

Abstract
The inositol 1,4,5-trisphosphate receptor (InsP3R) is up-regulated in patients with atrial fibrillation (AF) and InsP3-induced Ca2+ release (IICR) is linked to pro-arrhythmic spontaneous Ca2+ release events. Nevertheless, knowledge of the physiological relevance and regulation of InsP3Rs in atrial muscle is still limited. We hypothesize that InsP3R and NADPH oxidase 2 (NOX2) form a functional signaling domain where NOX2 derived reactive oxygen species (ROS) regulate InsP3R agonist affinity and thereby Ca2+ release. To quantitate the contribution of IICR to atrial excitation-contraction coupling (ECC) atrial myocytes (AMs) were isolated from wild type and NOX2 deficient (Nox2−/−) mice and changes in the cytoplasmic Ca2+ concentration ([Ca2+]i; fluo-4/AM, indo-1) or ROS (2’,7’-dichlorofluorescein, DCF) were monitored by fluorescence microscopy. Superfusion of AMs with angiotensin II (AngII: 1 μmol/L) significantly increased diastolic [Ca2+]i (F/F0, Ctrl: 1.00±0.01, AngII: 1.20±0.03; n=7; p<0.05), the field stimulation induced Ca2+ transient (CaT) amplitude (ΔF/F0, Ctrl: 2.00±0.17, AngII: 2.39±0.22, n=7; p<0.05), and let to the occurrence of spontaneous increases in [Ca2+]i. These changes in [Ca2+]i were suppressed by the InsP3R blocker 2-aminoethoxydiphenyl-borate (2-APB; 1 μmol/L). Concomitantly, AngII induced an increase in ROS production that was sensitive to the NOX2 specific inhibitor gp91ds-tat (1 μmol/L). In NOX2−/− AMs, AngII failed to increase diastolic [Ca2+]i, CaT amplitude, and the frequency of spontaneous Ca2+ increases. Furthermore, the enhancement of CaTs by exposure to membrane permeant InsP3 was abolished by NOX inhibition with apocynin (1 μM). AngII induced IICR in Nox2−/− AMs could be restored by addition of exogenous ROS (tert-butyl hydroperoxide, tBHP: 5 μmol/L). In saponin permeabilized AMs InsP3 (5 μmol/L) induced Ca2+ sparks that increased in frequency in the presence of ROS (InsP3: 9.65 ±1.44 sparks*s−1*(100 μm)−1; InsP3 + tBHP: 10.77 ± 1.5 sparks*s−1*(100 μm)−1; n=5; p<0.05). The combined effect of InsP3 + tBHP was entirely suppressed by 2-APB and Xestospongine C (XeC). Changes in IICR due to InsP3R glutathionylation induced by diamide could be reversed by the reducing agent dithiothreitol (DTT: 1 mmol/L) and prevented by pretreatment with 2-APB, supporting that the ROS-dependent post-translational modification of the InsP3R plays a role in the regulation of ECC. Our data demonstrate that in AMs the InsP3R is under dual control of agonist induced InsP3 and ROS formation and suggest that InsP3 and NOX2-derived ROS co-regulate atrial IICR and ECC in a defined InsP3R/NOX2 signaling domain.
Keywords: inositol 1,4,5-trisphosphate receptor dependent Ca2+ release; NADPH oxidase 2; atrial excitation contraction coupling; signaling domain; Angiotensin II
1. Introduction
Atrial fibrillation is the most common cardiac rhythm disorder. Its prevalence increases in conjunction with aging, obesity, diabetes, and other cardiovascular diseases (hypertension, diastolic dysfunction, heart failure) yet the mechanisms that promote atrial arrhythmia are incompletely understood. Atrial arrhythmia can be induced by the development of spontaneous, propagating trigger events that are generated in atrial muscle cells outside the sino-atrial node and/or by the development of re-entrant excitation due to a shortened refractory period, an attenuated conduction velocity, or obstacles in the conduction path (e.g. fibrosis) [1,2]. The mechanism for triggered events has been linked to the spontaneous release of Ca2+ from the sarcoplasmic reticulum (SR). The spontaneous rise in the intracellular Ca2+ concentration ([Ca2+]i) promotes the activation of the sodium-calcium exchanger (NCX) which extrudes Ca2+ from the cytoplasm to the extracellular space. Due to its electrogenicity (3 Na+ ions in exchange for 1 Ca2+), NCX activity can lead to a depolarization of the membrane potential and thereby can trigger action potentials (APs) [2]. The occurrence of spontaneous Ca2+ release events can increase due to an enhanced SR Ca2+ load, or an increased open probability of the ryanodine receptor (RyR) or the inositol 1,4,5-trisphosphate (InsP3) receptor (InsP3R), the two Ca2+ release channels in the SR. RyRs are the predominant Ca2+ release channels in atrial myocytes responsible for Ca2+ release during atrial excitation-contraction coupling (ECC). Ca2+ release from RyRs is triggered by Ca2+ induced Ca2+ release (CICR) through voltage dependent Ca2+ influx through L-type Ca2+ channels (LTCCs) [3,4].
In atrial tissue InsP3Rs, the second SR Ca2+ release channel of which the type 2 isoform is most prominently expressed, are out-numbered by RyRs [5]. Opening of the channel requires the second messenger InsP3 which together with diacyl-glycerol (DAG), is the product of the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) by phospholipase C (PLC) in response to an agonist dependent activation of inhibitory G-proteins (e.g. Gαq) [6,7]. Due to its dependence on InsP3 and low expression level, InsP3Rs are not believed to contribute directly to the elevation of [Ca2+]i in a substantial way during atrial ECC. Nevertheless, InsP3 induced Ca2+ release (IICR) [8] was shown to result in an increase in diastolic [Ca2+]i, Ca2+ transient (CaT) amplitude [9–11], nuclear [Ca2+]i [12,13], and transcription factor activation [14,15]. Most importantly, IICR has been linked to an increase in spontaneous Ca2+ release events [11,16,17] including spontaneous APs in isolated atrial, ventricular, and stem cell derived myocytes [9–11]. Due to the low density of InsP3Rs, IICR is believed to induce these events by sensitizing RyR channels to Ca2+ and thereby facilitating RyR mediated Ca2+ release events [11,18].
InsP3Rs were shown to be upregulated in atrial tissue of patients and animal models with AF [5,19], where agonists that activate signal transduction pathways linked to an increase in InsP3 production are found at higher levels [20]. Enhanced IICR therefore represents a potential target to attenuate triggered activity in the atrial muscle. Besides its activation through agonist induced second messenger production, little is known about the regulation of InsP3Rs through post-translational protein modifications and its relevance for IICR in atrial tissue under physiological and pathophysiological conditions. A regulation of InsP3Rs has been demonstrated through Ca2+ as well as the Ca2+-regulated proteins calmodulin (CaM) and CaM kinase II (CaMKII) [21–23]. Both exhibit a negative effect on the open probability of the InsP3R channel [22,23]. A reactive oxygen species (ROS) dependent regulation of InsP3R has been described in endothelial cells, platelets, and COS cells [24–26] and was linked to an increase in InsP3Rs affinity to InsP3 [27,28] through receptor glutathionylation [27,29,30], however such regulation has not been described for cardiac muscle.
In atrial myocytes, activators of IICR are agonists of the Gαq coupled receptors like Angiotensin II (AngII) and Endothelin-1 (ET-1), both of which increase the CaT amplitude and the propensity of arrhythmic Ca2+ release events [16]. Interestingly, both agonists also activate NADPH oxidase 2 (NOX2) and thereby promote an increase in the production of ROS [31,32]. ROS in itself are potent regulators of Ca2+ handling proteins and a ROS dependent increase in LTCC and RyR open probability as well as attenuation of SERCA activity have been described [32,33]. However, it remains unknown whether NOX2 dependent ROS production affects InsP3Rs in atrial myocytes and if there is an interplay between the agonist induced ROS production and IICR that affects ECC and Ca2+ release regulation. In this study we tested the hypothesis that NOX2 and InsP3Rs form a functional signaling domain where NOX2 derived ROS regulates InsP3R through post-translational modification and thereby represents a secondary control mechanism for IICR.
2. Materials and Methods
2.1. Cell isolation
Atrial myocytes (AMs) were isolated from 3 to 6 month old male WT (C57/BL6) and NOX2 deficient mice (gp91phox deficient: NOX2−/−; The Jackson Laboratory, Bar Harbor, ME USA [34]). The isolation was performed as previously described [31,35]. Isolated cells were plated on laminin (1 mg/ml, Sigma Aldrich) coated glass coverslips in standard tyrode solution (in mmol/L: NaCl 130, KCl 5.4, CaCl2 1, MgCl2 1.5, Glucose 10, HEPES 5; pH 7.4). Animal procedures were performed with the approval of the IACUC of Rush University and in accordance with the National Institute of Healths’ Guide for the Care and Use of Laboratory Animals.
2.2. Fluorescent imaging of [Ca2+]i and ROS production
To visualize changes of [Ca2+]i, AMs were incubated (15 min) at room temperature with fluo-4 acetoxymethyl ester (fluo-4/AM: 10 µmol/L; excitation/emission 494/506 nm). For ROS measurements cells were loaded with 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (DCF: 10 μmol/L for 30 min at 37°C; excitation/emission 494/506 nm). Changes in [ROS] are presented as F/F0 where F0 represents the DCF signal measured at the beginning of an experiments before agonist stimulation. Confocal [Ca2+]i and epifluorescent ROS measurements were performed and analyzed as previously described [31,35]. Ca2+ transients are presented as background-subtracted fluorescence normalized to the diastolic fluorescence (F0) at the beginning of the recording. CaT amplitudes were quantified as ΔF/F0, where ΔF=F-F0. AMs were field stimulated at 0.5 Hz for the duration of the experiments. Experiments were performed at room temperature (~22 °C). To compare Ca transient and Caffeine transient amplitudes, AMs were loaded (20 min) with the membrane permeable form of the ratiometric dye indo-1/AM (5 μM). After twenty minutes were allowed for de-esterification, field stimulated cells were excited at 360 nm and emission was collected at 410 nm (F410) and 485 nm (F485) using photomultiplier tubes. Fluorescence signals were background subtracted and [Ca2+]i changes expressed as changes in the fluorescent ratio R = F410/F485 [36].
2.3. Permeabilized cells and spark analysis
For membrane permeabilization freshly isolated AMs were exposed to saponin (0.005 %, 30 s) after which the cells were washed and maintained in an internal solution composed of (mmol/L): K aspartate 100, KCl 15, KH2PO4 5, MgATP 5, EGTA 0.35, CaCl2 0.12, MgCl2 0.75, phosphocreatine 10, HEPES 10, fluo-4 pentapotassium salt 0.03, creatine phosphokinase 5 U/ml, dextran (MW: 40,000) 8 %. The pH was adjusted to 7.2 (KOH) [37]. Experiments were performed at room temperature and free [Ca2+]i and [Mg2+]i were calculated to be 150 nmol/L and 1 mmol/L, respectively (Maxchelator, Stanford Univ. Standford, CA USA) [38]. Ca2+ spark events were detected and analyzed using SparkMaster at a threshold of 3.8 times the standard deviation of the background noise [39].
2.4. Quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from mouse atrial tissue using Trizol (Thermo Fisher Scientific)/chloroform and a Beadbug homogenizer. Extracted RNA was dissolved in diethylpyrocarbonate-treated water, stored at −80 °C and used as a template for cDNA synthesis within 24 h. Total RNA (1 μg) was used for cDNA synthesis with the iScript gDNA Clear, cDNA Synthesis Kit (Bio-Rad). The qPCR was performed using a Bio-Rad CFX96 qPCR Instrument. Primers were designed and tested for efficiency prior to quantitation experiments. PCR reactions consisted of first-strand cDNA template, forward and reverse primers (100 nmol/L final concentration) and iQ SYBR Green Supermix (BIO-RAD) in a total volume of 10 μl. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), hypoxanthine guanine phosphoribosyl transferase 1 (HPRT-1), and Peptidylprolyl Isomerase A (PPIA) transcript levels were used as housekeeping genes [31]. For every mRNA quantitation triplicates were obtained as well as a technical repeat. Standard control PCR reactions were carried out to test for contamination. Data analysis was performed using the relative expression software tool (REST 2009, Quiagen) for group-wise comparison and statistical analysis of relative expression levels. Primer sequences for InsP3R2 (Itpr2) are: forward: TGAGTCGGAGAACAGGAAAC; reverse: CTTGTTCACCGTCAGGTACT. Primers for GAPDH, PPIA, HPRT-1 have been previously published [31].
2.5. Chemicals
Saponin, 2-aminoethoxydiphenyl borate (2-APB), Xestospongine C (XeC), tert-butyl hydroperoxide (tBHP), AngII, apocynin (Apo) and dithiothreitol (DTT) were purchased from Sigma Aldrich. Fluo-4/AM, DCF and Indo-1/AM were purchased from Thermo Fisher Scientific (Waltham MA, USA) and diamide was purchased from Tokyo Chemical Industry Co., Tokyo, Japan. gp91ds-tat was purchased form Anaspec Inc, Fremont CA, USA.
2.6. Statistics
All summary data are presented as data cloud plots together with the mean plus standard error of the mean (SEM). The number of experiments (n) refers to the number of cells examined. For each experimental group, cells from at least 2 different cell isolations/animals were used. Significance was evaluated by paired and unpaired t-test or with one-way ANOVA with Dunnette’s or Tuckey’s multiple comparison test. The tests used are stated in the figure legend.
3. Results
3.1. AngII induced increase in Ca2+ transient amplitude and ROS production
Angiotensin II type 1 receptor (AT 1R) couples to Gαq which activates PLC and generates InsP3 and DAG through hydrolysis of PIP2 [40]. Through an alternative pathway AngII also stimulates NOX2 [31]. To determine the effect of AngII on [Ca2+]i, cellular ROS production, and the interdependence of these two signaling pathways, isolated single AMs were loaded with fluo-4 AM or DCF, respectively. Superfusion of atrial myocytes with AngII (1 μmol/L, Fig. 1A) induced a time dependent increase of diastolic [Ca2+]i and CaT amplitude, and an increase of the frequency of spontaneous rises of [Ca2+]i during the declining phase of the CaT (Fig. 1C–E). Treatment of the cells with the InsP3R blocker 2-APB (1 μmol/L) during (Fig. 1A) or prior to AngII superfusion (Fig. 1) reversed (A) or prevented (B) the increase in diastolic [Ca2+]i, CaT amplitude, and frequency of spontaneous [Ca2+]i release events (Fig. 1C–E). In DCF loaded AMs, AngII superfusion increased the production of ROS, reflected in the increase of the slope of the increase of DCF fluorescence (Fig. 2A, 2B). This AngII induced ROS production was prevented by the NOX2 specific inhibitor gp91ds-tat (1 μmol/L; Fig. 2A, 2C). The results support that AngII induces an increase in [Ca2+]i by stimulating IICR concomitant with increased ROS production through activation of NOX2.
Figure 1: AngII induced increase in [Ca2+]i depends on InsP3R.
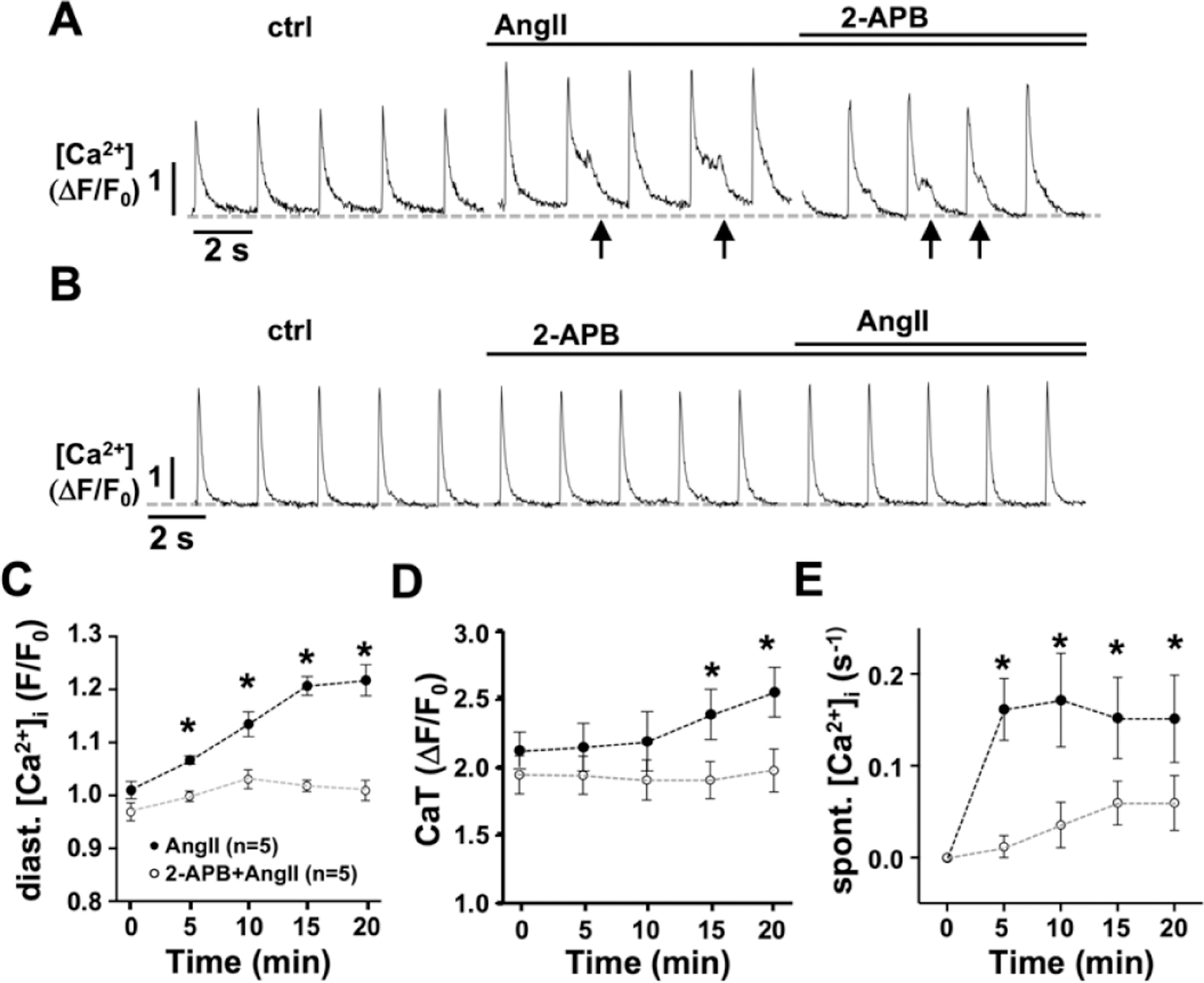
Representative ΔF/F0 plots of AMs during superfusion with AngII (1 μmol/L, 20’) before (A) or after (B) exposure to the InsP3R blocker 2-APB (1 μmol/L; 10’). Time dependent change in diastolic [Ca2+]i (C), the CaT amplitude (D), and the frequency of spontaneous Ca2+ release events (arrows; E) induced by AngII in absence (●) or presence (○) of 2-APB where time 0 is ctrl or ctrl + 2-APB before AngII superfusion. (*: p < 0.05 ANOVA, multiple comparison to time 0)
Figure 2: AngII induces NOX2 dependent ROS production independent of IICR.
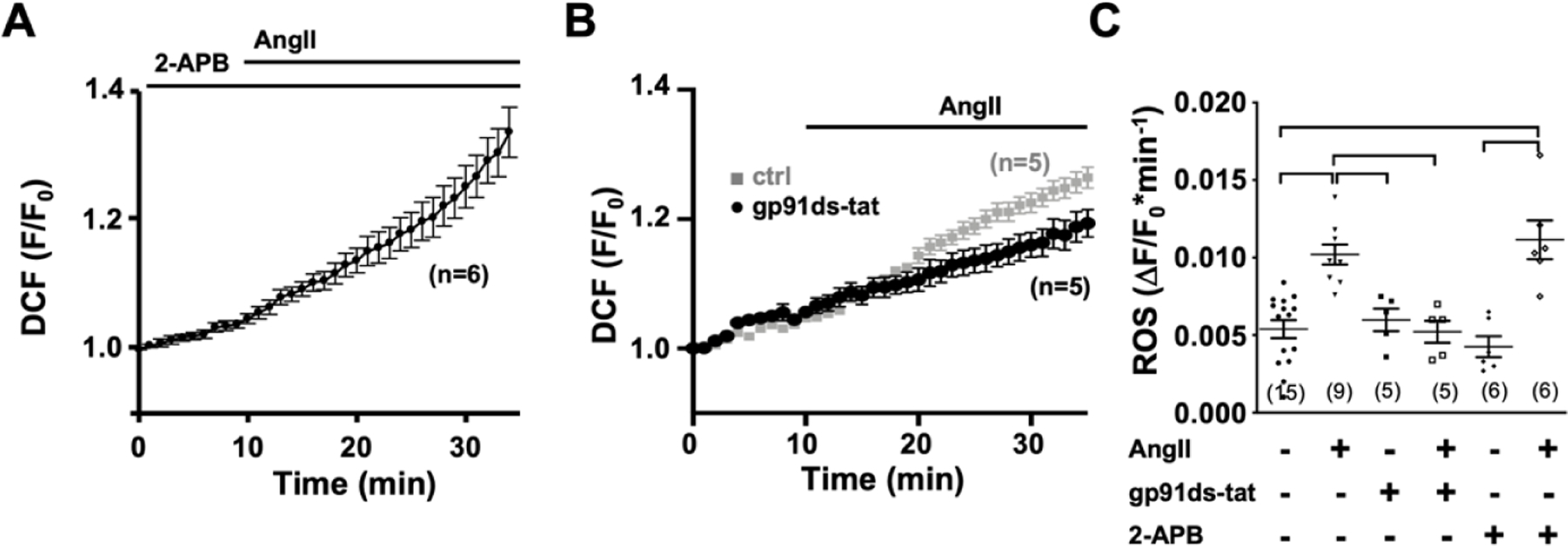
Time dependent change in DCF fluorescence during superfusion of WT cells with AngII in the presence of 2-APB (1 μmol/L; A) or with AngII (1 μmol/L) in presence (●) and absence (■) of gp91ds-tat (1 μmol/L; B). (C) Data cloud plot shows the rate of fluorescence change (obtained over a period of 10 min) in AMs under ctrl (●) conditions and after 10 min of AngII (▼), gp91-ds-tat (■) or 2-APB (◆) superfusion. Or after 10 min of AngII superfusion in the presence of gp91ds-tat (□,10 min) or 2-APB (◇, 10 min). (horizontal lines indicate statistical significance at p < 0.05; ANOVA, multiple comparison).
3.2. Interdependence of AngII induced ROS production and changes in [Ca2+]i
The ROS dependent modulation of cardiac ECC is well established [33] and increased [Ca2+]i can promote GTPase Rac1 (Ras-related C3 botulinum toxin substrate 1) and subsequently lead to NOX2 activation [41]. To determine if the AngII induced increase in [Ca2+]i is a modulator or prerequisite for NOX2 dependent ROS production, we suppressed IICR in atrial myocytes by 2-APB (1 μmol/L) before AngII stimulation (Fig. 2A, 2C). AngII induced ROS production was sustained in the presence of 2-APB suggesting that IICR is not a requirement. To determine if the AngII induced ROS production affects or amplifies AngII induced changes in [Ca2+]i, we used AMs isolated from NOX2 deficient (NOX2−/−) mice, that lack AngII-dependent ROS production [31]. In NOX2−/− AMs AngII (over 15 min) failed to induce an increase in diastolic [Ca2+]i and CaT amplitude, and to trigger spontaneous Ca2+ release events (Figs. 3A, 4E). In NOX2−/− AMs, the AngII induced changes in ECC could be recovered when AMs were superfused with low levels of the organic ROS compound tert-butyl hydroperoxide (tBHP: 5 μmol/L, 10 min) after (Fig. 3A) or prior (Fig. 3B) to AngII stimulation. Pretreatment of AMs with tBHP alone (10 min) did not have an effect on [Ca2+]i (Fig. 3D). The messenger RNA level for InsP3R2 (ITPR2) in NOX2−/− atrial tissue was comparable to that in WT atria (Fig. 4D) suggesting similar receptor expression levels. Superfusion of NOX2−/− AMs with the membrane permeable InsP3R agonist InsP3-AM (1 μmol/L) induced an increase in diastolic [Ca2+]i, CaT amplitude, and spontaneous Ca2+ release events (Fig. 4A–C,E), comparable to InsP3-AM effects on diastolic [Ca2+]i and CaT amplitude in WT AMs (Fig. 5). Interestingly, pre-treatment of WT cells with the NOX inhibitor apocynin (1 μM/L, 5 min) attenuated the InsP3-AM induced increase in [Ca2+]i (Fig. 5B–D). These experimental data support the conclusion that the lack of AngII induced IICR in NOX2−/− AMs was not due to an attenuation of InsP3R2 expression or impaired IICR machinery, rather that NOX2 dependent ROS formation is a prerequisite for an AngII- or InsP3 induced increase in [Ca2+]i through IICR.
Figure 3: In NOX2−/− AMs the AngII induced increase in [Ca2+]i requires ROS.
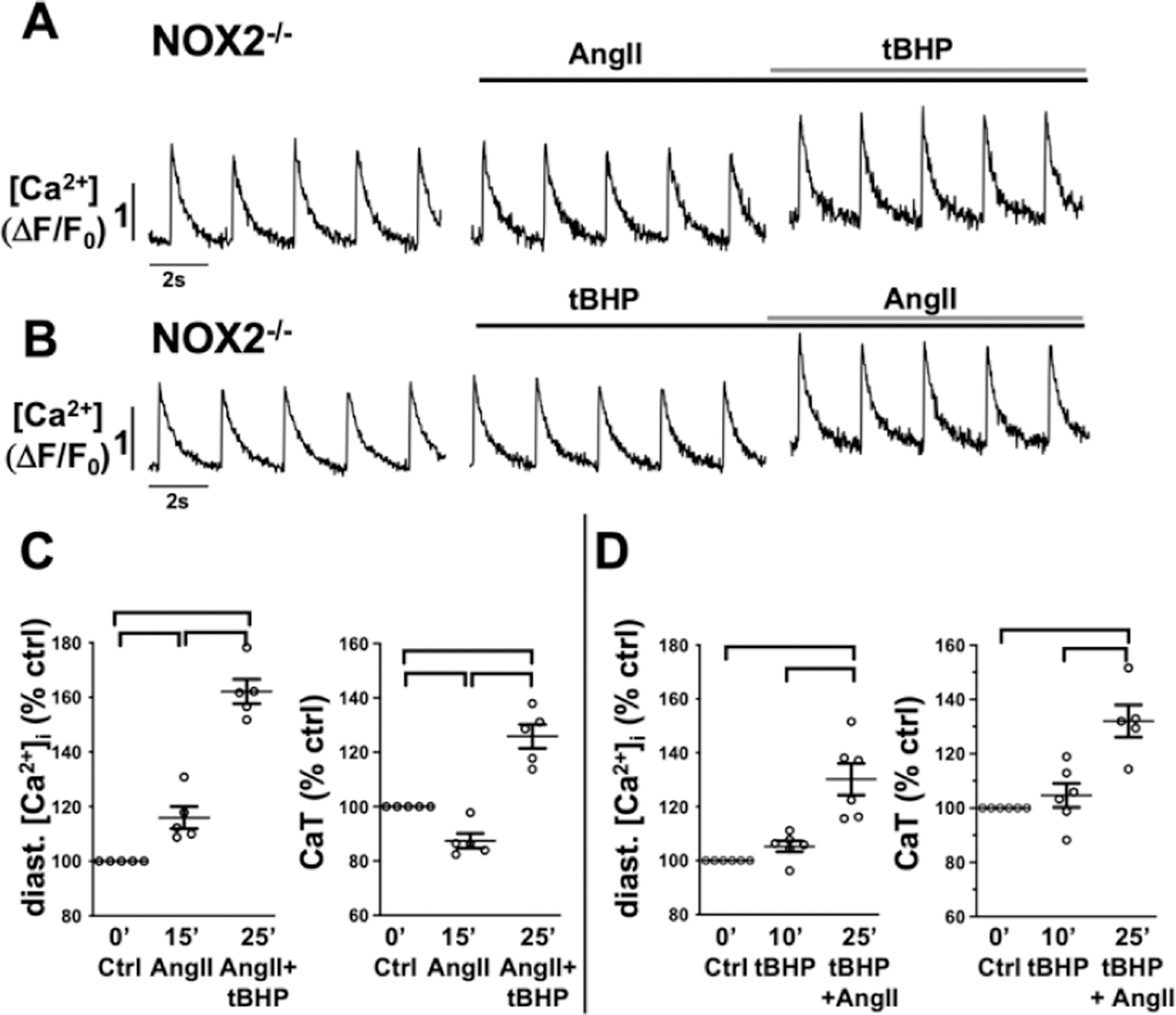
Representative ΔF/F0 plots obtained in AMs isolated from NOX2−/− mice after 15 min of superfusion with (A) AngII (1 μmol/L) and subsequent 10 min of tBHP (5 μmol/L) or (B) 10 min of tBHP and subsequent 15 min of AngII. Data cloud plots show the percent change in diastolic [Ca2+]i and CaT amplitude for (C) AngII + tBHP or (D) tBHP + AngII treated cells. (horizontal lines indicate statistical significance at p < 0.05; ANOVA, multiple comparison).
Figure 4: InsP3R induced Ca2+ release is functional in NOX2−/− AMs.
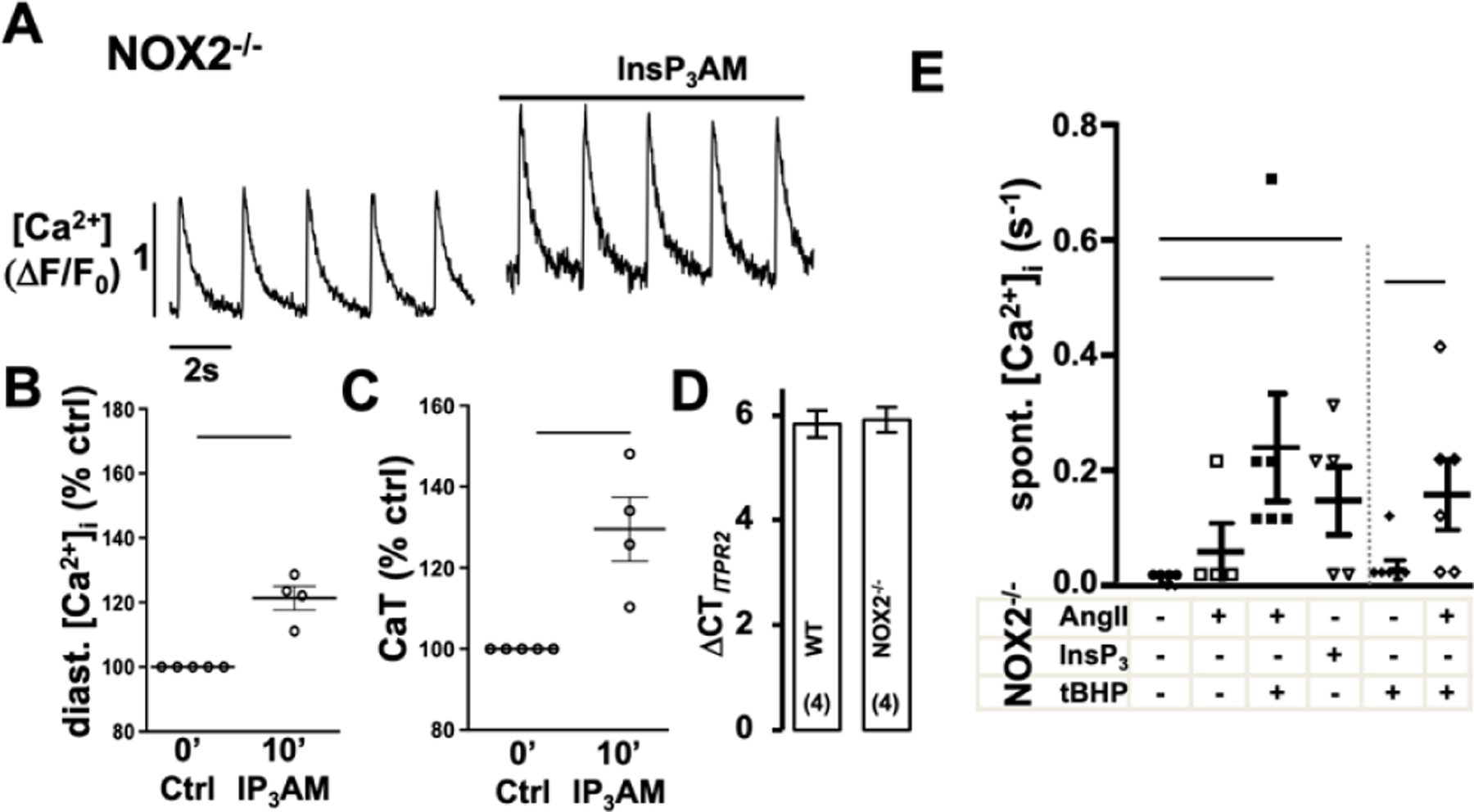
Representative ΔF/F0 plots obtained in AMs isolated from NOX2−/− mice after 10 min of superfusion with (A) InsP3AM (1 μmol/L). Data cloud plots show the percent change in diastolic [Ca2+]i (B) and CaT amplitude (C) after 10 min InsP3AM. (D) Atrial mRNA levels presented as the difference between the threshold cycles of ITPR2, and the average of the housekeeping genes for WT and NOX2−/− mice. (E) Frequency of spontaneous Ca2+ release events during control conditions (●) treatment with AngII (□), InsP3AM (▽) or tBHP (◆); or during treatment with AngII in presence (◇) or after pretreatment with tBHP (■) (horizontal lines indicate statistical significance at p < 0.05; paired t-test (B,C); ANOVA, multiple comparison to ctrl (E)).
Figure 5: In AMs IICR is facilitated by basal ROS production.
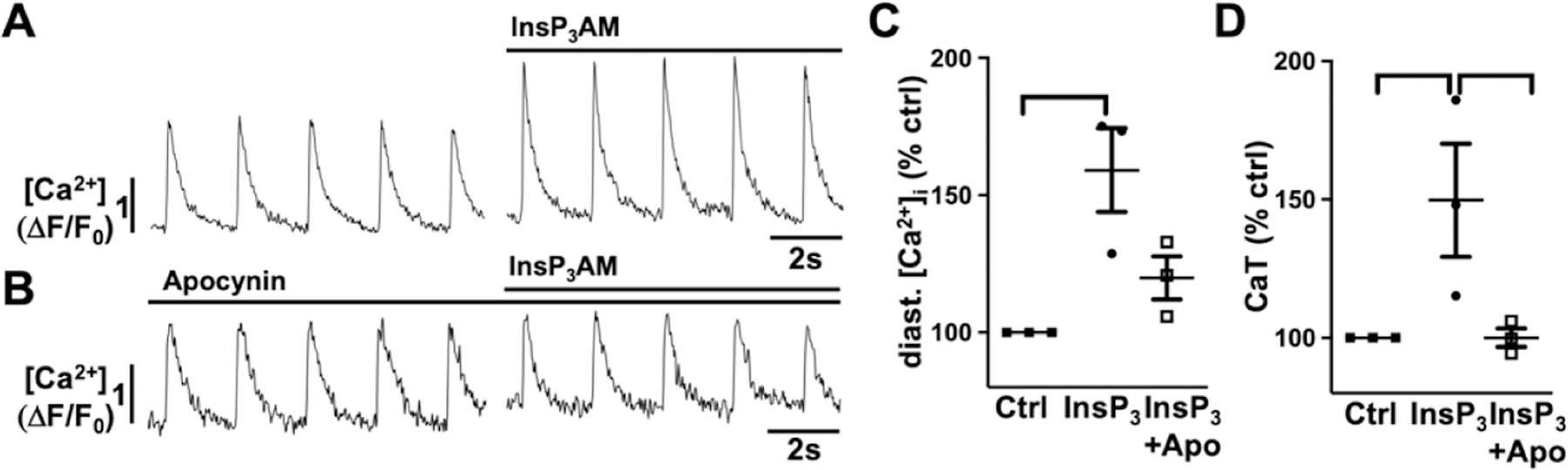
Representative ΔF/F0 plots obtained AMs isolated from control mice after InsP3AM superfusion (15 min; 1 μmol/L) in absence (A) and presence of the NOX inhibitor apocynin (1 μmol/L, B). Data cloud plots show the percent change in diastolic [Ca2+]i (C) and CaT amplitude (D). (horizontal lines indicate statistical significance at p < 0.05; ANOVA, multiple comparison).
3.3. Glutathionylation mimics IICR induced changes of [Ca2+]i
Previous reports demonstrated a regulation of RyRs and InsP3Rs through ROS-dependent glutathionylation [29]. To determine the effect of glutathionylation on atrial Ca2+ release and ECC we superfused AMs with the thiol-oxidizing agent diamide (100 μmol/L, 15 min) which increases protein S-glutathionylation in a concentration-dependent manner [42]. Diamide induced an increase in diastolic [Ca2+]i and the frequency of spontaneous Ca2+ release events (Fig. 6A,C–E). The CaT amplitude initially remained constant but significantly decreased after 15 min of superfusion (Fig. 6D), concomitant with the increase of diastolic [Ca2+]i. Application of the reducing agent dithiothreitol (DTT: 1 mmol/L, 5 min) reversed the effect (Fig. 6A). Diamide induced glutathionylation is unspecific and can affect multiple protein targets. To determine if InsP3R contributes to the diamide induced changes in ECC, AMs were treated with 2-APB (10 min) before diamide superfusion (Fig. 6B). Block of InsP3R prevented the diamide induced changes in ECC (Fig. 6C–E), supporting the conclusion that diamide induced effects are predominantly mediated by the InsP3R.
Figure 6: Diamide induced changes in atrial [Ca2+]i are mediated by IICR.
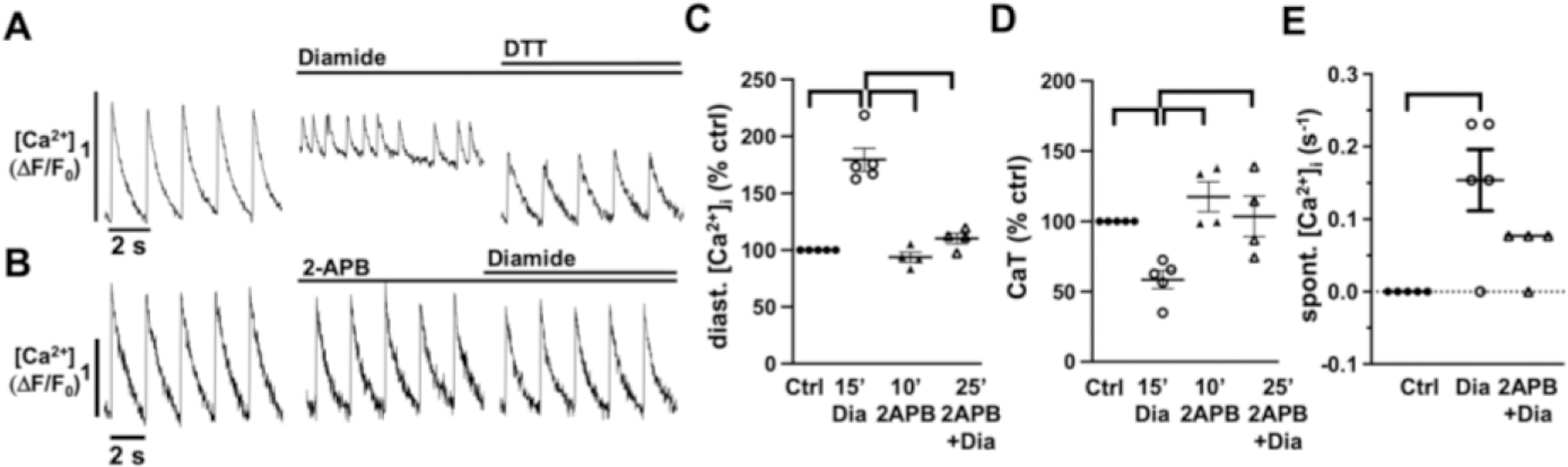
Representative ΔF/F0 plots obtained AMs isolated from control mice after after Diamide (100 μmol/L,15 min) and subsequent DTT (1 mmol/L) superfusion (A). Pretreatment with 2-APB (10 min) prevents the Diamide induced change in [Ca]i (B). Data cloud plots show the percent change in diastolic [Ca2+]i (C), CaT amplitude (D), and spontaneous Ca2+ release events. (horizontal lines indicate statistical significance at p < 0.05, ANOVA, multiple comparison).
3.4. IICR depends on basal cellular ROS production
To distinguish whether the ROS mediated regulation of IICR occurs at the level of PLC or InsP3R, we circumvented potential differences in InsP3 production and superfused permeabilized AMs directly with InsP3 (5 µmol/L) in the presence and absence of ROS (tBHP, 5 μmol/L; Fig. 7A). In permeabilized cells spontaneous spatially restricted Ca2+ release events were characterized by their frequency (Fig. 7B), amplitude, full width at half maximum (FWHM) and full duration at half maximum (FDHM) (Suppl. Fig 1). Based on their amplitude, kinetics, and sensitivity to tetracaine (not shown) these events were identified as Ca2+ sparks, i.e. as elementary Ca2+ release events originating from RyR clusters. InsP3 superfusion induced an increase in spark frequency which was further enhanced by exposure to tBHP (Fig. 7AB). The InsP3- and tBHP induced increase in Ca2+ spark frequency was completely reversed to control levels independently by two different InsP3R blockers, 2-APB or Xestospongin C (XeC: 5 µmol/L). The spark amplitude and kinetics did not change throughout the experiment and neither tBHP nor 2-APB at the concentrations used, had an impact on SR Ca2+ load, as determined by caffeine application (Suppl. Fig.1). The experimental results are in support of a ROS dependent regulation of IICR at the level of the InsP3R.
Figure 7: InsP3 induced spark frequency is enhanced by ROS.
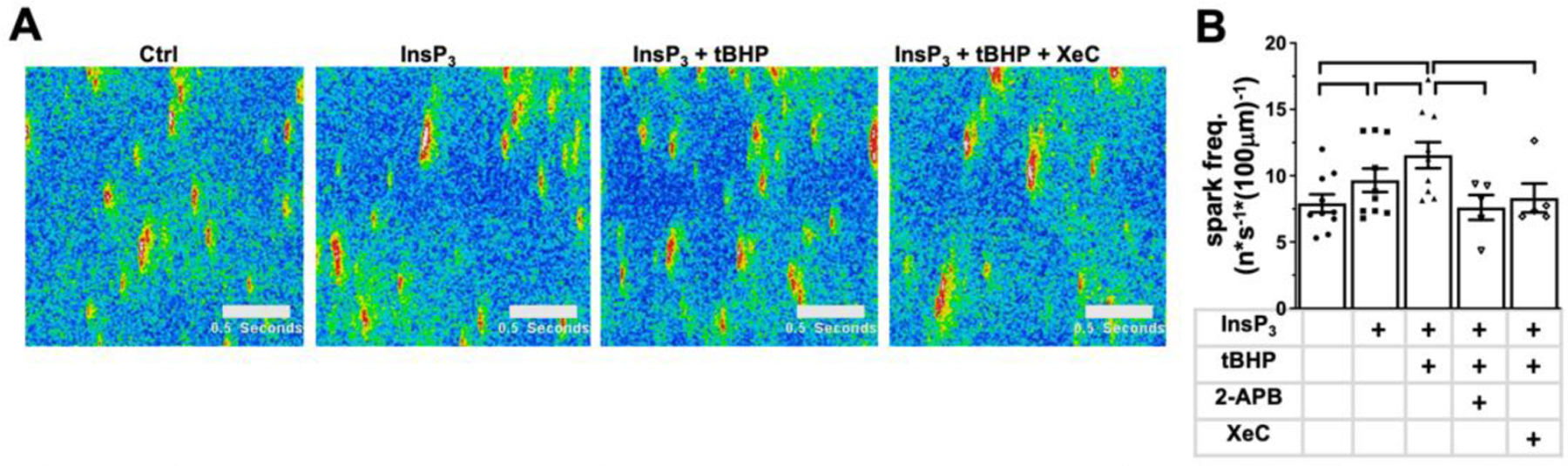
Representative line scan images under control conditions and the conditions indicated above the images (A). Data cloud plots show the spark frequency (B) induced by InsP3 (5 µmol/L), ROS (tBHP: 5 μmol/L), and InsP3R inhibitors 2-APB (5 μmol/L) and Xestospongin C (XeC: 5 μmol/L); number of experiments is listed in brackets. horizontal lines indicate statistical significance at p < 0.05; ANOVA, multiple comparison).
4. Discussion
Previous reports, including our work, demonstrated that AngII induces a NOX2 dependent increase in ROS production and an increase in [Ca2+]i in atrial and ventricular myocytes [31,35,43]. Our new data show a novel interdependence of these two trajectories of AngII induced signaling. While during AngII stimulation NOX2dependent ROS production persists in the absence of IICR, AngII failed to elicit changes in [Ca2+]i in the absence of NOX2/ROS. Here we propose that NOX2, in a functional signaling domain with InsP3Rs, is a prerequisite for AngII induced Ca2+ mobilization and amplifies IICR by increasing InsP3R open probability through ROS-dependent post-translational modification.
4.1. The AngII induced signal transduction pathway
AngII induced signaling in the heart has been linked to pathophysiologial conditions such as fibrosis, hypertrophy, as well as atrial and ventricular arrhythmia [31,44]. In cardiac muscle AngII binds to AT1R, the predominantly expressed receptor isoform [40,45]. AT1R activation leads to G protein and non-G protein-mediated signaling that results in the generation of second messengers such as InsP3, DAG, ROS, arachidonic acid, and phosphatidic acid [44]. InsP3 production depends on the activation of Gαq [45] and leads, as we and others have demonstrated, to an increase of [Ca2+]i (Fig. 1) [31,37]. The mechanism of AngII induced ROS generation is complex and can involve multiple parallel and complementary signaling pathways [46]. We and others have demonstrated that in atrial and ventricular myocytes AngII induces an increase in ROS production, diastolic [Ca2+]i and CaT amplitude, and facilitates spontaneous increase of [Ca2+]i (Fig. 1) [31,35,47]. However, different mechanisms have been proposed leading to the increase of ROS and [Ca2+]i.
4.2. Sources of ROS production
In mouse atrial myocytes we have established an AngII dependent activation of NOX2 by demonstrating a lack of ROS production in the presence of a NOX2 specific inhibitor (Fig. 2), or in the absence of NOX2 (NOX2−/− mice) [35]. An amplification of the AngII induced ROS production has been postulated through NOX4 and mitochondria [Ho:2014ds; 48]. We did not further explore sources of ROS downstream of NOX2 [46], because most relevant for the interpretation of our data was the observation of the complete suppression of AngII induced ROS production with the block or loss of NOX2 activity. Thus, any potential ROS sources downstream of NOX2 hinge entirely on NOX2 activity.
In cells overexpressing signaling components of the PLC pathway, stimulation of PLC contributed to the activation of NOX2 through a DAG-dependent increase in PKC activity [49]. We did not determine if PLC inhibition affects ROS production but tested experimentally ROS production in response to stimulation with the DAG analog 1-oleoyl-2-acetyl-sn-glycerol (OAG) (Suppl. Fig. 3). In line with the previous findings OAG increased cellular ROS production. However, the induced ROS production by itself did not affect [Ca2+]i, further supporting the notion that both ROS and InsP3 are required to induce changes of [Ca2+]i by AngII.
4.3. Effect of ROS on [Ca2+]i
AngII induced changes of [Ca2+]i have been proposed as a consequence of changes in Ca2+ influx as well as SR Ca2+ load and Ca2+ release [43]. As mechanisms underlying the increase of [Ca2+]i the activation of InsP3R, a ROS dependent increase in TRP channel activity, and a ROS dependent activation of the cAMP dependent protein kinase A (PKA) have been proposed [35,43,50]. The latter leads to an enhanced Ca2+ influx through LTCCs and enhanced Ca2+ release through RyRs. The ROS dependent activation of CaMKII could further amplify the AngII induced change of [Ca2+]i by activating the late Na+ current [47]. We reported earlier for mouse and canine atrial myocytes [31], that AngII superfusion increased diastolic [Ca2+]i, the CaT amplitude, SR Ca2+-load, and accelerated CaT decay. The sensitivity of the change of [Ca2+]i to 2-APB supports IICR as the cause of these [Ca2+]i changes. Interestingly, the elimination of NOX2/ROS in mouse AMs prevented also the AngII induced changes of [Ca2+]i. This could indicate that ROS i) increases PLC-dependent InsP3 and DAG production, ii) enhances InsP3R agonist affinity or open probability, and/or iii) modifies other Ca2+ handling proteins and their regulation downstream of IICR (e.g. CaM, CaMKII) [47].
In cardiac tissue the PLC isoforms PLCβ and PLCγ are implicated downstream of AngII stimulation, and a ROS-dependent stimulation of PLCγ has been reported in rat cardiomyocytes [51]. We rule out a significant increase in PLC activity through NOX2/ROS given that an increase in ROS production through the DAG analog OAG (Suppl. Fig. 3), or stimulation of cells with tBHP alone (Fig. 3B) failed to induce changes of [Ca2+]i. Also, the ROS induced increase in Ca2+ spark frequency depended on the presence of InsP3 (Fig. 7), suggesting a direct action of ROS on the InsP3R. A ROS dependent regulation of InsP3Rs has been demonstrated in unexcitable cells [27,30] and glutathionylation of thiol residues in the InsP3R type 1 and type 2 isoforms has been demonstrated [29,52]. Hu et al. [24] attributed the ROS dependent increase in IICR to an increase in InsP3R agonist affinity. In our experiments a regulation of InsP3R through glutathionylation is supported by the prevention of diamide induced changes of [Ca2+]i in the presence of an InsP3R inhibitor (Fig. 6) and the restoration of AngII induced IICR in NOX2−/− myocytes by tBHP superfusion (Fig. 3).
In atrial myocytes InsP3Rs are outnumbered by RyRs [17]. It is therefore believed that IICR rather than contributing directly to the CaT in a quantitatively significant way, sensitizes RyRs to Ca2+ induced Ca2+ release and thereby indirectly increases beat-to-beat changes in [Ca2+]i [9,11,53]. In our study we did not directly show Ca2+ release from InsP3Rs. IICR in cardiomyocytes is difficult to measure directly due to the low expression level of InsP3Rs and the low amplitude of the elementary InsP3R Ca2+ release events, also known as ‘Ca2+ puffs’ [11,37]. Overall this leaves the possibility that AngII induced changes of [Ca2+]i while initiated by IICR, are enhanced by a ROS dependent regulation of RyRs. ROS has been described to increase RyR open probability directly through glutathionylation and indirectly through CaMKII activation and subsequent RyR phosphorylation [54]. Our experiments do not support a direct regulation of RyR by ROS, given that tBHP alone (Fig. 3) did not induce changes in CaT amplitude, spontaneous Ca2+ release events, or SR Ca2+ load (Suppl. Fig.1, 4E) in the absence of IICR. We did not rule out an IICR- or ROS induced CaMKII activation and RyR phosphorylation, however a substantial CaMKII activation seems unlikely as it would through InsP3R phosphorylation attenuate IICR [23,55,56].
4.4. Localization of a InsP3R/NOX2/ROS signaling domain
ROS dependent post-translational modifications have been described for almost all proteins relevant to cardiac ECC [33]. These modifications are often induced experimentally by superfusion of cells with membrane permeable oxidizing agents (e.g. H2O2, tBHP or thimerosal) which can be expected to affect proteins throughout the cytoplasm [57]. However, given the fact that NOX2 and InsP3 production by PLC is restricted to the plasma membrane, and in atrial cells due to the lack or paucity of t-tubules further restricted to the cell periphery, we propose that InsP3Rs and NOX2 are organized in a circumscribed signaling domain where InsP3 and ROS co-regulate InsP3R activity and IICR. We might speculate that this putative signaling domain is located to caveolae given the fact that AngII dependent NOX2 signaling [58], stretch dependent NOX2 activation [59,60], as well as InsP3R [61] have been linked to caveolae signaling platforms that are implicated in numerous signaling activities [62,63].
5. Conclusion
We have demonstrated here for the first time that in atrial myocytes InsP3R is under dual control of InsP3 and ROS. AngII induced InsP3 production in this system is not sufficient to promote a significant inotropic response in the absence of NOX2 stimulation which can occur only through a concomitant activation of NOX2 and ROS production. This dual control mechanism would allow that at constant InsP3 concentrations, local changes in ROS and the redox environment can lead to a spatially variable activation of InsP3Rs through modulation of its agonist affinity. Our experimental data offer a new perspective into the mechanism of InsP3R regulation in atrial myocytes and the fine-tuned regulation of IICR in potential signaling domains.
Supplementary Material
Supplementary data
Highlights.
Modulation of atrial ECC by IICR enhances Ca2+ release and has positive inotropic effects, but also leads to pro-arrhythmic Ca2+ signaling
In atrial myocytes IICR is under a dual control by InsP3 and ROS
InsP3R and NOX2 form a functional signaling domain for the co-regulation of IICR by InsP3 and ROS
6. Acknowledgements:
This work was supported by funding from the National Institutes of Health (HL128330, HL132871 to KB; HL057832, HL132871, HL134781 to LAB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
The authors have nothing to disclose
References
- [1].Heijman J, Voigt N, Nattel S, Dobrev D, Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression, Circ Res 114 (2014) 1483–1499. doi: 10.1161/CIRCRESAHA.114.302226. [DOI] [PubMed] [Google Scholar]
- [2].Denham NC, Pearman CM, Caldwell JL, Madders GWP, Eisner DA, Trafford AW, et al. , Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure, Front Physiol 9 (2018) 1380. doi: 10.3389/fphys.2018.01380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Blatter LA, The intricacies of atrial calcium cycling during excitation-contraction coupling, J Gen Physiol 149 (2017) 857–865. doi: 10.1085/jgp.201711809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Eisner DA, Caldwell JL, Kistamás K, Trafford AW, Calcium and Excitation-Contraction Coupling in the Heart, 121 (2017) 181–195. doi: 10.1161/CIRCRESAHA.117.310230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Guo J-H, Liu Y-S, Zhang H-C, Li X-B, Xu Y, Zhang Y-Y, et al. , [Expression and function changes of ryanodine receptors and inositol 1,4,5-triphosphate receptors of atrial myocytes during atrial fibrillation], Zhonghua Yi Xue Za Zhi 84 (2004) 1196–1199. [PubMed] [Google Scholar]
- [6].Goutsouliak V, Rabkin SW, Angiotensin II-induced inositol phosphate generation is mediated through tyrosine kinase pathways in cardiomyocytes, Cell Signal 9 (1997) 505–512. doi: 10.1016/s0898-6568(97)00008-9. [DOI] [PubMed] [Google Scholar]
- [7].Luo D-L, Gao J, Lan X-M, Wang G, Wei S, Xiao R-P, et al. , Role of inositol 1,4,5-trisphosphate receptors in alpha1-adrenergic receptor-induced cardiomyocyte hypertrophy, Acta Pharmacol Sin 27 (2006) 895–900. doi: 10.1111/j.17457254.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- [8].Kockskamper J, Zima AV, Roderick HL, Pieske B, Blatter LA, Bootman MD, Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes, J. Mol. Cell. Cardiol 45 (2008) 128–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA, Blatter LA, IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes, Am. J. Physiol. Heart Circ. Physiol 294 (2008) H596–604. [DOI] [PubMed] [Google Scholar]
- [10].Kapur N, Banach K, Inositol-1,4,5-trisphosphate-mediated spontaneous activity in mouse embryonic stem cell-derived cardiomyocytes, J. Physiol. (Lond.) 581 (2007) 1113–1127. doi: 10.1113/jphysiol.2006.125955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zima AV, Blatter LA, Inositol-1,4,5-trisphosphate-dependent Ca(2+) signalling in cat atrial excitation-contraction coupling and arrhythmias, J. Physiol. (Lond.) 555 (2004) 607–615. doi: 10.1113/jphysiol.2003.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ljubojevic S, Radulovic S, Leitinger G, Sedej S, Sacherer M, Holzer M, et al. , Early remodeling of perinuclear Ca2+ stores and nucleoplasmic Ca2+ signaling during the development of hypertrophy and heart failure, Circulation 130 (2014) 244–255. doi: 10.1161/CIRCULATIONAHA.114.008927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zima AV, Bare DJ, Mignery GA, Blatter LA, IP3-dependent nuclear Ca2+ signalling in the mammalian heart, J. Physiol. (Lond.) 584 (2007) 601–611. doi: 10.1113/jphysiol.2007.140731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rinne A, Blatter LA, Activation of NFATc1 is directly mediated by IP3 in adult cardiac myocytes, Am. J. Physiol. Heart Circ. Physiol 299 (2010) H1701–7. doi: 10.1152/ajpheart.00470.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, et al. , Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling, J. Clin. Invest 116 (2006) 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li X, Zima AV, Sheikh F, Blatter LA, Chen J, Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice, 96 (2005) 1274–1281. [DOI] [PubMed] [Google Scholar]
- [17].Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W, et al. , Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart, Curr. Biol 10 (2000) 939–942. [DOI] [PubMed] [Google Scholar]
- [18].Wullschleger M, Blanch J, Egger M, Functional local crosstalk of inositol 1,4,5-trisphosphate receptor- and ryanodine receptor-dependent Ca2+ release in atrial cardiomyocytes, Cardiovasc. Res 113 (2017) 542–552. doi: 10.1093/cvr/cvx020. [DOI] [PubMed] [Google Scholar]
- [19].Liang X, Xie H, Zhu P-H, Hu J, Zhao Q, Wang C-S, et al. , Enhanced activity of inositol-1,4,5-trisphosphate receptors in atrial myocytes of atrial fibrillation patients, Cardiology 114 (2009) 180–191. doi: 10.1159/000228584. [DOI] [PubMed] [Google Scholar]
- [20].Cardin S, Li D, Thorin-Trescases N, Leung T-K, Thorin E, Nattel S, Evolution of the atrial fibrillation substrate in experimental congestive heart failure: angiotensin-dependent and -independent pathways, Cardiovasc. Res 60 (2003) 315–325. [DOI] [PubMed] [Google Scholar]
- [21].Bare DJ, Kettlun CS, Liang M, Bers DM, Mignery GA, Cardiac type 2 inositol 1,4,5-trisphosphate receptor: interaction and modulation by calcium/calmodulin-dependent protein kinase II, J. Biol. Chem 280 (2005) 15912–15920. [DOI] [PubMed] [Google Scholar]
- [22].Cardy TJ, Taylor CW, A novel role for calmodulin: Ca2+-independent inhibition of type-1 inositol trisphosphate receptors, Biochem J 334 (Pt 2) (1998) 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Maxwell JT, Natesan S, Mignery GA, Modulation of inositol 1,4,5-trisphosphate receptor type 2 channel activity by Ca2+/calmodulin-dependent protein kinase II (CaMKII)-mediated phosphorylation, J. Biol. Chem 287 (2012) 39419–39428. doi: 10.1074/jbc.M112.374058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hu Q, Zheng G, Zweier JL, Deshpande S, Irani K, Ziegelstein RC, NADPH oxidase activation increases the sensitivity of intracellular Ca2+ stores to inositol 1,4,5-trisphosphate in human endothelial cells, J. Biol. Chem 275 (2000) 15749–15757. doi: 10.1074/jbc.M000381200. [DOI] [PubMed] [Google Scholar]
- [25].Joseph SK, Nakao SK, Sukumvanich S, Reactivity of free thiol groups in type-I inositol trisphosphate receptors, Biochem J 393 (2006) 575–582. doi: 10.1042/BJ20050889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Redondo PC, Salido GM, Rosado JA, Pariente JA, Effect of hydrogen peroxide on Ca2+ mobilisation in human platelets through sulphydryl oxidation dependent and independent mechanisms, Biochem. Pharmacol 67 (2004) 491–502. doi: 10.1016/j.bcp.2003.09.031. [DOI] [PubMed] [Google Scholar]
- [27].Bootman MD, Taylor CW, Berridge MJ, The thiol reagent, thimerosal, evokes Ca2+ spikes in HeLa cells by sensitizing the inositol 1,4,5-trisphosphate receptor, J. Biol. Chem 267 (1992) 25113–25119. [PubMed] [Google Scholar]
- [28].Hu Q, Yu Z-X, Ferrans VJ, Takeda K, Irani K, Ziegelstein RC, Critical role of NADPH oxidase-derived reactive oxygen species in generating Ca2+ oscillations in human aortic endothelial cells stimulated by histamine, J. Biol. Chem 277 (2002) 32546–32551. doi: 10.1074/jbc.M201550200. [DOI] [PubMed] [Google Scholar]
- [29].Joseph SK, Young MP, Alzayady K, Yule DI, Ali M, Booth DM, et al. , Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells, J. Biol. Chem 293 (2018) 17464–17476. doi: 10.1074/jbc.RA118.005624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Joseph SK, Role of thiols in the structure and function of inositol trisphosphate receptors, Curr Top Membr 66C (2010) 299–322. doi: 10.1016/S1063-5823(10)66013-9. [DOI] [PubMed] [Google Scholar]
- [31].Desantiago J, Bare DJ, Varma D, Solaro RJ, Arora R, Banach K, Loss of p21-activated kinase 1 (Pak1) promotes atrial arrhythmic activity, Heart Rhythm 15 (2018) 1233–1241. doi: 10.1016/j.hrthm.2018.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zeng Q, Zhou Q, Yao F, O’Rourke ST, Sun C, Endothelin-1 regulates cardiac L-type calcium channels via NAD(P)H oxidase-derived superoxide, J Pharmacol Exp Ther 326 (2008) 732–738. doi: 10.1124/jpet.108.140301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zima AV, Blatter LA, Redox regulation of cardiac calcium channels and transporters, Cardiovasc. Res 71 (2006) 310–321. [DOI] [PubMed] [Google Scholar]
- [34].Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, et al. , Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production, Nat. Genet 9 (1995) 202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- [35].Desantiago J, Bare DJ, Xiao L, Ke Y, Solaro RJ, Banach K, p21-Activated kinase1 (Pak1) is a negative regulator of NADPH-oxidase 2 in ventricular myocytes, J. Mol. Cell. Cardiol 67 (2014) 77–85. doi: 10.1016/j.yjmcc.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Florea SM, Blatter LA, The role of mitochondria for the regulation of cardiac alternans, Front Physiol 1 (2010) 141. doi: 10.3389/fphys.2010.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hohendanner F, Walther S, Maxwell JT, Kettlewell S, Awad S, Smith GL, et al. , Inositol-1,4,5-trisphosphate induced Ca2+ release and excitation-contraction coupling in atrial myocytes from normal and failing hearts, J. Physiol. (Lond.) (2014). doi: 10.1113/jphysiol.2014.283226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Patton C, Thompson S, Epel D, Some precautions in using chelators to buffer metals in biological solutions, Cell Calcium 35 (2004) 427–431. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- [39].Picht E, Zima AV, Blatter LA, Bers DM, SparkMaster: automated calcium spark analysis with ImageJ, Am. J. Physiol., Cell Physiol 293 (2007) C1073–81. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- [40].Mehta PK, Griendling KK, Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system, Am. J. Physiol., Cell Physiol 292 (2007) C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- [41].Price LS, Langeslag M, ten Klooster JP, Hordijk PL, Jalink K, Collard JG, Calcium signaling regulates translocation and activation of Rac, J. Biol. Chem 278 (2003) 39413–39421. doi: 10.1074/jbc.M302083200. [DOI] [PubMed] [Google Scholar]
- [42].Mazurek SR, Bovo E, Zima AV, Regulation of sarcoplasmic reticulum Ca(2+) release by cytosolic glutathione in rabbit ventricular myocytes, Free Radic Biol Med 68 (2014) 159–167. doi: 10.1016/j.freeradbiomed.2013.12.003. [DOI] [PubMed] [Google Scholar]
- [43].Wagner S, Dantz C, Flebbe H, Azizian A, Sag CM, Engels S, et al. , NADPH oxidase 2 mediates angiotensin II-dependent cellular arrhythmias via PKA and CaMKII, J. Mol. Cell. Cardiol 75 (2014) 206–215. doi: 10.1016/j.yjmcc.2014.07.011. [DOI] [PubMed] [Google Scholar]
- [44].Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, et al. , Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology, Physiol Rev 98 (2018) 1627–1738. doi: 10.1152/physrev.00038.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Aplin M, Christensen GL, Schneider M, Heydorn A, Gammeltoft S, Kjølbye AL, et al. , The angiotensin type 1 receptor activates extracellular signal-regulated kinases 1 and 2 by G protein-dependent and -independent pathways in cardiac myocytes and langendorff-perfused hearts, Basic Clin. Pharmacol. Toxicol 100 (2007) 289–295. doi: 10.1111/j.1742-7843.2007.00063.x. [DOI] [PubMed] [Google Scholar]
- [46].Zhang M, Perino A, Ghigo A, Hirsch E, Shah AM, NADPH Oxidases in Heart Failure: Poachers or Gamekeepers? Antioxid Redox Signal 18 (2012) 1024–1041. doi: 10.1089/ars.2012.4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, et al. , Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload, 108 (2011) 555–565. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Steinhorn B, Sartoretto JL, Sorrentino A, Romero N, Kalwa H, Abel ED, et al. , Insulin-dependent metabolic and inotropic responses in the heart are modulated by hydrogen peroxide from NADPH-oxidase isoforms NOX2 and NOX4, Free Radic Biol Med 113 (2017) 16–25. doi: 10.1016/j.freeradbiomed.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kim EY, Anderson M, Wilson C, Hagmann H, Benzing T, Dryer SE, NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: essential role of podocin in formation of this complex, AJP: Cell Physiology 305 (2013) C960–71. doi: 10.1152/ajpcell.00191.2013. [DOI] [PubMed] [Google Scholar]
- [50].Satoh S, Tanaka H, Ueda Y, Oyama J-I, Sugano M, Sumimoto H, et al. , Transient receptor potential (TRP) protein 7 acts as a G protein-activated Ca2+ channel mediating angiotensin II-induced myocardial apoptosis, Mol Cell Biochem 294 (2007) 205–215. doi: 10.1007/s11010-006-9261-0. [DOI] [PubMed] [Google Scholar]
- [51].Mangat R, Singal T, Dhalla NS, Tappia PS, Inhibition of phospholipase C-gamma 1 augments the decrease in cardiomyocyte viability by H2O2, Am. J. Physiol. Heart Circ. Physiol 291 (2006) H854–60. doi: 10.1152/ajpheart.01205.2005. [DOI] [PubMed] [Google Scholar]
- [52].Bánsághi S, Golenár T, Madesh M, Csordás G, RamachandraRao S, Sharma K, et al. , Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species, J. Biol. Chem 289 (2014) 8170–8181. doi: 10.1074/jbc.M113.504159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kapur N, Banach K, IP3 Mediated Ca-Transients in Embryonic Stem Cell Derived Cardiomyocytes, Biophys J 90 (2006) 2540. [Google Scholar]
- [54].Nikolaienko R, Bovo E, Zima AV, Redox Dependent Modifications of Ryanodine Receptor: Basic Mechanisms and Implications in Heart Diseases, Front Physiol 9 (2018) 1775. doi: 10.3389/fphys.2018.01775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Aromolaran AAS, Blatter LA, Modulation of intracellular Ca2+ release and capacitative Ca2+ entry by CaMKII inhibitors in bovine vascular endothelial cells, Am. J. Physiol., Cell Physiol 289 (2005) C1426–36. doi: 10.1152/ajpcell.00262.2005. [DOI] [PubMed] [Google Scholar]
- [56].Aromolaran AS, Zima AV, Blatter LA, Role of glycolytically generated ATP for CaMKII-mediated regulation of intracellular Ca2+ signaling in bovine vascular endothelial cells, Am. J. Physiol., Cell Physiol 293 (2007) C106–18. doi: 10.1152/ajpcell.00543.2006. [DOI] [PubMed] [Google Scholar]
- [57].Travasso RDM, Sampaio Dos Aidos F, Bayani A, Abranches P, Salvador A, Localized redox relays as a privileged mode of cytoplasmic hydrogen peroxide signaling, Redox Biol 12 (2017) 233–245. doi: 10.1016/j.redox.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lobysheva I, Rath G, Sekkali B, Bouzin C, Feron O, Gallez B, et al. , Moderate caveolin-1 downregulation prevents NADPH oxidase-dependent endothelial nitric oxide synthase uncoupling by angiotensin II in endothelial cells, Arterioscler. Thromb. Vasc. Biol 31 (2011) 2098–2105. doi: 10.1161/ATVBAHA.111.230623. [DOI] [PubMed] [Google Scholar]
- [59].Noel J, Wang H, Hong N, Tao J-Q, Yu K, Sorokina EM, et al. , PECAM-1 and caveolae form the mechanosensing complex necessary for NOX2 activation and angiogenic signaling with stopped flow in pulmonary endothelium, Am J Physiol Lung Cell Mol Physiol 305 (2013) L805–18. doi: 10.1152/ajplung.00123.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang Y, Peng F, Gao B, Ingram AJ, Krepinsky JC, Mechanical strain-induced RhoA activation requires NADPH oxidase-mediated ROS generation in caveolae, Antioxid Redox Signal 13 (2010) 959–973. doi: 10.1089/ars.2009.2908. [DOI] [PubMed] [Google Scholar]
- [61].Fujimoto T, Miyawaki A, Mikoshiba K, Inositol 1,4,5-trisphosphate receptor-like protein in plasmalemmal caveolae is linked to actin filaments, J. Cell. Sci 108 (Pt 1) (1995) 7–15. [DOI] [PubMed] [Google Scholar]
- [62].Parton RG, Caveolae: Structure, Function, and Relationship to Disease, Annu Rev Cell Dev Biol 34 (2018) 111–136. doi: 10.1146/annurev-cellbio-100617-062737. [DOI] [PubMed] [Google Scholar]
- [63].Parton RG, Tillu VA, Collins BM, Caveolae, Curr. Biol 28 (2018) R402–R405. doi: 10.1016/j.cub.2017.11.075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data