

Abstract
Nonionic surfactant polysorbates, including PS-80 and PS-20, are commonly used in the formulation of biotherapeutic products for both preventing surface adsorption and acting as stabilizer against protein aggregation. Trace levels of residual host cell proteins (HCPs) with lipase or esterase enzymatic activity have been shown to degrade polysorbates in biologics formulation. The measurement and control of these low abundance, high-risk HCPs for polysorbate degradation are an industry-wide challenge to achieve desired shelf life of biopharmaceuticals in liquid formulation, especially for high-concentration formulation product development. Here, we reviewed the challenges, recent advances, and future opportunities of analytical method development, risk assessment, and control strategies for polysorbate degradation during formulation development with a focus on enzymatic degradation. Continued efforts to advance our understanding of polysorbate degradation in biologics formulation will help develop high-quality medicines for patients.
Keywords: analytical toolbox, control strategy, lipase or esterase, polysorbate degradation, risk assessment
Statement of Significance
Polysorbate degradation has been one of the biggest challenges for biotherapeutics formulation development. The challenges, recent advances, and future opportunities of analytical method development, risk assessment, and control strategies of high-risk host cell proteins for polysorbate degradation were discussed in this review.
INTRODUCTION
Polysorbate-80 (PS-80) and polysorbate-20 (PS-20) are common excipients used as shear protectants to stabilize proteins, prevent agitation-induced aggregation, and minimize surface adsorption of proteins [1–3]. PSs are composed of a hydrophilic head group of ethoxylated sorbitan or isosorbide and a lipophilic fatty acid (FA) tail connected via an ester bond (Figure 1). The degradation of PS could result in turbidity challenges due to the formation of visible or subvisible particles from FAs release and/or protein aggregation, and hence have a potential direct impact on product quality [1–3]. In addition, the concentrations of PS used in liquid formulations of protein drug products (DPs) are typically greater than the critical micelle concentration and have been reported in the range from 0.001% to 0.1% (w/v). Some degradation can occur without loss of excipient functionality, but the extent of PS degradation that can be accepted without impacting product quality can vary from product to product, which makes it challenging to set a universal specification limit. Hence, multiple potential outcomes of PS degradation can occur, leading to critical quality attributes out of specification/off limit, which ultimately lead to reduce the shelf life of a biological product or change its storage conditions.
Figure 1.
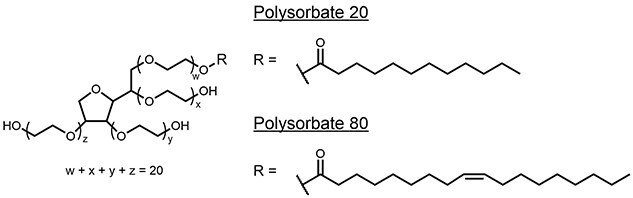
Structure of PS 20 and 80.
There are multiple routes for PS degradation, which can be grouped into primarily two categories: chemical degradation and enzymatic hydrolysis (Figure 2) [1–3]. Chemical degradation can occur via either hydrolysis route [cleavage of the ester bond releasing free FA (FFA)] with acid, base, or metal catalyst or oxidation and cleavage of other bonds in the PS. The enzymatic pathway is primarily ester bond cleavage by certain high-risk host cell proteins (HCPs), referred as polysorbate-degradation enzymes (PSDEs). PSDEs include lipases, esterases, and other process-related impurities which may not have known lipase or esterase activities. HCPs can copurify with therapeutic proteins via specific or nonspecific interaction [4–6]. High-risk HCPs are considered as those problematic HCPs that are immunogenic, biologically active, or enzymatically active with the potential to degrade either product molecules or excipients (such as PS) used in formulation [6]. The key to development of an appropriate control strategy of PS degradation is the ability to discern the mechanisms involved which can be inferred and ultimately confirmed with appropriate analytical tools (Figure 2). Enzymatic hydrolysis is often considered as the major root cause for PS degradation in biotherapeutics. However, it is generally recommended to consider several observations when specifying the predominant degradation pathway, such as the temperature-dependent PS degradation in formulated biologics compared with placebo controls, protein-concentration-dependent degradation, and the reduction of degradation rate by hydrolase inhibitors. The impact of PS degradation via oxidation pathways may be protein specific or buffer dependent, which have been extensively studied or reviewed in the literature [1–3, 7–8]. This review focuses on enzymatic degradation of PSs by PSDEs.
Figure 2.

Overall analytical and control strategy for PS degradation during biologics formulation development.
A typical target product profile for early clinical supplies is to achieve a 3-year DP shelf life (2–8 °C). The product stability is ideally demonstrated with research batches prior to initiation of toxicology (Tox) production. In certain cases, no process development occurs between Tox batch and First-In-Human (FIH) supplies production (owing to timeline constraints). The Tox process is effectively the FIH process, which can confirm the acceptable process performance and drug substance (DS) stability during scale up (Figure 2). Achieving this success metric will require investment of analytical and process development (both DS and DP) resources prior to Tox batch production.
ANALYTICAL TOOLBOX
To support the development of robust process control of PS degradation in biologics formulation, there needs to be a significant investment in analytical method development to accurately and robustly assess the challenges associated with residual PSDEs. Through these capable methods, it is essential to evaluate the type and amount of PSDEs for a molecule early in development so that appropriate process controls can be implemented to mitigate the problem. Multiple tools have been developed as part of an overall HCP control strategy for PS degradation. Orthogonal assays used to support the control strategy development are summarized in Table 1, which include PS content assay, PS purity method (can be applied to discern chemical from enzymatic degradation), free fatty acid (FFA) assay, total HCP and specific HCP enzyme-linked immunosorbent assay (ELISA) assay, Liquid Chromatography with tandem mass spectrometry (LC–MS/MS) [proteomics and multiple reaction monitoring (MRM) for identification and quantification of a specific PSDEs] [9], and enzyme activity assay.
Table 1.
Analytical toolbox for the measurements of HCPs for PS degradation
| Tool | Assay type | Pros | Cons |
|---|---|---|---|
| PS content | Absolute quantification of content, such as mixed-mode HPLC/CAD method | Simple, suitable for stability and release | Low sensitivity, not suitable for degradation pathway elucidation |
| PS purity | Mechanistic, such as RP-HPLC/CAD or MS | Good for degradation pathway elucidation | Sensitivity, semi-quantitative |
| FAA assay | Mechanistic, such as HPLC-MS | Absolute quantitation | Only single attribute analysis of the degraded products |
| ELISA (individual HCPs) | Absolute quantification of individual HCPs | Absolute quantitation, high throughput, suitable for stability and release | Time for assay development |
| abundance-based proteomics (TABP) | Global view of the entire HCP population | Individual HCPs and their relative abundances | Relatively high assay variability |
| ABPP | Active HCPs of certain enzyme classes based on chemical probes | Activity readout, high sensitivity for low-abundance PSDEs | Low throughput, single enzyme class |
| Targeted proteomics (individual HCPs) | Targeted MS quantification based on MRM or PRM | Absolute quantitation, multiplex, throughput | Time for assay development |
| Activity (PS-80 incubation) | Broad assay to determine lipase activity, semi-quantitative | Quicker turnaround compared with real-time stability studies | Low throughput |
| Activity (surrogate substrates) | Surrogate assay for activity | Higher throughput, automation | Sensitivity, specificity when compared with use PS |
PS content assays
The PS content method often uses high-performance liquid chromatography (HPLC) with a mixed-mode column in combination with an evaporative light scattering detector (ELSD) or a Corona charged aerosol detector (CAD) [10–12]. The Corona CAD is a mass sensitive detector that responds to essentially all nonvolatile and some semi-volatile compounds in the sample which elute from the column. For quantification purposes with a specific product, PS standard solutions are prepared at different concentration levels, e.g., from 0.05 mg/mL to 0.35 mg/mL. The range can cover 25% to 175% of the target PS concentration (e.g., 0.20 mg/mL). Samples are analyzed without any dilution. The content assay is typically qualified for both DS and DP matrices but the assay can also be applied to process intermediates [10]. However, the process intermediate matrix should first be evaluated to rule out any specific interferences from the buffer matrices and/or protein content in the sample. Due to its simplicity of the method and data analysis, the concentration of PS from the content assay in the final DS and DP is typically measured as part of release and/or stability testing panel. Because multiple PS species are eluted in a single peak from the PS content assay, this assay is not suitable to inform PS degradation pathway. Fluorescent micelle assay is another PS content assay, which has the potential for automation and high-throughput application [13]. However, it cannot be used for degradation investigation, because certain degraded PS species could still form micelles as interference factors, which results in over estimation of PS content by the fluorescent micelle assay [14].
PS purity assay
Compared with the PS content assays, the PS purity assay provides a fingerprint of PS degradation products. The enzymatic degradation pathway produces ester bond hydrolysis products and FAs. It often shows decreases in monoester peak and increase of peaks associated with the PS head group [10, 15]. The chemical degradation pathways produce the oxidation products, cleavage of both the polyoxethylene groups and esters of PS by autohydrolysis. The degraded PS products can then be directly detected/quantified by nuclear magnetic resonance (NMR) [16] or Matrix-Assisted Laser Desorption/Ionization - Mass Spectrometry (MALDI-MS). [17] If additional separation is needed, those degraded products can first be resolved on a reverse phase HPLC or two-dimensional liquid chromatography (2D-LC), and then detected with a universal mass detector such as CAD, ELSD, or MS. [11] The detected patterns, or monitoring individual degradation species, such as FFA, indicate the potential PS degradation pathway [18].
A novel analytical method for PS-80 characterization was recently developed using UPLC coupled with charge reduction high resolution MS. [19] Post column coinfusion of triethylamine was used to focus the signal into mainly singly charged molecular ions and reduce the extent of in-source fragmentation, resulting in a simpler ion map and enhanced measurement of PS-80 species with software-assisted composition analysis. With the help of a complete library of possible chemical formulas from PS-80, the method enabled quantification of many components in PS-80 with unprecedented detail and is a useful tool to study formulation and stability of pharmaceutical preparations [18].
FFA assay
PS content and purity assays described above detect the overall degradation of PS through both the oxidative and hydrolytic pathways, which often do not provide sensitive and reliable quantitative detection of low-level PS degradants. In recent years, the measurement of FFA production with MS provides a relatively simple, sensitive, and high-throughput approach for early readout of PS degradation in biologics formulation. Using the FFA assay, Cheng et al. accurately detected lauric acid produced from the degradation of <1% of PS20 in a 0.2 mg/mL formulation [20]. By further optimization of sample preparation and LC–MS, Zhang et al. enabled low detection limit of lauric acid (22 ng/mL) and oleic acid (211 ng/mL) for detecting the reduction of 0.000024% of spike-in PS-20 or 0.00016% of PS-80 [21]. This study also found glass vials were a better container for sample incubation, as these containers can also minimize FA adsorption compared with plastic containers. The FFA loss during sample preparation may be an interference factor for this assay. Using accelerated thermal stability testing as short as a few hours or 1 day, this FFA method identifies processes that exhibit fast PS degradation and consequently allows faster iterative optimization for process development.
ELISA assay for total HCP and individual HCP
Total HCP assay via traditional ELISA has been considered as the “gold standard” for HCP measurement since it has been employed for several decades [5]. ELISA provides one summed value for total HCP content and the ELISA release assay for HCP testing is relatively simple with high throughput producing a semi-quantitative value when applied to a range of HCPs often present and detectable by the antibody reagents used in the assay. The assay, however, has several limitations, such as coverage, sample dilution nonlinearity due to antigen excess, and the significant time required to develop the critical reagents for a given projects or platform (around 2 years) [5]. In some cases, specific antibodies were developed to measure and quantify known high-risk HCPs, such as phospholipase B-like 2 (PLBL2) presence in a sample [22, 23]. This targeted individual ELISA assay for high-risk PSDEs can be used to support high-throughput process development or characterization if certain PSDEs have been confirmed as the root cause for PS degradation.
LC–MS/MS-based traditional abundance-based proteomics
Some HCPs, including PSDEs, may not be detected or quantified by standard ELISA assays against total HCP due to the lack of antibody coverage [5]. Therefore, orthogonal tools to identify and quantify specific PSDEs or other high-risk HCPs are needed to support process development. The identification of individual HCPs is critical for Quality by Design-based process development (supports development of a control strategy) and risk assessment. Traditional abundance-based proteomics (TABP) has been demonstrated as a valuable tool to identify and track the clearance of PSDEs and other HCPs throughout a purification process, which supports process development efforts (both upstream and downstream) on key HCPs that can impact product quality and stability [5, 24].
Peptide-centric bottom-up TABP workflow starts with proteolytic digestion by recombinant enzymes, usually by trypsin, which produces peptides with C-terminally protonated amino acids [25]. The digested peptides are separated by one or more steps of HPLC and eluted into a mass spectrometer. The mass spectrum of peptides eluting at a specific time point is recorded as an MS1 spectrum. The computer algorithm associated with the MS system generates a prioritized list of these peptides or mass ranges for MS/MS fragmentation by collision gas depending on the MS scanning mode, such as data-dependent acquisition or data-independent acquisition (DIA). The MS and MS/MS spectra are then searched against protein sequence databases with theoretically digested proteomes from the host cell line using certain software, such as Mascot or Sequest [25]. The outcome of the experiment is the identity of the peptides (with certain probability) that make up the identified protein. This is a critically important step as the number of unique peptides is essential to the correct identification of the protein since the same peptides can be present in a range of proteins.
One of the unique challenges for HCP identification by TABP is the dynamic range issue of low-abundance HCPs compared with a DS. The dynamic range is a gauge of the signal available for detecting peptides or proteins. High dynamic range translates to detection of less abundant peptides in a milieu of more abundant ones. Most commercial MS instruments have a dynamic range of about 4 to 5 orders of magnitude; however, high-risk HCPs, such as PSDEs, could have a direct impact on PS degradation at ppb levels [26, 27]. This challenge is amplified in high concentration formulations, where the ppm level of HCP may not change, but the absolute concentrations of HCPs (ng/mL) significantly increase [28]. Despite their high biological activity, the concentrations of most PSDEs are so low that they are practically undetectable by MS without fractionation and enrichment. Thankfully, the field of HCP identification by proteomics benefits significantly from the accumulated knowledge on plasma or serum proteomics analysis, where the dynamic range of proteins is known to span more than 10 orders of magnitude [29].
Several sample preparation methods to reduce high-abundance proteins or enrich low-abundance proteins have been used in HCP proteomics analysis to expand the dynamic range and increase the identification sensitivity of the TABP approach, such as native digestion [30], ProteoMiner [31], DS depletion with Protein A [32], anti-HCP affinity chromatography [27, 33], molecular weight cutoff filtration [34], and hydrophilic interaction chromatography enrichment [35]. In addition to advancements in sample preparation, improvements in LC–MS analysis have also been made to improve sample dynamic range and improve the sensitivity of HCP identification. For LC improvements, longer chromatographic gradients or 2D-LC have been used [36, 37]. There are also improvements on the mass spectrometry side. Additional gas phase separation with ion mobility, such as high-field asymmetric waveform ion mobility spectrometry[32], and different mass spectrometry acquisition techniques, such as BoxCar and DIA [37, 38], have been employed to improve both HCP identification and quantification. Several PSDEs, including LPLA2 (PLA2G15, phospholipase A2, group XV), LPL (lipoprotein lipase), CES (liver carboxylesterase), LIPA (lysosomal acid lipase), PPT1 (palmitoyl-protein thioesterase 1), and SIAE (sialate o-acetylesterase) have been identified by TABP and have shown to have a direct impact on PS-80 or PS-20 degradation in biotherapeutics [15, 26, 27, 33, 39]. Drawbacks of these extensive sample preparation and LC–MS analytical approaches include reduced testing throughput and potential HCP loss, and hence impaired assay robustness. The generation of HCP databases using previous proteomics data will help establish the levels of individual HCPs (depending on the limit of quantification of the proteomics method, e.g., >10 ppm) with respect to changes in different process parameters to potentially guide future process development and risk assessment.
Based on the principle of the bottom-up TABP approach, there are several potential fundamental caveats for identification and quantification of HCPs by TABP, especially for low-abundance HCPs (below sub-ppm). First, every peptide is identified with certain confidence (e.g., >95%) from the proteomics approach. To assign the relevant confidence for each peptide, the search algorithm needs identification from a decoy database such as the reverse proteome database from the same host. In most process intermediates after the last polishing column from downstream purification, only a few HCPs (e.g., <5) are identified by proteomics. There may be challenges to assign the right false discovery rate for peptide identification due to limited identification from the decoy database. Most HCP proteomics studies use two or more unique peptides to increase the protein-level identification confidence. However, in some cases, low-abundance PSDEs may only be identified by one unique peptide. In these cases, it is important to manually check those potentially high-risk HCPs for identification and also verify their presents in less purified samples. Second, the bottom-up proteomics approach only directly identifies peptides but not proteins. Without majority of sequence coverage, it is impossible to know whether the peptide is part of a cleaved fragment or an intact protein while the database search may report it as the intact protein. This is particularly troublesome for enzymatic protein identification since the latent form and active form can share largely the same sequence with only the differences on propeptide presence. The assignment and confirmation of the correct protein sequence are critical for determination of HCP function and risk assessment beyond enzymatic activity. For example, most immunogenicity prediction algorithms are based on protein sequences which translate to specific motifs or epitopes. Because of this, incorrect protein sequences may yield totally different immunogenicity score. Third, the annotation of protein databases used for proteomics search (such as Uniprot, CHO-K1, or NCBI) is not complete and accurate. For example, the same protein name may have different protein sequences or the same protein sequence may have different protein descriptions. In summary, identifying an HCP by LC–MS-based proteomics is only the starting point and requires a holistic approach to fully understand both function and potential risks for the product and patients.
Activity-based protein profiling
The TABP approach mentioned above relies on measuring abundance and extensive work on sample preparation and LC–MS have been done to increase the abundance of high-risk HCPs, and therefore their probability to be identified by MS. This approach may fail to identify the true culprits in most cases due to the extremely low abundance of certain highly active PSDEs responsible for PS degradation. An approach which can enrich HCPs of low abundance while providing potential enzyme activity insight would therefore be very useful. Activity-based proteomics or activity-based protein profiling (ABPP) is a functional proteomic technology that uses chemical probes that react with mechanistically related classes of enzymes and subsequently identified them by LC–MS/MS. The basic unit of ABPP is a probe, which typically consists of a reactive group, a linker, and an enrichment tag, and the probe is used to capture targeted active enzymes. The reactive group labels the active site of mechanistically related enzymes to form a covalent linkage [40]. The covalently modified proteins can then be detected or purified by the enrichment tag (e.g., biotin-avidin or alkyne-azide reaction) and characterized by gel-based approaches or MS. ABPP has been adapted for activity detection of more than a dozen enzyme classes, such as serine hydrolases (SHs) [40], cysteine proteases [41], kinases, phosphatases, glycosidases, and oxidoreductases [40, 42]. One of the biggest advantages of the ABPP approach is that previously uncharacterized enzymes or proteins with known or unknown functions can be assigned to a new functional class by the ABPP approach.
The SH superfamily includes lipases, esterases, thioesterases, amidases, and peptidases [40]. The defining characteristic of SH superfamily members is the presence of a nucleophilic serine in their active site. Although not every lipase/esterase is from the SH superfamily, chemical probes against SHs have been demonstrated to block PS degradation in a time- and dose-dependent manner [10], which was also confirmed by other studies using similar chemical probes or SH inhibitors [15, 43]. Our previous study also indicated that chemical probes against SHs with different mechanisms of action may provide synergistic or complementary information for inhibition or enrichment of various classes of lipases/esterases [10]. All PSDEs that have been reported to impact PS degradation have been identified by our ABPP approach [10], such as LPLA2 [26], LPL [39], CES [15], LIPA [27], PPT1 [27], and SIAE [33]. In addition, PLA2G7 was identified and confirmed to directly contribute to PS degradation for the first time in our previous publication [10]. In contrast, PLBL2 was found to most likely not participate as an active lipase, esterase, amidase, or peptidase in the SH superfamily as it is not enriched and identified by ABPP [10]. This finding is consistent with several studies supporting that PLBL2 is not responsible for PS degradation at concentrations detected in biotherapeutics [23, 44–46], although the amount of PLBL2 in biologics still needs to be controlled because of its high immunogenicity risk [23].
The ABPP workflow for identifying active enzymes for PS degradation has a few technical challenges and limitations. First, certain lipases/esterases are not part of SH family [40]. For example, there are six groups of PLA2 enzymes, out of which four groups are considered as SHs, and the other two subtypes of PLA2 utilize His as the active site [47]. Interestingly, various chemical probes or inhibitors against SHs have been shown to almost completely block PS degradation [10, 15, 43], which indicates those lipases/esterases, which are not in the SH family, may not be expressed by CHO, or not secreted as HCPs. Second, not all SHs identified by ABPP have activities on PS degradation. There is a gap between active SHs and activities on degradation of a specific PS. One great examples is the active SHs SIAE, identified by our ABPP analysis as well [10], was found to specifically degrade PS-20 but not PS-80 [33]. Third, the sample preparation of ABPP workflow is generally laborious and not easy for high-throughput testing and automation. The method should be optimized for active PSDE identification from different biologics intermediates to increase its robustness.
LC–MS-based targeted HCP assays
Discovery proteomics approaches are mainly focused on driving the sensitivity limit to identify as many HCPs as possible for process understanding and risk assessment. There is a great interest to obtain relative or even absolute protein abundances estimation in reliable ways from the LC–MS-based proteomics approach [48–49]. With the implementation of accurately quantified reference proteins as a single calibrator or calibration curves from various concentrations of spike-in proteins, the total and individual HCPs levels can be estimated from the proteomics dataset [30, 49]. When analyzing a mixture of many HCPs, this quantification can give a good estimate for the total HCP amount in general considering MS response factor, and protein digest variation and recovery between spike-in proteins and total HCP populations can be averaged. This approach, however, may suffer the quantification accuracy for each individual HCP because an individual HCP can have no or much fewer tryptic cleavage sites especially when a native digestion is used. Beyond the accuracy limitation on HCP quantification, the proteomics workflow also has limitations on assay throughput and variability on identification and quantification of low-abundance HCPs. To support process characterization and qualification, targeted MS approaches, such as MRM or parallel reaction monitoring (PRM)-based MS methods are often employed [50, 51]. Targeted MS assays provide more accurate measurements of certain high-risk HCPs while maintaining relatively high analytical throughput [22]. Advantageously, targeted MS approaches do not require critical antibody reagent development like ELISA assays against individual HCPs, although an isotopically labeled HCP protein standard as a critical reagent will further expand the quantification accuracy compared with peptide standards. Ideally, the highly purified protein standard should be produced from the same host bearing the same types of post-translational modifications. Using PRM technology, endogenous LPLA2 was determined at ~0.3–0.6 ppm in three antibodies with PS degradation issues [26]. Following the finding from untargeted proteomics approach, we developed an 8-min LC-MRM method to simultaneously monitor three lipases (PLBL2, LPLA2, and LPL). This method was qualified (1 to 500 ng/mg protein) to support later phase process characterization [22]. Similarly, Chen et al. utilized a targeted LC–MS/MS with high-resolution multiple-reaction-monitoring quantitation method for three lipases PLBL2, LPL, and LIPA for the characterization of process intermediates [52]. This method demonstrated good sensitivity with lower limit of quantitation (LLOQ) around 1 ng/mL (translated to 0.8 to 2.2 ppm for the three lipases in a product matrix), and linear dynamic range of 3 orders of magnitude for the three lipase HCPs [52]. The LLOQ of those current targeted HCP quantification approaches is still relatively high compared with the low concentration for these active PSDE species to remain active. Reducing the LLOQ by 2–3 orders of magnitude via creative enrichment or improved innovative detection technology would be extremely beneficial to drive robust process development.
PSDE activity assay with accelerated PS stability
Considering the extended duration of measuring PS stability across the shelf life of biologics in real time or the equivalent of potentially several months during accelerated stability studies, a rapid readout of PS stability is desired for quick turnaround of process development and optimization. To accomplish this, an accelerated PS stability assessment (e.g., 7 days at 37 °C) is a key assay for predicting PS stability or total PSDE activity [10]. This approach assesses the potential levels of PSDE activity via a PS spike of process intermediates before final formulation combined with a readout from a PS content assay. Evaluation of stability trends across process intermediates is critical to defining the extent of degradation for risk assessments. Although the accelerated PS stability assay is capable of providing such information in a shorter timeframe, one major challenge is in defining the acceptable threshold of PS degradation that can then be translated to the out of specification of certain product quality attributes, such as subvisible or visible particle formation. Although the threshold may be specific to each program in terms of the extent of PS degradation that is acceptable for product quality, a general low threshold (e.g., 20% degradation compared to placebo control) should be set. The threshold limit also needs to take into consideration both oxidation-induced PS degradation and the method variability of the PS content assay (e.g., 10%).
PSDE activity assay with surrogate substrates
Because of the limitations of the accelerated PS stability assay using PS as the substrate for PSDEs owing to the heterogeneity of the substrate and the resulting reaction products and the associated challenges with quantification, a surrogate substrate-based enzyme activity assay for PSDEs is of high interest. The enzyme activity assay intends to provide a quick and sensitive alternative for the PS stability assay to track enzyme activity removal during process development, to build understanding regarding the correlation between residual PSDEs amounts and activity, and the impact on activity by sample matrix, pH, heat stress, etc. The selection of a suitable surrogate substrate is extremely important for this lipase activity assay to increase assay sensitivity and coverage, and several surrogate substrates have been tested as previously reported in the literature [45, 53]. For example, Jahn et al. developed a lipolytic activity assay based on the hydrolysis of the lipase substrate 4-methylumbelliferyl oleate to yield the fluorescent product 4-methylumbelliferone [53]. Bhargava et al. reported a fluorescence plate-based assay for quantifying esterase activity, utilizing 4-methylumbelliferyl caprylate (MU-C8) as the esterase substrate [45]. Several assay parameters, such as substrate, inhibitor and sample matrix (e.g., buffer salts and pH solvent), were evaluated for both enzyme activity assays using model enzymes or process intermediate samples in each of these examples [45, 53].
To compare different substrates, Glücklich et al. evaluated three different surrogate lipases with distinctively different degradation kinetics on various ester based substrates and high-purity PS-80 and PS-20 [54]. The evaluated ester based substrates included p-nitrophenyl butyrate and three 4-methylumbelliferon-based esters [capric acid (C10) in 4-methylumbelliferyl decanoate (MUD4), lauric acid (C12; main FA of PS-20) in 4-methylumbelliferyl laurate (MUL4), or oleic acid (C18:1; main FA of PS-80) in 4-methylumbelliferyl oleate (MUO4)]. The results indicated that the determined lipase activities varied not only from assay to assay, but also significantly for the lipase tested, thus showing a different degradation fingerprint in the RP-HPLC-CAD chromatogram [54]. This study highlighted that a comprehensive monitoring approach is essential to assess potential PSDEs contaminations.
Once demonstrated to be robust and operated in a high-throughput fashion, this enzymatic assay with surrogate substrates will significantly advance the analysis and control of PS degradation in process development. However, the sensitivities and specificities of these assays still need further evaluation to drive a meaningful outcome of PS degradation in biologics formulation, especially when compared with the FFA assay.
ANALYTICAL TESTING STRATEGY AND RISK ASSESSMENT
Risk assessment for potential PS degradation in biologics formulation will typically occur once the top candidate has been identified, or prior to process development when starting with a new cell line or clonal pool with increased titer or decreased cell viability where increased PSDEs secretion is possible. As shown in Figure 3, PSDEs enzymatic activity assays using PS or surrogate substrates should be applied for the 1st or 2nd polishing step intermediate to test PS degradation as a first step screening tool. Note that in the event of the presence of an active PSDE, the expectation is that PS degradation would be observed at both 25 °C and 37 °C, although the specific rate of PS degradation is anticipated to vary as a function of temperature. If an alarming level of PS degradation is observed, either TABP or ABPP or both can be employed to carefully investigate the presence of PSDEs. Once the problematic PSDEs are defined, a targeted quantitative and sensitive MS assay such as MRM or targeted ELISA assay (if available) can be used to drive process development to reduce and mitigate PS degradation (Figure 3) and to support an appropriate control strategy (including risk assessment for PS degradation and immunogenicity). If the early assessment does not show significant PS degradation, the stability of the pilot scale lot should also be confirmed as PSDE expression levels may change as a function of scale. An assessment of relative PSDE activity via the PS stability assay can also occur following Protein A purification to discriminate amongst clones/upstream processes for PSDE expression level assessment when necessary. Recommendations for additional investments in DP formulation screening studies include addition of PS content to pre-probe stability (4–8 weeks) and evaluation of high concentration (> 100 mg/mL) liquid formulation data available prior to process lock.
Figure 3.

Analytical testing strategy, risk assessment, and control strategy of PS degradation.
There are a few challenges for the overall analytical strategy of HCPs for PS degradation. First, it is challenging to set an acceptance limit for each assay, such as the limit for PS degradation in the enzymatic activity assay using PS as the substrates as discussed above. There are a wide range of PSDEs, which can have different impacts on PS stability at trace levels (e.g., a few ppm) and each PSDE may have different substrate selectivity against the PS mixture [54]. Hence, the risk assessment on the limit of each PSDE should be different, as well as the considerations that multiple active PSDEs may exist in a final product at the same time. As one example, a refined model suggested that DPs containing trace levels of HCPs that preferentially degrade higher order esters of PS-20 are at a higher risk of particle formation [55]. Therefore, determination of the PSDE limit from each assay needs to take into account the assay performance, platform knowledge, and the performance of the real-time stability data, such as PS degradation and subvisible particle formation from previous programs.
Second, each assay in the analytical toolbox for PS degradation has its unique advantages and disadvantages. In most cases, the data from each assay are highly correlated and show a similar trend. For example, the comparison of PLBL2-specific ELISA and MRM approaches for quantification of PLBL2 has been performed for a subset of process intermediates for several biologics programs, showing very comparable results [22]. If discrepancy was observed when comparing results from different assays, an investigation should be initiated to understand the cause of such difference, which includes but not limited to check assay coverage, sensitivity, specificity, and precision.
Third, even with the advancement of sample preparation and improved sensitivity of LC–MS-based proteomics, it may not always be possible to identify the true culprit for PS degradation in each program. To narrow in on potential root causes, the PS purity assay may inform which classes of enzymes are involved in the degradation based on the degradation patterns [56]. In this case, it may be helpful to leverage certain platform-based control strategies to mitigate the risk even when the root cause is not confirmed. Establishment and utilization of an activity assay under these circumstances helps obviate the risk to PS degradation.
CONTROL STRATEGY
Both chemical and enzymatic mechanisms can lead to formation of particulates owing to limited solubility of the PS degradants. Therefore, a control strategy to address both causes needs to be implemented accordingly, including specific PS handling procedures. The process toolbox deployed to control PS degradation spans the entire process development, including cell line development, upstream, downstream processing, and formulation strategies (Figure 4). In some cases, integrated control strategies may be required to achieve an acceptable level of PS stability.
Figure 4.

Process control strategies of HCPs for PS degradation.
Cell line and upstream processing
The expression and activity of PSDEs in harvested cell culture fluid (HCCF) are a function of cell line and upstream processing conditions. For the proof-of-concept study to determine the role of one critical lipase in PS stability, Chiu et al. generated LPL knockout CHO cells using CRISPR and TALEN technologies [39]. HCCF from this knockout cell line demonstrated significantly reduced PS degradation without significant impact on cell viability when compared with wild-type samples. PS-80 can be utilized to inactivate enveloped viruses as part of an overall adventitious agent control strategy. In an attempt to identify the components leading to oleic acid production from PS-80 for virus inactivation, Chen et al. evaluated CHO cell lines that had one to four lipase/esterase genes knocked out through genetic disruption [57]. It was observed that PS-80 was still able to inactivate virus, albeit to somewhat reduced efficiency, in the cell-free culture harvests from these genetically engineered CHO cell lines, including the quadruplet knockout CHO. These results indicate that many CHO-encoded enzymes (likely greater than four) are capable of hydrolyzing PS-80 [57]. In addition, Kol et al. created a “clean” CHO cell by disrupting up to 14 genes to eliminate HCPs, including LPL. These knockout CHO cell lines exhibit a substantial reduction in total HCP content (40%–70%), higher productivity, and improved growth characteristics in specific clones [58]. Interestingly, a complete deletion of a certain high-risk HCP may not be required when a cell line engineering approach was considered. For example, Dovgan et al. showed that in those engineered cell lines, even modest downregulation (≤50%) of the difficult-to-remove HCP cathepsin D is sufficient to greatly reduce levels of copurifying HCP [59]. This reduction led to improved product quality by reducing fragmentation of the DP in forced degradation studies to negligible levels, which significantly relieves the burden on downstream purification. Those studies demonstrated great potential of knocking out certain difficult-to-remove PSDEs for controlling PS degradation.
Not only depending on cell lines, PSDE expression can be viewed as a metabolic engineering challenge that may be impacted by clone, media, feeding strategy, and scale up. The type and extent of process development may be product-specific, owing to diversity of PSDEs, expression levels, and activity. Additional upstream process development (e.g., media optimization, cell culture density, harvest timing, and evaluation of backup clones) can also be pursued in parallel with PSDE investigations in the event that the stability criteria is not achieved.
Downstream processing
In addition to cell line and upstream processing efforts, high-throughput screening of chromatography conditions (resins and buffer systems) in downstream processing can be an effective tool to support process development. Clearance of an HCP may require dissociation of that HCP from the mAb followed by partitioning (e.g., chromatography, charged depth filtration). If the PSDE is associated with the mAb (or therapeutic protein in general) then process development may need to focus on the differential partitioning of the HCP/mAb complex versus the mAb on the chromatography media. There are multiple potential forms of associations between PSDE and biologics (e.g., charge, hydrophobicity, mixed mode) potentially leading to a diverse set of complexes that require removal in downstream processing. Given the diversity of PSDE and biologics in development, the approach to removal of these species in a downstream process could be molecule specific. For example, Tran et al. demonstrated that different therapeutic antibodies interact to varying degrees with host cell proteins in general, and PLBL2 specifically [60]. A high-throughput Protein A chromatography method was further used to examine the interaction between several antibodies and PLBL2. The results showed that the coelution of PLBL2 during Protein A chromatography is highly dependent on the individual antibody in addition to PLBL2 concentration in the chromatographic load [60]. Process parameters such as antibody resin load density and pre-elution wash conditions also influence the levels of PLBL2 in the Protein A eluate. Interestingly, PLBL2 was found to preferably interact with IgG4 subclass antibodies compared with IgG1 antibodies [60], although the sample set was small and there was limited diversity in the mAb evaluated.
Although multiple choices in the downstream toolbox, such as optimizing certain purification steps or adding an additional polish column, are available to remove certain PSDEs, the simple use of a heating approach was shown to inactivate a high-risk HCP cathepsin D. Lim et al. implemented a controlled temperature inactivation unit operation that enabled essentially complete inactivation of cathepsin D, resulting in the production of a stable DP that had not been possible using column chromatography alone [61]. The key was to provide heat to inactivate those high-risk HCPs without damaging the DS. The temperature 55 °C was selected based on CD thermal denaturation data that indicated the protease began to thermally unfold above 45 °C, with a Tm of 53 °C. In contrast, the Tm for the DS is 65 °C. This approach for PSDE deactivation during the downstream purification will be interesting to explore for PS control.
The targeted downstream development to remove PSDEs for each molecule relies on a high-throughput PSDE activity assay or targeted assays (e.g., individual ELISA or MRM) to quantify the known PSDE from each individual molecule as mentioned above. Significant efforts will be needed to enrich and identify the high-risk PSDEs by proteomics for downstream optimization. PLBL2, a putative lipase, is often detected at higher abundance compared with other PSDEs. However, accumulated evidence suggests that PLBL2 is most likely not responsible for PS degradation [10, 23, 44–46], and more work needs to be done to determine if PLBL2 can serve as a surrogate marker for active PSDE clearance. One recommendation to begin this effort is to evaluate a range of biophysical properties (e.g., pI, relative hydrophobicity) based on PSDE sequences identified. These biophysical properties may then be used as categories for possible activity correlation.
Formulation
Because most high-risk PSDEs may be present in DP at extremely low concentrations, direct detection is challenging and the slow destruction of PS and subsequent particle formation may take months or years to become significant. This means a problematic HCP population may remain undetected until after several key manufacturing steps have been locked and it has become difficult to implement process changes. In addition to the assessment of options regarding the cell line along with upstream and downstream processing steps to reduce PS degradation generated by PSDEs, it is worthwhile to consider formulation strategies to mitigate or reduce PS degradation to expand the shelf life of a biologic product. For example, when a liquid formulation fails to deliver the desired stability or shelf life, a frozen-liquid formulation can serve as a viable alternative approach.
In addition to changing storage temperature, there are several other formulation strategies that have been explored to mitigate PS degradation. One such formulation strategy starts with raw material control. PS is characterized as ethoxylated sorbitan esterified with FAs [2]. Higher concentrations of PS aid in the solubilization of FFAs but also provide a larger pool of substrate for the eventual generation of FAs via degradation. The distribution of different FAs in various grades and types of PS may also influence particle formation with less soluble, longer chain FAs contributing more to particle development.
PS-20 and PS-80 are available under several different grades: multicompendial (MC), super refined (SR), and Chinese Pharmacopia (ChP) for both PS-20 and PS-80. MC PS-20/80 typically used in formulations are heterogeneous compositions with different arrangements of ethylene oxides around a sorbitol/isosorbitan core, esterified with a range of FAs. SR PS-20 and PS-80 are like MC PSs in terms of their heterogeneous FA-ester composition but have undergone additional purification steps to reduce impurities such as primary and secondary oxidation products and unesterified FAs. ChP grade PSs have also undergone additional purification steps. They contain some heterogeneity in their ethylene oxide configuration but contain almost entirely (>98%) lauric acid (PS-20) and oleic acid (PS-80) as their hydrophobic components. Interestingly, no distinguishable differences in PS-80 functionality between MC and ChP grade were observed for a mAb subjected to three different mechanical stress conditions [62].
The sensitivity of different grades of PS-20 and PS-80 against enzymatic hydrolysis and oxidative degradation were compared [8, 18, 63]. No differences in the sensitivity toward enzymatic degradation were observed between MC and ChP grade PSs [8, 18]. ChP grade PS-80 was reportedly to be less prone to FFA particle formation [63]. Thus, visible and subvisible particles were significantly delayed [63]. However, ChP grade PS-20 and PS-80 have a higher predisposition for oxidative degradation as compared with MC PS-20 and PS-80, respectively [8, 18]. In this case, the buffer system also played a key role for oxidative degradation. Histidine showed a protective effect against hydrogen peroxide-induced oxidation, whereas hydrogen peroxide oxidation of PSs in acetate buffer was severe under the experimental conditions [8].
Certain PSDEs may have substrate preference of one PS over other PS. For example, Zhang et al. identified SIAE to have strong enzymatic activity for PS-20 degradation even at low concentrations (<5 ppm). Incubation of recombinant SIAE with PS-20 resulted in a unique degradation pattern where the hydrolysis of monoester with shorter FA chain (C12, C14) was observed but not the monoester with longer FA chain (C16, C18) or higher order esters. Interestingly, the lipase activity of SIAE is specific to PS-20, but not to PS-80. The specific esterase activity of SIAE on PS-20 suggests a possible solution of using PS-80 over PS-20 to eliminate surfactant degradation in mAb products with trace level of certain PSDEs.
To explore other formulation options for PS degradation, Roy et al. investigated a batch of high concentration (>100 mg/mL) mAb DP where subvisible particles formed abruptly after prolonged storage at 5 °C [43]. The work summarized the effectiveness of different formulation strategies for managing HCPs and FA particles and found that the concentration of FA composition of PSs was significant factors in particle development [43]. Solubilizers, such as Brij L23 and cyclodextrin (HPβCD), and alternative surfactants, such as poloxamer 188, were all shown to be effective means of preventing particle formation [43]. Lipase inhibitor, AA26–9 as a broad spectrum SH inhibitor, proved to be a simple means to prevent PS degradation. However, the inhibitor approach if used in DP would carry several concerns for patient safety. A more practical proposal for utilizing inhibitors is to incorporate them into an early DS purification step like the viral inactivation hold. This would lead to lipase inactivation due to covalent irreversible inhibition while allowing for the inhibitor itself removed with subsequent downstream purification steps [43].
CONCLUSION AND PERSPECTIVE
One critical aspect of biotherapeutics development is to control the stability of the DP during the manufacturing process as well as maintain its quality during shelf life. This often necessitates appropriate steps to help increase physical and chemical stability of those biotherapeutics throughout the different solution formulation conditions necessary for manufacturing and storage with minimal impact on product quality. Although PSs may help with these challenges, their degradation due to problematic residual HCPs may lead to products that become out of specification for several critical product quality attributes. Because of the complexity of PS structures and a potentially large, diversified, low-abundance mixture of HCPs existing in a product, PS degradation is one of the most challenging topics in the biopharma industry for analytical testing and process control. A significant amount of progress has been made on analytical methods and control strategies for PS degradation in the past few years, which significantly advances our knowledge in the field.
Due to its unique challenges across analytical, bioprocess, safety and regulatory, it often requires a collaborative effort across multiple disciplines to address PS degradation issues. There are also opportunities for the industry to work together to address this industry-wide challenges through external conferences or industry consortiums, such as National Institute for Innovation in Manufacturing Biopharmaceuticals (NIIMBL) or Biophorum. Standardized reagents (e.g., antibodies, recombinant proteins) and the use of harmonized methods through independent, scientific nonprofit organizations such as US Pharmacopeia (USP) or national institute of standards and technology (NIST) will help us compare data across different companies. Finally, the interaction and engagement with regulatory agencies and health authorities will be critical to the success of biologics development with potential CMC issues. The general guideline on how to manage high-risk HCPs and their impact on PS degradation during the life cycle of product development and commercialization will help deliver high-quality medicines to a broad community.
ACKNOWLEDGMENTS
The authors are grateful to the fruitful discussions from our internal working groups on HCP assay and polysorbate/lipase control strategy. We appreciate the feedback from Dr. Alyssa Stiving on the initial draft, and the help from Dr. Taku Tsukidate on the figures. The insightful feedback from the reviewers is highly appreciated.
FUNDING STATEMENT
None.
CONFLICT OF INTEREST STATEMENT
None declared.
DATA AVAILABILITY STATEMENTS
No new data were generated or analyzed in support of this research.
ETHICS AND CONSENT STATEMENT
Consent was not required.
ANIMAL RESEARCH STATEMENT
Not applicable.
References
- 1. Dwivedi, M, Blech, M, Presser, Iet al. Polysorbate degradation in biotherapeutic formulations: identification and discussion of current root causes. Int J Pharm 2018; 552: 422–36. [DOI] [PubMed] [Google Scholar]
- 2. Kerwin, BA. Polysorbates 20 and 80 used in the formulation of protein biotherapeutics: structure and degradation pathways. J Pharm Sci 2008; 97: 2924–35. [DOI] [PubMed] [Google Scholar]
- 3. Kishore, RS, Kiese, S, Fischer, Set al. The degradation of polysorbates 20 and 80 and its potential impact on the stability of biotherapeutics. Pharm Res 2011; 28: 1194–210. [DOI] [PubMed] [Google Scholar]
- 4. Vanderlaan, M, Zhu-Shimoni, J, Lin, Set al. Experience with host cell protein impurities in biopharmaceuticals. Biotechnol Prog 2018; 34: 828–37. [DOI] [PubMed] [Google Scholar]
- 5. Bracewell, DG, Francis, R, Smales, CM. The future of host cell protein (HCP) identification during process development and manufacturing linked to a risk-based management for their control. Biotechnol Bioeng 2015; 112: 1727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones, M, Palackal, N, Wang, Fet al. "high-risk" host cell proteins (HCPs): a multi-company collaborative view. Biotechnol Bioeng 2021; 118: 2870–85. [DOI] [PubMed] [Google Scholar]
- 7. Larson, NR, Wei, Y, Prajapati, Iet al. Comparison of polysorbate 80 hydrolysis and oxidation on the aggregation of a monoclonal antibody. J Pharm Sci 2020; 109: 633–9. [DOI] [PubMed] [Google Scholar]
- 8. Kranz, W, Wuchner, K, Corradini, Eet al. Factors influencing polysorbate's sensitivity against enzymatic hydrolysis and oxidative degradation. J Pharm Sci 2019; 108: 2022–32. [DOI] [PubMed] [Google Scholar]
- 9. Li, X, Richardson, D. Analysis of trace-level, high-risk HCPs: proteomics advances for preventing degradation of polysorbates in biotherapeutic formulations. Bioprocess Int 2021; 19: 8–13. [Google Scholar]
- 10. Li, X, Chandra, D, Letarte, Set al. Profiling active enzymes for polysorbate degradation in biotherapeutics by activity-based protein profiling. Anal Chem 2021; 93: 8161–69. [DOI] [PubMed] [Google Scholar]
- 11. Martos, A, Koch, W, Jiskoot, Wet al. Trends on analytical characterization of polysorbates and their degradation products in biopharmaceutical formulations. J Pharm Sci 2017; 106: 1722–35. [DOI] [PubMed] [Google Scholar]
- 12. Hewitt, D, Zhang, T, Kao, YH. Quantitation of polysorbate 20 in protein solutions using mixed-mode chromatography and evaporative light scattering detection. J Chromatogr A 2008; 1215: 156–60. [DOI] [PubMed] [Google Scholar]
- 13. Wenger, MD, Bowman, AM, Thorsteinsson, MVet al. An automated homogeneous method for quantifying polysorbate using fluorescence polarization. Anal Biochem 2005; 337: 48–54. [DOI] [PubMed] [Google Scholar]
- 14. Lippold, S, Koshari, SHS, Kopf, Ret al. Impact of mono- and poly-ester fractions on polysorbate quantitation using mixed-mode HPLC-CAD/ELSD and the fluorescence micelle assay. J Pharm Biomed Anal 2017; 132: 24–34. [DOI] [PubMed] [Google Scholar]
- 15. Zhang, S, Xiao, H, Molden, Ret al. Rapid polysorbate 80 degradation by liver carboxylesterase in a monoclonal antibody formulated drug substance at early stage development. J Pharm Sci 2020. [DOI] [PubMed] [Google Scholar]
- 16. Hvattum, E, Yip, WL, Grace, Det al. Characterization of polysorbate 80 with liquid chromatography mass spectrometry and nuclear magnetic resonance spectroscopy: specific determination of oxidation products of thermally oxidized polysorbate 80. J Pharm Biomed Anal 2012; 62: 7–16. [DOI] [PubMed] [Google Scholar]
- 17. Ayorinde, FO, Gelain, SV, Johnson, JHJret al. Analysis of some commercial polysorbate formulations using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 2000; 14: 2116–24. [DOI] [PubMed] [Google Scholar]
- 18. Liu, H, Jin, Y, Menon, Ret al. Characterization of polysorbate 80 by liquid chromatography-mass spectrometry to understand its susceptibility to degradation and its oxidative degradation pathway. J Pharm Sci 2021; S0022-3549(21)00418-4. [DOI] [PubMed] [Google Scholar]
- 19. Yang, RS, Bush, DR, DeGraan-Weber, Net al. Advancing structure characterization of PS-80 by charge-reduced mass spectrometry and software-assisted composition analysis. J Pharm Sci 2021; S0022-3549(21)00468-8. [DOI] [PubMed] [Google Scholar]
- 20. Cheng, Y, Hu, M, Zamiri, Cet al. A rapid high-sensitivity reversed-phase ultra high performance liquid chromatography mass spectrometry method for assessing polysorbate 20 degradation in protein therapeutics. J Pharm Sci 2019; 108: 2880–6. [DOI] [PubMed] [Google Scholar]
- 21. Zhang, S, Riccardi, C, Kamen, Det al. Monitoring polysorbate hydrolysis in therapeutic proteins using an ultrasensitive extraction-free fatty acid quantitation method. Anal Biochem 2021; 637: 114472. [DOI] [PubMed] [Google Scholar]
- 22. Gao, X, Rawal, B, Wang, Yet al. Targeted host cell protein quantification by LC-MRM enables biologics processing and product characterization. Anal Chem 2020; 92: 1007–15. [DOI] [PubMed] [Google Scholar]
- 23. Fischer, SK, Cheu, M, Peng, Ket al. Specific immune response to phospholipase B-like 2 protein, a host cell impurity in lebrikizumab clinical material. AAPS J 2017; 19: 254–63. [DOI] [PubMed] [Google Scholar]
- 24. Li, X, An, Y, Liao, Jet al. Identification and characterization of a residual host cell protein hexosaminidase B associated with N-glycan degradation during the stability study of a therapeutic recombinant monoclonal antibody product. Biotechnol Prog 2021; 37: e3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li, X, Pizarro, A, Grosser, T. Elective affinities – bioinformatic analysis of proteomic mass spectrometry data. Arch Physiol Biochem 2009; 115: 311–9. [DOI] [PubMed] [Google Scholar]
- 26. Hall, T, Sandefur, SL, Frye, CCet al. Polysorbates 20 and 80 degradation by group XV lysosomal phospholipase A2 isomer X1 in monoclonal antibody formulations. J Pharm Sci 2016; 105: 1633–42. [DOI] [PubMed] [Google Scholar]
- 27. Graf, T, Tomlinson, A, Yuk, IHet al. Identification and characterization of polysorbate-degrading enzymes in a monoclonal antibody formulation. J Pharm Sci 2021; 110: 3558–67. [DOI] [PubMed] [Google Scholar]
- 28. Labrenz, SR. Ester hydrolysis of polysorbate 80 in mAb drug product: evidence in support of the hypothesized risk after the observation of visible particulate in mAb formulations. J Pharm Sci 2014; 103: 2268–77. [DOI] [PubMed] [Google Scholar]
- 29. Anderson, NL, Anderson, NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics 2002; 1: 845–67. [DOI] [PubMed] [Google Scholar]
- 30. Huang, L, Wang, N, Mitchell, CEet al. A novel sample preparation for shotgun proteomics characterization of HCPs in antibodies. Anal Chem 2017; 89: 5436–44. [DOI] [PubMed] [Google Scholar]
- 31. Chen, IH, Xiao, H, Li, N. Improved host cell protein analysis in monoclonal antibody products through ProteoMiner. Anal Biochem 2020; 610: 113972. [DOI] [PubMed] [Google Scholar]
- 32. Johnson, RO, Greer, T, Cejkov, Met al. Combination of FAIMS, protein a depletion, and native digest conditions enables deep proteomic profiling of host cell proteins in monoclonal antibodies. Anal Chem 2020; 92: 10478–84. [DOI] [PubMed] [Google Scholar]
- 33. Zhang, S, Xiao, H, Li, N. Degradation of polysorbate 20 by sialate O-acetylesterase in monoclonal antibody formulations. J Pharm Sci 2021; 110: 3866–73. [DOI] [PubMed] [Google Scholar]
- 34. Chen, IH, Xiao, H, Daly, Tet al. Improved host cell protein analysis in monoclonal antibody products through molecular weight cutoff enrichment. Anal Chem 2020; 92: 3751–7. [DOI] [PubMed] [Google Scholar]
- 35. Wang, Q, Slaney, TR, Wu, Wet al. Enhancing host-cell protein detection in protein therapeutics using HILIC enrichment and proteomic analysis. Anal Chem 2020; 92: 10327–35. [DOI] [PubMed] [Google Scholar]
- 36. Yang, F, Walker, DE, Schoenfelder, Jet al. A 2D LC-MS/MS strategy for reliable detection of 10-ppm level residual host cell proteins in therapeutic antibodies. Anal Chem 2018; 90: 13365–72. [DOI] [PubMed] [Google Scholar]
- 37. Nie, S, Greer, T, O'Brien Johnson, Ret al. Simple and sensitive method for deep profiling of host cell proteins in therapeutic antibodies by combining ultra-low trypsin concentration digestion, Long chromatographic gradients, and BoxCar mass spectrometry acquisition. Anal Chem 2021; 93: 4383–90. [DOI] [PubMed] [Google Scholar]
- 38. Pythoud, N, Bons, J, Mijola, Get al. Optimized sample preparation and data processing of data-independent acquisition methods for the robust quantification of trace-level host cell protein impurities in antibody drug products. J Proteome Res 2021; 20: 923–31. [DOI] [PubMed] [Google Scholar]
- 39. Chiu, J, Valente, KN, Levy, NEet al. Knockout of a difficult-to-remove CHO host cell protein, lipoprotein lipase, for improved polysorbate stability in monoclonal antibody formulations. Biotechnol Bioeng 2017; 114: 1006–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Long, JZ, Cravatt, BF. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem Rev 2011; 111: 6022–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Greenbaum, D, Medzihradszky, KF, Burlingame, Aet al. Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem Biol 2000; 7: 569–81. [DOI] [PubMed] [Google Scholar]
- 42. Sanman, LE, Bogyo, M. Activity-based profiling of proteases. Annu Rev Biochem 2014; 83: 249–73. [DOI] [PubMed] [Google Scholar]
- 43. Roy, I, Patel, A, Kumar, Vet al. Polysorbate degradation and particle formation in a high concentration mAb: formulation strategies to minimize effect of enzymatic polysorbate degradation. J Pharm Sci 2021; 110: 3313–23. [DOI] [PubMed] [Google Scholar]
- 44. Repo, H, Kuokkanen, E, Oksanen, Eet al. Is the bovine lysosomal phospholipase B-like protein an amidase? Proteins 2014; 82: 300–11. [DOI] [PubMed] [Google Scholar]
- 45. Bhargava, AC, Mains, K, Siu, Aet al. High-throughput, fluorescence-based esterase activity assay for assessing polysorbate degradation risk during biopharmaceutical development. Pharm Res 2021; 38: 397–413. [DOI] [PubMed] [Google Scholar]
- 46. Zhang, S, Xiao, H, Goren, Met al. Putative phospholipase B-like 2 is not responsible for polysorbate degradation in monoclonal antibody drug products. J Pharm Sci 2020; 109: 2710–8. [DOI] [PubMed] [Google Scholar]
- 47. Dennis, EA, Cao, J, Hsu, YHet al. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev 2011; 111: 6130–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ahrne, E, Molzahn, L, Glatter, Tet al. Critical assessment of proteome-wide label-free absolute abundance estimation strategies. Proteomics 2013; 13: 2567–78. [DOI] [PubMed] [Google Scholar]
- 49. Schenauer, MR, Flynn, GC, Goetze, AM. Identification and quantification of host cell protein impurities in biotherapeutics using mass spectrometry. Anal Biochem 2012; 428: 150–7. [DOI] [PubMed] [Google Scholar]
- 50. Picotti, P, Aebersold, R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods 2012; 9: 555–66. [DOI] [PubMed] [Google Scholar]
- 51. Li, X, Fries, S, Li, Ret al. Differential impairment of aspirin-dependent platelet cyclooxygenase acetylation by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A 2014; 111: 16830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen, Y, Xu, CF, Stanley, Bet al. A highly sensitive LC-MS/MS method for targeted quantitation of lipase host cell proteins in biotherapeutics. J Pharm Sci 2021; 110: 3811–18. [DOI] [PubMed] [Google Scholar]
- 53. Jahn, M, Zerr, A, Fedorowicz, FMet al. Measuring lipolytic activity to support process improvements to manage lipase-mediated polysorbate degradation. Pharm Res 2020; 37: 118. [DOI] [PubMed] [Google Scholar]
- 54. Glucklich, N, Carle, S, Buske, Jet al. Assessing the polysorbate degradation fingerprints and kinetics of lipases – how the activity of polysorbate degrading hydrolases is influenced by the assay and assay conditions. Eur J Pharm Sci 2021; 166: 105980. [DOI] [PubMed] [Google Scholar]
- 55. Doshi, N, Martin, J, Tomlinson, A. Improving prediction of free fatty acid particle formation in biopharmaceutical drug products: incorporating Ester distribution during polysorbate 20 degradation. Mol Pharm 2020; 17: 4354–63. [DOI] [PubMed] [Google Scholar]
- 56. McShan, AC, Kei, P, Ji, JAet al. Hydrolysis of polysorbate 20 and 80 by a range of carboxylester hydrolases. PDA J Pharm Sci Technol 2016; 70: 332–45. [DOI] [PubMed] [Google Scholar]
- 57. Chen, D, Luo, W, Hoffman, Jet al. Insights into virus inactivation by polysorbate 80 in the absence of solvent. Biotechnol Prog 2020; 36: e2953. [DOI] [PubMed] [Google Scholar]
- 58. Kol, S, Ley, D, Wulff, Tet al. Multiplex secretome engineering enhances recombinant protein production and purity. Nat Commun 2020; 11: 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dovgan, T, Golghalyani, V, Zurlo, Fet al. Targeted CHO cell engineering approaches can reduce HCP-related enzymatic degradation and improve mAb product quality. Biotechnol Bioeng 2021; 118: 3821–31. [DOI] [PubMed] [Google Scholar]
- 60. Tran, B, Grosskopf, V, Wang, Xet al. Investigating interactions between phospholipase B-like 2 and antibodies during protein a chromatography. J Chromatogr A 2016; 1438: 31–8. [DOI] [PubMed] [Google Scholar]
- 61. Lim, A, Doyle, BL, Kelly, GMet al. Characterization of a cathepsin D protease from CHO cell-free medium and mitigation of its impact on the stability of a recombinant therapeutic protein. Biotechnol Prog 2018; 34: 120–9. [DOI] [PubMed] [Google Scholar]
- 62. Grabarek, AD, Bozic, U, Rousel, Jet al. What makes polysorbate functional? Impact of polysorbate 80 grade and quality on IgG stability during mechanical stress. J Pharm Sci 2020; 109: 871–80. [DOI] [PubMed] [Google Scholar]
- 63. Doshi, N, Giddings, J, Luis, Let al. A comprehensive assessment of all-oleate polysorbate 80: free fatty acid particle formation, interfacial protection and oxidative degradation. Pharm Res 2021; 38: 531–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analyzed in support of this research.