

ABSTRACT
Measles virus (MeV), an enveloped RNA virus in the family Paramyxoviridae, usually causes acute febrile illness with skin rash but in rare cases persists in the brain, causing a progressive neurological disorder, subacute sclerosing panencephalitis (SSPE). MeV bears two envelope glycoproteins, the hemagglutinin (H) and fusion (F) proteins. The H protein possesses a head domain that initially mediates receptor binding and a stalk domain that subsequently transmits the fusion-triggering signal to the F protein. We recently showed that cell adhesion molecule 1 (CADM1; also known as IGSF4A, Necl-2, and SynCAM1) and CADM2 (also known as IGSF4D, Necl-3, and SynCAM2) are host factors enabling cell-cell membrane fusion mediated by hyperfusogenic F proteins of neuropathogenic MeVs as well as MeV spread between neurons lacking the known receptors. CADM1 and CADM2 interact in cis with the H protein on the same cell membrane, triggering hyperfusogenic F protein-mediated membrane fusion. Multiple isoforms of CADM1 and CADM2 containing various lengths of their stalk regions are generated by alternative splicing. Here, we show that only short-stalk isoforms of CADM1 and CADM2 predominantly expressed in the brain induce hyperfusogenic F protein-mediated membrane fusion. While the known receptors interact in trans with the H protein through its head domain, these isoforms can interact in cis even with the H protein lacking the head domain and trigger membrane fusion, presumably through its stalk domain. Thus, our results unveil a new mechanism of viral fusion triggering by host factors.
IMPORTANCE Measles, an acute febrile illness with skin rash, is still an important cause of childhood morbidity and mortality worldwide. Measles virus (MeV), the causative agent of measles, may also cause a progressive neurological disorder, subacute sclerosing panencephalitis (SSPE), several years after acute infection. The disease is fatal, and no effective therapy is available. Recently, we reported that cell adhesion molecule 1 (CADM1) and CADM2 are host factors enabling MeV cell-to-cell spread in neurons. These molecules interact in cis with the MeV attachment protein on the same cell membrane, triggering the fusion protein and causing membrane fusion. CADM1 and CADM2 are known to exist in multiple splice isoforms. In this study, we report that their short-stalk isoforms can induce membrane fusion by interacting in cis with the viral attachment protein independently of its receptor-binding head domain. This finding may have important implications for cis-acting fusion triggering by host factors.
KEYWORDS: CADM1, CADM2, SSPE, subacute sclerosing panencephalitis, cis-acting fusion triggering, hemagglutinin, measles virus, membrane fusion, neuropathogenicity, splice isoform
INTRODUCTION
Measles is a highly contagious disease characterized by high fever, respiratory symptoms, conjunctivitis, and maculopapular rash (1). The disease is preventable by effective live-attenuated vaccines but is still an important cause of childhood morbidity and mortality worldwide, especially in developing countries (2). Measles virus (MeV), the causative agent of measles, is an enveloped RNA virus belonging to the genus Morbillivirus in the family Paramyxoviridae. In rare cases, MeV may persist in the central nervous system (CNS), causing a progressive neurological disorder, subacute sclerosing panencephalitis (SSPE), several years after acute infection (3, 4). Patients with SSPE show characteristic clinical symptoms such as personality changes, myoclonus, and dementia (4).The incidence of SSPE is 6.5 to 11 cases per 100,000 measles cases, but the risk of developing the disease is increased in children contracting measles below 5 years of age (5). No effective treatment is currently available for SSPE (6).
MeV has two envelope glycoproteins, the hemagglutinin (H) and fusion (F) proteins. These proteins cooperatively mediate membrane fusion required for virus entry and direct cell-to-cell spread (1). Recent studies have provided insights into molecular mechanisms of fusion triggering by viral envelope glycoproteins of MeV (7–14) and other paramyxoviruses (15–20). Paramyxovirus receptor attachment proteins, including the MeV H protein, consist of an N-terminal cytoplasmic tail, a transmembrane region, a membrane-proximal stalk region, and a C-terminal head domain. The head domain binds to its receptor, and the stalk region is involved in the interaction with the viral F proteins (11, 14–16, 18, 19). In the case of MeV, the head domain of the H protein initially binds to the known receptors, the signaling lymphocytic activation molecule family member 1 (SLAMF1; also known as SLAM and CD150) and nectin-4 (10, 21–24). Then, some conformational changes of the head domain are thought to occur, causing structural rearrangements of the stalk, which in turn trigger refolding of the F protein, leading to membrane fusion (9, 10).
Due to the lack of the known receptors in neurons (25, 26), wild-type (WT) MeV isolates from acute measles are not neurotropic. However, MeV mainly infects neurons in SSPE and is thought to propagate in neurons by transsynaptic cell-to-cell transmission (27–30). Recent studies have shown that hyperfusogenic mutations in the extracellular domain of the F protein (e.g., T461I), which are found in MeV isolates from SSPE, enable MeV spread in primary human neurons in vitro as well as in the brains of experimentally infected mice and hamsters (1, 6, 30–36). These hyperfusogenic mutant F proteins, which are structurally unstable, can be triggered even by the weak interaction between the SLAM- or nectin-4-blind mutant H protein and its corresponding receptor (37). More recently, we reported that cell adhesion molecule 1 (CADM1, also known as IGSF4A, Necl-2, and SynCAM1) and CADM2 (also known as IGSF4D, Necl-3, and SynCAM2) are host factors enabling MeV spread in neurons (38). While the known receptors interact in trans with the H protein on the envelope, CADM1 and CADM2 interact in cis with the H protein on the same membrane, triggering the mutant F protein-mediated membrane fusion (38). Thus, CADM1 and CADM2 are host factors utilizing a new mechanism for viral fusion triggering, named “receptor-mimicking cis-acting fusion triggering” (38).
CADM1 and CADM2 consist of an N-terminal immunoglobulin (Ig)-like V-type domain, two Ig-like C2-type domains, a membrane-proximal stalk region, a transmembrane region, and a C-terminal cytoplasmic tail. These molecules exist in multiple splice isoforms containing various lengths of the stalk regions (39–45), but their functional differences in fusion triggering have not been investigated. In this study, we show that specific isoforms of CADM1 and CADM2, which contain the short stalk regions, fully induce the mutant F protein-mediated membrane fusion. These short-stalk isoforms, but not long-stalk ones, interact with the H protein lacking its head domain and trigger membrane fusion, presumably through the H protein stalk region. These findings further our understanding of cis-acting fusion triggering by CADM1 and CADM2 and may have important implications for how viral attachment proteins can interact with their host factors.
RESULTS
Hamster CADM1 and CADM2 lacking specific exons induce hyperfusogenic F protein-mediated membrane fusion.
It has been shown that recombinant MeVs possessing hyperfusogenic mutant F proteins, but not WT MeV, spread in the brains of experimentally infected mice and hamsters in vivo (31–33, 35). We showed that mouse CADM1 and CADM2 induce hyperfusogenic mutant F protein-mediated membrane fusion as efficiently as human CADM1 and CADM2 (38), but whether hamster CADM1 and CADM2 also function similarly has not been determined.
The syncytium formation assay (fusion assay) was used to evaluate cell-to-cell membrane fusion (38). Briefly, expression plasmids encoding the H protein, the hyperfusogenic F(T461I) protein, enhanced green fluorescent protein (EGFP), and one of human, mouse, and hamster CADM1 and CADM2 were used to transfect 293FT cells, and the cells were observed 30 h after transfection under a fluorescence microscope. Although human and mouse CADM1 and CADM2 efficiently induced syncytium formation, confirming our previous findings (38), syncytium formation was not observed upon expression of hamster CADM1 and CADM2 (Fig. 1A).
FIG 1.

Hamster CADM1 having exon 8 and CADM2 having exon 9 do not induce membrane fusion. (A) Fusion assay with human, mouse, and hamster CADM1 (left) and CADM2 (right). pCA7 expression plasmids encoding the H protein, the F(T461I) protein, and EGFP with pCA7 encoding human, mouse, or hamster CADM1/2 or pCA7 alone as a negative control (NC) were used to transfect 293FT cells. Cells were observed by fluorescence microscopy 30 h after transfection. Exon organizations of CADM1 and CADM2 used in this experiment are also shown. The numbers above rectangles are exon numbers. The numbering of the exons follows previous studies (39, 44–46). The exons present in all isoforms are indicated by gray rectangles, and the exons that are present in only some isoforms are indicated by red, yellow, or magenta rectangles. Bar = 250 μm. (B) Fusion assay with different isoforms of hamster CADM1 (left) and CADM2 (right). pCA7 expression plasmids encoding the H protein, the F(T461I) protein, and EGFP with pCA7 encoding one of the hamster CADM1/2 isoforms or pCA7 alone (NC) were used to transfect 293FT cells. Cells were observed by fluorescence microscopy 30 h after transfection. Bar = 250 μm. (C and D) DSP assay with human (h), mouse (m), and hamster (ham) CADM1 (C) and CADM2 (D) isoforms. pCA7 expression plasmids encoding the H protein and the F(T461I) protein with pCA7 encoding one of the CADM1/2 isoforms or pCA7 alone were used to transfect cocultured 293FT/DSP1 and 293FT/DSP2 cells. Renilla luciferase activity was measured 24 h after transfection (n = 3, means ± standard deviations [SD]). (E and F) Cell surface biotinylation assay with hamster CADM1 (E) and CADM2 (F) isoforms. pCA7 encoding E-cadherin (a cell surface protein control) with pCA7 encoding Flag-tagged EGFP (an intracellular protein control), one of the Flag-tagged hamster CADM1/2 isoforms, or pCA7 alone was used to transfect 293FT cells. Precipitates of biotinylated cell surface proteins and cell lysates were detected by Western blotting using anti-E-cadherin (top) or anti-Flag Ab (bottom).
We initially thought that CADM1 and CADM2 do not serve as host factors for neuropathogenic MeV spread in hamsters. However, by examining sequence alignments of human, mouse, and hamster CADM1 and CADM2 genes, we noticed that hamster CADM1 and CADM2 used in the above-described experiment are in different splice isoforms, compared with the human and mouse CADM1 and CADM2 used (Fig. 1A). The hamster CADM1 used has exon 8, and the hamster CADM2 used lacks exon 2 but instead has exon 9. (The numbering of the exons in this paper follows that in previous studies [39, 44–46].) Therefore, we examined whether these isoform differences affect the fusion-triggering function of CADM1 and CADM2. While hamster CADM2 having exon 2 did not induce membrane fusion, hamster CADM1 lacking exon 8 and hamster CADM2 lacking exon 9 induced syncytium formation as efficiently as human and mouse CADM1 and CADM2 (Fig. 1B).
The levels of membrane fusion were also evaluated by the dual split protein (DSP) assay (37, 47–49). In this system, a pair of chimeric reporter proteins, DSP1 and DSP2, each comprising the split Renilla luciferase and split green fluorescent protein (GFP), are stably expressed in 293FT cells (293FT/DSP1 and 293FT/DSP2 cells). When cell-cell fusion is induced between 293FT/DSP1 and 293FT/DSP2 cells, Renilla luciferase and GFP activities are restored by the association of DSP1 and DSP2. 293FT/DSP1 and 293FT/DSP2 cells were cocultured and then transfected with plasmids encoding the H protein, the F(T461I) protein, and one of the CADM1 and CADM2 isoforms. Upon expression, hamster CADM1 lacking exon 8 and hamster CADM2 lacking exon 2 and exon 9 as well as human and mouse CADM1 and CADM2 induced cell-cell fusion, but the other hamster isoforms examined did not (Fig. 1C and D), consistent with the observations with the fusion assay (Fig. 1A and B).
We next examined expression levels of these hamster isoforms by the cell surface biotinylation. The presence or absence of exon 8 and exon 9 did not greatly affect the cell surface expression levels of hamster CADM1 (Fig. 1E) and CADM2 (Fig. 1F), respectively. The results indicate that the functional differences among hamster CADM1 and CADM2 isoforms are not caused by changes in their surface expression levels.
Specific isoforms of human CADM1 and CADM2 induce hyperfusogenic F protein-mediated membrane fusion.
Because only certain isoforms of hamster CADM1 and CADM2 exhibit the ability to induce hyperfusogenic F protein-mediated membrane fusion, we also examined different isoforms of human CADM1 and CADM2 for their fusion-triggering activity. Figure 2 shows the exon organization of the human CADM1 (Fig. 2A) and CADM2 (Fig. 2B) splice isoforms obtained from the Genotype-Tissue Expression (GTEx) consortium using the GTEx portal (https://www.gtexportal.org/home/gene/CADM1 and https://www.gtexportal.org/home/gene/CADM2). The canonical isoforms of human CADM1 and CADM2 used in the above-described experiments (Fig. 1) and our previous study (38) are referred to as CADM1 and CADM2, respectively, and the other isoforms are described by appending names of exons that the canonical isoforms do not have (e.g., CADM1+ex8). Four noncanonical CADM1 isoforms have different combinations of exon 8, exon 9, and exon 10 (Fig. 2A). These exons encode parts of the stalk region, endowing CADM1 isoforms with different lengths of the stalk. Two noncanonical CADM2 isoforms lack exon 2 but have exon 9, and one of the noncanonical isoforms has an exon 1 different from that of the canonical CADM2 (Fig. 2B). Exon 9 encodes part of the stalk region of CADM2.
FIG 2.

Various isoforms of human CADM1 and CADM2. Exon organizations of human CADM1 (A) and CADM2 (B) are shown. Exons present in all isoforms are indicated by gray rectangles, and exons present in only some isoforms are indicated by red, blue, green, yellow, magenta, or black rectangles. The canonical isoform is designated CADM1 or CADM2, and the other isoforms are designated by appending numbers of exons not present in the canonical isoform (e.g., CADM1+ex8). One of the noncanonical CADM2 isoforms, CADM2+ex9(1), has the same exon 1 as the canonical CADM2, whereas another noncanonical isoform, CADM2+ex9(2), has a different exon 1. Schematic domain organizations of CADM1/2 are shown below the corresponding exons. Amino acids encoded by exon 8, exon 9, and exon 10 of CADM1 and exon 9 of CADM2 in the stalk regions are also shown. V, Ig-like V-type domain; C, Ig-like C2-type domain; TM, transmembrane domain.
We used the fusion assay (Fig. 3A) and the DSP assay (Fig. 3B) to examine whether these different isoforms trigger membrane fusion. Although CADM1+ex9 and CADM1+ex10 are not found in the database, we also examined these possible isoforms in order to analyze the effect of exon 9 and exon 10 alone. While upon coexpression with the H protein and the F(T461I) protein, CADM1+ex9 induced syncytium formation as efficiently as the canonical CADM1 and CADM2, the other isoforms [CADM1+ex8, CADM1+ex10, CADM1+ex8/9, CADM1+ex9/10, CADM1+ex8/9/10, CADM2+ex9(1), and CADM2+ex9(2)] did not (Fig. 3A and B). Consistent results were obtained when the fusion assay was performed using two other hyperfusogenic F proteins, F(M94V) and F-triple(S103I N462S N465S) (Fig. 3C). These substitutions in the F protein have been detected in multiple MeV isolates from SSPE patients (33, 35). The isoforms that failed to induce hyperfusogenic F protein-mediated membrane fusion were expressed on the cell surface almost as abundantly as the canonical CADM1 and CADM2, as examined by the cell surface biotinylation (Fig. 3D). Thus, low levels of cell surface expression cannot account for their failure to induce membrane fusion.
FIG 3.

Specific isoforms of CADM1 and CADM2 induce membrane fusion. (A) Fusion assay with various isoforms of human CADM1 (left) and CADM2 (right). pCA7 plasmids encoding the H protein, the F(T461I) protein, and EGFP with pCA7 encoding human CADM1/2 [CADM1, CADM1+ex8, CADM1+ex9(*), CADM1+ex10(*), CADM1+ex8/9, CADM1+ex9/10, CADM1+ex8/9/10, CADM2, CADM2+ex9(1), or CADM2+ex9(2)] or pCA7 alone as a negative control (NC) were used to transfect 293FT cells. Cells were observed by fluorescence microscopy 30 h after transfection. Exon organizations of respective isoforms are also shown. The isoforms indicated with asterisks are not found in the GTEx database. Bar = 250 μm. (B) DSP assay with various isoforms of human CADM1/2. pCA7 expression plasmids encoding the H protein and the F(T461I) protein with pCA7 encoding one CADM1/2 isoform or pCA7 alone were used to transfect cocultured 293FT/DSP1 and 293FT/DSP2 cells. Renilla luciferase activity was measured 24 h after transfection (n = 3; means ± SD). (C) Fusion assay using different hyperfusogenic F proteins with isoforms of human CADM1/2. Bar = 200 μm. (D) Cell surface biotinylation assay with isoforms of human CADM1/2. pCA7 encoding E-cadherin (a cell surface protein control) with pCA7 encoding one of the Flag-tagged human CADM1/2 isoforms was used to transfect 293FT cells. Precipitates of biotinylated cell surface proteins and cell lysates were detected by Western blotting using anti-E-cadherin (top) or anti-Flag Ab (bottom).
trans-homophilic and heterophilic interactions of CADM1 and CADM2 are known to mediate cell-cell adhesion (50). Therefore, the long-stalk isoforms of CADM1 and CADM2 might have lost their fusion-inducing activity due to the inappropriate distance between membranes and resultant loss of cell-cell adhesion but not due to the loss of their cis-acting triggering function. We previously reported that cell-cell adhesion mediated by E-cadherin provides an optimal distance for cis-acting fusion triggering (37). Therefore, we performed the fusion and DSP assays by expressing E-cadherin together with the H protein, the F(T461I) protein, and one of the CADM1/2 isoforms. Nonfunctional isoforms of CADM1 and CADM2 did not trigger membrane fusion even when E-cadherin was coexpressed (Fig. 4). The results indicate that the insertion of extra sequences into the stalk region of human and hamster CADM1 and CADM2 disrupts their ability to trigger hyperfusogenic F protein-mediated membrane fusion.
FIG 4.

E-cadherin expression does not rescue the fusion-triggering function of long-stalk CADM1/2 isoforms. (A) Fusion assay with E-cadherin. pCA7 plasmids encoding the H protein, the F(T461I) protein, EGFP, and one of the isoforms of human CADM1/2 (CADM1, CADM1+ex8/9/10, CADM2, and CADM2+ex9) with pCA7 encoding E-cadherin or pCA7 alone as a negative control (NC) were used to transfect 293FT cells. Cells were observed by fluorescence microscopy 30 h after transfection. Bar = 200 μm. (B) DSP assay with E-cadherin. pCA7 plasmids encoding the H protein, the F(T461I) protein, and one of the isoforms of human CADM1/2 (CADM1, CADM1+ex8/9/10, CADM2, and CADM2+ex9) with pCA7 encoding E-cadherin or pCA7 alone were used to transfect cocultured 293FT/DSP1 and 293FT/DSP2 cells. Renilla luciferase activity was measured 24 h after transfection (n = 3; means ± SD).
Fusion-triggering isoforms of CADM1 and CADM2 are expressed in the brain.
We created graphs showing tissue distributions of the isoforms of CADM1 and CADM2 (Fig. 5A and B), based on the database of alternatively spliced mRNA isoforms in various human tissues provided by the GTEx Portal (http://www.gtexportal.org/home/). Although fusion-triggering isoforms of CADM1 and CADM2 (the canonical isoforms) are predominantly expressed, there exist some nontriggering isoforms (CADM1+ex8/9/10, CADM1+ex8/9, and CADM2+ex9), albeit at lower levels, in the brain. The expression of mouse and hamster short-stalk CADM1/2 isoforms in the brain was also confirmed by PCR and direct sequencing using cDNA templates derived from mouse primary neurons and hamster brain tissues, respectively (Fig. 5C and D). We then examined whether coexpression of nontriggering isoforms may inhibit the fusion-triggering function of the canonical isoforms. The fusion assay (Fig. 6A) and DSP assay (Fig. 6B) showed that the level of cell-cell fusion was not greatly affected by coexpression of CADM1+ex8/9/10 or CADM2+ex9, compared with that induced by the respective canonical isoform only.
FIG 5.

Tissue expression patterns of alternatively spliced mRNA isoforms of CADM1 and CADM2. (A and B) Tissue expression patterns of human CADM1/2 isoforms were obtained from the GTEx consortium using the GTEx Portal (https://www.gtexportal.org/home/gene/CADM1 and https://www.gtexportal.org/home/gene/CADM2). The bars in the graphs indicate the total expression levels of all isoforms of CADM1 (A) or CADM2 (B) (left y axis). The lines in the graphs indicate the expression levels of respective isoforms of human CADM1 and CADM2 (right y axis). TPM, transcripts per million. (C and D) Expression of mouse (C) or hamster (D) CADM1/2 isoforms in the brain was examined by PCR using cDNA templates derived from mouse primary neurons (m-cDNA) and hamster brains (ham-cDNA), respectively. DNA fragments encompassing exons 6 to 11 for CADM1 or exons 7 to 10 for CADM2 were amplified. pCA7 plasmids encoding mouse CADM1, mouse CADM1+ex8, mouse CAMD2, mouse CADM2+ex9, hamster CADM1, hamster CADM1+ex8, hamster CADM2, and hamster CADM2+ex9 were used as control templates for PCR. Nucleotide sequences of amplified PCR fragments were confirmed by direct sequencing. Red and black asterisks indicate fusion-triggering (short-stalk) isoforms and other isoforms of CADM1/2, respectively.
FIG 6.

Nontriggering isoforms of CADM1 and CADM2 do not affect membrane fusion triggered by the canonical isoforms. (A) Fusion assay. pCA7 plasmids encoding the H protein, the F(T461I) protein and EGFP with pCA7 encoding the canonical isoform, the nontriggering isoform, or both were used to transfect 293FT cells. Cells were observed by fluorescence microscopy 30 h after transfection. Bar = 200 μm. (B) DSP assay. pCA7 plasmids encoding the H protein and the F(T461I) protein with pCA7 encoding the canonical isoform, the nontriggering isoform, or both were used to transfect cocultured 293FT/DSP1 and 293FT/DSP2 cells. Renilla luciferase activity was measured 24 h after transfection (n = 3; means ± SD). The data were analyzed by the unpaired two-tailed Student's t test. *, P = 0.0098; ns, not significant (P = 0.4818).
Thus, fusion-triggering isoforms of CADM1 and CADM2 are expressed in the brain, which is consistent with our previous finding that CADM1 and CADM2 are host factors required for MeV transmission between neurons (38).
CADM1 and CADM2 can induce membrane fusion independently of the H protein head domain.
The head domain of the H protein binds to its receptors (10, 22, 24), which in turn triggers refolding of the F protein. Whether the head domain is also involved in the cis-acting fusion triggering by CADM1 and CADM2 remains to be determined. Because the fusion-triggering function of CADM1 and CADM2 is disrupted by the insertion of the extra sequences into their membrane-proximal stalk regions, we hypothesized that the stalk region, not the head domain, of the H protein is involved in its cis-interaction with CADM1 and CADM2. Namely, the stalk regions of both molecules (the H protein and CADM1/CADM2) may be critical for their interaction. To test this hypothesis, we examined whether the H protein lacking its head domain (headless H) induces membrane fusion with CADM1 and CADM2. First, the pCA7 expression plasmid (51) encoding the influenza virus hemagglutinin (HA)-tagged headless H protein (residues 1 to 180 of the H protein) [designated H(headless)] was generated (Fig. 7A). Furthermore, since the H protein forms a tetrameric structure, we also prepared pCA7 encoding the HA-tagged headless H protein with a tetramerization domain at the C terminus [H(headless)-TD] (Fig. 7A). The tetramerization domain is derived from a derivative of the leucine zipper domain of the yeast GCN4 transcriptional activator, which assembles into a parallel 4-helix bundle (13, 52). Western blot analysis confirmed that the H(headless) and H(headless)-TD proteins were both successfully expressed 24 h after transfection into 293FT cells (Fig. 7B). We then performed the fusion assay with these headless H proteins (Fig. 7C). Notably, CADM1 and CADM2 induced syncytium formation with the H(headless)-TD protein, but not with the H(headless) protein, when expressed together with the F(T461I) protein. The levels of cell-cell fusion supported by the H(headless)-TD protein, however, appeared to be somewhat lower than those supported by the full-length H protein [H(full)]. In contrast, SLAM and nectin-4 induced membrane fusion neither with the H(headless)-TD protein nor with the H(headless) protein. CADM1+ex8/9/10 and CADM2+ex9 did not induce membrane fusion with any of the H proteins examined. The fusion-inducing activity of the H(headless)-TD protein was further confirmed by the DSP assay (Fig. 7D). These results indicate that the tetramerized H protein stalk region alone can support the cis-acting fusion triggering by CADM1 and CADM2.
FIG 7.

The tetramerized headless H protein supports membrane fusion induced by fusion-triggering isoforms of CADM1 and CADM2. (A) Schematic diagrams of the HA-tagged full-length H protein [H(full)], the HA-tagged headless H protein (residues 1 to 180 of the H protein) [H(headless)] and the HA-tagged H(headless) protein with a tetramerization domain [H(headless)-TD]. CP, cytoplasmic domain; TM, transmembrane domain; TD, tetramerization domain. (B) Expression plasmids encoding H(headless) and H(headless)-TD were used to transfect 293FT cells. Cell lysates were detected by Western blotting using anti-beta-actin (top) or anti-HA Ab (bottom). (C) Fusion assay with various H constructs. pCA7 plasmids encoding the F(T461I) protein and one of the H constructs [H(full), H(headless), or H(headless)-TD] with pCA7 encoding one of the host proteins (CADM1, CADM1+ex8, CADM2, CADM2+ex9, SLAM, or nectin-4) or pCA7 alone (NC) were used to transfect 293FT cells. Cells were observed by fluorescence microscopy 48 h after transfection. Bar = 200 μm. (D) DSP assay with the H(headless)-TD protein. pCA7 plasmids encoding H(full) or H(headless)-TD and the F(T461I) protein with pCA7 encoding one of the host proteins (CADM1, CADM1+ex8, CADM2, CADM2+ex9, SLAM, or nectin-4) were used to transfect cocultured 293FT/DSP1 and 293FT/DSP2 cells. Renilla luciferase activity was measured 48 h after transfection (n = 3; means ± SD). The data were analyzed by the unpaired two-tailed Student's t test. *1, P < 0.0001; *2, P = 0.8059; *3, P < 0.0001; *4, P = 0.4118; *5, P = 0.001; *6, P = 0.2676.
Fusion-triggering isoforms of CADM1 and CADM2 interact with the headless H protein.
We next examined the interactions of HA-tagged H proteins [H(full), H(headless) and H(headless)-TD] with His-tagged host proteins (CADM1, CADM1+ex8/9/10, CADM2, and CADM2+ex9) by coimmunoprecipitation. The H(full) protein was found to interact with both fusion-triggering (CADM1 and CADM2) and nontriggering (CADM1+ex8/9/10 and CADM2+ex9) isoforms (Fig. 8A). The H(headless) protein interacted with CADM1 and CADM2 but not with CADM1+ex8/9/10 and CADM2+ex9 (Fig. 8B). The H(headless)-TD protein produced high background precipitates (even with negative control), so that no conclusion could be drawn (data not shown).
FIG 8.
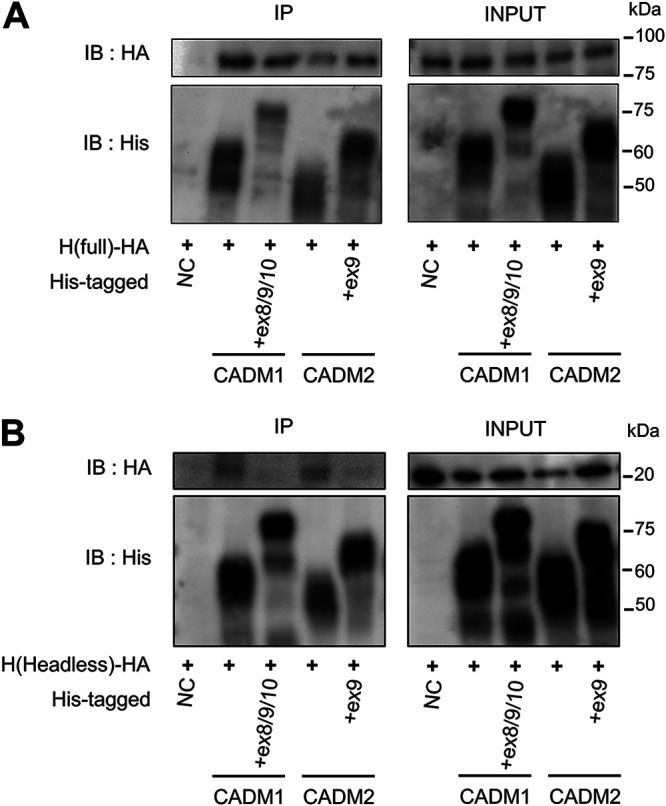
The canonical isoforms of CADM1/2 interact with the headless H protein. Interactions of the HA-tagged H(full) protein (A) or the HA-tagged H(headless) protein (B) with the His-tagged CADM1, CADM1+ex8/9/10, CADM2, and CADM2+ex9 proteins were examined by coimmunoprecipitation assay. Immunoprecipitates (IP) and total lysates (INPUT) were detected by immunoblotting (IB) using anti-HA (top) or anti-His Ab (bottom).
The results indicate that short-stalk isoforms of CADM1 and CADM2 can interact with the stalk region of the H protein. The interaction presumably allows cis-acting fusion triggering, in combination with a hyperfusogenic mutant F protein. The insertion of certain extra sequences into the stalk region in noncanonical CADM1 and CADM2 isoforms could interfere with the interaction, thereby negatively affecting cis-acting fusion triggering. Since the full-length H protein can interact not only with short-stalk isoforms of CADM1 and CADM2, but also with long-stalk isoforms, the head domain of the H protein might be able to interact with most, if not all, isoforms of CADM1 and CADM2.
Monoclonal Abs against the head domain of the MeV H protein do not inhibit membrane fusion induced by CADM1 and CADM2.
Next, we performed fusion and DSP assays in the presence of monoclonal antibodies (Abs) against the head domain of the MeV H protein, E81 and E103, which inhibit SLAM- and nectin-4-dependent MeV entry into cells (53, 54). As expected, E81 and E103 effectively inhibited membrane fusion induced by SLAM and nectin-4 but not that induced by CADM1 and CADM2 (Fig. 9).
FIG 9.

Monoclonal Abs against the H protein head domain, E81 and E103, do not inhibit membrane fusion induced by CADM1/2. (A) Fusion assay in the presence of the monoclonal Abs against the H head domain. pCA7 plasmids encoding the H protein, the F(T461I) protein and EGFP with pCA7 encoding SLAM, nectin-4, CADM1, or CADM2 or pCA7 alone were used to transfect 293FT cells. At 2 h after transfection, one monoclonal Ab (E81 or E103) (50 μg per mL) or mouse IgG (negative control) was added to culture media. Cells were observed under fluorescence microscopy 30 h after transfection. Bar = 200 μm. (B) The DSP assay in the presence of the monoclonal Abs against the H protein head domain. pCA7 plasmids encoding the H protein and the F(T461I) protein with pCA7 encoding CADM1, CADM2, SLAM, nectin-4, or pCA7 alone, were used to transfect cocultured 293FT/DSP1 and 293FT/DSP2 cells. At 2 h after transfection, one monoclonal Ab (E81 or E103) (50 μg per mL) or mouse IgG (negative control) was added to culture media. Renilla luciferase activity was measured 24 h after transfection (n = 3; means ± SD).
DISCUSSION
In this study, we show that only specific isoforms of CADM1 and CADM2 exhibit the cis-acting fusion-triggering function, causing hyperfusogenic F protein-mediated membrane fusion. Notably, these short-stalk isoforms can interact even with the H protein lacking the head domain (headless H) and trigger membrane fusion. A graphic summary of these findings is shown in Fig. 10.
FIG 10.

The graphical summary of interactions between the MeV H protein and trans- or cis-acting host factors. (A) Interactions of the MeV H protein with trans-acting viral receptors, SLAM and nectin-4, through the H protein head domain. (B) Interactions of the full-length and headless MeV H proteins with isoforms of cis-acting host factors, CADM1 and CADM2. The isoform containing a short stalk region functionally interacts with the full-length and headless H proteins. “YES” indicates the functional interaction leading to fusion triggering; “NO” indicates no fusion triggering. TD, tetramerization domain.
CADM1 and CADM2 belong to the Ig superfamily and function as intercellular adhesion molecules (39). Expression of CADM1 has been linked to diseases such as cancer, autism spectrum disorders, and venous thrombosis (55–57). Alternative splicing generates multiple isoforms of CADM1 and CADM2 (39). These isoforms could be functionally different in certain situations. For instance, specific CADM1 isoforms are functional for human mast cell adhesion (44), and a CADM2 isoform is found to be protective against psoriasis (40). The fusion-triggering (short-stalk) and nontriggering (long-stalk) isoforms of CADM1 and CADM2 are expressed at different levels in various tissues, including the brain (Fig. 5A and B). Notably, the fusion-triggering isoforms of CADM1 and CADM2 are expressed not only in the brain but also in several other tissues. Thus, factors other than the distribution of CADM1/2 isoforms, such as specific cell/tissue organization and immune responses, may play a role in MeV persistence in the brain.
The present study also shows that host factors could interact with the paramyxovirus attachment protein independently of its head domain. CADM1 and CADM2 functionally interact with the headless H protein, thereby triggering hyperfusogenic F protein-mediated membrane fusion (Fig. 7C and D and 10B). This is in contrast to the known MeV receptors, SLAM and nectin-4, which are known to interact in trans with the H protein through its head domain (10, 24) (Fig. 10A). It should be noted that SLAM also triggered membrane fusion with the headless H protein, although the level of membrane fusion induced was much lower than that induced by CADM1/2 (Fig. 7C and D). We showed previously that a mutant “SLAM-blind” H protein, which has a substitution in the SLAM binding site of its head domain and fails to use SLAM as its receptor, weakly interacts with SLAM in cis, leading to membrane fusion in combination with the hyperfusogenic F protein (37, 38). We suppose that the headless H protein, which also lacks the ordinary SLAM binding site in the head domain, may weakly interact with SLAM in cis and induce membrane fusion in conjunction with the hyperfusogenic F protein.
Although the headless H protein supported membrane fusion triggered by CADM1 and CADM2, the levels of membrane fusion induced by the full-length H protein were apparently higher than those by the headless H protein (Fig. 7C). The full-length H protein, but not the headless H protein, interacted with nontriggering (long-stalk) isoforms of CADM1 and CADM2 (Fig. 8), suggesting that the head domain also interacts with CADM1 and CADM2. Such interaction through the head domain may be required for CADM1 and CADM2 to exhibit the full activity of cis-acting fusion triggering. However, the stalk region of the H protein is functionally essential for cis-acting fusion triggering, because long-stalk isoforms of CADM1 and CADM2 cannot trigger membrane fusion despite their ability to interact with the head domain.
It is somewhat surprising that functional differences in fusion-triggering activity among different isoforms of CADM1 and CADM2 are caused by relatively small structural changes in their stalk regions. The insertion of extra sequences into the stalk region might disrupt the essential site directly interacting with the H protein. Alternatively, the height changes of CADM1 and CADM2 due to the extra sequences in the stalk region might simply make the binding sites on these molecules apart from the corresponding binding site on the H protein. Indeed, CADM1+ex9, which has a shorter insertion in the stalk region, compared with nontriggering CADM1+ex8 and CADM1+ex10, was found to maintain the fusion-triggering function (Fig. 3A and B). Further functional, biochemical, and structural studies will be required to determine exactly how CADM1 and CADM2 interact with the H protein.
High titers of anti-MeV Abs are generally detected in the cerebrospinal fluid and blood of patients with SSPE (1). The observation that CADM1 and CADM2 functionally interact with the headless H protein may provide insights into MeV immune evasion in the brain. The H protein is the main target of anti-MeV neutralizing Abs, and almost all of the neutralizing Abs are directed against epitopes on the head domain (58). MeV isolates from SSPE patients show antigenic properties similar to those of currently circulating MeVs (58). The present study indeed shows that monoclonal Abs against the H protein head domain inhibit the membrane fusion induced by SLAM and nectin-4 but not that induced by CADM1 and CADM2 (Fig. 9). Thus, anti-MeV Abs generated in SSPE patients may not be able to effectively inhibit the interaction of CADM1 and CADM2 with the H protein stalk region and MeV transmission, possibly contributing to MeV persistence in the brain.
Recombinant MeVs bearing hyperfusogenic F proteins have been shown to spread in the brains of experimentally infected mice and hamsters in vivo (31–33, 35). We have previously shown that mouse CADM1 and CADM2 induce hyperfusogenic F protein-mediated membrane fusion as efficiently as human CADM1 and CADM2 (38). The present study demonstrates that specific isoforms of hamster CADM1 and CADM2 also function in a similar manner (Fig. 1A and B). Furthermore, these fusion-triggering isoforms are expressed in mouse and hamster brains (Fig. 5C and D). These results indicate that MeVs carrying hyperfusogenic F proteins spread in the brains of these animals, through the same molecular mechanism as that in human brains. Thus, mice and hamsters should serve as good animal models to study MeV spread in the brain.
MATERIALS AND METHODS
Cells.
293FT cells (R700007; Invitrogen) were maintained in Dulbecco’s modified Eagle medium (DMEM) (Fujifilm Wako Pure Chemical Corporation, Japan) supplemented with 10% fetal bovine serum (FBS). 293FT cells stably expressing DSP1 and DSP2 (47–49), kindly provided by Z. Matsuda (University of Tokyo), were maintained in DMEM supplemented with 10% FBS and 1 μg/mL puromycin (InvivoGen, San Diego, CA).
Plasmids.
The eukaryotic expression vector pCA7 (51) is a derivative of pCAGGS (59). The pCA7 plasmids expressing MeV H (the IC-B strain), F(T461I), F(M94V), and F-triple(S103I N462S N465S), SLAMF1 (accession no. NM_003037.5), nectin-4 (NM_030916.3), human CADM1 (NM_001098517.2), mouse CADM1 (NM_001025600.1), human CADM2 (NM_001167675.2), and mouse CADM2 (NM_178721.4) were described previously (33, 37, 38, 60). The synthesized DNAs encoding hamster CADM1 (XM_005069479.4) and hamster CADM2 (XM_005082055.4) were purchased from Eurofins Genomics K.K. DNA fragments encoding H(headless) (residues 1 to 180 of the MeV H protein), H(headless)-TD [H(headless) protein with a tetramerization domain (GRMKQIEDKLEEILSKLYHIENELARIKKLLGER) derived from a derivative of the leucine zipper domain of the yeast GCN4 transcriptional activator (13, 52) at the C terminus], human CADM1+ex8, human CADM1+ex9, human CADM1+ex10, human CADM1+ex8/9, human CADM1+ex9/10, human CADM1+ex8/9/10, human CADM2+ex9(1), human CADM2+ex9(2), hamster CADM1, and hamster CADM2 were inserted into pCA7 predigested with EcoRI and NotI. For cell surface expression analysis, the Flag tag (DYKDDDDK) sequences were fused to the C termini of the EGFP, human CADM1, human CADM1+ex8, human CADM1+ex9, human CADM1+ex10, human CADM1+ex8/9, human CADM1+ex9/10, human CADM1+ex8/9/10, human CADM2, human CADM2+ex9, hamster CADM1, hamster CADM1+ex8, hamster CADM2, and hamster CADM2+ex9. For coimmunoprecipitation analysis, the HA tag (YPYDVPDYA) was fused to the C termini of H(full), H(headless), and H(headless)-TD, and the histidine (His) tag (HHHHHH) sequence was fused to the C termini of human CADM1, human CADM1+ex8/9/10, human CADM2, and human CADM2+ex9.
Abs.
Mouse monoclonal Abs (E81 and E103) were raised against the H protein of the Toyoshima MV strain (genotype A), as described previously (53, 54).
Fusion assay.
293FT cells cultured in 24-well plates were transfected with different combinations of pCA7 plasmids encoding MeV H [H(full), H(headless), or H(headless)-TD] (0.1 μg), MeV F [F(T461I), F(M94V), or F-triple(S103I N462S N465S)] (0.1 μg), and EGFP (0.1 μg) with pCA7 encoding one of the host molecules (SLAMF1, nectin-4, and isoforms of CADM1 and CADM2) or a vector plasmid alone (0.3 μg) using Lipofectamine LTX (Thermo Fisher Scientific). The cells were observed 30 to 48 h after transfection by fluorescence microscopy.
DSP assay.
DSP1 and DSP2 are split proteins of the Renilla luciferase and GFP which become functional when reassociated with each other after 293FT/DSP1 and 293FT/DSP2 cells are fused. The protocol of the DSP assay was previously described (37). Briefly, pCA7 expression plasmids encoding one MeV H protein [H(full), H(headless) or H(headless)-TD] (0.1 μg) and the MeV F(T461I) protein (0.1 μg) with a pCA7 plasmid encoding a host protein or pCA7 alone (0.3 μg) were used to transfect cocultured 293FT/DSP1and 293FT/DSP2 cells in 24-well plates using Lipofectamine LTX (Thermo Fisher Scientific). The Renilla luciferase activity was analyzed 24 to 48 h after transfection using the Renilla luciferase assay system (Promega).
Cell surface biotinylation assay.
Subconfluent monolayers of 293FT cells cultured on 12-well plates were transfected with 0.5 μg of pCA7 encoding one of the host molecules [Flag-tagged human CADM1, human CADM1+ex8/9/10, hamster CADM1, hamster CADM1+ex8, human CADM2, human CADM2+ex9, hamster CADM2, or hamster CADM2+ex9], the Flag-tagged EGFP (an intracellular protein control), or a pCA7 vector alone (a negative control) with 0.5 μg of pCA7 encoding E-cadherin (a cell surface protein control) using Lipofectamine LTX. At 24 h after transfection, cells were washed with phosphate-buffered saline (PBS) and then incubated with 200 μL of the biotin reagent solution (2 mM EZ-Link N-hydroxysulfosuccinimide [sulfo-NHS]–biotin [Thermo Scientific] in PBS) for 30 min at 4°C. After being washed three times with PBS containing 100 mM glycine for quenching, the cells were lysed in 200 μL of the immunoprecipitation (IP) lysis buffer (Thermo Fisher Scientific) containing the protease inhibitor cocktail (Sigma). The lysates were cleared by centrifugation for 30 min at 17,360 × g and 4°C. Then, 50 μL of each supernatant was mixed with an equal volume of 2× sodium dodecyl sulfate (SDS) loading buffer (125 mM Tris-HCl [pH 6.8], 10% 2-mercaptoethanol, 4% SDS, 0.1% bromophenol blue, and 20% glycerol), boiled for 5 min, and stored at −20°C as the cell lysate samples. The rest of the supernatant was incubated with avidin-agarose beads (A-9207; Sigma) for 3 h at 4°C. The samples were centrifuged and washed three times with immunoprecipitation (IP) lysis buffer. Pellets were mixed with 30 μL of 2× SDS loading buffer, boiled for 5 min, and stored at −20°C as biotinylated cell surface protein samples.
Coimmunoprecipitation assay.
Subconfluent monolayers of 293FT cells cultured on 6-well plates were transfected with 0.2 μg of pCA7 encoding MeV-H [H(full) or H(headless)] and 1 μg of pCA7 encoding His-tagged CADM1, CADM1+ex8/9/10, CADM2, or CADM2+ex9 or empty pCA7 as a control. At 48 h after transfection, the cells were washed with PBS and lysed in 500 μL of IP lysis buffer (Thermo Fisher Scientific) containing the protease inhibitor cocktail (EDTA free) (Nacalai). After incubation for 30 min at 4°C, the lysates were centrifuged at 17,360 × g for 30 min at 4°C. Then, 50 μL of each supernatant was mixed with an equal volume of 2× SDS loading buffer, boiled for 5 min, and stored as input samples. The rest of the supernatant was incubated for 10 min at 4°C with Dynabeads, which are magnetic beads coated with a cobalt-based immobilized-metal affinity chromatography (IMAC) chemistry and selectively bind His-tagged proteins (Invitrogen). The samples were then washed four times with binding/wash buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 0.01% Tween 20). His-tagged proteins were eluted from the magnetic beads with 100 μL of His elution buffer (50 mM sodium phosphate [pH 8.0], 300 mM imidazole, 300 mM NaCl, 0.01% Tween 20) for 5 min at 4°C. The magnetic beads were collected using a magnet, and the supernatant was mixed with 100 μL of 2× SDS loading buffer, boiled for 5 min, and stored as IP samples at −20°C.
Western blotting.
Proteins in samples were separated by SDS-polyacrylamide gel electrophoresis and then blotted onto polyvinylidene difluoride membranes (Hybond-P; Amersham Biosciences). The membranes were incubated with primary Abs for 1 h. Rabbit polyclonal Abs against the HA tag (GTX115044; GeneTex) and the His tag (PM 032; MBL) and mouse monoclonal Abs against E-cadherin (clone SHE78-7; TaKaRa Bio), Flag tag (F1804; Sigma-Aldrich), and β-actin (clone BA3R; BioVision) were used. The membranes were washed with Tris-buffered saline containing 0.05% Tween 20 (TBS-T) and incubated with horseradish peroxidase-conjugated goat anti-mouse (Bio-Rad) or anti-rabbit IgG Ab (Zymed) for 1 h at room temperature. After being washed with TBS-T, the membranes were treated with the ECL Plus reagent (Amersham Biosciences), and chemiluminescent signals were detected and imaged using a VersaDoc 5000 imager (Bio-Rad).
Tissue distribution analysis of CADM1 and CADM2 isoforms.
Tissue expression patterns of alternatively spliced mRNA isoforms of CADM1 and CADM2 in various human tissues were obtained from the GTEx consortium using the GTEx portal, which provides the database of isoform expressions for CADM1 (https://www.gtexportal.org/home/gene/CADM1) and CADM2 (https://www.gtexportal.org/home/gene/CADM2).
PCR was performed to investigate expressions of mouse and hamster CADM1/2 isoforms in the brain using cDNA templates derived from mouse primary neurons and hamster brain tissues. The mouse cDNA derived from mouse primary neurons was prepared as described previously (38). The hamster brain cDNA was purchased from Zyagen (AD-201). The primers used for this experiment were 5′-TGAAGGTGCACAAGGAGGAC-3′ and 5′-CTACGACGCCACCAATCACT-3′ for mouse CADM1, 5′-AGACCTTCACCGTTAGCAGC-3′ and 5′-CCTCCGATGAGAGCATGGTC-3′ for mouse CADM2, 5′-GAGTTGATGATGAAATGCCTCAACA-3′ and 5′-TTCTGTCCTCCTTCTGCATTGATTA-3′ for hamster CADM1, and 5′-GACAAGCATTAACTTTGACCTGTGA-3′ and 5′-CTCAGCACCTTTAGCTTCATTTGTT-3′ for hamster CADM2.
Measurements and statistical tests.
All measurements were taken from distinct samples. All statistical tests were performed using GraphPad Prism 8 software (unpaired two-tailed Student's t test).
ACKNOWLEDGMENTS
We thank Z. Matsuda for providing the DSP assay system, M. Suyama and C. Kikutake for supporting tissue distribution analysis of CADM1 and CADM2, and H. Harada for discussions. We also appreciate the technical assistance from The Research Support Center, Research Center for Human Disease Modeling, Kyushu University Graduate School of Medical Sciences.
This work was supported by JSPS KAKENHI grants JP20K07527 (to Y.S.) and JP20H00507 (to Y.Y.), Qdai-jump Research Program Wakaba Challenge FA79903505 (to Y.S.), and Japan Agency for Medical Research and Development (AMED) grant JP20wm0325002 (to T.H.). The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
Contributor Information
Yusuke Yanagi, Email: yanagi.yusuke.455@m.kyushu-u.ac.jp.
Yuta Shirogane, Email: shirogane.yuta.528@m.kyushu-u.ac.jp.
Stacey Schultz-Cherry, St. Jude Children's Research Hospital.
REFERENCES
- 1.Griffin DE. 2013. Measles virus, p 1042–1069. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Coughlin MM, Beck AS, Bankamp B, Rota PA. 2017. Perspective on global measles epidemiology and control and the role of novel vaccination strategies. Viruses 9:11. 10.3390/v9010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellini WJ, Rota JS, Lowe LE, Katz RS, Dyken PR, Zaki SR, Shieh WJ, Rota PA. 2005. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than was previously recognized. J Infect Dis 192:1686–1693. 10.1086/497169. [DOI] [PubMed] [Google Scholar]
- 4.Mekki M, Eley B, Hardie D, Wilmshurst JM. 2019. Subacute sclerosing panencephalitis: clinical phenotype, epidemiology, and preventive interventions. Dev Med Child Neurol 61:1139–1144. 10.1111/dmcn.14166. [DOI] [PubMed] [Google Scholar]
- 5.Schönberger K, Ludwig MS, Wildner M, Weissbrich B. 2013. Epidemiology of subacute sclerosing panencephalitis (SSPE) in Germany from 2003 to 2009: a risk estimation. PLoS One 8:e68909. 10.1371/journal.pone.0068909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe S, Shirogane Y, Sato Y, Hashiguchi T, Yanagi Y. 2019. New insights into measles virus brain infections. Trends Microbiol 27:164–175. 10.1016/j.tim.2018.08.010. [DOI] [PubMed] [Google Scholar]
- 7.Paal T, Brindley MA, St Clair C, Prussia A, Gaus D, Krumm SA, Snyder JP, Plemper RK. 2009. Probing the spatial organization of measles virus fusion complexes. J Virol 83:10480–10493. 10.1128/JVI.01195-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plemper RK, Brindley MA, Iorio RM. 2011. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog 7:e1002058. 10.1371/journal.ppat.1002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navaratnarajah CK, Oezguen N, Rupp L, Kay L, Leonard VHJ, Braun W, Cattaneo R. 2011. The heads of the measles virus attachment protein move to transmit the fusion-triggering signal. Nat Struct Mol Biol 18:128–135. 10.1038/nsmb.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hashiguchi T, Ose T, Kubota M, Maita N, Kamishikiryo J, Maenaka K, Yanagi Y. 2011. Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat Struct Mol Biol 18:135–142. 10.1038/nsmb.1969. [DOI] [PubMed] [Google Scholar]
- 11.Navaratnarajah CK, Negi S, Braun W, Cattaneo R. 2012. Membrane fusion triggering: three modules with different structure and function in the upper half of the measles virus attachment protein stalk. J Biol Chem 287:38543–38551. 10.1074/jbc.M112.410563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Apte-Sengupta S, Navaratnarajah CK, Cattaneo R. 2013. Hydrophobic and charged residues in the central segment of the measles virus hemagglutinin stalk mediate transmission of the fusion-triggering signal. J Virol 87:10401–10404. 10.1128/JVI.01547-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brindley MA, Suter R, Schestak I, Kiss G, Wright ER, Plemper RK. 2013. A stabilized headless measles virus attachment protein stalk efficiently triggers membrane fusion. J Virol 87:11693–11703. 10.1128/JVI.01945-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navaratnarajah CK, Kumar S, Generous A, Apte-Sengupta S, Mateo M, Cattaneo R. 2014. The measles virus hemagglutinin stalk: structures and functions of the central fusion activation and membrane-proximal segments. J Virol 88:6158–6167. 10.1128/JVI.02846-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bishop KA, Hickey AC, Khetawat D, Patch JR, Bossart KN, Zhu Z, Wang L-F, Dimitrov DS, Broder CC. 2008. Residues in the stalk domain of the hendra virus G glycoprotein modulate conformational changes associated with receptor binding. J Virol 82:11398–11409. 10.1128/JVI.02654-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bose S, Zokarkar A, Welch BD, Leser GP, Jardetzky TS, Lamb RA. 2012. Fusion activation by a headless parainfluenza virus 5 hemagglutinin- neuraminidase stalk suggests a modular mechanism for triggering. Proc Natl Acad Sci USA 109:E2625–E2634. 10.1073/pnas.1213813109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Q, Stone JA, Bradel-Tretheway B, Dabundo J, Benavides Montano JA, Santos-Montanez J, Biering SB, Nicola AV, Iorio RM, Lu X, Aguilar HC. 2013. Unraveling a three-step spatiotemporal mechanism of triggering of receptor-induced Nipah virus fusion and cell entry. PLoS Pathog 9:e1003770. 10.1371/journal.ppat.1003770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bose S, Song AS, Jardetzky TS, Lamb RA. 2014. Fusion activation through attachment protein stalk domains indicates a conserved core mechanism of paramyxovirus entry into cells. J Virol 88:3925–3941. 10.1128/JVI.03741-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Q, Bradel-Tretheway B, Monreal AI, Saludes JP, Lu X, Nicola AV, Aguilar HC. 2015. Nipah virus attachment glycoprotein stalk C-terminal region links receptor binding to fusion triggering. J Virol 89:1838–1850. 10.1128/JVI.02277-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navaratnarajah CK, Generous AR, Yousaf I, Cattaneo R. 2020. Receptor-mediated cell entry of paramyxoviruses: mechanisms, and consequences for tropism and pathogenesis. J Biol Chem 295:2771–2786. 10.1074/jbc.REV119.009961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. Slam (CDw150) is a cellular receptor for measles virus. Nature 406:893–897. 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 22.Tahara M, Takeda M, Shirogane Y, Hashiguchi T, Ohno S, Yanagi Y. 2008. Measles virus infects both polarized epithelial and immune cells by using distinctive receptor-binding sites on its hemagglutinin. J Virol 82:4630–4637. 10.1128/JVI.02691-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker pvrl4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7:e1002240. 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mühlebach MD, Mateo M, Sinn PL, Prüfer S, Uhlig KM, Leonard VHJ, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB, Cichutek K, Von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533. 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McQuaid S, Cosby SL. 2002. An immunohistochemical study of the distribution of the measles virus receptors, CD46 and SLAM, in normal human tissues and subacute sclerosing panencephalitis. Lab Invest 82:403–409. 10.1038/labinvest.3780434. [DOI] [PubMed] [Google Scholar]
- 26.Reymond N, Fabre S, Lecocq E, Adelaide J, Dubreuil P, Lopez M. 2001. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with nectin1/PRR1 through V domain interaction. J Biol Chem 276:43205–43215. 10.1074/jbc.M103810200. [DOI] [PubMed] [Google Scholar]
- 27.Iwasaki Y, Koprowski H. 1974. Cell to cell transmission of virus in the central nervous system. I. Subacute sclerosing panencephalitis. Lab Invest 31:187–196. [PubMed] [Google Scholar]
- 28.Allen IV, McQuaid S, McMahon J, Kirk JMR. 1996. The significance of measles virus antigen and genome distribution in the CNS in SSPE for mechanisms of viral spread and demyelination. J Neuropathol Exp Neurol 55:471–480. 10.1097/00005072-199604000-00010. [DOI] [PubMed] [Google Scholar]
- 29.Lawrence DM, Patterson CE, Gales TL, D'Orazio JL, Vaughn MM, Rall GF. 2000. Measles virus spread between neurons requires cell contact but not CD46 expression, syncytium formation, or extracellular virus production. J Virol 74:1908–1918. 10.1128/jvi.74.4.1908-1918.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato Y, Watanabe S, Fukuda Y, Hashiguchi T, Yanagi Y, Ohno S. 2018. Cell-to-Cell Measles virus spread between human neurons is dependent on hemagglutinin and hyperfusogenic fusion protein. J Virol 92:e02166-17. 10.1128/JVI.02166-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayata M, Takeuchi K, Takeda M, Ohgimoto S, Kato S, Sharma LB, Tanaka M, Kuwamura M, Ishida H, Ogura H. 2010. The F gene of the Osaka-2 strain of measles virus derived from a case of subacute sclerosing panencephalitis is a major determinant of neurovirulence. J Virol 84:11189–11199. 10.1128/JVI.01075-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shirogane Y, Watanabe S, Yanagi Y. 2012. Cooperation between different RNA virus genomes produces a new phenotype. Nat Commun 3:1235. 10.1038/ncomms2252. [DOI] [PubMed] [Google Scholar]
- 33.Watanabe S, Shirogane Y, Suzuki SO, Ikegame S, Koga R, Yanagi Y. 2013. Mutant fusion proteins with enhanced fusion activity promote measles virus spread in human neuronal cells and brains of suckling hamsters. J Virol 87:2648–2659. 10.1128/JVI.02632-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jurgens EM, Mathieu C, Palermo LM, Hardie D, Horvat B, Moscona A, Porotto M. 2015. Measles fusion machinery is dysregulated in neuropathogenic variants. mBio 6:e02528-14. 10.1128/mBio.02528-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe S, Ohno S, Shirogane Y, Suzuki SO, Koga R, Yanagi Y. 2015. Measles virus mutants possessing the fusion protein with enhanced fusion activity spread effectively in neuronal cells, but not in other cells, without causing strong cytopathology. J Virol 89:2710–2717. 10.1128/JVI.03346-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Angius F, Smuts H, Rybkina K, Stelitano D, Eley B, Wilmshurst J, Ferren M, Lalande A, Mathieu C, Moscona A, Horvat B, Hashiguchi T, Porotto M, Hardie D. 2019. Analysis of a subacute sclerosing panencephalitis genotype B3 virus from the 2009–2010 South African measles epidemic shows that hyperfusogenic F proteins contribute to measles virus infection in the brain. J Virol 93:e01700-18. 10.1128/JVI.01700-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shirogane Y, Hashiguchi T, Yanagi Y. 2020. Weak cis and trans interactions of the hemagglutinin with receptors trigger fusion proteins of neuropathogenic measles virus isolates. J Virol 94:e01727-19. 10.1128/JVI.01727-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shirogane Y, Takemoto R, Suzuki T, Kameda T, Nakashima K, Hashiguchi T, Yanagi Y. 2021. CADM1 and CADM2 triggers neuropathogenic measles virus-mediated membrane fusion by acting in cis. J Virol 95:e0052821. 10.1128/JVI.00528-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biederer T. 2006. Bioinformatic characterization of the SynCAM family of immunoglobulin-like domain-containing adhesion molecules. Genomics 87:139–150. 10.1016/j.ygeno.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 40.Hiruma A, Ikeda S, Terui T, Ozawa M, Hashimoto T, Yasumoto S, Nakayama J, Kubota Y, Iijima M, Sueki H, Matsumoto Y, Kato M, Akasaka E, Ikoma N, Mabuchi T, Tamiya S, Matsuyama T, Ozawa A, Inoko H, Oka A. 2011. A novel splicing variant of CADM2 as a protective transcript of psoriasis. Biochem Biophys Res Commun 412:626–632. 10.1016/j.bbrc.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 41.Kikuchi S, Iwai M, Sakurai-Yageta M, Tsuboi Y, Ito T, Maruyama T, Tsuda H, Kanai Y, Onizuka M, Sato Y, Murakami Y. 2012. Expression of a splicing variant of the CADM1 specific to small cell lung cancer. Cancer Sci 103:1051–1057. 10.1111/j.1349-7006.2012.02277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagara Y, Hagiyama M, Hatano N, Futai E, Suo S, Takaoka Y, Murakami Y, Ito A, Ishiura S. 2012. Tumor suppressor cell adhesion molecule 1 (CADM1) is cleaved by a disintegrin and metalloprotease 10 (ADAM10) and subsequently cleaved by γ-secretase complex. Biochem Biophys Res Commun 417:462–467. 10.1016/j.bbrc.2011.11.140. [DOI] [PubMed] [Google Scholar]
- 43.He W, Li X, Xu S, Ai J, Gong Y, Gregg JL, Guan R, Qiu W, Xin D, Gingrich JR, Guo Y, Chang G. 2013. Aberrant methylation and loss of CADM2 tumor suppressor expression is associated with human renal cell carcinoma tumor progression. Biochem Biophys Res Commun 435:526–532. 10.1016/j.bbrc.2013.04.074. [DOI] [PubMed] [Google Scholar]
- 44.Moiseeva EP, Leyland ML, Bradding P. 2013. CADM1 is expressed as multiple alternatively spliced functional and dysfunctional isoforms in human mast cells. Mol Immunol 53:345–354. 10.1016/j.molimm.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shirakabe K, Omura T, Shibagaki Y, Mihara E, Homma K, Kato Y, Yoshimura A, Murakami Y, Takagi J, Hattori S, Ogawa Y. 2017. Mechanistic insights into ectodomain shedding: susceptibility of CADM1 adhesion molecule is determined by alternative splicing and O-glycosylation. Sci Rep 7:46174. 10.1038/srep46174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pietri T, Easley-Neal C, Wilson C, Washbourne P. 2008. Six cadm/synCAM genes are expressed in the nervous system of developing zebrafish. Dev Dyn 237:233–246. 10.1002/dvdy.21397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kondo N, Miyauchi K, Meng F, Iwamoto A, Matsuda Z. 2010. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J Biol Chem 285:14681–14688. 10.1074/jbc.M109.067090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng Des Sel 25:813–820. 10.1093/protein/gzs051. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Li X, Nakane S, Liu S, Ishikawa H, Iwamoto A, Matsuda Z. 2014. Co-expression of foreign proteins tethered to HIV-1 envelope glycoprotein on the cell surface by introducing an intervening second membrane-spanning domain. PLoS One 9:e96790. 10.1371/journal.pone.0096790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fogel AI, Akins MR, Krupp AJ, Stagi M, Stein V, Biederer T. 2007. SynCAMs organize synapses through heterophilic adhesion. J Neurosci 27:12516–12530. 10.1523/JNEUROSCI.2739-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takeda M, Ohno S, Seki F, Nakatsu Y, Tahara M, Yanagi Y. 2005. Long untranslated regions of the measles virus M and F genes control virus replication and cytopathogenicity. J Virol 79:14346–14354. 10.1128/JVI.79.22.14346-14354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harbury PB, Zhang T, Kim PS, Alber T. 1993. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science 262:1401–1407. 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 53.Tahara M, Ito Y, Brindley MA, Ma X, He J, Xu S, Fukuhara H, Sakai K, Komase K, Rota PA, Plemper RK, Maenaka K, Takeda M. 2013. Functional and structural characterization of neutralizing epitopes of measles virus hemagglutinin protein. J Virol 87:666–675. 10.1128/JVI.02033-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takeda M, Tahara M, Hashiguchi T, Sato TA, Jinnouchi F, Ueki S, Ohno S, Yanagi Y. 2007. A Human Lung Carcinoma Cell Line Supports Efficient Measles Virus Growth and Syncytium Formation via a SLAM- and CD46-Independent Mechanism. J Virol 81:12091–12096. 10.1128/JVI.01264-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gomyo H, Arai Y, Tanigami A, Murakami Y, Hattori M, Hosoda F, Arai K, Aikawa Y, Tsuda H, Hirohashi S, Asakawa S, Shimizu N, Soeda E, Sakaki Y, Ohki M. 1999. A 2-MB sequence-ready contig map and a novel immunoglobulin superfamily gene IGSF4 in the LOH region of chromosome 11q23.2. Genomics 62:139–146. 10.1006/geno.1999.6001. [DOI] [PubMed] [Google Scholar]
- 56.Zhiling Y, Fujita E, Tanabe Y, Yamagata T, Momoi T, Momoi MY. 2008. Mutations in the gene encoding CADM1 are associated with autism spectrum disorder. Biochem Biophys Res Commun 377:926–929. 10.1016/j.bbrc.2008.10.107. [DOI] [PubMed] [Google Scholar]
- 57.Hasstedt SJ, Bezemer ID, Callas PW, Vossen CY, Trotman W, Hebbel RP, Demers C, Rosendaal FR, Bovill EG. 2009. Cell adhesion molecule 1: a novel risk factor for venous thrombosis. Blood 114:3084–3091. 10.1182/blood-2009-05-219485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muñoz-Alía MÁ, Muller CP, Russell SJ. 2018. Hemagglutinin-specific neutralization of subacute sclerosing panencephalitis viruses. PLoS One 13:e0192245. 10.1371/journal.pone.0192245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 60.Tahara M, Takeda M, Yanagi Y. 2007. Altered interaction of the matrix protein with the cytoplasmic tail of hemagglutinin modulates measles virus growth by affecting virus assembly and cell-cell fusion. J Virol 81:6827–6836. 10.1128/JVI.00248-07. [DOI] [PMC free article] [PubMed] [Google Scholar]