

Abstract
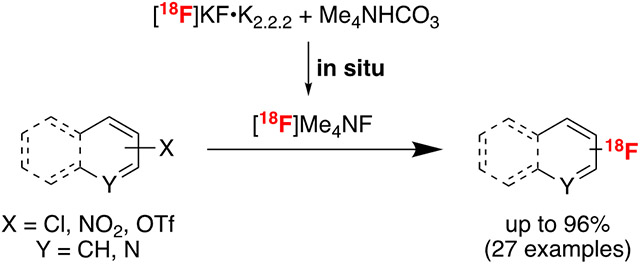
This report describes a method for the nucleophilic radiofluorination of (hetero)aryl chlorides, (hetero)aryl triflates, and nitroarenes using a combination of [18F]KF•K2.2.2 and Me4N HCO3 for the in situ formation of a strongly nucleophilic fluorinating reagent (proposed to be [18F]Me4NF). This method is applied to 24 substrates bearing diverse functional groups, and it generates [18F](hetero)aryl fluoride products in good to excellent radiochemical yields in the presence of ambient air/moisture. The reaction is applied to the preparation of 18F-labeled HQ-415 for potential (pre)clinical use.
Graphical Abstract
Introduction
(Hetero)aryl fluorides are common structural motifs in pharmaceutical scaffolds.1,2 These fluorine-containing biologically active molecules offer opportunities for the simultaneous development of imaging agents for positron emission tomography (PET) by preparation of 18F isotopologs. To capitalize on this, there is an urgent need for late-stage synthetic methods for C(sp2)–18F bond formation.1,3 In general, such methods remain challenging to develop due to the short half-life of 18F (~110 minutes), the low concentrations of 18F present during radiolabeling (typically nanomole scale), and the high sensitivity of many [18F]fluoride sources to water.4
One of the most common approaches for the formation of C(sp2)–18F bonds is nucleophilic aromatic (SNAr) radiofluorination, which involves the reaction of electron-deficient (hetero)aryl halide or nitroarene precursors with [18F]fluoride. These precursors are attractive starting materials that are widely used in PET radiochemistry due to their stability, ready availability, and ease of separation from 18F-labeled products by HPLC.4,5 Anhydrous alkali metal fluoride salts (e.g. [18F]KF or [18F]CsF) are common fluoride sources for SNAr radiofluorination. However, these salts are hydroscopic and become much less nucleophilic upon hydration.2b Moreover, the modest solubility of the salts as well as their tight ion pairing in organic solvents can result in low nucleophilicity and slow reaction rates. Consequently, these reactions can require elevated temperatures (often ≥150 °C), inclusion of additives like Kryptofix® 2.2.2 (K2.2.2), and relatively long reaction times to afford high radiochemical yields (RCYs).4c,d Such requirements often limit functional group compatibility, lead to the formation of side products, and result in undesirably long times of synthesis during 18F labeling.
An attractive approach to mitigate these issues is utilization of tetraalkylammonium [18F]fluoride derivatives ([18F]R4NF) for SNAr fluorination. The corresponding 19F reagents exhibit dramatically enhanced solubility and nucleophilicity in SNAr reactions relative to their alkali metal congeners (Scheme 1A).5-10 Quaternary alkylammonium [18F]fluoride salts (most commonly [18F]Bu4NF and [18F]Et4NF) have found some application in fluorine-18 radiochemistry, including both SNAr radiofluorination (Scheme 1B)11 and in other approaches to late-stage fluorination.12 For instance, in recent examples, Brichard and Aigbirhio13a and Packard13b independently reported the radiofluorination of nitro precursors with [18F]Et4NF, while the Scott group employed [18F]Et4NF to prepare [18F]FL2-b from a 2-chloropyridine precursor.14 While some of the newer approaches to late-stage radiofluorination have eliminated the need for azeotropic drying of quaternary alkylammonium [18F]fluorides,12g for SNAr [18F]R4NF salts are most commonly prepared via solid-phase extraction methods that use aqueous solutions of quaternary alkylammonium salts to elute [18F]fluoride from a quaternary methylammonium cartridge. As such, these sequences require azeotropic drying of the eluted [18F]R4NF prior to radiofluorination. However, the [18F]R4NF salts tend to be very hygroscopic, which impedes efficient drying. In addition, salts containing R groups with β-hydrogen atoms can undergo competing decomposition via Hoffmann elimination both during drying and during SNAr reactions at elevated temperatures. This problem is well-documented in 19F fluorination reactions.8,9,15 While its contribution to radiofluorination is less definitively established (and in certain instances [18F]Bu4NF has outperformed [18F]CsF and [18F]KF•K2.2.211h), the loss of volatile 18F-species can be difficult to detect and quantify.12l
Scheme 1.

SNAr fluorination and radiofluorination with tetraalkylammonium fluoride salts
This elimination problem can be alleviated by moving to tetramethylammonium fluoride (Me4NF), which lacks β-hydrogen atoms. Literature reports have shown that Me4NF is thermally stable and can be dried (albeit over days at elevated temperatures)6,16 to yield an anhydrous fluoride source that is highly nucleophilic in SNAr reactions.16 Despite these advantages, SNAr radiofluorination reactions with [18F]Me4NF remain rare,17,18 likely due to challenges with removal of [18O]H2O (a process that is known to be slow and poorly reproducible in the 19F analogue16,19). In this report, we describe the development of a convenient and reproducible in situ preparation of [18F]Me4NF from [18F]KF and tetramethylammonium salts that circumvents the requirement for rigorous drying of [18F]Me4NF (Scheme 1C). We employ this reagent for the SNAr radiofluorination of a variety of electron deficient (hetero)aryl precursors, including HQ-415, a potential radioligand for metal-protein aggregates implicated in neurodegeneration. We show that the in situ procedure offers key advantages, including relatively low reaction temperatures (≤110 °C), short synthesis times, and compatibility with ambient moisture.
Results and Discussion
Our initial studies explored the development of a simple and reliable method for the preparation of [18F]Me4NF. We first evaluated solid phase extraction approaches involving the direct elution of [18F]Me4NF from a quaternary methylammonium cartridge using Me4NHCO3.13 However, there are key problems with such procedures. Elution with aqueous Me4NHCO3 using standard approaches13,20 results in hydrated [18F]Me4NF which, given the literature precedent of drying nonradioactive Me4NF for hours to days,4a is not compatible with azeotropic drying of short-lived 18F (8-12 min for [18F]KF20). In an attempt to shorten the azeotropic drying time, we examined elution with non-aqueous solutions of tetramethylammonium salts. However, this resulted in poor recovery of fluoride [just 21–80% from [18F]fluoride in 18O target water, decay-corrected (see Supplementary Information, Table S2)].
These initial results suggested a need for an alternative synthetic approach to [18F]Me4NF that circumvents the requirement for elution and azeotropic drying of this salt. Thus, we next pursued the in situ formation of [18F]Me4NF from the combination of [18F]KF and a tetramethylammonium (Me4N+) salt (Scheme 1c). This approach offers the advantages that the elution and azeotropic drying of [18F]KF is already highly optimized,21 allowing short (8-12 min) drying times and high (>95%) recovery of 18F from [18F]fluoride in 18O target water.
2-Chloroquinoline was employed as a model substrate for SNAr radiofluorination with in situ generated [18F]Me4NF. A [18F]KF•K2.2.2 stock solution in DMF was prepared via azeotropic drying under our standard mild conditions (see Experimental Section). Briefly, [18F]fluoride was eluted into the reaction vessel using aqueous KOTf (5 mg in 0.5 mL water, 0.05 M) and K2.2.2 in acetonitrile (15 mg, 1 mL, 0.04 M). [18F]KF•K2.2.2 was then azeotropically dried by heating the reaction vessel at 100 °C while drawing vacuum for 6 min. The reaction vessel was subsequently subjected to an Ar stream and simultaneous vacuum draw for an additional 6 min to produce anhydrous [18F]KF•K2.2.2. Notably, the use of KOTf for elution (as opposed to eluting with stronger bases like K2CO3) allows us to precisely control the amount of base present within a reaction mixture, and such customization has proven useful for other methodology development in our labs.12d An aliquot of this stock solution [92.5-129.5 MBq (2500–3500 μCi) in 100 μL] was then added to a solution containing 2-chloroquinoline (50 μmol) and a tetramethylammonium salt (Me4NX, 100 μmol) in a 4 mL vial. Reactions were heated at 100 °C for 30 min and then analyzed by radio-TLC. To benchmark reactivity in this system, we first carried out control reactions in the absence of Me4NX additives (Table 1, entries 1–6). With just [18F]KF•K2.2.2 as the [18F]fluoride source, no radiofluorinated product was detected under these conditions (Table 1, entry 1). Low radiochemical yields of 2-[18F]fluoroquinoline were obtained upon the addition of KHCO3 or K2CO3 (<10% RCY, in each case; entries 2 and 3) or extra K2.2.2 (8% RCY; entry 6).23 In contrast, the addition of Me4NHCO3 resulted in 91% radiochemical yield of 2-[18F]fluoroquinoline (entry 8), indicating the formation of a dramatically more reactive [18F]fluoride source, which is proposed to be [18F]Me4NF.
Table 1.
In situ formation of [18F]Me4NF for the radiofluorination of 2-chloroquinolinea
|
|||
|---|---|---|---|
| Entry | Me4NX | Additive | RCY (%)f |
| 1 | none | none | 0 |
| 2 | none | KHCO3 | 6 |
| 3 | none | K2CO3 | <1 |
| 4 | none | NaHCO3 | <1 |
| 5 | none | KOTf | <1 |
| 6 | none | K2.2.2 | 8 |
| 722 | Me4NBF4 | none | <1 |
| 8 | Me4NOAc | none | 17 |
| 9b | Me4NHCO3e | none | 91 |
| 10b | Et4NHCO3 | none | 85 |
| 11c | Me4NHCO3, H2O | none | 70 |
| 12d | Me4NHCO3 | none | 96 |
Azeotropically dried [18F]K18F with K2.2.2 in DMF prepared in an GE TRACERlab FXFN. 100 μL of the [18F]K18F/K2.2.2 was used for each manual reaction (total reaction volume = 100 μL).
Me4NX was added to the reaction vial on the bench top.
2 equiv of H2O (1.8 μL, 100 μmol) added compared to approx. 0.00001 equiv of fluoride (92.5 MBq (2.5 mCi) assuming molar activity of 185 GBq/μmol (5 mCi/nmol), 0.5 nmol)
110 °C
Up to 6 equiv of Me4NHCO3 could be added without significant effect on RCY (see Table S4 in Supporting Information)
Radiochemical yield (RCY) was determined by radio-TLC, and the identity of the product was confirmed by radio-HPLC.
We next investigated a series of different Me4NX salts, with X = Cl, Br, I, BF4, OAc, and HCO3). Among these, only Me4NOAc (entry 8) and Me4NHCO3 (entry 9) afforded >10% radiochemical yield of 2-[18F]fluoroquinoline (Tables 1 and S5). Me4NBF4 gave poor (<1%) yield with this substrate (entry 7); however, for some other substrates it proved an effective tetramethylammonium source (see Supporting Information).24 The radiofluorination of 2-chloroquinoline with [18F]KF•K2.2.2/Me4NHCO3 proceeded in high radiochemical yield at 100 °C. An analogous effect was observed by adding Et4NHCO3 (presumably to form [18F]Et4NF in situ), although in slightly diminished RCY (entry 10).The reactions proceed in the presence of ambient air/moisture. Indeed, the radiochemical yield remained >70% even upon the addition of 2 equiv of H2O to the mixture (entry 11), indicating only modest sensitivity to moisture. Further optimization of the reaction temperature, time, concentration, and solvent (see Supporting Information) afforded optimal conditions as 50 μmol of substrate, 100 μmol of Me4NHCO3, 100 μL of DMF, 110 °C, resulting in 96% radiochemical yield of 2-[18F]fluoroquinoline (entry 12).
The scope of this transformation was next evaluated with a series of (hetero)aryl precursors containing standard chloride, nitro, or triflate leaving groups.25 In all cases, the results with [18F]KF•K2.2.2 (control, conditions A) were compared to those with [18F]KF•K2.2.2 and Me4NHCO3 (conditions B) under otherwise identical conditions (at 100-100 °C).23,24 As summarized in Figure 1-i, for 118F–1518F the control reaction with [18F]KF•K2.2.2 afforded low yield at 100-110 °C (ranging from 0–27% yield, Figure 1-i). Conversely, moderate to excellent RCY (up to 97%) was obtained upon addition of Me4NX, even with the precursors that afforded low (or no) RCY with [18F]KF•K2.2.2 (Figure 1-i). In other cases (1618F–2418F) the addition of Me4NHCO3 led to comparable yields as the control reactions with [18F]KF•K2.2.2 (Figure 1-ii).
Figure 1. Substrate scope27, a,b.

aRCY was determined by radio-TLC and identity of final product was confirmed by HPLC. N.a. = not available. Reaction temperature was optimized to 110 °C for 618F, 1218F, 1318F, and 2118F. All other reactions were conducted at 100 °C; b For data using Me4NBF4 as an additive, see Supporting Information.
This study also revealed that for pyridines bearing both nitro and chloro groups, 18F-fluorination at the 2-position is favored over the 3- and 5-positions, regardless of leaving group. For example, 2-chloropyridines were converted to 2-[18F]fluoropyridines in the presence of 3-nitro or 5-nitro substituents (918F, and 2318F, respectively), while a 2-nitro group was substituted in the presence of a 3-chloro (418F). In these cases, there was no evidence of 18F-labeled side products from competing substitution reactions. Conversely, in the case of pyridines containing both chloro and fluoro substituents, isotopic exchange was the dominant reaction pathway. Thus, 2-chloro-3-fluoropyridine and 2-chloro-6-fluoropyridine underwent isotopic exchange (6-91% RCY), while minimal quantities (≤9%) of the 18F-difluoropyridines were observed (Supporting Information, Figure S2).
Substituted pyridine, pyrazine, quinoline, and isoquinoline precursors all underwent efficient radiofluorination using this method. Less activated substrates, such as 2-chlorobenzonitrile and 4-chlorobenzophenone afforded modest yields of the radiofluorinated products 1018F and 1218F (16 and 9%, respectively). However, moving from the chloro- to the nitro- precursor led to a dramatic increase in the yield of 1218F (to 77%). It is also noteworthy that 2-chlorobenzonitrile was significantly more reactive than 4-chlorobenzonitrile, which afforded <5% yield of the radiofluorinated product 1118F under conditions A or B. A similar trend was observed for chloropyridine derivatives bearing electron withdrawing groups either ortho- or para- to the chlorine (for instance compare 818F to 2418F and 1618F to 1818F). These results are consistent with the previously documented ortho effect in nucleophilic aromatic substitution.26 A variety of less electrophilic precursors, including 2-chloro-4-aminopyridine, 3-chloroquinoline, and 3-chlorobenzonitrile, were also examined in this transformation. As expected based on the SNAr literature, these all showed low reactivity, affording <1% yield of the targeted radiofluorinated products (Supplementary Information, Figure S3).
This method was next applied to the labeling of the 2-chloroquinoline moiety in Pre-HQ415 to form [18F]HQ415, a molecule of interest for PET imaging of the toxic TDP43 protein deposits implicated in amyotrophic lateral sclerosis (ALS).28,29 As shown in Scheme 2, a deprotection of the Boc groups with 2 M HCl was incorporated into the manual one-pot synthesis. Using the combination of [18F]KF•K2.2.2 and Me4NHCO3, the radiofluorination proceeded in 82 ± 2% (n = 3) radiochemical yield, and [18F]HQ415 was obtained in 56 ± 3% (n = 3) yield from the two step sequence. In contrast, the radiofluorination proceeded in just 10% RCY with [18F]KF•K2.2.2 alone, again highlighting the advantages of in situ-generated [18F]Me4NF for enhancing reactivity and yields in SNAr fluorination reactions conducted at lower temperatures.
Scheme 2.

Synthesis of [18F]HQ415
Finally, the SNAr radiofluorination of 2-chloroquinoline was translated to an automated method using 63 GBq (1.7 Ci) of [18F]KF•K2.2.2 (Scheme 3). The reaction was upscaled with respect to the amount of the precursor, amount of Me4NHCO3, and reaction volume to simplify automation, as smaller volumes are not compatible with our automated synthesis modules). Conditions for automated runs were as follows: 100 μmol precursor, 500 μmol Me4NHCO3, and 0.5 mL reaction volume. The fully automated SNAr radiofluorination with [18F]KF•K2.2.2 and Me4NHCO3 afforded 2-[18F]fluoroquinoline (118F) in 18 ± 4% radiochemical yield, with molar activity of 155.5 ± 66.4 GBq/μmol (4.2 ± 1.8 Ci/μmol) and radiochemical purity of greater than 99% (n = 4). Automated RCYs of late-stage fluorination reactions are frequently lower than those for the corresponding manual reactions,30 and the present work is no exception. The reasons for this are currently unknown, but efforts are underway in our laboratories to investigate the problem via a systems approach using machine learning (ML).31 The data from this study will be incorporated into ML models and findings reported in due course. Lastly, one of the automated production batches was reformulated into 10% ethanol in saline using a C18 cartridge. We analyzed the formulated product for residual tetramethylammonium salts using recently developed TLC methods.32 This analysis shows levels below the accepted regulatory limits (not more than 2.6 mg/10 mL patient dose), promising results for translation of this methodology to the production of PET radiotracers for clinical use.
Scheme 3.

Automation of SNAr radiofluorination with in situ Me4N18F
Conclusions
In conclusion, this report describes a method for the SNAr radiofluorination of (hetero)aryl chlorides and triflates as well as nitroarenes using a combination of [18F]KF•K2.2.2 and Me4NHCO3 for the in situ formation of a strongly nucleophilic fluorination reagent (presumed to be [18F]Me4NF). This method circumvents the need to independently synthesize and dry [18F]Me4NF, steps that can be inefficient and low yielding. Instead, this approach enables rapid nucleophilic aromatic radiofluorination using a well-optimized [18F]fluoride source at moderate temperature (100-110 °C) in the presence of ambient air/moisture, offering a valuable approach for labeling temperature sensitive molecules. This method is operationally simple and proceeds reliably without rigorous exclusion of water to provide radiolabeled products in high RCYs. Furthermore, it is readily translated to automation. The method is effective for the late-stage nucleophilic radiofluorination of biologically active molecules, as highlighted by the synthesis of [18F]HQ415, a molecule of interest for PET imaging of the TDP43 deposits implicated in ALS. Overall, this work makes [18F]Me4NF readily accessible to the PET community, enabling exploitation of its unique reactivity profile for the development of new radiochemistry methodology and the application to the synthesis of new PET radiotracers.
Experimental Section
Chemistry
Materials and Methods
All commercial products were used as received and reagents were stored under ambient conditions unless otherwise stated. Quinoline, pyridine, pyrazine, benzophenone, and benzonitrile precursors for the substrate scope as well as their fluorinated reference standards were purchased from commercial sources (Synthonix, Frontier Scientific, Oakwood Products, Acros Organics, Synthonix, Chem Impex, TCI America, Matrix Scientific, Alfa Aesar, Ark Pharm, or Sigma Aldrich) unless otherwise stated. Isopropyl 5-chloro-6-phenylpicolinate were prepared using previously described methods.10 Isopropyl 5-fluoro-6-phenylpicolinate and 4-fluoro-7-(trifluoromethyl)quinoline was prepared using previously described methods.16 3-Ethoxy-4-methoxybenzaldehyde was purchased from Sigma Aldrich. The manipulation of solid reagents was conducted on the benchtop unless otherwise stated. Reactions were conducted under an ambient atmosphere unless otherwise stated. Reaction vessels were sealed with either a septum (flask) or a Teflon-lined cap (4 mL or 20 mL vial). Reactions conducted at elevated temperatures were heated on a hot plate using an aluminum heating block. The temperatures was regulated using an external thermocouple. TLC analyses were performed using TLC Silica gel 60 F254 MS-grade plates. RF values are reported based on standard silica gel plates with fluorescent indicator.
NMR spectra were obtained on a Varian vnmrs700 (699.76 MHz for 1H; 175.95 MHz for 13C), a Varian vnmr500 (500.09 MHz for 1H; 470.56 MHz for 19F; 125.75 MHz for 13C), or a Varian MR400 (400.53 MHz for 1H; 376.87 MHz for 19F) spectrometer. All 13C NMR data presented are proton-decoupled 13C NMR spectra, unless noted otherwise. 1H and 13C NMR chemical shifts (δ) are reported in parts per million (ppm) relative to TMS with the residual solvent peak used as an internal reference. 1H and 19F NMR multiplicities are reported as follows: singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m). Melting point (mp) data were collected on an OptiMelt Automated Melting Point System and are uncorrected. High performance liquid chromatography (HPLC) was performed using a Shimadzu LC-2010A HT system equipped with a Bioscan B-FC-1000 radiation detector. High resolution mass spectroscopy (HRMS) was performed on an Agilent Technologies 6520 Accurate Mass Q-TOF LC/MS using electrospray ionization (ESI+).
Preparation and characterization of aryl chloride and aryl triflate precursors
Quinolin-2-yl trifluoromethanesulfonate.
In a modification of a published method,33 triflic anhydride (3.10 g, 11 mmol, 1.1 equiv) was added dropwise to the corresponding phenol (1.45 g, 10 mmol, 1 equiv) in pyridine (10 mL) at 0 °C. The resulting solution was stirred for 10 min at 0 °C. The reaction was then allowed warm to ambient temperature and was stirred for an additional 12 h. The solution was diluted with diethyl ether (25 mL) and extracted with a 1 M solution of CuSO4 (3 x 25 mL). The organic layer was washed with water (1 x 25 mL), dried over MgSO4, and concentrated in vacuo. The aryl triflate was then purified via column chromatography on silica gel (95 : 5 hexanes/ethyl acetate). The product was isolated as a pale yellow oil (2.69 g, 97%).
Rf: 0.50 (9 : 1 hexanes/ethyl acetate).
1H NMR (400 MHz, CDCl3): δ 8.34 (d, J = 8.7 Hz, 1H), 8.05 (d, J = 8.5 Hz, 1H), 7.90 (dd, J = 8.2, 1.4 Hz, 1H), 7.80 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H), 7.64 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.24 (d, J = 8.8 Hz, 1H).
13C{1H} NMR (176 MHz, CDCl3): δ 153.8, 146.0, 142.0, 131.3, 129.0, 127.9, 127.8, 127.7, 118.8 (q, J = 320.5 Hz), 113.3.
19F NMR (377 MHz, CDCl3): δ –73.00.
HRMS: (ESI) m/z [M+H]+ calcd. for C10H7F3NO3S+ 278.0093; found 278.0085.
8-(Benzyloxy)-2-chloroquinoline.
2-Chloro-4-hydroxyquinoline (121.2 mg, 0.67 mmol, 1.0 equiv), potassium carbonate (185.1 mg, 1.34 mmol, 2.0 equiv), potassium iodide (11.1 mg, 0.067 mmol, 0.1 equiv), and benzyl bromide (96 μL, 0.80 mmol, 1.2 equiv) were combined in acetonitrile (3 mL). The reaction mixture was heated at reflux for 24 h. After cooling to room temperature, solids were removed by filtration, and the filtrate was concentrated under reduced pressure. Purification via column chromatography on silica (85 : 15 hexanes/ethyl acetate) afforded the product as a white solid (142.4 mg, 79% yield).
Rf: 0.54 (8 : 2 hexanes/ethyl acetate).
m.p. 90.2-91.1 °C.
1H NMR (700 MHz, CDCl3): δ 8.06 (dd, J = 8.6, 1.4 Hz, 1H), 7.51 (s, 1H), 7.50 (s, 1H), 7.41 (dd, J = 8.6, 1.4 Hz, 1H), 7.39-7.34 (multiple peaks, 4H), 7.30 (m, 1H), 7.06 (dt, J = 6.6, 1.9 Hz, 1H), 5.45 (s, 2H).
13C{1H} NMR (176 MHz, CDCl3): δ 153.7, 150.0, 140.1, 138.9, 137.0, 128.7, 128.3, 127.9, 127.2, 127.0, 123.1, 119.7, 111.8, 71.0.
HRMS: (ESI) m/z [M+H]+ calcd. for C16H13ClNO+ 270.0680; found 270.0680.
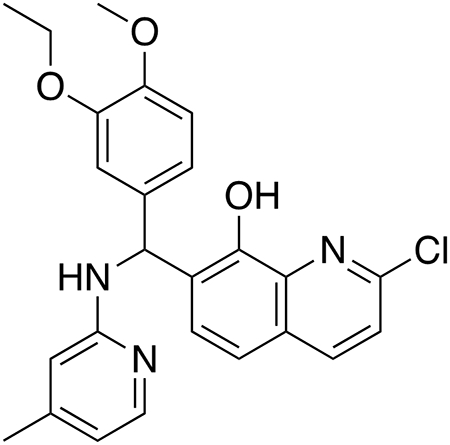
2-Chloro-7-((3-ethoxy-4-methoxyphenyl)((4-methylpyridin-2-yl)amino)methyl)quinolin-8-ol (HQ415).
3-Ethoxy-4-methoxy-benzaldehyde (0.620 g, 3.45 mmol, 1 equiv) and 2-amino-4-methyl pyridine (0.373 g, 3.45 mmol, 1 equiv) was dissolved in EtOH (25 mL) at room temperature. 2-Chloro-5-hydroxyquinoline (0.622 g, 3.45 mmol, 1 equiv) was added. The reaction was stirred at room temperature for 14 d. The solvent was removed under vacuum. The crude product was diluted with DCM (30 mL). The organic solution was extracted with water (30 mL), the organic layer was collected, and the aqueous fraction was washed with dichloromethane (3 x 30 mL). The combined organic extracts were washed with brine (30 mL), dried over anhydrous magnesium sulfate, and concentrated under vacuum. The product was purified by column chromatography on silica gel (60 : 40 hexanes/ethyl acetate) afforded the product as a white solid (713 mg, 46%).
Rf: 0.29 (4 : 6 hexanes/ethyl acetate).
m.p. 77.6-79.6 °C.
1H NMR (400 MHz, CDCl3): δ 8.03 (d, J = 8.6 Hz, 1H), 7.96 (d, J = 5.2 Hz, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.35 (d, J = 8.5 Hz, 1H), 7.28 (d, J = 8.6 Hz, 1H), 6.99 (d, J = 2.1 Hz, 1H), 6.92 (dd, J = 8.4, 2.1 Hz, 1H), 6.79 (d, J = 8.3 Hz, 1H), 6.43 (d, J = 5.2 Hz, 1H), 6.33 (d, J = 6.4 Hz, 1H), 6.21 (s, 1H), 5.42 (d, J = 6.4 Hz, 1H), 4.02 (qd, J = 7.0, 2.3 Hz, 2H), 3.82 (s, 3H), 2.15 (s, 3H), 2.04 (s, 1H), 1.39 (t, J = 7.0 Hz, 3H).
13C{1H} NMR (126 MHz, CDCl3): δ 158.2, 149.4, 148.9, 148.6, 148.5, 148.4, 147.8, 139.0, 138.2, 134.4, 126.5, 126.3 122.8, 119.1, 118.1, 115.3, 112.1, 111.4, 107.5, 64.4, 56.0, 54.7, 21.4, 14.8.
HRMS: (ESI) m/z [M+H]+ calcd. for C25H25ClN3O3+ 450.1579; found 450.1576.
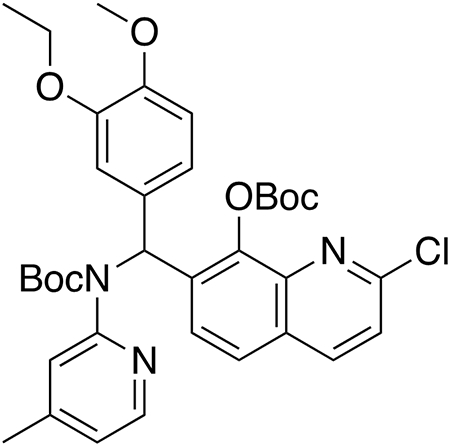
Tert-butyl ((8-((tert-butoxycarbonyl)oxy)-2-chloroquinolin-7-yl)(3-ethoxy-4-methoxyphenyl)methyl)(4-methylpyridin-2-yl)carbamate (pre-HQ415).
2-Chloro-7-((3-ethoxy-4-methoxyphenyl)((4-methylpyridin-2-yl)amino)methyl)quinolin-8-ol (31.4 mg, 0.0697 mmol, 1.0 equiv), Boc2O (56.0 μL, 0.244 mmol, 3.5 equiv), and 4-dimethylaminopyridine (17.0 mg, 0.139 mmol, 2.0 equiv) were dissolved in tetrahydrofuran (0.5 mL). The reaction was stirred for 24 h at room temperature and then diluted with dichloromethane (10 mL). The organic solution was extracted with brine (10 mL), the organic layer were collected, and the aqueous fraction was washed with dichloromethane (3 x 10 mL). The combined organic extracts were washed again with brine (10 mL), dried over anhydrous magnesium sulfate, and concentrated under vacuum. Purification by column chromatography on silica gel (3 : 2 hexanes/ethyl acetate) afforded the product as a white solid (30.9 mg, 68% yield).
Rf: 0.89 (2 : 3 hexanes/ethyl acetate).
m.p. 80.3-82.4 °C.
1H NMR (400 MHz, CDCl3): δ 8.18 (d, J = 5.1 Hz, 1H), 8.02 (d, J = 8.6 Hz, 1H), 7.88 (d, J = 8.7 Hz, 1H), 7.55 (d, J = 8.7 Hz, 1H), 7.34 (d, J = 8.6 Hz, 1H), 7.12 (s, 1H), 6.99 (s, 1H), 6.95 (d, J = 2.1 Hz, 1H), 6.87 (dd, J = 8.4, 2.1 Hz, 1H), 6.77 (d, J = 4.6 Hz, 1H), 6.72 (d, J = 8.3 Hz, 1H), 3.96 (p, J = 6.9 Hz, 2H), 3.81 (s, 3H), 2.17 (s, 3H), 1.54 (s, 1H), 1.45 (s, 9H), 1.37 (t, J = 6.9 Hz, 3H), 1.31 (s, 9H).
13C{1H} NMR (176 MHz, CDCl3): δ 154.6, 154.0, 151.0, 150.9, 148.2, 147.7, 147.5, 144.4, 140.6, 138.5, 134.9, 131.6, 129.2, 126.9, 124.4, 123.7, 122.9, 122.4, 121.3, 113.7, 110.7, 83.4, 81.4, 64.2, 60.4, 56.0, 28.3, 27.6, 20.9, 14.8 (d, J = 2.3 Hz).
HRMS: (ESI) m/z [M+H]+ calcd. for C35H41ClN3O7+ 650.2628; found 650.2619.
Preparation and characterization of fluorinated standards
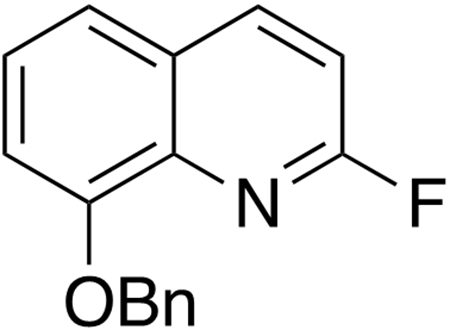
8-(Benzyloxy)-2-fluoroquinoline.
4-(Benzyloxy)-2-chloroquinoline (135.6 mg, 0.50 mmol, 1.0 equiv) and Me4NF•t-AmylOH (1:0.95) (132.6 mg, 0.75 mmol, 1.5 equiv) were dissolved in anhydrous DMSO (2.5 mL). The reaction was stirred for 24 h at 80 °C. After cooling to room temperature, the mixture was diluted with dichloromethane (10 mL), and the organic solution was extracted with brine (10 mL). The organic layer was collected, and the aqueous fraction was washed with dichloromethane (3 x 10 mL). The combined organic extracts were then combined, washed with brine (10 mL), dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. Purification by column chromatography on silica gel (4 : 1 hexanes/ethyl acetate) afforded the product as a light yellow solid (85.0 mg, 67% yield).
Rf: 0.71 (7 : 3 hexanes/ethyl acetate).
m.p. 68.6-69.7 °C.
1H NMR (400 MHz, CDCl3): δ 8.22 (t, J = 8.4 Hz, 1H), 7.53–7.49 (multiple peaks, 2H), 7.44–7.33 (multiple peaks, 4H), 7.30 (t, J = 7.3 Hz, 1H), 7.10 (td, J = 8.4, 2.2 Hz, 2H), 5.42 (s, 2H).
13C{1H} NMR (126 MHz, CDCl3): δ 160.7 (dd, J = 241.9, 3.7 Hz), 153.6 (d, J = 3.2 Hz), 142.1 (dd, J = 9.7, 3.6 Hz), 137.7 (dd, J = 15.3, 3.7 Hz), 136.9 (d, J = 3.9 Hz), 128.7 (d, J = 3.8 Hz), 128.2, 127.9 (d, J = 3.7 Hz), 127.1 (d, J = 3.8 Hz), 126.2 (d, J = 4.1 Hz), 119.7 (d, J = 3.7 Hz), 111.7 (d, J = 3.7 Hz), 110.6 (dd, J = 42.5, 3.7 Hz), 70.9 (d, J = 3.7 Hz).
19F NMR (377 MHz, CDCl3): δ –61.07 (dd, J = 8.1, 3.1 Hz).
HRMS: (ESI) m/z [M+H]+ calcd. for C16H13FNO+ 254.0976; found 254.0982.
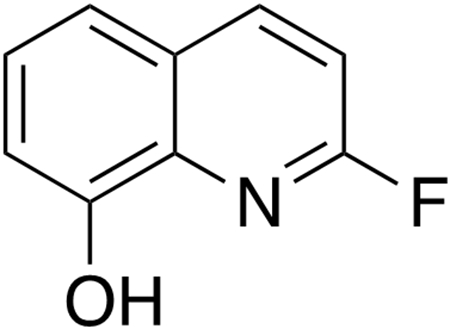
2-Fluoroquinolin-8-ol.
4-(benzyloxy)-2-fluoroquinoline (93.2mg, 0.367mmol, 1.0 equiv) was added to a 20mL vial and dissolved in DCM (1.5mL). A 1M solution of BBr3 in DCM (0.147mL, 1.47mmol, 4.0 equiv) was added to the reaction dropwise. The reaction was capped and stirred for 24 hours at room temperature. After 24 hours, the reaction was diluted with DCM (10 mL) and brine (10 mL). The organic layer was collected and the aqueous fraction was washed 3 times with DCM (10 mL). The combined organic phases were then washed with brine (10 mL), dried over anhydrous magnesium sulfate, and concentrated under reduced pressure using a rotary evaporator. Purification by column chromatography (85 : 15 hexanes/ethyl acetate) afforded the product as a white solid (49.7 mg, 83% yield).
Rf: 0.64 (8 : 2 hexanes/ethyl acetate).
m.p. 54.6-55.4 °C.
1H NMR (700 MHz, CDCl3): δ 8.26 (dd, J = 8.8, 7.7 Hz, 1H), 7.45 (t, J = 7.9 Hz, 1H), 7.40 – 7.34 (multiple peaks, 2H), 7.23 (dd, J = 7.6, 1.3 Hz, 1H), 7.10 (dd, J = 8.8, 2.3 Hz, 1H).
13C{1H} NMR (176 MHz, CDCl3): δ 160.5 (d, J = 246.0 Hz), 151.3 (d, J = 2.1 Hz), 142.5 (d, J = 9.6 Hz), 135.3 (d, J = 13.3 Hz), 127.2 (d, J = 2.5 Hz), 127.1 (d, J = 2.0 Hz), 118.1, 112.1, 110.6 (d, J = 40.9 Hz).
19F NMR (377 MHz, CDCl3): δ –64.33 (d, J = 7.5 Hz).
HRMS: (ESI) m/z [M+H]+ calcd. for C9H7FNO+ 164.0507; found 164.0506.
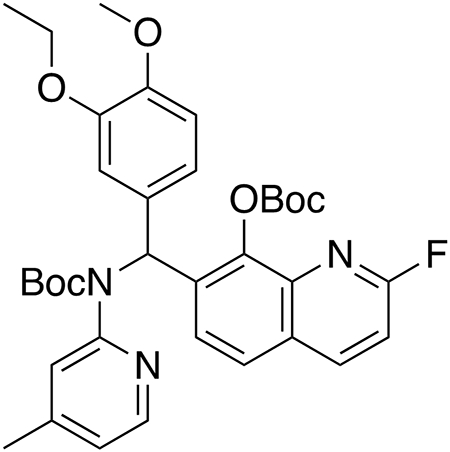
Tert-butyl ((8-((tert-butoxycarbonyl)oxy)-2-fluoroquinolin-7-yl)(3-ethoxy-4-methoxyphenyl)methyl)(4-methylpyridin-2-yl)carbamate (F-inter-HQ415).
7-((3-Ethoxy-4-methoxyphenyl)((4-methylpyridin-2-yl)amino)methyl)-2-fluoroquinolin-8-ol (23.6 mg, 0.054 mmol, 1.0 equiv), Boc2O (124 μL, 0.54 mmol, 10.0 equiv), and 4-dimethylaminopyridine (26.4 mg, 0.216 mmol, 4.0 equiv) were dissolved in tetrahydrofuran (0.5 mL). The reaction was stirred for 48 h at room temperature and then diluted with DCM (10 mL). The organic solution was extracted with water (10 mL), the organic layer was collected, and the aqueous fraction was washed with dichloromethane (3 x 10 mL). The combined organic extracts were washed with brine (10 mL), dried over anhydrous magnesium sulfate, and concentrated under vacuum. Purification by column chromatography on silica gel (7 : 3 hexanes/ethyl acetate) afforded the product as a white solid (11.4 mg, 33% yield).
Rf: 0.62 (6 : 4 hexanes/ethyl acetate).
m.p. 67.6-68.9 °C.
1H NMR (400 MHz, CDCl3): δ 8.22–8.13 (multiple peaks, 2H), 7.86 (d, J = 8.6 Hz, 1H), 7.58 (d, J = 8.7 Hz, 1H), 7.10 (s, 1H), 7.04 (dd, J = 8.9, 2.6 Hz, 1H), 6.98 (s, 1H), 6.96 (d, J = 1.9 Hz, 1H), 6.88 (d, J = 9.2 Hz, 1H), 6.77 (d, J = 5.1 Hz, 1H), 6.72 (d, J = 8.3 Hz, 1H), 3.96 (p, J = 6.9 Hz, 2H), 3.81 (s, 3H), 2.16 (s, 3H), 1.57 (s, 1H), 1.43 (s, 9H), 1.37 (t, J = 6.9 Hz, 3H), 1.30 (s, 9H).
13C{1H} NMR (176 MHz, CDCl3): δ 161.2 (d, J = 243.5 Hz), 154.5, 154.0, 150.9, 148.2 (d, J = 6.0 Hz), 147.6, 147.4, 144.2, 141.7 (d, J = 9.7 Hz), 135.2, 131.6, 128.3, 126.99 (d, J = 1.4 Hz, 124.4, 123.7, 122.4, 121.4, 113.8, 110.8, 110.7, 110.5, 83.4, 81.3, 64.2, 60.4, 56.0, 28.4, 28.3, 27.6, 20.9, 14.8.
19F NMR (377 MHz, CDCl3): δ –60.57 (d, J = 7.7 Hz).
HRMS: (ESI) m/z [M+H]+ calcd. for C35H41FN3O7+ 634.2923; found 643.2912.
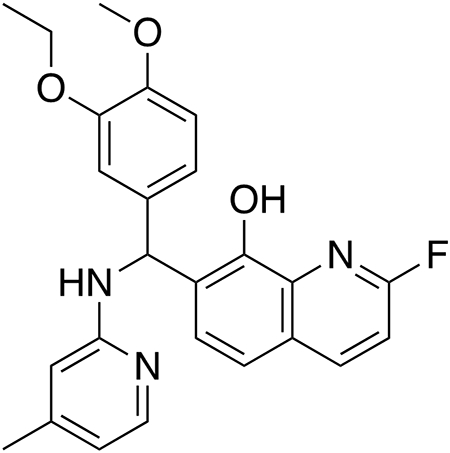
7-((3-Ethoxy-4-methoxyphenyl)((4-methylpyridin-2-yl)amino)methyl)-2-fluoroquinolin-8-ol (F-HQ415).
3-Ethoxy-4-methoxybenzaldehyde (119.3 mg, 0.662 mmol, 1.0 equiv), 2-amino-4-methylpyridine (71.6 mg, 0.662 mmol, 1.0 equiv), and 2-fluoroquinolin-8-ol (108.0 mg, 0.662 mmol, 1.0 equiv) were dissolved in ethanol (2.6 mL). The reaction was stirred for 380 d at room temperature and then diluted with DCM (10 mL). The organic solution was extracted with brine (10 mL), the organic layer was collected, and the aqueous fraction was washed with dichloromethane (3 x 10 mL). The combined organic extracts were washed again with brine (10 mL), dried over anhydrous magnesium sulfate, and concentrated under vacuum. Purification by column chromatography on silica gel (3 : 2 hexanes/ethyl acetate) afforded the product as a white solid (44.4 mg, 15% yield).
Rf: 0.29 (2 : 3 hexanes/ethyl acetate).
m.p. 66.7-68.5 °C.
1H NMR (400 MHz, CDCl3): δ 8.19 (t, J = 8.3 Hz, 1H), 7.92 (d, J = 5.4 Hz, 1H), 7.53 (d, J = 8.5 Hz, 1H), 7.33 (d, J = 8.5 Hz, 1H), 7.06 (dd, J = 8.7, 2.2 Hz, 1H), 7.01 (d, J = 2.1 Hz, 1H), 6.94 (dd, J = 8.3, 2.1 Hz, 1H), 6.80 (d, J = 8.3 Hz, 1H), 6.43 (d, J = 5.4 Hz, 1H), 6.34 (d, J = 6.3 Hz, 1H), 6.25 (s, 1H), 5.91 (s, 1H), 4.03 (qd, J = 6.9, 2.2 Hz, 2H), 3.83 (s, 3H), 2.16 (s, 3H), 1.40 (t, J = 6.9 Hz, 3H).
13C{1H} NMR (126 MHz, CDCl3): δ 160.7 (d, J = 245.6 Hz), 158.2, 149.0, 148.6, 148.5, 148.4, 147.6, 142.2 (d, J = 9.5 Hz), 135.7 (d, J = 13.4 Hz), 134.4, 126.9, 126.3, 126.2 (d, J = 2.2 Hz), 119.0 118.2, 115.3, 112.1, 111.4, 110.3 (d, J = 41.1 Hz), 107.6, 64.4, 56.0, 54.6, 21.4, 14.8.
19F NMR (377 MHz, CDCl3): δ –63.86 (dd, J = 7.5, 2.5 Hz).
HRMS: (ESI) m/z [M+H]+ calcd. for C25H25FN3O3+ 434.1874; found 434.1879.
Radiochemistry
Materials and methods.
HPLC grade acetonitrile, potassium trifluoromethanesulfonate, and anhydrous dimethylformamide were purchased from Fisher Scientific. Sodium bicarbonate, kryptofix® 2.2.2 (K2.2.2), and anhydrous acetonitrile were purchased from Sigma-Aldrich. Tetramethylammonium bicarbonate (Me4NHCO3) was purchased from Enamine. Tetramethylammonium tetrafluoroborate (Me4NBF4) was purchased from TCI America. Sterile product vials (10 mL) were purchased from Hollister-Stier. QMA-light and C18 Lite Sep-Paks were purchased from Waters Corporation. QMA-light Sep-Paks were flushed with isopropanol (10 mL), 0.5 M aqueous sodium bicarbonate (10 mL), and MilliQ water (10 mL) prior to use for the generation of K18F and in situ Me4N18F. C18 Sep-Paks were flushed with ethanol (10 mL) and sterile water for injection (10 mL) prior to use. High performance liquid chromatography (HPLC) was performed using a Shimadzu LC-2010A HT system equipped with a Bioscan B-FC-1000 radiation detector. Radio-TLC analyses were performed using a Bioscan AR 2000 Radio-TLC scanner with EMD Millipore TLC silica gel 60 plates (3.0 cm wide x 6.5 cm long). Residual TMA levels were determined using TLC spot tests as previously described.32 Chromatography was performed using the following conditions:
Chromatography Conditions
HPLC Conditions A.
Solvent conditions: Gradient
| 0 – 4 min | 5 % acetonitrile in water, 0.1 % (v/v) trifluoroacetic acid |
| 4 – 6 min | 25 % acetonitrile in water, 0.1 % (v/v) trifluoroacetic acid |
| 6 – 19 min | 95 % acetonitrile in water, 0.1 % (v/v) trifluoroacetic acid |
| 19 – 25 min | 5 % acetonitrile in water, 0.1 % (v/v) trifluoroacetic acid |
Flow rate: 2 mL/min, running time: 25 min
Column: Phenomenex® Kinetex PFP column 250 × 4.6 mm, 5 μm
HPLC Conditions B.
Solvent: Isocratic 60 % acetonitrile in water, 0.1 % (v/v) trifluoroacetic acid
Flow rate: 2 mL/min, running time: 20 min
Column: Phenomenex® Luna PFP(2) column 250 × 4.6 mm, 5 μm
HPLC Conditions C.
Solvent: Isocratic 50 % acetonitrile in water, 0.1 % (v/v) trifluoroacetic acid
Flow rate: 5 mL/min, running time: 30 min
Column: Phenomenex® Luna PFP(2) column 250 × 10.0 mm, 5 μm
Radio-TLC method
Mobile phase: hexane/ethyl acetate (1/1)
Scanning time: 1 min
Scanning range: 60 mm – 125 mm
Spotting position of radioactive product: 65 mm
Generation of [18F]Fluoride
Generation of [18F]KF.
The [18F]fluoride was delivered to the automated synthesis module (TRACERLab FXFN, GE) in a 1.5 mL bolus of 18O target water and was trapped on a preconditioned QMA-light Sep-Pak to remove 18O target water and other aqueous impurities. [18F]Fluoride was eluted into the reaction vessel using aqueous potassium trifluoromethanesulfonate (5 mg in 0.5 mL water, 0.05 M) with potassium carbonate (0.5 mg) and K2.2.2 in acetonitrile (15 mg, 1 mL, 0.04 M). Azeotropic drying/evaporation was achieved by heating the reaction vessel at 100 °C while drawing vacuum for 6 min. The reaction vessel was then subjected to an argon stream and simultaneous vacuum draw for an additional 6 min to produce anhydrous [18F]KF•K2.2.2. The reaction vessel was cooled to room temperature under an argon stream, and anhydrous DMF (6 mL) was added. The mixture was heated to 50 °C with stirring for 5 min to suspend the [18F]KF•K2.2.2. The resulting solution was cooled to room temperature and was transferred to a sterile vial.
Generation of in situ [18F]Me4NF.
[18F]KF•K2.2.2 was prepared by the same protocol described above. A aliquot of [18F]KF•K2.2.2 in DMF was added to a vial containing a tetramethylammonium salt (e.g. Me4NHCO3 or Me4NBF4, 100-300 μmol) and the substrate (20-50 μmol) in DMF.
Radiosynthesis of 18F-labeled molecules
Manual synthesis general procedure.
A 4 mL vial was charged with chloro-, nitro-, or triflate substrate (50 μmol) and tetramethylammonium salt (Me4NHCO3 or Me4NBF4) (100-300 μmol) on the bench top. An aliquot of [18F]KF•K2.2.2 in DMF (100 μL, 92.5 – 129.5 MBq (2500–3500 μCi) of radioactivity) was used for each manual reaction. Total reaction volume was 100μL. The reaction vial was sealed with a Teflon-lined cap and heated in an aluminum block at 100-110 °C for 30 min. After 30 min, the reaction was cooled to room temperature, and the radiochemical yield (RCY, %) was determined by radio-TLC analysis using the following procedure. The crude reaction mixture was spotted onto a standard silica-coated glass TLC plate (EMD Millipore Corporation, TLC Silica-gel 60 F254) and developed with hexanes:ethyl acetate (1:1) in a glass TLC chamber. The RCY was determined by dividing the integrated area of radiation under the fluorinated product spot by the total integrated area of radiation on the TLC plate. In reactions where radio-HPLC traces show multiple peaks, the RCY (determined by radio-TLC) was corrected by dividing the integrated area of radiation under the desired 18F-labeled product peak by the total integrated area of radiation on the analytical radio-HPLC (HPLC conditions A). To prepare samples for HPLC analysis, 80 μL of the reaction mixture was spiked with 20 μL of 2 mg/mL fluorinated standard solution in DMF. Eluent systems and columns used for HPLC analysis are described below.
Automated synthesis of 2-[18F]fluoroquinoline.
All loading operations were conducted under ambient atmosphere. Argon gas and vacuum were used for automated sample transfers. [18F]Fluoride was produced via the 18O(p, n)18F nuclear reaction using a General Electric (GE) PETTrace cyclotron (55 μA beam for 3 min generated ca. 63 GBq (1.7 Ci) of [18F]fluoride). [18F]KF•K2.2.2 was prepared in a GE TRACERLab FXFN automated synthesis module as described above. Dried [18F]KF•K2.2.2 was prepared in the reactor pre-charged with tetramethylammonium bicarbonate (67.5 mg, 500 μmol, 5 equiv). A solution containing 2-chloroquinoline (16.4 mg, 100 μmol, 1 equiv) in 0.5 mL of anhydrous DMF was added to the reactor by applying Ar gas through the valve containing the anhydrous DMF. The open valves leading out of the reactor were closed, and the reaction mixture was stirred at 110 °C for 30 min. The mixture was cooled to 30 °C with compressed air cooling, and the resulting mixture was diluted with 1 mL of DI water and then with 3 mL of semi-preparative HPLC buffer (50% acetonitrile in water, 0.1 % (v/v) trifluoroacetic acid). The resulting solution was then loaded onto the HPLC injection loop and injected onto the semi-prep HPLC for purification by HPLC Conditions C described in Section 4.2.5. The peak for the 18F-labeled organic product was collected for 1 min (tR = 8.4 min, collected volume: 5 mL) in a 10 mL sterile product vial. The dose vial was transferred out of the synthesis module. Final radiochemical yield and product identity were then determined using a Capintec dose calibrator and analytical HPLC (see Supplementary Information, Table S1).
Reformulation of 2-[18F]fluoroquinoline.
2-[18F]fluoroquinoline in HPLC mobile phase (5 mL) was diluted with water (40 mL) and passed through a C18 Lite Sep-Pak. The cartridge was washed with water (10 mL) and 2-[18F]fluoroquinoline was eluted into a sterile dose vial with ethanol (1 mL). Sterile saline (9 mL) was added to provide product formulated in 10 mL.
Molar activity of 2-[18F]fluoroquinoline.
A 10 μL aliquot was analyzed by HPLC, using HPLC Conditions B, and the area of the UV peak (280 nm) corresponding to the 2-fluoroquinoline standard (tR = 8.9 min) was determined. The molar concentration (μmol/μL) of 2-fluoroquinoline in the sample was then determined by linear regression analysis against a standard curve generated from injection of identical volumes of solutions of known concentration of 2-fluoroquinoline. The concentration of activity was determined by dividing the total activity (4.588 – 7.474 GBq 12.4 – 20.2 ×10−2 Ci) by the volume of the solution (5 mL). The end of synthesis (EOS) molar activity (Ci/μmol) is given by dividing the concentration of activity for 2-[18F]fluoroquinoline (GBq/μL or Ci/μL) by the molar concentration of the product (μmol/μL). EOS molar activity was found to be 155.5 ± 66.4 GBq/μmol (4.2 ± 1.8 Ci/μmol) for the high activity runs (n = 4, Table S1).
Synthesis of [18F]Inter-HQ415 followed by deprotection to provide [18F]HQ415.
A 4 mL vial was charged with pre-HQ415 (20 μmol) and tetramethylammonium bicarbonate (100 μmol) on the bench top. An aliquot of [18F]KF•K2.2.2 in DMF (100 μL, 92.5 – 129.5 MBq (2500–3500 μCi) of radioactivity) was used for each manual reaction. Total reaction volume was 100μL. The reaction vial was sealed and heated in an aluminum block at 110 °C for 30 min. After 30 min, the reaction was cooled to rt, and 10 μL of the crude mixture was used for analysis [radiochemical yield (RCY, %) determined by radio-TLC analysis and analytical HPLC (HPLC Conditions A)]. Deprotection of the Boc groups was achieved by the addition of 2M aqueous HCl (200 μL) to the crude reaction mixture. The reaction was heated at 120 °C for 15 min to yield [18F]HQ415.
Supplementary Material
Acknowledgements
This work was supported by the NIH (Award Number R01EB021155 to M.S.S. and P.J.H.S.). The authors also thank Dr. Sean Tanzey for supporting chemistry on related projects.
Footnotes
The authors declare no competing financial interest.
Supporting Information
- Materials and methods; preparation of precursors and reference standards; radiofluorination details; screening information; NMR spectra; HPLC traces.
References
- 1.Wang J; Sańchez-Roselló M; Aceña JL; delPozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001-2011). Chem. Rev 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]
- 2.(a) Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in medicinal chemistry. Chem. Soc. Rev 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]; (b) Haupt A Organic and inorganic fluorine chemistry, Berlin, Boston: De Gruyter, 2021. [Google Scholar]
- 3.Gillis EP; Eastman KJ; Donnelly DJ; Meanwell NA Applications of fluorine in medicinal chemistry. J. Med. Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- 4.(a) Adams DJ; Clark JH Nucleophilic routes to selectively fluorinated aromatics. Chem. Soc. Rev 1999, 28, 225–231. [Google Scholar]; (b) Tredwell M; Gouverneur V 18F-Labeling of arenes. Angew. Chem., Int. Ed 2012, 51, 11426–11437. [DOI] [PubMed] [Google Scholar]; (c) Dolci L; Dolle F; Jubeau S; Vaufrey F; Crouzel C 2-[18F]Fluoropyridines by no-carrier-added nucleophilic aromatic substitution with [18F]FK-K222—a comparative study. J Label Compd Radiopharm 1999, 42, 975–985. [Google Scholar]; (d) Naumiec GR; Cai L; Lu S; Pike VW Quinuclidine and DABCO enhance the radiofluorination of 5-substituted 2-halopyridines. Eur. J. Org. Chem 2017, 6593–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Champagne PA; Desroches J; Hamel JD; Vandamme M; Paquin JF Monofluorination of organic compounds: 10 years of innovation. Chem. Rev 2015, 115, 9073–9174. [DOI] [PubMed] [Google Scholar]
- 6.(a) See YY; Morales-Colón MT; Bland DC; Sanford MS Development of SNAr nucleophilic fluorination: A fruitful academia-industry collaboration. Acc. Chem. Res 2020, 53, 2372–2383. [DOI] [PubMed] [Google Scholar]; (b) Morales-Colón MT; See YY; Lee SJ; Scott PJH; Bland DC; Sanford MS Tetramethylammonium fluoride alcohol adducts for SNAr fluorination. Org. Lett, 2021, 23, 4493–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sasson Y; Negussie S; Royz M; Mushkin N Tetramethylammonium chloride as a selective and robust phase transfer catalyst in a solid–liquid halex reaction: the role of water. Chem. Commun 1996, 23, 297–298. [Google Scholar]
- 8.Sun HR; DiMagno SG Anhydrous tetrabutylammonium fluoride. J. Am. Chem. Soc 2005, 127, 2050–2051. [DOI] [PubMed] [Google Scholar]
- 9.Sun H; DiMagno SG Room-temperature nucleophilic aromatic fluorination: Experimental and theoretical studies. Angew. Chem. Int. Ed 2006, 45, 2720–2725. [DOI] [PubMed] [Google Scholar]
- 10.Allen LJ; Muhuhi JM; Bland DC; Merzel R; Sanford MS Mild fluorination of chloropyridines with in situ generated anhydrous tetrabutylammonium fluoride. J. Org. Chem 2014, 79, 5827–5833. [DOI] [PubMed] [Google Scholar]
- 11.(a) Uddin J; Wilson AJ; Crews BC; Malerba P; Uddin I; Kingsley PJ; Ghebreselasie K; Daniel CK; Nickels ML; Tantawy MN; Jashim E; Manning HC; Khabele D; Marnett LJ Discovery of furanone-based radiopharmaceuticals for diagnostic targeting of COX-1 in ovarian cancer. ACS Omega. 2019, 5, 9251–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brugarolas P; Freifelder R; Cheng SH; DeJesus O Synthesis of meta-substituted [18F]3-fluoro-4-aminopyridine via direct radiofluorination of pyridine N-oxides. 2016, 52, 7150–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Meleán JC; Humpert S; Ermert J; Coenen HH Stereoselective radiosynthesis of l- and d-3-[18F]fluoro-α-methyltyrosine. J. Fluor. Chem 2015, 178. 202–207. [Google Scholar]; (d) Khan NH; Lee BC; Lee SY; Choe YS; Jun CH; Chi DY Hydroacylation of 4-[18F]fluorobenzaldehyde: a novel method for the preparation of 4′-[18F]phenylketones. J. Labelled. Comp. Radiopharm 2002, 45, 1045–1053. [Google Scholar]; (e) Herman LW; Fischman AJ; Tompkins RG; Hanson RN; Byon C; Strauss HW; Elmaleh DR The use of pentafluorophenyl derivatives for the 18F labelling of proteins. Nucl. Med. Biol 1994, 21, 1005–1010. [DOI] [PubMed] [Google Scholar]; (f) Maeda M; Fukumura T; Kojima M The dimethylsulfonium moiety as a leaving group in aromatic radiofluorination using tetra-n-butylammonium [18F]fluoride. Int. J. Rad. Appl. Instr. A 1987, 38, 307–310. [Google Scholar]; (g) Kilbourn MR; Welch J; M. J; Dence CS; Tewson TJ; Saji H; Maeda M Carrier-added and no-carrier-added syntheses of [18F]spiroperidol and [18F]haloperidol. Int. J. Rad. Appl. Instr 1984, 35, 591–598. [DOI] [PubMed] [Google Scholar]; (h) Wagner FM; Ermert J; Coenen HH Three-step, "one-pot" radiosynthesis of 6-fluoro-3,4-dihydroxy-L-phenylalanine by isotopic exchange. J Nucl Med. 2009, 50, 1724–1729. [DOI] [PubMed] [Google Scholar]
- 12.(a) Wright JS; Sharninghausen Liam S., L. S; Preshlock S; Brooks AF; Sanford Melanie S., M. S; Scott PJH Sequential Ir/Cu-mediated method for the meta-selective C–H radiofluorination of (hetero)arenes. J. Am. Chem. Soc 2021, 143, 6915–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang L; White AR; Chen W; Wu Z; Nicewicz DA; Li Z Direct radiofluorination of arene C─H bonds via photoredox catalysis using a peroxide as the terminal oxidant. Org. Lett 2020, 22, 7971 – 7975. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mossine AV; Tanzey SS; Brooks AF; Makaravage KJ; Ichiishi N; Miller JM; Henderson BD; Erhard T; Bruetting C; Skaddan MB; Sanford MS; Scott PJH Synthesis of high-molar-activity [18F]6-fluoro-L-DOPA suitable for human use via Cu-mediated fluorination of a BPin precursor. Nat. Prot 2020, 15, 1742–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mossine AV; Brooks AF; Ichiishi N; Makaravage KJ; Sanford MS; Scott PJH Development of customized [18F] fluoride elution techniques for the enhancement of copper-mediated late-stage radiofluorination. Sci. Rep 2017, 7, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yuan G; Guehl NJ; Zheng B; Qu X; Moon SH; Dhaynaut M; Shoup TM; Afshar S; Kang HJ; Zhang Z; El Fakhri G; Normandin MD; Brownell AL Synthesis and characterization of [18F]JNJ-46356479 as the first 18F-labeled PET imaging ligand for metabotropic glutamate receptor 2. Mol Imaging Biol. 2021, 23, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lai TH; Schröder S; Toussaint M; Dukić-Stefanović S; Kranz M; Ludwig FA; Fischer S; Steinbach J; Deuther-Conrad W; Brust P; Moldovan RP Development of 18F-labeled radiotracers for PET imaging of the adenosine A2A receptor: synthesis, radiolabeling and preliminary biological evaluation. Int J Mol Sci 2021. 22, 2285. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zischler J; Kolks N; Modemann D; Neumaier B; Zlatopolskiy BD Alcohol-enhanced Cu-mediated radiofluorination. Chem. Eur. J 2017, 23, 3251–3256. [DOI] [PubMed] [Google Scholar]; (h) Zlatopolskiy BD; Zischler J; Schäfer D; Urusova EA; Guliyev M; Bannykh O; Endepols H; Neumaier B Discovery of 7-[18F]Fluorotryptophan as a novel positron emission tomography (PET) probe for the visualization of tryptophan metabolism in vivo. J Med Chem. 2018, 61, 189–206. [DOI] [PubMed] [Google Scholar]; (i) Orlovskaya V; Fedorova O; Kuznetsova O; Krasikova R Cu-mediated radiofluorination of aryl pinacolboronate esters: alcohols as solvents with application to 6-L-[18F]FDOPA synthesis. Eur. J. Org. Chem 2020, 7079–7086. [Google Scholar]; (j) Rotstein BH; Stephenson NA; Vasdev N; Liang SH Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat Commun. 2014, 5, 4365. [DOI] [PubMed] [Google Scholar]; (k) Cortés González MA; PaNordeman P; Gómez AB; Meyer DN; Antoni G; Schou M; Szabó KJ [18F]Fluoro-benziodoxole: a no-carrier-added electrophilic fluorinating reagent. Rapid, simple radiosynthesis, purification and application for fluorine-18 labelling. Chem. Commun 2018, 54, 4286 – 4289. [DOI] [PubMed] [Google Scholar]; (l) Tay NES; Chen W; Levens A; Pistritto VA; Huang Z; Wu Z; Li Z; Nicewicz DA 19F- and 18F-Arene deoxyfluorination via organic photoredox-catalysed polarity-reversed nucleophilic aromatic substitution. Nat Catal 2020, 3, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Brichard L; Aigbirhio FI An efficient method for enhancing the reactivity and flexibility of [18F]fluoride towards nucleophilic substitution using tetraethylammonium bicarbonate. Euro. J. Org. Chem 2014, 28, 6145–6149. [Google Scholar]; (b) Inkster JAH; Akurathi V; Sromek AW; Chen Y; Neumeyer JL; Packard AB A non-anhydrous, minimally basic protocol for the simplification of nucleophilic 18F-fluorination chemistry. Sci Rep. 2020, 10, 6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brooks A; Tanzey S; Shao X; Scott P Binding potential of radioligand [18F]FL2-b by autoradiography in amyotrophic lateral sclerosis and Lewy body dementia. J. Nucl. Med 2018, 59 (supplement 1), 613. [Google Scholar]
- 15.(a) Cox DP; Terpinski J; Lawrynowics W "Anhydrous" tetrabutylammonium fluoride: a mild but highly efficient source of nucleophilic fluoride ion. J. Org. Chem 1984, 49, 3216–3119. [Google Scholar]; (b) Upon drying of tetrabutylammonium fluoride, such decomposition occurs even at room temperature. [Google Scholar]
- 16.Schimler SD; Ryan SJ; Bland DC; Anderson JE; Sanford MS Anhydrous tetramethylammonium fluoride for room temperature SNAr fluorination. J. Org. Chem 2015, 80, 12137–12145. [DOI] [PubMed] [Google Scholar]
- 17.(a) Feliu AL Synthetic studies with [18F]p-fluorobenzenediazonium chloride application to the synthesis of a radiolabelled glucocorticoid: [18F]WIN 44577. J. Labelled. Comp. Radiopharm 1988, 25, 1245–1254. [Google Scholar]; (b) Rosenthal MS; Bosch AL; Nickles RJ; Gatley SJ Synthesis and some characteristics of no-carrier added [18F]fluorotrimethylsilane. Int. J. Rad. Appl. Instr. A 1985, 36, 318–319. [DOI] [PubMed] [Google Scholar]
- 18.Feliu AL; Rottenberg DA Synthesis and evaluation of fluorine-18 21-fluoroprednisone as a potential ligand for neuro-PET studies. J. Nucl. Med 1986, 28, 998–1005. [PubMed] [Google Scholar]
- 19.(a) Christe KO; Wilson WW; Wilson RD; Bau R; Feng J Syntheses, properties, and structures of anhydrous tetramethylammonium fluoride and its 1:1 adduct with trans-3-amino-2-butenenitrile. J. Am. Chem. Soc 1990, 112, 7619–7625. [Google Scholar]; (b) Gnann RZ; Wagner RI; Christe KO; Bau R; Olah GA; Wilson WW Naked fluoride ion sources: synthesis, characterization, and coupling reaction of 1-methylhexamethylenetetramine fluoride. J. Am. Chem. Soc 1997, 119, 112–115. [Google Scholar]
- 20.Evaporation can be achieved by heating the reaction vessel to 100 °C and drawing vacuum for 4-6 min. After this time, the reaction vessel is subjected to an argon stream and simultaneous vacuum draw for an additional 4-6 min. Cary BP; Brooks AF; Fawaz MV; Shao X; Desmond TJ; Carpenter GM; Sherman P; Quesada CA; Albin RL; Scott PJH Targeting metal-Aβ aggregates with bifunctional radioligand [11C]L2-b and a fluorine-18 analogue [18F]FL2-b. ACS Med. Chem. Lett 2015, 6 (2), 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.For the seminal paper reporting the first use of K2CO3 and K2.2.2, see: Hamacher K; Coenen HH; Stöcklin G Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-D-glucose using aminopolyether supported nucleophilic substitution. J Nucl Med. 1986, 27, 235–238. [PubMed] [Google Scholar]; (b) For a recent optimization campaign, see: Hassan H; Bakar SA; Halim KNCA; Idris J; Saad FFA; Nordin AJ Optimising the azeotropic drying of 18F-fluorine way to improve the 18F-fluorocholine radiochemical yield. Curr. Radiopharm 2016, 9, 121–127. [DOI] [PubMed] [Google Scholar]
- 22.Although 2-[18F]-quinoline was not detected when using Me4NBF4, this salt was an effective additive for promoting the radiofluorination of other (hetero)aryl precursors, unlike other quaternary ammonium salts tested. Further investigation using both Me4NHCO3 and Me4NBF4 was conducted for a wide range of substrates. See Figure S2 for more information.
- 23.[18F]KF can be effective, but frequently requires higher temperatures and inclusion of other additives. This limits compatibility with, for example, heat sensitive molecules. See, for example references 4c and 4d. We have limited the current study to low temperature reactions to minimize this issue.
- 24.For some substrates Me4NHCO3 or Me4NBF4 afforded similar enhancements in RCY, while for others Me4NBF4 afforded higher RCY than Me4NHCO3 [but comparable to [18F]KF (see Supporting Information)]. Given that the mechanistic origin of this effect remains unclear at this time and there is potential for isotopic exchange with BF4−, we have focued on the use of Me4NHCO3 in this work. However, the results do suggest that different tetramethylammonium salts should be screened when evaluating new substrates in this chemistry. For more information on isotopic exchange with BF4− salts, see: Modemann DJ; Zlatopolskiy BD; Urusova EA; Zischler J; Craig A; Ermert J; Guliyev M; Endepols H; Neumaier B 2-[18F]Fluorophenylalanine: synthesis by nucleophilic 18F-fluorination and preliminary biological evaluation. Synthesis 2019, 51, 664–676. [Google Scholar]
- 25.Dollé F [18F]Fluoropyridines: From conventional radiotracers to the labeling of macromolecules such as proteins and oligonucleotides. Ernst Schering Res Found Workshop. 2007, 62, 113–157. [DOI] [PubMed] [Google Scholar]
- 26.For examples of the ortho effect, see: (a) Ogata Y; Tsuchida. Ortho effect in the aromatic nucleophilic substitution. Nippon Kagaku Zassi, 1953, 74, 1000–1003. [Google Scholar]; (b) An ortho-directing effect in the nucleophilic aromatic substitution reactions of primary and secondary 2,4-dichloro- and 2,3,4-trichlorobenzamides with ethanethiolate. Synth Commun, 1995, 25, 899–906. [Google Scholar]; (c) Drennen B; Scheenstra JA; Yap JL; Chen L; Lanning ME; Roth BM; Wilder PT; Fletcher S Structural re-engineering of the α-helix mimetic jy-1-106 into small molecules: disruption of the Mcl-1-Bak-BH3 protein-protein interaction with 2,6-di-substituted nicotinates. ChemMedChem. 2016, 11, 827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hirowaka Y; Horikawa T; Kato S An efficient synthesis of 5-bromo-2-methoxy-6-methylaminopyridine-3-carboxylic acid. Chem. Pharm. Bull 2000, 48, 1847–1853. [DOI] [PubMed] [Google Scholar]
- 27.RCY (%) of control reactions using K18F in presence of K2.2.2 under these conditions are shown as (A) in Figure 1.
- 28.Tanzey S; Thompson S; Shao X; Brooks A; Scott P Preparation of metal-protein aggregate radioligand [11C]HQ415 and evaluation by small animal PET imaging. J. Nucl. Med 2018, 59 (supplement 1), 1019. [Google Scholar]
- 29.Tardiff DF; Tucci ML; Caldwell KA; Lindquist S Different 8-hydroxyquinolines protect models of TDP-43 protein, α-synuclein, and polyglutamine proteotoxicity through distinct mechanisms. J. Biol. Chem 2021, 287, 4107–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mossine AV; Brooks AF; Bernard-Gauthier V; Bailey JJ; Ichiishi N; Schirrmacher R; Sanford MS; Scott PJH Automated synthesis of PET radiotracers by copper-mediated 18F-fluorination of organoborons: Importance of the order of addition and competing protodeborylation. J Label Comp Radiopharm. 2018, 61, 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webb EW; Scott PJH Potential applications of artificial intelligence and machine learning in radiochemistry and radiochemical engineering. PET Clinics 2021, doi: 10.1016/j.cpet.2021.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Halvorsen NE; Kvernenes OH A Fast and simple method for the determination of TBA in 18F-labeled radiopharmaceuticals. Pharmaceuticals 2020, 13, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tanzey SS; Mossine AV; Sowa AR; Torres J; Brooks AF; Sanford MS; Scott PJH A spot test for determination of residual TBA levels in 18F-radiotracers for human use using Dragendorff reagent. Anal. Methods 2020, 12, 5004–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seganish WM; DeShong P Preparation and palladium-catalyzed cross-coupling of aryl triethylammonium bis(catechol) silicates with aryl triflates. J. Org. Chem 2004, 69, 1137–1143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.