

Abstract
Efficient biomarker-driven randomized clinical trials are a key tool for implementing precision oncology. A commonly used biomarker phase III design is focused on testing the treatment effect in biomarker-positive and overall study populations. This approach may result in recommending new treatments to biomarker-negative patients when these treatments have no benefit for these patients.
Advancing treatment in the era of precision medicine requires randomized clinical trial (RCT) designs that can identify biomarkers that can select patients for whom new therapies have favorable risk-to-benefit ratios. These biomarker RCT designs range from enrichment trials, which restrict eligibility to the biomarker-positive patient subgroup, to biomarker-stratified trials, which are designed to perform formal evaluation of the treatment effect in the biomarker-positive and biomarker-negative subgroups separately. The choice of design should generally ensure a rigorous validation of a biomarker’s clinical utility, that is, the biomarker’s ability to accurately distinguish patients who benefit from the experimental treatment vs patients who do not benefit (1). Unfortunately, one commonly used biomarker design is flawed in that respect: the biomarker-positive/overall design is based on formal evaluation of the treatment effect in the biomarker-positive subgroup as well as in the overall population (combining biomarker-positive and biomarker-negative subgroups). The noted problem with biomarker-positive/overall design is that a treatment effect present only in a biomarker-positive subgroup can lead to an observed positive treatment effect in the overall population (2-4). This is due to the fact that when the treatment effect in biomarker-positive subgroup is sufficiently large and/or the biomarker-positive population is sufficiently prevalent, estimates of the treatment effect in the overall population are driven by the biomarker-positive treatment effect. The design could then erroneously recommend the new therapy for the biomarker-negative subgroup when it is not beneficial or is even harmful for these patients. For example, this design was used to compare letrozole plus lapatinib vs letrozole plus placebo in breast cancer in the HER2-positive and overall population (5). The trial reported statistically significant results in the HER2-positive and overall populations, thus allowing formal recommendation of the therapy to the HER2-negative population. However, because there was no clinically meaningful benefit observed in the HER2-negative population, the investigators overruled their prespecified statistical trial design and rightfully concluded that the benefit was limited to HER2-positive patients.
Many biomarkers are measured on an ordinal or continuous scale, with the magnitude of the treatment benefit expected to increase with higher biomarker values. For practical applications where the biomarker is used to guide patient treatment, one needs to define and validate biomarker cutoffs that correspond to distinct treatment decisions. For these biomarkers, the biomarker-positive/overall designs typically evaluate several nested biomarker subgroups defined by increasing values of biomarker cutoffs. For example, for a checkpoint inhibitor, programmed death-ligand 1 (PD-L1) combined positive score (CPS)≥10 and CPS≥1 subgroups as well as the overall population could be tested; often these analyses are done sequentially starting with the highest biomarker subgroup. That is, first the treatment effect is evaluated in the CPS≥10 subgroup. If this is statistically significant (at level α), then the treatment effect is evaluated in the CPS≥1 subgroup; and if this is statistically significant (at level α), then the treatment effect is evaluated in the overall population (at a statistical significance level α).
For a graphical illustration of the potential problem with biomarker-positive/overall designs, consider the KEYNOTE-119 trial (6) of pembrolizumab vs chemotherapy in metastatic triple-negative breast cancer, which was designed to sequentially test CPS≥10, CPS≥1, and the overall populations, with exploratory analysis performed for CPS≥20. Overall survival outcomes from KEYNOTE-119 are summarized in Table 1. Although the formal trial results were negative , the following thought experiment can be useful to demonstrate the problem with the biomarker-positive/overall trial design. Suppose that the true benefit of pembrolizumab was limited to the CPS≥20 subgroup (and was equal to the benefit observed in KEYNOTE-119 in that subgroup). That is, the hazard ratio (HR) is 0.58 for patients with CPS≥20 and 1 (no benefit) for patients with CPS<20 (Figure 1). Then using the observed proportions of deaths in each of the subgroups in the trial, we can approximately estimate what the hazard ratios would be in each subgroup (see Figure 1 legend for details). We estimate that the hazard ratios would have been 0.74, 0.87, and 0.91 for the CPS≥10, CPS≥1, and overall populations, respectively, consistent with the hazard ratios observed in KEYNOTE-119: 0.78, 0.86, and 0.97. The formal results of the KEYNOTE-119 did not reach statistical significance because the P values for the CPS≥10 and CPS≥1 subgroups were not sufficiently small (0.057 and 0.073, respectively). However, one can imagine that with a slightly larger sample size, the trial would have concluded that pembrolizumab was beneficial for patients with CPS≥1, even though the results could be explained by a model where pembrolizumab had no benefit in the 20 > CPS ≥ 1 subgroup.
Table 1.
| PD-L1 CPS subgroup | Sample size | No. of deaths | Overall survival HR (95% CI) |
|---|---|---|---|
| CPS ≥ 20 | 109 | 88 | 0.58 (0.38 to 0.88) |
| CPS ≥ 10 | 194 | 161 | 0.78 (0.57 to 1.06) |
| CPS ≥ 1 | 405 | 354 | 0.86 (0.69 to 1.06) |
| CPS < 1 | 217 | 185 | 1.27 (0.95 to 1.70) |
| Overall | 622 | 539 | 0.97 (0.82 to 1.15) |
CI = confidence interval; PD-L1= programed death-ligand 1; CPS = PD-L1 combined positive score; HR = hazard ratio.
Figure 1.
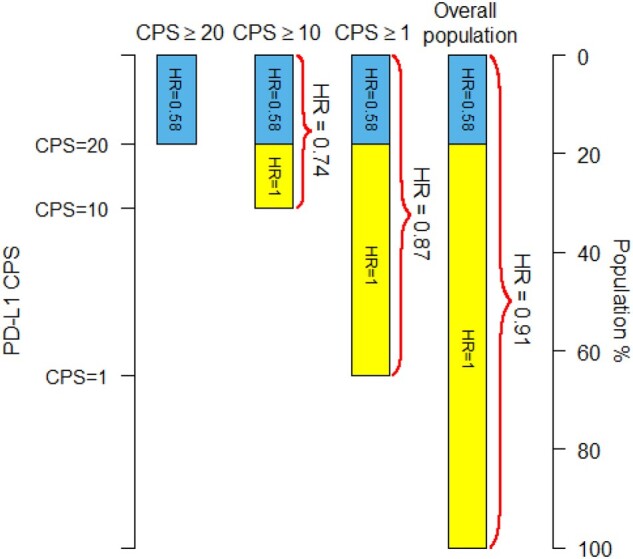
Estimated hazard ratios (HR) for three PD-L1 subgroups and the overall population under the model that the new over standard treatment hazard ratio is 0.58 for the PD-L1 combined positive score (CPS)≥20 subpopulation (blue in the bars) and the hazard ratio is 1.00 (no effect) in the CPS<20 subpopulation (yellow in the bars). The number of patients and number of deaths in each CPS subgroup as well as the value of the hazard ratio for the CPS≥20 subpopulation are taken from the KEYNOTE-119 trial (see Table 1). Hazard ratios are estimated as weighted averages on the log scale of the individual subgroup hazard ratios weighted by the number deaths in each subgroup. The PD-L1 CPS values on the left-hand vertical axis are scaled to the percentiles of the PD-L1 CPS distribution.
Potentially misleading conclusions drawn from a biomarker-positive/overall design are not only a theoretical concern. For example, the investigators of the IMpassion130 trial concluded “Atezolizumab plus nab-paclitaxel prolonged progression-free survival among patients with metastatic triple-negative breast cancer in both the intention-to-treat population and the PD-L1–positive subgroup,” even though the hazard ratio for the PD-L1–negative subgroup was 0.95 (95% confidence interval [CI] = 0.79 to 1.15) (7). Another example is given by the report (8) of IMpower110 assessing atezolizumab in non-small-cell lung cancer (NSCLC), which initially did not report the hazard ratios for the low or intermediate PD-L1 expression subgroups, but only for these subgroups combined with the high PD-L1 subgroup where the treatment was very effective (9). A third example is given by KEYNOTE-042 (10), which assessed pembrolizumab vs chemotherapy in metastatic NSCLC, where the potential pembrolizumab benefits were seen in the sequentially tested PD-L1 tumor proportion score (TPS)≥ 50%, TPS≥20%, and TPS≥1% populations. The conclusion that pembrolizumab should be recommended for all patients with TPS≥1% was questioned (11) because the hazard ratio was 0.92 (95% CI = 0.77 to 1.11) for the TPS 1%–49% subgroup. These trials are not isolated examples: the biomarker (PD-L1)–positive/overall design has been used 20 times in trials published within the last 5 years evaluating pembrolizumab (KEYNOTE-series) and atezolizumab (IM-series).
If the biomarker-positive/overall design is not to be used, what should be done instead? At a minimum, the treatment effect must be assessed in the biomarker-negative subgroup, even if not a properly powered part of the formal statistical design. However, this informal approach can lead to an unfortunate disconnect between the formal statistical conclusion (“the new treatment works for all”) and the informal assessment (“the new treatment works only in the biomarker-positive subgroup”) as illustrated by the lapatinib example above. Another example is given by conflicting regulatory decisions in NSCLC following KEYNOTE-042, with the US Food and Drug Administration expanding the first-line pembrolizumab indication to patients with TSP in the 1%-49% range while the European Medicines Agency declined to do so (12).
Definitive biomarker RCTs should be formally designed to provide adequate assessment of the treatment effect in each relevant biomarker subgroup. In practice, what constitutes adequate assessment and the corresponding design would depend on the overall incidence of the specific cancer type and the prevalence of the relevant biomarker subgroups. With the exception of rare disease settings, a biomarker-stratified trial designed for independent assessment of the treatment effect in each biomarker subgroup should be used. Unlike the biomarker-positive/overall design, the biomarker-stratified design ensures rigorous, adequately powered treatment assessment in each relevant biomarker subgroup by specifying separate sample sizes and analysis plans for each subgroup (note that this may require keeping accrual and/or follow-up open to some subgroups after other subgroups are completed). For example, this approach should be feasible in relatively high-incidence diseases such as breast cancer for biomarkers with subgroup prevalences above 10%-15%. In the lapatinib example where the HER2-positive subpopulation constituted 17.0% of the population, instead of the 1280-patient trial, a biomarker-stratified design of approximately one-half that size could have been used (Table 2). The design would enroll 218 patients in the HER2-postive subgroup targeting a hazard ratio of 0.645 (same target and sample size as used in the actual trial design) and 430 patients in the HER2-negative subgroup targeting a hazard ratio of 0.7. This would have allowed a much more efficient elucidation of the role of lapatinib and the HER2 biomarker. Note that the proposed design targets a hazard ratio of 0.7 in the HER2-negative population (instead of HR = 0.769 targeted for overall population in the actual design) because a hazard ratio of 0.7 represents a more meaningful clinical benefit in this population with a median progression free survival (PFS) of approximately 4 months; targeting a hazard ratio of 0.769 in the HER2-negative subgroup would have required 800 HER2-negative patients but could arguably lead to a statistically significant result that is not clinically meaningful, a different important clinical trial issue.
Table 2.
Actual and alternative biomarker-stratified trial design of lapatinib trial (5)
| Patient population | Actual designa |
Biomarker-stratified designb |
||||
|---|---|---|---|---|---|---|
| Actual sample size | Target HR (power, %) | Formally tested | Target sample size | Target HR (power, %) | Formally tested | |
| Biomarker-positive: HER2-positive | 218 | 0.645 (80) | Yes | 218 | 0.645 (80) | Yes |
| Biomarker-negative: HER2-negative | — | — | No | 430 | 0.700 (90) | Yes |
| Overall | 1280 | 0.769 (90) | Yes | 648 | — | No |
Sequential biomarker-positive/overall design; sequential testing (α = 0.025): first test the biomarker-positive subgroup at the statistical significance threshold level α (with the treatment declared ineffective if the test is not statistically significant). If the biomarker-positive subgroup test is statistically significant then test the overall population at the same statistical significance level α; the treatment is recommended for both biomarker-positive and biomarker-negative subgroups if the overall test is statistically significant and only for the biomarker-positive subgroup otherwise. (This procedure controls the overall type I error of the design at level α). CPS = PD-L1 combined positive score; HR = hazard ratio.
Sequential biomarker-stratified design; sequential testing (α = 0.025): first test the biomarker-positive subgroup at the statistical significance threshold level α (with the treatment declared ineffective if the test is not statistically significant). If the biomarker-positive subgroup test is statistically significant, then test the biomarker-negative subgroup at the same statistical significance level α; the treatment is recommended for both biomarker-positive and biomarker-negative subgroups if the biomarker-negative test is statistically significant and only for the biomarker-positive subgroup otherwise. (This procedure controls the overall type I error of the design at level α).
In the KEYNOTE-119 setting where the potential role of the CPS score had been known before trial initiation (13), a biomarker-stratified trial designed to provide separate evaluations in the CPS≥20, 20 > CPS ≥ 1, and CPS<1 subgroups could have been used. For example, a trial that randomly assigns 194, 420, and 360 patients in the CPS≥20, 20 > CPS ≥ 1, and CPS<1 subgroups, respectively, would have been feasible (Table 3). This 974-patient biomarker-stratified design, although larger than the KEYNOTE-119 622-patient biomarker-positive/overall design, would have provided rigorous validation of the clinical utility for the CPS-score–guided treatment in this setting.
Table 3.
Actual and alternative biomarker-stratified trial design of KEYNOTE-119 (6)
| Patient population | Actual designa |
Biomarker-stratified designb |
||||
|---|---|---|---|---|---|---|
| Actual sample size | Target HR (power, %) | Formally tested | Target sample size | Target HR (power, %) | Formally tested | |
| CPS ≥ 10 | 194 | 0.6 (85) | Yes | — | — | No |
| CPS ≥ 1 | 405 | 0.7 (90) | Yes | — | — | No |
| CPS ≥ 20 | — | — | No | 194 | 0.6 (85) | Yes |
| 20 > CPS ≥ 1 | — | — | No | 420 | 0.7 (90) | Yes |
| CPS < 1 | — | — | No | 360 | 0.7 (85) | Yes |
| Overall | 622 | 0.78 (80) | Yes | 974 | — | No |
Sequential biomarker-positive/overall design: initial α allocation 0.017 and 0.008 to CPS≥10 and CPS≥1 subgroups, respectively, with sequential testing of the overall population if the CPS≥1 subgroup is statistically significant. (This procedure controls the overall type I error of the design at level α). CPS = PD-L1 combined positive score; HR = hazard ratio.
Sequential biomarker-stratified design; fully sequential testing with α = 0.025: first test CPS≥20 subgroup; if statistically significant, then test the 20 > CPS ≥ 1 subgroup, and if statistically significant, then test the CPS<1 subgroup. (This procedure controls the overall type I error of the design at level α).
For practical reasons, in rare disease settings and/or for biomarkers with low prevalence, some compromises in evidentiary standards are necessary to have RCT designs that minimize the probability of incorrectly recommending a new treatment for a biomarker-negative subgroup (3). This can be achieved by using a relaxed statistical significance threshold, for example, with a 1-sided statistical significance level α of 0.05, 0.10, or even 0.15 (instead of the typical α = 0.025), allowing reduction of the required subgroup sample size by 18%, 37%, and 49%, respectively. Furthermore, for settings where the lowest prevalence subgroup(s) is the subgroup(s) with the highest expected benefit (ie, a biomarker-positive subgroup), the required sample size can be reduced by targeting higher treatment effects: for example, targeting a hazard ratio equal to 0.6 (0.5) instead of 0.7 would reduce the required sample size by 50% (74%). In some circumstances, model-based approaches (14) could further assist in elucidating the biomarker and treatment–effect association with reasonable sample sizes.
To sustain precision medicine in oncology, properly powered biomarker-stratified trials designed to assess treatment effects across relevant biomarker subgroups should be used. This would enable validation of the biomarker’s clinical utility and minimize the probability of incorrectly recommending ineffective treatments to patients for whom these treatments are not beneficial.
Funding
Not applicable.
Notes
Role of the funder: Not applicable.
Disclosures: The authors do not have any conflicts of interest to disclose.
Author contributions: Conceptualization: all authors. Data Curation: all authors. Formal Analysis: all authors. Investigation: all authors. Writing-original draft: all authors. Writing-review and edit: all authors.
Data Availability
Data were taken from available publications and as such can be widely accessed.
References
- 1. Freidlin B, Korn EL.. Biomarker enrichment strategies: matching trial design to biomarker credentials. Nat Rev Clin Oncol. 2014;11(2):81–90. [DOI] [PubMed] [Google Scholar]
- 2. Rothmann MD, Zhang JJ, Lu L, Fleming TR.. Testing in a prespecified subgroup and the intent-to-treat population. Drug Inf J. 2012;46(2):175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Freidlin B, Korn EL, Gray R.. Marker Sequential Test (MaST) design. Clin Trials. 2014;11(1):19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fundytus A, Booth CM, Tannock IF.. How low can you go? PD-L1 expression as a biomarker in trials of cancer immunotherapy. Ann Oncol. 2021;32(7):833–836. [DOI] [PubMed] [Google Scholar]
- 5. Johnston S, Pippen J Jr, Pivot X, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2009;27(33):5538–5546. [DOI] [PubMed] [Google Scholar]
- 6. Winer EP, Lipatov O, Im SA, et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(4):499–511. [DOI] [PubMed] [Google Scholar]
- 7. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–2121. [DOI] [PubMed] [Google Scholar]
- 8. Herbst RS, Giaccone G, de Marinis F, et al. Atezolizumab for first-line treatment of PD-L1-selected patients with NSCLC. N Engl J Med. 2020;383(14):1328–1339. [DOI] [PubMed] [Google Scholar]
- 9. Horita N, Fukuda N, Kaneko T.. Atezolizumab for PD-L1-selected patients with NSCLC. N Engl J Med. 2021;384(6):583–584. [DOI] [PubMed] [Google Scholar]
- 10. Mok TSK, Wu YL, Kudaba I, et al. ; KEYNOTE-042 Investigators. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393(10183):1819–1830. [DOI] [PubMed] [Google Scholar]
- 11. Smit EF, de Langen AJ.. Pembrolizumab for all PD-L1-positive NSCLC. Lancet. 2019;393(10183):1776–1778. [DOI] [PubMed] [Google Scholar]
- 12. Doroshow DB, Bhalla S, Beasley MB, et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18(6):345–362. [DOI] [PubMed] [Google Scholar]
- 13. Mittendorf EA, Philips AV, Meric-Bernstam F, et al. PD-L1 expression in triple-negative breast cancer. Med Oncol. 2014;35(1):13–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin R, Thall PF, Yuan Y.. BAGS: a Bayesian adaptive group sequential trial design with subgroup-specific survival comparison. J Am Stat Assoc. 2021;116(533):322–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data were taken from available publications and as such can be widely accessed.