

Abstract
Investigations of major mevalonate pathway enzymes have demonstrated the importance of local isoprenoid synthesis in cardiac homeostasis. Farnesyl diphosphate synthase (FPPS) synthesizes isoprenoid precursors needed for cholesterol biosynthesis and protein prenylation. Wang et al., in a recently published article in the Journal of Pathology, elegantly elucidated the pathological outcomes of FPPS deficiency in cardiomyocytes, which paradoxically resulted in increased prenylation of the small GTPases, Ras and Rheb. Cardiomyocyte FPPS depletion caused severe dilated cardiomyopathy that was associated with enhanced GTP-loading and abundance of Ras and Rheb in lipidated protein-enriched cardiac fractions and robust activation of downstream hypertrophic ERK1/2 and mTOR signaling pathways. Cardiomyopathy and activation of ERK1/2 and mTOR caused by loss of FPPS were ameliorated by inhibition of farnesyltransferase, suggesting that impairment of FPPS activity results in promiscuous activation of Ras and Rheb through non-canonical actions of farnesyltransferase. Here, we discuss the findings and adaptive signaling mechanisms in response to disruption of local cardiomyocyte mevalonate pathway activity, highlighting how alteration in a key branch point in the mevalonate pathway affects cardiac biology and function and perturbs protein prenylation, which might unveil novel strategies and intricacies of targeting the mevalonate pathway to treat cardiovascular diseases.
Keywords: farnesyl diphosphate synthase, mevalonate pathway, prenylation, statins, farnesylation, geranylgeranylation, small GTPases, CAAX motif, Ras, Rheb
The mevalonate pathway is critical for cholesterol biosynthesis as well as the generation of lipid precursors for protein prenylation, a post-translational modification essential for the processing, trafficking, and function of many essential signaling transduction proteins, including heterotrimeric G protein γ-subunits and most Ras superfamily small GTPases, as well as nuclear lamins [1,2]. The mevalonate pathway has long been targeted clinically for the treatment of cardiovascular diseases with statins that inhibit HMG-CoA reductase, the rate-limiting enzyme in the pathway (Figure 1). Although predominantly thought to reduce cardiovascular risk through their cholesterol-lowering effects, statins are also cardioprotective in part through inhibition of protein prenylation and consequent diminishment of the activity of Rho family small GTPases, particularly Rac1 and its promotion of myocardial oxidative stress in cardiac hypertrophy and heart failure [1,3,4].
Figure 1:
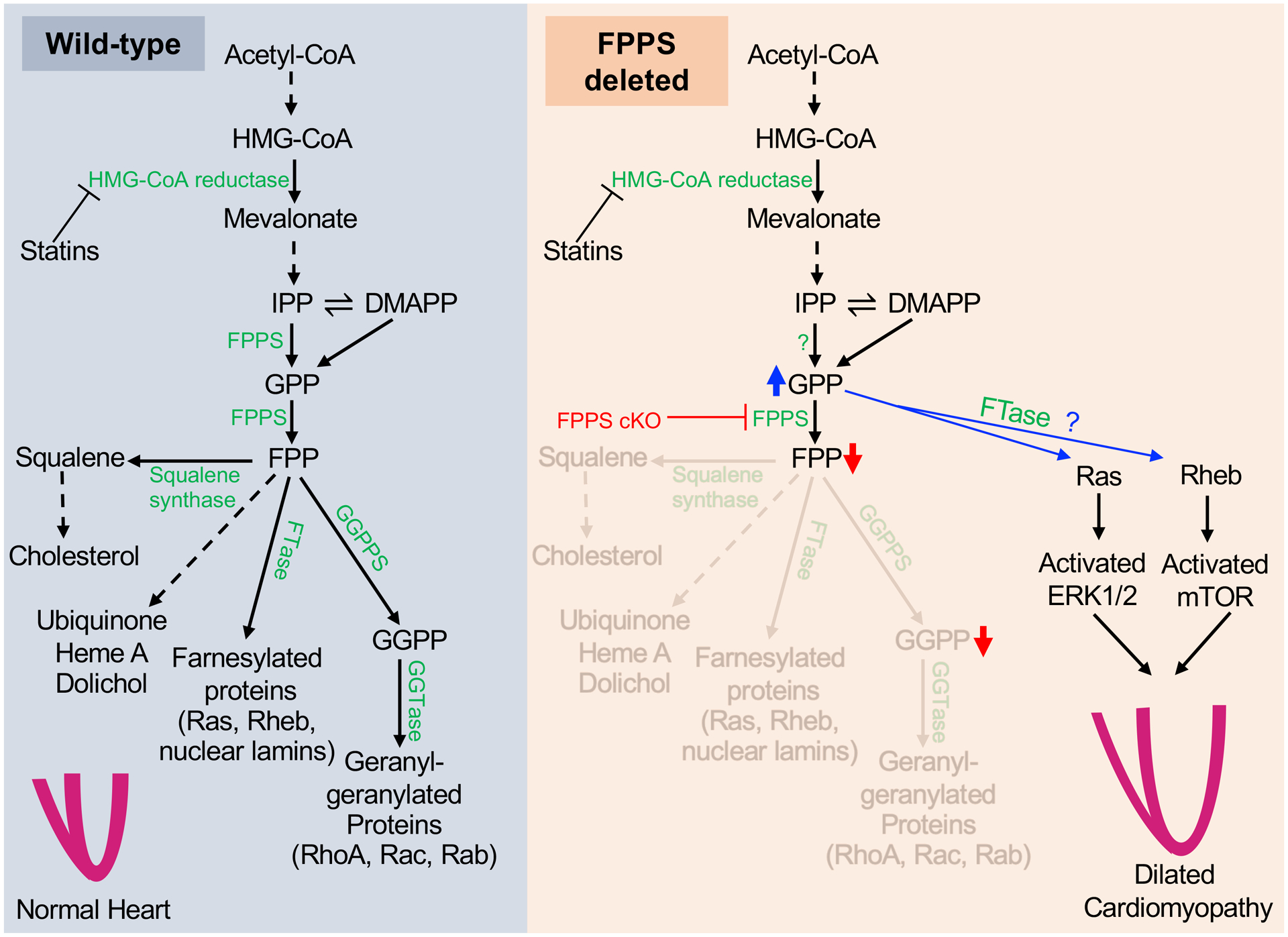
Mevalonate pathway dysregulation and compensatory mechanisms in cardiomyocytes in the absence of farnesyl diphosphate synthase (FPPS). (Left) In wild-type hearts, physiologic expression of major enzymes involved in the mevalonate pathway facilitates normal synthesis of isoprenoid precursors needed for steroid synthesis and protein prenylation, leading to preserved heart development and function. (Right) In FPPS-depleted hearts, FPP and GGPP levels are reduced due to absence of FPPS, while GPP levels are substantially elevated. Accumulated GPP may serve as a substrate for the FTase-mediated prenylation of Ras and Rheb resulting in downstream activation of the ERK1/2 and mTOR pathways, respectively. Activated ERK1/2 and mTOR induce pathological remodeling that contributes to lethal dilated cardiomyopathy in FPPS-deficient mice. HMG-CoA: hydroxymethylglutaryl-CoA; IPP: Isopentenyl pyrophosphate; DMAPP: Dimethylallyl pyrophosphate; GPP: Geranyl pyrophosphate; FPP: Farnesyl pyrophosphate; GGPP: Geranylgeranyl pyrophosphate; FPPS: Farnesyl pyrophosphate synthase; GGPPS: Geranylgeranyl pyrophosphate synthase; FTase: Farnesyl transferase; GGTase: Geranylgeranyl transferase; ERK: Extracellular signal-regulated kinase; mTOR: mammalian target of rapamycin.
In contrast to the cardioprotective effects derived from pharmacological inhibition of HMG-CoA reductase, Wang et al. recently reported that mice with cardiomyocyte-specific deletion of farnesyl pyrophosphate synthase (FPPS), a nodal branch point enzyme in the mevalonate biosynthetic pathway (Figure 1), develop lethal dilated cardiomyopathy [5]. This finding highlights critical homeostatic roles for local mevalonate pathway activity and FPPS in cardiomyocytes that are indispensable for proper cardiac physiology and function.
FPPS converts geranyl pyrophosphate (GPP, 10-carbon) into the 15-carbon unsaturated isoprenyl lipid moiety, farnesyl pyrophosphate (FPP), that is used for protein farnesylation or to generate squalene for cholesterol production (Figure 1). FPP can be further converted into geranylgeranyl pyrophosphate (GGPP, 20-carbon isoprenoid) by GGPP synthase (GGPPS). Geranylgeranyl (20-carbon) or farnesyl (15-carbon) isoprenoids are covalently added to cysteines on proteins by the action of geranylgeranyl transferases and farnesyltransferase, respectively (collectively termed protein prenylation) [2].
The liver is the major systemic regulator of cholesterol homeostasis and generates sufficient cholesterol for the entire body, suggesting that cardiac maladaptation in mice lacking cardiomyocyte FPPS (FPPS cKO) likely originates from deficits or dysfunction in protein prenylation as a consequence of loss of local generation of isoprenoid pyrophosphates necessary for geranylgeranylation and farnesylation. Specifically, disruption of prenylation of small GTPases that require these modifications for proper signaling activity and are essential for cardiac physiology and adaptation [2,3,6,7] could promote dilated cardiomyopathy in FPPS cKO mice. Indeed, hearts lacking FPPS have normal cholesterol levels [5], suggesting absence of cardiomyocyte FPPS does not impair cholesterol homeostasis. Consistent with loss of FPPS-mediated catalysis, FPPS-deleted hearts exhibit accumulation of the FPPS substrate GPP (10-carbon) and reduced levels of FPP (15-carbon) and GGPP (20-carbon) [5] needed for canonical protein prenylation. Importantly, data from FPPS cKO mice and other animal models indicate that impairment of protein prenylation in cardiomyocytes results in severe cardiac dysfunction even in the absence of pathological stimuli [5,8,9].
Interestingly, the same group previously found that partial loss of FPPS activity with siRNA-mediated knockdown or pharmacological inhibition ameliorated cardiac hypertrophy and fibrosis in response to pressure overload [10] or angiotensin-II [11], respectively. These data, taken together with the cardiovascular protection afforded by statin drugs, indicate that dampening flux through the mevalonate pathway reduces adverse cardiac remodeling, whereas complete abolishment of this pathway using a clean genetic strategy is deleterious, resulting in cardiac decompensation and lethality [5]. Interestingly, enhanced generation of isoprenoids in cardiomyocytes can also be maladaptive, as transgenic mice with cardiomyocyte-specific overexpression of FPPS similarly develop dilated cardiomyopathy and exhibit exacerbated cardiac damage in response to ischemia reperfusion injury [12,13]. Although this result is not surprising given the heart failure phenotypes of RhoA [6] and Rac1 [7] transgenic mice and established functions of Rho family GTPases in heart failure [3,4], it underscores the necessity for proper homeostatic isoprenoid biosynthesis in cardiac physiology.
Wang et al. found accumulation of GPP and enhanced abundance of membrane-associated Rheb and Ras in FPPS cKO hearts [5]. How could Rheb and Ras accumulate in the membrane without farnesylation? The authors hypothesize the rogue prenylation of Rheb and Ras using the abundant 10-carbon GPP substrate rather than the 15-carbon FPP typically used to modify the C-terminus of these proteins (Figure 1). Concomitant with increased membrane association, the authors observed profound enhancement of GTP-loading on Rheb and Ras and downstream phosphorylation of mammalian target of rapamycin (mTOR) and extracellular signal-regulated kinases 1 and 2 (ERK1/2) in FPPS cKO hearts (Figure 1). Wang et al. propose that, under the conditions of elevated GPP levels in hearts lacking FPPS, farnesyltransferase transfers the geranyl moiety to Rheb and Ras, resulting in hyperactivation of mTOR and ERK1/2 signaling that promotes cardiac hypertrophy and maladaptation [5]. This has some plausibility given that recombinant farnesyltransferase is capable of transferring the 10-carbon GPP as an alternate substrate in a purified in vitro system [14]. More importantly, treatment of FPPS cKO mice with a farnesyltransferase inhibitor ameliorated cardiac hypertrophy and dysfunction as well as activation of the mTOR and ERK1/2 pathways [5].
It is notable that cardiomyocyte-specific deletion of GGPPS, the enzyme downstream of FPPS (Figure 1), results in a very similar dilated cardiomyopathy phenotype to FPPS cKO mice that is also associated with elevated levels of membrane-localized Rheb and activation of mTOR signaling [8]. GGPPS produces GGPP that is used for geranylgeranylation (20-carbon prenylation) of the CAAX cysteine on the C-terminus of proteins such as Rho family small GTPases, while Rheb is typically farnesylated (15-carbon prenylation) [2,5]. While depletion of FPPS or GGPPS in cardiomyocytes both enhance membrane-associated Rheb and cause cardiomyocyte hypertrophy that is mitigated by pharmacological inhibition of farnesyltransferase, specific inhibition of mTOR signaling with rapamycin improves cardiomyopathy in mice lacking GGPPS but not in mice lacking FPPS [5,8]. Thus, while activation of Rheb/mTOR signaling is responsible for cardiac hypertrophy and maladaptive remodeling elicited by loss of GGPPS, cardiac decompensation in FPPS cKO mice requires additional pathogenic signaling mechanisms such as Ras/ERK.
Elevation of GPP levels in FPPS-deleted hearts is unexpected, given that FPPS catalyzes not just conversion of GPP to FPP, but also the prior step in the mevalonate pathway that generates GPP (Figure 1). These data suggest that some other enzyme can compensate and synthesize GPP (but not FPP) in the absence of FPPS. Therefore, there appear to be redundancies in the mevalonate pathway to maintain adequate protein lipidation and synthesis of metabolites (i.e. dolichols, ubiquinone, heme A) when a nodal enzyme is lacking or malfunctional. In the FPPS cKO heart, this mechanism might allow compensatory production of both GPP as well as presumably farnesyltransferase-mediated transfer of GPP onto small GTPases, which hijacks prenyltransferases when deficiencies in FPP and GGPP prevent protein farnesylation and geranylgeranylation, respectively (Figure 1). Although compensatory mechanisms exist in the absence of mevalonate pathway enzymes, the process of protein prenylation seems particularly prone to dysfunction and promiscuous transfer of isoprenoids to small GTPases, which results in aberrant signaling and cardiac dysfunction. Indeed, gain-of-function or loss of an individual mevalonate pathway enzyme in cardiomyocytes can result in severe cardiomyopathy that is often associated with enhanced small GTPase signaling [5,8,9,12,13].
Hyperactive Rheb/mTOR and Ras/ERK signaling due to aberrant protein prenylation likely account for or contribute significantly to cardiomyopathy in FPPS cKO mice [5]. However, the mevalonate pathway also has essential functions in cardiac development including promotion of cardiomyocyte proliferative capacity [15] and G protein γ1 signaling necessary for cardiac morphogenesis [9]. Thus, defective mevalonate pathway function in the early postnatal period following α-myosin heavy chain-promoter-driven deletion of FPPS [5] could manifest in cardiac maturation defects that could also participate in deterioration of cardiac function. Moreover, FPPS is also needed to generate metabolites such as dolichols necessary for synthesis of oligosaccharides used for protein N-glycosylation [16] and ubiquinone (coenzyme Q) that participates in the mitochondrial electron transport chain [17] (Figure 1). Thus, potential impairment of glycoprotein function and mitochondrial bioenergetics, respectively, could also play a role in the pathogenesis of FPPS cKO mice.
Overall, findings in FPPS cKO mice highlight the necessity of mevalonate pathway enzymes in cardiomyocytes and adaptive mechanisms that maintain lipidation of small GTPases in the absence of nodal biosynthetic enzymes, although this often results in aberrant hyperactivation of small GTPase signaling that induces cardiomyopathy. In the future, metabolic labeling studies such as with bioorthogonal lipid probes amenable to click chemistry could clarify the promiscuity of protein prenylation machinery, including the role of farnesyltransferase and other enzymes in transferring the 10-carbon geranyl group, the kinetics of this modification on Rheb, Ras, and other small GTPases, and if this occurs at the canonical cysteine on the C-terminal CAAX motif.
Notably, FPPS mRNA and protein levels were substantially reduced in hearts of patients with heart failure with reduced ejection fraction (HFrEF) [5], implicating FPPS in the molecular etiology of human heart failure. Collectively, studies by Wang et al. [5] and others [8,10–13] have established that proper mevalonate pathway activity and protein prenylation are imperative for cardiac homeostasis but also dysregulated in heart failure. Thus, further investigation of the cell-type specific functions of mevalonate pathway enzymes in vivo will facilitate greater understanding of the nuances of local mevalonate pathway function and dysregulation in disease. This knowledge could aid the identification of next-generation drug targets and the development of strategies to fill unmet needs for the treatment of heart failure.
Acknowledgements
This work was supported by grants from the National Institutes of Health to MJB (R00HL136695) and the American Heart Association to KE (827440).
Footnotes
Invited Commentary for Wang et al. Cardiac-specific deletion of FDPS induces cardiac remodeling and dysfunction by enhancing the activity of small GTP-binding proteins. J Pathol 2021; doi: 10.1002/path.5789
Conflict of Interest: The authors declare no conflicts of interest
References
- 1.Werner N, Nickenig G, Laufs U. Pleiotropic effects of HMG-CoA reductase inhibitors. Basic Res Cardiol 2002; 97: 105–116. [DOI] [PubMed] [Google Scholar]
- 2.Jiang H, Zhang X, Chen X, et al. Protein lipidation: Occurrence, mechanisms, biological functions, and enabling technologies. Chem Rev 2018; 118: 919–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maack C, Kartes T, Kilter H, et al. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation 2003; 108: 1567–1574. [DOI] [PubMed] [Google Scholar]
- 4.Takemoto M, Node K, Nakagami H, et al. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest 2001; 108: 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Zhang X, Chen Y, et al. Cardiac-specific deletion of FDPS induces cardiac remodeling and dysfunction by enhancing the activity of small GTP-binding proteins. J Pathol 2021; doi: 10.1002/path.5789 [DOI] [PubMed] [Google Scholar]
- 6.Sah VP, Minamisawa S, Tam SP, et al. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest 1999; 103: 1627–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sussman MA, Welch S, Walker A, et al. Altered focal adhesion regulation correlates with cardiomyopathy in mice expressing constitutively active rac1. J Clin Invest 2000; 105: 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu N, Guan S, Chen Z, et al. The alteration of protein prenylation induces cardiomyocyte hypertrophy through Rheb-mTORC1 signalling and leads to chronic heart failure. J Pathol 2015; 235: 672–685. [DOI] [PubMed] [Google Scholar]
- 9.Yi P, Han Z, Li X, et al. The mevalonate pathway controls heart formation in Drosophila by isoprenylation of Ggamma1. Science 2006; 313: 1301–1303. [DOI] [PubMed] [Google Scholar]
- 10.Zhao CZ, Zhao XM, Yang J, et al. Inhibition of farnesyl pyrophosphate synthase improves pressure overload induced chronic cardiac remodeling. Sci Rep 2016; 6: 39186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Zhu HH, Chen GP, et al. Inhibition of farnesyl pyrophosphate synthase attenuates angiotensin II-induced cardiac hypertrophy and fibrosis in vivo. In J Biochem Cell Biol 2013; 45: 657–666. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Mou Y, Wu T, et al. Cardiac-specific overexpression of farnesyl pyrophosphate synthase induces cardiac hypertrophy and dysfunction in mice. Cardiovasc Res 2013; 97: 490–499. [DOI] [PubMed] [Google Scholar]
- 13.Dai D, Sun X, Ding J, et al. Overexpression of farnesyl pyrophosphate synthase increases myocardial ischemia/reperfusion injury in mice. Gene 2018; 672: 72–78. [DOI] [PubMed] [Google Scholar]
- 14.Micali E, Chehade KA, Isaacs RJ, et al. Protein farnesyltransferase isoprenoid substrate discrimination is dependent on isoprene double bonds and branched methyl groups. Biochemistry 2001; 40: 12254–12265. [DOI] [PubMed] [Google Scholar]
- 15.Mills RJ, Parker BL, Quaife-Ryan GA, et al. Drug screening in human PSC-cardiac organoids identifies pro-proliferative compounds acting via the mevalonate pathway. Cell Stem Cell 2019; 24: 895–907 e896. [DOI] [PubMed] [Google Scholar]
- 16.Siddals KW, Marshman E, Westwood M, et al. Abrogation of insulin-like growth factor-I (IGF-I) and insulin action by mevalonic acid depletion: synergy between protein prenylation and receptor glycosylation pathways. J Biol Chem 2004; 279: 38353–38359. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Oxer D, Hekimi S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat Commun 2015; 6: 6393. [DOI] [PMC free article] [PubMed] [Google Scholar]