

Abstract
Context
Youth with classical congenital adrenal hyperplasia (CAH) exhibit abnormal adrenomedullary function with decreased epinephrine levels noted in newborns and young infants. Little is known about how this relates to morbidity during the first year of life.
Objective
This work aimed to study plasma epinephrine levels in infants with classical CAH and examine the clinical significance of epinephrine deficiency in the first year of life.
Methods
This prospective cohort study comprised participants recruited from a pediatric tertiary care center: 36 infants with classical CAH due to 21-hydroxylase deficiency and 27 age-matched unaffected controls with congenital hypothyroidism. Main outcome measures included plasma epinephrine levels (N = 27), CYP21A2 genotype (N = 15), and incidence of acute illnesses from birth to age 1 year (N = 28).
Results
Epinephrine levels in CAH infants independently predicted illness incidence in the first year of life (β = –0.018, R = –0.45, P = .02) and were negatively correlated with 17-hydroxyprogesterone at diagnosis (R = –0.51, P = .007). Infants with salt-wasting CAH exhibited lower epinephrine levels as newborns than simple-virilizing infants (P = .02). CAH patients had lower epinephrine as newborns than did controls (P = .007) and showed decreases in epinephrine from birth to age 1 year (P = .04). Null genotype was associated with lower newborn epinephrine and more illness in the first year of life, compared to less severe mutation categories.
Conclusion
Lower epinephrine levels are associated with increased risk of illness among CAH infants. While not currently part of clinical standard of care, measuring epinephrine levels and assessing genotype may help predict acute illness in the first year of life.
Keywords: congenital adrenal hyperplasia, pediatrics, adrenal medulla, illness, catecholamines, epinephrine
Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency (21-OHD) is a potentially life-threatening disorder caused by pathogenic variations in the CYP21A2 gene. It is the most common cause of primary adrenal insufficiency in children, affecting 1:15 000 newborns in its classical, severe form (1). Youth with classical (severe) CAH exhibit cortisol deficiency, often with decreased aldosterone synthesis, that can result in adrenal crisis during illness (2, 3). Rates of illness, hospital admissions, and mortality are highest in the first few years of life and are largely driven by adrenal crisis following infection (4-10), with the mechanisms underlying increased illness remaining unknown.
Epinephrine deficiency may play an important role in illness in very young patients with classical CAH, in conjunction with the cortisol deficiency. Epinephrine and cortisol levels typically rise in response to hypoglycemia in healthy infants and toddlers as counterregulatory hormones; this places infants and toddlers with CAH who are deficient in these hormones at risk for hypoglycemia during illness (11, 12); multiple studies have shown that hypoglycemia is a significant issue for youth with CAH (13-16). Intra-adrenally produced cortisol regulates the differentiation of adrenal chromaffin cells and catecholamine synthesis through paracrine signaling (17), resulting in abnormal adrenomedullary morphology and function in youth with CAH (14). As well, CYP21A2 genotypes that predict a more severe cortisol deficiency are also associated with a greater epinephrine deficiency (18-23).
As early as the newborn period, infants with CAH exhibit significantly lower levels of plasma epinephrine as compared to unaffected age-matched controls (24). Adolescent and adult patients with CAH exhibit lower plasma and urinary epinephrine in resting states and during mild-, moderate-, and high-intensity exercise (18, 23, 25-28), with inadequate production of epinephrine thought to cause glucose deficiency during illness and exercise. In addition, undetectable or low levels of epinephrine have been found to be associated with increased emergency room (ER) visits during childhood and increased illness during adulthood (2).
To our knowledge, the clinical significance of epinephrine deficiency has yet to be demonstrated early in life in patients with CAH, when they are at highest risk for severe illnesses and life-threatening adrenal crises. Therefore, we proposed to study epinephrine levels in the first year of life, along with CYP21A2 genotype, in infants with classical CAH to better understand the biochemical and genetic factors contributing to higher incidences of illness during infancy.
Materials and Methods
This study was approved by the Children’s Hospital Los Angeles Institutional Review Board (IRB). Parents of participating infants gave written informed consent (IRB No. CHLA09-00260). Study data were collected and managed using REDCap (Research Electronic Data Capture) housed at the Southern California Clinical and Translational Science Institute (29).
Study Participants
Newborns were recruited from the pediatric endocrinology clinic or hospital ward on referral by the California Department of Health Services for a positive newborn screen. All CAH youth had 21-OHD as confirmed by biochemical testing (elevated 17-hydroxyprogesterone; 17-OHP) and/or genotyping of the CYP21A2 gene. The control group consisted of newborns with congenital primary hypothyroidism as diagnosed by a positive newborn screen (elevated thyrotropin) and confirmatory serum testing, with no other medical problems. Control newborns were not acutely ill, became euthyroid after initiation of levothyroxine therapy, and were selected as controls as the IRB did not allow for blood draws in healthy infants unless clinically indicated. Additionally, previous research has indicated no confounding effects of levothyroxine treatment on plasma catecholamine levels (24). Exclusion criteria included infants who had a different type of CAH than 21-hydroxylase deficiency, and newborns with medical conditions other than congenital hypothyroidism.
Thirty-six infants with classical CAH due to 21-OHD and 27 controls were enrolled in the study. The groups were similar for sex (P = .4), age at newborn blood draw (P = .8), and age at 1-year blood draw (P = .2) (Table 1). Within the CAH group, 28 infants had salt-wasting (SW) CAH and 8 had simple-virilizing (SV) CAH. The average value of the highest 17-OHP level at the time of diagnosis (see “Hormone Analyte Measurements”) was 19 653 ± 13 095 ng/dL (SI unit conversion * 0.0302 = nanomoles per liter).
Table 1.
Study participant characteristics
| Median (IQR), mean ± SD, or n (%) | P | ||
|---|---|---|---|
| CAH (N = 36) | Controls (N = 27) | ||
| Sex (female) | 22 (63.9) | 21 (77.8) | .4 |
| Age, y | |||
| At newborn draw | 0.02 (0.02-0.06) | 0.02 (0.02-0.03) | .8 |
| At 1-y draw | 1.3 (1.1-1.4) | 1.4 (1.3-1.9) | .2 |
| Type of CAH | |||
| Salt-wasting | 28 (77.8) | – | |
| Simple-virilizing | 8 (22.2) | – | |
| 17-OHP at birth, ng/dL | 19 653 ± 13 095 | – | |
| Epinephrine, pg/mL | 77.06 ± 51.61 | 127.69 ± 54.02 | .004 |
Abbreviations: 17-OHP, 17-hydroxyprogesterone; CAH, congenital adrenal hyperplasia; IQR, interquartile range.
Hormone Analyte Measurements
Serum 17-OHP (liquid chromatography–tandem mass spectrometry; LC/MS-MS), and plasma epinephrine levels (quantified by high-performance LC, Quest Diagnostics Nichols Institute) were obtained at the time of a clinically indicated blood draw. Interassay and intra-assay coefficients of variation were less than 5%. Draws between age 0 and 0.12 years (0-6 weeks) were categorized as 0 year (birth) and draws occurring between age 0.87 and 1.92 years (45-100 weeks) were categorized as 1 year. Interassay and intra-assay coefficients of variation were less than 5%. Some levels of epinephrine were under the commercial clinical assay limit of detection or LOD (40 pg/mL or 80 pg/mL, depending on sample volume, SI unit conversion * 5.454 = picomoles per liter). In these scenarios, the laboratory reported values when a distinct nonblank peak could be detected in the high-performance LC system whereas undetectable results were reported as one one-hundredth less than the LOD (eg, < 40 pg/mL was recorded as 39.99 pg/mL) for analysis. Within CAH youth, 17-OHP was collected at newborn screen and/or confirmatory draw at the time of diagnosis as a newborn. The highest 17-OHP between the newborn screen filter-paper, dried blood specimen (California Department of Health Services; conversion nanomolar [nM] whole blood units × 66 for conversion to nanogram per deciliter [ng/dL] serum concentration) (30) and confirmatory serum draw at diagnosis (LC/MS-MS, Quest Diagnostics Nichols Institute) was used for analysis.
Genotyping
Fifteen of 36 infants with CAH also had CYP21A2 gene testing performed. Following previously described methodology, large gene rearrangements (deletions and large gene conversions) were identified through Multiplex Ligation-Dependent Probe Amplification methodology, and known and novel variants through the entire CYP21A2 gene sequencing (31). Genotypes were then categorized based on predicted residual 21-OHD enzyme activity (20, 31, 32). Null included those patients with variants (eg, homozygous large gene deletions, severe point mutations) on both alleles that were predicted to abolish the enzymatic activity. Group A included those with less than 2% residual predicted enzymatic activity, most commonly due to homozygosity for aberrant splicing at intron 2 (In2/In2) or in compound heterozygosity with a null variant (Null/In2). Group B included those with 3% to 7% residual predicted enzymatic activity, most often caused by a homozygous I172N variant (I172N/I172N) considered to be a less severe variant, or in compound heterozygosity with a more severe variant (I172N/Null or I172N/In2). The genotypes categorized as Null are expected to result in the SW form of CAH. Group A is associated with either the SW and SV form of CAH, and Group B is associated with the SV form.
Medical Chart Review
Medical history and physical examination data for each participant were collected from the electronic medical record. In a subset of 28 CAH and 17 control youth, medical charts were available to retrospectively track frequency of acute illness (ie, fever, vomiting, and/or diarrhea), details of stress-dosing, and number of ER visits and hospital admission during their first year of life.
Statistical Analysis
Data were analyzed using R version 3.6.1 (R Foundation for Statistical Computing). Sample characteristics were reported as count and percentage, median and interquartile range, or mean and SDs. Group comparisons were assessed using chi-square and Mann-Whitney U tests. Mann-Whitney U tests were conducted for cross-sectional comparison of plasma epinephrine between CAH and control groups. Pearson correlations were used to assess associations between continuous variables, with a sensitivity analysis used to examine the effect of uncertain left-censored values. P values of less than .05 were considered statistically significant for all analyses.
Results
Hormone Analytes and Acute Illness
Among newborns with CAH, the newborn epinephrine was negatively correlated with the highest 17-OHP level at the time of diagnosis (N = 27, R = –0.51, P = .007; Fig. 1A). Six patients without newborn 17-OHP data were removed from the original 36 for this cohort, and 3 patients with 17-OHP greater than 40 000 ng/dL were excluded as outliers for the newborn analysis because of adrenal crises and acute illness at birth.
Figure 1.

Plasma epinephrine and 17-hydroxyprogesterone (17-OHP) levels in newborns with classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. A, Newborn epinephrine was negatively correlated with 17-OHP at the time of diagnosis (N = 27, R = –0.51, P = .007). B, Newborn epinephrine was negatively correlated with number of acute illnesses in the first year of life (N = 28, R = –0.45, P = .02). As well, newborn epinephrine was predictive of number of acute illnesses in the first year of life (β = –0.018, P = .02).
Controlling for 17-OHP, newborn epinephrine significantly predicted the number of acute illnesses in the first year of life (β = –0.018, P = .02). The number of acute illnesses in the first year of life was not correlated with highest newborn 17-OHP (R = 0.33, P = .1), but was significantly negatively correlated with newborn epinephrine (N = 28, R = –0.45, P = .02; Fig. 1B). Among these patients, 18 were recorded to have acute illness in the first year of life (64%), 7 were admitted to the ER (25%), 4 were admitted to the hospital (14%), 7 had stress doses administered at home before coming to the ER (25%), and 2 had intramuscular cortisol dosing pre-ER (7%). There was no correlation between average glucocorticoid daily dose from 6 to 12 months and illness episodes within the first year of life (N = 27, R = –0.18, P = .36).
Considering the large subset of epinephrine samples that were approximated below the commercial LOD (33%), we ran a sensitivity analysis with other approximations of these values, including LOD/2, LOD/sqrt(2), and by complete case analysis removing the approximated data points entirely. The highest 17-OHP at the time of diagnosis was still significantly correlated with newborn epinephrine in these analyses (LOD/2: R = –0.56, P = 0.003; LOD/sqrt(2): R = –0.54, P = .003, complete case: R = –0.57, P = .01), as was the negative correlation between acute illnesses in the first year of life with newborn epinephrine (LOD/2: R = –0.39, P = .04; LOD/sqrt(2): R = –0.42, P = .03, complete case: R = –0.54, P = .02). Finally, to consider the possibility that all values of epinephrine, both measured and approximated, under the highest LOD were uncertain, we ran a Tobit regression to estimate the linear relationship between highest 17-OHP value and left-censored newborn epinephrine. With a lower bound of 80 pg/mL to newborn epinephrine, the linear relationship was still significant as was with approximated values at one one-hundredth under the LOD (Tobit: β = –0.0050, P = .007, Approximation: β = –0.0031, P = .002).
Genotype, Hormone Analytes, and Acute Illness
There were 15 infants with CAH who had both a newborn epinephrine level and CYP21A2 genotype (Table 2). We compared the Null genotype category with group A or B and found that the Null group was associated with higher 17-OHP values at diagnosis, lower newborn epinephrine, and more illnesses in the first year of life. Four infants classified as Null had a mean 17-OHP at diagnosis of 22 371 ± 14 024 ng/dL, a mean newborn epinephrine of 60.3 ± 29.4 pg/mL, and 4.0 ± 2.2 illnesses in the first year of life. Five infants classified as group A had a mean 17-OHP of 17 951 ± 2538 ng/dL, an epinephrine of 56.0 ± 21.9 pg/mL (entirely approximated), and 2.2 ± 2.7 illnesses. Finally, 6 infants classified as group B had a mean 17-OHP of 10 724 ± 8782 ng/dL, an epinephrine of 116.0 ± 58.4 pg/mL, and 1.3 ± 1.2 illnesses.
Table 2.
CYP21A2 genotype, newborn analytes, and illness in infants with congenital adrenal hyperplasia
| Infant | Genotype | Group | Predicted phenotype | Clinical phenotype | 17-OHP, ng/dL | Epinephrine, pg/mL | Illness events |
|---|---|---|---|---|---|---|---|
| 1 | Deletion/deletion | Null | SW | SW | 41 970 | 84 | 2 |
| 2 | Deletion/deletion | Null | SW | SW | 20 151 | 39 | 3 |
| 3 | p.R356W/deletion | Null | SW | SW | 18 678 | 87 | 4 |
| 4 | p.G110EfsX21/p.G110EfsX21 | Null | SW | SW | 11 220 | 31 | 7 |
| 5 | g.5774A/C > G/p.G110EfsX21 | A | SW/SV | SW | 19 934 | 79.99 | 0 |
| 6 | g.5774A/C > G/p.P453S/deletion | A | SW | SW | 14 442 | 39.99 | 0 |
| 7 | g.5774A/C > G/c.1451-1452 del | A | SW/SV | SW | 16 796 | 39.99 | 4 |
| 8 | p.R356W/g.5774A/C > G | A | SW/SV | SW | 17 777 | 79.99 | 1 |
| 9 | p.Leu307fs/c.1451-1452 del | A | SW | SW | 20 808 | 39.99 | 6 |
| 10 | p.I172N,p.P453S/p.G110EfsX21 | B | SV | SW | 26 695 | 79.99 | 2 |
| 11 | p.I172N/p.I172N | B | SV | SV | 5219 | 221 | 1 |
| 12 | p.I172N/p.I172N | B | SV | SV | 13 444 | 84 | 3 |
| 13 | p.I172N/p.I172N | B | SV | SV | 10 406 | 83 | 0 |
| 14 | p.I172N/p.I172N | B | SV | SV | 2062 | 78 | 2 |
| 15 | p.172N/g.5774A/C > G | B | SV | SV | 6519 | 150 | 0 |
Abbreviations: 17-OHP, 17-hydroxyprogesterone; SV, simple-virilizing; SW, salt-wasting.
Congenital Adrenal Hyperplasia vs Control Newborns and Infants
Newborns with CAH had significantly lower levels of plasma epinephrine at birth (Fig. 1B; 80.0 pg/mL [range, 40.0-93.5 pg/mL]) as compared to control newborns (101.0 pg/mL [range, 89.0-152.0 pg/mL]; P = .007). There was insufficient statistical power to examine epinephrine group differences at age 1 year.
Infants with CAH had significantly more illnesses in the first year of life when compared to non-CAH controls (CAH: 2 [0-3], controls: 0 [0-0], P = .003]. Stratified by CAH form and genotype category (SW, SV, Null, A, and B), relative risk ratio of illness incidence in the first year of life when compared to controls were all significantly higher, except for the SV subgroup (Table 3).
Table 3.
Relative risk ratio for illness incidence in patients with congenital adrenal hyperplasia vs controls
| Patient group | Relative risk ratio (95% CI) | z score | P |
|---|---|---|---|
| CAH | 4.5 (1.4745-13.7338) | 2.642 | .008 |
| SW | 4.8214 (1.5712-14.7952) | 2.750 | .006 |
| SV | 3.3750 (0.8387-13.5817) | 1.712 | .09 |
| Null | 9 (3.0967-26.1569) | 4.037 | <0.001 |
| A | 6 (1.7934-20.0734) | 2.908 | .004 |
| B | 5.4 (1.4944-19.5134) | 2.573 | .01 |
Abbreviations: 17-OHP, 17-hydroxyprogesterone; CAH, congenital adrenal hyperplasia; SV, simple-virilizing; SW, salt-wasting.
Congenital Adrenal Hyperplasia Form and Epinephrine
Stratified by clinical phenotype, newborns with SW CAH had significantly lower levels of epinephrine (51.0 pg/mL [range, 39.7-84.8 pg/mL]) compared to newborns with SV CAH (87.0 pg/mL [range, 83.5-137.0 pg/mL]; P = .02).
In cross-sectional analysis, plasma epinephrine did not significantly decrease between 0 and 1 year among infants with CAH (P = .1). However, in a paired analysis of 11 infants with CAH followed longitudinally, plasma epinephrine significantly decreased from birth (Fig. 2; 84.0 pg/mL [range, 60.0-155.0 pg/mL]) to age 1 year (40.0 pg/mL [range, 40.0-70.0 pg/mL]; P = .04).
Figure 2.
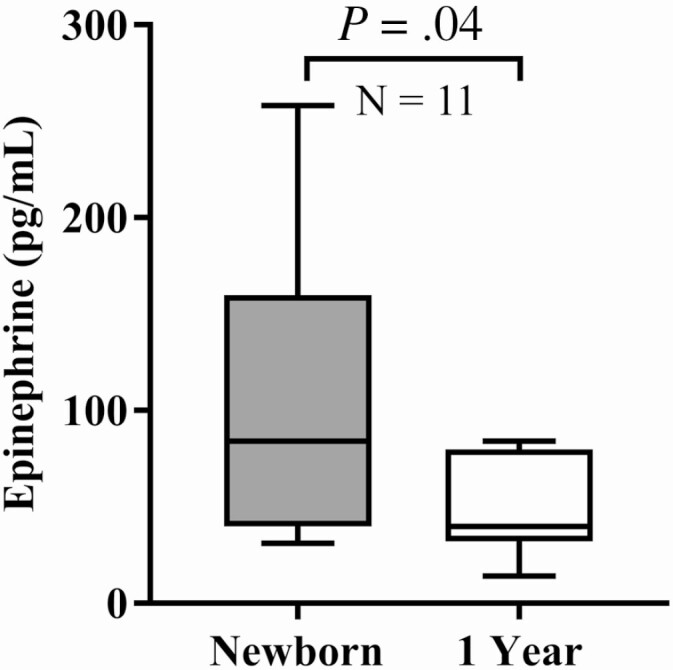
Plasma epinephrine levels at newborn and age 1 year in patients with classical congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency. In 11 infants with CAH, plasma epinephrine decreased longitudinally from newborn 84.0 pg/mL (range, 60.0-155.0 pg/mL) to age 1 year: 40.0 pg/mL (range, 40.0-70.0 pg/mL), P = .04.
Discussion
Our study demonstrates the potential clinical significance of epinephrine deficiency in infants with classical CAH due to 21-OHD during the first year of life. The main finding of our study was that a lower newborn epinephrine level predicted a greater number of illnesses from birth to age 1 year, independent of newborn 17-OHP levels. This suggests that epinephrine deficiency may be a more direct predictor than 17-OHP for frequency of acute illness during infancy for those with CAH. As well, newborn epinephrine levels were significantly lower at age 1 year in the same infants with CAH, suggesting that epinephrine levels are low in infancy and adrenomedullary function remains low in patients with classical CAH (26), even with glucocorticoid replacement therapy.
As a marker of severity, the 17-OHP at presentation may not reflect the situation at times of illness; indeed, our findings show that the number of acute illnesses in the first year of life was not correlated with highest newborn 17-OHP. Resting epinephrine, measured in a newborn with CAH, may not inherently increase throughout their lifespan, and may therefore be another useful indicator of disease severity. Of note, in the 3 newborn outliers with elevated 17-OHP due to adrenal crisis, measured epinephrine values were above the average of patients with CAH, but below the average for control participants (84, 79.99, and 104 pg/mL). This suggests that patients with CAH may be able to mount an epinephrine response in times of illness and stress, albeit a lower response than that of their non-CAH peers. Careful scrutiny of CAH epinephrine draws is thus needed in future studies to ensure that “normal” epinephrine values, coupled with high 17-OHP, are not mistaken as physiological responses similar to controls.
In terms of severity by CYP21A2 genotype and acute illness, we found that infants in the Null category had higher 17-OHP levels at diagnosis, lower newborn epinephrine, and more illnesses in the first year of life. Although the number of genotyped patients was too small to perform statistically significant group analyses, further study is merited with larger sample sizes to better understand the relationship between genotype and acute illness. Similarly, CAH youth with Null- or group A–categorized mutations have been found to exhibit the highest frequency of hypoglycemia accompanying illness (32), and both cortisol and epinephrine deficiencies are likely factors in the impaired glucose metabolism reported in CAH infants during illness (12-16).
Similar to our previous findings, but in a larger cohort, we found that CAH newborns and young infants have reduced epinephrine levels as newborns compared to controls, suggesting that impaired development of the adrenal medulla occurs in utero in these patients (24). The driving mechanism could be deficient production of intra-adrenal glucocorticoids, a necessary cofactor for the action of phenylethanolamine N-methyl transferase (33, 34). Assessment of epinephrine levels is not currently part of the standard-of-care management of patients with CAH. However, our research indicates that baseline epinephrine measurements may help predict those infants who are at highest risk of illness and need closer monitoring. Epinephrine is thought to help mediate the short-term human stress response and can stimulate hepatic glycogenolysis, hepatic gluconeogenesis, adipose tissue lipolysis, and hepatic ketogenesis (35, 36). More studies are needed to explore the causal relationships between cortisol and epinephrine deficiencies, and acute illness, in young patients with CAH.
There are several limitations to our study. First, the sample size is small, although larger than earlier studies in this field. With clinically indicated analytes prioritized over research analytes, only infants who gave more blood than was clinically needed could participate in our study. Obtaining sufficient blood specimens for clinical purposes is challenging in newborns and infants, making the collection of additional volume for research testing and genotyping even more difficult. Owing to small blood volume sample sizes, some of the epinephrine levels were recorded below the clinical limit of detection, diluted, and/or had to be approximated. This led to potential bias in some analyses, especially in certain subsets of patients, such as those with group A mutations, in which all epinephrine samples were below the limit of clinical detection. Using a conventional regression approach does not address the complexity of covariate interactions that may exist between disease severity, 17-OHP at presentation, newborn epinephrine, and other variables (such as free metanephrine or cortisol), all of which will eventually be important in determining causation. Causation of an observed outcome from one event would likely need a study with higher power and mediation models. Finally, the retrospective chart review for illness-related information could have involved several biases including information, recall, and rumination biases as potential influences on the collected data.
We conclude that patients with classical CAH due to 21-OHD have an epinephrine deficiency from early in life, as newborns, that is likely due to in utero alterations to adrenomedullary structure and function, and epinephrine levels remain low throughout infancy. This epinephrine deficiency independently predicts acute illness in the first year of life. Further longitudinal studies are merited, with the need for more robust genotyping of CAH patients in clinical settings. Finally, epinephrine replacement therapy during acute illness and adrenal crises could be assessed as a means to reduce associated morbidity and mortality in patients with CAH.
Acknowledgments
We gratefully thank our patients and families who participated in the study. We also thank Sandy Hall, RN, Norma Martinez, the CHLA Clinical Research Specimen Processing Team, Trevor Pickering, PhD, Jon Nakamoto, MD, and Quest Diagnostics Nichols Institute for their assistance.
Financial Support: This work was supported by the Abell Foundation (to M.E.G.), CARES Foundation Congenital Adrenal Hyperplasia Comprehensive Care Center award (to M.E.G. and M.S.K.), and the National Institutes of Health/National Institute of Child Health and Human Development (grant Nos. 1K23HD084735-01A1 and 1R03HD101718-01 to M.S.K.), and the CHLA Clinical and Translational Science Institute Clinical Trials Unit (grant No. RGP010501 to M.S.K.). REDCap was supported by Southern California Clinical and Translational Science Institute. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Glossary
Abbreviations
- 17-OHP
17-hydroxyprogesterone
- 21-OHD
21-hydroxylase deficiency
- CAH
congenital adrenal hyperplasia
- ER
emergency room
- In2
intron 2
- LC/MS-MS
liquid chromatography–tandem mass spectrometry
- LOD
limit of detection
- SV
simple-virilizing
- SW
salt-wasting
Contributor Information
Jonathan Weber, Center for Endocrinology, Diabetes and Metabolism, Children’s Hospital Los Angeles, Los Angeles, California 90027, USA.
Veeraya K Tanawattanacharoen, Center for Endocrinology, Diabetes and Metabolism, Children’s Hospital Los Angeles, Los Angeles, California 90027, USA.
Amy Seagroves, Center for Endocrinology, Diabetes and Metabolism, Children’s Hospital Los Angeles, Los Angeles, California 90027, USA.
Mark C Liang, Center for Endocrinology, Diabetes and Metabolism, Children’s Hospital Los Angeles, Los Angeles, California 90027, USA.
Christina M Koppin, Center for Endocrinology, Diabetes and Metabolism, Children’s Hospital Los Angeles, Los Angeles, California 90027, USA.
Heather M Ross, Center for Endocrinology, Diabetes and Metabolism, Children’s Hospital Los Angeles, Los Angeles, California 90027, USA.
Tania A S S Bachega, Laboratory of Hormones and Molecular Genetics-LIM 42, Division of Endocrinology, Clinics Hospital, School of Medicine, São Paulo University, São Paulo 05508-220, Brazil.
Mitchell E Geffner, Center for Endocrinology, Diabetes and Metabolism, Children’s Hospital Los Angeles, Los Angeles, California 90027, USA; Keck School of Medicine of University of Southern California, Los Angeles, California 90033, USA; The Saban Research Institute at Children’s Hospital Los Angeles, Los Angeles, California 90027, USA.
Monica Serrano-Gonzalez, Keck School of Medicine of University of Southern California, Los Angeles, California 90033, USA; Division of Pediatric Endocrinology, Hasbro Children’s Hospital, Warren Alpert Medical School of Brown University, Providence, Rhode Island 02903, USA.
Gagandeep Bhullar, Center for Endocrinology, Diabetes and Metabolism, Children’s Hospital Los Angeles, Los Angeles, California 90027, USA.
Mimi S Kim, Center for Endocrinology, Diabetes and Metabolism, Children’s Hospital Los Angeles, Los Angeles, California 90027, USA; The Saban Research Institute at Children’s Hospital Los Angeles, Los Angeles, California 90027, USA.
Additional Information
Disclosures: M.E.G. has a research contract with Novo Nordisk; receives consultant fees from Adrenas, Daiichi Sankyo, Eton Pharmaceuticals, Neurocrine Biosciences, Novo Nordisk, Pfizer, and QED; receives royalties from McGraw-Hill and UpToDate; and serves on a data safety monitoring board for Ascendis. M.S.K. receives research support from Neurocrine Biosciences. The other authors have nothing to disclose.
Data Availability
Restrictions apply to the availability of some or all data generated or analyzed during this study to preserve patient confidentiality or because they were used under license. The corresponding author will on request detail the restrictions and any conditions under which access to some data may be provided.
References
- 1. Pang SY, Wallace MA, Hofman L, et al. Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics. 1988;81(6):866-874. [PubMed] [Google Scholar]
- 2. El-Maouche D, Hargreaves CJ, Sinaii N, Mallappa A, Veeraraghavan P, Merke DP. Longitudinal assessment of illnesses, stress dosing, and illness sequelae in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2018;103(6):2336-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet. 2005;365(9477):2125-2136. [DOI] [PubMed] [Google Scholar]
- 4. Falhammar H, Frisén L, Norrby C, et al. Increased mortality in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2014;99(12):E2715-E2721. [DOI] [PubMed] [Google Scholar]
- 5. Donaldson MD, Thomas PH, Love JG, Murray GD, McNinch AW, Savage DC. Presentation, acute illness, and learning difficulties in salt wasting 21-hydroxylase deficiency. Arch Dis Child. 1994;70(3):214-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frisch H, Waldhauser F, Lebl J, et al. ; MEWPE-CAH Study Group . Congenital adrenal hyperplasia: lessons from a multinational study. Horm Res. 2002;57(Suppl 2):95-101. [DOI] [PubMed] [Google Scholar]
- 7. Reisch N, Willige M, Kohn D, et al. Frequency and causes of adrenal crises over lifetime in patients with 21-hydroxylase deficiency. Eur J Endocrinol. 2012;167(1):35-42. [DOI] [PubMed] [Google Scholar]
- 8. Rushworth RL, Falhammar H, Munns CF, Maguire AM, Torpy DJ. Hospital admission patterns in children with CAH: admission rates and adrenal crises decline with age. Int J Endocrinol. 2016;2016:5748264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Swerdlow AJ, Higgins CD, Brook CG, et al. Mortality in patients with congenital adrenal hyperplasia: a cohort study. J Pediatr. 1998;133(4):516-520. [DOI] [PubMed] [Google Scholar]
- 10. Tseng T, Seagroves A, Koppin CM, et al. Increased frequency of acute illness and hospitalizations in infants and toddlers with congenital adrenal hyperplasia. medRxiv. 2019:19005462. doi: 10.1101/19005462, September 5, 2019, preprint: not peer reviewed. [DOI] [Google Scholar]
- 11. Kim MS, Ryabets-Lienhard A, Geffner ME. Management of congenital adrenal hyperplasia in childhood. Curr Opin Endocrinol Diabetes Obes. 2012;19(6):483-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jackson L, Williams FL, Burchell A, Coughtrie MW, Hume R. Plasma catecholamines and the counterregulatory responses to hypoglycemia in infants: a critical role for epinephrine and cortisol. J Clin Endocrinol Metab. 2004;89(12):6251-6256. [DOI] [PubMed] [Google Scholar]
- 13. Odenwald B, Nennstiel-Ratzel U, Dörr HG, Schmidt H, Wildner M, Bonfig W. Children with classic congenital adrenal hyperplasia experience salt loss and hypoglycemia: evaluation of adrenal crises during the first 6 years of life. Eur J Endocrinol. 2016;174(2):177-186. [DOI] [PubMed] [Google Scholar]
- 14. Hinde FR, Johnston DI. Hypoglycaemia during illness in children with congenital adrenal hyperplasia. Br Med J (Clin Res Ed). 1984;289(6458):1603-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Keil MF, Bosmans C, Van Ryzin C, Merke DP. Hypoglycemia during acute illness in children with classic congenital adrenal hyperplasia. J Pediatr Nurs. 2010;25(1):18-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mackinnon J, Grant DB. Hypoglycaemia in congenital adrenal hyperplasia. Arch Dis Child. 1977;52(7):591-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schinner S, Bornstein SR. Cortical-chromaffin cell interactions in the adrenal gland. Endocr Pathol. 2005;16(2):91-98. [DOI] [PubMed] [Google Scholar]
- 18. Merke DP, Chrousos GP, Eisenhofer G, et al. Adrenomedullary dysplasia and hypofunction in patients with classic 21-hydroxylase deficiency. N Engl J Med. 2000;343(19):1362-1368. [DOI] [PubMed] [Google Scholar]
- 19. Bachega TA, Billerbeck AE, Marcondes JA, Madureira G, Arnhold IJ, Mendonca BB. Influence of different genotypes on 17-hydroxyprogesterone levels in patients with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol (Oxf). 2000;52(5):601-607. [DOI] [PubMed] [Google Scholar]
- 20. Charmandari E, Eisenhofer G, Mehlinger SL, et al. Adrenomedullary function may predict phenotype and genotype in classic 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2002;87(7):3031-3037. [DOI] [PubMed] [Google Scholar]
- 21. Falhammar H, Filipsson Nyström H, Wedell A, Thorén M. Cardiovascular risk, metabolic profile, and body composition in adult males with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Eur J Endocrinol. 2011;164(2):285-293. [DOI] [PubMed] [Google Scholar]
- 22. Torres N, Mello MP, Germano CMR, Elias LLK, Moreira AC, Castro M. Phenotype and genotype correlation of the microconversion from the CYP21A1P to the CYP21A2 gene in congenital adrenal hyperplasia. Braz J Med Biol Res. 2003;36(10):1311-1318. [DOI] [PubMed] [Google Scholar]
- 23. Tutunculer F, Saka N, Arkaya SC, Abbasoglu S, Bas F. Evaluation of adrenomedullary function in patients with congenital adrenal hyperplasia. Horm Res. 2009;72(6):331-336. [DOI] [PubMed] [Google Scholar]
- 24. Kim MS, Ryabets-Lienhard A, Bali B, et al. Decreased adrenomedullary function in infants with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2014;99(8):E1597-E1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Charmandari E, Weise M, Bornstein SR, et al. Children with classic congenital adrenal hyperplasia have elevated serum leptin concentrations and insulin resistance: potential clinical implications. J Clin Endocrinol Metab. 2002;87(5):2114-2120. [DOI] [PubMed] [Google Scholar]
- 26. Green-Golan L, Yates C, Drinkard B, et al. Patients with classic congenital adrenal hyperplasia have decreased epinephrine reserve and defective glycemic control during prolonged moderate-intensity exercise. J Clin Endocrinol Metab. 2007;92(8):3019-3024. [DOI] [PubMed] [Google Scholar]
- 27. Weise M, Mehlinger SL, Drinkard B, et al. Patients with classic congenital adrenal hyperplasia have decreased epinephrine reserve and defective glucose elevation in response to high-intensity exercise. J Clin Endocrinol Metab. 2004;89(2):591-597. [DOI] [PubMed] [Google Scholar]
- 28. Zuckerman-Levin N, Tiosano D, Eisenhofer G, Bornstein S, Hochberg Z. The importance of adrenocortical glucocorticoids for adrenomedullary and physiological response to stress: a study in isolated glucocorticoid deficiency. J Clin Endocrinol Metab. 2001;86(12):5920-5924. [DOI] [PubMed] [Google Scholar]
- 29. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mei J, Williams I (eds.). Centers for Disease Control and Prevention. Newborn Screening Quality Assurance Program, second-tier congenital adrenal hyperplasia proficiency testing program (CAHPT) quarterly report. Vol. 7, No.1. February 2017. https://www.cdc.gov/labstandards/pdf/nsqap/nsqap_CAHFeb2017.pdf
- 31. de Carvalho DF, Miranda MC, Gomes LG, et al. Molecular CYP21A2 diagnosis in 480 Brazilian patients with congenital adrenal hyperplasia before newborn screening introduction. Eur J Endocrinol. 2016;175(2):107-116. [DOI] [PubMed] [Google Scholar]
- 32. Bachega TA, Billerbeck AE, Madureira G, et al. Molecular genotyping in Brazilian patients with the classical and nonclassical forms of 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1998;83(12):4416-4419. [DOI] [PubMed] [Google Scholar]
- 33. Bornstein SR, Tajima T, Eisenhofer G, Haidan A, Aguilera G. Adrenomedullary function is severely impaired in 21-hydroxylase-deficient mice. FASEB J. 1999;13(10):1185-1194. [DOI] [PubMed] [Google Scholar]
- 34. Wong DL. Epinephrine biosynthesis: hormonal and neural control during stress. Cell Mol Neurobiol. 2006;26(4-6):891-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4(1):177-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Deibert DC, DeFronzo RA. Epinephrine-induced insulin resistance in man. J Clin Invest. 1980;65(3):717-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Restrictions apply to the availability of some or all data generated or analyzed during this study to preserve patient confidentiality or because they were used under license. The corresponding author will on request detail the restrictions and any conditions under which access to some data may be provided.