

ABSTRACT
The Pim family of serine/threonine kinases promote tumorigenesis by enhancing cell survival and inhibiting apoptosis. Three isoforms exist, Pim-1, -2, and -3, that are highly expressed in hematological cancers, including Pim-1 in adult T-cell leukemia (ATL). Human T-cell leukemia virus type-1 (HTLV-1) is the etiological agent of ATL, a dismal lymphoproliferative disease known as adult T-cell leukemia. The HTLV-1 virally encoded oncogene Tax promotes CD4+ T-cell transformation through disruption of DNA repair pathways and activation of survival and cellular proliferation pathways. In this study, we found Tax increases the expression of Pim-1 and Pim-3, while decreasing Pim-2 expression. Furthermore, we discovered that Pim-1, -2, and -3 bind Tax protein to reduce its expression thereby creating a feedback regulatory loop between these two oncogenes. The loss of Tax expression triggered by Pim kinases led to loss in Tax-mediated transactivation of the HTLV-1 long terminal repeat (LTR) and reductions in HTLV-1 virus replication. Because Tax is also the immunodominant cytotoxic T cell lymphocytes (CTL) target, our data suggest that Pim kinases may play an important role in immune escape of HTLV-1-infected cells.
IMPORTANCE The Pim family of protein kinases have established pro-oncogenic functions. They are often upregulated in cancer; especially leukemias and lymphomas. In addition, a role for Pim kinases in control of virus expression and viral latency is important for Kaposi sarcoma-associated herpesvirus (KSHV) and human immunodeficiency virus type 1 (HIV-1). Our data demonstrate that HTLV-1 encodes viral genes that promote and maintain Pim kinase activation, which in turn may stimulate T-cell transformation and maintain ATL leukemic cell growth. HTLV-1 Tax increases expression of Pim-1 and Pim-3, while decreasing expression of Pim-2. In ATL cells, Pim expression is maintained through extended protein half-life and heat shock protection. In addition, we found that Pim kinases have a new role during HTLV-1 infection. Pim-1, -2, and -3 can subvert Tax expression and HTLV-1 virus production. This may lead to partial suppression of the host immunogenic responses to Tax and favor immune escape of HTLV-1-infected cells. Therefore, Pim kinases have not only pro-oncogenic roles but also favor persistence of the virus-infected cell.
KEYWORDS: ATL, HBZ, Pim kinase, STAT signaling, Tax, human T-cell leukemia virus, leukemia, replication, transformation, virus
INTRODUCTION
Human T-cell leukemia virus type-1 (HTLV-1) is the only known human retrovirus associated with cancer in humans, adult T-cell leukemia (ATL) (1). The most recent epidemiological surveys for HTLV-1 infections, suggests that 5 to 10 million people worldwide are carriers of HTLV-1. However, the HTLV-1 infections are likely higher, because this estimate is nearly a decade old and based upon a smaller worldwide population, and does not consider newly endemic areas (2). Therefore, the true amount of global HTLV-1 carriers is likely much higher. It is projected that 5% of HTLV-1 infected individuals, and 25% of perinatal HTLV-1 carriers, have a lifetime risk of developing ATL (3). HTLV-1 encodes regulatory and accessory proteins from the pX region of the viral genome. These include Tax, the main viral transactivating protein, and Rex, involved with viral mRNA export, and the accessory proteins p30, p12, p13, and p8. In addition, HTLV-1 encodes the hemoglobin subunit zeta (HBZ) gene, which can suppress Tax expression and is important for the immunophenotype of infected cells (4). Despite the central role of Tax as an transducer of HTLV-1 transcription, and as an oncogene and co-activator of cellular pathways, Tax is often downregulated in ATL patient’s cells due to the high Tax-specific cytotoxic T-cell response (CTL) response (5). Consequently, Tax expression is low in vivo in most ATL patient’s cells and is sporadically expressed during the cell cycle (6, 7). In contrast, HBZ is expressed in ATL patient’s cells and some HTLV-1 carriers’ cells; and may have a role in leukemic cell persistence and viral latency (8, 9). This interplay between Tax and HBZ likely accounts for the transformation and persistence of ATL cells.
Numerous cellular pathways are de-regulated in ATL patients infected cells including the JAK/STAT, PI3K/AKT, NF-κB, AP-1, NF-AT, Rho-GTPases pathways, Notch/FBXW7, telomerase, and p53 (10–17). Pim-1 is also altered in ATL cells. The Pim-1 kinase is a serine/threonine kinase that is pro-oncogenic. Pim-1 functions in proliferation and pro-survival activities by phosphorylating targets such as c-Myc, BAD, Bcl-2, p21, p27, p70SK6, and EIF4B (18). Pim-1 is often upregulated in cancer, and has been linked to drug resistance (19). We have previously shown that ATL patient samples express high levels of Pim-1 kinase (20). We found that Pim-1 expression strongly correlates with the activation of the JAK/STAT pathway, especially expression of the transcription factor, STAT3, in ATL patients. We have also found that treatment with pan-Pim kinase inhibitors leads to decreased ATL cell proliferation and ATL tumor burden in vivo; clearly demonstrating the importance of Pim kinases in ATL tumor growth (20). Further studies have found pan Pim kinase inhibitors could also decrease TSP cell growth (21). In addition to Pim-1, the Pim kinase family consists of two other proto-oncogenes, Pim-2, and Pim-3. These isoforms have similar, yet distinct roles in promoting cellular proliferation, viability, translation, and migration (22). Pim-3 has been shown to be expressed in HTLV-1 infected cells (23). In addition to their proto-oncogenic role, the Pim kinases have garnered attention for their roles in viral latency. Pim-1 has been shown to be a key regulator in human immunodeficiency virus type 1 (HIV-1) latency control; able to re-activate HIV-1 in T-cell lines and primary CD4+ T-cells through alteration in transcription factor(s) binding to the HIV-1 LTR (24). Pim-1 is upregulated in Kaposi sarcoma-associated herpesvirus (KSHV) expressing cells, partially through latency-associated nuclear antigen (LANA) induced transcriptional activation (25). It was found that Pim-1 and Pim-3 were bound to LANA only in activated, non-latent, cells; and that Pim-1 and Pim-3, in turn, could phosphorylate LANA, leading to KSHV re-activation (26). In addition, infection of primary bone marrow CD34+ cells with KSHV, leads to significant upregulation of Pim-2 as part of the cellular signature of virally infected cells (27). Epstein-Barr virus (EBV) infected cells can upregulate Pim-1 and Pim-2 expression and were able to increase EBV nuclear antigen (EBNA) transcriptional activity (28). It was also recently shown that Pim-1 and Pim-3 can phosphorylate viral protein X (Vpx) of the lentiviruses, HIV-2, and Simian immunodeficiency virus (SIV) (29). This phosphorylation leads to enhanced binding and degradation of the lentiviral restriction factor, sterile alpha motif, and histidine-aspartate domain-containing protein 1 (SAMHD1); and in turn Pim kinase inhibitors could block lentiviral replication.
Given the dynamic interplay between Pim kinases and oncogenic viruses, and the fact that Pim-1 kinase is expressed in ATL patient samples, we sought to determine whether HTLV-1 viral genes play a role in Pim kinase regulation. Alternatively, we also examined whether Pim kinases may interact with the HTLV-1 virus or viral genes, thereby altering HTLV-1 gene expression, activation, or infectivity. Our results demonstrated that ATL cells and HTLV-1 derived cells expressed Pim-1, -2, and -3. We found Tax increases the expression of Pim-1 and Pim-3, while decreasing Pim-2 expression. However, we also found that Pim-1, -2, and -3 could bind Tax, and decrease Tax expression, thereby dampening its transactivation activities. We further demonstrate that Pim kinases decrease HTLV-1 replication and infectivity, suggesting that Pim kinases are important in maintaining HTLV-1 viral latency in ATL patients that over-express Pim kinases. In ATL cells that do not express high levels of Tax, we found Pim kinase expression to be stabilized, and regulated by heat shock proteins. These data suggest that targeting Pim kinases, in combination with anti-retroviral therapy, may be a viable strategy aimed to reactivate viral expression and unmask virally infected cells to the host immune response.
RESULTS
A feedback loop between Pim kinases and the HTLV-1 Tax protein.
We have previously shown that in uncultured ATL patient samples and ATL cell lines, Pim-1 is highly expressed (20). Importantly, use of several pan-Pim kinases inhibitors leads to decreased proliferation in ATL-derived cells and HTLV-1 transformed cells and a loss in ATL tumor growth (20, 23). pan-Pim kinase inhibitors can inhibit not only Pim-1, but also Pim-1 isoforms, Pim-2, and Pim-3. Few studies have examined the expression of all three isoforms in HTLV-1 cells. To expand upon this data, we looked at the protein and RNA expression levels of Pim-1, -2, and -3 in ATL-derived cells and HTLV-1 cell lines. We found all three Pim kinases were expressed in cell lines; however, ATL, patient derived cell lines in general displayed higher RNA levels compared with HTLV-1 cell lines (Fig. 1A). Several ATL-derived lines expressed very high amounts of Pim-1 and Pim-2 and are cultured in media supplemented with IL-2 (ATL43T, ATL55T, KK1, LM-Y1, and KOB). IL-2 has been shown to increase Pim-1 and Pim-2 expression (30). However, IL-2 is unlikely to be the sole cause for high Pim expression in these cell lines, as several HTLV-1 cell lines (1185, LAF, MUO4, and SP) are also cultured in IL-2 containing media. Unlike ATL lines, these cell lines are immortalized by HTLV-1 and are dependent upon exogenous IL-2; however, they express Pim-1 and Pim-2 at levels comparable with transformed HTLV-1 lines. ATL-derived cell lines displayed higher expression of Pim kinases compared with normal peripheral blood mononuclear cells (PBMCs), and HL60 and Jurkat (data not shown) cell lines. To compare Pim kinases expression with other cancers, we took advantage of cell line expression data for Pim-1, -2, and -3 from lymphoid and myeloid cells lines deposited in the human protein atlas (31–33). Pim-1 is generally expressed higher in lymphoid cell lines, while Pim-2 is expressed higher in lymphoid and myeloid cell lines compared with other cell/tissue origins. Using Jurkat and HL-60 as a common reference, ATL-derived cell lines were enriched in expression for all three Pim kinases (Fig. 1A and data not shown). Pim-3 generally has less tissue specificity and was expressed at a higher level in all ATL, and HTLV-1 infected cell lines and lymphoid/myeloid cell lines. Examination of Pim kinase protein levels displayed similar findings to the RNA expression data (Fig. 1B). The higher expression of Pim kinases in ATL lines suggests higher Pim kinase activity because Pim kinases lack a regulatory domain and are constitutively active once transcribed (34). K562 was used a reference because Pim-1 is highly enriched in this cell line.
FIG 1.
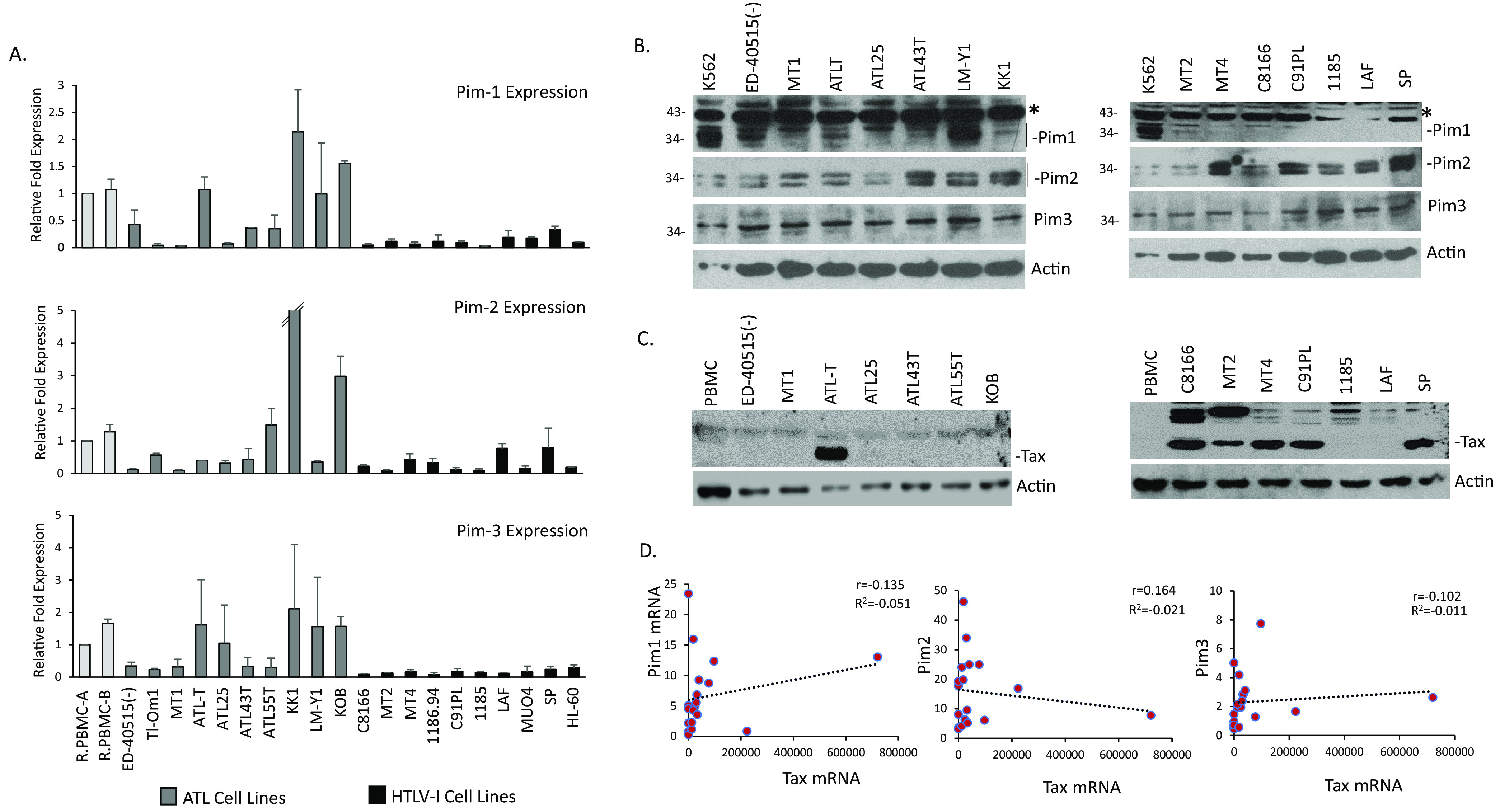
ATL-derived cell lines express high levels of Pim kinases. (A) Real-time PCR expression of Pim-1, Pim-2, and Pim-3 from whole cell extracts of ATL-derived (Gray graphs: Tl-Om1, ED-40515(-), MT1, ATLT, ATL25, ATL43T, ATL55T, KK1, LM-Y1, and KOB) and HTLV-1 (Black graphs: MT2, MT4, C91PL, 1186.94, C8166, 1185, LAF, MUO4, and SP) cell lines. Resting PBMCs (RPBMC) and HL-60 were used as controls. Gene expression was normalized to GAPDH expression and performed at least twice. (B) Protein expression data for Pim kinases in ATL-derived and HTLV-1-derived cell lines. Actin served as a loading control. A nonspecific band was detected for Pim-1. Fisher EZRun protein ladder is marked, when needed. (C) Tax expression in ATL-derived (left blot) and HTLV-1 (right blot) cell lines. Actin served as loading control. (D) Correlation between Tax/Rex gene expression and Pim kinases in ATL-derived and HTLV-1 cell lines. Gene expression was normalized to GAPDH expression; and correlations between genes were determined using Pearson’s correlation coefficient (r).
Unlike most ATL-derived cell lines, HTLV-1 cell lines often express higher levels of the viral protein, Tax (Fig. 1C). Therefore, we hypothesized that HTLV-1 Tax may be responsible for the lower levels of Pim kinases expressed in these cell lines. First, we compared RNA expression data of Pim kinases compared with Tax/Rex mRNA expression. We found a weak, positive correlation between Pim-1/Pim-3 and Tax/Rex, and a similar negative correlation between Pim-2 and Tax/Rex (Fig. 1D). This was unsurprising given that cell lines, in addition to Tax, have de-regulated expression of strong Pim kinase regulators, such as IL-2, IL-6, and the JAK/STAT pathway that may also lead to expression differences in Pim kinases. Therefore, to determine if an association exists between HTLV viral regulators and Pim kinases, we transfected 293T cells with two HTLV-1 viral proteins involved in leukemogenesis, Tax and HBZ. We found that Tax expression substantially increased Pim-1 and Pim-3 protein expression; however, Tax decreased Pim-2 protein expression (Fig. 2A). We did not find any differences in Pim kinase expression following HBZ transfection in our experimental conditions. Because Tax was over-expressed in a non-T-cell line (293T), we compared the level of transfected Tax with the level of endogenous Tax in HTLV-1 cell lines, MT4 and C91PL (Fig. 2B). The level of Tax was higher in Tax-expressing 293T cells; therefore, we decided to use Jurkat TET-Tax cells that express the Tax gene under a doxycycline inducible promoter. We first examined the level of Pim kinases in Jurkat cells and found they expressed low to undetectable amounts of Pim-1 and Pim-2 protein expression, compared to cell lines, such as K562 (Fig. 2C). However, doxycycline-induced Tax expression led to an increase in endogenous Pim-1 and Pim-3 gene levels, while decreasing Pim-2 expression (Fig. 2D). We also found strong increases in Pim-1 and Pim-3 protein expression (Fig. 2E). These results demonstrate that Tax can increase Pim kinases at the RNA level. Similar results between Tax and Pim-3 have been previously found; however, Tax was shown not to appreciably alter Pim-1 or Pim-2 (23). However, further examination did show that Pim-1 gene expression increased slightly, and Pim-2 decreased slightly after short-term and long-term Tax induction, respectively. The discrepancy could be due to Tax specifically altering RNA/protein levels, the experimental conditions (including length and concentration of induction), or the use of a different Tax expressing Jurkat, T-cell lines (in our manuscript we used Jurkat TET-Tax cells, where the other study used Jurkat, JPX9). To further validate our results, we knocked-down Tax expression with Tax shRNA in ATLT cells (Fig. 2F). Tax expression was decreased at the RNA and protein level and led to decreased expression of both Pim-1 and Pim-3, while Pim-2 expression remained unchanged. Further knock-down of Tax may be required to increase Pim-2 in ATLT. Our results demonstrate that Tax can alter the transcriptional level and protein level of Pim kinases; and Tax can regulate the expression of Pim kinases by increasing Pim-1 and Pim-3 levels and decreasing Pim-2 levels. This was found in several different cell lines and under different conditions of Tax expression.
FIG 2.
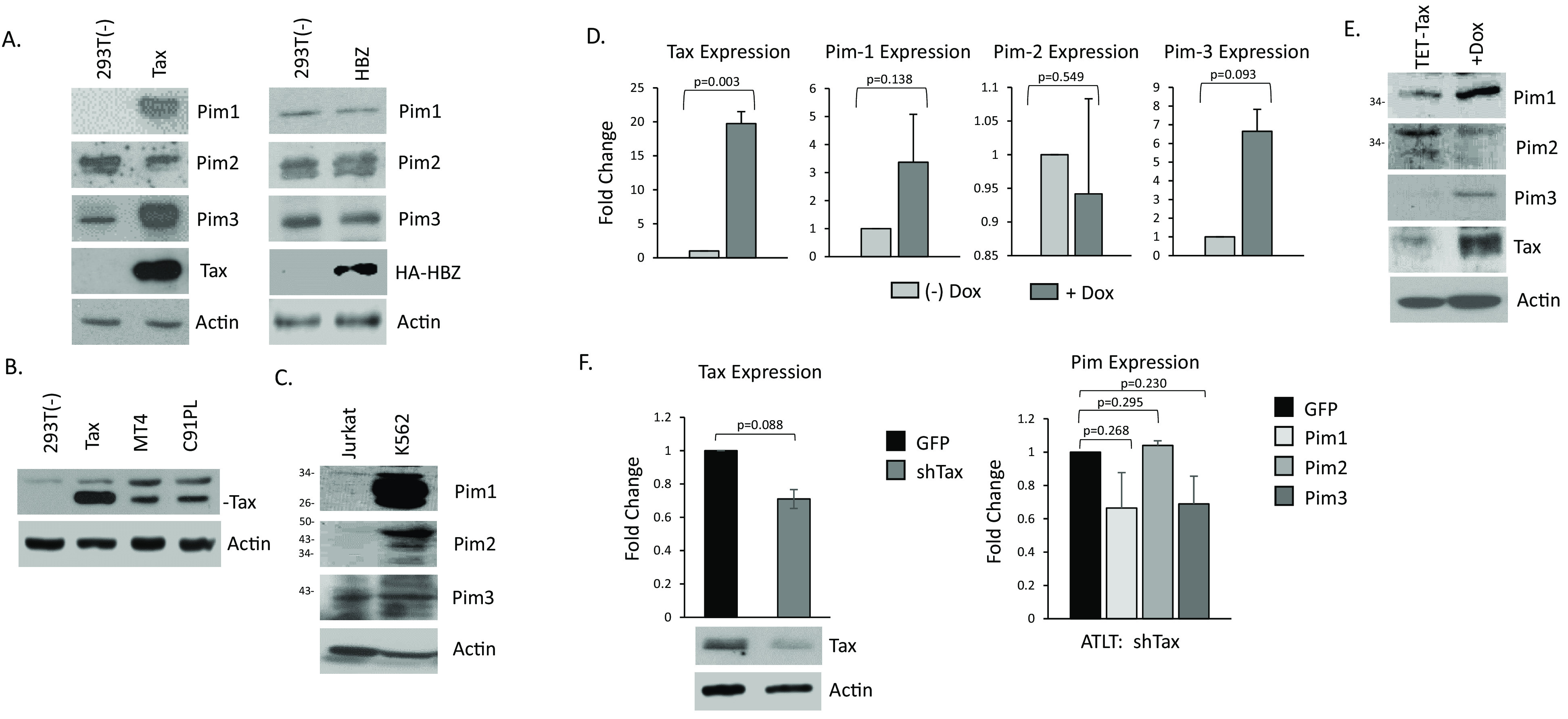
Tax increases Pim-1 and Pim-3 kinase expression, while inhibiting Pim-2. (A) 293T cells were transfected for 48 h with Tax or HBZ expression vectors and then probed for Pim kinases expression. Actin served as a loading control. (B) Comparison of Tax expression between 293T-Tax expressing cells and Tax-positive, HTLV-1 cell lines, MT4 and C91PL. Actin served as a loading control. (C) Western blot analysis of whole cell lysate of Jurkat and K562 cells for Pim expression. Actin served as a loading control. (D) Jurkat TET-Tax lines were induced with or without doxycycline for 6 days. Representation of at least two independent experiments is shown. Gene expression was normalized to GAPDH expression and performed at least twice. (E) Western blots for Pim kinase expression following induction of Jurkat TET-Tax lines. Cells were induced with or without doxycycline for 6 days. Actin served as a control. (F) Knock-down of Tax expression in ATLT cells. Cells were transduced with a Tax shRNA and 72 h later, cells were analyzed for Tax and Pim kinase expression. Experiments were performed at least twice. Gene expression was normalized to GAPDH.
ATL develops over a long latency period of 20 years. Tax is believed to be the main driver of ATL progression; however, due to its high immunogenicity, Tax expression is appreciably altered in late ATL. To examine if Tax could alter Pim kinase expression in newly immortalized T-cells, we transduced normal, healthy PBMCs with a Tax expressing lentivirus. These cells continued to grow for over 4 weeks in IL-2 supplemented media; unlike non-Tax expressing cells which stopped proliferating (35). Analysis of several Tax transduced cells demonstrated that Tax induced the expression of Pim-1 and Pim-3 (Fig. 3A). This was despite the low level of Tax expressed (Fig. 3B). Corresponding to previous data, Pim-2, did not consistently increase in all the Tax-transduced cells. We then examined a mixed population of PBMCs transduced with Tax at even earlier time points of 4 and 7 weeks. Pim kinase expression increased in Tax-transduced PBMCs, even as early as 4-weeks post-transduction (Fig. 3C). The level of Pim kinase induction was comparable with the level of Tax expression, as cells expressing more Tax, expressed more Pim kinases (Fig. 3D). However, even at 7-weeks post-transduction, Pim kinase expression was elevated in most cells expressing Tax. This suggests that Tax induction of Pim kinase expression occurs in newly infected cells and is an early event in HTLV-1 carcinogenesis. This also correlates with Pim kinases as drivers of tumorigenesis.
FIG 3.
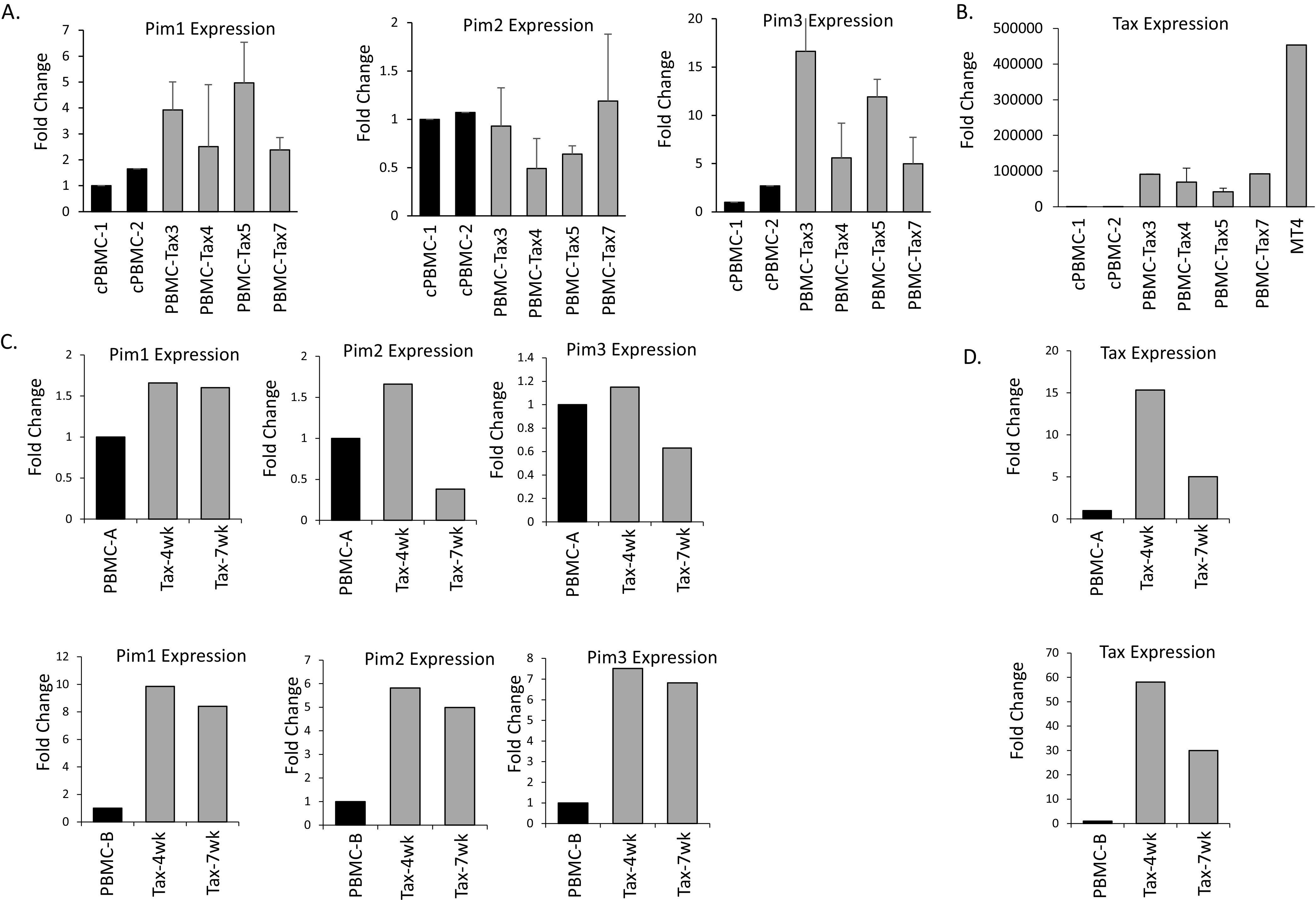
Tax transduction of normal PBMCs induces Pim kinases early after infection. (A–B) Four Tax transduced cultures of PBMCs and two cycling PBMCs (cPBMCs) were compared for Pim kinase expression (A) or Tax expression (B). Gene expression was normalized to GAPDH expression and performed at least twice. For Tax expression, Tax transduced cultures were compared with the MT4 cell line. (C–D) Tax transduced PBMCs express Pim kinases in as little as 4- or 7-weeks post-transduction. Two separate, healthy donor PBMCs were transduced with Tax and grown for 4 or 7 weeks. Gene expression of Pim kinases (C) and Tax (D) were examined at both time points and compared with non-transduced PBMCs at 4 weeks. Gene expression was normalized to GAPDH.
To determine if Pim kinases have a role in regulating HTLV-1 viral genes, we examined the effect of Pim kinases and Tax when both proteins were co-expressed. We transfected 293T cells with expression vectors for Tax and HBZ, along with expression vectors for Pim-1, -2, and -3. In agreement with of our previous results, Tax increased the expression of Pim-1 and Pim-3 expression vectors, while decreasing Pim-2 (Fig. 4A). Despite the positive relationship between Tax and Pim-1 and -3, all three Pim kinases led to strong decreases in Tax expression. This was especially true of Pim-1 and Pim-3 over-expression, which led to a dramatic loss of Tax protein. Examination of HBZ and Pim kinase co-expression showed no change in Pim kinases or HBZ expression (Fig. 4B) suggesting these effects are specific to Tax. To further substantiate these results, we used three shRNA directed toward the Pim-1 gene. All three Pim-1 shRNA led to decreases in Pim-1 expression, albeit it at varying levels (Fig. 4C and D). Importantly, shRNA targeting Pim-1 specifically knocked-down Pim-1 expression and did not affect Pim-2 and Pim-3 expression. 293T cells express very low levels of Pim-1 protein; however, a decrease in Pim-1 could still be seen (Fig. 4D). Because Pim-1 shRNA’s (A) and (B) knocked-down Pim-1 expression the most, we co-transfected these shRNA along with Tax. In cells transfected with Pim-1 shRNA, Tax expression was no longer inhibited at the RNA or protein level (Fig. 4E and F). Knock-down of Pim-1 expression led to increases in Tax expression, suggesting that loss of endogenous Pim-1 is enough to de-repress Tax expression. These results also uncover a novel feedback loop between Tax and Pim kinase, whereby Tax increases Pim-1 and Pim-3, which leads to overall Pim kinase-initiated loss of Tax expression.
FIG 4.
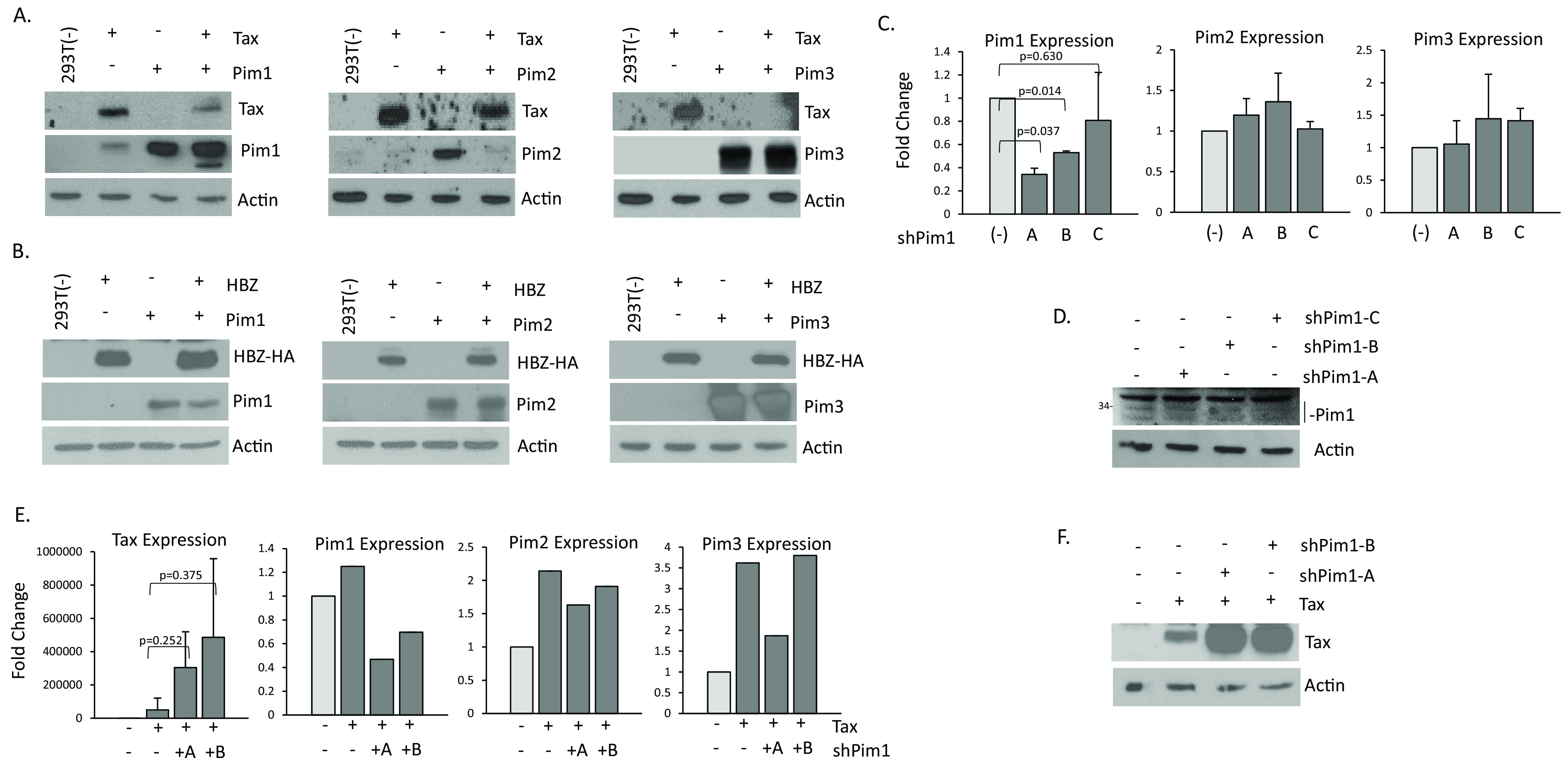
Pim kinases inhibit Tax expression. (A) 293T cells were co-transfected with pcTax and pCDNA-Pim kinases, -1, -2, or -3 for 48 h and probed for Tax or Pim kinase expression. Actin served as an internal control. (B) 293T cells were co-transfected with HBZ-HA and pCDNA-Pim kinases, -1, -2, or -3 for 48 h and probed for HA (HBZ) or Pim kinase expression. Actin served as an internal control. (C–D) 293T cells were transfected for 72 h with three individual shRNA to Pim-1 to determine the level of individual Pim-1 shRNA. Pim kinase gene expression was normalized to GAPDH expression from at least two independent experiments. Western blots for Pim-1 following Pim-1 knock-down are shown. A nonspecific band for Pim-1 is present (D). (E–F) 293T cells were transfected with pcTax and two individual shRNA to Pim-1. Pim kinase gene expression was normalized to GAPDH expression from at least two independent experiment (D). Elevation of Tax expression following knock-down of Pim-1 was also seen at the protein level (F). Actin served as a loading control.
Pim kinases bind Tax and delocalize Tax to the insoluble nuclear fraction.
Because a feedback loop exists between Tax and Pim isoforms, we examined whether Tax and Pim kinases interacted in vivo. We cloned all three Pim kinases with an HA tag into the PMH vector and performed co-immunoprecipitation experiments in the presence of Tax. We found that immunoprecipitated HA-Pim-1 could pulldown bound Tax protein (Fig. 5A). This was true for all three Pim kinase isoforms. Western blots of lysates confirmed 1- Pim kinases and Tax were expressed, 2- Tax could still increase Pim-1 and Pim-3 expression, and 3- HA-tagged-Pims could still inhibit Tax expression. Attempts at demonstrating endogenous Pim kinase interaction with Tax in established HTLV-1 cell lines were unsuccessful. This was likely due to the feedback loop between Tax and Pim kinases, which made it difficult to find a cell line that expressed high levels of both Tax and Pim. Because Pim kinases decrease Tax protein expression, it suggests Pim kinases may directly or indirectly mediate proteasomal or lysosomal degradation of Tax. Transfection of cells with Tax, in the presence of Pim-1 led to decreased Tax expression (Fig. 5B). However, addition of the proteasomal inhibitor MG132 did not rescue Tax induced degradation by Pim-1. To verify that this was not due to MG132, we also treated cells with lactacystin. Tax expression was not rescued by either proteasomal inhibitor (Fig. 5C). To examine if Tax was targeted for lysosomal degradation, we also treated Tax and Pim-1 co-expressing cells with lysosomal inhibitors, chloroquine and leupeptin but these did not rescue Tax expression from Pim-1 (Fig. 5D). In addition, neither of these inhibitors was able to rescue Tax expression following co-transfection of cells with Tax and/or Pim-2 or -3 (Fig. 5E and F).
FIG 5.
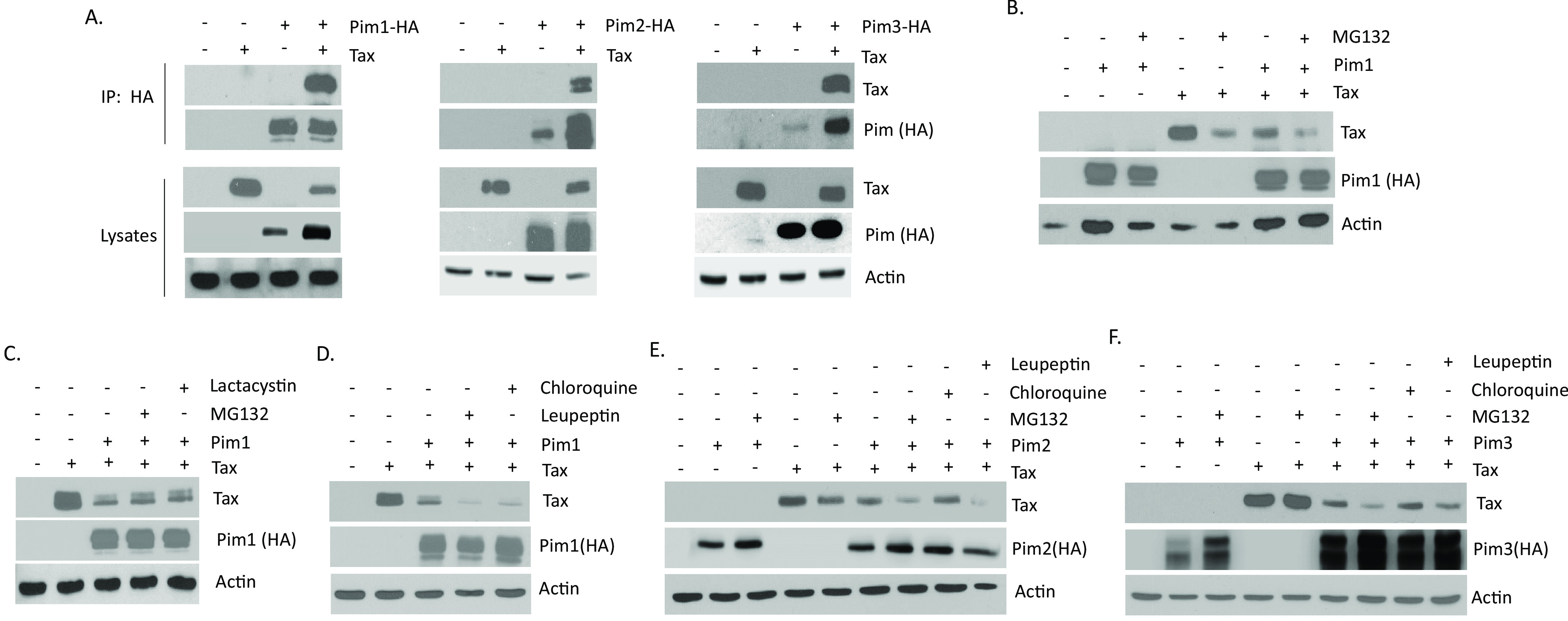
Pim kinases bind Tax. (A) 293T cells were co-transfected with pcTax and PMH-Pim kinases for 48 h. Lysates were subject to co-immunoprecipitation with HA antibody and probed with anti-Tax and anti-HA antibodies. Whole cell lysates were probed for Tax and HA-Pim expression to verify expression. Actin demonstrates the amount of protein in each lysate. (B) 293T cells were transfected with pcTax and HA-tagged Pim-1 expression plasmids for 48 h. Cells were treated with or without proteosome inhibitor, MG132 (10 μM) for the last 8 h. (C) 293T cells were transfected with pcTax and HA-tagged Pim-1 expression plasmids for 48 h. Cells were treated with or without proteosome inhibitors, MG132 (10 μM) and lactacystin (10 μM) for the last 8 h. (D–F) 293T cells were transfected with pcTax and HA-tagged Pim-1, -2, or-3 expression plasmids for 48 h. Cells were treated with or without proteosome inhibitor, MG132 (10 μM), or lysosomal inhibitors, chloroquine (100 μM) or leupeptin (100 μM) for the last 8 h. For (B–F), whole cell lysates were probed with Tax and anti-HA (Pim) antibodies. Actin served as a loading control.
Pim kinases could be (i) re-localizing Tax protein, (ii) inhibiting Tax translation, and/or (iii) degrading Tax in a proteosome/lysosome independent manner. Tax contains an N-terminal nuclear localization domain that allows Tax to reside predominantly in the nucleus, in nuclear speckles (36–38). We co-expressed HA-Pims and Tax in 293T cells and fractionated lysates into nuclear and cytoplasmic extracts. Tax localization was not impaired, because Tax expression decreased, yet remained mostly nuclear in the presence of Pim kinases (Fig. 6A). Previous reports suggest that Tax can be regulated by ubiquitination and subsequently localized to the nuclear matrix where it is degraded (39, 40). Furthermore, proteasomal inhibitors could rescue Tax expression in these insoluble fractions. To examine the level of Tax in the insoluble fraction, we co-transfected Tax and Pim kinases and lysed the cells in concentrated urea. We found Tax was no longer decreased in the presence of Pim-1 and Pim-2 in the insoluble fraction (Fig. 6B). However, co-expression of Pim-3 still led to a significant loss of Tax expression, suggesting Pim-3 degrades Tax. We co-expressed Tax and Pim isoforms and fractionated lysates into cytoplasmic and soluble or insoluble nuclear fractions. All three Pim kinases decreased Tax expression in the soluble nuclear fraction (Fig. 6C). However, upon Pim-1 and Pim-2 co-expression, Tax accumulated in the insoluble fraction. This data is consistent with observations made using the lysis in urea buffer, whereby Tax expression was no longer decreased in the insoluble portion of the nucleus upon Pim-1 and Pim-2 expression. Furthermore, when cells were treated with MG132 or leupeptin, Tax levels did not appreciably change in the insoluble nuclear fraction with MG132 treatment (Fig. 6D). This suggests that Pim kinases re-route Tax to the insoluble nuclear fraction; however, Tax is not degraded. In contrast, Tax was strongly decreased in all fractions when co-expressed with the Pim-3 (Fig. 6C). This suggests that Tax is degraded by Pim-3.
FIG 6.
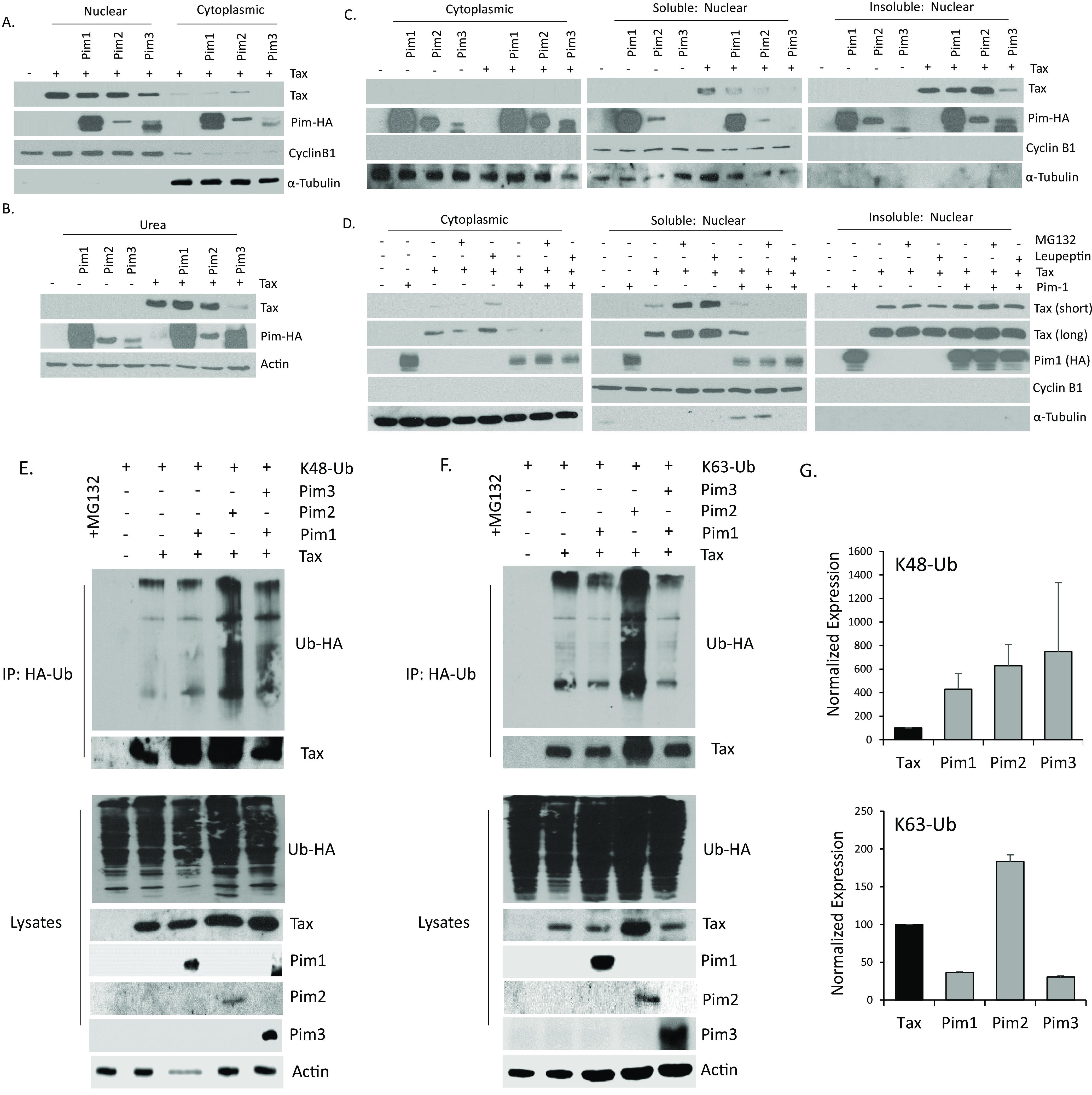
(A–F) 293T cells were transfected for 48 h with pcTax and HA-Pim kinases. (A) Cell lysates were fractionated into nuclear and cytoplasmic lysates and probed with anti-Tax, anti-HA, and anti-Cyclin B1 and α-Tubulin, control antibodies. (B) Cell lysates were lysed directly in urea buffer and whole cell lysates were probed with anti-Tax and anti-HA antibodies. Actin served as a loading control. (C–D) Cell lysates were fractionated into soluble and insoluble nuclear and cytoplasmic lysates and probed with anti-Tax, anti-HA, anti-Cyclin B1, and anti-α-Tubulin antibodies. In (D) cell lysates were treated for 7 h with MG132 (10 μM) or leupeptin (100 μM). Long and short exposures of tax are presented. (E–F) 293T cells were transfected for 48 h with pcTax, pcDNA-Pim-1, Pim-2, or Pim-3, and/or HA-tagged K48 (E) or K63 (F). All cells were treated with MG132 (10 μM) for 6 h, prior to collection. Cell lysates were immunoprecipitated overnight with anti-Tax antibody. Due to Pim repression of Tax, prior to Western blot analysis, additional lysate was run for extracts containing Pim kinases. Western blots were performed on immunoprecipitates and lysates with anti-Tax, anti-HA (Ub), and Pim kinase antibodies. Actin served as a loading control. (G) Quantification of K48 and K63 immunoprecipitation. Bands were quantified from several HA-immunoprecipitates and normalized to lysates.
We then examined if Tax is ubiquitinated in the presence of Pim kinases. K48 ubiquitination is often viewed as proteasome-dependent degradation mark, whereas K63 is proteosome-independent, such as with signal transduction and endocytosis pathways. However, K63 has also been tied to some proteosome-dependent processes, by seeding the assembly of K63/K48 ubiquitin chains (41). Therefore, we co-expressed lysine-K48 or K63 and treated cells with MG132. Because Pim kinases strongly reduce Tax expression, we increased the amount of lysate in Western blots expressing Pim kinases. Using these Tax-normalized lysates, we immunoprecipitated Tax and performed Western blots for HA-tagged K48 and K63 (Fig. 6E and F). In the presence of all three Pim kinases, there was an increase in K48 ubiquitination of Tax (for Pim-1 lysates, less Tax was pulled down and expressed) (Fig. 6E and G). We found Tax to be conjugated to K63 only in the presence of Pim-2 (Fig. 6F and G). Quantification of the pulldowns to normalize for protein lysates confirmed these results (Fig. 6G). These results demonstrate that Pim-1 and Pim-2 localize Tax to the insoluble nuclear matrix where it ubiquitinated, but not degraded. This occurs through K48-mediated and, in the case of Pim-2, K63-mediated ubiquitination. Tax is known to be ubiquitinated and interact with the proteosome, but does not undergo massive degradation (42). In fact, Tax ubiquitination is known to affect subcellular localization and activities and has a role in activation of NF-κB activity (43, 44). In contrast to Pim-1 and -2, Pim-3 binds to Tax, causing Tax ubiquitination and subsequent degradation.
Pim kinases alter Tax transactivation activity of the HTLV-1 LTR.
HTLV-1 Tax protein is the main viral transcriptional activator. Tax indirectly binds to the HTLV-1 LTR and stimulates viral transcription. Because Pim kinases can substantially decrease Tax expression, it stands to reason that Tax transactivation activity would also likely be reduced. We transfected 293T cells with an HTLV-1-LTR luciferase reporter construct along with Tax and increasing amounts of Pim kinase (Fig. 7A). As expected, the addition of Tax significantly elevated HTLV-1-LTR luciferase activity. Co-transfection of Pim kinases led to a dose-dependent loss in Tax mediated HTLV-1-LTR luciferase activity. The effect was strongest for Pim-1 and Pim-3; however, Pim-2 was also capable of decreasing Tax-induced HTLV-1 LTR luciferase. Western blot on non-normalized lysates confirmed the expression of Pim kinases in 293T cells (Fig. 7B). To determine if the effect of Pim kinases on Tax transactivation was due to Pim kinases directly affecting Tax transcription or affecting solely Tax protein level, we examined the Tax gene expression following transfection of Pim kinases. Unlike Tax protein, Tax gene levels were not decreased in the presence of Pim kinases (Fig. 7C). This demonstrates that Pim kinases affect Tax protein levels, and therefore affect Tax transcriptional targets downstream. To determine the effect of endogenous Pim kinases on Tax transcriptional activity, we transfected 293T cells with Tax and Pim-1 shRNA. Knock-down of endogenous Pim-1 led to rescue of Tax activity (Fig. 7D). Furthermore, loss of endogenous Pim-1 seemed to increase Tax activity and protein (Fig. 7D). These results were also seen when cells were treated with AZD1208, a pan-Pim kinase inhibitor (Fig. 7E). These results demonstrate that Pim kinases decrease HTLV-1 transactivation mediated by Tax.
FIG 7.
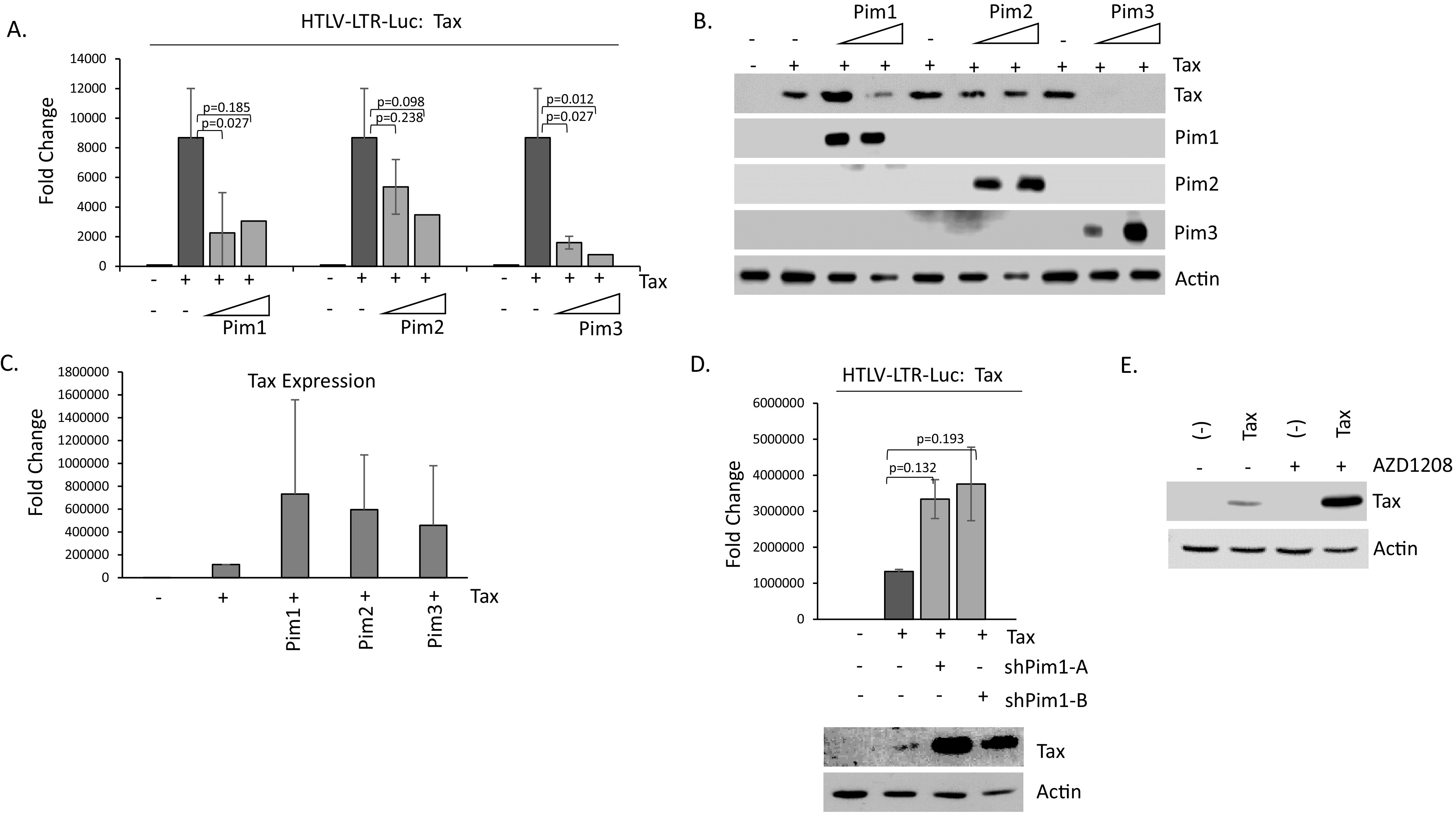
Pim kinases inhibit Tax transactivation activity. (A) 293T cells were transfected with the HTLV-1 LTR luciferase construct along with pcTax and increasing amounts of pcDNA-Pim-1, -2, and -3 kinases. Fold change was compared with 293T-pCDNA transfected controls. Luciferase activity was normalized to RL-TK renilla expression. Results represent the average of at least two experiments. (B) Verified expression of Pim kinases, -1, -2, and -3 in 293T transfected cells. Actin served as a loading control. (C) 293T cells were transfected with pcTax along with individual Pim kinases. 48-h post-transfection, gene expression was performed for Tax expression. Experiments were performed at least twice, and GAPDH served as a control. (D) 293T cells were transfected with HTLV-1 LTR luciferase construct along with pcTax and/or Pim-1 shRNA A and B. Fold change was compared with 293T-pCDNA transfected controls. Luciferase activity was normalized to RL-TK renilla expression. Results represent the average of at least two results. Western blots confirming Tax over-expression are provided. (E) 293T cells were transfected for 48 h with Tax. The pan-Pim kinase inhibitor, AZD1208 (10 μM), was added during the last 30 h. Actin served as a loading control.
Because AZD1208 led to an increase in Tax transfected 293T cells, we tested whether AZD1208 could also affect Tax expression in established HTLV-1 cell lines. This is important because, unlike 293T and Jurkat cells, most HTLV-1 cells express higher levels of all three Pim kinases and express the entire HTLV-1 provirus, in addition to Tax. We treated four ATL cell lines with increasing doses of AZD1208 and examined the level of expression of Tax (Fig. 8A and B). While Tax expression did increase slightly upon AZD1208 treatment, it was not statistically significant. To verify these results, we used two additional Pim inhibitors. Pim IV is a Pim-1 kinase inhibitor and M-100 is a pan-Pim kinase inhibitor. M-100 behaved like AZD1208 in ATLT cells, producing a small increase in Tax gene expression (Fig. 8C), while inhibition of only one Pim kinase (with Pim IV inhibitor) did not lead to an increase in Tax expression at all. Treatment of Jurkat TET-Tax cells with AZD1208 showed similar results, whereby there was a small increase in Tax RNA expression (Fig. 8D). However, treatment of Jurkat TET-Tax cells with AZD1208 led to a large increase in Tax expression at the protein level (Fig. 8E). We then treated HTLV-1 cells with AZD1208 and examined Tax protein expression. Treatment with AZD1208 could only increase Tax protein levels in MT4 cells, which was further increased in the presence of MG132. However, in ATLT, an ATL-like cell line that expresses lower levels of Tax, Tax was not rescued in whole cell lysates (Fig. 8F). Because Pim kinases re-distribute Tax in the nucleus, we decided to fractionate ATLT into cytoplasmic, nuclear, and insoluble cell fractions. Upon fractionation, we found AZD1208 led to profound increases in Tax expression in the soluble and insoluble nuclear fractions of ATLT (Fig. 8G). We performed the same experiments in MT4 cells and found increased Tax expression in the presence of AZD1208 in all three fractions. These results confirm that blocking Pim kinase activity leads to increases in Tax expression in HTLV-1 cell lines. Furthermore, when HTLV-1 cells were treated with shRNA to Pim-1, no differences in Tax expression were seen at the RNA or protein level (data not shown). This suggests that all three Pim kinases must be subject to knock-down to increase Tax expression in HTLV-1 cells.
FIG 8.
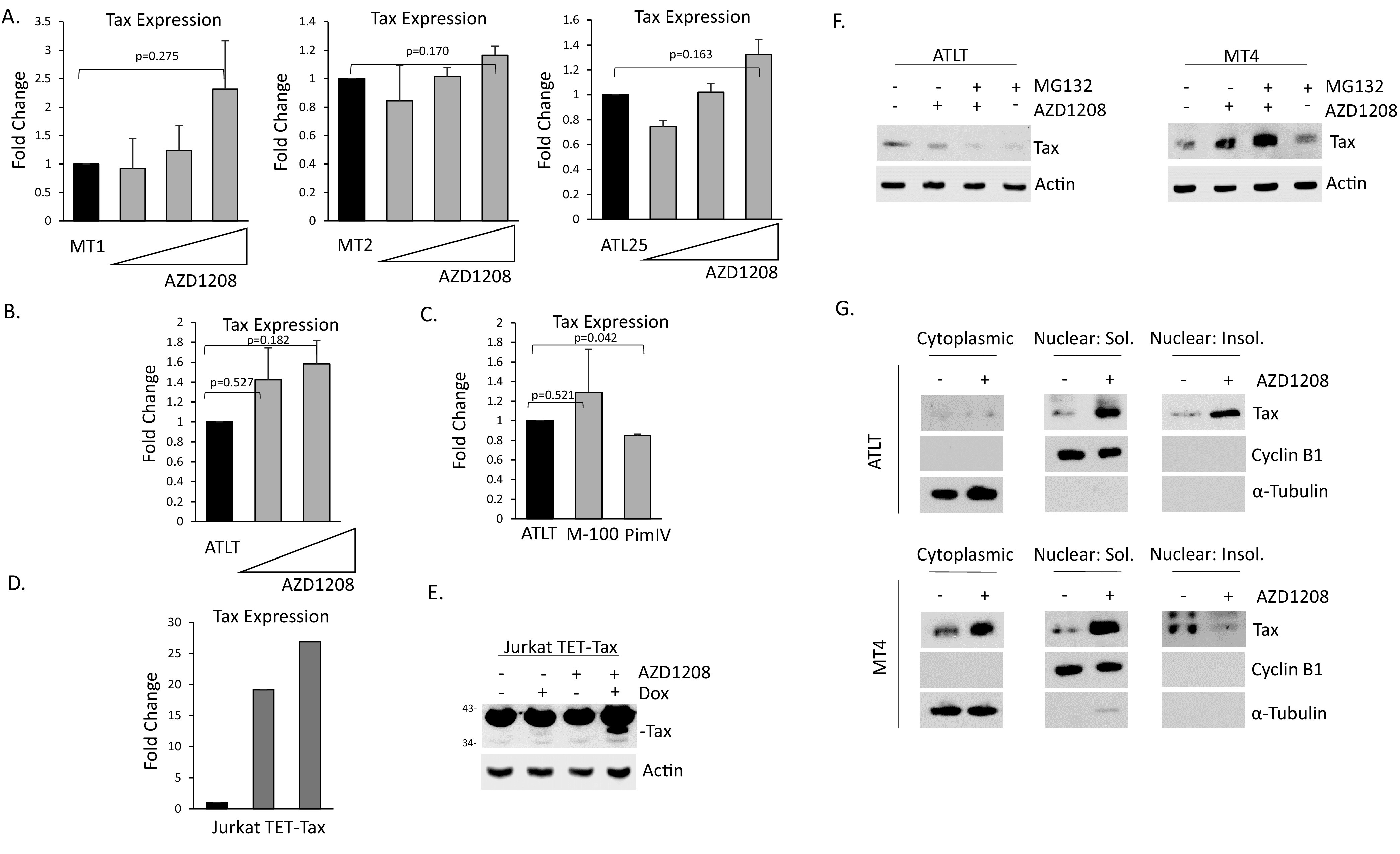
Inhibition of Pim kinases leads to elevation of Tax expression in HTLV-1 lines. (A–C) Treatment of MT1, MT2, ATL25 (A), and ATLT (B–C) with 5, 10, or 20 μM AZD1208, or 10 μM M-100 or PimIV inhibitor. Treatments were performed for 48 h or 72 h. Representation of at least two independent experiments is shown. Gene expression was normalized to GAPDH expression and performed at least twice. (D–E) Jurkat TET-Tax cells were induced with doxycycline for 72 h. 10 μM AZD1208 was added for the final 18 h for gene expression (D) or 72 h for protein expression (E). Actin served as a loading control. (F) ATLT and MT4 cells were treated with 10 μM AZD1208 and/or 10 μM MG132. Total cell lysate was probed for Tax and actin served as loading control. (G) ATLT and MT4 were treated with 10 μM AZD1208. Cytoplasmic, soluble nuclear, and insoluble nuclear fractions were probed with Tax antibody. Purity of fractions was confirmed with Cyclin B1 and α-Tubulin antibodies.
Pim kinases inhibit HTLV-1 transactivation and viral replication.
Because inhibition of Pim kinase activity led to increased Tax expression in MT4 cells, which express low levels of HTLV-1 proteins, we next examined the effect of Pim kinases in the context of the entire HTLV-1 virus. Cells were transfected with the HTLV-1 LTR luciferase construct along with PBST, a full molecular clone of HTLV-1 (45). Pim kinases had little effect on HTLV-1 luciferase activity in the absence of PBST (Fig. 9A). Transfection of PBST led to a substantial increase in HTLV-1 activity. However, the addition of Pim-1, -2, or -3 completely ablated PBST activation (Fig. 9A). This decrease in PBST transactivation of the viral LTR was comparable with Pim kinase inhibition of Tax transactivation, as Pim-1 and Pim-3 had stronger inhibition than Pim-2. The PBST molecular clone is derived from a TSP patient infected with HTLV-1B. To verify the results, and to see if Pim kinases could interfere with other strains of HTLV-1, we repeated the experiments using two additional HTLV-1 molecular clones, ACH and X1MT (46, 47). The ACH molecular clone is derived from a Japanese patient infected with HTLV-1A, that presented with ATL. X1MT is a modified HTLV-1 molecular clone derived from an American ATL patient in which open reading frames (ORFs) X-I and X-II were replaced with the corresponding ORFs from the cell line, MT2. Like PBST, Pim kinases were able to inhibit ACH and X1MT transactivation of the HTLV-1 LTR luciferase in a dose-dependent manner (Fig. 9B and data not shown). This suggests that Pim inhibition of HTLV is universal to various HTLV subtypes.
FIG 9.
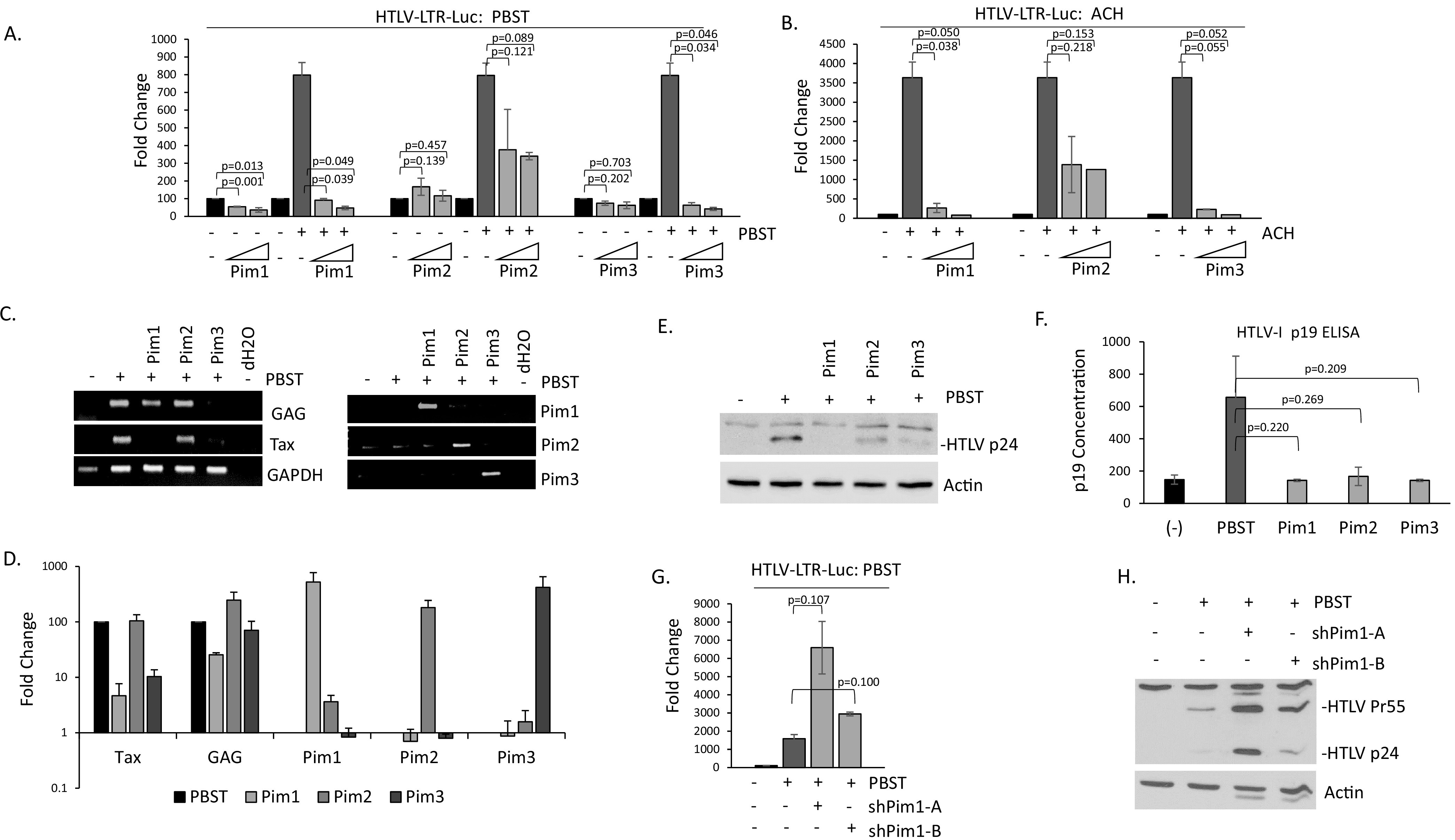
Pim kinases inhibit HTLV-1 replication and virus production. (A–B) 293T cells were transfected with the HTLV-1 LTR luciferase construct along with the HTLV-1 molecular clones PBST (A) or ACH (B), along with increasing amounts of Pim-1, -2, and -3 kinases. Fold change was compared with 293T-pCDNA transfected controls. Luciferase activity was normalized to RL-TK renilla expression. Results represent the average of at least two experiments. (C–D) 293T cells were transfected with or without PBST along with Pim-1, -2, or -3. cDNA was analyzed by (C) PCR or (D) real-time PCR for HTLV-1 and Pim gene expression. Fold change is compared with PBST transfected cells, with GAPDH serving as an internal control. Results represent the average of two experiments. (E–F) 293T cells were transfected with PBST with or without Pim-1, -2, or -3. Whole cell lysates were probed with anti-HTLV p24 antibody (E). Actin expression served as a loading control. Supernatant was analyzed for HTLV p19 antigen by ELISA (F). Results were performed at least twice. (G–H) 293T cells were transfected with PBST with or without Pim-1 shRNA A or B. In (G) HTLV-1 luciferase plasmid was transfected, and Luciferase activity was normalized to RL-TK renilla expression. Results were performed at least twice. For (H) whole cell lysates were probed with anti-HTLV p24 antibody and demonstrate the precursor (Pr55) and p24. Actin expression served as a loading control.
The expression of HTLV-1 viral genes from the PBST molecular clone were then examined. PCR analysis validated Pim kinases were expressed (Fig. 9C and D). Pim-1 and Pim-3 were able to inhibit Tax and GAG expression from the PBST clone, in contrast to Pim-2 (Fig. 9D). Intracellular p24 Gag expression also decreased in the presence of Pim-1, -2, and -3 (Fig. 9E). p24 declined even in the presence of Pim-2 kinase, suggesting that GAG expression is affected in the presence of all three Pim kinases. The reason Pim-2 is unable to fully inhibit Tax expression, Tax transactivation, and gene induction from the HTLV-1 molecular clones could be due to differing functions of Pim-2 and/or because Tax inhibits expression of Pim-2. Tax increases Pim-1 and Pim-3 expression, which could lead to further reductions in Tax expression and activity, whereas Tax decreases Pim-2 expression leading to less repression of Tax transactivation activities. In fact, expression analysis confirmed less Pim-2 was being expressed than Pim-1 and Pim3 (Fig. 9D). HTLV-1 virus production significantly declined in the presence of Pim kinases, as there was a substantial loss of extracellular p19 expression from PBST as detected by ELISA (Fig. 9F). Finally, inhibition of endogenous Pim-1, led to increased HTLV-1 luciferase activity from the PBST molecular clone (Fig. 9G). This was confirmed by increased expression of GAG p24 and precursor, p55 (Fig. 9H).
In ATL-derived cells Hsp90 protects Pim kinases from proteasomal degradation.
Finally, we examined the correlation between viral genes and Pim kinases in ATL-derived cell lines that in general express little to no Tax. Despite little Tax expression, ATL-derived cell lines have higher levels of Pim kinases (Fig. 1A and B). We first examined the localization of Pim kinases in ATL-derived cells. Pim protein isoforms have been known to reside in different subcellular locations. We found this to be true of Pim-1, in which isoforms were expressed in the cytoplasm and nucleus (Fig. 10A). Nuclear Pim-1 has been found to be essential for Pim-1 activity in Burkitt’s lymphoma cells and suggests that nuclear Pim-1 in ATL cells is also important (48). We also found strong Pim-2 and Pim-3 nuclear expression in all tested ATL cells. Treatment of two Tax, protein negative cell lines, ED-40515(-) and MT1 cells, with cycloheximide demonstrated that Pim kinases have long half-lives in ATL-derived cell lines (Fig. 10B). This was analogous to the long protein half-life seen in K562 cells (49). We then treated ATL-derived cells with two proteosome inhibitors, MG132 and lactacystin. We found that proteasomal inhibition could increase the amount of Pim-1 and Pim-3 in ATL-derived cell lines (Fig. 10C). Further testing on a panel of ATL and HTLV-1 cell lines confirmed these results (Fig. 10D). In fact, Pim-1 expression was significantly increased in HTLV-1 cell lines (C91PL and C8166) that expressed little Pim-1 kinase. These results are also in agreement with studies demonstrating that heat shock protein, Hsp70, can target Pim-1 for ubiquitin-mediated proteasomal degradation (50). In nearly all cell lines tested, Pim-2 was not significantly altered by inhibition of the proteosome. Pim-1 has been shown to be protected from Hsp70 ubiquitin-mediated proteasomal degradation by binding to the molecular chaperone, Hsp90 (51). Therefore, we tested whether Hsp90 could protect Pim kinases from degradation in ATL-derived cells. ATL-derived cells were treated with geldanamycin, a specific inhibitor of Hsp90. Expression of Pim-1 and Pim-3 (and to a much lesser extent Pim-2) were significantly decreased when Hsp90 was inhibited (Fig. 10E). These results indicate the Pim kinase proteins are protected from proteasomal degradation by binding to Hsp90 in ATL-derived cells. Treatment of ATL and HTLV-1 cell lines with geldanamycin significantly inhibited cell growth (Fig. 10F). Loss of proliferation was like that seen in K562 cells, a cell line with some of the highest reported Pim-1 kinase levels and with that seen when HTLV-1 lines were treated with Hsp90 inhibitors (39, 50, 52). In support of our observations, use of AUY922, an Hsp90 inhibitor, led to inhibition of the PI3K/AKT, NF-κB, and Pim kinases in ATL and HTLV-1 cells (53). Our results suggest that Pim kinases are tightly regulated in ATL and HTLV-1 cell lines (Fig. 10G). Our current understanding is that Tax can increase the expression of Pim-1 and Pim-3, while decreasing Pim-2. Tax alters Pim kinases at the gene level. All three Pim kinases can then engage in a negative feedback loop, whereby they bind Tax and inhibit Tax activities. In cells with low Tax expression, Pim kinases are stabilized through Hsp90 and possibly HBZ induction of Pim-1 kinase expression.
FIG 10.
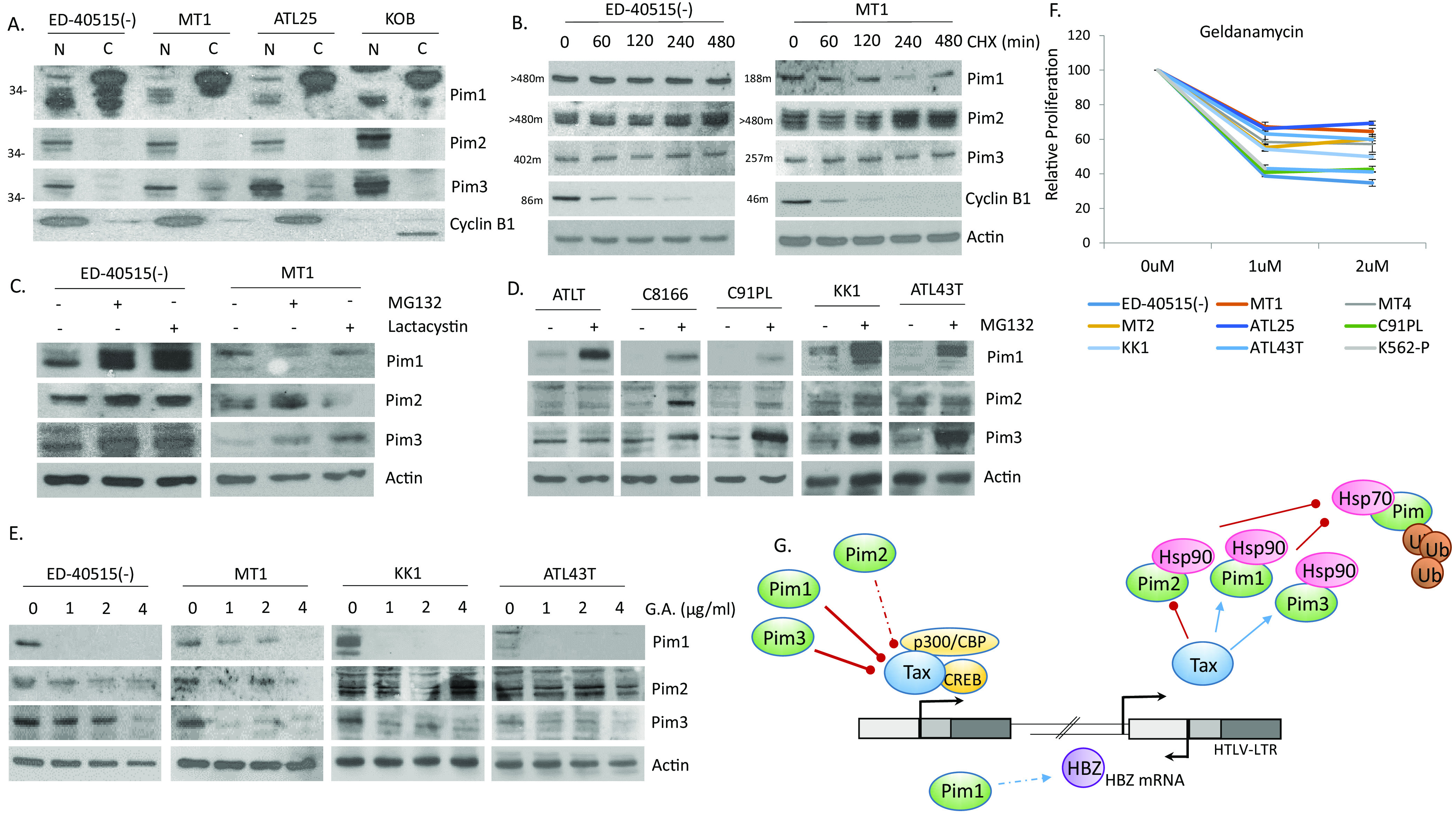
Pim kinase expression is stabilized in ATL cells through Hsp90. (A) Nuclear and cytoplasmic extracts of ATL lines, ED-40515(-), MT1, ATL25, and KOB. Cyclin B1 served as control for subcellular fractionation. (B–D) Analysis of Pim kinase protein stabilization following treatments with (B) cycloheximide (100 μg/mL CHX for 0, 2, 4, 6, or 8 h) in ED-40515(-) and MT1 ATL-derived cells, with (C) proteases (10 μM MG132 and 1 μM Lactacystin for 6 h) in ED-40515(-) and MT1 ATL-derived cells, and with (D) protease (10 μM MG132 for 6 h) in ATLT, C8166, C91PL, KK1, and ATL43T cells. For (B–D) actin expression served as a loading control. For (B) Cyclin B1 degradation confirmed that CHX treatment was working. Half-life of each protein is labeled next to the gel. (E–F) Pim kinases are stabilized by heat shock proteins in ATL-derived lines. ED-40515(-), MT1, KK1, and ATL43T cells were treated with the heat shock inhibitor, geldanamycin (1, 2, 4, or 6 μg/mL GA for 24 h). Whole cell lysates were analyzed for Pim protein expression (E). Actin served as a loading control. Proliferation, after GA treatment, was measured with XTT assays (F). Results were carried out in duplicate and calculated as a fold change compared with cells treated without GA. (G) Illustration of feedback loops between Tax/HBZ proteins and Pim kinases. Tax can drive the expression of Pim-1 and Pim-3, possibly contributing to the cellular transformation of T-cells. However, Pim-1, -2, and -3 and HBZ can also inhibit Tax expression, leading to loss of HTLV-1 transactivation and viral replication. This may allow for immune escape and viral latency. Pim kinase levels are stabilized in ATL cells through interaction with heat shock protein, Hsp90, which protects Pim kinases from degradation.
DISCUSSION
Tax and HBZ are critical for controlling leukemic cell transformation, proliferation, viability, and latency. Tax is only expressed in a fraction of the infected cells which is inducible and critical for the survival of the entire population of leukemic cells (54). In contrast, HBZ is expressed in all ATL patient’s cells and plays a role in viral latency. Therefore, their contribution to ATL development and persistence is critical. We have previously shown that ATL patient samples express Pim-1 kinase and, moreover, require Pim kinase for tumor growth in vivo (20). Given the fact that Pim kinases and Tax/HBZ are critical for the development of ATL leukemia, we sought to examine a link between these cellular and viral genes. Our results demonstrate a unique feedback loop between Pim kinases and Tax. All three Pim kinase family members can significantly inhibit Tax transactivation, virus replication, and particle(s) production. However, there are differences within the Pim kinase family when it comes to Tax degradation and the level of repression. In addition, Tax could only increase Pim-1 and Pim-3, and not Pim-2; and appeared to have the greatest influence on Pim-3 levels. This demonstrates a lot of variety in the degree Pim kinases can prevent Tax and HTLV-1 activity. Pim-1 and Pim-3 provide much stronger Tax repression, than Pim-2, though this could be due to less Pim-2 being produced from the plasmid. However, when larger amounts of Pim-2 were transfected, HTLV-1 transactivation did not change appreciably, suggesting a real difference in Pim-2 effects on Tax. Pim-2 is known to have distinct roles in suppressing T-cell responses. Some of this could be due to structural differences within the Pim isoforms. While sharing about 60% structural similarities, there are disparities between the Pim kinases and their affinity for ATP (55). Unlike Pim-1 and Pim-3, Pim-2 negatively regulates T cell responses in transplantation and tumor immunity (56). Pim-2 is also highly expressed in multiple myeloma (MM) cells where it has a unique role in cell survival, activating the apoptotic pathway and repressing the DNA damage response (57). Of the three Pim kinases, Pim-2 also has the most restricted tissue and disease expression pattern. While Pim-3 is expressed in nearly all tissues, Pim-2 is largely confined to bone marrow and lymphoid tissues and the blood, especially B-cells. The fact that Pim-2 expression is elevated in ATL-derived cell lines to that seen in high Pim-2 expressing lymphoid cell lines, suggests that Pim-2 has an important, and perhaps distinct, role in ATL disease. Further examination of Pim-2, through knock-down and expression studies in ATL cells will be warranted.
While HBZ was not the focus of this study, we did find small increases in Pim-1 expression, following HBZ over-expression. We also found small gene correlations between HBZ and Pim-1 and HBZ and Pim-3 in HTLV-1 and ATL cell line cDNA (data not shown). This suggests HBZ may play a part in Pim regulation and requires further study; and proposes a dynamic feedback loop between HBZ/Tax and Pim kinases. We have recently shown that ATL patients carry methylation of the fragile histidine triad (FHIT) gene, which, together with Tax, can promote an oncogenic environment by altering the DNA damage response (58, 59). This produces a favorable environment for the development of leukemia. Our data suggests that during early infection, Tax can also increase the protein expression of Pim-1 and Pim-3, taking advantage of their proto-oncogenic functions to increase T-cell proliferation, viability, and invasion. HBZ may partner with Tax to sustain these increases. However, Pim kinases, along with HBZ, can negatively regulate Tax expression and transactivation. This would lead to a diminished Tax CTL response and favor viral latency. Pim kinase expression may then be preserved in ATL cells through Tax, HBZ, and constitutive activation of the STAT3 pathway that allows for ATL cells to take advantage of the pro-oncogenic activities of Pim kinases. We have already demonstrated that Pim-1 expression can be maintained in ATL patients through downregulation of the miR-124a/STAT3 pathway (20). However, a detailed analysis of Pim kinase expression in primary, ATL patient samples is lacking. Given that Pim kinases are regulated by the IL-2/IL-6 and JAK/STAT pathways, it is very likely that Pim kinases are highly expressed in ATL patient samples (30, 60). ATL patients display high levels of these pathways, which would likely maintain Pim expression even in the absence of Tax (12, 61). Furthermore, an examination of the individual roles of Pim kinases in ATL persistence could lead to novel functions of individual Pim kinases. Current Pim kinase therapies in clinical trial are pan-Pim kinase inhibitors that target all three Pim kinases. However, several studies have been shuttered due to observed toxicity. The use of individual Pim kinases inhibitors may show more favorable results; and single kinases inhibitors are currently being developed (62). This prompts the demand for a more thorough examination of the expression and role of individual Pim kinases in ATL patient samples.
MATERIALS AND METHODS
Cell lines and culture conditions.
The CML line, K562, and T-ALL line, Jurkat, the AML line, HL-60, and ATL and HTLV-1 lines, Tl-Om1, ED-40515(-), MT1, ATLT, ATL25, MT2, MT4, C91PL, C8166, and 1186.94 were grown in RPMI with 10% fetal bovine serum (FBS). Immortalized, IL-2 dependent lines, ATL43T, ATL55T, LM-Y1, KK1, KOB, 1185, LAF, MUO4, and SP, were grown in RPM1, supplemented with 20% FBS and 50U/mL IL-2. Jurkat TET-Tax cells were grown in RPMI with 10%, TET tested serum and induced with doxycycline. 293T cells were grown in Dulbecco’s modified Eagle medium (DMEM) with 10% FBS. The creation of Tax transduced PBMCs was previously described (35).
Transfections and plasmids.
293T cells were transfected with Polyfect (Qiagen). HTLV-1 DNA vectors were pcTax and PMH-HBZ. HA-tagged K48 and K63 plasmids were cloned into the PMH vector. Pim kinases were cloned into pCDNA or HA-tagged in the PMH vector. All cloned genes were verified by sequencing. Three individual shRNA sequences to Pim-1 were obtained from Sigma-Aldrich. An siRNA sequence targeting the Tax gene of HTLV-1 was cloned into the shRNA, lentiviral pSIH1-H1-GFP vector. shPim-1 and shTax virus was obtained by co-expressing individual shRNA with VSV-G and pDLN plasmids in 293T cells. Viral containing supernatant was ultra-centrifuged to obtain high titer shRNA Pim-1 and Tax virus, which was subsequently used to infect cells. HTLV-1 molecular clones, PBST, ACH, and X1MT, have been described (45–47).
Drug studies.
For RNA and protein half-life studies, cells were treated with cycloheximide (100 μg/mL) or actinomycin D (10 μg/mL). For protein stability and drug studies, 293T transfected cells were treated with MG132 (10 μM), lactacystin (1 μM), leupeptin (100 μM), chloroquine (100 μM), AZD1208 (5-20μM) (Selleckchem), PIM1 kinase inhibitor IV (10 μM) (Calbiochem), and Pim kinase inhibitor VII, M-100 (10 μM) (Calbiochem). Protein half-life was calculated by treating cell lines with 100 μg/mL cycloheximide, while Pim kinase protein stability and proliferation were measured by treating cells with geldanamycin (1, 2, 4, or 6 μg/mL), according to figure legends. Cell proliferation assays were carried out in duplicate and measured with the XTT cell viability kit (Trevigen).
Luciferase assays and enzyme-linked immunosorbent assays.
For luciferase assays, 293T were transfected with the HTLV-1 LTR firefly luciferase construct and the RL-TK internal control plasmid. Cells were lysed in passive lysis buffer and assayed using the Dual Luciferase-Reporter kit (Promega). Cells were normalized to renilla expression (RL-TK) and fold change was compared with pCDNA control transfected cells. Expression of p19 in the supernatant of transfected cells was carried out using the HTLV-1 p19 Antigen ELISA kit (ZeptoMetrix), according to the manufacturer’s instructions.
Western blots.
Total protein was extracted from cell lines with RIPA lysis buffered and separated by SDS-PAGE electrophoresis. The following antibodies were used: Santa-Cruz (Actin (sc-47778), Pim-1 (sc-13513), Pim-2 (sc-13514), Cyclin B1 (sc-245), HTLV-1 Tax (sc-57872), and HTLV-1 p24 (sc-53891), Cell Signaling, Pim-1 (D8D7Y), Pim-2 (D1D2), Pim-3 (D17C9), Abcam (α-Tubulin), and Roche (HA-High Infinity 3F10). Protein extracts were normalized to Actin expression. Soluble nuclear and cytoplasmic extracts were obtained using hypertonic (5 mM HEPES, pH 7.9, 5 mM MgCl2, 0.1 mM EDTA, 0.4M NaCl, and 1 mM DTT) and hypotonic lysis buffers (10 mM HEPES, pH 7.9, 1 mM MgCl2, 0.5 mM NaCl, and 0.5% NP-40), respectively. Briefly, cell pellets were lysed in hypotonic buffer, spun down, and the supernatant was collected (cytoplasmic fraction). The cell pellet was then washed and lysed in hypertonic buffer. A series of freeze/thaw cycles were performed, and the lysates were spun down for 30 min at 4°C. The supernatant was then collected containing the nuclear fraction. Insoluble nuclear fractions were obtained after nuclear/cytoplasmic fractionation and lysed in buffer (150 mM NaCl, 50 mM HEPES, pH 7.4, 0.5% NaDeoxycholate, 1% SDS, and 100 mM DTT. For insoluble urea extracts, cells were lysed directly in 8M Urea and diluted in 2x loading dye for gel electrophoresis. For ubiquitin immunoprecipitations and cycloheximide treatment, blots were quantified using ImageJ software.
Protein immunoprecipitation.
293T cells were transfected with pcTax and HA-tagged Pim kinases. Cell lysates were collected, washed, and lysed in NP-40 lysis buffer. Equal amounts of protein were immunoprecipitated overnight at 4°C with HA antibody (Sigma: anti-HA [12CA5]). The next day, Protein A/G agarose was added for 2 h at 4°C. Immunoprecipitates were then washed three times, spun down, and resuspended in 2x loading dye. Protein lysates were then run on SDS-PAGE electrophoresis gels and probed with appropriate Pim kinase and Tax antibodies. Non-immunoprecipitated protein lysates were served as loading and expression controls. For ubiquitin pulldowns, 293T cells were transfected with pcTax, pcDNA-Pim kinases, and K48- or K63-UB-HA tagged plasmids for 48 h. Cells were treated with 10 μM MG132 for 6 h prior to collection, lysed in NP-40 lysis buffer, and immunoprecipitated overnight with anti-Tax antibody.
RNA expression.
RNA was extracted through TRIzol (Invitrogen) and equal amounts of RNA were treated with DNase I (Thermofisher). cDNA was generated using the RNA-to-cDNA kit (Applied Biosystems). RNA expression was analyzed using the StepOnePlus instrument (Thermofisher), using iTaq Universal SYBR green or iTaq Universal Probes Supermix (Bio-Rad). Primers used in the study: GAPDH (GAAGGTGAAGGTCGGAGTC/GAAGATGGTGATGGGATTTC), Pim-1 (GAGTCGCAGTACCAGGTG/GCTCACCTTCTTCAGCAGGAC), Pim-2 (GAAGGCCCAAGCCGCTGCTTC/GTAGGTCTATCAGGATGTTCTCATC), Pim-3 (CAGGACCTCTTCGACTTTATCAC/GTCCTTAATGTCGCGGTGCACGAC), Tax/Rex (ATCCCGTGGAGACTCCTCAA/AACACGTAGACTGGGTATCC), and GAG (CCTCTCAGGCCCCCTTTCAGGC/GCCTCAGTTGTGGTTGCCCC). GAPDH expression was used as an internal control and fold change was calculated as the ratio of normalized expression of the target gene divided by the normalized expression of the control sample.
ACKNOWLEDGMENTS
M.B. performed experiments, analyzed results, made the figures, and wrote the manuscript; and C.N. designed analyzed results.
We declare no competing financial interests.
This work was supported by grant R01CA201309 to Christophe Nicot.
Contributor Information
Marcia Bellon, Email: mbellon@kumc.edu.
Guido Silvestri, Emory University.
REFERENCES
- 1.Tagaya Y, Matsuoka M, Gallo R. 2019. 40 years of the human T-cell leukemia virus: past, present, and future. F1000Res 8:228. 10.12688/f1000research.17479.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gessain A, Cassar O. 2012. Epidemiological aspects and world distribution of HTLV-1 Infection. Front Microbiol 3:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malik B, Taylor GP. 2019. Can we reduce the incidence of adult T-cell leukaemia/lymphoma? Cost-effectiveness of human T-lymphotropic virus type 1 (HTLV-1) antenatal screening in the United Kingdom. Br J Haematol 184:1040–1043. 10.1111/bjh.15234. [DOI] [PubMed] [Google Scholar]
- 4.Matsuoka M, Mesnard JM. 2020. HTLV-1 bZIP factor: the key viral gene for pathogenesis. Retrovirology 17:2. 10.1186/s12977-020-0511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kannagi M, Matsushita S, Harada S. 1993. Expression of the target antigen for cytotoxic T lymphocytes on adult T-cell-leukemia cells. Int J Cancer 54:582–588. 10.1002/ijc.2910540411. [DOI] [PubMed] [Google Scholar]
- 6.Kinoshita T, Shimoyama M, Tobinai K, Ito M, Ito S, Ikeda S, Tajima K, Shimotohno K, Sugimura T. 1989. Detection of mRNA for the tax1/rex1 gene of human T-cell leukemia virus type I in fresh peripheral blood mononuclear cells of adult T-cell leukemia patients and viral carriers by using the polymerase chain reaction. Proc Natl Acad Sci USA 86:5620–5624. 10.1073/pnas.86.14.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeda S, Maeda M, Morikawa S, Taniguchi Y, Yasunaga J-I, Nosaka K, Tanaka Y, Matsuoka M. 2004. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int J Cancer 109:559–567. 10.1002/ijc.20007. [DOI] [PubMed] [Google Scholar]
- 8.Satou Y, Yasunaga J-i, Yoshida M, Matsuoka M. 2006. HTLV-1 basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci USA 103:720–725. 10.1073/pnas.0507631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giam CZ, Semmes OJ. 2016. HTLV-1 infection and adult T-cell leukemia/lymphoma-a tale of two proteins: Tax and HBZ. Viruses 8:161. 10.3390/v8060161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hall WW, Fujii M. 2005. Deregulation of cell-signaling pathways in HTLV-1 infection. Oncogene 24:5965–5975. 10.1038/sj.onc.1208975. [DOI] [PubMed] [Google Scholar]
- 11.Qu Z, Xiao G. 2011. Human T-cell lymphotropic virus: a model of NF-kappaB-associated tumorigenesis. Viruses 3:714–749. 10.3390/v3060714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takemoto S, Mulloy JC, Cereseto A, Migone TS, Patel BK, Matsuoka M, Yamaguchi K, Takatsuki K, Kamihira S, White JD, Leonard WJ, Waldmann T, Franchini G. 1997. Proliferation of adult T cell leukemia/lymphoma cells is associated with the constitutive activation of JAK/STAT proteins. Proc Natl Acad Sci USA 94:13897–13902. 10.1073/pnas.94.25.13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pancewicz J, Taylor JM, Datta A, Baydoun HH, Waldmann TA, Hermine O, Nicot C. 2010. Notch signaling contributes to proliferation and tumor formation of human T-cell leukemia virus type 1-associated adult T-cell leukemia. Proc Natl Acad Sci USA 107:16619–16624. 10.1073/pnas.1010722107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeh C-H, Bellon M, Pancewicz-Wojtkiewicz J, Nicot C. 2016. Oncogenic mutations in the FBXW7 gene of adult T-cell leukemia patients. Proc Natl Acad Sci USA 113:6731–6736. 10.1073/pnas.1601537113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bellon M, and, Nicot C. 2008. Central role of PI3K in transcriptional activation of hTERT in HTLV-1-infected cells. Blood 112:2946–2955. 10.1182/blood-2008-01-134692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellon M, Nicot C. 2015. Multiple pathways control the reactivation of telomerase in HTLV-1-associated leukemia. Int J Cancer Oncol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pise-Masison CA, Mahieux R, Radonovich M, Jiang H, Duvall J, Guillerm C, Brady JN. 2000. Insights into the molecular mechanism of p53 inhibition by HTLV type 1 Tax. AIDS Res Hum Retroviruses 16:1669–1675. 10.1089/08892220050193128. [DOI] [PubMed] [Google Scholar]
- 18.Panchal NK, Sabina EP. 2020. A serine/threonine protein PIM kinase as a biomarker of cancer and a target for anti-tumor therapy. Life Sci 255:117866. 10.1016/j.lfs.2020.117866. [DOI] [PubMed] [Google Scholar]
- 19.Isaac M, Siu A, Jongstra J. 2011. The oncogenic PIM kinase family regulates drug resistance through multiple mechanisms. Drug Resist Updat 14:203–211. 10.1016/j.drup.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 20.Bellon M, Lu L, Nicot C. 2016. Constitutive activation of Pim1 kinase is a therapeutic target for adult T-cell leukemia. Blood 127:2439–2450. 10.1182/blood-2015-11-685032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jean-Baptiste D, Belrose G, Meniane JC, Lézin A, Jeannin S, Mesnard JM, Olindo S, Peloponese JM, Jr, Césaire R. 2017. Differential effects of AZD-1208 and SMI-4a, two Pim-1 kinase inhibitors on primary HAM/TSP and ATL cells. Annals of Carcinogenesis 2. [Google Scholar]
- 22.Mondello P, Cuzzocrea S, Mian M. 2014. Pim kinases in hematological malignancies: where are we now and where are we going? J Hematol Oncol 7:95. 10.1186/s13045-014-0095-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishikawa C, Senba M, Hashimoto T, Imaizumi A, Mori N. 2017. Expression and significance of Pim-3 kinase in adult T-cell leukemia. Eur J Haematol 99:495–504. 10.1111/ejh.12940. [DOI] [PubMed] [Google Scholar]
- 24.Duverger A, Wolschendorf F, Anderson JC, Wagner F, Bosque A, Shishido T, Jones J, Planelles V, Willey C, Cron RQ, Kutsch O. 2014. Kinase control of latent HIV-1 infection: PIM-1 kinase as a major contributor to HIV-1 reactivation. J Virol 88:364–376. 10.1128/JVI.02682-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bajaj BG, Verma SC, Lan K, Cotter MA, Woodman ZL, Robertson ES. 2006. KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology 351:18–28. 10.1016/j.virol.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 26.Cheng F, Weidner-Glunde M, Varjosalo M, Rainio E-M, Lehtonen A, Schulz TF, Koskinen PJ, Taipale J, Ojala PM. 2009. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog 5:e1000324. 10.1371/journal.ppat.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mikovits J, Ruscetti F, Zhu W, Bagni R, Dorjsuren D, Shoemaker R. 2001. Potential cellular signatures of viral infections in human hematopoietic cells. Dis Markers 17:173–178. 10.1155/2001/896953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rainio E-M, Ahlfors H, Carter KL, Ruuska M, Matikainen S, Kieff E, Koskinen PJ. 2005. Pim kinases are upregulated during Epstein-Barr virus infection and enhance EBNA2 activity. Virology 333:201–206. 10.1016/j.virol.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Miyakawa K, Matsunaga S, Yokoyama M, Nomaguchi M, Kimura Y, Nishi M, Kimura H, Sato H, Hirano H, Tamura T, Akari H, Miura T, Adachi A, Sawasaki T, Yamamoto N, Ryo A. 2019. PIM kinases facilitate lentiviral evasion from SAMHD1 restriction via Vpx phosphorylation. Nat Commun 10:1844. 10.1038/s41467-019-09867-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aho TLT, Lund RJ, Ylikoski EK, Matikainen S, Lahesmaa R, Koskinen PJ. 2005. Expression of human Pim family genes is selectively up-regulated by cytokines promoting T helper type 1, but not T helper type 2, cell differentiation. Immunology 116:82–88. 10.1111/j.1365-2567.2005.02201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Pontén F. 2015. Tissue-based map of the human proteome. Science 347:1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 32.Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, Bäckström A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lindskog C, Mulder J, Mulvey CM, Nilsson P, Oksvold P, Rockberg J, Schutten R, Schwenk JM, Sivertsson Å, Sjöstedt E, Skogs M, Stadler C, Sullivan DP, Tegel H, Winsnes C, Zhang C, Zwahlen M, Mardinoglu A, Pontén F, von Feilitzen K, Lilley KS, Uhlén M, Lundberg E. 2017. A subcellular map of the human proteome. Science 356. 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- 33.Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnström H, Glimelius B, Sjöblom T, Edqvist P-H, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F. 2017. A pathology atlas of the human cancer transcriptome. Science 357. 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- 34.Alvarado Y, Giles FJ, and, Swords RT. 2012. The PIM kinases in hematological cancers. Expert Rev Hematol 5:81–96. 10.1586/ehm.11.69. [DOI] [PubMed] [Google Scholar]
- 35.Bellon M, Baydoun HH, Yao Y, Nicot C. 2010. HTLV-1 Tax-dependent and -independent events associated with immortalization of human primary T lymphocytes. Blood 115:2441–2448. 10.1182/blood-2009-08-241117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fryrear KA, Durkin SS, Gupta SK, Tiedebohl JB, Semmes OJ. 2009. Dimerization and a novel Tax speckled structure localization signal are required for Tax nuclear localization. J Virol 83:5339–5352. 10.1128/JVI.00232-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith MR, and, Greene WC. 1992. Characterization of a novel nuclear localization signal in the HTLV-1 tax transactivator protein. Virology 187:316–320. 10.1016/0042-6822(92)90320-O. [DOI] [PubMed] [Google Scholar]
- 38.Tsuji T, Sheehy N, Gautier VW, Hayakawa H, Sawa H, Hall WW. 2007. The nuclear import of the human T lymphotropic virus type I (HTLV-1) tax protein is carrier- and energy-independent. J Biol Chem 282:13875–13883. 10.1074/jbc.M611629200. [DOI] [PubMed] [Google Scholar]
- 39.Gao L, Harhaj EW. 2013. HSP90 protects the human T-cell leukemia virus type 1 (HTLV-1) tax oncoprotein from proteasomal degradation to support NF-kappaB activation and HTLV-1 replication. J Virol 87:13640–13654. 10.1128/JVI.02006-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan P, Fu J, Qu Z, Li S, Tanaka T, Grusby MJ, Xiao G. 2009. PDLIM2 suppresses human T-cell leukemia virus type I Tax-mediated tumorigenesis by targeting Tax into the nuclear matrix for proteasomal degradation. Blood 113:4370–4380. 10.1182/blood-2008-10-185660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohtake F, Tsuchiya H, Saeki Y, Tanaka K. 2018. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc Natl Acad Sci USA 115:E1401–E1408. 10.1073/pnas.1716673115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiari E, Lamsoul I, Lodewick J, Chopin C, Bex F, Pique C. 2004. Stable ubiquitination of human T-cell leukemia virus type 1 tax is required for proteasome binding. J Virol 78:11823–11832. 10.1128/JVI.78.21.11823-11832.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shirinian M, Kfoury Y, Dassouki Z, El-Hajj H, Bazarbachi A. 2013. Tax-1 and Tax-2 similarities and differences: focus on post-translational modifications and NF-kappaB activation. Front Microbiol 4:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shembade N, Harhaj NS, Yamamoto M, Akira S, Harhaj EW. 2007. The human T-cell leukemia virus type 1 Tax oncoprotein requires the ubiquitin-conjugating enzyme Ubc13 for NF-kappaB activation. J Virol 81:13735–13742. 10.1128/JVI.01790-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicot C, Astier-Gin T, Edouard E, Legrand E, Moynet D, Vital A, Londos-Gagliardi D, Moreau JP, Guillemain B. 1993. Establishment of HTLV-1-infected cell lines from French, Guianese and West Indian patients and isolation of a proviral clone producing viral particles. Virus Res 30:317–334. 10.1016/0168-1702(93)90099-9. [DOI] [PubMed] [Google Scholar]
- 46.Kimata JT, Wong FH, Wang JJ, Ratner L. 1994. Construction and characterization of infectious human T-cell leukemia virus type 1 molecular clones. Virology 204:656–664. 10.1006/viro.1994.1581. [DOI] [PubMed] [Google Scholar]
- 47.Derse D, Mikovits J, Ruscetti F. 1997. X-I and X-II open reading frames of HTLV-1 are not required for virus replication or for immortalization of primary T-cells in vitro. Virology 237:123–128. 10.1006/viro.1997.8781. [DOI] [PubMed] [Google Scholar]
- 48.Ionov Y, Le X, Tunquist BJ, Sweetenham J, Sachs T, Ryder J, Johnson T, Lilly MB, Kraft AS. 2003. Pim-1 protein kinase is nuclear in Burkitt's lymphoma: nuclear localization is necessary for its biologic effects. Anticancer Res 23:167–178. [PubMed] [Google Scholar]
- 49.Meeker TC, Loeb J, Ayres M, Sellers W. 1990. The human Pim-1 gene is selectively transcribed in different hemato-lymphoid cell lines in spite of a G + C-rich housekeeping promoter. Mol Cell Biol 10:1680–1688. 10.1128/MCB.10.4.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shay KP, Wang Z, Xing P-X, McKenzie IFC, Magnuson NS. 2005. Pim-1 kinase stability is regulated by heat shock proteins and the ubiquitin-proteasome pathway. Mol Cancer Res 3:170–181. 10.1158/1541-7786.MCR-04-0192. [DOI] [PubMed] [Google Scholar]
- 51.Mizuno K, Shirogane T, Shinohara A, Iwamatsu A, Hibi M, Hirano T. 2001. Regulation of Pim-1 by Hsp90. Biochem Biophys Res Commun 281:663–669. 10.1006/bbrc.2001.4405. [DOI] [PubMed] [Google Scholar]
- 52.Yan P, Qing G, Qu Z, Wu C-C, Rabson A, Xiao G. 2007. Targeting autophagic regulation of NFkappaB in HTLV-1 transformed cells by geldanamycin: implications for therapeutic interventions. Autophagy 3:600–603. 10.4161/auto.4761. [DOI] [PubMed] [Google Scholar]
- 53.Taniguchi H, Hasegawa H, Sasaki D, Ando K, Sawayama Y, Imanishi D, Taguchi J, Imaizumi Y, Hata T, Tsukasaki K, Uno N, Morinaga Y, Yanagihara K, Miyazaki Y. 2014. Heat shock protein 90 inhibitor NVP-AUY922 exerts potent activity against adult T-cell leukemia-lymphoma cells. Cancer Sci 105:1601–1608. 10.1111/cas.12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mahgoub M, Yasunaga J-I, Iwami S, Nakaoka S, Koizumi Y, Shimura K, Matsuoka M. 2018. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc Natl Acad Sci USA 115:E1269–E1278. 10.1073/pnas.1715724115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keane NA, Reidy M, Natoni A, Raab MS, O’Dwyer M. 2015. Targeting the Pim kinases in multiple myeloma. Blood Cancer J 5:e325. 10.1038/bcj.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daenthanasanmak A, Wu Y, Iamsawat S, Nguyen HD, Bastian D, Zhang M, Sofi MH, Chatterjee S, Hill EG, Mehrotra S, Kraft AS, Yu X-Z. 2018. PIM-2 protein kinase negatively regulates T cell responses in transplantation and tumor immunity. J Clin Invest 128:2787–2801. 10.1172/JCI95407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramachandran J, Santo L, Siu KT, Panaroni C, Raje N. 2016. Pim2 is important for regulating DNA damage response in multiple myeloma cells. Blood Cancer J 6:e462. 10.1038/bcj.2016.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bellon M, Bialuk I, Galli V, Bai X-T, Farre L, Bittencourt A, Marçais A, Petrus MN, Ratner L, Waldmann TA, Asnafi V, Gessain A, Matsuoka M, Franchini G, Hermine O, Watanabe T, Nicot C. 2021. Germinal epimutation of Fragile Histidine Triad (FHIT) gene is associated with progression to acute and chronic adult T-cell leukemia diseases. Mol Cancer 20:86. 10.1186/s12943-021-01370-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baydoun HH, Bai XT, Shelton S, Nicot C. 2012. HTLV-1 tax increases genetic instability by inducing DNA double strand breaks during DNA replication and switching repair to NHEJ. PLoS One 7:e42226. 10.1371/journal.pone.0042226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Warfel NA, Kraft AS. 2015. PIM kinase (and Akt) biology and signaling in tumors. Pharmacol Ther 151:41–49. 10.1016/j.pharmthera.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maeda M, Tanabe-Shibuya J, Miyazato P, Masutani H, Yasunaga J-i, Usami K, Shimizu A, Matsuoka M. 2020. IL-2/IL-2 receptor pathway plays a crucial role in the growth and malignant transformation of HTLV-1-infected T cells to develop adult T-cell leukemia. Front Microbiol 11:356. 10.3389/fmicb.2020.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nair JR, Caserta J, Belko K, Howell T, Fetterly G, Baldino C, Lee KP. 2017. Novel inhibition of PIM2 kinase has significant anti-tumor efficacy in multiple myeloma. Leukemia 31:1715–1726. 10.1038/leu.2016.379. [DOI] [PMC free article] [PubMed] [Google Scholar]