

Abstract
Progress has been made using B-form DNA duplex strands to template chromophores in ordered molecular aggregates known as J-aggregates. These aggregates can exhibit strong electronic coupling, extended coherent lifetimes, and long-range exciton delocalization under appropriate conditions. Certain cyanine dyes such as pseudoisocyanine (PIC) dye have shown a proclivity to form aggregates in specific DNA sequences. In particular, DX-tiles containing nonalternating poly(dA)−poly(dT) dinucleotide tracks (AT-tracks), which template noncovalent PIC dye aggregates, have been demonstrated to exhibit interesting emergent photonic properties. These DNA-based aggregates are referred to as J-bits for their similarity to J-aggregates. Here, we assemble multifluorophore DX-tile scaffolds which template J-bits into both contiguous and noncontiguous linear arrays. Our goal is to understand the relay capability of noncontiguous J-bit arrays and probe the effects that orientation and position have on the energy transfer between them. We find that linearly contiguous J-bits can relay excitons from an initial AlexaFluor 405 donor to a terminal AlexaFluor 647 acceptor across a distance of up to 16.3 nm. We observed a maximum increase in energy transfer of 41% in the shortest scaffold and an 11% increase in energy transfer across the maximum distance. However, in nonlinear arrays, exciton transfer is not detectable, even when off-axis J-bit-to-J-bit transfer distances were <2 nm. These results, in conjunction with the previous work on PIC−DNA systems, suggest that PIC−DNA-based systems may currently be limited to simple 1-D designs, which prevent isolating J-bits for enhanced energy-transfer characteristics until further understanding and improvements to the system can be made.
Graphical Abstract
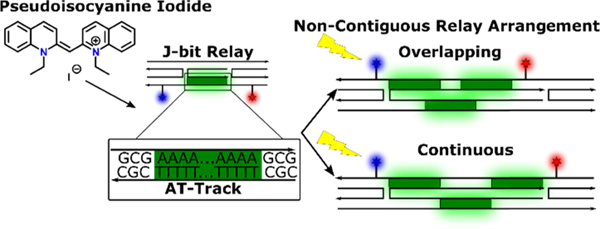
INTRODUCTION
Over millennia of evolution, low-light photosynthetic organisms have developed directional energy-transfer pathways that are able to engage in exciton migration over long distances with extremely low loss. This feat of optimized energy transfer is thought to be mediated through both Förster resonance energy transfer (FRET) and quantum mechanical processes.1–7 FRET occurs when an excited-state donor chromophore (D) nonradiatively transfers its energy to an acceptor chromophore (A) in the ground state through dipole−dipole interactions.4–7 Although potentially quite useful for nanoscale engineering, FRET pathways are known to suffer from certain limits in efficiency.5–7 For example, due to the nature of the Stokes shifts, in hetero-FRET (FRET between different chromophores with distinct energy levels), the A chromophore’s excitation is necessarily red-shifted compared to the D chromophore’s emission, leading to descendent energy levels with each transfer step. Alternatively, homo-FRET (FRET between identical molecules, i.e., the D−A are indistinguishable) recovers this loss by keeping D emission and A excitation at constant energy levels, it instead suffers from increased transfer steps and nondirectionality due to the random walk nature of the exciton transfer.8–11 Furthermore, the efficiency of any FRET step inherently decays by the D−A separation distance (r) raised to the inverse sixth power, thus imposing tight spatial constraints on exciton migration which limits FRET system scalability.12,13 As a result, more recent devices have sought to emulate a natural photosynthetic approach that appears to manifest both FRET and quantum mechanical processes into a single nanoscale architecture.14 It has been shown that under certain conditions, cyanine dyes, such as pseudoisocyanine (PIC), are able to form noncovalent aggregates exhibiting evidence of coherence in aqueous solutions.15–22 Intermolecular van der Waals forces enable monomers to be densely packed (<2 nm separation) and rigidly lock molecular transition dipoles in a favorable head-to-tail orientation. This polymeric state, known as a J-aggregate, allows for strong coupling between the monomers and the manifestation of a coherent excited state (e.g., super-radiance). In this state, the exciton can be partially delocalized throughout the length of the aggregate.
In establishing both a stable and finely tuned energy-transfer landscape, several different scaffolding motifs have been proposed to arrange chromophores. For artificial systems, these alternatives include, but are not limited to, metal organic frameworks,23 recombinant proteins,24 viral scaffolds,25 and synthetic polymers.26,27 The right-handed B-form DNA double helix has also been heavily relied on to construct directional hetero- and homo-FRET energy-transfer cascades.28 However, as previously mentioned, scaling up complexity to larger, more robust light-harvesting architectures is constrained by FRET’s inherent limits.8,11 One possible means to overcome these limitations is the integration of coherently coupled dye aggregates into a canonical DNA scaffold-based FRET cascade.19–21 In originating work pursuing this goal, PIC monomers have been demonstrated to spontaneously form ordered supramolecular aggregates with J-like properties (J-bits) in nonalternating poly(dA)−poly(dT) dinucleotide sequences (AT-tracks) of B-form DNA duplex.19–22,29 These J-bits exhibit favorable optical properties such as bathochromically shifted absorbance bands, enhanced quantum yield (QY), super-radiant emission, and long-range coherent exciton delocalization with enhanced oscillator strength. By book-ending the AT-track with covalently conjugated D and A chromophores, photonic relays can be potentially created, which have both FRET and coherent energy-transfer characteristics. Though promising, the design parameters of these systems are still not fully understood. For example, Boulais et al. demonstrated energy transfer along parallel AT-tracks in a DNA double-crossover tile (DX-tile).20 However, Mandal and colleagues demonstrated that a two-base pair (bp) GC spacer in linear 1-D J-bits attenuated excitonic relay capabilities by∼50%.21
Here, we aim to further probe and understand the limits of J-bit interactions by manipulating the placement and orientations of J-bits in a modified DX-tile architecture that presents noncontiguous, nonlinear transfer pathways. Fundamentally, we intend to determine if independent PIC aggregates function as molecular relay units capable of multidirectional communication. The DX-tile used as the basis for our experimental sample platform self-assembles to enable energy transfer in a stepwise pathway (D-R-A) from an initial Donor (AlexaFluor 405, AF405, D405) to a PIC-based J-bit Relay and then to the final Acceptor (AlexaFluor 647, AF647, A647), see Figure 1. We observe a maximum increase in energy transfer of 41% in the shortest scaffold, a distance of 9.5 nm. Furthermore, an 11% increase in energy transfer across a maximum distance of 16.3 nm was determined by exploiting the properties of J-bits as energy-transfer relays. In both instances, scaffolds arranged J-bits in contiguous and directionally linear arrays. When we arrange J-bits into multidirectional arrays, forcing trans-helical exciton migration, we find that J-bits efficiently form in these AT-tracks, but off-axis separation between J-bits (even when the distances are <2 nm from center to center) nullifies exciton transfer through the system.
Figure 1.
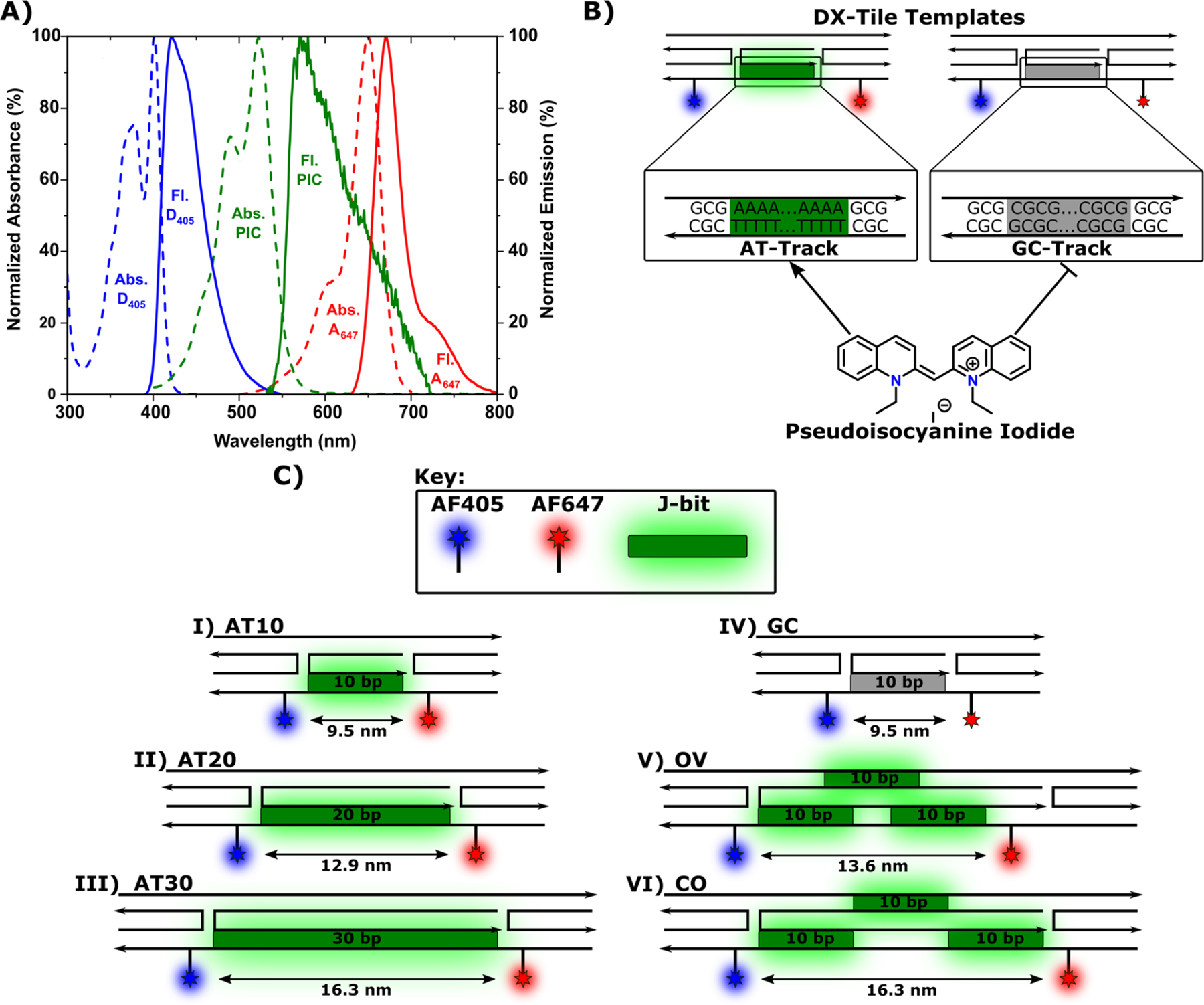
Design of the DNA DX-tile with J-bits. (A) Spectra of AF405 (blue), PIC dye (green), and AF647 (red) normalized to maximum absorbance (dashed) and maximum emission (solid). (B) Illustration of pseudoisocyanine iodide interactions with DX-tile AT-track (green) and GC-track region (gray). The arrow indicates preferential J-bit formation, while the line indicates no J-bit formation. (C) Structural schematics of (I) AT10, (II) AT20, (III) AT30, (IV) GC, (V) OV, and (VI) CO DNA DX-tiles utilized including estimated D−A separation distances. Labels represent D405 (blue), AT-track regions (green), GC-track region (gray), and A647 (red). DNA strand arrows indicate 5′−3′ sequence directionality. The “Top” helix and the “Bottom” helix are indicated in reference to scaffold orientations displayed on OV and CO structures.
METHODS
Materials.
Single-stranded DNA oligonucleotides (ssDNA) labeled with AF405 were purchased from Bio-Synthesis Inc., while unlabeled ssDNA and AF647-labeled ssDNA were obtained from Integrated DNA Technologies Inc. The quantum yield reference 5-(ethylimino)-10-methyl-5H-benzo-[a]phenoxazin-9-ethylamine perchlorate (Oxazine 720) was sourced from the Exciton division of Luxottica Group S.p.A. Ninety-eight percent sulfuric acid and Optima LC/MS grade methanol were sourced from Fisher Scientific. All other chemicals including (R)-(6-methoxyquinolin-4-yl)-[(1S,2S,4S,5R)-5-vinylquinuclidin-2-yl]methanol sulfate dihydrate (Quinine sulfate, CAS No: 207671–44-1); 97% 1,1′-diethyl-2,2′-cyanine iodide (PIC, CAS No: 977–96-8);molecular biology-grade water; NaCl; MgCl2; 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 7.0); GelRed nucleic acid stain 10 000× DMSO; and tris-(hydroxymethyl) aminomethane (TRIS; pH 7.0) were sourced from Sigma-Aldrich.
DX-Tile Preparation.
Upon receipt and without any further purification, ssDNA oligonucleotide stocks were fully rehydrated in molecular biology-grade water. Their concentration was determined by UV−vis absorbance at 260 nm using the manufacturer-reported molar extinction coefficient. To assemble DX-tile structures (Figure 1A), ssDNA oligonucleotides (see Figures S2−S7 and accompanying tables) were added to a HEPES-Mg buffer (10 mM HEPES and 5 mM MgCl2 at pH 7.0) in 1:1 stoichiometry with each strand at 2.5 μM. In total, six unique DX-tile structures designs were explored. The first three DX-tile designs positioned a contiguous AT-track (Figure 1B) along a single duplex in the DX-tile extending 10, 20, and 30 bps in length and therefore named AT10, AT20, and AT30, respectively (Figure 1C). The fourth design was similar to the AT10; however, the AT-track sequence was modified to a GC-track (Figure 1B), to serve as a sequence-specific control and hence referred to as the GC structures (Figure 1C). The fifth and sixth structures were similar to the AT30 structure except that the 30 bp AT-track was split into three separate 10 bp AT-tracks positioned on alternate strands in the DX-tile. The difference between the two structures was that one structure overlapped the AT-tracks by 3 bps in the x-direction (referred to as overlapping or OV structure) and the other structure maintained continuity without any bp gaps or overlaps in the x-direction (referred to as continuous or CO structure) (Figure 1C). Each structure was annealed in a thermocycler by heating the reaction mixture to 85 °C for 10 min, then decreasing to 80 °C for 1 min; from 80 °C, the temperature was decreased by 1 °C per minute until 20 °C, where it hovered for 3 min, after which the temperature was decreased from 20 °C by 1 °C per minute until reaching 4°C. Final DX-tile concentrations were assumed to be 2.5 μM based on the 1:1 stoichiometry. Fully formed sample aliquots were taken directly from the thermocycler and analyzed via 10% polyacrylamide gel electrophoresis (PAGE). PAGE gels were prerun for 15 min with HEPES-Mg buffer. After prerunning, 1 pmol of DX-tile was loaded and the gel was run on ice for 10 min at 70 V after which the voltage was increased to 120 for 40 min. Upon completion, 1× GelRed was used to visualize DNA band migration, see the Supporting Information (SI), Figure S1, for a representative gel electro-pherogram image.
PIC Dye Stock Preparation.
200 μM PIC dye stock solution was prepared from the powder by dissolving in Tris−HCl buffer (10 mM NaCl, 5 mM Tris HCl, pH 7.0). To ensure fully solvated dye, the dye solution was sonicated for 1 h at 60 °C. The solution was then allowed to cool to room temperature and filtered through a 0.2 μm syringe filter. The final PIC concentration was calculated by UV−vis absorbance using the PIC monomer’s molar absorption coefficient of 53 500 M−1 cm−1 at 523 nm.
DNA-PIC Sample Preparation.
Unless otherwise noted, samples were prepared in Tris−HCl buffer by adding the necessary volumes to reach a final DX-tile concentration of 400 nM and PIC dye concentration of 52 μM, a 130-fold excess necessary to drive PIC aggregation into AT-tracks.19–21
Dye Photophysical and FRET Properties.
AF405-(D405) and AF647- (A647) labeled oligonucleotides were purchased and assumed to have a 100% labeling since they underwent post-synthesis high-performance liquid chromatog-raphy (HPLC) purification. The AlexaFluor dyes were chemically attached to DNA through amide bonds via six-carbon linkers, and the specific DNA sequences used in each construct can be found in Figures S2−S7. PIC dye was used to form noncovalent supramolecular aggregates and therefore had no molecular modifications. The measured photophysical properties of each dye species including quantum yield (QY), extinction coefficient, excitation-, and emission maxima are presented in Table 1. Additional information on the QY, spectral overlap, and Förster distance (R0, D−A separation corresponding to 50% FRET efficiency) calculations is available in the SI.
Table 1.
Selected Photophysical Properties of Each Component Fluorophore
| QY | ε (M−1 cm−1) | λ Absmax (nm) | λ Emmax (nm) | |
|---|---|---|---|---|
| AF405 | 72 ± 5.1%a | 35 000 | 401 | 421 |
| PIC (monomer/J-bit) | 0.012/0.18%b | 53 500 | 525 | 584 |
| AF647 | 41 ± 2.6%c | 270 000 | 650 | 665 |
| calculated spectral overlap integral (J, nm4 M−1 cm−1)/R0 (nm) | ||||
| PIC (monomer) | AF647 | |||
| AF405 | 4.3 × 1014/3.8 | 7.8 × 1012/1.9 | ||
| PIC (monomer) | 6.9 × 1013/0.7 | 1.3 × 1016/2.4 | ||
Absorbance and Fluorescence Measurements.
Absorbance spectra were collected using a TECAN Spark plate reader with samples loaded into a white Corning clear bottom 384 flat well plate. Each 50 μL sample consisted of 400 nM DNA and 52 μM PIC. Absorbance spectra were collected from 300 to 800 nm at 20 °C with an n = 3 replicates, and the presented spectra are the average of the three. To qualitatively observe the differences in J-band shoulder spectral shape, each scaffold spectra were normalized to the 0−0 transition (∼525 nm), followed by subtracting the GC control DX-tile absorbance as a baseline, see Figure 1 for different configurations of the DX-tile that were tested. To quantitatively observe the differences in J-band shoulder area, each scaffold’s absorbance shoulder was integrated. The integration indices were determined using the PIC only absorbance as a baseline correction. The integration range was defined as beginning where the baseline-corrected spectra first yielded a positive absorbance value and ending at 580 nm in all cases. The integration was performed on the uncorrected, nonnormalized spectra to get an unbiased quantification of the relative shoulder area.
Fluorescence emission spectra were collected using a TECAN Spark plate reader with a white Corning clear bottom 384 flat well plate. The spectra were recorded from 420 to 800 nm at 20 °C with an n = 3, and the presented spectra are the average of the three repeats. The excitation wavelength was set to 395 nm with a slit width of 10 nm. After the spectra were collected, they were analyzed using the A|E UV−vis−IR Spectral Software.32 Spectra were deconvoluted as linear combinations of component emissions; for more details, see the SI.
Circular Dichroism Measurements.
Circular dichroism (CD) spectra ranging from 400 to 700 nm were collected using a J-1500 JASCO spectrometer. Each presented spectrum was the average of three independent measurements. Each measurement was made at 20 °C unless otherwise specified. For PIC/DX-tile ratio experiments, the DNA concentration was held constant at 400 nM while the initial PIC concentration was 20 μM (50×). After initial measurement, samples with PIC concentrations ranging up to 100 μM (250×) were measured in 20 μM increments. During J-bit temperature stability experiments, the sample contained 400 nM DNA and 100 μM PIC (250×). The initial measurement was made at 10 °C, and then the sample temperature was sequentially increased to 60 °C with spectra taken after each 5 °C increment.
Energy-Transfer Quantification.
As an estimate of energy transfer between a given D and A pair, we consider both D405 quenching (EQ), A647 sensitization (As), and end-to-end transfer efficiency (Eee). D quenching can be calculated following eq 1
| (1) |
Here, EQ is the percent energy transferred by estimation of the D quenching, ΨD is the integrated fluorescence spectra of the D405 only scaffold in the absence of PIC dye (+D405, −J-bit,−A647). A similar approach is not feasible in the PIC to AF647 transfer step due to the low and environmentally sensitive fluorescent QY of the PIC dye. However, as an overall measure of the PIC’s relay capability, we calculate energy transfer as a relative A sensitization (As) as defined in eq 2
| (2) |
As compares the A647 sensitization with and without the presence of D405 while taking into account the direct excitation of the PIC relay component. Therefore, any additional sensitization as measured by an increase in As is necessarily relayed through the J-bit(s). Where ΨDRA is the integrated spectral peak associated with A647 obtained from the spectral decomposition fit to the measured spectra of each scaffold with all dyes present (+D405, +J-bit, +A647), ΨRA is the same spectral peak as measured for each scaffold with only the J-bit relay and A present (−D405, +J-bit, +A647).
End-to-end transfer efficiency, Eee, is an alternative representation of the As value that takes into account the number of photons absorbed by the initial D and fluorescent QYs of D and A dyes.10,33–35 This metric is useful for allowing comparison between disparate transfer systems. Eee is determined using eq 3
| (3) |
With similar parameters as eq 2, ΨD is the area of the spectral peak associated with D405 (+D405, −J-bit, −A647) in the absence of the PIC relay and A647. The terms QA and QD refer to the QY of A647 and D405, respectively.
RESULTS AND DISCUSSION
DX-Tile and Structural Design.
Our motivation here is to build upon the results demonstrated by Banal, Boulais, and Mandal in their initial work and to better understand PIC− DNA J-bit constraints for energy transfer.19–21 J-bits have demonstrated transfer between directionally linear arrange-ments but have yet to be fully tested in multidirectional arrays, which may potentially be achieved through the use of the DNA DX-tile. Structurally, the DNA DX-tile is composed of two parallel DNA helices constrained by antiparallel double crossovers giving them a planar rigidity.36–39 Exploiting this DX-tile motif, we designed a total of six unique configurations, each with the same underlying architecture (Figure 1). All six DNA scaffolds have covalently attached D405 and A647 dyes bookending a sequence-specific J-bit relay region enabling at a minimum a two-step energy-transfer cascade. The variation between the scaffolds revolves around differences in the design of the J-bit relay. The first three scaffolds, based on previously published systems,20 were self-assembled from five single-stranded oligonucleotides, which positioned J-bits along a single contiguous AT-track extending 10, 20, and 30 bps in length, respectively. Hence, these scaffolds are referred to as AT10, AT20, and AT30 (Figure 1C). In solution, J-aggregates form when the molecular transition dipoles of monomers are arranged in a head-to-tail orientation. Therefore, J-aggregates tend to innately favor a linear, fiberlike composite structure. DNA-based J-aggregates are estimated to aggregate with 8−12 PIC molecules per turn of B-form DNA (∼10 bps) and with an expected exciton coherence length of up to three dyes.20 Given preference for AT-track sequences, a linearly contiguous design should be a relatively favorable guide for inter-J-bit coupling. This fact is supported by available literature where PIC-based J-bits efficiently form in duplex AT-tracks of up to 48 bps in length.21
Two novel scaffolds exhibiting noncontiguous AT-tracks were also designed. These two scaffolds, self-assembled from six ssDNA oligonucleotides, positioned J-bits along three separated 10 bp AT-tracks, which alternate between helices within each DX-tile. To avoid confusion, we refer to the two helices as the “top” helix and the “bottom” helix, in reference to scaffold orientations displayed in Figure 1. The nomenclature is arbitrary, based merely on the system’s illustration. D405 and A647 dyes are conjugated to the bottom helix. The first and third AT-tracks are also located on the bottom helix, while the second AT-track is located on the top helix. The principal difference between the scaffolds is that the overlapping (OV) scaffold positions the second AT-track partially overlapping with the end of the first AT-track and the beginning of the third AT-track. The continuous (CO) scaffold shifts the ATtracks such that there is no overlap, but there is also no spacing between them (Figure 1C).
Considering the distance from 1 bp to the next in a B-DNA duplex is on average 0.34 nm,40 separations between different dyes are reported for all scaffolds in Table 2. It should be noted that these distances are approximated by scaling 0.34 nm proportionally by the number of bases in a given sequence; dye attachment linkers are not considered in these approximations. The separation distances are important to consider as they affect dipole coupling and FRET efficiency. Therefore, to enable direct comparison, scaffolds were designed to keep dye positions as consistent as possible. As such, D405 is always positioned 9 bps away from the first dA−dT bp in an AT-track equating to ∼3.0 nm in distance. A647 was also positioned 9 bps from the final dA−dT bp except in the OV scaffold, where the overlapping sequence is shifted, thus decreasing the separation distance to ∼2.4 nm. The overall separation distance of D405 → A647 (→ = FRET step) is therefore the sum of these two distances plus the length of the extendable AT-track length, which is the D405 → A647 value reported in Table 2. Overall, the D405−A647 separation distance ranges from ∼9.5 nm in the AT10 to a maximum of ∼16.3 nm in the AT30 scaffold.
Table 2.
Predicted Dye Separation Distances (nm)/Estimated FRET Efficiencya
| name | D405 → A647 | D405 → PIC | PIC → A647b | extension of AT-trackc |
|---|---|---|---|---|
| AT10 | 9.5/0% | 3.0/82% | 3.0/26.8% | 3.4 |
| AT20 | 12.9/0% | 3.0/82% | 3.0/26.8% | 6.8 |
| AT30 | 16.3/0% | 3.0/82% | 3.0/26.8% | 10.2 |
| OV | 13.6/0% | 3.0/82% | 2.4/62.4% | 8.1 |
| CO | 16.3/0% | 3.0/82% | 3.0/26.8% | 10.2 |
| GC | 9.5/0% | N/A | N/A | N/A |
N/A = Not applicable (GC-tracks do not specifically bind PIC leading to uncertainty in the placement of the initial or final PIC monomers). → = FRET step.
Estimated FRET efficiency based on using PIC QY measured in DNA from ref 18.
PIC homo-FRET through AT-track discussed in the Results and Discussion section.
Based on reported trans-helical exciton transfer, as well as molecular dynamics simulations introducing non-AT bp spacers, we speculate J-bits may be able to retain their excitonic coupling under these conditions.20,21 Thus, OV and CO are novel scaffolds intended to probe the boundaries of directionally nonlinear J-bit to J-bit transfer, a potential requisite for the synthesis of higher-order light harvesting and energy-transfer devices. In addition to the above structures, a fourth scaffold was designed (referred to as the GC scaffold) as a negative control identical in sequence to AT10, except the AT-track was replaced by an alternating poly(dG)−poly(dC) sequence (GC-track). The GC sequence is known to disrupt PIC intercalation and, in theory, should prevent J-bit formation (Figure 1B,C).20,21
AT-Track’s Ability to Template J-bits.
Assembly of all of the DX-tile structures was analyzed via 10% PAGE after thermal annealing (Figure S1). From examining band intensities and lack of other bands, we estimate that all of the structures had ∼80−90% formation efficiency, and these were used as is for PIC studies without further purification. Initial J-bit formation of PIC dye in the DNA was confirmed via steady-state absorbance (Figure S8) and CD spectrometry (Figure S9). PIC dyes form DNA-based aggregates in AT-tracks when in sufficient excess concentration. Dissociation constants of PIC to AT double strand (ds) DNA have been reported ranging from 25 to 60 μM,20 which means that in our system an estimated 42−65% of AT bps should have at least one PIC bound to them, depending on which dissociation constant we assume as being viable. Our experimental range, which is show in the titration experiments discussed below, is in line with the values found in the previous work of 40− 73%.20 As mentioned, there is nonspecific PIC binding to other bps, which means that these are likely upper bounds for the values, and as such, the discussed values are used merely as estimates. In simple DNA duplexes, J-bits can be identified relative to the PIC monomer by the emergence of a sharp red-shifted absorption band located at approximately ∼550 nm in the absorbance profile.19–21,38 In DX-tiles, the rigidity constrains the J-bits resulting in a broad shoulder at 550 nm as opposed to a sharp peak.20 A corresponding decrease in monomer absorption has also been previously observed and attributed to PIC dimerization upon interaction with the DNA scaffold.38,41 Similarly, J-bits can be identified in CD spectra by an induced positive Cotton effect, where the CD maximum signal appears at ∼550 nm overlapping the J-bit steady-state absorption band.19,20 These spectral features become more exaggerated with the extension of the AT-track length. Therefore, in preliminary characterization, we utilized the AT30 scaffold for its high signal-to-noise relationship. We do note that due to the multistate nature of the PIC dye it is extremely difficult to extract the explicit or relative amount of each PIC binding species (free monomer, DNA bound monomer, or J-bit) from absorbance spectra alone. The low quantum yield of monomer PIC means that these populations should contribute little background fluorescence to the resulting measurements. However, it is possible for monomer species to engage in exciton transfer without J-bit formation, which is a challenge further addressed in the sections below.
To determine a preferential concentration ratio of DX-tile to PIC dye, CD measurements were made spanning a range of concentrations. During each of these measurements, the DX-tile concentration was kept constant at 400 nM. For reference, this DX-tile concentration was lower compared to reference literature19–21 in hopes of mitigating potential inner filtering effects during fluorescence measurements. Upon measurement, no CD signal was observed with 400 nM DNA and 20 μM dye (50×). Upon increasing PIC concentration, we observe the appearance of a positive Cotton effect with a peak maximum initially at ∼550 nm, overlapping the J-bit absorption band (Figure 2A). When increasing the dye concentration from 80 μM (200×) to 100 μM (250×), a slight decrease in peak intensity was observed. Using the 25 μM value for the AT dsDNA-PIC dissociation constant, we observe that J-bit signals appear once ∼50% binding site saturation is reached; furthermore increases from 200× to 250× would only increase binding from a predicted 74−78%. To assay J-bit thermo-stability, a sample of the AT30 scaffold at 400 nM with 100 μM excess dye was prepared at 10 °C. An initial CD spectrum was captured at 10 °C, and then the temperature was increased to 60 °C while measuring spectra at every 5 °C change (Figure 2B). Upon completion of the temperature cycle, we observe a linear decrease in the CD peak intensity as the temperature increases. Compared to GC bps, AT bps have significantly lower thermos stability as a result of nonideal base stacking and decreased hydrogen bonding.42 Though it has been postulated that J-bit formation increases the melting temperature of DNA,21 it is possible that the reason behind thermal destabilization of J-bits may be twofold. On the one hand, the increased temperature destabilizes J-bits through disruption of intermolecular forces. While on the other hand, DNA regions with high AT content begin unzipping prior to full denaturation of the entire DX-tile. However, in melting temperature curves ranging from 10 to 80 °C, we observe a single sigmoidal curve with a Tm around 60 °C for the AT30 scaffold (Figure S10). This suggests that J-bit destabilization occurs primarily through intermolecular force disruption rather than DNA denaturing.
Figure 2.
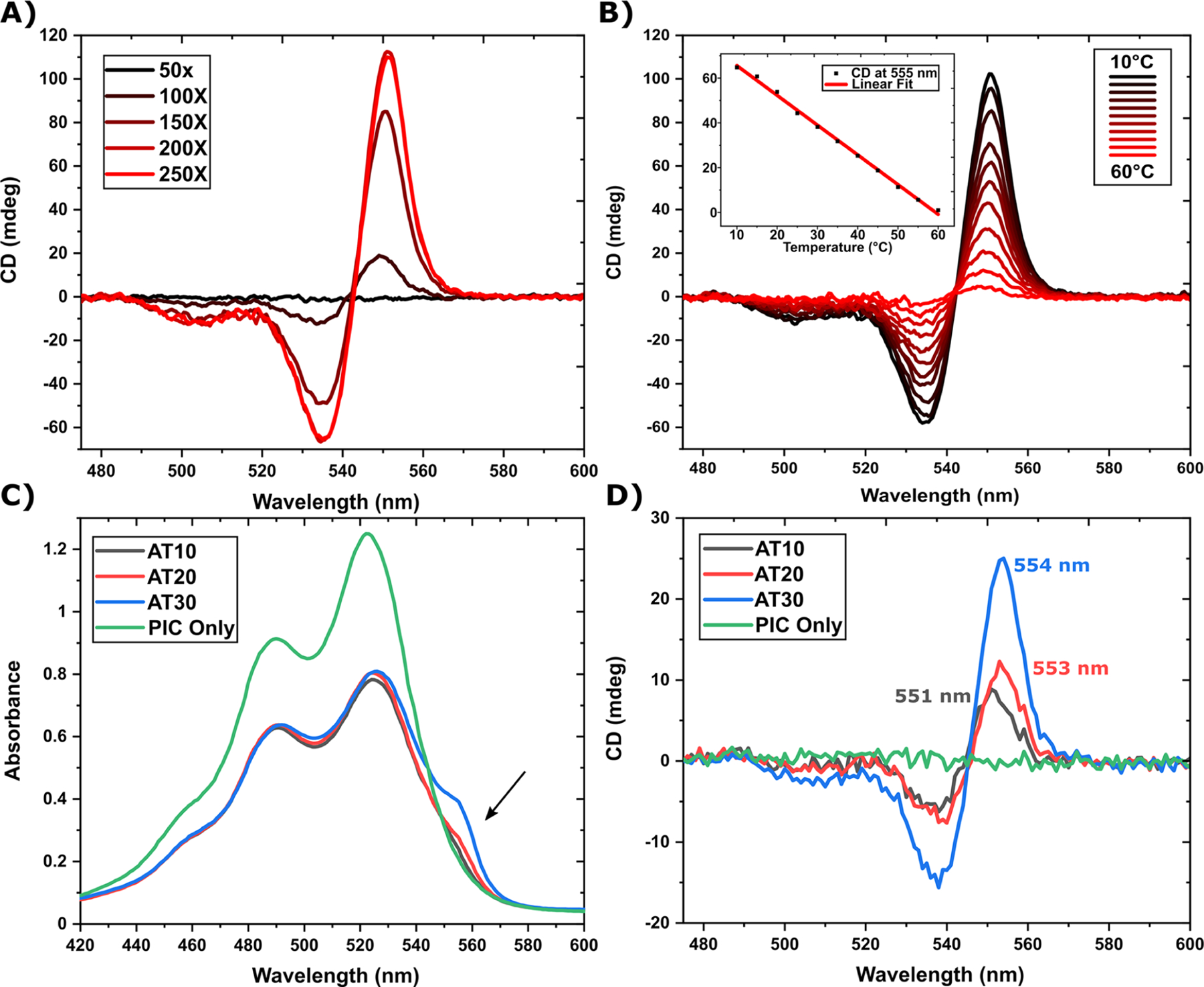
CD and absorbance measurements of J-bit in contiguous AT-tracks. (A) CD spectra of 400 nM AT30 DNA in the presence of PIC dye at various concentration ratios. Fresh samples were made at each Dye/DNA ratio to avoid DNA dilution by PIC titration. (B) CD spectra of the AT30 scaffold at 400 nM in the presence of 100 μM PIC dye. Each spectrum follows increasing temperature in 5 °C increments. The inset graph displays a scatterplot of the 555 nm CD intensity as a function of temperature with a corresponding linear regression. (C) Linear absorbance spectra measured in parallel. The arrow indicates that the ∼550 nm J-shoulder red-shifted from the monomer 0−0 transition peak at 523 nm. (D) CD spectra comparing contiguous AT-track scaffolds with a PIC only control. Each sample DNA concentration is at 400 nM, while the PIC dye is 130× excess (52 μM).
J-bit Formation on Contiguous AT-Tracks.
Based on the results of the above experimental characterization, J-bits were formed at 18 °C and a concentration ratio of 400 nM DNA to 52 μM PIC dye (130×). Once combined, PIC−DNA reaction mixtures immediately exhibit a clear red-shifted absorption shoulder relative to the monomer 0−0 absorption band at 523 nm, see Figure 2C. This shoulder increases in correspondence with increasing AT-track length. In addition, the shoulder’s peak absorption in AT20 and AT30 is red-shifted relative to AT10. The shorter AT-track has a peak shoulder absorption at 551 nm, while AT20 and AT30 have absorption max at 555 nm (see Figure 3C), suggesting an increase in exciton delocalization as a function of track length rather than simply more PIC chromophores binding to scaffolds.19 In the CD spectra, we see a characteristic induced positive Cotton effect of increasing intensity as track length increases (Figure 2D). The wavelength of the positive peak maximum can be seen at 551, 553, and 554 nm for AT10, AT20, and AT30 respectively, continuing the red shift trend and further supporting the aforementioned speculation of a potential increase in exciton delocalization. DNA negative controls lack any J-shoulder formation (Figure S8) or deviation from CD baseline (Figure S9), indicating that no solution-based J-aggregates form in this operative concentration regime. These results are interesting in comparison to other investigations, which report a single stable 550 nm J-aggregate peaks seen in AT-tracks with lengths greater than 4 bps.19,21
Figure 3.
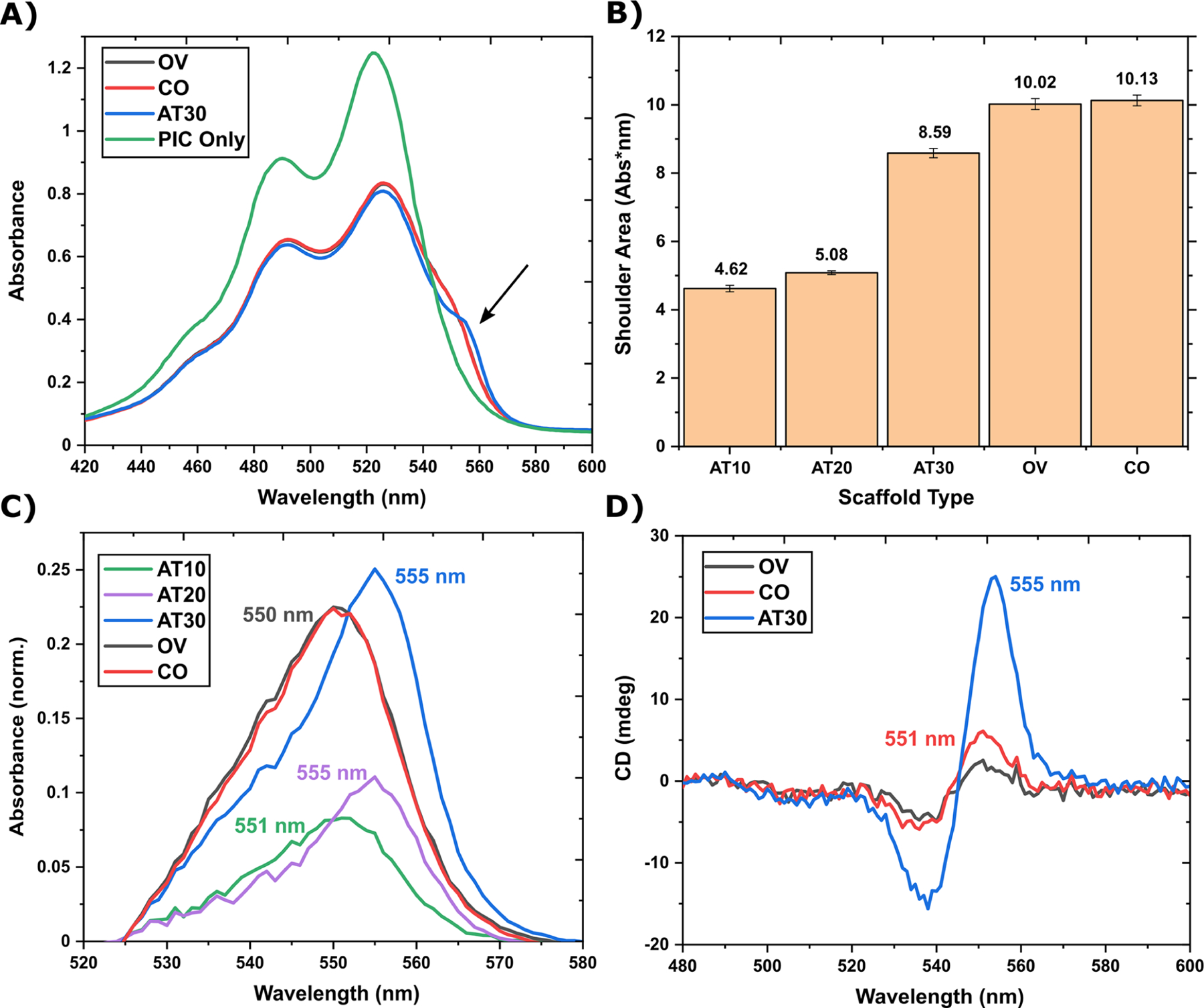
J-bit formation in noncontiguous AT-tracks. (A) Absorbance spectra comparing the AT30 (blue) scaffold to OV (gray) and CO (red) scaffolds (OV overlaps with CO). The arrow indicates J-shoulder location. (B) Quantification of the area under the J-shoulder. J-shoulder area is not statistically different between OV and CO structures (i.e., within 1 standard deviation). (C) Absorbance spectra comparing AT10 (gray), AT20 (red), AT30 (blue), OV (green), and CO (purple) J-shoulders. The shown spectra were the result of average spectra normalized to the 0−0 transition (∼523 nm), and then the GC spectra were subtracted as a baseline. Labels indicate λmax. (D) CD spectra comparing the same data set in panel (A). Labels indicate λmax. In all spectra, DNA is 400 nM and the PIC dye is 52 μM.
J-bit Formation in Noncontiguous Structures.
The noncontiguous OV and CO scaffolds were prepared and measured following the same procedures as the previous contiguous samples. Both OV and CO exhibit a nearly overlapping J-shoulder in the linear absorbance spectra (Figure 3A). This is logical as AT30, OV, and CO all have the same relative concentrations of AT bp’s. Closer analysis, however, reveals some interesting differences between the three shoulders. First, the AT30 shoulder has a slightly higher maximum amplitude than OV and CO shoulders, but a smaller area under the curve indicates that there might be more overall PIC monomers binding to the noncontiguous DNA (Figure 3B). Second, the AT30 J-shoulder has a ca. 5 nm red-shifted peak relative to OV and CO, suggesting a lower energy exciton and increased delocalization length (Figure 3C). Third, the full width at half-maximum (FWHM) was calculated to be 20 nm for AT30 and 21 nm for OV and CO scaffolds. Peak broadening has been speculated to result from a static disorder in aggregate formation, limiting the coherence length and subsequently exciton delocalization. The spectral line width of the J-band exhibits an N−1/2 dependence, where N is correlated with the coherence length of the aggregate.39 Thus, a broader shoulder directly implies a shorter exciton delocalization length. This broadening is consistent with results incorporating GC-tracks and/or alternating poly(AT)−poly(TA) spacers as well.19−21 The differences between AT30 and the two noncontiguous scaffolds are also apparent in their respective CD spectra (Figure 3D). Although OV and CO scaffolds both exhibit an induced positive Cotton effect, positive peaks are much weaker than the AT30’s. These results support the hypothesis that inhomogeneous coupling or static disorder is limiting the exciton coherence length in OV and CO scaffolds in comparison to longer AT-tracks such as in AT30.
Energy-Transfer Predictions.
Using Förster theory, we estimated the probability of exciton transfer between each dye species in each of the structures, see the SI for more details on predicted FRET calculations. The base FRET efficiencies from D405 → A647, D405 → PIC, PIC → PIC, and PIC → A647 were estimated for each scaffold. The calculated spectral overlap J-integral and R0 values utilized in these calculations are available in Table 1. Dye species separation distances and predicted FRET efficiency are available in Table 2. For each scaffold, we expect no FRET transfer directly from D405 → A647 as their separation distance is significantly greater than the correspond in the AT-track, which is within the calculated Förster distance leading to a predicted energy transfer of 82%.
PIC’s relatively low and environmentally sensitive QY causes difficulties in predicting PIC’s ability to act as an energy D. The monomeric PIC chromophore has an extremely low QY reported to be 0.012 ± 0.002%.20 As a result, transfer from PIC to A647 is dramatically reduced even with suitable spectral overlap (Table 1). Assuming that transfer is strictly from the last PIC molecule in the AT-track, acting as if it is monomeric, PIC → A647 R0 is approximately 1.6 nm. Under these conditions, the energy transfer from the terminal bp in the AT-track to A647 is predicted to be 2.4% for all scaffolds except the OV scaffold, which has a decreased dye separation with a subsequent ∼9.9% transfer, expected. However, PIC has at least an order of magnitude increase in QY simply upon the addition of nonspecific DX-tile DNA with reports of up to 0.18 ± 0.03%20 in DX-tiles with AT-track sequences. This increase in QY is speculated to be a result of DNA interactions constraining the available excited-state relaxation pathway by restricting the free rotation of quinolinic rings.43 Assuming this elevated QY, R0 increases to 2.6 nm and the predicted energy transfer increases to 62.4% in the OV scaffold and 26.8% transfer in all other scaffolds. These values however assume no energy loss from transfer through D and relay. Alternate factors, such as energy traps or misdirected pathways, may result in the exciton decaying before it reaches A647.
The final FRET step that must be addressed is the potential PIC → PIC homo-FRET through the AT-track. Again, we consider PIC in either the monomeric state or in the DNA-based state. There is the additional assumption of the separation distance between the PIC monomers. As previously mentioned, molecular docking simulations estimate PIC to pack into the DNA minor groove with a density of 8−12 molecules per turn.20 Since each helical turn of DNA is approximately 10.5 bps in length, we assume 10 equally spaced PIC molecules per turn of DNA. Using this, we can set a lower bound on the number of transfer steps through a given AT-track and a potential upper bound on efficiency. If we assume that the exciton is transferred through each intercalated PIC monomer in a strictly linear direction, the minimum number of transfer steps would be equal to n − 1, where n is the number of bases in the AT-track. In reality, this will not be the case because the nature of homo-FRET eliminates the directionality associated with the natural energy gradient of hetero-FRET. However, assuming linear transfer and applying Förster theory, we can calculate the monomeric PIC → PIC R0 to be approximately 0.7 nm. Assuming 10 equally spaced PIC molecules per turn, the PIC → PIC separation distance would be 0.34 nm, thus leading to a predicted transfer efficiency of 98% per transfer step. Assuming optimized directional transfer along the dye cascade and that the other FRET assumptions held, ∼88% of the input energy would be able to propagate through the full length of a 10 bp AT-track requiring nine transfer steps. If instead calculations are made with an enhanced QY of DNA-based PIC, ∼99% of the energy is predicted to arrive at the terminal end of the 10 bp AT-track. Once the nondirectionality of homo-FRET11 is taken into account (using the PIC lifetimes reported in the literature of 35 and 60 ps, for monomeric and AT J-bit PIC, respectively),20 the predicted efficiency falls to 31 and 63%, respectively. This theoretical homo-FRET relay capability, due to the tight packing of PIC molecules, likely explains the observed transfer in the GC control structure (see below for details).
Experimentally Observed Energy Transfer.
The first method we used to approximate energy transfer was to observe the D405 quenching. As previously mentioned, we concluded direct D405 → A647 transfer to be negligible, so any quenching is assumed to be D405 to PIC transfer (see Figure S11). Given the complementary spectral overlap and minimized D−A distance, we expect near 100% quenching, and this was indeed confirmed experimentally. In every sample measured, we observed approximately 96−98% D405 quenching (EQ) (Figure S11). However, this result must be understood with the caveat that EQ is known to be environmentally sensitive to reabsorption in the presence of an excess bulk dye that is present throughout our experiments and evident by the near-complete quenching observed in the GC control scaffold. To our knowledge, this same phenomenon is also reported in all other DNA-based PIC templates.19–21 As an alternative measurement strategy, others have assayed EQ using photo-luminescence lifetimes. However, reported donor to acceptor transfer efficacies using this method similarly approach 100% in both AT and non-AT controls.19,20
Since there is near-complete quenching of D405 in each sample, the J-bit relay capacity could be characterized through sensitization of the terminal A647, see Table 3, Figures 4, and S12. Due to the previously mentioned inherently low photoluminescence of PIC dye, measuring PIC quenching or sensitization is difficult and subject to high contribution from noise. Furthermore, since the QY changes dynamically based on the J-bit coherence length, which is not constant, it is difficult to derive a reliable energy-transfer metric using PIC fluorescence. Therefore, similar to previous approaches,18–20 we utilized relative enhancement of A (As) upon inclusion of D405 as compared to direct excitation of the J-bit and A647 components at the same wavelength. Considering contiguous scaffolds, we expected to see the highest enhancement in the scaffold with the shortest distance path from D405 to A647, i.e., AT10. The As and Eee values would be predicted to decrease as the length of the AT-track is extended from 10 to 20 and then 30 bps in length, which also matched what was seen experimentally (Figure 4A−C). We observe As values of ∼41, ∼20, and ∼11% (with uncertainties of ∼1%) for AT10, AT20, and AT30 scaffolds, respectively. Eee calculated for AT10, AT20, and AT30 scaffolds displayed ∼2, ∼0.6, and ∼0.4% transfer efficiencies.
Table 3.
Estimated Energy Transfer from EQ and Asa
| DX-tile structure | E Q | A s |
|---|---|---|
| AT10 | 97 ± 1% | 41.5 ± 1.3% |
| AT20 | 98 ± 1% | 19.6 ± 0.7% |
| AT30 | 97 ± 1% | 11.4 ± 0.6% |
| OV | 96 ± 0% | N.D. |
| CO | 96 ± 1% | N.D. |
| GC | 97 ± 2% | 34.1 ± 0.5% |
N.D.—not detectable. EQ from D405 quenching and As from A647 sensitization as described in the experiments. Spectra for the GC system are shown in Figure S12.
Figure 4.
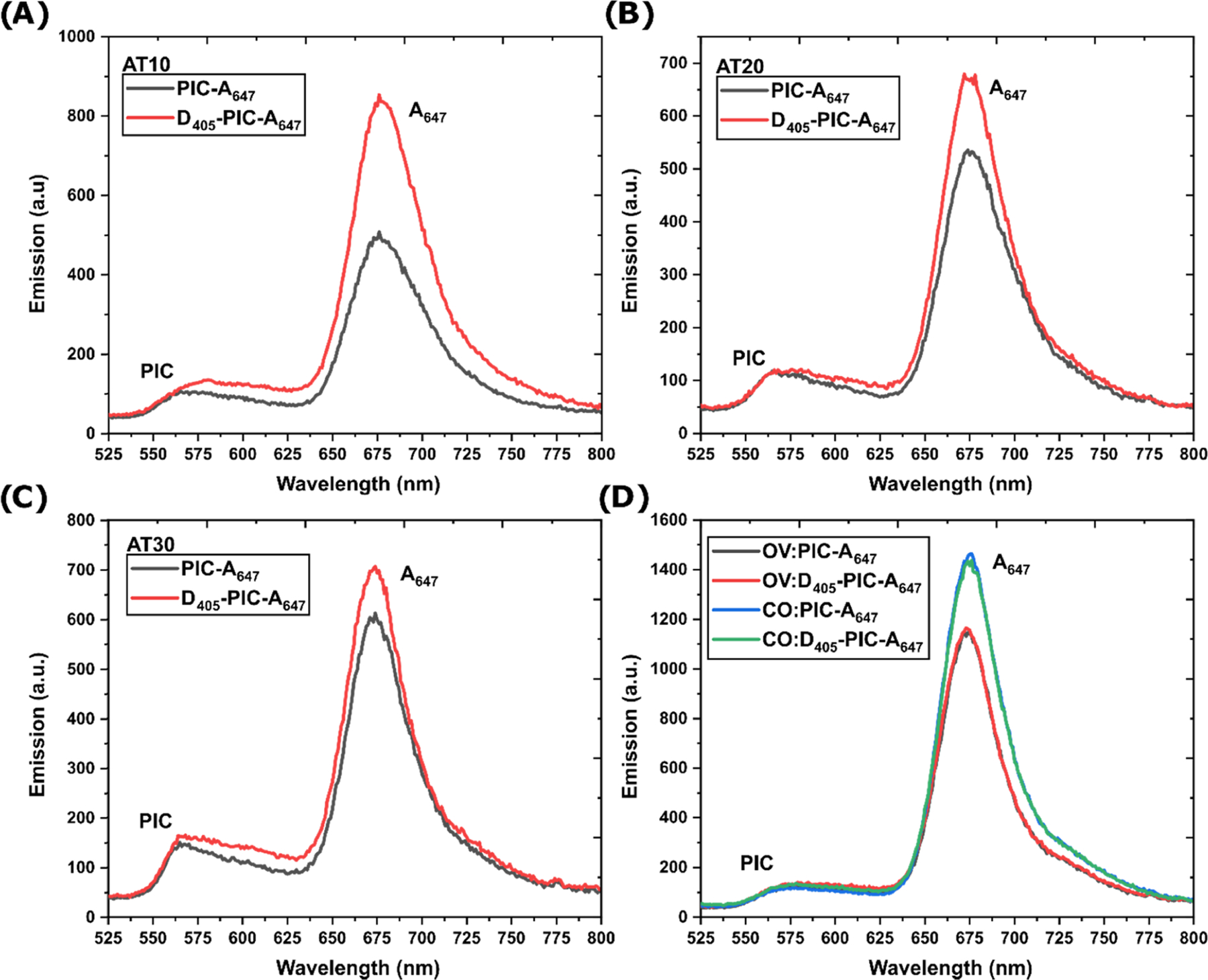
Energy transfer through the PIC relay and fluorescence emission. Fluorescence emission spectra comparing (A) AT10, (B) AT20, (C) AT30, and (D) OV and CO scaffolds with and without the AF405 donor present. Each sample contains 400 nM DNA and 52 μM. PIC. Full spectra including the D405 portion are in Figure S13.
Using the same experimental approach, we determined that the energy transfer in noncontiguous OV and CO scaffolds was undetectable (Figure 4D, Table 3). As discussed above, the structures were capable of forming the requisite J-bit structures as designed, and PIC was capable of acting as an antennae to A647, evident by a 461 ± 23 and 681 ± 67% increase in A647 emission (see Figures S12−S14) upon addition of the PIC dye compared to direct A647 excitation for OV and CO structures, respectively. Yet the addition of D405 did not result in appreciable As values. The structures were designed to minimize J-bit separation, with OV and CO scaffolds having less than 2 nm distance between AT-tracks, yet the result suggests that J-bit units are inherently inefficient (homo)FRET relays. Further dialog on physicochemical reasons and implications of this result are provided in subsequent paragraphs. If we consider a 2 nm interhelical distance in terms of bps, this would be equivalent to a distance manifest by a 6 bp GC spacer, which could further offset the J-bit coupling. It should further be noted that the GC control, which is equivalent to the shortest AT10 scaffold, had a significant amount of observed A647 sensitization. A result of the monomeric PIC interacting with nonspecific DNA sites has been previously seen in nonpoly(AT) sequences which template the monomer and demonstrate energy relay capabilities.19,21 Seen consistently in literature and confirmed here in our work, for the same overall length, J-bit structures integrated into AT-tracks result in higher As values presumably through their coherently coupled properties.
PIC aggregates have previously shown promise as 1-D photonic nanostructures both within free-standing fibers as well as when templated within nanoconstructs.44–47 The current report expands from previous studies examining self-assembled PIC J-bit aggregates that form within the AT-tracks of DNA sequences.44–47 As such, we put our results into that context and compare our findings to what has been observed in these assemblies previously and then discuss how this could influence future design criteria and potentially improve these devices.
The first point of comparison is on the formation efficiency of the self-assembled J-bits within AT-tracks. In Boulais et al., it was shown that J-bit features appear in AT-tracks as small as 8 bps long,20 with follow-up work in Banal et al. demonstrating that, although longer AT-tracks do increase the average PIC J-bit length, i.e., the coherence length, this is in no way linear.19 In fact, a 3-fold increase in AT-track length, from 8 to 24 bps, resulted in only a ca. 33% increase in coherence length. Mandal et al. looked at the coherence length when the AT-track was interrupted by a CG bp and observed a decrease of ∼50% when going from a 24 bp AT-track to 11AT:2CG:11AT sequence.21 We provide CD-based evidence that J-bits form in AT-tracks when ≥50% of AT sites are binding a PIC molecule. Due to the large dissociation constant (≥25 μM), this requires a large excess of PIC to be present in the solution as seen in all experiments. Calculations based on the values reported in the literature as well as our own show that all of the efforts realize their studies with values approaching 50−60% PIC loading of potential AT binding sites.19–21,44–47 Expanding on the above, we compare the J-bit formation and its respective spectro-scopic signals when the AT-track is contiguous as compared to splitting into distinct bits on alternating DNA helixes. We find that splitting a 30 bp AT-track into three 10 bp tracks results in shorter coherence length J-bits, as evidenced by a marked decrease in CD signals as well as a blue shift and broadening of the absorbance shoulder at 550 nm. Of particular note is the observation that the integrated intensity of the absorbance shoulder for AT30 J-bits was less than the two non-contiguous structures. The latter suggests that validating the presence of correct J-bit aggregate formation should not depend solely on absorbance spectra. We also confirm that J-bits are quite temperature sensitive, with a linear decrease from 10 to 60 °C, until a complete loss of CD signal at the latter temperature (Figure 2B).
The capability of J-bits to function as enhanced energy-transfer systems was also examined here in comparison to previous results. Here, we distinguish the work of Mandal21 and Banal19 that focused on simple dsDNA from our work and that of Boulais,20 which specifically incorporated a DX-tile-based DNA structure. Banal showed enhanced transfer from an ATTO390 D to an AF647 A if the relay was a continuous AT-track of 8−16 bps in length as compared to a control non-J-bit forming dsDNA, which still templated monomeric PIC.19 If the AT-tract was extended to 24 bps, the enhancement over the control was null. For the shorter 8 bps relay (total wire length of ∼9 nm), they report a ∼35% As value, while for the longer 16 bps relay (wire length of ∼11.5 nm), the As value was ∼15%. These values are in line with 41 and 19% values we report for similar length systems. Mandal utilized a QD D with 545 nm emission and the same AF647 as the final A.21 Due to the QD inherent brightness, they were able to manifest enhanced transfer in J-bit forming AT-tracks with lengths of 24 bps (As = 40%). Of greater relevance to the current work, the CG interrupted AT-track also reported an As value of 15%. The values in their case are greater than what we report, likely due to the choice of a bright QD D as the starting point of the transfer system. Similar to others, when they attempt to extend their system further with a 48 bps AT-tracks, they find null transfer.21
In Boulais et al., a DX-tile having two parallel 16 bps AT-tracks, with a discontinuity caused by the crossover strand, is used as a relay between ATTO390 dye D on one helix and AF647 as an A on the other DNA helix.20 The design was intended to force the transfer to occur from one J-bit to the J-bit on the parallel helix. However, the authors do not report particular As nor Eee values; they provide fluorescence lifetime spectroscopic data that demonstrate transfer through the relay system as well as enhanced FRET from the PIC to AF647 when the DX-tile is assembled with AT-tracks as compared to CG-tracks, which are only meant to bind monomeric PIC. Our system was similarly designed to have J-bits on the two DNA helices of the DX-tile; however, our J-bits were fully discontinuous, and we thus did not determine any transfer through the whole system. We speculate that the observed transfer in the Boulais system with the relatively small As values may potentially be accounted for by the flexibility of D and A dyes due to the extended linkers that serve to attach them to DNA (∼1 nm length).20 This linker flexibility, in turn, would allow A and D dyes to interact directly with the J-bit on the opposite helix. Furthermore, as demonstrated by Mandal et al., J-bits formed in partially interrupted AT-tracks, as the Boulais design has, are capable of acting as enhanced transfer relays.21 The general concordance seen between the results discussed above suggests that the PIC−DNA system based on AT aggregating sites is tractable and reproducible.
Altogether, the results suggest generalized design criteria that should be considered when working with this experimental system. Though PIC has the particular advantage of forming aggregates in a site-specific manner in AT-tracks, as compared to other dyes such as the K21 dye reported by Zhou et al., which are nonspecific binders,48 the overall binding is not efficient, thereby requiring large excesses of PIC dye. Furthermore, even within J-aggregates, the PIC coherent length (two to three PICs per coupled aggregates)19 and fluorescent QY (0.18 highest reported value)20 are extremely limited. A simplified model of the system is quite helpful in illustrating some of these limitations. This model assumes that AT-tracks that have J-bits do not have monomeric PIC absorbed within the tract and that coupled aggregates are of a consistent length and evenly spaced throughout the AT-track. We can provide a range of separations for the coupled aggregates within the AT-tracks as being between 3.4 and 10 Å apart (the 8 bps AT-track reported by Banal19 and the 48 bps AT-track reported by Mandal,21 respectively); this includes our own AT systems with 4.7, 5.2, and 6.6 Å separation for AT10, AT20, and AT30, respectively. We further note that enhanced transfer is quantifiable when the separation is <7.5 Å, which roughly aligns with a 2 bp separation between coupled aggregates. This estimate aligns quite well with the computational determinations realized in Boulais et al. that similarly reported separations of 2 bps retained substantial excitonic coupling.20 There is one more component that plays a role in the transfer efficiency; at greater lengths, the number of coupled aggregates necessarily increased (estimates go from 3 to 10 aggregates in each AT-track, again the 8 bps AT-track reported by Banal19 and the 48 bps AT-track reported by Mandal,21 respectively). As demonstrated in other DNA-based photonic wire systems, the random walk excitonic hopping present in homo-FRET systems results in decreased transfer as the number of individual components increases.11,35,49 There-fore, longer AT-tracks, which a priori would seem to be the most interesting and useful, have both greater distance between coupled aggregates as well as a greater number of aggregates two components, which cumulatively greatly diminish the expected transfer efficiency.
Within our OV and CO DX-tile designs, we aimed to clearly separate the J-bit systems but maintain their capability to function as relays. Though we designed systems where the minimal distance of the J-bit was <20 Å, this value appears to surpass the range of functional homo-FRET transfer. Not previously discussed, but perhaps more limiting to the design is the dipole orientation component (κ2).50,51 In a simple linear dsDNA system, the dipoles of any D−A pair within the J-bits are almost optimally aligned, the κ2 component of the FRET equation trending toward 4.0 and therefore maximal transfer, while in our parallel off-helix transfer systems, κ2 will be distributed around 1.0 with far less efficient transfer expected. This value is still higher than the random distribution κ2 = 2/3 value most commonly used in FRET calculations5–7,12,13,50–54 but still considerably inferior to the interaction on the same DNA helix. Given the sheer excess of PIC monomer present, even if it does not bind and form the J-bit, it still can clearly engage in some level of association with the DNA regardless of the sequence and facilitate energy transfer. The amount of this monomer present in these samples may also serve to cumulatively overcome its inherently low QY. This confirms that the system is not binary between on and off states and makes parsing the results, confirming what is due to J-bit and what is due to monomer nonspecificity, and then interpreting the function of the overall system that much harder.
In conclusion, we find that the self-assembly PIC:AT-track systems present interesting and motivating bottom-up photonic properties that are somewhat consistent in the hands of different groups (not a minor detail given the inherent delicacy and complexity of achieving assembly) and that are relevant for doing proof-of-concept experiments. Yet the relatively low binding constant (≥25 μM) that requires considerable excess PIC, the limited coherence length (∼two PIC moieties), and unexceptional QY (maximum estimated value of 0.18)20 makes the application space for the systems in its current form quite limited. As is, templated J-bits are limited to continuous 1-D wires of about 30 bps in length with minimal ∼1% transfer efficiency, values of merit that are surpassed by other pre-existing purely classical FRET systems.8,11 For such materials to reach fruition beyond just an interesting experimental system, the directed self-assembly of PIC:AT-tracks into J-bit aggregates will need to be tailored with improved DNA binding characteristics and photophysical properties. The large library of as-yet untested organic dyes, DNA binding dyes, and cyanine-based fluorophores along with their analogues suggests that there are many more dyes that can potentially be harnessed to accomplish this.55–58 Complementary approaches that combine both DNA binding sequence design and synthesis of new dyes to match that binding motif may offer the most promise in this regard.20
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge the George Mason University Department of Bioengineering, the Office of Naval Research, the U.S. Naval Research Laboratory (NRL), and the NRL Nanosciences Institute. D.M. was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under Award Number K99EB030013. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Matthew Chiriboga, Center for Bio/Molecular Science & Engineering Code 6900, U.S. Naval Research Laboratory, Washington, District of Columbia 20375, United States; Volgenau School of Engineering, Department of Bioengineering, Institute for Advanced Biomedical Research George Mason University, Manassas, Virginia 22030, United States.
Sebastian A. Diaz, Center for Bio/Molecular Science & Engineering Code 6900, U.S. Naval Research Laboratory, Washington, District of Columbia 20375, United States.
Divita Mathur, Center for Bio/Molecular Science & Engineering Code 6900, U.S. Naval Research Laboratory, Washington, District of Columbia 20375, United States; College of Science, George Mason University, Fairfax, Virginia 22030, United States.
David A. Hastman, Center for Bio/ Molecular Science & Engineering Code 6900, U.S. Naval Research Laboratory, Washington, District of Columbia 20375, United States; A. James Clark School of Engineering, Fischell Department of Bioengineering, University of Maryland College Park, College Park, Maryland 20742, United States.
Joseph S. Melinger, Electronics Science and Technology Division Code 6800, U.S. Naval Research Laboratory, Washington, District of Columbia 20375, United States.
Remi Veneziano, Volgenau School of Engineering, Department of Bioengineering, Institute for Advanced Biomedical Research George Mason University, Manassas, Virginia 22030, United States.
Igor L. Medintz, Center for Bio/Molecular Science & Engineering Code 6900, U.S. Naval Research Laboratory, Washington, District of Columbia 20375, United States.
REFERENCES
- 1.Engel GS; Calhoun TR; Read EL; Ahn T-K; Mancǎl T; Cheng Y-C; Blankenship RE; Fleming GR Evidence for Wavelike Energy Transfer Through Quantum Coherence in Photo-synthetic Systems. Nature 2007, 446, 782–786. [DOI] [PubMed] [Google Scholar]
- 2.Collini E; Wong CY; Wilk KE; Curmi PM; Brumer P; Scholes GD Coherently Wired Light-Harvesting in Photosynthetic Marine Algae at Ambient Temperature. Nature 2010, 463, 644–647. [DOI] [PubMed] [Google Scholar]
- 3.Panitchayangkoon G; Hayes D; Fransted KA; Caram JR; Harel E; Wen J; Blankenship RE; Engel GS Long-Lived Quantum Coherence in Photosynthetic Complexes at Physiological Temperature. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 12766–12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Förster T Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys 1948, 437, 55–75. [Google Scholar]
- 5.Lakowicz JR Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Boston, 2013. [Google Scholar]
- 6.Clegg RM Laboratory Techniques in Biochemistry and Molecular Biology; Gadella TWJ, Ed.; Elsevier, 2009; Vol. 33, pp 1–57. [Google Scholar]
- 7.Van der Meer BW FRET-Förster Resonance Energy Transfer: From Theory to Applications; Medintz IL; Hildebrandt N, Eds.; Wiley VCH: Weinheim, Germany, 2013; pp 23–62. [Google Scholar]
- 8.Hannestad JK; Sandin P; Albinsson B Self-Assembled DNA Photonic Wire for Long-Range Energy Transfer. J. Am. Chem. Soc 2008, 130, 15889–15895. [DOI] [PubMed] [Google Scholar]
- 9.Hannestad JK; Gerrard SR; Brown T; Albinsson B Self-Assembled DNA-Based Fluorescence Waveguide With Selectable Output. Small 2011, 7, 3178–3185. [DOI] [PubMed] [Google Scholar]
- 10.Díaz SA; Buckhout-White S; Ancona MG; Spillmann CM; Goldman ER; Melinger JS; Medintz IL Extending DNA-Based Molecular Photonic Wires with Homogeneous Förster Resonance Energy Transfer. Adv. Opt. Mater 2016, 4, 399–412. [Google Scholar]
- 11.Klein WP; Rolczynski BS; Oliver SM; Zadegan R; Buckhout-White S; Ancona MG; Cunningham PD; Melinger JS; Vora PM; Kuang W; et al. DNA Origami Chromophore Scaffold Exploiting HomoFRET Energy Transport to Create Molecular Photonic Wires. ACS Appl. Nano Mater 2020, 3, 3323–3336. [Google Scholar]
- 12.Dale RE; Eisinger J; Blumberg W The Orientational Freedom of Molecular Probes. The Orientation Factor in Intramolecular Energy Transfer. Biophys. J 1979, 26, 161–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Algar WR; Hildebrandt N; Vogel SS; Medintz IL FRET as a Biomolecular Research Tool Understanding Its Potential While Avoiding Pitfalls. Nat. Methods 2019, 16, 815–829. [DOI] [PubMed] [Google Scholar]
- 14.Rolczynski BS; Yeh S-H; Navotnaya P; Lloyd LT; Ginzburg AR; Zheng H; Allodi MA; Otto JP; Ashraf K; Gardiner AT; et al. Time-Domain Line-Shape Analysis from 2D Spectroscopy to Precisely Determine Hamiltonian Parameters for a Photosynthetic Complex. J. Phys. Chem. B 2021, 125, 2812–2820. [DOI] [PubMed] [Google Scholar]
- 15.Seifert JL; Connor RE; Kushon SA; Wang M; Armitage BA Spontaneous Assembly of Helical Cyanine Dye Aggregates on DNA Nanotemplates. J. Am. Chem. Soc 1999, 121, 2987–2995. [Google Scholar]
- 16.Wang M; Silva GL; Armitage BA DNA-Templated Formation of a Helical Cyanine Dye J-Aggregate. J. Am. Chem. Soc 2000, 122, 9977–9986. [Google Scholar]
- 17.Garoff RA; Litzinger EA; Connor RE; Fishman I; Armitage BA Helical Aggregation of Cyanine Dyes on DNA Templates: Effect of Dye Structure on Formation of Homo-and Heteroaggregates. Langmuir 2002, 18, 6330–6337. [Google Scholar]
- 18.Hannah KC; Armitage BA DNA-Templated Assembly of Helical Cyanine Dye Aggregates: A Supramolecular Chain Polymerization. Acc. Chem. Res 2004, 37, 845–853. [DOI] [PubMed] [Google Scholar]
- 19.Banal JL; Kondo T; Veneziano R; Bathe M; Schlau-Cohen GS Photophysics of J-Aggregate-Mediated Energy Transfer On DNA. J. Phys. Chem. Lett 2017, 8, 5827–5833. [DOI] [PubMed] [Google Scholar]
- 20.Boulais É; Sawaya NP; Veneziano R; Andreoni A; Banal JL; Kondo T; Mandal S; Lin S; Schlau-Cohen GS; Woodbury NW; et al. M. Programmed Coherent Coupling in a Synthetic DNA-Based Excitonic Circuit. Nat. Mater 2018, 17, 159–166. [DOI] [PubMed] [Google Scholar]
- 21.Mandal S; Zhou X; Lin S; Yan H; Woodbury N Directed Energy Transfer Through DNA-Templated J-Aggregates. Bioconjugate Chem 2019, 30, 1870–1879. [DOI] [PubMed] [Google Scholar]
- 22.Kamalakshan A; Ansilda R; Mandal S Nanotube Template-Directed Formation of Strongly Coupled Dye Aggregates with Tunable Exciton Fluorescence Controlled by Switching between J- and H-Type Electronic Coupling. J. Phys. Chem. B 2021, 125, 7447–7455. [DOI] [PubMed] [Google Scholar]
- 23.Wang J-L; Wang C; Lin W Metal–Organic Frameworks for Light Harvesting and Photocatalysis. ACS Catal 2012, 2, 2630–2640. [Google Scholar]
- 24.Zou Q; Liu K; Abbas M; Yan X Peptide-Modulated Self-Assembly of Chromophores toward Biomimetic Light-Harvesting Nanoarchitectonics. Adv. Mater 2016, 28, 1031–1043. [DOI] [PubMed] [Google Scholar]
- 25.Miller RA; Presley AD; Francis MB Self-Assembling Light-Harvesting Systems From Synthetically Modified Tobacco Mosaic Virus Coat Proteins. J. Am. Chem. Soc 2007, 129, 3104–3109. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y-J; Yang S-H; Hsu C-S Synthesis of Conjugated Polymers for Organic Solar Cell Applications. Chem. Rev 2009, 109, 5868–5923. [DOI] [PubMed] [Google Scholar]
- 27.Stefik M; Guldin S; Vignolini S; Wiesner U; Steiner U Block Copolymer Self-Assembly for Nanophotonics. Chem. Soc. Rev 2015, 44, 5076–5091. [DOI] [PubMed] [Google Scholar]
- 28.Bui H; Díaz SA; Fontana J; Chiriboga M; Veneziano R; Medintz IL Utilizing the Organizational Power of DNA Scaffolds for New Nanophotonic Applications. Adv. Opt. Mater 2019, 7, No. 1900562. [Google Scholar]
- 29.Bricker WP; Banal JL; Stone MB; Bathe M Molecular Model of J-Aggregated Pseudoisocyanine Fibers. J. Chem. Phys 2018, 149, No. 024905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ware WR; Rothman W Relative Fluorescence Quantum Yields Using an Integrating Sphere. The Quantum Yield of 9, 10-Diphenylanthracene in Cyclohexane. Chem. Phys. Lett 1976, 39, 449–453. [Google Scholar]
- 31.Sens R; Drexhage KH Fluorescence Quantum Yield of Oxazine and Carbazine Laser Dyes. J. Lumin 1981, 24–25, 709–712. [Google Scholar]
- 32.A|E - UV-Vis-IR Spectral Software 1.2, Fluortools http://www.fluortools.com/ (accessed August 11, 2021).
- 33.Aissaoui N; Moth-Poulsen K; Käll M; Johansson P; Wilhelmsson LM; Albinsson B FRET Enhancement Close to Gold Nanoparticles Positioned in DNA Origami Constructs. Nanoscale 2017, 9, 673–683. [DOI] [PubMed] [Google Scholar]
- 34.Dutta PK; Lin S; Loskutov A; Levenberg S; Jun D; Saer R; Beatty JT; Liu Y; Yan H; Woodbury NW Reengineering the Optical Absorption Cross-Section of Photosynthetic Reaction Centers. J. Am. Chem. Soc 2014, 136, 4599–4604. [DOI] [PubMed] [Google Scholar]
- 35.Klein WP; Díaz SA; Buckhout-White S; Melinger JS; Cunningham PD; Goldman ER; Ancona MG; Kuang W; Medintz IL Utilizing HomoFRET to Extend DNA-Scaffolded Photonic Networks and Increase Light-Harvesting Capability. Adv. Opt. Mater 2018, 6, No. 1700679. [Google Scholar]
- 36.Jones MR; Seeman NC; Mirkin CA Programmable Materials and the Nature of the DNA Bond. Science 2015, 347, No. 1260901. [DOI] [PubMed] [Google Scholar]
- 37.Li X; Yang X; Qi J; Seeman NC Antiparallel DNA Double Crossover Molecules as Components for Nanoconstruction. J. Am. Chem. Soc 1996, 118, 6131–6140. [Google Scholar]
- 38.Nordén B; Tjerneld F Optical Studies on Complexes Between DNA and Pseudoisocyanine. Biophys. Chem 1976, 6, 31–45. [DOI] [PubMed] [Google Scholar]
- 39.Knapp E Lineshapes of Molecular Aggregates, Exchange Narrowing and Intersite Correlation. Chem. Phys 1984, 85, 73–82. [Google Scholar]
- 40.Mandelkern M; Elias JG; Eden D; Crothers DM The Dimensions of DNA in Solution. J. Mol. Biol 1981, 152, 153–161 [DOI] [PubMed] [Google Scholar]
- 41.Cooper W Multiplet Structure of Aggregated States in 1, 1′- Diethyl-2, 2′-Cyanine Dye. Chem. Phys. Lett 1970, 7, 73–77 [Google Scholar]
- 42.Yakovchuk P; Protozanova E; Frank-Kamenetskii MD Base-Stacking and Base-Pairing Contributions Into Thermal Stability of the DNA Double Helix. Nucleic Acids Res 2006, 34, 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Menegussi LR; Valandro SR; Poli AL; Schmitt CC; Neumann MG Behaviour of Pseudoisocyanine in Macromolecular and Hydrotropic Solutions. J. Braz. Chem. Soc 2014, 25, 1455–1461. [Google Scholar]
- 44.Gadde S; Batchelor EK; Weiss JP; Ling Y; Kaifer AE Control of H-and J-Aggregate Formation via Host–Guest Complexation Using Cucurbituril Hosts. J. Am. Chem. Soc 2008, 130, 17114–17119. [DOI] [PubMed] [Google Scholar]
- 45.Sorokin AV; Zabolotskii AA; Pereverzev NV; Bespalova II; Yefimova SL; Malyukin YV; Plekhanov AI Metal-Enhanced Fluorescence of Pseudoisocyanine J-Aggregates Formed in Layer-By-Layer Assembled Films. J. Phys. Chem. C 2015, 119, 2743–2751. [Google Scholar]
- 46.Würthner F; Kaiser TE; Saha-Möller CR J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials. Angew. Chem., Int. Ed 2011, 50, 3376–3410. [DOI] [PubMed] [Google Scholar]
- 47.Sorokin AV; Pereverzev NV; Grankina II; Yefimova SL; Malyukin YV Evidence of Exciton Self-Trapping in Pseudoisocyanine J-Aggregates Formed in Layered Polymer Films. J. Phys. Chem. C 2015, 119, 27865–27873. [Google Scholar]
- 48.Zhou X; Mandal S; Jiang S; Lin S; Yang J; Liu Y; Whitten DG; Woodbury NW; Yan H Efficient Long-Range, Directional Energy Transfer Through DNA-Templated Dye Aggregates. J. Am. Chem. Soc 2019, 141, 8473–8481. [DOI] [PubMed] [Google Scholar]
- 49.Díaz SA; Oliver SM; Hastman DA; Medintz IL; Vora PM Increased Transfer Efficiency From Molecular Photonic Wires on Solid Substrates and Cryogenic Conditions. J. Phys. Chem. Lett 2018, 9, 3654–3659. [DOI] [PubMed] [Google Scholar]
- 50.Kashida H; Kurihara A; Kawai H; Asanuma H Orientation-Dependent FRET System Reveals Differences in Structures and Flexibilities of Nicked and Gapped DNA Duplexes. Nucleic Acids Res 2017, 45, No. e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van der Meer BW; Van der Meer DM; Vogel SS FRET-Förster Resonance Energy Transfer: From Theory to Applications; Medintz IL; Hildebrandt N, Eds.; Wiley VCH: Weinheim, Germany, 2013; pp 63–104. [Google Scholar]
- 52.Massey M; Algar WR; Krull UJ Fluorescence Resonance Energy Transfer (FRET) For DNA Biosensors: FRET Pairs and Förster Distances for Various Dye–DNA Conjugates. Anal. Chim. Acta 2006, 568, 181–189. [DOI] [PubMed] [Google Scholar]
- 53.Didenko VV DNA Probes Using Fluorescence Resonance Energy Transfer (FRET): Designs and Applications. Biotechniques 2001, 31, 1106–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Preus S; Wilhelmsson LM Advances in Quantitative FRET-Based Methods for Studying Nucleic Acids. ChemBioChem 2012, 13, 1990–2001. [DOI] [PubMed] [Google Scholar]
- 55.Shindy H Fundamentals in the Chemistry of Cyanine Dyes: A Review. Dyes Pigm 2017, 145, 505–513. [Google Scholar]
- 56.Biver T; De Biasi A; Secco F; Venturini M; Yarmoluk S Cyanine Dyes as Intercalating Agents: Kinetic and Thermodynamic Studies on the DNA/Cyan40 and DNA/CCyan2 Systems. Biophys. J 2005, 89, 374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bricks JL; Slominskii YL; Panas ID; Demchenko AP Fluorescent J-Aggregates of Cyanine Dyes: Basic Research and Applications Review. Methods Appl. Fluoresc 2018, 6, No. 012001. [DOI] [PubMed] [Google Scholar]
- 58.Dsouza RN; Pischel U; Nau WM Fluorescent Dyes and Their Supramolecular Host/Guest Complexes With Macrocycles in Aqueous Solution. Chem. Rev 2011, 111, 7941–7980. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.