

Abstract
Viruses are traditionally thought to be under selective pressure to maintain compact genomes and thus depend on host cell translational machinery for reproduction. However, some viruses encode abundant tRNA and other translation related genes, potentially optimizing for codon usage differences between phage and host. Here, we systematically interrogate selective advantages that carrying 18 tRNAs may convey to a T4-like Vibriophage. Host DNA and RNA degrade upon infection, including host tRNAs, which are replaced by those of the phage. These tRNAs are expressed, at levels slightly better adapted to phage codon usage, especially that of late genes. The phage is unlikely to randomly acquire as diverse an array of tRNAs as observed (p = 0.0016). Together, our results support that the main driver behind phage tRNA acquisition is pressure to sustain translation as host machinery degrades, a process resulting in a dynamically adapted codon usage strategy during the course of infection.
eTOC blurb
Yang, et al., systematically interrogate selective advantages that carrying 18 tRNAs may convey to a T4-like Vibriophage and find that a main driver behind phage tRNA acquisition is pressure to sustain translation as host machinery degrades, a process resulting in a dynamically adapted codon usage strategy during the infection course.
Graphical Abstract
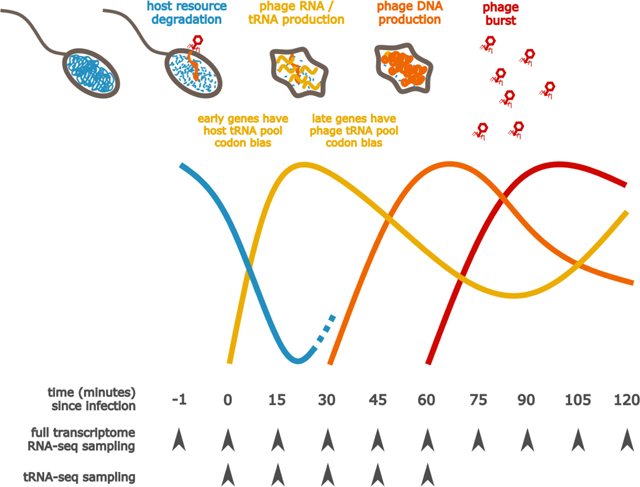
Introduction
The question of why some bacteriophages encode their own transfer RNAs (tRNAs) has been of interest since the late 1960s, when tRNAs were discovered to be carried by T4 (Daniel et al., 1968; Weiss et al., 1968). This finding countered the notion that bacteriophage should be under selective pressure to maintain compact genomes. Most phage simply make use of the hosts’ translational machinery, and thus tRNA genes and other translation-related genes are often considered a hallmark of cellular life (Abergel et al., 2015; La Scola et al., 2003; Raoult et al., 2004; Schulz et al., 2017). Why then do some phage carry tRNAs?
There are currently several hypotheses for why phage carry tRNA. For the T4 phage, almost all of its eight tRNAs correspond to codons that it uses more frequently than its host (Scherberg and Weiss, 1972), leading to the proposal that bacteriophage typically carry tRNA in order to bias translation toward their own genes. Additionally, Cowe, et al. found evidence suggesting that the codon usage bias introduced by T4 tRNAs is especially pronounced toward its late genes (Cowe and Sharp, 1991). Experimentally, tRNA mutants of T4 are still able to replicate and lyse their hosts but show a moderate decrease in burst size under some experimental conditions (Wilson, 1973). However, for another broad host-range T4-like phage KVP40, which also carries 25 tRNAs (Matsuzaki et al., 1992; Miller et al., 2003), the signal for codon usage bias optimization by phage tRNAs is less clear (Miller et al., 2003). In fact, this signal may be an artifact because bacterial tRNA levels are often highly optimized for their codon utilization (Ikemura, 1981; Sharp et al., 2010), and codon usage tends to be very species-specific (Botzman and Margalit, 2011; Grantham et al., 1980). Hence, even phage that do not carry tRNAs commonly have noticeably different codon usage distributions than that of their hosts. It therefore remains an open question whether codon bias optimization is a strong enough driving force to explain phage carriage of tRNA genes. Moreover, other correlations have been described, such as larger phage carrying more tRNAs, and lytic phage being more likely to carry tRNA than temperate phage (Bailly-Bechet et al., 2007). And through a process of elimination, Delesalle, et al. (Delesalle et al., 2016) hypothesized that tRNAs help to sustain growth during infection or to expand host range of the phage.
To address these hypotheses, we systematically explored the selective advantages that carrying tRNA might confer by performing an in-depth genomics and transcriptomics analysis on the infection characteristics of a broad host-range (T4-like Vibriophage 2.275.O., carrying 18 tRNA genes) in its host of isolation (Vibrio cyclitrophicus, strain 10N.286.54.E11). We find that the phage tRNA participate in translation but while there may be some codon optimization, this may not be the most important factor at play. Instead, the infection phenotype is all-destructive in that within approximately the first 15 minutes of infection, the host genome is degraded, as is the host transcriptome. There is therefore little host RNA left to optimize codon usage bias against. Rather, because the host tRNA are degraded as well, the phage must supply its own translational machinery in order to sustain its reproduction cycle. Finally, we show that the main factor optimized for by the phage tRNA is the diversity of the tRNA array, which allows the large phage to sustain a longer replication cycle amid the decaying pool of host resources that result from the lytic infection cycle. This illustrates the potential for a positive feedback loop: large phage must degrade host machinery for parts, so bringing its own machinery allows it to gain a competitive edge - which selects for even larger phage.
Results
Genomic analysis of phage and host reveals differences in genomic codon usage patterns
In order to dissect the selective advantages that tRNA carriage may convey, we used phage 2.275.O [NCBI:txid1881285] as a model. This phage is part of the Nahant Collection, an extensive collection of Vibriophage previously described by Kauffman, et al. (Kauffman, 2014; Kauffman et al., 2018). At 348,911 bp, it is among the largest known bacteriophage (Figure 1, see also Figure S1), and it is capable of infecting hosts from at least two different species, Vibrio cyclitrophicus and V. lentus. And finally, its genome encodes for 18 tRNAs that correspond to 15 amino acids (missing are alanine, tryptophan, aspartic acid, histidine, and lysine), as well as another seven tRNA-like sequences with putative introns.
Figure 1. Phage 2.275.O carries 18 tRNA genes and is a large phage in both capsid size (120 nm) and genome size (348,911 bp).

(A) Estimation of transcriptional units and their timing of expression. Top plot shows genome position of KEGG annotated genes, and bottom plot shows time to reach half the maximum expression of that gene (bottom). Blue and red bars indicate genes on the positive and negative strand, respectively. Early genes tend to be polymerases and sigma factors, while late genes tend to be structural proteins. More detailed annotations can be found in Supplementary Figure 1. (B) Electron microscopy image of phage 2.275.O.
To test whether the codon usage hypothesis is plausible, we first conducted preliminary analyses to verify that a codon usage difference between the phage and host exists. To this end, we applied multidimensional scaling (Figure 2a) and multinomial discriminant analysis (Figure 2b) to the codon usage for each gene from the genomes of the phage and its host of isolation. We observed that there is indeed a codon usage difference between the two organisms, allowing us to next ask, do the phage tRNA bias translation in the direction of this difference? Previously, tRNA copy number (Figure 3a) in the genomes of phage and their hosts was used to assess whether phage tRNAs may optimize codon usage differences (Bailly-Bechet et al., 2007). And in examining the odds ratio for each codon in the phage vs. host genome (Figure 3b), the codons that can be recognized by an anticodon from a host tRNA (according to extended wobble rules summarized by dos Reis, et al. (Reis et al., 2004; Watanabe and Osawa, 1995; Yokoyama and Nishimura, 1995)) appears to be more commonly used by host genes than by phage genes. On the other hand, codons that can be recognized by both phage and host tRNAs span the range of usage preferences. Instead of selectively acquiring tRNAs that are more beneficial to it than its host, it appears the phage seeks to acquire diverse tRNAs, but places lower priority on those that benefit mainly its host.
Figure 2. Codon usage bias introduced by the phage tRNA pool is more pronounced in late genes than early genes.

(A) A multidimensional scaling (MDS) plot of phage and host proteins using Shannon-Jensen Divergence of the codon distributions shows that codon usage difference between phage and host. Points representing the codon recognition capacities of the tRNA pool for each organism are overlaid. Points representing the average codon usage for each organism are also overlaid. (B) Preference for the phage tRNA vs. the host tRNA pool (slant). (Zero signifies ambivalence.) (C) Mean codon usage for host and phage. (D) Slant toward the phage tRNA pool vs. timing of expression depicted as center of mass of RNA expression for the first round of infection. Note that this is different from the expression timing described in figure 1. The center of mass in this plot indicates how quickly the RNA transcript is degraded, while the time to half maximum expression shown in figure 1 summarizes transcription timing.
Figure 3. Analysis of the phage and host genome supports the codon usage hypothesis.

(A) tRNA content in the genomes of the phage and host. Less saturated bars indicate putative tRNA with introns. (B) Differences between the codon usages of phage and host. Darker-hued bars indicate codons that cannot be recognized by phage tRNA, given the wobble rules summarized by dos Reis, et al.
However, this analysis does not account for RNA modifications, which can often be found at the wobble base and may thus change tRNA specificity; and furthermore, tRNA expression level may be more relevant information for assessing any translational bias that may be introduced.
tRNA sequencing suggests that phage tRNA actively participate in translation
The phage tRNA are both expressed and modified, suggesting they are involved in translation. We performed tRNA sequencing on an infection time course, sampled at 15-minute intervals, which enabled us to infer post-transcriptional modifications on the phage tRNA transcripts, as required for translation. These modifications include the CCA tail, which allows for amino acid attachment as well as for successful interaction with the ribosome, and synthesis of the CCA tail is thought to be a step in tRNA quality control (Dupasquier et al., 2008; Hou, 2010; Korostelev et al., 2006). We observed that the tails of the five phage tRNA whose genomic sequences do not end in CCA (Cys-GCA ends in CTA, Gly-TCC ends in CTA, Ile-GAT ends in CAA, Leu-TAA ends in CCG, and Tyr-GTA ends in CAA), are modified into CCA upon transcription (see also Figure S2 and STAR Methods). On the other hand, the genomic sequences of all host tRNA end with a CCA tail. Additionally, while the host carries a CCA modification protein in its genome (Genbank locus tag NVP2275O_348); the phage carries its own CCA modification protein as well. Hence, the phage tRNA appear to be processed such that they can participate in translation.
We are also able to infer putative addition of similar base modifications on the phage and host tRNAs. In our tRNA sequencing protocol, we used the group II intron reverse transcriptase TGIRT (Clark et al., 2016), which can read through RNA modifications but may leave DNA base substitution signatures evident as a multinomial mixture of A, C, G, T, and/or a decrease in read density downstream of the modification (see also Figure S3 and STAR Methods). For example, in comparing one of the phage CAU tRNAs (Genbank genome location: 182648–18272) with a host CAU tRNA (Genbank locus tag: BCV12_11325), both are modified with 4-thiouridine on the 8th base, 3-(3-amino-3-carboxypropyl)uridine on the 47th base, and, importantly, 2-lysidine on the 34th base (see also Figure S3 and STAR Methods) putatively changing them to AUA-recognizing isoleucine tRNA (Harada and Nishimura, 1974). Other similar putative modifications can be observed between phage tRNA and their host analogs, for example, 5-carboxymethylaminomethyl-2-thiouridine on base 34 of Glutamine tRNA, 1-methylguanosine on base 37 of (most) Leucine tRNA (Supplementary Materials). These similarities suggest that phage tRNAs may be recognized and processed by the same enzymes as corresponding host tRNAs.
Interestingly, we also observed seven intron-containing tRNA-like sequences. These, while not recognized by tRNAscan-SE (Lowe and Eddy, 1997), were recognized by another tRNA caller, Aragorn (Laslett and Canback, 2004). These sequences are only expressed at levels as low as 0.003 times (as in the case of threonine tRNA) to 0.2 times (as in the case of serine tRNA) the abundance of an isoacceptor phage tRNA without an intron. Although a small fraction of the reads did appear to be spliced as called, many did not, and the aligned anticodon loop was fairly heterogeneous for these species. In addition, many of these sequences do not end in CCA and did not appear to receive CCA tails. These intron-containing tRNA-like sequences may serve non-canonical functions, and so were therefore not used beyond the initial preliminary analyses.
Codon usage bias is present but not pronounced
Having found evidence supporting the idea that the 18 phage tRNAs without introns likely participate in translation, we tested the most common hypothesis as to why phage carry tRNA - to increase the translational efficiency of their own genes over that of their hosts’ (Bailly-Bechet et al., 2007; Enav et al., 2012; Scherberg and Weiss, 1972; Wilson, 1973). For each gene, we calculated a value representing the efficiency with which it can be translated by the phage tRNA pool, relative to the efficiency with which it can be translated by the host tRNA pool (see Materials and Methods), for simplicity, we will refer to this value as the “slant” of a gene.
We observed that the slant of the phage genes was slightly more in the direction of the phage tRNA pool than was the slant of the host genes (Figure 2C); however, this effect was weak compared to a more optimal axis of discrimination, which is defined by the average gene codon usage for each organism (Figure 2B). The statistical significance of this small effect (KS-test: p < 2.2e-16) seems unreasonable; and in fact, we must re-evaluate this test under the context of the problem at hand. Specifically, we already know that a codon usage bias exists between the host and phage genes, and we also know that the host tRNA are closely matched to the host codon usage (Figure 2A). Therefore, almost any randomly chosen set of tRNA will betray a codon usage difference between the phage and host genes. It is simply then a coin flip as to whether the difference is in the direction of the host tRNA pool or the phage tRNA pool.
We therefore instead asked whether the slant values for phage vs. host genes are different given the known difference in codon usage for the two organisms. Conditioning appropriately, the probability of seeing as high or a higher difference in slant between the phage proteins and host proteins in the direction of the phage tRNA pool was approximately 0.08. (see STAR Methods and Figure S4) This probability is, at best, suggestive, but we cannot be confident that codon usage bias optimization has been the main factor driving tRNA acquisition.
We next calculated the codon usage profiles for other Vibrios in the Nahant Collection for which we have completely sequenced genomes, including Vibrio lentus and additional Vibrio cyclitrophicus strains that are known hosts of 2.275.O. As expected, we find that codon usage profiles cluster by species (see STAR Methods and Figure S5, top panel). V. cyclitrophicus hosts are more similar in codon usage to 2.275.O than V. lentus hosts. (see STAR Methods and Figure S5, bottom panel). As V. cyclitrophicus was the host of isolation, one could posit that V. cyclitrophicus might be a “preferred” host for 2.275.O; however we note that we cannot exhaustively identify all potential hosts for this phage.
Host genomic DNA and RNA degradation as the driving factor for tRNA acquisition
One potential explanation for the low overall signal for codon optimization is that only a subset of the phage genes is affected, in particular the late genes, as previously shown for T4 (Cowe and Sharp, 1991). Codon usage optimization toward the late genes might be advantageous for a few reasons: (1) mRNAs from the earliest genes might already be undergoing translation and degradation as the phage tRNAs are transcribed, and therefore must utilize mainly the host tRNA pool; and (2) the host tRNA pool might degrade, in which case translation during the late stages of infection might heavily rely on phage tRNA.
Some evidence in the literature supports the latter hypothesis. During T4 infection of E. coli, degradation of host DNA is initiated by Endo II and Endo IV (Miller et al., 2003), in part to help supply the nucleotide pool for phage replication. This comes with a consequence: although tRNAs tend to be more stable than other RNAs (Davis et al., 1986), they can undergo rapid degradation under stress conditions (Svenningsen et al., 2017; Zhong et al., 2015). In fact, during T4 infection, E. coli uses nucleases to deplete its own lysine tRNA, dialing down translation, seemingly in defense while, as a “rebuttal,” T4 RNA ligase is able to repair damaged tRNA (Amitsur et al., 1987). Evidence for this all-destructive infection phenotype suggests that supplying translational components might help the phage to fill the growing gaps in host machinery and thereby prolong the replication period.
To test whether phage 2.275.O infection is similarly all-destructive, we used qPCR to check whether host DNA was degraded upon infection. We found that the genomic copy number of the host genes probed for (GroEL and CTP Synthetase) dropped by approximately 80% within the first 15 minutes of infection, which is in line with expectation given the number of infected cells in our assay (Figure 4A and B, additional validation is also presented in Figure S6). On the other hand, the phage production does not occur until approximately 30 minutes into the infection.
Figure 4. Time course of phage 2.275.O. infection.

Approximately a tenth of the hosts remain uninfected and begin to grow again. A second round of infection starts at approximately 90 minutes. (A) Phage plaques begin to appear 30 minutes into infection, and peak at approximately 90 minutes. (B) qPCR results to quantify bacterial chromosome copies show that the host genome is degraded rapidly upon infection. (C) The tRNA subset of RNA-seq shows that host tRNA, shown in purple, are degraded upon infection while phage tRNA, shown in orange, increase. An average has been taken over all tRNA species, and the errors shown are 1.96*standard dev of the log mean. Reads are normalized to a firefly luciferase spike-in for each sample.
Because the host genome is degraded, tRNA can no longer be produced from the host genome, and if host tRNA are degraded as well, tRNA might become a limiting resource for translation during the late stages of infection.
When examining the tRNA expression from transcriptome sequencing data, we found that the host tRNA were indeed degraded rapidly, reaching a minimum value around 15 minutes, whereas phage tRNA were continually produced (Figure 4C, see Figure S7, related to STAR Methods for individual tracts of host and phage tRNA by amino acid). The increase in host RNA after 15 minutes is likely due to regrowth of uninfected cells in the culture. We note that the presence of uninfected cells in the culture renders it difficult to reliably infer the levels of host tRNA in infected cells.
These observations support the hypothesis that, as the host tRNA pool is degraded, the phage tRNA allow translation to be sustained; this may especially benefit the late genes, which do not reach half their maximum expression until 40–45 minutes into the infection (Figure 1).
Having found that the host genome and transcriptome (including the tRNA) were indeed degraded during infection, we next utilized the full transcriptome sequencing data to quantify 2.275.O gene expression timing in order to assess whether late genes, which have a greater necessity for relying on phage translational machinery, were more adapted to the phage tRNA pool than early genes. We did in fact observe that the slant of the late phage genes was further in the direction of the phage tRNA pool than the slant of the early genes (Figure 2D). However, the absolute slant of even the late genes was closer toward the host tRNA pool than the phage tRNA pool, implying that while suggestive, codon usage bias optimization might not be the driving force for phage acquisition of tRNAs.
Prolonging the replication period amidst host cell shutdown
If the phage tRNAs optimize for the ability to sustain translation in the absence of the host tRNAs, we would expect the phage to carry as diverse an array of tRNA as possible. In fact, it is striking that the 18 phage tRNAs without introns each represent different anticodons (there are two CAU anticodons, however, one of these is likely modified by 2-lysidine, making it an AUA-recognizing leucine as opposed to an AUG-recognizing methionine). In simulating draws of tRNA from the host genome, we find that the tRNA carried by 2.275.O is more diverse in anticodons encoded than would be expected at random (Figure 5A, p=0.0016).
Figure 5. tRNA carried by the phage may supplement the degrading pool of host tRNA.

(A) The probability of selecting, uniformly at random from the host genome, a tRNA array that is able to encode as many anticodons as that carried by the phage is 0.0016. In addition, contiguous stretches of tRNA in the host genome, which are typically thought to be the result of duplication events, encode very lowly diverse anticodons. The phage’s tRNA collection, therefore, appears to be the result of multiple acquisition and selection events. (B) Of the tRNA not carried by the phage, most are highly expressed by the host, and others correspond to codons not very highly used by the phage genome.
This observation was even more striking when considering that neighboring tRNAs within the host genome generally have very low diversity, as they are likely the result of gene duplication events, and tend to code for the same amino acid (Figure 5A). This indicates that picking up as diverse an array of tRNAs as observed in the 2.275.O genome was not a matter of a few simple recombination events, but many. Thus, the diversity of this tRNA array appears to be under high selective pressure.
And what of the tRNAs that the phage does not carry? We found that the host expresses of many of these tRNAs more highly than the tRNAs with phage analogs (Figure 5B). Assuming similar rates of degradation for each tRNA, the more highly expressed tRNAs may persist longer during infection, thereby reducing the selective pressure for the phage to acquire its own copies. The tRNAs without phage analogs that are expressed lowly by the host recognize codons that are used very infrequently by the phage. These two types of tRNA may confer less of a selective advantage to the phage than the tRNA already present in the phage, implying that for these tRNAs the phage has reduced its dependence on their codons rather than acquire its own copies of the tRNAs. Taken together, the observations presented in this paper strongly imply that the primary function for phage tRNAs is to supplement degrading host translational machinery, which results from an all-destructive lytic infection phenotype.
Discussion
Our results indicate that the main role of the phage 2.275.O tRNAs is to support translation of this large lytic phage as the host cell shuts down. Upon 2.275.O infection, host genomic DNA is degraded, as are mRNA transcripts. Degradation reaches a baseline around 15 minutes; however, phage particles are only released starting around 30 minutes after the onset of the infection, implying that without phage tRNA production, late genes might experience resource limitation during translation. Although the tRNA array of phage 2.275.O does not appear to optimize tRNA/codon usage bias toward its genes on the whole, we do observe that the codon usage of the phage genes expressed late during the infection are more in the direction of the phage tRNA pool than that of the early genes. Additionally, the diversity of the phage tRNA array appears to be optimized, implying that the main selective force at play is a drive toward self-sufficiency in the wake of host degradation.
This simple line of logic unifies many observations previously made in the literature, either through deep interrogations of the T4 infection cycle or broad analyses of tRNA-carrying phage: First, the presence of many tRNAs is more often found in genomes of lytic phage than those of temperate phage (Bailly-Bechet et al., 2007). Aggressively lytic phage often degrade the host genome as the phage can then use the nucleotides to increase its burst size. But because translation is required for phage particle production, these phage presumably benefit from shuttling their own translation machinery by extending the replication period beyond the time at which the host resources are depleted.
A second observation is that phage tRNAs appear to optimize codon usage bias toward phage genes and away from host genes (Scherberg and Weiss, 1972). According to our findings, tRNAs absent from the phage correspond to those that tend to be more highly expressed in the host than tRNAs present in the phage, and tRNAs highly expressed in the host correspond to codons that are most commonly used by the host, or are most biased toward the host (Figure S7, related to STAR Methods). This backdoor correlation may explain a large part of the observed codon usage bias effect. Moreover, once the phage acquire tRNA, an adaptive feedback loop can form between the phage genome codon usage and the tRNA array that it carries. This feedback loop explains a third observation, which is that codon usage bias is more pronounced in late phage genes than in the early genes (Cowe and Sharp, 1991; Kunisawa, 1992; Kunisawa et al., 1998). Early mRNAs require use of host tRNAs, as the bulk of phage tRNAs are still in the process of being transcribed and processed, whereas late mRNAs are expected to be more dependent on the phage tRNAs, as the bulk of the host tRNA might be degraded later in the infection.
And finally, a fourth observation is that phage with tRNAs generally have larger genomes than those without (Bailly-Bechet et al., 2007). For phage with large genomes, it is more important to degrade the host genome to free up nucleotides, leading again to the quandary presented by the first point. In addition, larger phage might require longer periods of replication and more resources for supporting translation in the wake of degrading host tRNA. The longer the infection persists past host resource degradation, the stronger the selective pressure for phage to encode their own machinery, which in turn selects for larger phage. A tRNA deletion mutant of phage 2.275.O could address the key questions of how tRNAs change the latent period and burst size during infection. We note that this experiment may prove very difficult to do in a wild, non-model system such as we utilize here but could potentially be carried out in a more tractable model system.
Using this lens, it is interesting to compare 2.275.O against its foil in the same phage sampling collection, the Autolykiviridae (Kauffman et al., 2018). In contrast with this 349kb phage, Autolykiviridae carry a particularly small, streamlined genome, at only 10kb. With such small genomes, there may be less selective pressure to free up the nucleotide pool. In fact, Autolykiviridae do not degrade the host genome. The infection cycle of these viruses can last on the order of weeks, and with only 20 genes in its genome, having no known translation supporting functions, they must rely entirely on the host translation machinery. Although a whole spectrum of strategies might co-exist, we can see here two extreme and contrasting strategies.
This type of phage infection might have convenient applications for studying tRNA regulation. Although translation is fundamental to all of life, many aspects are still unknown. For example, while some tRNA modifications have been shown to be necessary for correct folding, synthetase recognition, degradation, and translation regulation (Lorenz et al., 2017), the functions of most modifications are unknown (Kirchner and Ignatova, 2015). Many recent findings about tRNA are conducted in systems in which a cellular stress response involving tRNA can be triggered (Kirchner and Ignatova, 2015). Lytic phage infection offers a similar convenience in that it can be synchronized through a one-step-growth experiment (Ellis and Delbrück, 1939). Because of this, tRNA can be “tracked” from the newly synthesized nascent form to processed intermediates and degraded products. In fact, this can be seen in our tRNA sequencing timecourse (Figures S8–11, related to STAR Methods). Exciting new technologies for probing translation now exist, such as ribosomal footprint profiling (Ingolia et al., 2009), tRNA-ribo-seq (Chen and Tanaka, 2018). Work in combining these techniques with phage growth experiments may be a promising future direction for uncovering further insights into tRNA processing and use in translation.
Star Methods
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Libusha Kelly (libusha.kelly@einsteinmed.org).
Materials Availability:
Phage and host are available from Martin Polz (martin.f.polz@univie.ac.at).
Data and Code Availability:
Source data statement.
Raw sequencing reads are available on the NCBI Sequenced Reads Archive under BioProject numbers PRJNA524872 (tRNA-seq) and PRJNA524877 (full transcriptome RNA-seq).
Code statement.
This paper does not report original code.
Scripts statement.
Scripts to generate the figures reported in this paper are available in the (https://github.com/ratatstats/tRNAbias) R package and their use is described in vignettes included with the package. Please first install the package devtools, and then from R, run following command: devtools::install_github(“ratatstats/tRNAbias”, build_vignettes = TRUE, force = TRUE)
Any additional information required to reproduce this work is available from the Lead Contact.
METHOD DETAILS
Exploratory genome-based codon usage bias analysis
In order to assess the plausibility of the codon usage bias hypothesis, we conducted a preliminary analysis of the phage and host genomes. The tRNA carried by each organism was called using tRNAscan-SE (Lowe and Eddy, 1997) and Aragorn (Laslett and Canback, 2004). The multidimensional scaling analysis uses Shannon-Jensen divergence between codon distributions for each protein as the distance metric. The odds ratio for each codon is defined as
tRNA sequencing timecourse
In order to explore the shift in tRNA abundance throughout the course of infection, we conducted a one-step-growth experiment and collected samples at 15 minute intervals. Cells from the same culture were split in two (control vs. infection) then centrifuged to pellets. The control sample was resuspended in 200 μL of Difco 2216 Marine Broth, and the infection sample was resuspended in 150μL 2216 and 50 μL of phage lysate. The samples were left to sit for 5 minutes to allow for adsorption of the phage, then diluted to a volume of 15 mL in order to deter further infection. This total volume was split in 5, one sample from each set was immediately centrifuged and flash frozen as a “time 0” sample, and the rest were placed on a shaker, then centrifuged down and flash frozen at 15 minute intervals. 500 μL aliquots were taken from each sample prior to centrifugation to be plated as a spot check of phage concentration. A schematic of this experiment is presented in Figure S12, and the resulting plaque data is presented in Figure S13.
RNA sequencing timecourse
To further hone in on particular genes that the codon usage bias may favor, a second phage growth experiment was collected for full-transcriptome RNA sequencing. In this experiment, an aliquot of a culture was centrifuged and flash frozen as a preinfection timepoint, the remainder of the culture was then centrifuged to a pellet then resuspended with 400 μL of a phage lysate. The samples were left to sit for 5 minutes to allow for adsorption of the phage, then diluted to a total volume of 45 mL in order to deter further infection. This total volume was split into 9 samples of 5 mL each, to be taken in 15 minute intervals from time 0 to 120 minutes. The sampling procedure involved flash freezing 3 pellets spun down from 1.5 mL aliquots of each sample, then immediately doing serial dilutions of unfiltered and filtered viruses to assess phage growth and stage of infection. A schematic of this experiment is presented in Figure S14, and the resulting plaque data is presented in Supplementary Figure S15
Total RNA extraction
Total RNA from the infection time series (flash-frozen pellets) was extracted by the hot phenol method. Briefly, cell pellets were resuspended in TE (10 mM Tris pH 7.0, 1 mM EDTA) and treated with 0.5 mg/ml lysozyme at room temperature for 5 minutes. Then NaOAc (100 mM final concentration) and SDS (1% final concentration) were added, followed by an equal volume of acid phenol:chloroform pH 4.5 (ThermoFisher). The mixture was shaken at 65°C for 10 minutes using a thermomixer, and centrifuged at 20000 × g for 5 minutes. The upper phase was washed by chloroform and centrifuged at 20000 × g for 5 minutes. The phenol/chloroform extraction was repeated once, and the upper phase was precipitated with isopropanol and 300 mM NaOAc. Precipitated RNA was washed with 75% ethanol, air dried and resuspended in water.
High-throughput sequencing of tRNAs
tRNAs were gel purified from total RNA on a 10% urea polyacrylamide gel (size selected between 70 and 100 nt). Gel pieces were macerated and soaked in 0.3 M NaCl overnight with rotation at 4°C for elution. tRNAs were precipitated with isopropanol using linear acrylamide as the carrier. RNA pellets were resuspended in 100 mM Tris-Cl pH 9.5 and incubated at 37°C for 1.5 hours for deacylation. After deacylation RNA was purified using Oligo Clean & Concentrator (Zymo Research) and eluted in 10 mM Tris pH 8.0. Purified RNA was ligated to a 3’ preadenylated adapter AppNNNNAGATCGGAAGAGCACACGTCT/iBiodT/iBiodT/3ddC/ (final concentration 10 uM) using RNL2 truncated KQ (NEB) with 10% PEG8000 at room temperature overnight. After ligation RNA was purified using the MinElute PCR Purification kit (Qiagen) and reverse transcribed using TGIRT™-III enzyme (InGex) under manufacturer’s instructions. Briefly, RNA was incubated with an RT primer AGACGTGTGCTCTTCCGATCT (0.1 μM final concentration) and RT buffer at 85°C for 5 minutes, and cooled to 25°C at 0.1°C per second. DTT and TGIRT™-III were added and the mixture was incubated at room temperature for 30 minutes. dNTPs were added and the reaction was incubated at 60°C for 30 minutes. RNA was hydrolyzed by NaOH, neutralized by HCl and purified using MinElute PCR purification kit. cDNA was ligated to a preadenylated DNA adapter AppNNNNGATCGTCGGACTGTAGAACTCTGA/3ddC/ (preadenylated by 5’ DNA adenylation kit (NEB)) using thermostable 5’ App DNA/RNA ligase (NEB) following manufacturer’s protocol (ligated at 65°C for 5 hours and heated inactivated at 90°C for 3 minutes). cDNA was purified using MinElute PCR purification kit, and amplified using KAPA HiFi HotStart PCR kit (Roche). PCR primers were AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGACGATC and CAAGCAGAAGACGGCATACGAGATBBBBBBGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT (BBBBBB stands for barcode sequence). PCR products were size selected between 150 nt and 260 nt using Pippin Prep (Sage Science), and sequenced on an Illumina HiSeq under 100 bp by 100 bp paired-end mode.
Full transcriptiome RNA-seq
For quantitative RNA-seq, 1.5 ng Firefly luciferase mRNA and 0.015 ng of Renilla luciferase mRNA was added to each cell pellet before hot phenol extraction. As each sample was derived from the same infection batch, which was distributed in equal volumes, these two luciferase mRNA spike-ins are proportional to the starting quantity of infected cells. Total RNA was treated by TURBO DNase (ThermoFisher) according to manufacturer’s protocol and purified using RNA Clean & Concentrator kit (Zymo Research). mRNA was isolated using the Illumina Ribozero prokaryote kit (gram positive and negative, Illumina) and was prepared into libraries using the Kapa Hyperprep kit (Roche) following manufacturer’s protocols. Libraries were sequenced on an Illumina HiSeq under 40 bp single-end mode. Host gene expression data are displayed and clustered in Figures S16 and S17.
Calling modifications on tRNA sequencing data
For the dual purposes of 1. assessing whether the phage tRNA are functional tRNA that participate in translation and 2. more accurately assigning wobble base affinities, RNA modifications were called based on reverse transcriptase substitutions. First, a reference alignment of phage and host tRNA was made. LocaRNA (Will et al., 2007), which accounts for RNA secondary structure, was used to make a first-pass multiple alignment. Phage tRNA called with introns often aligned poorly, and so the alignments were fixed using the putative secondary structures provided by Aragorn. The variable loops were aligned separately using MUSCLE (Edgar, 2004), then stitched back into the tRNA alignment. The final multiple alignment of the unique tRNA is shown in Figure S18. Next, E. coli tRNA from the MODOMICS database (Boccaletto et al., 2018; Dunin-Horkawicz et al., 2006) were aligned to this reference in order to identify what types of post-transcriptional modifications may be present. Reads from tRNA sequencing were then aligned to this reference using the affine gap penalty method “gotoh” provided by the align. seqs() function in mothur, version 1.34.4 (Schloss et al., 2009), with the following scoring: match=2, mismatch=0, gapopen=−5, gapextend=−1. Phage and host tRNA are sufficiently different such that reads can be mapped back to the originating organism (Figure S19). And finally, the base distribution at each position was used to fit a model for how modifications correspond to “missequenced” reads (as many of these are likely the result of base substitutions inserted by the reverse transcriptase upon encountering a modified base).
Calculating phage RNA expression timing
Instead of classifying phage genes into distinct categories of expression timing, continuous scales were defined. Because the host RNA expression level climbs until 75 minutes, this is taken to be the timepoint before the second round of infections begin. In order to get a sense of transcription and degradation for the purposes of the analyses presented in Figure 2, the phage RNA expression timing was then defined as the center of mass of the expression levels over time. Each RNA is color coded by its expression timing in Figure S20. For the purposes of visually identifying what may be transcriptional units within the genome (Figure 1, Supplementary Figure 1), another measure of expression timing - the time taken to reach half the maximum level of expression - was defined.
Assessing Genome Degradation
Two genes were selected for qPCR to assess whether the host genome is degraded upon infection: GroEL (Genbank locus tag BCV12_01410, primers: CAATGGATCTTAAGCGCGGC and CAGAGATAACCGTACCGCCC) and CTP synthetase (Genbank locus tag BCV12_03025, primers: CTTTGGCGATCGTGGTGTTG and TTTTCTAATTCGCCGCGCTG). The phage genes, GroEL (Genbank locus tag NVP2275O_355, primers: CTTTGAAGACATGGGCGCAC and AACGACTAGGGTTGCAAGCA) and the Major Capsid Protein (Genbank locus tag NVP2275O_445, primers: TGAAGGTGTTATGGGTCGCC and ATACGGGCAGTAGAACGCAG), was also assayed for contrast. Each sample was prepared using the Kapa SYBR Fast kit according to the manufacturer’s instructions. A standard curve for each primer was made on 10 two-fold serial dilutions of each genome. This was then used to convert the CT values to copy number.
Codon Usage Bias Analysis
In order to assess whether the phage tRNA pool may introduce translational bias toward its own genes, a summary statistic for each gene that represents the efficiency with which it can be translated by the phage tRNA pool, relative to the efficiency with which it can be translated by the host tRNA pool was calculated. This value, essentially a likelihood ratio, is referred to as the “slant” of a gene for brevity. The calculation is as follows:
To set up the analysis, the codon usage preference of a tRNA pool must first be calculated. In particular, it is defined as an estimated probability of a random codon being bound given the tRNA pool:
Here, P(tRNA = x|pool = host) is defined as the read abundances from tRNA sequencing, normalized to each organism. P(codon|tRNA = x) is defined according to revised wobble rules noted by Murphy, et al. (Murphy and Ramakrishnan, 2004; Watanabe and Osawa, 1995; Yokoyama and Nishimura, 1995), accounting for wobble base modifications inferred through tRNA sequencing results. Low affinity pairings receive a third the weight of high affinity parings.
These two points define a path that the codon distribution for each protein can, essentially, be projected upon in order to calculate a tRNA pool preference for each protein (here, referred to in short as the “slant”). Specifically, for each protein, the slant for a given gene with codon counts y, is calculated as the log likelihood ratio of observing the codons from that gene given the coding capacity of the phage tRNA pool vs. given the coding capacity of the host tRNA pool (divided by the total number of codons, to make the value more easily comparable among proteins):
where pc = P(codon = c|pool = host) and hc = P(codon = c|pool = phage). The slant is 0 if the two multinomial probabilities are equal, which can be interpreted intuitively as the gene having equal efficiencies of coding by the pool of phage tRNA and the pool of host tRNA. A nice property of this calculation is that if y is exactly np (if the codon usage of a gene matches exactly the coding efficiency of the phage tRNA pool), then the slant is KL(p||h), or the Kullback-Leibler divergence between p and h. And if y is exactly nh (if the codon usage preference of a gene matches exactly the coding efficiency of the host tRNA pool), then the slant is ―KL(h||p), or the negative Kullback-Leibler divergence between h and p. However, it is possible for the slant to be less than ―KL(h||p) if the codon distribution for a given protein is even further away from the coding efficiency of the phage tRNA pool than that of the host tRNA pool. And likewise, the slant can be greater than KL(p||h) if the codon distribution for a given protein is further away from the coding efficiency of the host tRNA pool than that of the phage tRNA pool.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments and all software used can be found in the results section. Details of assessing the statistical significance of codon usage bias and tRNA diversity analysis are provided below.
Assessing the statistical significance of codon usage bias
A one-sided Kolmogorov-Smirnov test was used to assess the difference in distributions of slant values for phage genes vs. slant values for host genes. This resulted in a suspiciously low p-value (less than 1e-22), which was taken as a signal that the null model used may not have been fair in the context of this problem, and more relevant assumptions should be specified. In this case, we already know from preliminary analyses that the distributions of codon usage between phage and host genes is different (Figures 2C and 4B), so any randomly chosen vector of phage tRNA expression is likely to betray this difference.
Instead, it is more appropriate to ask whether the slant values for phage genes vs. the slant values for host genes are different conditioning on the known codon usage distribution of the two organisms. This was done using the following resampling scheme: First, 18 tRNA (the number of phage tRNA) are randomly sampled with replacement from the host genome, then a random expression vector for these 18 tRNA was generated by normalizing exponentially distributed random variables. This expression vector was used as a random phage tRNA expression vector. Then, based on this random phage tRNA expression vector and the known host tRNA expression vector, slant values were calculated for phage and host genes, and a one-sided KS-test was conducted on these slant values. This procedure was replicated 200 times, and these test results formed the null distribution against which the original test result was compared.
tRNA array diversity analysis
The diversity of the tRNA pools was defined as the Shannon entropy of the amino acids encoded by the tRNA for each organism. The simulated randomly acquired diversity depicted in Figure 5 was calculated by sampling 18 tRNA (the number of tRNA in the phage genome) with replacement from the host genome.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| T4-like Vibrio phage 2.275.O._10N.286.54.E11 | Martin Polz Lab | MG592671.1 |
| Vibrio cyclitrophicus, strain 10N.286.54.E11 | Martin Polz Lab | NZ_MCTE00000000.1 |
| Critical commercial assays | ||
| Ribo-Zero Bacteria kit (gram negative and positive) | Illumina | discontinued |
| Kapa Hyperprep | Roche | KK8504 |
| Pippin Prep | Sage Science | No catalog # |
| RNA Clean & Concentrator | Zymo Research | R1080 |
| TGIRTTM-III enzyme | InGex | TGIRT50 |
| RNA Ligase 2, truncated KQ | New England Biolabs | M0373S |
| 5´ DNA Adenylation Kit | New England Biolabs | E2610L |
| Oligo Clean & Concentrator | Zymo Research | D4060 |
| KAPA SYBR FAST | Roche | KK4600 |
| MinElute PCR Purification Kit | Qiagen | 28004 |
| Deposited data | ||
| tRNA-seq | Raw sequencing reads | PRJNA524872 |
| RNA-seq | Raw sequencing reads | PRJNA524877 |
| Oligonucleotides | ||
| 3’ preadenylated adapter AppNNNNAGATCGGAAGAGCACACGTCT/iBiodT/iBiodT/3ddC/ | This paper | N/A |
| qPCR primers, GroEL, host genome: CAATGGATCTTAAGCGCGGC and CAGAGATAACCGTACCGCCC | This paper | Genbank locus tag BCV12_01410 |
| qPCR primers, CTP synthetase, host genome: CTTTGGCGATCGTGGTGTTG and TTTTCTAATTCGCCGCGCTG | This paper | Genbank locus tag BCV12_03025 |
| qPCR primers, GroEL, phage genome: CTTTGAAGACATGGGCGCAC and AACGACTAGGGTTGCAAGCA | This paper | Genbank locus tag NVP2275O_355 |
| qPCR primers, Major Capsid Protein, phage genome: TGAAGGTGTTATGGGTCGCC and ATACGGGCAGTAGAACGCAG | This paper | NVP2275O_445 |
| preadenylated DNA adapter AppNNNNGATCGTCGGACTGTAGAACTCTGA/3ddC/ | This paper | N/A |
| Sequencing primer AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGACGATC | This paper | N/A |
| Sequencing primer CAAGCAGAAGACGGCATACGAGATBBBBBBGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT (BBBBBB indicates barcode sequence) | This paper | N/A |
| Software and algorithms | ||
| LocaRNA | Will S, Reiche K, Hofacker IL, et al. (2007) | http://www.bioinf.uni-freiburg.de/Software/LocARNA/ |
| tRNAscan-SE | Lowe TM, Eddy SR (1997) | http://lowelab.ucsc.edu/tRNAscan-SE/ |
| Aragorn | Laslett D, Canback B (2004) | http://lowelab.ucsc.edu/tRNAscan-SE/ |
| MUSCLE | Edgar RC (2004) | https://www.drive5.com/muscle/ |
Highlights.
We ask what selective pressures drive tRNA carriage in a T4-like Vibriophage.
Phage tRNAs are expressed at levels adapted to phage codon usage in late genes.
Random acquisition of the diverse array of 18 phage tRNAs observed is unlikely.
The phage has a dynamically adapted codon usage strategy.
Acknowledgements
We thank Uttam RajBhandary for poring over the tRNA structures with us and for pointing us toward relevant literature. We would additionally like to thank Jan-Christian Hütter for thoughtful discussion and comments about the analysis methods. J. Y. Y. was supported by the Department of Energy Computational Science Graduate Fellowship Program of the Office of Science and National Nuclear Security Administration in the Department of Energy under contract DE-FG02–97ER25308. This work was supported by NSF OCE 1435993 and 1435868 to M.P. and L.K., respectively. L. K. is supported in part by a Peer Reviewed Cancer Research Program Career Development Award from the United States Department of Defense (CA171019). M.P. was supported by the Simons Foundation (LIFE ID 284 572792). D.P.B. is supported by GM118135 from the NIH. W.F. was supported by the Damon Runyon Cancer Research Foundation (DRG-2174–13) and the NIH (K99GM123230).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Supplementary spreadsheets.
Table S1 related to Figure 1. KEGG hits to Vibriophage 2.275.O.
Table S2 related to Figure 1. BLAST hits to Vibriophage 2.275.O.
Table S3 related to Figure S17. Genes representing “outlier” clusters from Figure S17.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abergel C, Legendre M, Claverie J-M, 2015. The rapidly expanding universe of giant viruses: Mimivirus, Pandoravirus, Pithovirus and Mollivirus. FEMS Microbiology Reviews 39, 779–796. 10.1093/femsre/fuv037 [DOI] [PubMed] [Google Scholar]
- Amitsur M, Levitz R, Kaufmann G, 1987. Bacteriophage T4 anticodon nuclease, polynucleotide kinase and RNA ligase reprocess the host lysine tRNA. The EMBO journal 6, 2499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly-Bechet M, Vergassola M, Rocha E, 2007. Causes for the intriguing presence of tRNAs in phages. Genome research 17, 1486–95. 10.1101/gr.6649807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaletto P, Machnicka MA, Purta E, Piątkowski P, Bagiński B, Wirecki TK, Crécy-Lagard V. de, Ross R, Limbach PA, Kotter A, Helm M, Bujnicki JM, 2018. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Research 46, D303–D307. 10.1093/nar/gkx1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botzman M, Margalit H, 2011. Variation in global codon usage bias among prokaryotic organisms is associated with their lifestyles. Genome biology 12, R109. 10.1186/gb-2011-12-10-r109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-W, Tanaka M, 2018. Genome-wide Translation Profiling by Ribosome-Bound tRNA Capture. Cell Reports 23, 608–621. 10.1016/j.celrep.2018.03.035 [DOI] [PubMed] [Google Scholar]
- Clark WC, Evans ME, Dominissini D, Zheng G, Pan T, 2016. tRNA base methylation identification and quantification via high-throughput sequencing. RNA 22, 1771–1784. 10.1261/rna.056531.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowe E, Sharp PM, 1991. Molecular evolution of bacteriophages: Discrete patterns of codon usage in T4 genes are related to the time of gene expression. Journal of Molecular Evolution 33, 13–22. 10.1007/BF02100191 [DOI] [Google Scholar]
- Daniel V, Sarid S, Littauer UZ, 1968. Amino acid acceptor activity of bacteriophage T4 transfer RNA. FEBS Letters 2, 39–41. 10.1016/0014-5793(68)80095-X [DOI] [PubMed] [Google Scholar]
- Davis BD, Luger SM, Tai PC, 1986. Role of ribosome degradation in the death of starved Escherichia coli cells. Journal of bacteriology 166, 439–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delesalle VA, Tanke NT, Vill AC, Krukonis GP, 2016. Testing hypotheses for the presence of tRNA genes in mycobacteriophage genomes. Bacteriophage 6, e1219441. 10.1080/21597081.2016.1219441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunin-Horkawicz S, Czerwoniec A, Gajda MJ, Feder M, Grosjean H, Bujnicki JM, 2006. MODOMICS: a database of RNA modification pathways. Nucleic Acids Research 34, D145–D149. 10.1093/nar/gkj084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupasquier M, Kim S, Halkidis K, Gamper H, Hou Y-M, 2008. tRNA integrity is a prerequisite for rapid CCA addition: implication for quality control. Journal of molecular biology 379, 579–88. 10.1016/j.jmb.2008.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research 32, 1792–7. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis EL, Delbrück M, 1939. THE GROWTH OF BACTERIOPHAGE. The Journal of general physiology 22, 365–84. 10.1085/JGP.22.3.365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enav H, Béjà O, Mandel-Gutfreund Y, 2012. Cyanophage tRNAs may have a role in cross-infectivity of oceanic Prochlorococcus and Synechococcus hosts. The ISME journal 6, 619–28. 10.1038/ismej.2011.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R, Gautier C, Gouy M, Mercier R, Pavé A, 1980. Codon catalog usage and the genome hypothesis. Nucleic acids research 8, r49–r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada F, Nishimura S, 1974. Purification and characterization of AUA specific isoleucine transfer ribonucleic acid from Escherichia coli B. Biochemistry 13, 300–307. 10.1021/bi00699a011 [DOI] [PubMed] [Google Scholar]
- Hatfull GF, 2008. Bacteriophage genomics. Current opinion in microbiology 11, 447–53. 10.1016/j.mib.2008.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y-M, 2010. CCA addition to tRNA: implications for tRNA quality control. IUBMB life 62, 251–60. 10.1002/iub.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T, 1981. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: A proposal for a synonymous codon choice that is optimal for the E. coli translational system. Journal of Molecular Biology 151, 389–409. 10.1016/0022-2836(81)90003-6 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS, 2009. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science 324, 218–223. 10.1126/SCIENCE.1168978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman AKM, 2014. Demographics of lytic viral infection of coastal ocean vibrio. [Google Scholar]
- Kauffman KM, Hussain FA, Yang J, Arevalo P, Brown JM, Chang WK, VanInsberghe D, Elsherbini J, Sharma RS, Cutler MB, Kelly L, Polz MF, 2018. A major lineage of non-tailed dsDNA viruses as unrecognized killers of marine bacteria. Nature 554, 118–122. 10.1038/nature25474 [DOI] [PubMed] [Google Scholar]
- Kirchner S, Ignatova Z, 2015. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nature Reviews Genetics 16, 98–112. 10.1038/nrg3861 [DOI] [PubMed] [Google Scholar]
- Korostelev A, Trakhanov S, Laurberg M, Noller HF, 2006. Crystal Structure of a 70S Ribosome-tRNA Complex Reveals Functional Interactions and Rearrangements. Cell 126, 1065–1077. 10.1016/j.cell.2006.08.032 [DOI] [PubMed] [Google Scholar]
- Kunisawa T, 1992. Synonymous codon preferences in bacteriophage T4: a distinctive use of transfer RNAs from T4 and from its host Escherichia coli. Journal of theoretical biology 159, 287–98. [DOI] [PubMed] [Google Scholar]
- Kunisawa T, Kanaya S, Kutter E, 1998. Comparison of synonymous codon distribution patterns of bacteriophage and host genomes. DNA research : an international journal for rapid publication of reports on genes and genomes 5, 319–26. [DOI] [PubMed] [Google Scholar]
- La Scola B, Audic S, Robert C, …L.J., 2003, U., 2003. A giant virus in amoebae. science.sciencemag.org 299, 2033. [DOI] [PubMed] [Google Scholar]
- Laslett D, Canback B, 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic acids research 32, 11–6. 10.1093/nar/gkh152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz C, Lünse CE, Mörl M, 2017. tRNA Modifications: Impact on Structure and Thermal Adaptation. Biomolecules 7. 10.3390/biom7020035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR, 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research 25, 955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki S, Tanaka S, Koga T, Kawata T, 1992. A Broad-Host-Range Vibriophage, KVP40, Isolated from Sea Water. Microbiology and Immunology 36, 93–97. 10.1111/j.1348-0421.1992.tb01645.x [DOI] [PubMed] [Google Scholar]
- Miller ES, Heidelberg JF, Eisen JA, Nelson WC, Durkin AS, Ciecko A, Feldblyum TV, White O, Paulsen IT, Nierman WC, Lee J, Szczypinski B, Fraser CM, 2003. Complete genome sequence of the broad-host-range vibriophage KVP40: comparative genomics of a T4-related bacteriophage. Journal of bacteriology 185, 5220–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy FV, Ramakrishnan V, 2004. Structure of a purine-purine wobble base pair in the decoding center of the ribosome. Nature Structural & Molecular Biology 11, 1251–1252. 10.1038/nsmb866 [DOI] [PubMed] [Google Scholar]
- Raoult D, Audic S, Robert C, Abergel C, Renesto P, Ogata H, La Scola B, Suzan M, Claverie J-M, 2004. The 1.2-megabase genome sequence of Mimivirus. Science (New York, N.Y.) 306, 1344–50. 10.1126/science.1101485 [DOI] [PubMed] [Google Scholar]
- R Core Team, 2018. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Reis M. dos, Savva R, Wernisch L, 2004. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic acids research 32, 5036–44. 10.1093/nar/gkh834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherberg NH, Weiss SB, 1972. T4 transfer RNAs: codon recognition and translational properties. Proceedings of the National Academy of Sciences of the United States of America 69, 1114–8. 10.1073/PNAS.69.5.1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF, 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology 75, 7537–41. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz F, Yutin N, Ivanova NN, Ortega DR, Lee TK, Vierheilig J, Daims H, Horn M, Wagner M, Jensen GJ, Kyrpides NC, Koonin EV, Woyke T, 2017. Giant viruses with an expanded complement of translation system components. Science (New York, N.Y.) 356, 82–85. 10.1126/science.aal4657 [DOI] [PubMed] [Google Scholar]
- Sharp PM, Emery LR, Zeng K, 2010. Forces that influence the evolution of codon bias. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 365, 1203–12. 10.1098/rstb.2009.0305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsen SL, Kongstad M, Stenum TS, Muñoz-Gómez AJ, Sørensen MA, 2017. Transfer RNA is highly unstable during early amino acid starvation in Escherichia coli. Nucleic acids research 45, 793–804. 10.1093/nar/gkw1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Osawa S, 1995. tRNA sequences and variation in the genetic code, in: Söll D, Rajbhandary U. (Eds.), TRNA: Structure, Biosynthesis, and Function. ASM Press, Washington, DC, pp. 225–250. [Google Scholar]
- Weiss SB, Hsu WT, Foft JW, Scherberg NH, 1968. Transfer RNA coded by the T4 bacteriophage genome. Proceedings of the National Academy of Sciences of the United States of America 61, 114–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will S, Reiche K, Hofacker IL, Stadler PF, Backofen R, 2007. Inferring Noncoding RNA Families and Classes by Means of Genome-Scale Structure-Based Clustering. PLoS Computational Biology 3, e65. 10.1371/journal.pcbi.0030065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JH, 1973. Function of the bacteriophage T4 transfer RNA’s. Journal of Molecular Biology 74, 753–757. 10.1016/0022-2836(73)90065-X [DOI] [PubMed] [Google Scholar]
- Yokoyama S, Nishimura S, 1995. Modified nucleosides and codon recognition, in: Söll D, RajBhandary UL (Eds.), TRNA: Structure, Biosynthesis, and Function. ASM Press, Washington, DC, pp. 207–223. [Google Scholar]
- Zhong J, Xiao C, Gu W, Du G, Sun X, He Q-Y, Zhang G, 2015. Transfer RNAs Mediate the Rapid Adaptation of Escherichia coli to Oxidative Stress. PLOS Genetics 11, e1005302. 10.1371/journal.pgen.1005302 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data statement.
Raw sequencing reads are available on the NCBI Sequenced Reads Archive under BioProject numbers PRJNA524872 (tRNA-seq) and PRJNA524877 (full transcriptome RNA-seq).
Code statement.
This paper does not report original code.
Scripts statement.
Scripts to generate the figures reported in this paper are available in the (https://github.com/ratatstats/tRNAbias) R package and their use is described in vignettes included with the package. Please first install the package devtools, and then from R, run following command: devtools::install_github(“ratatstats/tRNAbias”, build_vignettes = TRUE, force = TRUE)
Any additional information required to reproduce this work is available from the Lead Contact.