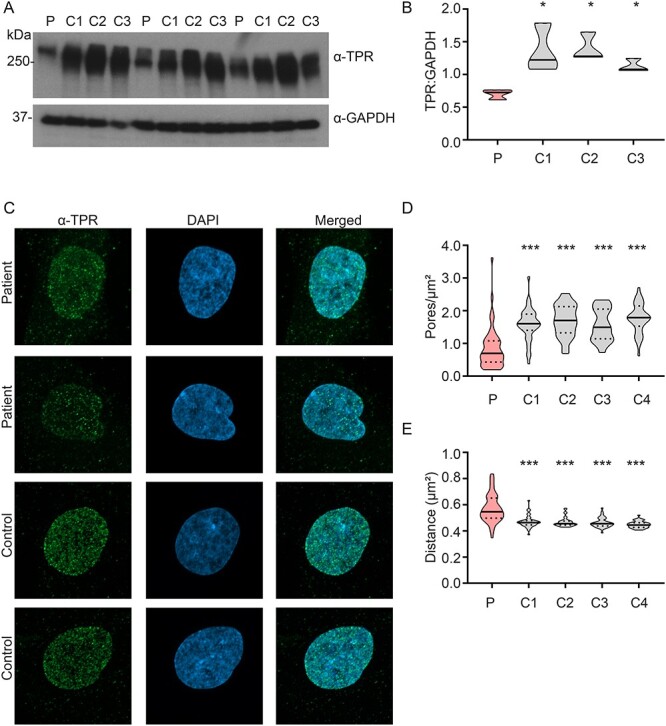
Decreased TPR steady-state levels and decreased TPR-containing NPC density in patient fibroblasts. (A) Western blot of TPR in patient fibroblasts (P) and three controls (C1-C3). (B) Densitometry shows significantly reduced TPR levels in patient fibroblasts compared to controls. N = 3 biological samples/cell line, two technical repeats. One-way ANOVA with Sidak’s multiple comparisons test; *P < 0.05. (C) Compressed Z-stack representative immunostained images of TPR (green spots) and nucleus (DAPI; blue) in patient fibroblasts and controls. (D) Quantification of TPR-containing NPC pore density (pores per μm2 of nuclear surface area) in patient fibroblasts compared to controls shows a significant decrease in patient fibroblasts. N = 3 biological samples/cell line, pooled data from 38–50 nuclei/cell line. One-way ANOVA with Sidak’s multiple comparisons test; ***P < 0.001. (E) Distance to the nearest TPR-immunostained pore neighbour measurement shows a significant increase in distance in patient fibroblasts compared to controls, indicating more sparsely distributed TPR-containing NPCs in patient fibroblasts. N = 3 biological samples/cell line, pooled data from 38–50 nuclei/cell line. One-way ANOVA with Sidak’s multiple comparisons test; ***P < 0.001.
Firstly, we examined if there were any gross abnormalities in nuclear shape or size. Blinded samples were assessed for abnormal nuclear morphology including blebbing, pinched nuclei, multi-nuclear cells or flower-shaped nuclei. The percentage of abnormal nuclei ranged from 0.6–3.1% of the total population in control cells, and 1.4% of nuclei in patient cells were scored as abnormal. There was no significant difference between the patient cells and any of the control cells.
We then determined whether there were any changes in the TPR-containing nuclear pore density in patient fibroblasts by immunostaining with an antibody against TPR. We utilized super resolution microscopy imaging of the nuclear region defined by DAPI stain, and captured z-stack images of control and patient fibroblasts (Fig. 2C, Supplementary Material, Fig. S2). We then batch-processed the images to quantify the number of TPR-containing pores in the nuclear region, and then TPR-containing pore density was normalized to the nuclear surface area. Cells from the affected individual showed a significant reduction in TPR-containing pore density in the nuclear envelope when compared to four control fibroblast lines (Fig. 2D). We also determined the proximity to the nearest TPR-containing pore using the ‘nearest neighbour’ algorithm acquired during ‘spot analysis’ in the Imaris image processing pipeline. This revealed a significant increase in distance to the nearest neighbour in patient fibroblasts when compared to four control fibroblast lines (Fig. 2E). Together these results indicate reduced number, and a more sparsely distributed TPR-containing pore density in patient fibroblasts.
A subset of NPCs lack TPR protein
Given the significant reduction in TPR protein levels (Fig. 2A) and decrease in TPR pore density (Fig. 2C), we sought to determine whether some NPCs had a full complement of TPR, or whether TPR was distributed to all NPCs at a lower concentration. We immunostained fibroblasts with both an antibody against mAB414 and TPR, and counterstained with DAPI. We used the same spot detection algorithm described above to determine the TPR- and mAB414-containing pores and added in an additional function to identify co-stained pores within a close proximity, which were classified as colocalized. This demonstrated a clear reduction in the percentage of colocalization in patient cells compared to controls (Fig. 3B). We then examined a subset of the colocalization data, specifically only the spots that were classed as being colocalized. We used Imaris to extract the raw fluorescent intensity of the individual TPR spots that were scored as colocalized. Analysis of the intensity of fluorescent spots shows that even the subset of spots that colocalize with the nuclear pore have a significantly decreased fluorescent intensity (Fig. 3C).
Figure 3.
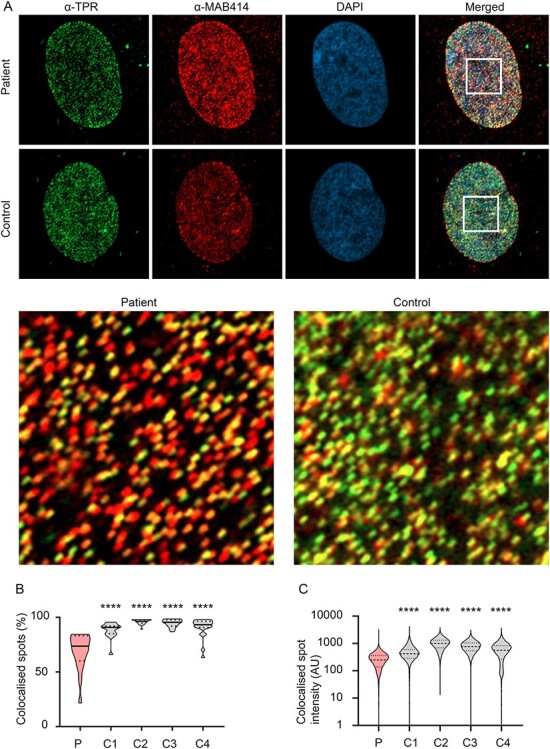
Decreased colocalization of TPR to nuclear pores in patient fibroblasts. (A) Compressed Z-stack representative immunostained images of TPR (green spots), mAB414 (red spots) and nucleus (DAPI; blue) in patient fibroblasts and controls. Yellow sections indicate colocalized regions. (B) Quantification of TPR colocalizing with mAB414 in patient fibroblasts compared to controls (C1—C4) shows a significant decrease in TPR-occupied pore density in patient fibroblasts compared the four controls. N = 3 biological samples/cell line, pooled data from 15–22 nuclei/cell line. One-way ANOVA with Sidak’s multiple comparisons test; ** P < 0.01, ***P < 0.001. (C) Colocalized TPR spot intensity shows a significant decrease in intensity in patient fibroblasts compared to controls, indicating that a subset of spots that colocalize with the nuclear pore have a significantly decreased fluorescent intensity. N = 3 biological samples/cell line, pooled data from >20 000 individual spots/cell line. One-way ANOVA with Sidak’s multiple comparisons test; ***P < 0.001.
Depletion of TPR modestly increases total NPC numbers
We then determined whether there were any changes in NPC density. Patient fibroblasts were immunostained with an antibody (mAb414, Fig. 4A) that recognizes the conserved FxFG repeats present in a number of nuclear pore complex proteins including p62, Nup153 and Nup214. mAb414 is commonly used to determine NPC density and study changes in nuclear structure and composition (11). We captured z-stack images of control and patient fibroblasts (Fig. 4A, Supplementary Material, Fig. S3). While mAB414 protein levels were broadly unchanged in patients compared to controls (data not shown), we did observe a modest but significant increase in nuclear pore complex (NPC) density in the nuclear envelope of patient fibroblasts compared to three of the four controls used in this study (Fig. 4B). We also determined the ‘distance to nearest neighbour’ for mAB414 stained NPCs as described above. The ‘nearest neighbour’ algorithm showed a closer proximity to neighbouring NPCs in patient fibroblasts when compared to three of the four control fibroblast lines (Fig. 4C). Together these results suggest a more densely populated NPC density in patient fibroblasts.
Figure 4.
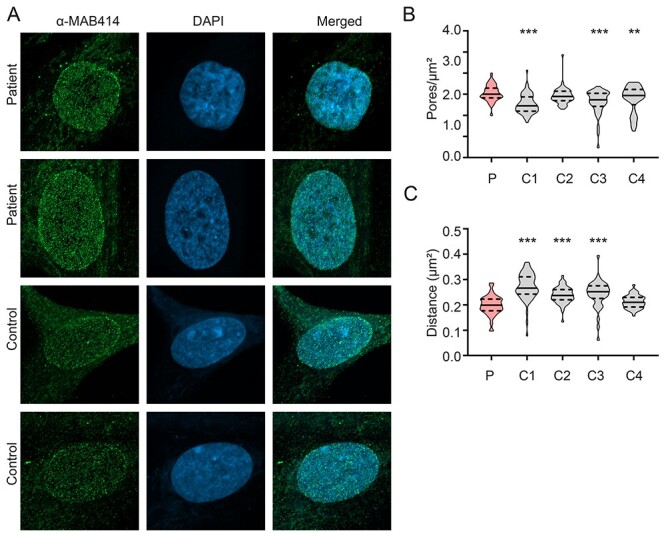
Increased global nuclear pore density in patient fibroblasts. (B) Compressed Z-stack representative immunostained images of mAB414 (green spots) and nucleus (DAPI; blue) in patient (P) fibroblasts and controls. (C) Quantification of mAB414 immunostained NPC pore density (pores per μm2 of nuclear surface area) in patient fibroblasts compared to controls (C1—C4) shows a significant increase in NPC pore density in patient fibroblasts compared to three of the four controls. N = 3 biological samples/cell line, pooled data from 40–79 nuclei/cell line. One-way ANOVA with Sidak’s multiple comparisons test; ** P < 0.01, ***P < 0.001. (D) Distance to the nearest mAB414 immunostained pore neighbour measurement shows a significant decrease in distance in patient fibroblasts compared to controls, indicating more densely populated NPCs in patient fibroblasts. N = 3 biological samples/cell line, pooled data from 40–79 nuclei/cell line. One-way ANOVA with Sidak’s multiple comparisons test; ***P < 0.001.
Depletion of TPR reduces nuclear mRNA content
Given the role of TPR in mRNA export through its interaction with the TREX-2 mRNA export complex (12,13), we next assessed the distribution of poly (A) + RNA between the cytoplasm and nucleus by in situ hybridization (FISH) with a biotinylated oligo-dT 50mer (14) in patient fibroblasts (Fig. 5A). Interestingly, the total intensity of RNA in the nucleus was significantly reduced in patient fibroblasts compared to a panel of control fibroblasts (Fig. 5B). However, cytoplasmic RNA levels were unchanged in both patient fibroblasts compared to control cells (Fig. 5C). Additionally, the total intensity of RNA-FISH in the whole cell was much lower in patient fibroblasts compared to three of the four controls (Fig. 5D), due to the difference in nuclear intensity (Fig. 5B).
Figure 5.
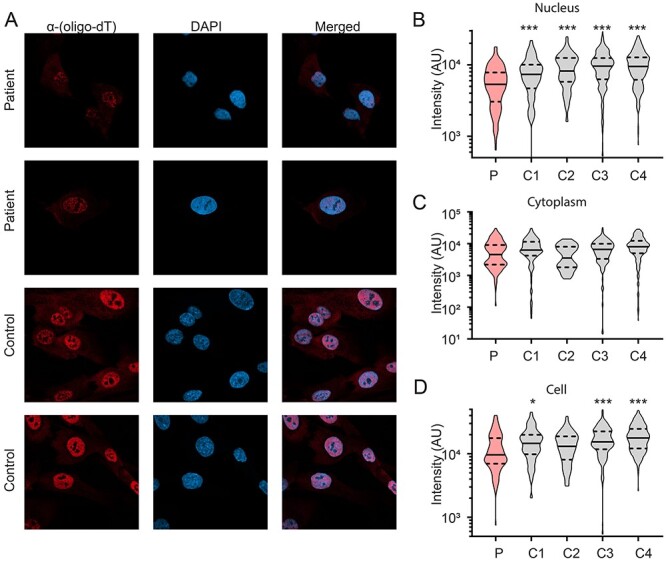
Decreased nuclear poly(A) + RNA intensity in patient fibroblasts. (A) Representative images of the distribution of poly (A) + RNA determined by in situ hybridization with a biotinylated oligo-dT 50-mer in patient fibroblasts (red) and nucleus (DAPI; blue). (B) Quantification of RNA-FISH intensity in the nucleus revealed a significant reduction in patient fibroblasts (P) compared to control fibroblasts (C1—C4). N = 3 biological samples/cell line, pooled data from 39–223 cells/cell line. One-way ANOVA with Sidak’s multiple comparisons test; **P < 0.01, ***P < 0.001. (C) RNA was distributed evenly in the cytoplasm between patient fibroblasts compared to control cells. (D) The total intensity of RNA-FISH in the whole cell was much lower in patient fibroblasts compared to three of the four controls, likely due to the difference in nuclear intensity described in B. N = 3 biological samples/cell line, pooled data from 39–223 cells/cell line. One-way ANOVA with Sidak’s multiple comparisons test; *P < 0.05, ***P < 0.001.
Discussion
In this report we demonstrate the critical importance of TPR in humans. We report biallelic variants in TPR, discovered by genetic analysis of a family with two affected children with microcephaly, ataxia and severe intellectual disability. Both children carried a stop-gain variant and a pathogenic intronic variant located 5 bp into intron 20, resulting in a splicing defect.
The first TPR variant causes a premature stop codon in exon 47 in the TPR gene, which induces NMD, as seen in the RNAseq data. The second splice variant in TPR is located 5 bp into the start of intron 20 and caused an in-frame 12 amino acid deletion from the TPR transcript, and only 15% of reads supported canonical splicing. The reads with canonical splicing are predicted to be derived from the TPR alternative allele that have yet to undergo NMD. The deleted 12 amino acids (VEQRHTLTRNLD) are highly conserved and form part of the coiled coil domain, a structural motif that forms alpha-helices and provides structural rigidity to the nuclear basket. Alpha helices are a common structural element of proteins and have a right-handed helix with a repeat length of 3.6 amino acids per helical turn. Therefore, the deletion of 12 amino acids would remove 3 full turns of the helix and rotate the remaining alpha helix by 120°. The structural integrity of an alpha helix is dependent on correct steric configuration, and any disruptions to the regular geometry or rotation of the helix may have destabilizing effects on the three-dimensional structure of the protein. It is predicted that the removal of 12 amino acids destabilizes regions of the protein, and thus impairs function of the TPR protein, but it likely that some residual function still exists, as TPR knockout is embryonic lethal in mouse models (15) and is lethal in some Drosophila knockouts of the homologue, Mtor (16). TPR is the 257th most intolerant gene to loss of function mutations in the human genome, based on gnomAD analysis (17) and before now has had no reported disease association.
The overall consequence of both TPR mutations is that the two affected siblings are predicted to have no wild type TPR, with one allele undergoing NMD and the other expressed but missing a 12-amino acid region of a crucial structural domain. We provide evidence that the presence of these two variants in patient fibroblast led to a significant reduction of both TPR steady-state levels by western blotting, and TPR-containing NPC density by immunofluorescence.
Our analysis clearly demonstrates that TPR depletion in patient cells results in 1) a significant reduction in the total number of nuclear pores occupied by TPR and 2) the subset of colocalized TPR spots are lower in fluorescent intensity in patients compared to controls. This potentially indicates a dual mechanism of action whereby there is insufficient TPR to occupy all nuclear pores, and that a subset of nuclear pores do not contain the full complement of TPR protein.
Deficiency of other NUP proteins including NUP80 (18), NUP153 (19) and NUP214 (20,21) have been shown to result in highly abnormal nuclear morphology; however, we did not quantify any difference in nuclear abnormalities in patient fibroblasts.
Interestingly, the total NPC density was significantly elevated in patient fibroblasts (mAB414 staining), and NPCs were more closely distributed. Previous studies demonstrated that TPR depletion increases the total NPC numbers, and this phenotype was consistent across a number of cell types, including human fibroblasts (22). Furthermore depletion of X. laevis Nup188, a core component of the NUP inner ring, also led to an increase in the density of nuclear pores per nucleus (23). The number of NPCs per cell are believed to be critical for cell differentiation (24), specifically for neural cells. NPC numbers change throughout differentiation, such as for neural progenitor cells, which give rise to multiple neuronal cell types with significantly different NPC numbers (25,26). It is therefore plausible that a significant disruption to TPR function, as observed in our patient fibroblasts, is likely to have implications for other cell types, particularly neural cells. This may explain the predominantly neurological phenotype observed in patients with partial impairment of TPR function. Other nucleoporins, such as M. musculus Nup210, Nup98 and Nup133, have also been implicated in the regulation of neural differentiation (27), so it would be intriguing to test if these nucleoporins perhaps interact with distinct key transcription factors to regulate cell fate. It is worth noting that M. musculus Nup153 interacts with the chromatin architectural proteins, CTCF and cohesion to regulate chromatin architecture (28) and since TPR is a nuclear basket protein that is tethered to the nuclear pore complexes via Nup153, is possible that TPR also regulates chromatin architecture.
Human TPR is a highly conserved structural component of the nuclear basket and forms a homo-or heteropolymetric architectural element. TPR consists of an N-terminal region that interacts with the NPC directly, followed by an alpha helical coiled-coil region that spans over 1500 residues, and provides structural integrity (29). The C-terminal region of TPR is a highly disorganized acidic domain that forms fibrillary extensions that protrude into the nucleus (30). TPR is a key component of the TREX-2 mRNA export pathway, and depletion of TPR disrupts the association of TREX-2 components to the NPC and causes profound changes to RNA transcription and export (12,13). Indeed, TPR levels are important for mRNA export, as either overexpression or depletion of TPR leads to a block in mRNA export (13,31,32), Interestingly, analysis of cellular mRNA distribution of patient fibroblasts demonstrated a significant decrease in RNA intensity in the nucleus compared to controls, while RNA levels in the cytoplasm were unchanged, indicative of a potential defect in transcription. Importantly, a recent study uncovered an important role for TPR in regulating transcription. Specifically, acute depletion of TPR resulted in a reduction in mRNA abundance, and also a reduction in the synthesis of RNA transcripts (11). Given that transcription is upstream of mRNA export and the processes are coupled (9), affecting transcription will reduce mRNA export. Therefore, this may explain why we observed a reduction in nuclear levels of mRNA in our patient fibroblasts, potentially reflecting a long-term adaptation of these cells to low levels of TPR. Collectively, our data indicates a critical role of TPR in human health, and disturbances to TPR levels in patient fibroblasts impair NPC abundance and nuclear levels of RNA, with downstream effects on mRNA export.
Given the central and diverse role of the NPC, it is not surprising that pathogenic variants in protein subunits of the NPC, nuclear basket and TREX-2 mRNA export process are causative of human disease. Neurological diseases can be caused broadly by disruptions to RNA metabolism (33), nucleocytoplasmic transport defects in amyotrophic lateral sclerosis (ALS) and Huntington disease (34,35), and direct blockage of NPCs from the interaction of neurofibrillary tangles with the nuclear pore complex in Alzheimer disease (36). More specifically, pathogenic variants in NUP subunits and protein subunits of the TREX-2 complex (Supplementary Material, Table S1) are associated with neurological disorders (37), associated with disrupted NUP structure, enlarged nuclear size and impaired mRNA export (38,39). Specifically, pathogenic variants have been reported in the TREX components and THOC2 (40), THOC6 (41–44), as well as TREX-2 component, MCM3AP/GANP (45–48) and cytoplasmic mRNA export factor and nucleoporin GLE1 (49–52), and the nuclear pore complex subunits NUP37 (53), NUP62 (54), NUP95 (53), NUP107 (53,55,56) and NUP188 (39,57). A range of phenotypes in individuals with variants in these genes are observed (Table 1), most commonly intellectual disability, frequently associated with microcephaly and sometimes ataxia, seizures and peripheral neuropathy.
Table 1.
Summary of clinical phenotype of patients with pathogenic variants in TPR and protein subunits of the TREX-2 and NPC complexes
| Gene | Protein complex | Intellectual disability | Microcephaly | Inheritance | Brain MRI and/or CT abnormalities |
|---|---|---|---|---|---|
| TPR (Subject 1) | Nuclear pore basket | Severe | Y | Non-progressive white matter changes with delayed myelination | |
| TPR (Subject 2) | Nuclear pore basket | Severe | Y | Prominent optic sheaths with relative thinning of the optic nerves, but an otherwise normal brain structure | |
| THOC2 | TREX complex | Mild–Severe | Some | XLR | Mild cervical cord compression, gliosis, ventriculomegaly, cranial hyperostosis, inferior cerebellar vermis dysplasia |
| THOC6 | TREX complex | Severe in some | Y | AR | Y (not described) |
| MCM3AP/GANP | TREX-2 complex | Mild–Severe | NR | AR | Mostly normal, Middle fossa arachnoid cysts, Minimal colpocephaly |
| GLE1 * | Cytoplasmic-associated NPC | Severe | Y | AR | Generalized parenchymal volume loss and a small brainstem; diffuse cerebral atrophy, with prominent CSF spaces and dilated ventricles |
| NUP37 | NPC | Mild | Y | AR | Cerebellar vermis hypoplasia |
| NUP62 | NPC | Moderate–severe | Y | AR | Infantile bilateral striatal necrosis, horizontal pendular nystagmus, gradual disappearance of the basal ganglia |
| NUP85 | NPC | Reported in one family | NR | AR | No structural abnormalities reported |
| NUP107 | NPC | Moderate | Y | AR | Normal brain structures with simplified gyri and sulci, and reduced frontal cortex volume with well-preserved volumes of corpus callosum, brainstem and cerebellum |
| NUP188 | NPC | Severe in some | Some | AR | Sandestig-Stefanova Syndrome (congenital cataracts, hypotonia, prenatal-onset ventriculomegaly, white-matter abnormalities, hypoplastic corpus callosum, congenital heart defects, and central hypoventilation, characteristic dysmorphic features) |
*Phenotypes described for GLE1 are for patients who survived beyond the newborn period. NR = not recorded, Y = present, N = not present, NPC = nuclear pore complex, XLR = X-linked recessive, AR = autosomal recessive. Further clinical information and referencing can be found in Supplementary Material, Table S1.
Given our findings of pathogenicity for the variants in two affected individuals, that NPC integrity is critical for neurons, and that pathogenic variation in genes for other NPC complex proteins result in neurological diseases, we suggest that the TPR variants we have identified are the cause of the severe neurological disorder in these individuals.
Materials and Methods
Ethics
All procedures followed were in accordance with the ethical standards and approved by the Human Research Ethics Committee of the Royal Children’s Hospital (HREC/67401/RCHM-2020) and in accordance with the Helsinki Declaration of 1975, as revised in 2000. Written informed consent was obtained from the parents individually and on behalf of their children. Two siblings (Family 1; individuals 1 and 2) and their parents were recruited as part of a gene discovery program in children with undiagnosed neurological disease.
Biological samples
Genomic DNA from whole blood cells of reported cases and their parents was isolated using the QIAamp DNA mini kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions and used for whole genome sequencing. RNA from skin fibroblasts cultured from Individual 1 was extracted using a commercially available kit (Qiagen RNeasy kit) for RNA sequencing. Confirmatory Sanger sequencing was completed in a genomic DNA sample from the individual’s fibroblasts to confirm the presence of both TPR variants in the established fibroblast line.
Whole genome sequencing
Whole genome sequencing and data processing were performed by the Genomics Platform at the Broad Institute of MIT and Harvard. PCR-free preparation of sample DNA (350 ng input at > 2 ng/ul) using Illumina HiSeq X Ten v2 chemistry. Libraries sequenced to a mean target coverage of >30×. Data were processed through a pipeline based on Picard, using base quality score recalibration and local realignment at known indels. The BWA aligner mapped reads to the human genome build 38. Single nucleotide variants (SNVs) and insertions/deletions (indels) were jointly called across all samples in the cohort using the Genome Analysis Toolkit (GATK) HaplotypeCaller package version 4.0. Default filters were applied to SNV and indel calls using the GATK Variant Quality Score Recalibration (VQSR) approach. Annotation was performed using Variant Effect Predictor (VEP). Lastly, the variant call set was uploaded to Seqr for collaborative analysis between the CMG and MCRI/VCGS.
RNA sequencing (RNAseq)
Human whole transcriptome sequencing was performed by the Genomics Platform at the Broad Institute of MIT and Harvard. The transcriptome product combines poly(A)-selection of mRNA transcripts with a strand-specific cDNA library preparation, with a mean insert size of 550 bp. Libraries were sequenced on the HiSeq 2500 platform to a minimum depth of 50–75 million STAR-aligned reads. Reads were aligned to the human genome build 38 using STAR, 2-pass. Alignment was visualized using IGV.
Variant analysis
Genomic variants were uploaded into Seqr (https://seqr.broadinstitute.org/) for analysis. Variants were filtered on a number of factors, including inheritance pattern, potential pathogenicity of variant and population frequency of variant utilizing GnomAD (Exome and Genome) (17), Topmed (58) and 1000 genomes (59) databases. Only variants with a population frequency < 0.001 for de novo dominant searches and < 0.03 for recessive searches were considered. Additional searches looking for variable penetrance variants were also performed.
Because the two siblings were both similarly affected and different gender, the most likely model of pathogenicity was a biallelic autosomal recessive disorder. Only two genes were found to contain biallelic variants in the proband, TPR and MUC22. The disease phenotype was inconsistent with what is known about the biology of MUC22, and so this gene was excluded from further study.
TPR was submitted to Matchmaker Exchange, an international database to match subjects of similar phenotype and potential pathogenic variants, but no matches were identified as of 06/06/21 (60).
Candidate variants were validated in RNA-seq and any potential effect on TPR splicing and nonsense mediate decay determined.
Cell culture
Primary cultures of fibroblasts from Individual 1 and unrelated control fibroblasts in Family 1 were established from skin biopsies as previously described (61). Fibroblasts were cultured in basal medium (high-glucose DMEM (Gibco) with 10% foetal bovine serum (Gibco), 100 units/mL penicillin, and 100 μg/mL streptomycin) at 37°C with 5% CO2. All fibroblast control cell lines were established in-house and were established from paediatric individuals without any suspected genetic disorders.
Immunoblotting
For denaturing gels, fibroblasts were extracted in RIPA buffer (10 mM Tris–Cl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, 1 mM PMSF and protease inhibitor cocktail (Roche)) with gentle sonication then incubated on ice for 30 minutes prior to centrifugation at 20 000 g for 20 minutes at 4°C. Protein concentration was determined using the Pierce BCA Protein Assay Kit. SDS-PAGE was performed on 10–30 μg of cell lysate per sample using standard techniques on Tris-glycine-SDS polyacrylamide gels (Biorad).
Antibodies were specific to TPR (Cat. # sc-101 294, Santa Cruz; 1:2000), mAB-414 (Cat. # ab24609, Abcam; 1:1000), and GAPDH (Cat. #G9545, Sigma Aldrich; 1:10000). GAPDH was used as a loading control. Primary antibodies were detected with appropriate anti-mouse or anti-rabbit horseradish peroxidase-conjugated antibodies (GE Healthcare, NJ, USA), using enhanced chemiluminescence reagents (GE Healthcare, NJ, USA) and Amersham Hyperfilm. Protein band intensities were quantified using ImageJ software, and band intensity determined in the linear range was normalized to band intensity of GAPDH.
Immunofluorescence microscopy
Patient and control fibroblasts were fixed in 4% paraformaldehyde for 20 minutes and washed in PBST (PBS + 0.5% Triton X-100). Cells were blocked and permeabilized with 1% BSA in PBST for 1 hour at room temperature. Cells were probed with primary antibody mAB414 (Cat. # ab24609) and/or anti-TPR antibody (Rabbit, 1:500, Ab84516) diluted with 1% BSA in PBST (1:500) for 1 hour at room temperature and washed in PBS. Secondary incubation was with Alexa Fluor 488 conjugated goat anti-mouse antibody (ThermoFisher Scientific) in 1% BSA in PBST (1:500) for 1 hour at room temperature and washed in PBS. Coverslips were mounted on a microscopy slide using ProLongTM Gold Antifade with DAPI (Life Technologies).
Fluorescence in situ hybridisation with immunofluorescence
Fibroblast cells grown on round coverslips were fixed in 4% PFA before placing in ice cold, 100% ethanol and stored for a minimum 16 hours at −20°C. Cells were permeablized in 0.5% Triton-X100 in PBS and blocked in pre-hyb buffer (1 mg/ml yeast tRNA, 2× SSC, 0.2% BSA and 20% deionized formamide) prior to staining in hyb buffer (prehyb buffer +10% dextran sulfate, 100uM Cy5-oligodT 50 (i.e. 50 repeats of T primer labelled with Cy3, Sigma) and anti-TPR antibody (Rabbit, 1:500, Ab84516). After incubation at 37°C for 3 hours, coverslips were subjected to stringency washes of 2 × 5 minutes with 20% formamide +2× SSC, followed by 2 × SSC before application of an anti-rabbit 488 secondary antibody diluted in 1 x SSC buffer to detect TPR protein for 30 minutes at 37°C. Coverslips were washed in 1 X SSC prior to DAPI staining (0.2 mg/mL in PBS) and a final PBS wash. Coverslips were mounted onto superfrost plus slides using Dako aqueous mounting medium.
Microscopy and analysis
Images were captured with a Zeiss LSM 900 confocal microscope with Airyscan 2 super resolution imaging. TPR and mAB414 immunostained-cells were captured with a 63× oil immersion lens, and Z-stacks with a series of 30 slices. For TPR and mAB414 quantification, images were loaded in Imaris ×64 (Version 9.7.1) software and an analysis pipeline was generated for batch processing. A 3D surface was created from the DAPI channel using the ‘Surfaces’ function. This was used to mask the channel containing TPR or mAB414 staining, to exclude any instances of cytoplasmic staining. The masked channel was used for quantification of pore density using the ‘Spots’ function and setting the average spot size to 120 μm. The total number of spots per nucleus was normalized to the surface area of the nucleus to calculate the pores/μm2. Additionally, the ‘average distance to the nearest neighbour’ was quantified from each nucleus. For colocalization analysis, Imaris software was used to filter spots from images co-stained with both mAB414 and TPR. After spot identification, colocalized spots were filtered as a subgroup of TPR-stained spots filtered based on an average distance of <360 μm from the nearest mAB414 spots, and the number of spots was scored. Colocalized spots were expressed as a percentage of the total mAB414 spots per nucleus.
RNA-FISH-stained cells were captured with a 40× oil immersion lens. RNA-FISH images were processed with a pipeline function generated in CellProfiler (Version 4.0.7, Broad Institute) (62). Masks were generated for the nucleus and cell, to measure the intensity. Primary objects (nuclei) were identified on the DAPI channel, then masked on the RNA-FISH channel. Secondary objects (cell) were masked using the propagation function on the RNA-FISH channel, propagating from the primary object (nucleus). Tertiary objects (cytoplasm) were identified by subtracting the mask generated from the DAPI (nucleus) from the mask generated by the cell. The mean intensity was measured for each masked region.
Statistical analysis
Statistical analyses were carried out using either a two-tailed Student’s t-test or one-way ANOVA corrected for multiple comparisons as appropriate (GraphPad Prism® Software). Error bars represent the standard deviation of the mean (± SD). A P-value <0.05 was considered statistically significant.
Author contributions
Conceptualization : Nicole J Van Bergen, Katrina M Bell, Kirsty Carey, Cas Simons, Vihandha O. Wickramasinghe, Susan M. White, John Christodoulou.
Methodology: Nicole J Van Bergen, Katrina M Bell, Kirsty Carey, Russell Gear, Sean Massey, Edward K Murrell, Lyndon Gallacher, Kate Pope, Andrew Kornberg, Lynn Pais, Marzena Walkiewicz, Cas Simons.
Formal analysis and investigation: Nicole J Van Bergen, Katrina M Bell, Kirsty Carey, Russell Gear, Sean Massey, Edward K Murrell, Lyndon Gallacher, Kate Pope, Andrew Kornberg, Lynn Pais, Marzena Walkiewicz, Cas Simons.
Writing—original draft preparation: Nicole J Van Bergen, Katrina M Bell, Susan M. White, John Christodoulou.
Writing—review and editing: Nicole J Van Bergen, Katrina M Bell, Kirsty Carey, Russell Gear, Sean Massey, Edward K Murrell, Lyndon Gallacher, Kate Pope, Paul Lockhart, Andrew Kornberg, Lynn Pais, Marzena Walkiewicz, Cas Simons, RDnow Flagship, Vihandha O. Wickramasinghe, Susan M. White, John Christodoulou.
Funding acquisition: Paul Lockhart, Cas Simons, RDnow Flagship, Vihandha O. Wickramasinghe, Susan M. White, John Christodoulou.
Resources: Cas Simons, RDnow Flagship, Vihandha O. Wickramasinghe, Susan M. White, John Christodoulou.
Supervision: Nicole J Van Bergen, Katrina M Bell, Paul Lockhart, Cas Simons, RDnow Flagship, Vihandha O. Wickramasinghe, Susan M. White, John Christodoulou.
Supplementary Material
Acknowledgements
We thank Matthew Burdon at the MCRI imaging facility for his assistance in establishing imaging protocols.
The authors wish it to be known that, in their opinion, the first three authors should be regarded as joint first authors, and the last three authors as joint last-authors.
Contributor Information
Nicole J Van Bergen, Brain and Mitochondrial Research Group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia; Department of Paediatrics, University of Melbourne, Melbourne, Australia.
Katrina M Bell, Bioinformatics Methods group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia; Victorian Clinical Genetics Services, Royal Children’s Hospital, VIC, Australia.
Kirsty Carey, RNA Biology and Cancer Laboratory, Peter MacCallum Cancer Centre, Melbourne, VIC, Australia.
Russell Gear, Victorian Clinical Genetics Services, Royal Children’s Hospital, VIC, Australia.
Sean Massey, Brain and Mitochondrial Research Group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia.
Edward K Murrell, Brain and Mitochondrial Research Group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia.
Lyndon Gallacher, Department of Paediatrics, University of Melbourne, Melbourne, Australia; Victorian Clinical Genetics Services, Royal Children’s Hospital, VIC, Australia.
Kate Pope, Bruce Lefroy Centre for Genetic Health Research, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia.
Paul J Lockhart, Bruce Lefroy Centre for Genetic Health Research, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia.
Andrew Kornberg, Department of Paediatrics, University of Melbourne, Melbourne, Australia; Neurology Department, Royal Children's Hospital, Melbourne, Australia; Neurosciences Research, Murdoch Children’s Research Institute, Victoria, Australia.
Lynn Pais, Center for Mendelian Genomics, Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
Marzena Walkiewicz, Translational Genomics Research Group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia.
Cas Simons, Victorian Clinical Genetics Services, Royal Children’s Hospital, VIC, Australia; Translational Genomics Research Group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia.
MCRI Rare Diseases Flagship, Brain and Mitochondrial Research Group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia; Bioinformatics Methods group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia; Victorian Clinical Genetics Services, Royal Children’s Hospital, VIC, Australia; Bruce Lefroy Centre for Genetic Health Research, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia; Neurosciences Research, Murdoch Children’s Research Institute, Victoria, Australia; Translational Genomics Research Group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia.
Vihandha O Wickramasinghe, RNA Biology and Cancer Laboratory, Peter MacCallum Cancer Centre, Melbourne, VIC, Australia; Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Parkville, VIC, Australia.
Susan M White, Department of Paediatrics, University of Melbourne, Melbourne, Australia; Victorian Clinical Genetics Services, Royal Children’s Hospital, VIC, Australia.
John Christodoulou, Brain and Mitochondrial Research Group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia; Department of Paediatrics, University of Melbourne, Melbourne, Australia; Discipline of Child & Adolescent Health, Sydney Medical School, University of Sydney, Sydney, NSW, Australia.
Funding
The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian Government’s Operational Infrastructure Support Program. The Chair in Genomic Medicine awarded to J.C. is generously supported by the Royal Children’s Hospital Foundation. UDP-Vic acknowledges financial support from the Murdoch Children’s Research Institute and the Harbig Foundation. The Rare Disease Flagship acknowledges financial support from the Royal Children’s Hospital Foundation, the Murdoch Children’s Research Institute and the Harbig Foundation. Sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 and in part by National Human Genome Research Institute grant R01 HG009141. VOW is supported by a Veski Innovation Fellowship and a mid-career fellowship from the Victorian Cancer Agency and funding from NHMRC (GNT1127745, 2003542 and 2003545). Conflict of Interest statement: All authors declare that they have no conflict of interest.
Data Availability
All raw data and code will be made available upon reasonable request to the corresponding author.
Ethics
All procedures followed were in accordance with the ethical standards and approved by the Human Research Ethics Committee of the Royal Children’s Hospital (HREC/67401/RCHM-2020) and in accordance with the Helsinki Declaration of 1975, as revised in 2000.
Consent to participate and publish
Written informed consent was obtained from the parents individually and on behalf of their children. Two siblings (Family 1; individuals 1 and 2) and their parents were recruited as part of a gene discovery program in children with undiagnosed neurological disease.
References
- 1. Lin, D.H. and Hoelz, A. (2019) The structure of the nuclear pore complex (an update). Annu. Rev. Biochem., 88, 725–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoelz, A., Debler, E.W. and Blobel, G. (2011) The structure of the nuclear pore complex. Annu. Rev. Biochem., 80, 613–643. [DOI] [PubMed] [Google Scholar]
- 3. Maul, G.G. and Deaven, L. (1977) Quantitative determination of nuclear pore complexes in cycling cells with differing DNA content. J. Cell Biol., 73, 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bayliss, R., Littlewood, T., Strawn, L.A., Wente, S.R. and Stewart, M. (2002) GLFG and FxFG nucleoporins bind to overlapping sites on importin-β. J. Biol. Chem., 277, 50597–50606. [DOI] [PubMed] [Google Scholar]
- 5. Jani, D., Lutz, S., Hurt, E., Laskey, R.A., Stewart, M., and Wickramasinghe, V.O. (2012) Functional and structural characterization of the mammalian TREX-2 complex that links transcription with nuclear messenger RNA export. Nucleic Acids Res., 40, 4562–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wickramasinghe, V.O., McMurtrie, P.I., Mills, A.D., Takei, Y., Penrhyn-Lowe, S., Amagase, Y., Main, S., Marr, J., Stewart, M., and Laskey, R.A. (2010) mRNA export from mammalian cell nuclei is dependent on GANP. Curr. Biol., 20, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Umlauf, D., Bonnet, J., Waharte, F., Fournier, M., Stierle, M., Fischer, B., Brino, L., Devys, D. and Tora, L. (2013) The human TREX-2 complex is stably associated with the nuclear pore basket. J. Cell Sci., 126, 2656–2667. [DOI] [PubMed] [Google Scholar]
- 8. Fischer, T., Strasser, K., Racz, A., Rodriguez-Navarro, S., Oppizzi, M., Ihrig, P., Lechner, J. and Hurt, E. (2002) The mRNA export machinery requires the novel Sac3p-Thp1p complex to dock at the nucleoplasmic entrance of the nuclear pores. EMBO J., 21, 5843–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wickramasinghe, V.O. and Laskey, R.A. (2015) Control of mammalian gene expression by selective mRNA export. Nat. Rev. Mol. Cell Biol., 16, 431–442. [DOI] [PubMed] [Google Scholar]
- 10. Schwarz, J.M., Cooper, D.N., Schuelke, M. and Seelow, D. (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods, 11, 361–362. [DOI] [PubMed] [Google Scholar]
- 11. Kubitscheck, U., Wedekind, P., Zeidler, O., Grote, M. and Peters, R. (1996) Single nuclear pores visualized by confocal microscopy and image processing. Biophys. J., 70, 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aksenova, V., Smith, A., Lee, H., Bhat, P., Esnault, C., Chen, S., Iben, J., Kaufhold, R., Yau, K.C., Echeverria, C.et al. (2020) Nucleoporin TPR is an integral component of the TREX-2 mRNA export pathway. Nat. Commun., 11, 4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wickramasinghe, V.O., Andrews, R., Ellis, P., Langford, C., Gurdon, J.B., Stewart, M., Venkitaraman, A.R. and Laskey, R.A. (2014) Selective nuclear export of specific classes of mRNA from mammalian nuclei is promoted by GANP. Nucleic Acids Res., 42, 5059–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stolz, L.E. and Tuan, R.S. (1996) Hybridization of biotinylated oligo(dT) for eukaryotic mRNA quantitation. Mol. Biotechnol., 6, 225–230. [DOI] [PubMed] [Google Scholar]
- 15. Dickinson, M.E., Flenniken, A.M., Ji, X., Teboul, L., Wong, M.D., White, J.K., Meehan, T.F., Weninger, W.J., Westerberg, H., Adissu, H.et al. (2016) High-throughput discovery of novel developmental phenotypes. Nature, 537, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Larkin, A., Marygold, S.J., Antonazzo, G., Attrill, H., Dos Santos, G., Garapati, P.V., Goodman, J.L., Gramates, L.S., Millburn, G., Strelets, V.B.et al. (2021) FlyBase: updates to the Drosophila melanogaster knowledge base. Nucleic Acids Res., 49, D899–D907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P.et al. (2020) Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. Nature, 581, 434–443. [Google Scholar]
- 18. Hashizume, C., Nakano, H., Yoshida, K. and Wong, R.W. (2010) Characterization of the role of the tumor marker Nup88 in mitosis. Mol. Cancer, 9, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lussi, Y.C., Shumaker, D.K., Shimi, T. and Fahrenkrog, B. (2010) The nucleoporin Nup153 affects spindle checkpoint activity due to an association with Mad1. Nucleus, 1, 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bhattacharjya, S., Roy, K.S., Ganguly, A., Sarkar, S., Panda, C.K., Bhattacharyya, D., Bhattacharyya, N.P. and Roychoudhury, S. (2015) Inhibition of nucleoporin member Nup214 expression by miR-133b perturbs mitotic timing and leads to cell death. Mol. Cancer, 14, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shamseldin, H.E., Makhseed, N., Ibrahim, N., Al-Sheddi, T., Alobeid, E., Abdulwahab, F. and Alkuraya, F.S. (2019) NUP214 deficiency causes severe encephalopathy and microcephaly in humans. Hum. Genet., 138, 221–229. [DOI] [PubMed] [Google Scholar]
- 22. McCloskey, A., Ibarra, A. and Hetzer, M.W. (2018) Tpr regulates the total number of nuclear pore complexes per cell nucleus. Genes Dev., 32, 1321–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Theerthagiri, G., Eisenhardt, N., Schwarz, H. and Antonin, W. (2010) The nucleoporin Nup188 controls passage of membrane proteins across the nuclear pore complex. J. Cell Biol., 189, 1129–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jacinto, F.V., Benner, C. and Hetzer, M.W. (2015) The nucleoporin Nup153 regulates embryonic stem cell pluripotency through gene silencing. Genes Dev., 29, 1224–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia-Segura, L.M., Lafarga, M., Berciano, M.T., Hernandez, P. and Andres, M.A. (1989) Distribution of nuclear pores and chromatin organization in neurons and glial cells of the rat cerebellar cortex. J. Comp. Neurol., 290, 440–450. [DOI] [PubMed] [Google Scholar]
- 26. Toda, T., Hsu, J.Y., Linker, S.B., Hu, L., Schafer, S.T., Mertens, J., Jacinto, F.V., Hetzer, M.W. and Gage, F.H. (2017) Nup153 interacts with Sox2 to enable bimodal gene regulation and maintenance of neural progenitor cells. Cell Stem Cell, 21, 618–634.e7. [DOI] [PubMed] [Google Scholar]
- 27. D'Angelo, M.A., Gomez-Cavazos, J.S., Mei, A., Lackner, D.H. and Hetzer, M.W. (2012) A change in nuclear pore complex composition regulates cell differentiation. Dev. Cell, 22, 446–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kadota, S., Ou, J., Shi, Y., Lee, J.T., Sun, J. and Yildirim, E. (2020) Nucleoporin 153 links nuclear pore complex to chromatin architecture by mediating CTCF and cohesin binding. Nat. Commun., 11, 2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hase, M.E., Kuznetsov, N.V. and Cordes, V.C. (2001) Amino acid substitutions of coiled-coil protein Tpr abrogate anchorage to the nuclear pore complex but not parallel, in-register homodimerization. Mol. Biol. Cell, 12, 2433–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krull, S., Thyberg, J., Bjorkroth, B., Rackwitz, H.R. and Cordes, V.C. (2004) Nucleoporins as components of the nuclear pore complex core structure and Tpr as the architectural element of the nuclear basket. Mol. Biol. Cell, 15, 4261–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bangs, P., Burke, B., Powers, C., Craig, R., Purohit, A. and Doxsey, S. (1998) Functional analysis of Tpr: identification of nuclear pore complex association and nuclear localization domains and a role in mRNA export. J. Cell Biol., 143, 1801–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee, E.S., Wolf, E.J., Ihn, S.S.J., Smith, H.W., Emili, A. and Palazzo, A.F. (2020) TPR is required for the efficient nuclear export of mRNAs and lncRNAs from short and intron-poor genes. Nucleic Acids Res., 48, 11645–11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nussbacher, J.K., Tabet, R., Yeo, G.W. and Lagier-Tourenne, C. (2019) Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron, 102, 294–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jovičić, A., Mertens, J., Boeynaems, S., Bogaert, E., Chai, N., Yamada, S.B., Paul, J.W., 3rd, Sun, S., Herdy, J.R., Bieri, G.et al. (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci., 18, 1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grima, J.C., Daigle, J.G., Arbez, N., Cunningham, K.C., Zhang, K., Ochaba, J., Geater, C., Morozko, E., Stocksdale, J., Glatzer, J.C.et al. (2017) Mutant huntingtin disrupts the nuclear pore complex. Neuron, 94, 93–107.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sheffield, L.G., Miskiewicz, H.B., Tannenbaum, L.B. and Mirra, S.S. (2006) Nuclear pore complex proteins in Alzheimer disease. J. Neuropathol. Exp. Neurol., 65, 45–54. [DOI] [PubMed] [Google Scholar]
- 37. Nofrini, V., Di Giacomo, D. and Mecucci, C. (2016) Nucleoporin genes in human diseases. Eur. J. Hum. Genet., 24, 1388–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fichtman, B., Harel, T., Biran, N., Zagairy, F., Applegate, C.D., Salzberg, Y., Gilboa, T., Salah, S., Shaag, A., Simanovsky, N.et al. (2019) Pathogenic variants in NUP214 cause "plugged" nuclear pore channels and acute febrile encephalopathy. Am. J. Hum. Genet., 105, 48–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sandestig, A., Engstrom, K., Pepler, A., Danielsson, I., Odelberg-Johnsson, P., Biskup, S., Holz, A. and Stefanova, M. (2020) NUP188 Biallelic loss of function may underlie a new syndrome: nucleoporin 188 insufficiency syndrome? Mol Syndromol, 10, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kumar, R., Corbett, M.A., vanBon, B.W., Woenig, J.A., Weir, L., Douglas, E., Friend, K.L., Gardner, A., Shaw, M., Jolly, L.A.et al. (2015) THOC2 mutations implicate mRNA-export pathway in X-linked intellectual disability. Am. J. Hum. Genet., 97, 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mattioli, F., Isidor, B., Abdul-Rahman, O., Gunter, A., Huang, L., Kumar, R., Beaulieu, C., Gecz, J., Innes, M., Mandel, J.L.et al. (2019) Clinical and functional characterization of recurrent missense variants implicated in THOC6-related intellectual disability. Hum. Mol. Genet., 28, 952–960. [DOI] [PubMed] [Google Scholar]
- 42. Beaulieu, C.L., Huang, L., Innes, A.M., Akimenko, M.A., Puffenberger, E.G., Schwartz, C., Jerry, P., Ober, C., Hegele, R.A., McLeod, D.R.et al. (2013) Intellectual disability associated with a homozygous missense mutation in THOC6. Orphanet J. Rare Dis., 8, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Amos, J.S., Huang, L., Thevenon, J., Kariminedjad, A., Beaulieu, C.L., Masurel-Paulet, A., Najmabadi, H., Fattahi, Z., Beheshtian, M., Tonekaboni, S.H.et al. (2017) Autosomal recessive mutations in THOC6 cause intellectual disability: syndrome delineation requiring forward and reverse phenotyping. Clin. Genet., 91, 92–99. [DOI] [PubMed] [Google Scholar]
- 44. Casey, J., Jenkinson, A., Magee, A., Ennis, S., Monavari, A., Green, A., Lynch, S.A., Crushell, E. and Hughes, J. (2016) Beaulieu-Boycott-Innes syndrome: an intellectual disability syndrome with characteristic facies. Clin. Dysmorphol., 25, 146–151. [DOI] [PubMed] [Google Scholar]
- 45. Karakaya, M., Mazaheri, N., Polat, I., Bharucha-Goebel, D., Donkervoort, S., Maroofian, R., Shariati, G., Hoelker, I., Monaghan, K., Winchester, S.et al. (2017) Biallelic MCM3AP mutations cause Charcot-Marie-tooth neuropathy with variable clinical presentation. Brain, 140, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kennerson, M.L., Corbett, A.C., Ellis, M., Perez-Siles, G. and Nicholson, G.A. (2018) A novel MCM3AP mutation in a Lebanese family with recessive Charcot-Marie-tooth neuropathy. Brain, 141, e66. [DOI] [PubMed] [Google Scholar]
- 47. Schuurs-Hoeijmakers, J.H., Vulto-van Silfhout, A.T., Vissers, L.E., vanDe, V., II, vanBon, B.W., deLigt, J., Gilissen, C., Hehir-Kwa, J.Y., Neveling, K., delRosario, M.et al. (2013) Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J. Med. Genet., 50, 802–811. [DOI] [PubMed] [Google Scholar]
- 48. Ylikallio, E., Woldegebriel, R. and Tyynismaa, H. (2018) Reply: a novel MCM3AP mutation in a Lebanese family with recessive Charcot-Marie-tooth neuropathy. Brain, 141, e67. [DOI] [PubMed] [Google Scholar]
- 49. Nousiainen, H.O., Kestila, M., Pakkasjarvi, N., Honkala, H., Kuure, S., Tallila, J., Vuopala, K., Ignatius, J., Herva, R. and Peltonen, L. (2008) Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat. Genet., 40, 155–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smith, C., Parboosingh, J.S., Boycott, K.M., Bönnemann, C.G., Mah, J.K., Care4Rare Canada Consortium, Lamont, R.E., Micheil Innes, A. and Bernier, F.P. (2017) Expansion of the GLE1-associated arthrogryposis multiplex congenita clinical spectrum. Clin. Genet., 91, 426–430. [DOI] [PubMed] [Google Scholar]
- 51. Said, E., Chong, J.X., Hempel, M., Denecke, J., Soler, P., Strom, T., Nickerson, D.A., Kubisch, C., University of Washington Center for Mendelian, G, Bamshad, M.J.et al. (2017) Survival beyond the perinatal period expands the phenotypes caused by mutations in GLE1. Am. J. Med. Genet. A, 173, 3098–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tan, Q.K., McConkie-Rosell, A., Juusola, J., Gustafson, K.E., Pizoli, C.E., Buckley, A.F. and Jiang, Y.H.et al. (2017) The importance of managing the patient and not the gene: expanded phenotype of GLE1-associated arthrogryposis. Cold spring Harb Mol case study, 3. doi: 10.1101/mcs.a002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Braun, D.A., Lovric, S., Schapiro, D., Schneider, R., Marquez, J., Asif, M., Hussain, M.S., Daga, A., Widmeier, E., Rao, J.et al. (2018) Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J. Clin. Invest., 128, 4313–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Basel-Vanagaite, L., Muncher, L., Straussberg, R., Pasmanik-Chor, M., Yahav, M., Rainshtein, L., Walsh, C.A., Magal, N., Taub, E., Drasinover, V.et al. (2006) Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann. Neurol., 60, 214–222. [DOI] [PubMed] [Google Scholar]
- 55. Rosti, R.O., Sotak, B.N., Bielas, S.L., Bhat, G., Silhavy, J.L., Aslanger, A.D., Altunoglu, U., Bilge, I., Tasdemir, M., Yzaguirrem, A.D.et al. (2017) Homozygous mutation in NUP107 leads to microcephaly with steroid-resistant nephrotic condition similar to Galloway-Mowat syndrome. J. Med. Genet., 54, 399–403. [DOI] [PubMed] [Google Scholar]
- 56. Alazami, A.M., Patel, N., Shamseldin, H.E., Anazi, S., Al-Dosari, M.S., Alzahrani, F., Hijazi, H., Alshammari, M., Aldahmesh, M.A., Salih, M.A.et al. (2015) Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep., 10, 148–161. [DOI] [PubMed] [Google Scholar]
- 57. Muir, A.M., Cohen, J.L., Sheppard, S.E., Guttipatti, P., Lo, T.Y., Weed, N., Doherty, D., DeMarzo, D., Fagerberg, C.R., Kjaersgaard, L.et al. (2020) Bi-allelic loss-of-function variants in NUP188 cause a recognizable syndrome characterized by neurologic, ocular, and cardiac abnormalities. Am. J. Hum. Genet., 106, 623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Taliun, D., Harris, D.N., Kessler, M.D., Carlson, J., Szpiech, Z.A., Torres, R., Taliun, S.A.G., Corvelo, A., Gogarten, S.M., Kang, H.M.et al. (2021) Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature, 590, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Genomes Project, C., Auton, A., Brooks, L.D., Durbin, R.M., Garrison, E.P., Kang, H.M., Korbel, J.O., Marchini, J.L., McCarthy, S., McVean, G.A.et al. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Philippakis, A.A., Azzariti, D.R., Beltran, S., Brookes, A.J., Brownstein, C.A., Brudno, M. et al. (2015) The Matchmaker Exchange: a platform for rare disease gene discovery. Hum. Mutat., 36, 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fowler, K.J. (1984) Storage of skin biopsies at −70 degrees C for future fibroblast culture. J. Clin. Pathol., 37, 1191–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jones, T.R., Kang, I.H., Wheeler, D.B., Lindquist, R.A., Papallo, A., Sabatini, D.M., Golland, P. and Carpenter, A.E. (2008) CellProfiler analyst: data exploration and analysis software for complex image-based screens. BMC Bioinformatics, 9, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data and code will be made available upon reasonable request to the corresponding author.