

Abstract
Background and Objective
Patients with earlier age at onset of sporadic Alzheimer disease (AD) are more likely than those with later onset to present with atypical clinical and cognitive features. We sought to determine whether this age-related clinical and cognitive heterogeneity is mediated by different topographic distributions of tau-aggregate neurofibrillary tangles (NFTs) or by variable amounts of concomitant non-AD neuropathology.
Methods
The relative distribution of NFT density in hippocampus and midfrontal neocortex was calculated, and α-synuclein, TAR DNA binding protein 43 (TDP-43), and microvascular copathologies were staged, in patients with severe AD and age at onset of 51–60 (n = 40), 61–70 (n = 41), and >70 (n = 40) years. Regression, mediation, and mixed effects models examined relationships of pathologic findings with clinical features and longitudinal cognitive decline.
Results
Patients with later age at onset of AD were less likely to present with nonmemory complaints (odds ratio [OR] 0.46 per decade, 95% confidence interval [CI] 0.22–0.88), psychiatric symptoms (β = −0.66, 95% CI −1.15 to −0.17), and functional impairment (β = −1.25, 95% CI −2.34 to −0.16). TDP-43 (OR 2.00, 95% CI 1.23–3.35) and microvascular copathology (OR 2.02, 95% CI 1.24–3.40) were more common in later onset AD, and α-synuclein copathology was not related to age at onset. NFT density in midfrontal cortex (β = −0.51, 95% CI −0.72 to −0.31) and midfrontal/hippocampal NFT ratio (β = −0.18, 95% CI −0.26 to −0.10) were lower in those with later age at onset. Executive function (β = 0.48, 95% CI 0.09–0.90) and visuospatial cognitive deficits (β = 0.97, 95% CI 0.46–1.46) were less impaired in patients with later age at onset. Mediation analyses showed that the effect of age at onset on severity of executive function deficits was mediated by midfrontal/hippocampal NFT ratio (β = 0.21, 95% CI 0.08–0.38) and not by concomitant non-AD pathologies. Midfrontal/hippocampal NFT ratio also mediated an association between earlier age at onset and faster decline on tests of global cognition, executive function, and visuospatial abilities.
Discussion
Worse executive dysfunction and faster cognitive decline in people with sporadic AD with earlier rather than later age at onset is mediated by greater relative midfrontal neocortical to hippocampal NFT burden and not by concomitant non-AD neuropathology.
There is age-related heterogeneity in the clinical and cognitive presentation of Alzheimer disease (AD).1-4 Patients with earlier age at symptom onset are more likely than those with later age at onset to report noncognitive (i.e., behavioral) or nonmemory cognitive decline as their initial symptom5 and to have atypical clinical presentations with prominent nonmemory cognitive deficits,1,5,6 greater psychiatric involvement,7 and more rapid cognitive decline.8-10 These atypical features result in greater misattribution of the underlying etiology to non-AD causes that display these symptom profiles, such as frontotemporal dementia, dementia with Lewy bodies (DLB) or vascular dementia, despite the fact that at autopsy patients with early age at onset have less concomitant non-AD vascular and nonvascular (e.g., α-synuclein, TAR DNA binding protein 43 [TDP-43]) pathology than those with late age at onset.4 One neuropathologic feature that could contribute to age-related clinical heterogeneity is variation in the distribution of AD pathology.11,12 Murray et al.13 reported substantially different average ages at symptom onset in distinct neuropathologic subtypes of AD defined by tau-containing neurofibrillary tangle (NFT) density in hippocampal vs neocortical brain regions. Patients with disproportionally high neocortical tangle densities (i.e., hippocampal sparing) had an estimated age at onset of 63 compared to age 76 for those with disproportionally high hippocampal tangle densities (i.e., limbic predominant) and were more likely to have exhibited an atypical clinical presentation of AD.
To determine whether distinct distributions of NFT mediate age-related heterogeneity in the clinical presentation of AD, we examined their relative distribution in hippocampus and middle frontal gyrus in patients with sporadic, autopsy-confirmed severe AD with wide variation in reported age at onset. We also assessed concomitant α-synuclein (i.e., Lewy body), TDP-43, and microvascular pathologies. We then examined the relationship between age at onset and clinical and cognitive features (including longitudinal decline) and performed mediation analyses to test whether observed relationships were mediated by distribution of NFT pathology or non-AD copathology.
Methods
Participants
Participants with sporadic, pathologically confirmed, severe AD by National Institute on Aging–Alzheimer Association (NIA-AA) criteria14 were selected from the autopsy series of the University of California, San Diego (UCSD) Shiley-Marcos Alzheimer's Disease Research Center without regard to concomitant pathologies or clinical diagnoses. Participants had to have clinical data available for a baseline visit and at least 1 subsequent follow-up. Participants were excluded if they had a known dominantly inherited mutation for AD (e.g., PSEN1), a family history of such a mutation, or a reported age at onset (estimated from clinical interviews with the patient and an informant) younger than 50. To generate demographically matched sampling throughout the age range, equal numbers of participants with ages at onset of 61–70 and 70+ years were matched to all available patients with onset ages of 50–60 years on sex, years of education, and year of autopsy using the R MatchIt package (random selection blind to clinical, cognitive, and neuropathologic data). When performing retrospective immunostaining for TDP-43 and α-synuclein pathology, it was discovered that tissue was missing or not of adequate quality for immunohistochemistry for 6 individuals with onset before 60, 5 individuals with onset of 61–70, and 6 individuals with onset after 70. These participants were dropped, resulting in the final study sample.
Neuropathologic Evaluation
Autopsy was performed using a previously described protocol.15 Brains were divided sagittally and the left hemibrain was fixed in 10% buffered formalin. After 14 days, the formalin-fixed hemibrain was cut serially into 1 cm slices for paraffin embedding. Sections taken and stained with hematoxylin & eosin (H&E) for histopathologic examination were middle frontal cortex (Brodmann areas 8/9), rostral superior temporal cortex, inferior parietal cortex, hippocampus (CA1-CA4 and dentate gyrus), entorhinal cortex, basal ganglia, midbrain with substantia nigra, pons with locus coeruleus, and cerebellar cortex with dentate nucleus.
AD Pathology
Neuritic plaques, diffuse plaques, and NFTs were identified either with 1% thioflavin-S stain on 10-µm-thick sections viewed with ultraviolet illumination and a 440 µm bandpass wavelength excitation filter or with immunohistochemical staining using antibodies to β-amyloid (Ab 69D, rabbit polyclonal from Edward Koo, 1:1,200) and paired helical filament (PHF) tau (PHF1 from Peter Davies, 1:600) on 5-µm-thick sections. Neuritic plaque density was estimated using methods recommended by the Consortium to Establish a Registry for AD (CERAD),16 and Braak stage for NFT pathology was determined.17 Pathologic diagnosis of AD was made using NIA-AA consensus criteria for the postmortem diagnosis of AD,14 wherein Thal phase 4–5, Braak stage V–VI, and moderate to severe neuritic plaque density corresponds to “high likelihood” of AD.
Hippocampal and neocortical NFT densities were approximated utilizing a modified version of the counting method of Terry et al.18 In brief, NFTs were stained using either thioflavin-S (90 cases) or PHF-1 tau immunohistochemistry (31 cases) as described above. The areas of heaviest pathologic burden in the CA1 sector of the hippocampus and the midfrontal gyrus of the neocortex were counted for each case. Counts were performed in 3 high-magnification fields per region and averaged to provide a single NFT count per 0.1 mm2 microscopic field. Regional NFT counts differed in absolute value by staining method (thioflavin-S midfrontal mean 5.5, SD 3.6; thioflavin-S hippocampal mean 21.6, SD 12.8; PHF-1 midfrontal mean 10.4, SD 5.7; PHF-1 hippocampal mean 39.6, SD 17.2). Thus, regional NFT counts were z-transformed separately for each method, centering the mean to 0 and the SD to 1. Furthermore, a midfrontal/hippocampal tangle ratio was calculated from raw counts resulting in a unitless variable that is a single continuous measure that approximates the relative distribution of pathology regardless of staining procedure. Subgroups of patients with each staining method did not differ in average age at onset, age at death, disease duration, sex, education, or APOE genotype distribution (all p > 0.25 by t test for continuous and Fisher exact test for discrete variables). Figure 1 shows examples of 2 participants with relatively high and low midfrontal/hippocampal tangle ratios.
Figure 1. Sample Micrographs of Neurofibrillary Tangle Pathology Distribution With PHF-1 Tau Immunostaining in 2 Participants.
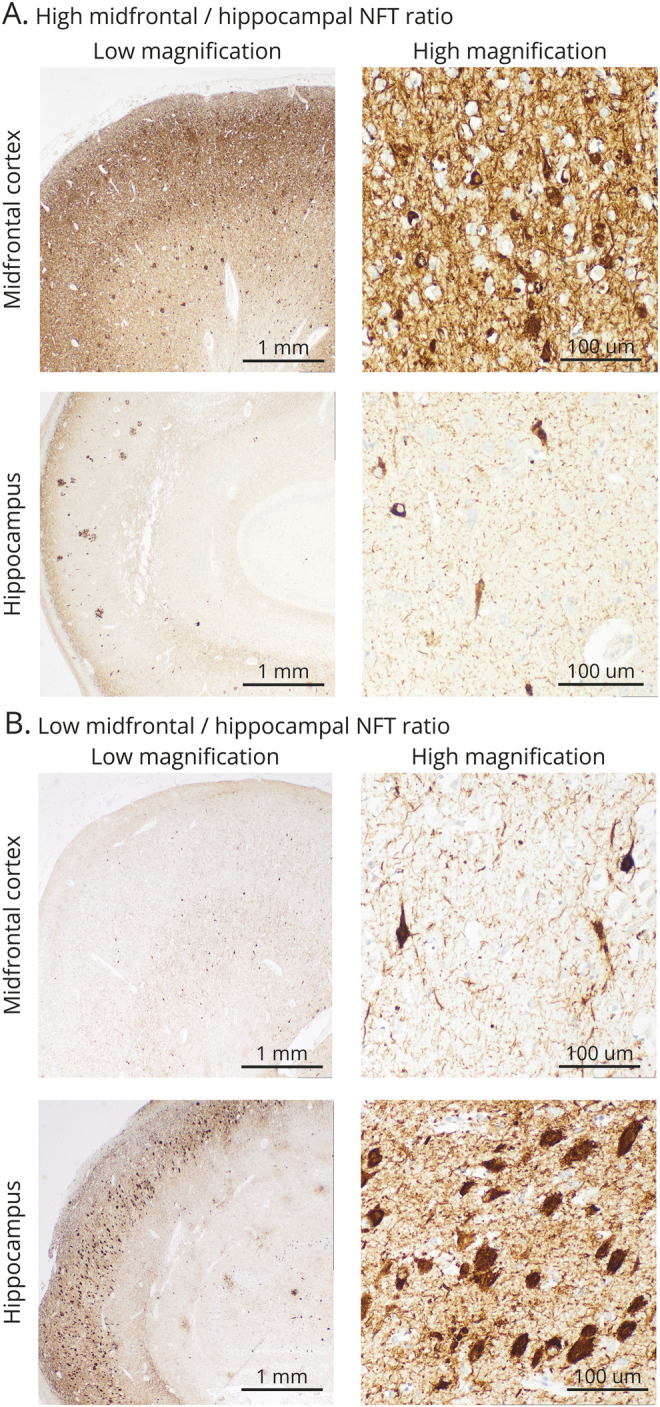
Example A corresponds with a relatively high midfrontal/hippocampal tangle ratio; example B corresponds with a relatively low midfrontal/hippocampal tangle ratio. Midfrontal = midfrontal cortex; NFT = neurofibrillary tangle.
Patients were classified into hippocampal sparing, limbic predominant, or typical neuropathologic subtypes using an approximation of the Murray et al.13 criteria. Hippocampal sparing was defined as (1) midfrontal tangle density above the median of the sample, (2) hippocampal tangle density below the median of the sample, and (3) midfrontal/hippocampal tangle ratio above the 75th percentile of the sample. Limbic predominant was defined as (1) midfrontal tangle density below the median of the sample, (2) hippocampal tangle density above the median of the sample, and (3) midfrontal/hippocampal tangle ratio below the 25th percentile of the sample. All other participants were considered typical.
Non-AD Pathology
Lewy body pathology identified by H&E staining and immunostaining with antibodies against α-synuclein (phospho-synuclein 81A, from Virginia Lee, 1:15,000) was staged according to consensus DLB guidelines19 into brainstem, limbic (transitional), or diffuse (neocortical) subtypes. Individuals with amygdala-predominant Lewy bodies were not included, given the low likelihood of a clinical diagnosis of DLB in this group.19,20 In all cases, the amygdala was screened for TDP-43 pathology by immunohistochemical staining (Proteintech 10782-2-AP polyclonal, 1:12,000). When positive, further staining was completed to allow staging according to Limbic-predominant Age-related TDP-43 Encephalopathy consensus guidelines21 into amygdala, hippocampal, or neocortical stages. Hippocampal sclerosis (HS) was diagnosed independent of TDP-43 pathology when neuronal loss in the CA1 and subiculum was out of proportion with the degree of AD pathology. Vascular pathology was assessed by examining the brain for large arterial and lacunar infarcts, microinfarcts, and hemorrhages. Arteriolosclerosis, atherosclerosis of the circle of Willis, and amyloid angiopathy (in parenchymal or leptomeningeal vessels) were each rated as none, mild, moderate, or severe using a semiquantitative scale.
Clinical and Neuropsychological Evaluation
Participants had annual standardized clinical, neurologic, and neuropsychological evaluations as previously described.22,23 The clinical evaluation included review of history with the patient or informant, mental status testing, assessment of psychiatric symptoms using the Neuropsychiatric Inventory (NPI), and assessment of functional impairment using the Pfeffer Outpatient Disability (POD) scale24 or the Functional Assessment Questionnaire (converted to corresponding POD scores). Clinical Dementia Rating (CDR) and the sum of its 6 subdomain scores (i.e., CDR sum of boxes) were computed. Neuropsychological assessment included tests of global cognition (Dementia Rating Scale [DRS]), memory (Visual Reproduction Test, Logical Memory Test, California Verbal Learning Test, CERAD Word List Learning), language (Boston Naming Test, Letter Fluency Test, Category Fluency Test, Vocabulary Test), executive functions (modified Wisconsin Card Sorting Test, Trail-Making Test Parts A and B, Digit Symbol Substitution Test), and visuospatial abilities (Block Design Test, Visual Reproduction Test copy, Clock Drawing Test, Cube Drawing Test).
Consensus clinical diagnoses were made according to published criteria by 2 or more board-certified neurologists blind to individual cognitive test scores but told whether the neuropsychological assessment identified deficits in 2 or more cognitive domains. Probable or possible AD or mild cognitive impairment (MCI) was diagnosed according to National Institute of Neurologic and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA)25 or NIA-AA criteria.26 Probable DLB was diagnosed clinically based on presence of dementia and at least 2 of 3 core features of mild parkinsonism, well-formed visual hallucinations, and fluctuations in consciousness or attention.19 Primary progressive aphasia (PPA) was diagnosed based on predominant language impairment thought to be the principal cause of impaired daily living activities and the presenting deficit at symptom onset.27 No participant met clinical criteria for behavioral variant frontotemporal dementia28 or posterior cortical atrophy.29
Standard Protocol Approvals, Registrations, and Patient Consents
The research protocol was reviewed and approved by the UCSD Human Subjects Review Board. Informed consent was obtained from all patients or their caregivers consistent with California state law.
Statistical Analysis
Associations of age at onset with demographic and clinical features were examined by linear regression for continuous variables and logistic regression for categorical variables. Standard regression diagnostics, including Cook distance and residuals vs predicted value plots, were examined for consistency with model assumptions. Age at onset was treated as a continuous variable in all analyses, with coefficients standardized for a 10-year (decade) change in age. Means and SDs are presented by age at onset tercile in demographic tables to illustrate the results of these analyses. Neuropathologic outcomes were analyzed using linear or logistic regression with terms for age at onset, sex, and APOE genotype (ε4+ or ε4−). Beta coefficients for linear regression, or odds ratios (ORs) for logistic regression, were calculated with 95% confidence intervals (CIs).
Cognitive domain scores were created from the neuropsychological test battery using previously described methods.30 Principal component analysis with varimax rotation identified 4 orthogonal components conceptually labeled as visuospatial, memory, executive, and language. Baseline component scores were derived and transformed to z scores using reference values from an independent pool of 497 robust controls diagnosed as cognitively normal on their first and all subsequent annual evaluations (average 5.2 ± 5.0 evaluations). Effects of age at onset on baseline cognitive domain scores were examined using linear regression models adjusting for sex, education, and APOE genotype. When an effect was significant, mediation analysis was performed to determine whether the effect was mediated by tangle pathology distribution (i.e., midfrontal/hippocampal tangle ratio) or by presence (any level vs none) of a concomitant pathology. First, a full linear model was fit for the cognitive domain score with terms for age at onset, sex, education, APOE genotype, and 1 of the pathologic measures (midfrontal/hippocampal tangle ratio or presence of copathology). Second, a mediation model31 was fit for the pathologic measure predicted by age at onset, sex, education, and APOE genotype. The mediation R package was used to evaluate the direct effect of age at onset on the cognitive domain score and the portion of the effect mediated by the pathologic measure. 95% CIs were determined using 10,000 nonparametric bootstrap simulations.
Linear mixed effects models were used to extract subject-specific slopes (i.e., rate of decline) across 2–3 annual evaluations for a subset of cognitive tests administered throughout more severe stages of disease: DRS, Mini-Mental State Examination (MMSE), CERAD Word List, Digit Symbol Substitution, Block Design, Verbal Fluency, and Boston Naming Test. Effects of age at onset on slopes of decline were examined using models adjusting for sex, education, APOE genotype, and baseline test score. When a significant age at onset effect was observed for a cognitive test, mediation analysis for NFT distribution or presence of a concomitant pathology was performed using random effects slope as the dependent measure.32
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results
Participant Demographics at Baseline
The overall sample had a mean ± SD age of 70.3 ± 7.7 years at baseline, 14.5 ± 2.7 years of education, and 36% were female. Sex distribution and years of education did not differ by age at onset, in accordance with our matching procedure (Table 1). The average baseline MMSE score was 22.1 ± 4.9, DRS score was 112.7 ± 17.1, and CDR sum of boxes was 5.7 ± 2.5. None of these global measures varied by age at onset. The average baseline basic activities of daily living (ADL) score was 6.9 ± 1.5 and instrumental ADL score (i.e., POD) was 9.4 ± 5.0. Both measures were less impaired with later onset (β = −0.35 per decade of age [95% CI −0.68 to −0.02] and β = −1.25 [−2.34 to −0.16], respectively). The APOE genotype distribution was 63% ε4+ and did not differ by age at onset.
Table 1.
Participant Demographics

The average reported age at onset was 66.0 ± 8.1 years, age at death was 76.7 ± 8.3 years, interval from age at onset to baseline visit was 4.2 ± 2.7 years, interval from baseline visit to death was 6.5 ± 3.1 years, and total duration of illness was 10.7 ± 3.7 years. Intervals from age at onset to baseline visit (β = −1.06 [95% CI −1.63 to −0.49]) or death (β = −0.78 [−1.60 to −0.04]) were shorter in those with later onset.
The first cognitive symptom reported at onset was usually memory (84%), but nonmemory presenting symptoms were less likely with later onset (OR 0.46 per decade [95% CI 0.22–0.88]). Psychiatric symptoms at baseline reflected by NPI scores were less common in those with later onset (β = −0.66 [95% CI −1.15 to −0.17]). Use of antidepressants, antipsychotics, Food and Drug Administration (FDA)–approved medications for the treatment of AD, or other medications with CNS activity did not differ by age at onset.
The most frequent baseline clinical diagnosis was probable (73%) or possible (12%) AD. The probability of receiving an AD diagnosis did not vary with age. A clinical diagnosis of MCI was rare (7%), but more likely with later age at onset (OR 2.49 [95% CI 1.09–6.29]). A clinical diagnosis of Lewy body disease was also relatively rare (6%), and less likely with later age at onset (OR 0.24 [95% CI 0.06 to 0.71]). Other non-AD diagnoses (n = 1 PPA; n = 1 depression) were very rare (2%) and unrelated to age at onset.
Concomitant Non-AD Neuropathology
The proportion of participants with various copathologies is shown as a function of age at onset in Figure 2, A–C. ORs with 95% CI for the effects of age at onset, sex, and APOE genotype are shown for each pathology and stage in Table 2. Concomitant α-synuclein (i.e., Lewy) pathology was present in 21% of the overall sample, but the likelihood of its presence or severity/stage did not differ by age at onset, APOE genotype, or sex.
Figure 2. Pathologies by Age at Onset.

Age at onset effects on presence and stage of Lewy body (α-synuclein) pathology (A), TAR DNA binding protein 43 (TDP-43) pathology (B), and arteriolosclerosis (C). The p values correspond to effects for age at onset from logistic regression models predicting presence of any level of the pathology (vs none), with terms for age at onset, APOE genotype, and sex. (D) Association of hippocampal tangle density (R2 = 0.002), midfrontal tangle density (R2 = 0.17), and the midfrontal/hippocampal tangle ratio (R2 = 0.15) with age at onset. (E) Scatterplot of midfrontal tangles against hippocampal tangles, in which colder (blue) colors correspond with earlier onset, while warmer (red) colors correspond with later ages at onset. Delineations of the approximation of the Murray et al.13 hippocampal sparing, limbic predominant, or typical Alzheimer disease (AD) neuropathologic subtypes are shown on the plot. Full model results, and tests for each level of pathology individually, can be found in Tables 2 and 3. Missing data: Lewy pathology (n = 3 [2%]), TDP-43 (n = 1 [<1%]), hippocampal tangle density (n = 2 [2%]), midfrontal/hippocampal tangle ratio (n = 2 [2%]). Midfrontal = midfrontal cortex.
Table 2.
Concomitant Pathology Outcomes Models

TDP-43 pathology was present in 41% of the overall sample and was more likely in those with later onset (OR 2.00 [95% CI 1.23–3.35]) as previously reported.33,34 This effect was driven by neocortical TDP-43 (OR 1.94 [95% CI 1.1–3.57]). TDP-43 copathology was also more likely in those with an APOE ε4 allele (OR 2.46 [95% CI 1.10–5.78]), driven by TDP-43 in the amygdala (OR 4.41 [95% CI 1.37–19.82]). There was no effect of sex on the likelihood of TDP-43 pathology. Hippocampal sclerosis (diagnosed independently of TDP-43 pathology) was present in 7% of the overall sample and its likelihood did not differ by age at onset, APOE genotype, or sex.
Infarcts, microinfarcts, and hemorrhages were rare and did not differ by age at onset, APOE genotype, or sex. Arteriolosclerosis was present in 36% of the overall sample and more likely in those with later onset (OR 2.02 [95% CI 1.24–3.40]), driven by moderate severity arteriolosclerosis (OR 2.50 [95% CI 1.37–4.91]). Atherosclerosis of the circle of Willis was present in 73% of the overall sample and also more likely in those with later onset (OR 3.02 [95% CI 1.70–5.74]), driven by moderate severity atherosclerosis (OR 2.17 [95% CI 1.3–3.8]). Mild, moderate, or severe amyloid angiopathy was present in 90% of the overall sample and was not related to age at onset. There was no effect of APOE genotype or sex on the likelihood of any vascular copathology.
Distribution of NFT Neuropathology
The midfrontal/hippocampal NFT ratio declined with increasing age at onset (β = −0.18 [95% CI −0.26 to −0.10]), indicating a greater relative neocortical burden with earlier onset (Figure 2D). This effect was driven by less NFT pathology in the midfrontal cortex with increasing age at onset (β = −0.51 [95% CI −0.72 to −0.31]). Density of NFT pathology in the hippocampus was not associated with age at onset. APOE genotype was not related to the midfrontal/hippocampal tangle ratio or density of NFT in midfrontal cortex; however, hippocampal NFT pathology was greater in those with an APOE ε4 allele (β = 0.49 [95% CI 0.13–0.86]). Sex was not associated with the midfrontal/hippocampal tangle ratio or density of NFT pathology in midfrontal cortex or hippocampus. When patients were classified into neuropathologic subtypes,13 limbic predominant was associated with later age at onset (OR 6.11 [95% CI 2.61–18.14]), while hippocampal sparing was associated with earlier age at onset (OR 0.46 [95% CI 0.24–0.85]) (Figure 2E, Table 3). Subtypes were not associated with sex or APOE genotype. Both concomitant TDP-43 and arteriolosclerosis were more common in the limbic predominant than hippocampal sparing subtype, but the subtypes did not differ in degree of concomitant Lewy pathology.
Table 3.
Neurofibrillary Tangle Pathology Outcomes Models

Effect of Age at Onset on Cognition and Its Mediation by Neuropathology
While age at onset was not related to baseline memory or language scores, executive (β = 0.48 [95% CI 0.09–0.90]) and visuospatial (β = 0.97 [95% CI 0.46–1.46]) scores were higher in those with later onset (Figure 3). Mediation analyses showed that the midfrontal/hippocampal tangle ratio mediated the effect of age at onset on baseline executive domain scores (β = 0.21 [95% CI 0.08–0.38], Figure 3B), suggesting that age at onset produces indirect effects on executive cognitive abilities via its direct effects on distribution of NFT pathology (which, in turn, has a direct effect: worse executive domain performance in those with higher ratios). There was a strong direct relationship between age at onset and visuospatial domain scores, with lower age at onset associated with lower scores, but this effect was not mediated by the distribution of NFT pathology (β = 0.05 [95% CI −0.16–0.27], Figure 3C). No concomitant non-AD pathology mediated the effect of age at onset on executive or visuospatial scores.
Figure 3. Age at Onset Effects on Cognition and Mediation by Pathology.

(A) Baseline cognitive performance in each cognitive domain in plotted against age at onset. The p values correspond to effects for age at onset from regression models predicting each cognitive domain outcome, with terms for age at onset, APOE genotype, sex, and education. A significant mediation effect of the midfrontal/hippocampal tangle ratio on executive function score was observed (B). However, the mediation effect of the midfrontal/hippocampal tangle ratio on visuospatial scores was not significant (C). Age at onset coefficients are standardized per 10 years (decade) increase in age. Midfrontal = midfrontal cortex.
Longitudinal analyses controlling for sex, APOE genotype, and baseline cognitive test score showed there was faster decline with earlier age at onset on several specific neuropsychological tests: MMSE, DRS, Digit Symbol Substitution, and Block Design (Figure 4). Mediation analyses showed that no concomitant non-AD pathology mediated the effect of age at onset on rate of decline on any of these cognitive measures. In contrast, the relationship between age at onset and rate of decline on each of these 4 tests was mediated by the distribution of NFT pathology (all p < 0.01; faster decline in those with higher ratios).
Figure 4. Age at Onset Effects on Longitudinal Cognitive Decline and Implications for Clinical Trials.

(A–D) Annualized rates of decline calculated from 2–3 longitudinal evaluations on each measure are plotted against age at onset. The p values correspond to effects for age at onset from regression models predicting each test’s rate of decline, with terms for age at onset, APOE genotype, sex, and education. All 4 significant effects of age at onset were found to be mediated by the neurofibrillary tangle ratio, but not any concomitant pathologies. Digit symbol substitution data truncated due to a higher level of missing data at the top of the age range. DRS = Dementia Rating Scale; MMSE = Mini-Mental State Examination.
Discussion
We identified age-related heterogeneity in neuropathologic and clinical features of patients with sporadic, pathologically confirmed, severe AD. Those with earlier age at onset were less likely than those with later age at onset to have concomitant non-AD neurodegenerative or vascular pathology and to report nonmemory cognitive impairment as their initial presenting symptom, have more functional impairment in ADLs, report more psychiatric symptoms, and have worse deficits in executive and visuospatial cognitive domains. This paradoxical pattern of more atypical clinical features in patients with early onset AD, despite less concomitant non-AD neurodegenerative or vascular pathology, suggests that these differences may result from age-related differences in the distribution of AD pathology. We support previous findings by showing that NFT density in midfrontal neocortex, and midfrontal/hippocampal NFT ratio, are strongly inversely related to estimated age at onset. When we categorized patients into hippocampal sparing, limbic predominant, and typical NFT subtypes proposed by Murray et al.,13 we replicated their result that the hippocampal sparing subtype is associated with earlier age at onset and less copathology than the limbic predominant subtype. We extend their findings to show that differences in distribution of NFT pathology mediate distinct cognitive deficit profiles and rates of decline across the age at onset spectrum. These findings coincide with imaging studies demonstrating greater tau-PET tracer uptake in neocortical regions with earlier ages at symptom onset35 or atypical clinical presentations.36 We also show that hippocampal NFT density is not related to age at onset, but is related to APOE genotype with greater density in those with the ε4 risk allele. This finding is consistent with studies showing fluorodeoxyglucose (FDG) hypometabolism37 or greater tau-PET tracer uptake38 in limbic regions in APOE ε4+ patients with AD.
Mediation analyses demonstrated that midfrontal/hippocampal tangle ratio, but not concomitant Lewy body, TDP-43, or microvascular pathology, was a significant mediator of the effect of age at onset on cognition in the executive domain. This finding suggests that age at onset produced indirect effects on executive function primarily through its direct effects on distribution of NFT pathology which, in turn, directly affected executive function. Any effects of TDP-43 and vascular copathologies are relatively minor and are likely overshadowed by the presence of severe (Braak stage V–VI) AD pathology. In contrast, neither the distribution of NFT pathology nor concomitant non-AD pathology significantly mediated the effect of age at onset on visuospatial cognition. This latter finding suggests that age-related effects on visuospatial abilities is not attributable to concomitant Lewy body pathology that can cause disproportionate visuospatial impairment.39 Consistent with previous studies,4,39,40 concomitant Lewy pathology was present in approximately 20%–25% of individuals, but did not vary by age at onset. The lack of mediation of visuospatial impairment by NFT distribution could be related to our choice of midfrontal neocortex in calculating the distribution of pathology. Deficits in visuospatial abilities may better map onto NFT pathology in posterior temporo-occipital and parieto-occipital cortical regions that show the greatest hypometabolism and atrophy on imaging of posterior cortical atrophy due to AD.41,42 Longitudinally, earlier age at onset was associated with faster decline on global cognitive measures and specific tests of executive function that have a visuospatial component (e.g., Digit Symbol Substitution, Block Design). Mediation analyses showed that these relationships were mediated by the midfrontal/hippocampal NFT ratio, but not by concomitant pathologies.
The higher midfrontal/hippocampal NFT ratio observed with earlier age at onset suggests that either the neocortex is more vulnerable, or the hippocampus is less vulnerable, to NFT pathology in younger individuals who develop severe AD. Selective neocortical vulnerability could result from greater loss of cortical cholinergic innervation, as suggested by a recent study demonstrating greater NFT pathology and neuron loss in the nucleus basalis of Meynert in patients with earlier age at onset of AD.43 Alternatively, the hippocampus may be relatively less vulnerable to NFT pathology in patients with early onset AD given that increasing age leads to selective vulnerability of the hippocampus to ischemia, hypoglycemia, hyperexcitability, metabolic stresses, and neurodegenerative diseases.44,45 Age-related microglial senescence46 and increased proinflammatory signaling in the hippocampus may be mechanisms that lead to impaired homeostasis47 and development of AD neuropathology.48 Another possibility is that individuals with later age at onset of AD had more time to develop age-related NFT pathology that is usually restricted to the medial temporal lobe (i.e., primary age-related tauopathy49) prior to the development of abnormal amyloid and acceleration of tau spread to the neocortex.
The use of strict pathologic definitions of AD (assessment of microvascular, TDP-43, and Lewy pathology) that cannot be detected during life and detailed cognitive phenotyping are strengths of our study. Several limitations also should be considered. First, there were changes in clinical and neuropathologic practices and criteria over the 35 years during which this clinical–pathologic cohort was established. We minimized potential bias this may have caused by conducting recently developed immunohistochemical staining for concomitant pathologies (e.g., α-synuclein, TDP-43) on all participants and retrospectively applying current consensus neuropathologic criteria for AD and related disorders. Second, our study was confined to those with severe AD pathology and this may have attenuated our ability to perceive the effect of copathologies on clinical and cognitive features. In addition, this made it difficult to neuropathologically diagnose HS as neuronal loss and gliosis out of proportion to AD pathology, a problem that is likely accentuated in patients with later onset who have a higher hippocampal tangle burden. Third, we compared NFT counts from only the hippocampus (1 area) and frontal cortex to classify individuals as hippocampal sparing, limbic predominant, or typical, in contrast to the 2 hippocampal and 3 cortical regions used by Murray et al.13 This may have slightly reduced our classification accuracy compared to theirs, but it should be noted that we identified comparable proportions of each subtype and replicated their finding that hippocampal sparing is associated with early-onset AD. The lack of NFT counts in additional cortical regions also makes it difficult to interpret the lack of mediation of visuospatial deficits by NFT distribution and this needs to be examined in future research. Finally, our mediation analyses utilized cognitive measures collected several years before the pathologic mediators, conflicting with the temporal order implied by this method. However, even if the pathologic burden grew between cognitive testing and death, recent research using tau PET has shown that patterns of tau distribution diverge early in the course of disease and proceed along distinct trajectories,36,50 suggesting that the distribution of pathology at death is representative of its distribution at the time of symptom onset.
Our results are consistent with the idea that age-related differences in the distribution of a key pathologic feature of AD, NFT, drives heterogeneity in the clinical presentation of the disorder. Why the distribution of NFT pathology varies with age remains unknown, but we speculate that it may have to do with age-related differences in hippocampal or neocortical vulnerability, and does not indicate that early- and late-onset AD are nosologically distinct diseases. It may be prudent, however, to consider age at onset in the design and selection of cognitive outcome measures for tau-targeting therapeutic trials given its relationship to clinical and cognitive heterogeneity related to tau distribution. Specifically, a cognitive outcome measure weighted towards visuoexecutive function may be more effective (i.e., require less power and smaller sample size) in detecting a tau treatment effect in patients with early-onset compared to late-onset AD.
Acknowledgment
The authors thank Jeffrey Metcalf and Sara Shuldberg for technical assistance.
Glossary
- AD
Alzheimer disease
- ADL
activities of daily living
- CDR
Clinical Dementia Rating
- CERAD
Consortium to Establish a Registry for AD
- CI
confidence interval
- DLB
dementia with Lewy bodies
- DRS
Dementia Rating Scale
- H&E
hematoxylin & eosin
- HS
hippocampal sclerosis
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- NFT
neurofibrillary tangle
- NIA-AA
National Institute on Aging–Alzheimer Association
- NPI
Neuropsychiatric Inventory
- OR
odds ratio
- PHF
paired helical filament
- POD
Pfeffer Outpatient Disability
- PPA
primary progressive aphasia
- TDP-43
TAR DNA binding protein 43
- UCSD
University of California, San Diego
Appendix. Authors
Study Funding
This work was supported by NIH P30-AG062429, F30-AG063440, and the D.H. Chen Foundation.
Disclosure
D.S. Smirnov, V.S. Goodwill, L.A. Hansen, Y. Zhao, G.C. Leger, G.M. Peavy, D.M. Jacobs, R.A. Rissman, D.P. Pizzo, and A. Hiniker have no financial or nonfinancial disclosures. S.D. Edland is a paid consultant for Eli Lilly and Johnson & Johnson. D. Galasko is a paid consultant for Biogen, Inc.; Fujirebio, Inc.; Cognition Therapeutics; Amprion; vTv Pharmaceuticals; and GE Healthcare. D.P. Salmon is a consultant for Aptinyx, Inc.; and Biogen, Inc. Go to Neurology.org/N for full disclosures.
References
- 1.Koedam ELGE, Lauffer V, van der Vlies AE, van der Flier WM, Scheltens P, Pijnenburg YAL. Early- versus late-onset Alzheimer's disease: more than age alone. J Alzheimers Dis. 2010;19(4):1401-1408. [DOI] [PubMed] [Google Scholar]
- 2.van der Flier WM, Pijnenburg YA, Fox NC, Scheltens P. Early-onset versus late-onset Alzheimer's disease: the case of the missing APOE ɛ4 allele. Lancet Neurol. 2011;10(3):280-288. [DOI] [PubMed] [Google Scholar]
- 3.Rossor MN, Fox NC, Mummery CJ, Schott JM, Warren JD. The diagnosis of young-onset dementia. Lancet Neurol. 2010;9(8):793-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smirnov DS, Galasko D, Hiniker A, Edland S, Salmon DP. Age-at-onset and APOE-related heterogeneity in pathologically confirmed sporadic Alzheimer disease. Neurology. 2021;96(18):e2272-e2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnes J, Dickerson BC, Frost C, Jiskoot LC, Wolk D, Flier WM. Alzheimer's disease first symptoms are age dependent: evidence from the NACC dataset. Alzheimers Dement. 2015;11(11):1349-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendez MF, Lee AS, Joshi A, Shapira JS. Nonamnestic presentations of early-onset Alzheimer's disease. Am J Alzheimers Dis Other Dement. 2012;27(6):413-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baillon S, Gasper A, Wilson-Morkeh F, Pritchard M, Jesu A, Velayudhan L. Prevalence and severity of neuropsychiatric symptoms in early- versus late-onset Alzheimer's disease. Am J Alzheimers Dis Other Dement. 2019;34(7-8):433-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnes J, Bartlett JW, Wolk DA, van der Flier WM, Frost C. Disease course varies according to age and symptom length in Alzheimer's disease. J Alzheimers Dis. 2018; 64(2):631-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wattmo C, Wallin ÅK. Early- versus late-onset Alzheimer's disease in clinical practice: cognitive and global outcomes over 3 years. Alzheimers Res Ther. 2017;9(1):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobs D, Sano M, Marder K, et al. Age at onset of Alzheimer's disease: relation to pattern of cognitive dysfunction and rate of decline. Neurology. 1994;44(7):1215. [DOI] [PubMed] [Google Scholar]
- 11.Petersen C, Nolan AL, Resende EdePF, et al. Alzheimer's disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol. 2019;138(4):597-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10(9):785-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8(1):1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terry RD, Katzman RK. Senile dementia of the Alzheimer type. Ann Neurol. 1983;14(5):497-506. [DOI] [PubMed] [Google Scholar]
- 16.Mirra S, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41(4):479. [DOI] [PubMed] [Google Scholar]
- 17.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Tredici KD. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terry RD, Hansen LEA, Deteresa R, Davies P, Tobias H, Katzman R. Senile dementia of the Alzheimer type without neocortical neurofibrillary tangles. J Neuropath Exp Neur. 1987;46(3):262-268. [DOI] [PubMed] [Google Scholar]
- 19.McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies. Neurology. 2017;89(1):88-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uchikado H, Lin W-L, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of α-synucleinopathy. J Neuropathol Exp Neurol. 2006;65(7):685-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galasko D, Hansen LA, Katzman R, et al. Clinical-neuropathological correlations in Alzheimer's disease and related dementias. Arch Neurol. 1994;51(9):888-895. [DOI] [PubMed] [Google Scholar]
- 23.Salmon D, Butters N. Neuropsychological assessment of dementia in the elderly. In: Katzman R, Rowe J, eds. Principles of Geriatric Neurology. FA Davis; 1992:144. [Google Scholar]
- 24.Pfeffer R, Kurosaki T, Harrah C, et al. A survey diagnostic tool for senile dementia. Am J Epidemiol. 1981;114(4):515-527. [DOI] [PubMed] [Google Scholar]
- 25.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34(7):939. [DOI] [PubMed] [Google Scholar]
- 26.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crutch SJ, Schott JM, Rabinovici GD, et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017;13(8):870-884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smirnov DS, Galasko D, Edland SD, Filoteo JV, Hansen LA, Salmon DP. Cognitive decline profiles differ in Parkinson disease dementia and dementia with Lewy bodies. Neurology. 2020;94(20):e2076-e2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imai K, Keele L, Tingley D. A general approach to causal mediation analysis. Psychol Methods. 2010;15(4):309-334. [DOI] [PubMed] [Google Scholar]
- 32.Zhao Y, Edland SD. Power formulas for mixed effects models with random slope and intercept comparing rate of change across groups. Int J Biostat. ePub January 18, 2021. doi: 10.1515/ijb-2020-0107. [DOI] [PMC free article] [PubMed]
- 33.Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and TDP‐43 pathology in aging and Alzheimer disease. Ann Neurol. 2015;77(6):942-952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uchino A, Takao M, Hatsuta H, et al. Incidence and extent of TDP-43 accumulation in aging human brain. Acta Neuropathol Commun. 2015;3:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schöll M, Ossenkoppele R, Strandberg O, et al. Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer's disease. Brain. 2017;140(9):2286-2294. [DOI] [PubMed] [Google Scholar]
- 36.Ossenkoppele R, Schonhaut DR, Schöll M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016;139(Pt 5):1551-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jagust WJ, Landau SM, Alzheimer's Disease Neuroimaging Initiative. Apolipoprotein E, not fibrillar β-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci. 2012;32(50):18227-18233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Therriault J, Benedet AL, Pascoal TA, et al. Association of apolipoprotein E ε4 with medial temporal tau independent of amyloid-β. JAMA Neurol. 2020;77(4):470-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansen L, Salmon D, Galasko D, et al. The Lewy body variant of Alzheimer's disease: a clinical and pathologic entity. Neurology. 1990;40(1):1-8. [DOI] [PubMed] [Google Scholar]
- 40.White L. Brain lesions at autopsy in older Japanese-American men as related to cognitive impairment and dementia in the final years of life: a summary report from the Honolulu-Asia Aging Study. J Alzheimers Dis. 2009;18(3):713-725. [DOI] [PubMed] [Google Scholar]
- 41.Montembeault M, Brambati SM, Lamari F, et al. Atrophy, metabolism and cognition in the posterior cortical atrophy spectrum based on Alzheimer's disease cerebrospinal fluid biomarkers. Neuroimage Clin. 2018;20:1018-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Day GS, Gordon BA, Jackson K, et al. Tau-PET binding distinguishes patients with early-stage posterior cortical atrophy from amnestic Alzheimer disease dementia. Alzheimer Dis Assoc Disord. 2017;31(2):87-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Shaikh FSH, Duara R, Crook JE, et al. Selective vulnerability of the nucleus basalis of Meynert among neuropathologic subtypes of Alzheimer disease. JAMA Neurol. 2020;77(2):225-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lalonde CC, Mielke JG. Selective vulnerability of hippocampal sub-fields to oxygen–glucose deprivation is a function of animal age. Brain Res. 2014;1543:271-279. [DOI] [PubMed] [Google Scholar]
- 45.Morrison JH, Hof PR. Chapter 37 selective vulnerability of corticocortical and hippocampal circuits in aging and Alzheimer's disease. Prog Brain Res. 2002;136:467-486. [DOI] [PubMed] [Google Scholar]
- 46.Ojo JO, Rezaie P, Gabbott PL, Stewart MG. Impact of age-related neuroglial cell responses on hippocampal deterioration. Front Aging Neurosci. 2015;7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hajieva P, Kuhlmann C, Luhmann HJ, Behl C. Impaired calcium homeostasis in aged hippocampal neurons. Neurosci Lett. 2009;451(2):119-123. [DOI] [PubMed] [Google Scholar]
- 48.Streit WJ, Braak H, Xue Q-S, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118(4):475-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vogel JW, Young AL, Oxtoby NP, et al. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021;27(5):871-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.