

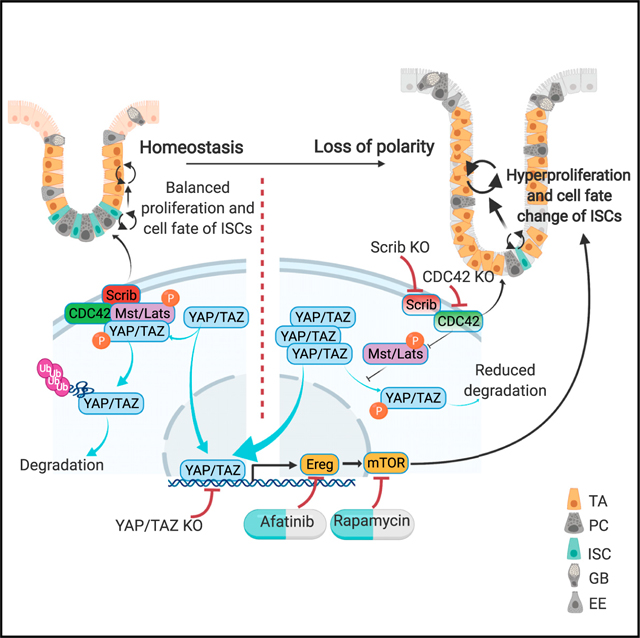
SUMMARY
Epithelial polarity is controlled by a polarity machinery that includes Rho GTPase CDC42 and Scribble/PAR. By using intestinal stem cell (ISC)-specific deletion of CDC42 in olfactomedin-4 (Olfm4)-internal ribosome entry site (IRES)-EGFP/CreERT2;CDC42flox/flox mice, we find that CDC42 loss initiated in the ISCs causes a drastic hyperproliferation of transit amplifying (TA) cells and disrupts epithelial polarity. CDC42-null crypts display expanded TA cell and diminished ISC populations, accompanied by elevated Hippo signaling via YAP/TAZ-Ereg (yes-associated protein/WW domain-containing transcription regulator protein 1-epiregulin) and mechanistic target of rapamycin (mTOR) activation, independent from canonical Wnt signaling. YAP/TAZ conditional knockout (KO) restores the balance of ISC/TA cell populations and crypt proliferation but does not rescue the polarity in CDC42-null small intestine. mTOR or epidermal growth factor receptor (EGFR) inhibitor treatment of CDC42 KO mice exhibits similar rescuing effects without affecting YAP/TAZ signaling. Inducible ablation of Scribble in intestinal epithelial cells mimics that of CDC42 KO defects, including crypt hyperplasia and Hippo signaling activation. Mammalian epithelial polarity regulates ISC/TA cell fate and proliferation via a Hippo-Ereg-mTOR cascade.
In brief
Zhang et al. discover that CDC42-dependent polarity signaling regulates ISC and TA cell fate and proliferation via a YAP-Ereg-mTOR cascade in the small intestine. This study shows that mammalian epithelial polarity-controlled Hippo signaling is central to cell fate balance between ISC and TA cells and intestinal crypt proliferation.
Graphical Abstract
INTRODUCTION
The mammalian intestinal epithelium is one of the most vigorously self-renewing tissues in adults, with complete turnover occurring every 4–5 days (van der Flier and Clevers, 2009). This columnar epithelium is composed of villi structures, finger-like projections that extend into the lumen of the small intestine where nutrients are absorbed, and proliferative crypts in between villi where intestinal stem cells (ISCs) reside. At the bottom of each crypt is the stem cell niche, an area composed of crypt-based columnar cells with stem cell activity and intercalating stem cell-supporting Paneth cells (Sato et al., 2011; Snippert et al., 2010). During homeostasis, ISCs in the stem cell niche proliferate constantly, giving rise to rapidly proliferating transit amplifying (TA) cells. The TA cells migrate upward to exit the crypt and differentiate further into specialized epithelial cell lineages: absorptive enterocytes, mucus-secreting goblet cells, antimicrobial Paneth cells, and hormone-secreting enteroendocrine cells (Tian et al., 2015). This cycle of cell genesis and differentiation is tightly regulated by multiple signaling pathways.
Among the known ISC regulatory signaling pathways, the Wnt pathway was the first to be discovered as a key regulator for ISC maintenance and differentiation (Gehart and Clevers, 2019). The Hippo-YAP (yes-associated protein) pathway is a critical regulator for ISC regeneration after injury, during intestinal tumorigenesis, or following chemical or irradiation-induced injury (Barry et al., 2013; Gregorieff et al., 2015). YAP and TAZ (WW domain-containing transcription regulator protein 1) were initially found to promote ISC proliferation (Imajo et al., 2015); in fact, YAP and TAZ activation, by deletion of their endogenous suppressors MST1 and MST2, causes increased crypt proliferation and tumorigenicity (Zhou et al., 2011). Alternatively, no effect was observed following loss of YAP and TAZ in the small intestine under normal homeostasis (Azzolin et al., 2014; Barry et al., 2013; Cai et al., 2015). Further studies showed that YAP signaling is crucial for ISC regeneration following chemical or irradiation-induced injury (Barry et al., 2013; Gregorieff et al., 2015). Multiple studies indicate a close connection between the Hippo-YAP pathway and Wnt/β-catenin pathway to regulate the ISC function (Azzolin et al., 2014; Barry and Camargo, 2013; Cai et al., 2015; Imajo et al., 2012).
The epidermal growth factor receptor (EGFR) ligand epiregulin (EREG) is a known YAP1 target for ISC proliferation and regeneration (Gregorieff et al., 2015) and, as discussed in a recent Drosophila study, indicates that EGFR signaling regulates ISC proliferation (Zhang et al., 2019). EGFR signaling is also one of the intracellular and extracellular cues influencing mechanistic target of rapamycin (mTOR), a highly conserved eukaryotic serine-threonine kinase that regulates maintenance of the olfactomedin-4 (Olfm4)+ ISC/progenitor population and crypt regeneration after injury (Sampson et al., 2016).
The role played by apical-basal polarity and cell adhesion junctions, two mechanisms important to intestinal epithelium morphogenesis, in mammalian ISC regulation is incompletely understood. The Rho GTPase family member CDC42 maintains apical-basolateral polarity by regulating tight junctions and membrane protein traffic (Martin-Belmonte et al., 2007; Rojas et al., 2001). In addition, CDC42 interacts with Scribble, a master regulator of planar polarity and adhesion, during key events such as cell polarity regulation and proliferation (Zhan et al., 2008). Loss of CDC42 in all intestinal epithelial cells induces broad tissue hyperplasia accompanied by hyperproliferation, crypt enlargement, and defective Paneth cell differentiation and localization (Melendez et al., 2013).
Multiple molecular markers have been identified in ISCs. Leucine-rich repeat-containing G protein-coupled receptor 5 (Lgr5) marks an actively cycling and long-lived stem cell pool that continuously gives rise to all four epithelial lineages (Sato et al., 2011; Snippert et al., 2010). An Lgr5-EGFP-internal ribosome entry site (IRES)-CreERT2 allele was initially designed to mark and characterize the function of ISCs (Barker et al., 2007); however, the mosaic expression levels of the Lgr5-driven GFP and CreERT2 proteins in the crypts make it a suboptimal genetic tool for ISC-specific manipulation, as well as a bottleneck for consistent exploration of ISC-specific gene functions. Olfm4, another highly specific and robust gene marker for ISCs (Itzkovitz et al., 2011), is the foundation for an Olfm4-IRES-EGFP/CreERT2 (Olfm4-CreER) mouse line, generating better visualization of ISCs and, importantly, more robust gene modification (Schuijers et al., 2014).
In this study, we perform ISC-specific deletion of CDC42 by Olfm4-CreER, which induces intestinal hyperplasia and loss of polarity, mimicking whole intestinal epithelium loss-of-function effects. Surprisingly, in the Olfm4-CreER CDC42 knockout (KO) mice, the ISC population is severely diminished, and the TA cell population is expanded. Single-cell RNA-sequencing (scRNA-seq), genetic rescue, and pharmacologic inhibition assays reveal that loss of CDC42 drives the hyperproliferative phenotype and the ISC-TA cell fate switch via the Hippo-YAP pathway and downstream Ereg and mTOR signaling. Our findings suggest that CDC42-mediated polarity represses YAP/TAZ-Ereg-mTOR signaling, which is required to maintain ISC-TA cell fate balance.
RESULTS
ISC-specific deletion of CDC42 causes crypt hyperproliferation
Previous studies using a Villin-Cre model found that constitutive loss of CDC42 in all intestinal epithelium cells caused significant hyperplasia, crypt enlargement, and permeability and barrier functional defects (Melendez et al., 2013). To study the ISC-intrinsic function of CDC42 in adult intestine, we generated an inducible ISC-specific CDC42 ablation line by breeding CDC42flox/flox mice with Olfm4-CreER knockin mice (Schuijers et al., 2014), in which the ISC-specific Olfm4 promoter delivers tamoxifen (TAM)-inducible Cre activity. While Olfm4-IRES-EGFP/CreERT2;CDC42flox/flox (also known as CDC42 ISC-KO) mice only showed a slight reduction in CDC42 in whole-crypt lysates on day 1 compared to Olfm4-IRES-EGFP/CreERT2; CDC42+/flox (control) mice, loss of CDC42 was complete by day 4 (Figure 1A, upper panel; Figure S1A). We then used flow cytometry to separate the Olfm4-CreER-driven GFP+ cell population (ISCs) and the GFP cell population (non-ISCs) from whole crypts harvested on day 1 and found that CDC42 was depleted in the ISCs as early as day 1 (Figure 1A, lower panel; Figure S1B), suggesting that loss of CDC42 starts in the ISCs and then the progenies of ISCs. On day 4, H&E and immunostaining of duodenum from CDC42 ISC-KO mice (versus controls) revealed a severe disruption of crypt-villus morphology, enlarged crypts, and an increased number of proliferating phosphorylated histone H3 (pH3)+ crypt cells (M-phase cells) (Figures 1B–1E; Figures S1C–S1H), which mimics the whole intestine epithelium deletion of CDC42 (Melendez et al., 2013). Importantly, the enlarged crypts are evident throughout the whole small intestine of CDC42 ISC-KO mice, including duodenum, jejunum, and ileum, with the proximal duodenum exhibiting the most significant size increase. We focused our study on the duodenum due to the abundant literature on the function of ISCs in this region.
Figure 1. ISC-specific deletion of CDC42 causes crypt hyperproliferation.

Two- to 3-month-old CDC42 ISC-KO (Olfm4-CreER, CDC42flox/flox) and control (Olfm4-CreER, CDC42flox/+) mice were injected with TAM once per day for 3 days, and then were sacrificed at 12 h (day 0.5), 24 h (day 1), 72 h (day 3), or 96 h (day 4) after the third TAM injection.
(A) Small intestinal crypts were isolated, lysed, and western blotted for CDC42. Actin is the loading control. (Upper) Day 1 and day 4 whole-crypt cell lysates. (Lower) Day 1 GFP sorted crypt cells.
(B and C) Representative images of H&E staining of duodenal sections; one crypt is circled in each image based on morphology.
(D–M) Representative images of immunofluorescence staining of duodenal sections.
(D and E) Anti-pH3 and anti-E-cadherin; one crypt is circled in each image.
(F–K) Lineage tracing tdTomato, DAPI, E-cadherin, as well as lysozyme, Ki67, and Olfm4; one crypt is circled in each day 3 image.
(L and M) anti-αE-catenin; enlargements of villi (Ľ and M’) and crypt (Ľ’ and M”).
Data are representative of at least three independent experiments. Scale bars, 50 μm. n = 4 for each genotype.
To test the potential of CDC42-null ISCs to differentiate into specialized cell types of the intestinal epithelium, CDC42 ISC-KO mice were crossed with Rosa26tdTomato reporter mice. With a single injection of TAM at day 0, the EGFP/CreERT2 enzyme is activated to excise the STOP sequence and turn on the tdTomato reporter, thereby labeling the Olfm4-CreER+ ISCs and their progeny. In day 0.5 CDC42 ISC-KO epithelium, tdTomato+ cells were detectable in some crypts and at the bottom of villi–versus controls, where only some ISCs at the bottom of crypts were tdTomato+ (Figures 1F–1K, red cells). By day 1, most crypts and villi of CDC42 ISC-KO epithelium were tdTomato+–versus controls, in which tdTomato+ cells were only visible in crypts. Co-staining for the Paneth cell marker lysozyme, proliferating cell marker Ki67 (non-G0 cycling cells), and the ISC marker Olfm4 revealed hyperproliferation in CDC42 ISC-KO crypts by day 3, and diminished Olfm4 signal starting at day 1 (Figures 1F–1K, green cells; Figures S1I and S1J). The loss of Olfm4 corresponds to the loss of CDC42 protein in the ISC population (Figure 1A). Thus, in CDC42 ISC-KO intestinal epithelium, loss of CDC42 specifically in ISCs appears to first drive an increase in overall cell turnover rates, repopulating crypts and villi faster than control ISCs, followed by a reduction in ISC numbers.
To examine the disrupted morphology in both CDC42 ISC-KO and CDC42 KO intestinal epithelium, we examined expression of E-cadherin and αE-catenin (α-cat), two key components of adhesion junctions. Immunolabeling revealed a drastic disruption of villi and crypts in CDC42 ISC-KO epithelium, with distorted cell alignment and epithelial polarity (Figures 1L and 1M; Figure S1K). Alternatively, expression levels of E-cadherin and αE-catenin were not significantly altered, suggesting that adherent junctions are largely not impacted by the induced CDC42 loss starting from ISCs (Figure S1L).
Loss of CDC42 in ISCs increases proliferation in TA cells at the expense of ISCs
ISCs are the reservoir and driving force for the proliferative activity of crypts (van der Flier and Clevers, 2009). Surprisingly, Olfm4+ ISCs were reduced in CDC42 ISC-KO crypts (Figures 1J and 1K), concomitant with over-proliferation and hyperplasia. One possible source of the increased proliferation and enlarged crypt size is an expanded population of TA cells, the active cycling progenitor cells in the transit amplifying zone located just above the ISCs at the base of the crypts.
To test this, we measured Olfm4 expression in intestinal epithelium using in situ hybridization, western blotting, and immunofluorescence. This ISC marker was significantly decreased in both mRNA and protein levels in CDC42 ISC-KO crypts on day 4 post-TAM induction (Figures 2A and 2B; Figures S2A and S2C). Moreover, quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) confirmed that two other ISC markers achaete scute-like 2 (Ascl2) and Lgr5 were also significantly reduced (Figure 2F). Flow cytometry analysis of cells harvested from crypts also showed that the Olfm4-CreER-driven GFP+ cell population was reduced by ∼6-fold (Figure S2B). These data all point to a repression of the ISC population by CDC42 deletion.
Figure 2. Loss of CDC42 in ISCs increases proliferation in TA cells at the expense of ISCs.

(A) Representative images of in situ staining of duodenal sections; one crypt is circled in each image.
(B–E) Representative images of immunofluorescence staining of duodenal sections: anti-Olfm4, anti-β-catenin, anti-Pcna, anti-Ki67, and anti-lysozyme.
(F and G) Relative mRNA levels of ISC markers, TA cell markers, and Wnt pathway markers in isolated small intestinal crypts. Data are mean ± SD. *p < 0.05, **p < 0.01; ns, not significant.
Data are representative of at least three independent experiments. Scale bars, 50 μm. n = 4 for each genotype.
In contrast to ISCs, immunostaining revealed that two other proliferating cell populations expanded in CDC42 ISC-KO intestinal epithelia, i.e., Pcna+ active-cycling TA cells (Figure 2C; Figure S2D) and Ki67+ cells (Figure 2D; Figure S2D), which is reflected in the drastically enlarged crypts (Figure 1B). Consistently, qRT-PCR showed that four out of five TA cell markers were significantly elevated in CDC42 ISC-KO crypts (Figure 2F). In addition, lysozyme+ Paneth cells were ectopic in day 4 CDC42 ISC-KO epithelium (Figure 2E; Figure S2E), especially villi, similar to that noted in constitutive whole epithelium deletion of CDC42 (Melendez et al., 2013). This defect in mislocalization was not apparent in the tdTomato lineage tracing experiment (Figures 1F and 1G), suggesting that CDC42-null ISCs need longer than 3 days after the final TAM injection to differentiate into CDC42-null Paneth cells. Notably, no excessive apoptosis was detected in CDC42 ISC-KO epithelium (Figures S2F and S2G).
Since canonical Wnt signaling is critical for ISC self-renewal (Pinto et al., 2003; van der Flier and Clevers, 2009), we measured canonical Wnt pathway activity by qRT-PCR analysis of canonical Wnt effectors Axin2 and cyclin D1. Neither was significantly different in CDC42 ISC-KO versus control crypts (Figure 2G). Thus, the observed decrease in ISC number appears independent from canonical Wnt signaling changes.
Activities of YAP, Ereg, and mTOR signaling are increased in CDC42-depleted TA cells and ISCs
To understand the mechanistic underpinnings of the observed CDC42 ISC-KO defects, we analyzed crypt cells by performing high-throughput scRNA-seq, followed by UMAP, a nonlinear dimensionality-reduction technique recently developed as an alternative for t-distributed stochastic neighbor embedding (Becht et al., 2019) (Figure 3A). In crypt cells isolated from CDC42 ISC-KO mice, our data revealed seven clusters with distinct gene expression signatures. Cluster annotation based on cell marker expression (Haber et al., 2017) revealed at least seven cell types: endocrine cells, enterocytes, goblet cells, Paneth cells, ISCs, TA cells, and tuft cells. The data are consistent with the immunofluorescence, fluorescence-activated cell sorting (FACS), and RT-PCR results above showing a relative reduction in the ISC population and an expansion of the TA cell population in CDC42 ISC-KO crypts (Figure S3A).
Figure 3. YAP and mTOR signaling activities are increased in CDC42-depleted TA cells and ISCs.

(A) Single-cell RNA-seq analysis of isolated single cells from duodenal crypts by UMAP clustering.
(B) Heatmap of most differentially expressed genes in the YAP pathway (10 genes), mTOR pathway (7 genes), proliferation pathways (15 genes), and apical polarity pathways (8 genes) in control and CDC42 ISC-KO ISC cluster. Columns indicate individual cells; rows indicate genes. n refers to gene numbers.
(C) GSEA pathway enrichment map for YAP and proliferation pathways from cells in the ISC cluster.
(D) Expression level of Ereg in ISC and TA cell cluster.
Data from one experiment. n = 1 for each genotype.
We next performed gene set enrichment analysis (GSEA) analysis for YAP, mTOR, and Wnt signaling pathways in the ISCs cluster, in which each plays known critical roles in ISC function, as well as apical polarity and proliferation-related genes. The YAP signaling pathway is critical for regeneration of the ISC population after injury (Gregorieff et al., 2015; Taniguchi et al., 2015), and the YAP1 target, Ereg (an EGFR ligand), modulates ISC proliferation and regeneration (Gregorieff et al., 2015). GSEA analysis shows that a conserved YAP pathway signature is elevated in CDC42-null ISCs (Figures 3B and 3C). Consistently, sequencing data revealed a drastically elevated Ereg expression (Figure 3D; Figure S3B). In addition, ISCs isolated from CDC42 ISC-KO mice showed elevated mTOR, apical polarity, adhesion signaling, and genes involved in proliferation (Figures 3B and 3C; Figures S3C–S3E), whereas CDC42-depleted TA cells had a similar increase in YAP signaling, mTOR signaling, and proliferation (Figures S3F–S3H). Interestingly, canonical Wnt signaling was not significantly changed in either CDC42-null (versus control) ISCs or TA cells (Figures S3I and S3J). These pathway analysis data suggest that YAP, Ereg, and mTOR pathways are potential targets of CDC42 in ISCs and TA cells.
YAP KO/TAZ Het rescues ISCs and reduces TA cell proliferation
Our high-throughput scRNA-seq analysis suggests that YAP signaling is elevated in CDC42-null ISCs (Figures 3B and 3C). To confirm that transcription levels of YAP pathway targets are also increased in crypts isolated from CDC42 ISC-KO mice, we used qRT-PCR. While mRNA levels of three targets (Ctgf, Cyr61, and Ereg) were dramatically elevated, YAP and TAZ mRNA levels were unchanged (Figure 4A). Alternatively, western blotting showed massive accumulation of YAP protein in CDC42 ISC-KO crypts but reduced levels of phospho-YAP, which undergoes degradation (Figure 4B). Consistently, the YAP pathway suppressors pLats1/2 and pMST1/2, which phosphorylate YAP and Lats1/2, respectively, were reduced (Figure 4B; Figure S4A). In addition, ubiquitin K48 is significantly reduced in immunoprecipitated YAP protein from CDC42 ISC-KO crypts (Figure 4C). These data suggest that in intestinal epithelium, CDC42 depresses YAP signaling.
Figure 4. YAP KO/TAZ Het rescues ISCs and reduces TA cell proliferation in CDC42 KO intestine.

YAP KO rescue (Olfm4-CreER;CDC42flox/flox, YAPflox/flox) and YAP KO/TAZ Het rescue (Olfm4-CreER;CDC42flox/flox, YAPflox/flox, TAZflox/+) were generated to compare with control and CDC42 ISC-KO.
(A) Relative mRNA levels of YAP pathway components and effectors in isolated small intestinal crypts. Data are mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.005; ns, not significant.
(B) Small intestinal crypts were isolated, lysed, and western blotted for YAP and YAP pathway repressor MST1/2 and Lats1/2. Actin represents the loading control.
(C) Small intestinal crypts were isolated, lysed, and immunoprecipitated with anti-YAP antibody, then western blotted for ubiquitin K48.
(D) Relative mRNA levels of ISC markers, YAP pathway markers and TA cell markers in isolated small intestinal crypts. *p < 0.05, **p < 0.01, ***p < 0.005.
(E) UMAP clusters of isolated single cells from duodenal crypts.
(F) Representative images of H&E staining of duodenal sections; one crypt is circled in each image.
(G–K) Representative images of immunofluorescence staining of duodenal sections: anti-YAP, anti-Ki67, anti-Pcna, anti-b-catenin, anti-pH3, anti-αE-catenin, and anti-Olfm4.
(L) Heatmap of most differentially altered genes in the YAP pathway (10 genes), mTOR pathway (7 genes), and proliferation pathways (15 genes) in control, CDC42 ISC-KO, and YAP KO/TAZ Het rescue ISC cluster. Columns indicate individual cells; rows indicate genes. n refers to gene numbers.
For (A)–(D) and (F)–(K), data are representative of at least three independent experiments. Scale bars, 50 μm. n = 4 for each genotype. For (E) and (L), data are from one experiment. n = 1 for each genotype.
To test whether repressing YAP signaling rescues CDC42 ISC-KO defects, we generated Olfm4-CreER;CDC42flox/flox, YAPflox/flox mice (also known as YAP KO rescue mice), by crossing YAPflox/flox and Olfm4-CreER;CDC42flox/flox mice. Due to the redundancy of YAP and TAZ in mammals (Hong et al., 2016), we crossed YAP KO rescue mice with TAZflox/flox mice to obtain Olfm4-CreER;CDC42flox/flox, YAPflox/flox, TAZflox/+ mice (also known as YAP KO/TAZ Het rescue mice). Efficiency of YAP loss of function in both YAP KO and YAP KO/TAZ Het rescue mice was confirmed in intestinal crypts by western blot (Figure S4B) demonstrating loss of YAP and by qRT-PCR demonstrating that YAP signaling targets Ctgf and Ereg were significantly reduced by YAP KO and even more so by YAP KO/TAZ Het (versus CDC42 ISC-KO) mice (Figure 4D). The ability of these genetic crosses to rescue ISCs was demonstrated first by western blots showing that the cross rescued the ISC marker Olfm4 (Figure S4B), and second by qRT-PCR analysis demonstrating that each rescued ISC markers Ascl2, Lgr5, and Olfm4 (Figure 4D); in both experiments, rescue was greater in crypts from YAP KO/TAZ Het rescue mice. Similarly, compared to CDC42 ISC-KO mice, the TA cell marker Pcna was significantly reduced in crypts by either YAP or YAP KO/TAZ Het mice (Figure 4D). We next performed scRNA-seq analyses using crypt cells from YAP KO/TAZ Het rescue mice and found that the relative ISC percentage appears to be restored compared with that in crypts from CDC42 ISC-KO mice (Figure 4E; Figure S3A). These data suggest that reduction of YAP/TAZ signaling restores ISC fate at the expense of TA cells, driving both cell populations toward wild-type levels.
The size of crypts in YAP KO/TAZ Het rescue epithelium was drastically decreased, compared to CDC42 ISC-KO epithelium, but the epithelial structures remained disorganized (Figure 4F; Figure S4C). Anti-YAP immunostaining of intestinal epithelium found mainly a cytoplasm localization in control crypts; in CDC42 ISC-KO crypts it was significantly increased in the cytoplasm and also in the nucleus (Figure 4G; Figure S4D) where it is known to activate downstream pathways (Hong et al., 2016). As expected, YAP KO and YAP KO/TAZ Het crypts showed a clear reduction of YAP immunostaining, and they also reduced Ki67 (Figure 4H; Figure S4E), Pcna (Figure 4I; Figure S4E), and pH3 (Figure 4J; Figure S4E). In addition, Olfm4, which was reduced in CDC42 ISC-KO crypt, was elevated in YAP KO/TAZ Het rescue crypt (Figure 4K; Figure S4D), validating our biochemical data (Figure 4D; Figure S4B). While some of the lysozyme+ Paneth cells remained ectopically localized in intestinal epithelium in the YAP KO and YAP KO/TAZ Het rescue mice (Figures S4F and S4F’), the number of lysozyme+ Paneth cells was partially rescued. GSEA analysis confirmed that YAP KO/TAZ Het reduced YAP, Ereg, mTOR, and proliferation pathways in ISCs and TA cells (Figure 4L; Figures S4G–S4K), suggesting that YAP/TAZ signaling is upstream of Ereg signaling and mTOR signaling that regulate the proliferation and cell fate balance of ISCs/TA cells.
Inhibition of mTOR by rapamycin rescues ISCs and reduces TA cell proliferation
The mTOR pathway is dispensable for ISC homeostasis but is required for ISC maintenance and regeneration after injury (Sampson et al., 2016). Our scRNA-seq data show that mTOR signaling is elevated in CDC42 ISC-KO ISCs and TA cells (versus WT controls) (Figure 3B; Figure S3C). Consistently, we found that the mTOR pathway effectors p4EBP and pS6 were both increased in CDC42 ISC-KO crypts (versus control) and reduced in YAP KO and YAP KO/TAZ Het rescue crypts (versus CDC ISC-KO) (Figure 5A; Figure S5A), suggesting that the mTOR pathway is downstream of and the YAP pathway. To test this, CDC42 ISC-KO mice were injected with the mTOR inhibitor rapamycin. The mTOR inhibitor treatment significantly reduced p4EBP and pS6 levels in CDC42 ISC-KO crypts, while having no noticeable influence on YAP protein expression or localization (Figures 5B and 5E; Figures S5B and S5D). Furthermore, Ascl2, Lgr5, and Olfm4 levels in the KO crypts were restored to WT levels (Figures 5C and 5H; Figure S5D). Alternatively, the disrupted epithelial structure of the duodenum was still evident, although the size of the crypts was reduced (Figure 5D; Figure S5C). Hyperproliferation, marked by pH3 and Ki67, was reduced by rapamycin (Figures 5F and 5G; Figure S5E); however, Paneth cell mislocalization still persisted (Figure 5I; Figure S5F). This pharmacological reversal of defects via rapamycin, combined with genetic rescue in YAP KO/TAZ Het rescue mice, indicates that the mTOR pathway is downstream of CDC42-suppressed YAP signaling and regulates ISC/TA cell proliferation and cell fate decision.
Figure 5. Inhibition of mTOR signaling by rapamycin rescues ISCs and reduces TA cell population.

CDC42 ISC-KO and controls were injected with TAM once per day for 3 consecutive days, followed by 3 consecutive days of PBS or rapamycin injection, then sacrificed 24 h after the third rapamycin injection.
(A and B) Small intestinal crypts were isolated, lysed, and western blotted for CDC42 and mTOR pathway effector S6 and 4EBP. Actin represents the loading control.
(C) Relative mRNA levels of ISC markers in isolated small intestinal crypts. Data are mean ± SD. *p < 0.05, **p < 0.01.
(D) Representative images of H&E staining of duodenal sections; one crypt is circled in each image.
(E–I) Representative images of immunofluorescence staining of duodenal sections: anti-YAP, anti-E-cad, anti-pH3, anti-Ki67, anti-b-catenin, anti-Olfm4, anti-E-cad, and anti-lysozyme.
Data are representative of at least three independent experiments. Scale bars, 50 μm. n = 4 for each genotype.
Ereg/EGFR signaling, downstream of CDC42 and YAP, activates the mTOR pathway in intestinal crypts
To identify the mechanistic link between YAP and the mTOR pathway, we chose to focus first on Ereg/EGFR signaling, given that Ereg, an EGFR family member, is a known target of YAP in small intestine and EGFR can stimulate the mTOR pathway (Meng et al., 2018). In these experiments, we injected the EGFR inhibitor afatinib into CDC42 ISC-KO and control mice and found, as expected, reduced levels of pEGFR in lysates of intestinal crypts from both mice (versus untreated CDC42 ISC-KO mice) (Figure 6A). Afatinib reduced the mTOR pathway effector p4EBP while it did not affect YAP expression (Figures 6A and 6D; Figures S6A and S6B). Other features of intestinal epithelium from CDC42 ISC-KO mice were “rescued” by afatinib treatment: ISC markers (Figures 6B and 6G; Figure S6B), crypt sizes (Figure 6C; Figure S6C), proliferation marked by Ki67 (Figure 6E; Figure S6D) and pH3 (Figure 6F; Figure S6D), and TA cell population size (e.g., Pcna marker, Figure 6E; Figure S6D). Alternatively, the structure of the duodenal epithelium remained disorganized (Figure 6C), and the ectopic Paneth cells remained dislocated (Figure 6H; Figure S6E). These data show that Ereg/EGFR signaling, acting downstream of YAP, activates the mTOR pathway in intestinal crypts to regulate ISC/TA cell fate shift and proliferation.
Figure 6. Ereg/EGFR signaling, acting downstream of CDC42-YAP, activates the mTOR pathway in crypts.

CDC42 ISC-KO and controls were injected with TAM once per day for 3 consecutive days, followed by 3 consecutive days of PBS or afatinib injection, then sacrificed 24 h after the third afatinib injection.
(A) Small intestinal crypts were isolated, lysed, and western blotted for EGFR, YAP, and the mTOR pathway effector 4EBP. Actin represents the loading control.
(B) Relative mRNA levels of ISC markers in isolated small intestinal crypts. Data are mean ± SD. *p < 0.05, **p < 0.01.
(C) Representative images of H&E staining of duodenal sections; one crypt is circled in each image.
(D–H) Representative images of immunofluorescence staining of duodenal sections: anti-YAP anti-Ki67, anti-Pcna, anti-b-catenin, anti-pH3, anti-E-cad, anti-Olfm4, anti-αE-catenin, and anti- lysozyme.
Data are representative of at least three independent experiments. Scale bars, 50 μm. n = 4 for each genotype.
Loss of CDC42 disrupts polarity and binding of Scribble and YAP, while loss of Scribble mimics CDC42 loss-of-function defects
To study the interactions of CDC42 and YAP and the consequences for cell polarity, we performed GSEA, comparing crypts from CDC42 ISC-KO and YAP KO/TAZ Het rescue mice. The defective apical pathway in ISCs and TA cells from CDC42 ISC-KO intestinal epithelium was not rescued in YAP KO/TAZ Het mice (Figures S7A and S7B). To examine the effects of CDC42 on polarity of the intestinal columnar cells, we took advantage of the cell polarity marker Na+K+-ATPase, which is concentrated at the basal surface of villi and crypt cells. Na+K+-ATPase immunolabeling was less polarized and slightly reduced in CDC42 ISC-KO villi cells (versus control), and the crypts also lost the surrounding Na+K+-ATPase basal lateral layer (Figures 7A and 7B). Importantly, YAP KO/TAZ Het could not rescue the epithelium polarization defects (i.e., disrupted villi structure and loss of Na+K+-ATPase enrichment) (Figure 7C), suggesting that YAP is downstream of CDC42-regulated polarity in the small intestine.
Figure 7. Effects by loss of CDC42 are mimicked by a loss of Scribble in disrupting epithelial polarity and ISC-TA cell fate.

Scribble het (villin-CreERT2;Scribbleflox/flox) and Scribble KO (villin-CreERT2;Scribbleflox/+) mice were injected with TAM once per day for 3 days, and then sacrificed 1 day after the third TAM injection.
(A–C) Representative images of immunofluorescent staining of duodenal sections: anti-Na+K+-ATPase; enlargements of villi (A’, B’, and C’) and crypt (A”, B”, and C”).
(D and E) Small intestinal crypts were isolated, lysed, and immunoprecipitated with (D) anti-YAP and (E) anti-Scribble, then western blotted for (D) Scribble and (E) MST1/2 and Lats1/2. (representative of two independent repeats)
(F) Representative images of H&E staining of duodenal sections; one crypt is circled in each image.
(G–K) Representative images of immunofluorescence staining of duodenal sections: anti-E-cad, anti-Na+K+-ATPase, anti-YAP, anti-Ki67, anti-Pcna, αE-catenin, and anti-Olfm4.
(L and M) qRT-PCR analysis of (L) ISC marker, TA cell marker, and YAP pathway, and (M) Wnt pathway in isolated small intestinal crypts. Data are mean ± SD. *p < 0.05, **p < 0.01; ns, not significant.
For (A)–(C), data are representative of at least three independent experiments. Scale bars, 50 μm. n = 4 for each genotype. For (D) and (E), data are representative of two independent experiments. Scale bars, 50 μm. n = 3 for each genotype. For (F)–(M), data are representative of at least three independent experiments. Scale bars, 50 μm. n = 3 for each genotype.
To determine whether CDC42 elicited effects through its function on basal-lateral polarity, we examined the role of Scribble, a tumor suppressor gene and a key component of epithelial basal lateral polarity (Bonello and Peifer, 2019). Together with Lgl (lethal giant larvae) and Dlg (discs large) proteins, Scribble directs formation of basolateral membranes (Zhan et al., 2008). In vitro, Scribble binds multiple YAP signaling pathway components, including MST1/2, Lats1/2, YAP, and TAZ (Bonello and Peifer, 2019; Clattenburg et al., 2015). We tested whether Scribble physically interacts with the YAP/MST/Lats complex in crypts: a co-immunoprecipitation (coIP) test revealed that binding was significantly weaker in CDC42 ISC-KO crypts when compared to strong Scribble-YAP binding in control crypts (Figure 7D; Figure S7C), and the Scribble to MST1/2 and Scribble to Lats1/2 bindings were similarly reduced (Figure 7E; Figure S7C).
To determine whether depleting Scribble in intestinal epithelium mimics CDC42 loss-of-function defects, we bred villin-CreERT2 (villin-CreER) and Scribbleflox/flox mice (Hartleben et al., 2012) to delete Scribble in the whole intestinal epithelium, under TAM control. Western blots of isolated crypts confirmed loss of Scribble in villin-creERT2;Scribbleflox/flox (i.e., Scribble KO) compared to villin-creERT2;Scribble+/flox (i.e., Scribble Het) and revealed in the epithelium effects that mimicked those seen in epithelium of CDC42 ISC-KO mice even though the villin-CreER model differs from the Olfm4-CreER model used in the above CDC42 ISC-KO studies. Such differences included elevated mTOR pathway effectors p4EBP and pS6 (Figures S7D and S7D’), increased crypt size (Figure 7F; Figure S7E), defective morphology (Figure 7G), disrupted apical-basal polarity (Figure 7H), and increased levels of YAP protein (Figure 7I; Figure S7G), and increased YAP pathway effectors Ctgf and Ereg (Figure 7L), all further confirming that loss of Scribble disrupts apical basal polarity functioning. Importantly, mRNA levels of ISC markers Ascl2 and Lgr5 decreased, and the TA cell marker Pcna increased (Figure 7L; Figure S7F). We further observed an expansion of TA cell population with increased proliferation (Figure 7J; Figure S7F) and a loss of ISC population (Figure 7K; Figure S7G), also mimicking the effects of CDC42 KO. These results suggest that loss of a polarity complex component is sufficient to elevate YAP-mTOR signaling and trigger the ISC-to-TA cell fate shift. Wnt is not involved during this process, as canonical Wnt targets were not significantly altered (Figure 7M). Thus, in intestinal crypts, CDC42/Scribble-mediated apical-basal cell polarity is a key regulator of YAP/EGFR/mTOR signaling, which, in turn, regulates ISC and TA cell fate balance with regard to proliferation.
DISCUSSION
CDC42 regulates ISC/TA cell proliferation and fate balance in intestinal crypts
Cdc42 is known for its critical role in establishing cell polarity in eukaryotes. To study the function of CDC42-regulated polarity specifically in mammalian ISCs in homeostasis, we used mice in which CDC42 is inducibly knocked out specifically in adult ISCs (Schuijers et al., 2014). Similar to the constitutive deletion of CDC42 in the whole intestinal epithelium (Melendez et al., 2013), CDC42 ISC-KO led to significant hyperplasia and crypt enlargement. Conversely, ISC proliferation and differentiation increased in the CDC42 ISC-KO (versus controls), and ISC number and marker gene expression decreased while TA cells, which grow almost twice as quickly as ISCs in the cell cycle (Carroll et al., 2018; Schepers et al., 2011), increased. From these observations, we begin to construct a hypothesis in which CDC42 functions as a switch between the two pluripotent cell populations. Specifically, when CDC42 is reduced, apical polarity is disrupted, proliferation accelerates, and ISC-to-TA cell transition is favored over ISC self-renewal. As a result, the slower cycling stem cell pool is depleted, while the faster cycling TA cell pool is expanded. These changes in proliferation and differentiation lead to the tissue hyperplasia observed in the CDC42 ISC-KO mice. Our results support the notion that intestinal tissue is highly plastic, driven by proliferation that may affect the balance between ISCs and TA cells.
Canonical Wnt signaling is a key regulator for ISC renewal and proliferation (Pinto et al., 2003). Indeed, a recent study found that in YAP gain-of-function crypts, ISCs are diminished due to decreased Wnt signaling (Li et al., 2020). In contrast, in our study, Wnt signaling does not appear to be altered in either ISC or TA cell populations, indicating that CDC42-mediated polarity controls ISC/TA cell fate switch and TA cell proliferation independent of changes in canonical Wnt signaling. These two apparently disparate findings could suggest that, in crypts, YAP signaling can be fine-tuned in different cell populations to drive diverse functional responses.
CDC42/Scribble-mediated apical/basal-lateral polarity controls Hippo-YAP signaling
The Hippo-YAP signaling cascade is highly conserved and is a critical regulator of organ size, tissue homeostasis, and regeneration (Hong et al., 2016). A previous study suggested that YAP signaling, elevated in CDC42 ISC-KO crypts, is differentially regulated by adhesion junctions and apical-basal polarity (Yang et al., 2015). In addition to enlarged crypts and increased intestinal size, CDC42 ISC-KO leads to disrupted epithelial cell morphology, suggesting underlying defects in cell adhesion and/or apical/basal polarity. The observed abnormal polarity marker staining in the CDC42 ISC-KO epithelium indicates that apical polarity is compromised by loss of CDC42. The effects on epithelial disruption and ectopic localization of Paneth cells could not be rescued by downstream YAP/TAZ KO or EREG/mTOR inhibition, suggesting that additional cellular function regulated by CDC42 such as cell-cell or cell-matrix adhesion is at play.
Loss of the basal polarity protein Scribble in cell line and Drosophila experiments causes YAP accumulation and activation in the nucleus, possibly due to reduced binding of Scribble to YAP suppressors MST1/2 and Lats1/2 (Mohseni et al., 2014; Yang et al., 2015). We show that in CDC42 ISC-KO crypts, Scribble binding to the MST1/2-Lats1/2-YAP complex is reduced, leading to reduced phosphorylation of MST1/2, Lats1/2, and YAP, reduced degradation of YAP, and increased nuclear localization of YAP. Interestingly, in dental epithelium, loss of CDC42 decreased YAP nuclear localization via PP1A (Hu et al., 2017), suggesting that the CDC42-YAP relationship varies between different tissues. Our whole epithelium Scribble KO mouse model provides initial evidence that a loss of apical/basal polarity is sufficient to elevate YAP signaling in the crypts including ISCs, thereby driving over-proliferation and ISC/TA cell fate change. The fact that YAP/TAZ loss of function reverses the CDC42 ISC-KO crypt proliferation, except for the epithelial cell polarity, further supports that YAP and its effectors are downstream of polarity. Although whole epithelial KO of Scribble phenocopies CDC42 ISC-KO in general, the defects are milder, suggesting that, in ISCs, CDC42 performs more functions than just regulating apical/basal polarity. Future study of ISC-specific loss of polarity molecules such as Scribble, Dlg, and Lgl may further confirm whether their ISC-intrinsic function is sufficient to impact YAP signaling.
Our finding that CDC42-mediated polarity directly regulates YAP signaling in small intestine will inform studies of YAP signaling during intestinal homeostasis and in diseased conditions such as colon cancer. A recent publication shows that YAP overexpression, driven by genetic loss of Lats1/2, triggers a wound healing signature (Cheung et al., 2020), which is also observed in ISCs and TA cells in our CDC42 ISC-KO mice (data not shown). However, increased YAP activity appeared to serve as a tumor suppressor in the context of a defined oncogenic driver (Cheung et al., 2020), differing from our observation that significantly elevated YAP signaling in CDC42 ISC-KO is sufficient to drive hyperproliferation in intestinal crypts. Thus, this work, together with previous studies, suggests that loss of polarity-induced over-activation of YAP signaling may cause intestinal hyperplasia and, thereby, contribute to intestinal transformation.
CDC42 regulates proliferation and ISC/TA cell fate via a YAP-EGFR-mTOR signaling cascade
While the role of EGFR signaling in the mammalian intestine is largely unknown, EGFR regulates stem and progenitor cell proliferation and differentiation in Drosophila midgut (Jiang et al., 2016). Furthermore, EGFR ligand Ereg is a YAP target in intestinal crypts (Gregorieff et al., 2015). Indeed, in CDC42 ISC-KO ISC and TA cell populations we see, in addition to elevated YAP signaling, a significant increase in Ereg and also EGFR signaling, and recent observations indicate that Neuregulin 1, one of four proteins in the neuregulin family that act on the EGFR family of receptors, can promote ISC proliferation and epithelial regeneration after injury (Abud et al., 2019). Using the EGFR inhibitor afatinib, we demonstrate that EGFR signaling regulates proliferation and ISC/TA cell fate in crypts. Our high-throughput scRNA-seq analysis revealed that mTOR signaling, which is known to be transduced by EGFR activity (Wee and Wang, 2017), was also increased in CDC42 KO ISCs and TA cells and was required for the proliferation phenotype.
Thus, our results present a model in which ISC maintenance and TA progenitor proliferation and cell fate are regulated by an apical/basal lateral polarity-mediated Hippo-EGFR-mTOR signaling cascade that does not require canonical Wnt signaling. This work raises an interesting possibility that defective ISC/TA cell polarity may play a role in driving normal cells toward hyperproliferation.
Limitations of the study
For the loss-of-polarity experiment, we used villin-CreERT2 to achieve Scribble deletion in the whole intestinal epithelium. While the initial observations from this model showed consistent crypt and ISC/TA cell phenotypes to those in the model of ISC-specific deletion of CDC42 by Olmf4-CreER, future studies using ISC-specific loss of polarity molecules including Scribble are needed to further dissect the role of polarity in ISC regulation. In addition, UMAP is a non-linear dimensionality reduction method that is limited in drawing quantitative conclusions. We have annotated the clusters using marker gene sets for each intestinal cell type, and also treated the data based on the marker gene sets from the raw data and relied on other analyses by morphologies, immunofluorescence, FACS, and RT-PCR to reach a consistent conclusion.
STAR*METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yi Zheng (yi.zheng@cchmc.org).
Materials availability
This study did not generate new unique reagents. All reagents involved in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit anti- YAP1 (D8H1X) | Cell Signaling | Cat# 14074 |
| Rabbit anti- cleaved caspase 3 | Cell Signaling | Cat# 9661 |
| Rabbit anti- p-histone H3 | Cell Signaling | Cat# 3377 |
| Rabbit anti- Olfm4 | Cell Signaling | Cat# 39141 |
| Mouse anti- E-cadherin | BD Biosciences | Cat# 610182 |
| Mouse anti- beta-catenin | BD Biosciences | Cat# 610153 |
| Mouse anti- alpha E-catenin | Santa Cruz | Cat# SC-9988 |
| Rabbit anti- Pcna | Santa Cruz | Cat# SC-56 |
| Rabbit anti- Na+K+ATPase | MBL | Cat# MC-9784 |
| Rabbit anti- Ki67 | Abcam | Cat# AB16667 |
| Rabbit anti- Lysozyme | Dako | Cat# A0099 |
| Rabbit anti- Cdc42 | Cell Signaling | Cat# 2466 |
| Rabbit anti- phospho-YAP (S127) | Cell Signaling | Cat# 13008 |
| Rabbit anti-phospho-MST1 (Thr183)/MST2 (Thr180) | Cell Signaling | Cat# 49332 |
| Rabbit anti- MST 1/2 | Bethyl Laboratories | Cat# A300-468A |
| Rabbit anti- phospho-S6 Ribosomal Protein (S235/236) | Cell Signaling | Cat# 4858 |
| Mouse anti- S6 Ribosomal Protein | Cell Signaling | Cat# 2317 |
| Rabbit anti- phospho-4E-BP1 (T37/46) | Cell Signaling | Cat# 2855 |
| Rabbit anti- 4E-BP1 | Cell Signaling | Cat# 9644 |
| Rabbit anti- K48-linkage polyubiquitin | Cell Signaling | Cat# 8081 |
| Rabbit anti- phosphor-EGFR (Y1068) | Cell Signaling | Cat# 3777 |
| Rabbit anti- EGFR | Cell Signaling | Cat# 2232 |
| Rabbit anti- beta-actin | Cell Signaling | Cat# 4970 |
| Rabbit anti- phosphor-Lats-1/2 (Ser909, Ser872) | Invitrogen | Cat# PA5-64591 |
| Rabbit anti- Lats 1/2 | Bethyl Laboratories | Cat# A300-479A |
| Mouse anti- Scribble | Santa Cruz | Cat# SC-55543 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling | Cat# 7076 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling | Cat# 7074 |
| Alexa Fluor 488, Goat anti Rabbit IgG | Invitrogen | Cat# A32731 |
| Alexa Fluor 568, Goat anti Rabbit IgG | Invitrogen | Cat# A11011 |
| Alexa Fluor 633, Goat anti Rabbit IgG | Invitrogen | Cat# A21070 |
| Alexa Fluor 488, Goat anti Mouse IgG | Invitrogen | Cat # A32723 |
| Alexa Fluor 568, Goat anti Mouse IgG | Invitrogen | Cat# A11004 |
| Alexa Fluor 633, Goat anti Mouse IgG | Invitrogen | Cat# A21052 |
| Normal Rabbit IgG | Cell Signaling | Cat #2729 |
| Normal Mouse IgG | Sigma | Cat #12-371 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Tamoxifen | Cayman Chemical | 13258 |
| Rapamycin | ApexBio | A8167 |
| Afatinib | ChemieTek | CT-BW2992 |
| Pierce Phosphatase Inhibitor Mini Tablets | Thermo Fisher | A32957 |
| Simple Stop 1 Phosphatase Inhibitor Cocktail | Gold Biotechnology | GB450 |
| complete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 |
| Alcian Blue | Sigma | A3157 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | Invitrogen | D1306 |
|
| ||
| Critical commercial assays | ||
|
| ||
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | 4368814 |
| TaqMan Gene Expression Master Mix | Applied Biosystems | 4369514 |
| TaqMan Gene Expression Master Mix | Applied Biosystems | 4369514 |
|
| ||
| Deposited data | ||
|
| ||
| Single Cell RNA-seq data | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166736 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Olfm4-IRES-eGFPCreERT2 | Schuijers et al., 2014 | N/A |
| Mouse: CDC42flox/flox | Melendez et al., 2013 | N/A |
| Mouse: villin-creERT2 | Melendez et al., 2013 | N/A |
| Mouse: Rosa26tdTomato | Madisen et al., 2010 | JAX # 007909 |
| Mouse: Yapflox/flox; Tazflox/flox | Xin et al., 2013 | N/A |
| Mouse: Scribbleflox/flox | Hartleben et al., 2012 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers for qPCR (Table S1) | N/A | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | National institutes of Health and the Laboratory for Optical and Computational Instrumentation | https://imagej.nih.gov/ij/ |
| Prism 8.1 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Cell Ranger 1.3.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/1.3 |
| R | R Foundation for Statistical Computing | https://www.r-project.org/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
CDC42flox/flox (Melendez et al., 2013), Olfm4-IRES-eGFPCreERT2 (Schuijers et al., 2014), Yapflox/flox, Tazflox/flox (Xin et al., 2013), and Scribbleflox/flox mice (Hartleben et al., 2012) have been previously described. Villin-CreERT2 mice were provided by Dr. Helen Piwnica-Worms (The University of Texas MD Anderson Cancer Center). Rosa26tdTomato reporter mice (Madisen et al., 2010) were purchased from Jackson Lab. All mice were generated on or extensively backcrossed to the C57/BL6 background.
All mice were bred at the Animal Resources Center at Cincinnati Children’s Hospital Medical Center. Animal protocols were approved by the CCHMC Institutional Animal Care and Use Committee. Mice were housed in a specific pathogen-free breeding barrier. Both male and female mice were used in this study; age-matched 2- to 3-month-old mice were used for in vivo studies. Mice were maintained with a 12 h light/dark cycle and were fed standard chow.
Mice breeding and tissue harvesting
CDC42flox/flox mice (Melendez et al., 2013) were crossed with either Villin-CreERT2 mice or Olfm4-IRES-eGFPCreERT2 mice (Schuijers et al., 2014). Littermate controls were generated by standard pairings. To induce Cre recombinase, tamoxifen (TAM) was injected (TAM; 2 mg in corn oil, IP; Sigma-Aldrich) once per day for 3 consecutive days. Animals were then sacrificed at 1 of 4 time points post- TAM injection: 12 hours (day 0.5), 24 hours (day 1), 72 hours (day 3) or 96 hours (day 4). For lineage tracing experiment, Olfm4-IRES-eGFPCreERT2; CDC42flox/flox and littermate control Olfm4-IRES-eGFPCreERT2; CDC42flox/+ were crossed with Rosa26tdTomato reporter mice. For genetic rescue, Olfm4-IRES-eGFPCreERT2; CDC42flox/flox were crossed with Yapflox/flox, Tazflox/flox mice (Xin et al., 2013). For Scribble deletion in intestinal epithelium, Scribbleflox/flox mice (Hartleben et al., 2012) were crossed with Villin-CreERT2 mice. For the rapamycin rescue experiment, rapamycin (8 mg/kg/day) was injected for 3 consecutive days post-3 days of TAM injection. For EGFR inhibitor rescue test, Afatinib (20 mg/kg/day) was injected for 3 consecutive days 3 days after TAM injection.
Euthanasia was performed with CO2 followed by cervical dislocation. Intestinal tissue was isolated and flushed with PBS. For some experiments, crypt tissue was isolated from the intestinal epithelium, as previously described (Liu et al., 2017).
METHOD DETAILS
H&E staining, in situ hybridization, and immunofluorescence
Intestinal tissue was fixed in 10% formalin or 4% paraformaldehyde overnight at 4°C, embedded in paraffin (H&E and in situ) or cold optimum cutting temperature embedding medium (H&E). Tissues were processed for paraffin-embedding and H&E staining by the CCHMC Digestive Health Center Pathology Core. In situ hybridization was performed in the lab following the published protocol (Gregorieff and Clevers, 2010). Both H&E and in situ images were captured using Nikon scientific microscope AX series.
Intestinal tissue was fixed in 4% paraformaldehyde overnight at 4°C, cryoprotected in 30% sucrose, embedded in OCT compound, and sectioned while frozen at 12 μm.
Images were acquired on a Nikon A1R GaAsP Inverted Confocal Microscope, with signal intensity optimized for control samples and the same settings (laser power/gain/offset) used for KO and rescue tissues. p-histone H3+ cell numbers were counted (20 crypts per mouse in 3 mice) to get the average p-histone H3+ cells per crypt. Results are expressed as the mean ± standard deviation, and significance calculated by Student’s t test (p < 0.05 was considered significant).
Western blot and co-immunoprecipitation (coIP)
Intestinal epithelium from crypts was processed for western blot, as previously described (Liu et al., 2017). For co-IP, lysates (1.5 mg total protein) were incubated at 4°C overnight with protein A/G magnetic beads (20 μl; Thermofisher) conjugated with anti-YAP1 or anti-Scribble antibody. Beads were then washed 3 times in lysis buffer, once with pure water, heated at 95°C for 10 minutes in loading buffer, and applied to 4%–15% SDS-PAGE gels (Biorad) for YAP and Scribble western blot analysis, or MST1/2, Lats1/2 and Scribble western blot analysis, respectively.
RNA extraction and quantitative real-time PCR
mRNA was isolated from intestinal crypts using an RNA mini kit (QIAGEN), concentration was measured by a Nanodrop, and cDNA produced with a cDNA synthesis kit (ABI). Quantitative PCR was performed using SYBR or Taqman qPCR master mix (ABI) on an ABI700 qRT-PCR instrument, and relative expression levels determined with the ΔΔCt method standardized to gapdh. Primer sequences for SYBR reaction were obtained from RTPrimerDB or https://pga.mgh.harvard.edu/primerbank.
Single-cell suspension preparation and RNA library preparation and sequencing
Intestinal epithelium from crypts was resuspended in medium (30% 10x TrypLE and 70% Advanced DMEM/F12), incubated at 37°C for 30 to 45 minutes, and then mixed and passaged through a glass pipette every 10 min while monitoring suspensions with a microscope. Cells were then filtered (40-μm), pelleted (5 min at 300–100 g), washed once with Advanced DMEM/F12, and resuspended in the same medium. The CCFIMC DNA Genotyping and Sequencing Core prepared a 3’ gene expression library for scRNA-seq to obtain gene expression profiles for 4000–5000 cells per sample, using Chromium Single Cell 3’ Reagent Kit (v3; 10x Genomics). The manufacturer protocol found in the CG000183 Rev B user guide was strictly followed, using 14 cycles during section 2.2 cDNA amplification. The NovaSeq 6000 Sequencer was used with the sequencing read parameters designated in the user guide.
Sample demultiplexing, barcode processing, and UMI counting were performed using Cell Ranger 1.3.0 (10x Genomics). Because single-cell data were derived from intestinal tissue samples from 3 different genotypes, BEER (PMID:31636959) and BBKNN (PMID:31400197) were used to remove batch effects from the various runs that included three different genotypes. UMAP was used to visualize all cells in a 2-dimensional plot. Cells were annotated using published marker genes (Haber et al., 2017). Briefly, 200 small cell clusters were identified using k-means method based on UMAP coordinates, the cell type of each cluster annotated by checking the marker genes’ expression value, and small clusters sharing the same cell type merged. This method revealed a total of 7 cell type clusters: enterocyte (Alpi, Apoa1, Apoa4), stem (Lgr5, Olfm4), TA (Pcna, Mki67), goblet (Muc2, Clca3), endocrine (Chga, Chgb), tuft (Dclk1, Trpm5), and immune (Cd3g). The FeaturePlot function in Seurat (https://doi.org/10.1016/j.cell.2019.05.031) was used to visualize the expression value of individual genes (eg, Ereg), and Complex Heatmap (PMID:27207943) was used to generate heatmaps of multiple genes in different samples. Expression values in each row of a heatmap were smoothed using an R function named “smooth.spline,” and GSEA (https://doi.org/10.1073/pnas.0506580102) was used to conduct gene set enrichment analysis (Subramanian et al., 2005).
QUANTIFICATION AND STATISTICAL ANALYSIS
Most of the statistical details can be found in Figure legends. GraphPad Prism 8.0 and Microsoft Excel software was used for statistical analysis. Data were represented as the mean ± SD. Statistical significance was determined by t tests (two-tailed) for two groups. p < 0.05 was considered statistically significant (*** p < 0.001, ** p < 0.005, * p < 0.05, ns: not significant, p > 0.05).
Supplementary Material
Highlights.
Cdc42 deletion in mouse ISCs causes defective polarity and hyperplasia of crypts
CDC42 regulates polarity-Hippo signaling independent of canonical Wnt signaling
Ereg and mTOR mediate CDC42-YAP signaling in the crypts
Apical polarity-YAP signaling maintains ISC and TA cell fate balance
ACKNOWLEDGMENTS
We thank James Johnson for professional assistance with the animal care. We acknowledge Dr. Jose Cancelas for his critical reading of the manuscript and sharing important reagents. The graphical abstract was drafted in BioRender. com with confirmed publication and licensing rights (agreement no. VB22ZGOCGE). This work was supported in part by NIH R01 CA204895, R01 AG063967, and P30 DK078392, the German Research Foundation (DFG) (German Excellence Strategy, CIBSS_EXC-Project 390939984), and Else Kröner-Fresenius-Stiftung.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110009.
REFERENCES
- Abud H, Jarde T, Rosello F, Kurian Arackal T, Chan E, Donoghue J, Abe S, Flores T, Giraud M, Prasko M, et al. (2019). Neuregulin1 promotes intestinal stem cell proliferation and epithelial regeneration following injury. In ISSCR 2019 Annual Meeting. https://pubhtml5.com/oekz/whzq/basic/151-200. [Google Scholar]
- Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V, et al. (2014). YAP/TAZ incorporation in the b-catenin destruction complex orchestrates the Wnt response. Cell 158, 157–170. [DOI] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, and Clevers H (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Barry ER, and Camargo FD (2013). The Hippo superhighway: Signaling crossroads converging on the Hippo/Yap pathway in stem cells and development. Curr. Opin. Cell Biol. 25, 247–253. [DOI] [PubMed] [Google Scholar]
- Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, Fuchs CS, Magness ST, Smits R, Ogino S, et al. (2013). Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 493, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Bonello TT, and Peifer M (2019). Scribble: A master scaffold in polarity, adhesion, synaptogenesis, and proliferation. J. Cell Biol. 218, 742–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Maitra A, Anders RA, Taketo MM, and Pan D (2015). β-Catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 29, 1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll TD, Newton IP, Chen Y, Blow JJ, and Näthke I (2018). Lgr5+ intestinal stem cells reside in an unlicensed G1 phase. J. Cell Biol. 217, 1667–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung P, Xiol J, Dill MT, Yuan WC, Panero R, Roper J, Osorio FG, Maglic D, Li Q, Gurung B, et al. (2020). Regenerative reprogramming of the intestinal stem cell state via Hippo signaling suppresses metastatic colorectal cancer. Cell Stem Cell 27, 590–604.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clattenburg L, Wigerius M, Qi J, Rainey JK, Rourke JL, Muruganandan S, Sinal CJ, and Fawcett JP (2015). NOS1AP functionally associates with YAP to regulate Hippo signaling. Mol. Cell. Biol. 35, 2265–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehart H, and Clevers H (2019). Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 16, 19–34. [DOI] [PubMed] [Google Scholar]
- Gregorieff A, and Clevers H (2010). In situ hybridization to identify gut stem cells. Curr. Protoc. Stem Cell Biol. Chapter 2, Unit 2F.1. [DOI] [PubMed] [Google Scholar]
- Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, and Wrana JL (2015). Yap-dependent reprogramming of Lgr5+ stem cells drives intestinal regeneration and cancer. Nature 526, 715–718. [DOI] [PubMed] [Google Scholar]
- Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y, et al. (2017). A single-cell survey of the small intestinal epithelium. Nature 551, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartleben B, Widmeier E, Wanner N, Schmidts M, Kim ST, Schneider L, Mayer B, Kerjaschki D, Miner JH, Walz G, and Huber TB (2012). Role of the polarity protein Scribble for podocyte differentiation and maintenance. PLoS ONE 7, e36705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong AW, Meng Z, and Guan KL (2016). The Hippo pathway in intestinal regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 13, 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JK, Du W, Shelton SJ, Oldham MC, DiPersio CM, and Klein OD (2017). An FAK-YAP-mTOR signaling axis regulates stem cell-based tissue renewal in mice. Cell Stem Cell 21, 91–106.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imajo M, Miyatake K, Iimura A, Miyamoto A, and Nishida E (2012). A molecular mechanism that links Hippo signalling to the inhibition of Wnt/β-catenin signalling. EMBO J. 31, 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imajo M, Ebisuya M, and Nishida E (2015). Dual role of YAP and TAZ in renewal of the intestinal epithelium. Nat. Cell Biol. 17, 7–19. [DOI] [PubMed] [Google Scholar]
- Itzkovitz S, Lyubimova A, Blat IC, Maynard M, van Es J, Lees J, Jacks T, Clevers H, and van Oudenaarden A (2011). Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat. Cell Biol. 14, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Tian A, and Jiang J (2016). Intestinal stem cell response to injury: Lessons from Drosophila. Cell. Mol. Life Sci. 73, 3337–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Sun Y, Jarugumilli GK, Liu S, Dang K, Cotton JL, Xiol J, Chan PY, DeRan M, Ma L, et al. (2020). Lats1/2 sustain intestinal stem cells and Wnt activation through TEAD-dependent and independent transcription. Cell Stem Cell 26, 675–692.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Zhang Z, Sampson L, Zhou X, Nalapareddy K, Feng Y, Akunuru S, Melendez J, Davis AK, Bi F, et al. (2017). RHOA GTPase controls YAP-mediated EREG signaling in small intestinal stem cell maintenance. Stem Cell Reports 9, 1961–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, and Mostov K (2007). PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell 128, 383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez J, Liu M, Sampson L, Akunuru S, Han X, Vallance J, Witte D, Shroyer N, and Zheng Y (2013). Cdc42 coordinates proliferation, polarity, migration, and differentiation of small intestinal epithelial cells in mice. Gastroenterology 145, 808–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng D, Frank AR, and Jewell JL (2018). mTOR signaling in stem and progenitor cells. Development 145, dev152595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohseni M, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL, Wei C, Frazier M, Samson O, Wong KK, et al. (2014). A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat. Cell Biol. 16, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Gregorieff A, Begthel H, and Clevers H (2003). Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 17, 1709–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas R, Ruiz WG, Leung SM, Jou TS, and Apodaca G (2001). Cdc42-dependent modulation of tight junctions and membrane protein traffic in polarized Madin-Darby canine kidney cells. Mol. Biol. Cell 12, 2257–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson LL, Davis AK, Grogg MW, and Zheng Y (2016). mTOR disruption causes intestinal epithelial cell defects and intestinal atrophy postinjury in mice. FASEB J. 30, 1263–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, and Clevers H (2011). Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers AG, Vries R, van den Born M, van de Wetering M, and Clevers H (2011). Lgr5 intestinal stem cells have high telomerase activity and randomly segregate their chromosomes. EMBO J. 30, 1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuijers J, van der Flier LG, van Es J, and Clevers H (2014). Robust Cre-mediated recombination in small intestinal stem cells utilizing the Olfm4 locus. Stem Cell Reports 3, 234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, and Clevers H (2010). Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143, 134–144. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, et al. (2015). A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 519, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Biehs B, Chiu C, Siebel CW, Wu Y, Costa M, de Sauvage FJ, and Klein OD (2015). Opposing activities of Notch and Wnt signaling regulate intestinal stem cells and gut homeostasis. Cell Rep. 11, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier LG, and Clevers H (2009). Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 71, 241–260. [DOI] [PubMed] [Google Scholar]
- Wee P, and Wang Z (2017). Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel) 9, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, et al. (2013). Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 110, 13839–13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CC, Graves HK, Moya IM, Tao C, Hamaratoglu F, Gladden AB, and Halder G (2015). Differential regulation of the Hippo pathway by adherens junctions and apical-basal cell polarity modules. Proc. Natl. Acad. Sci. USA 112, 1785–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, Allred C, and Muthuswamy SK (2008). Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 135, 865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Holowatyj AN, Roy T, Pronovost SM, Marchetti M, Liu H, Ulrich CM, and Edgar BA (2019). An SH3PX1-dependent endocytosis-autophagy network restrains intestinal stem cell proliferation by counteracting EGFR-ERK signaling. Dev. Cell 49, 574–589.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Zhang Y, Wu H, Barry E, Yin Y, Lawrence E, Dawson D, Willis JE, Markowitz SD, Camargo FD, and Avruch J (2011). Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc. Natl. Acad. Sci. USA 108, E1312–E1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit anti- YAP1 (D8H1X) | Cell Signaling | Cat# 14074 |
| Rabbit anti- cleaved caspase 3 | Cell Signaling | Cat# 9661 |
| Rabbit anti- p-histone H3 | Cell Signaling | Cat# 3377 |
| Rabbit anti- Olfm4 | Cell Signaling | Cat# 39141 |
| Mouse anti- E-cadherin | BD Biosciences | Cat# 610182 |
| Mouse anti- beta-catenin | BD Biosciences | Cat# 610153 |
| Mouse anti- alpha E-catenin | Santa Cruz | Cat# SC-9988 |
| Rabbit anti- Pcna | Santa Cruz | Cat# SC-56 |
| Rabbit anti- Na+K+ATPase | MBL | Cat# MC-9784 |
| Rabbit anti- Ki67 | Abcam | Cat# AB16667 |
| Rabbit anti- Lysozyme | Dako | Cat# A0099 |
| Rabbit anti- Cdc42 | Cell Signaling | Cat# 2466 |
| Rabbit anti- phospho-YAP (S127) | Cell Signaling | Cat# 13008 |
| Rabbit anti-phospho-MST1 (Thr183)/MST2 (Thr180) | Cell Signaling | Cat# 49332 |
| Rabbit anti- MST 1/2 | Bethyl Laboratories | Cat# A300-468A |
| Rabbit anti- phospho-S6 Ribosomal Protein (S235/236) | Cell Signaling | Cat# 4858 |
| Mouse anti- S6 Ribosomal Protein | Cell Signaling | Cat# 2317 |
| Rabbit anti- phospho-4E-BP1 (T37/46) | Cell Signaling | Cat# 2855 |
| Rabbit anti- 4E-BP1 | Cell Signaling | Cat# 9644 |
| Rabbit anti- K48-linkage polyubiquitin | Cell Signaling | Cat# 8081 |
| Rabbit anti- phosphor-EGFR (Y1068) | Cell Signaling | Cat# 3777 |
| Rabbit anti- EGFR | Cell Signaling | Cat# 2232 |
| Rabbit anti- beta-actin | Cell Signaling | Cat# 4970 |
| Rabbit anti- phosphor-Lats-1/2 (Ser909, Ser872) | Invitrogen | Cat# PA5-64591 |
| Rabbit anti- Lats 1/2 | Bethyl Laboratories | Cat# A300-479A |
| Mouse anti- Scribble | Santa Cruz | Cat# SC-55543 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling | Cat# 7076 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling | Cat# 7074 |
| Alexa Fluor 488, Goat anti Rabbit IgG | Invitrogen | Cat# A32731 |
| Alexa Fluor 568, Goat anti Rabbit IgG | Invitrogen | Cat# A11011 |
| Alexa Fluor 633, Goat anti Rabbit IgG | Invitrogen | Cat# A21070 |
| Alexa Fluor 488, Goat anti Mouse IgG | Invitrogen | Cat # A32723 |
| Alexa Fluor 568, Goat anti Mouse IgG | Invitrogen | Cat# A11004 |
| Alexa Fluor 633, Goat anti Mouse IgG | Invitrogen | Cat# A21052 |
| Normal Rabbit IgG | Cell Signaling | Cat #2729 |
| Normal Mouse IgG | Sigma | Cat #12-371 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Tamoxifen | Cayman Chemical | 13258 |
| Rapamycin | ApexBio | A8167 |
| Afatinib | ChemieTek | CT-BW2992 |
| Pierce Phosphatase Inhibitor Mini Tablets | Thermo Fisher | A32957 |
| Simple Stop 1 Phosphatase Inhibitor Cocktail | Gold Biotechnology | GB450 |
| complete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 |
| Alcian Blue | Sigma | A3157 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | Invitrogen | D1306 |
|
| ||
| Critical commercial assays | ||
|
| ||
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | 4368814 |
| TaqMan Gene Expression Master Mix | Applied Biosystems | 4369514 |
| TaqMan Gene Expression Master Mix | Applied Biosystems | 4369514 |
|
| ||
| Deposited data | ||
|
| ||
| Single Cell RNA-seq data | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166736 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Olfm4-IRES-eGFPCreERT2 | Schuijers et al., 2014 | N/A |
| Mouse: CDC42flox/flox | Melendez et al., 2013 | N/A |
| Mouse: villin-creERT2 | Melendez et al., 2013 | N/A |
| Mouse: Rosa26tdTomato | Madisen et al., 2010 | JAX # 007909 |
| Mouse: Yapflox/flox; Tazflox/flox | Xin et al., 2013 | N/A |
| Mouse: Scribbleflox/flox | Hartleben et al., 2012 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers for qPCR (Table S1) | N/A | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | National institutes of Health and the Laboratory for Optical and Computational Instrumentation | https://imagej.nih.gov/ij/ |
| Prism 8.1 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Cell Ranger 1.3.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/1.3 |
| R | R Foundation for Statistical Computing | https://www.r-project.org/ |