

Abstract
Worldwide, nasopharyngeal carcinoma (NPC) is a rare head and neck cancer; however, it is a common malignancy in southern China. Radiotherapy is the most important treatment strategy for NPC. However, although radiotherapy is a strong tool to kill cancer cells, paradoxically it also promotes aggressive phenotypes. Therefore, we mimicked the treatment process in NPC cells in vitro. Upon exposure to radiation, a subpopulation of NPC cells gradually developed resistance to radiation and displayed cancer stem-cell characteristics. Radiation-induced stemness largely depends on the accumulation of the antiapoptotic myeloid cell leukemia 1 (MCL-1) protein. Upregulated MCL-1 levels were caused by increased stability and more importantly, enhanced protein synthesis. We showed that repeated ionizing radiation resulted in persistently enhanced reactive oxygen species (ROS) production at a higher basal level, further promoting protein kinase B (AKT) signaling activation. Intracellular ROS and AKT activation form a positive feedback loop in the process of MCL-1 protein synthesis, which in turn induces stemness and radioresistance. AKT/MCL-1 axis inhibition attenuated radiation-induced resistance, providing a potential target to reverse radiation therapy-induced radioresistance.
Subject terms: Experimental models of disease, Radiotherapy, Head and neck cancer
Introduction
Nasopharyngeal carcinoma (NPC) is a kind of head and neck cancer arising from the nasopharyngeal epithelium [1]. NPC has a unique pattern of geographical distribution, and newly diagnosed cases are most common in South China and Southeast Asia [2, 3]. Nonkeratinizing undifferentiated NPC cells are highly sensitive to radiation therapy (RT) [4]; therefore, RT (with or without chemotherapy) is the standard curative treatment for NPC [5]. Ionizing radiation (IR) induces cell death via direct and indirect effects. Direct effects include chromosomal aberrations, such as nuclear DNA damage and mutagenesis. IR also induces indirect damage through reactive oxygen species (ROS) produced by water radiolysis [6]. However, low levels of ROS also promote secondary cancer or tumor progression [7, 8]. Cancer cells that survive IR treatment display more aggressive phenotypes (including epithelial to mesenchymal transition (EMT)) [9, 10] and enriches the fraction of cancer stem cells [11–13]. Both are closely related to metastasis and therapeutic resistance.
Cancer stem cells (CSCs) are a small subpopulation of cells displaying properties such as self-renewal, differentiation, tumorigenic capabilities, and increased resistance to treatment [14, 15]. In many cancers, the existence of CSCs has been found to interfere with treatment success [16]. Radiation induces CSC generation or enriches their subpopulation from non-stem cancer cells [17, 18]. Intracellular ROS levels are critical to maintain CSC properties in breast cancer and Head and Neck Cancer cells [19]. In contrast to cancer cells that obtain energy from glycolysis, CSCs preferentially use mitochondrial respiration to obtain sufficient energy for self-renewal [20, 21]. Mitochondrial membrane potential and levels of ROS are reported as elevated in stem-like triple-negative breast cancer cells [22].
Apoptosis induced by mass ROS production is the major method of cell death caused by radiation therapy; therefore, regulators of apoptosis are important factors the response to IR [23]. BCL-2 apoptosis regulator (BCL-2) family proteins play an integral role in apoptosis by governing mitochondrial outer membrane potential, consisting of pro-apoptotic proteins (Bim, Bid, Puma, and Noxa) and antiapoptotic proteins (MCL-1, BCLl-2, and BCL-XL). All family member share a BCL-2 homology (BH) domain; however, MCL-1 is distinguished from the others by its fast turnover [24]. The half-life of MCL-1 may be shortened or lengthened significantly depending on the cellular conditions or environmental stress [25]. In response to IR-induced DNA damage, the balance between pro-apoptotic and antiapoptotic BCL-2 proteins determines cell fate [26–28]. Cancer cells may escape apoptosis by expressing high level of antiapoptotic proteins. The overexpression of antiapoptotic proteins, including BCL-2, BCL-XL and MCL-1, contributes to treatment resistance in stem cell-like cancer cells. Inhibition of BCL-2 family proteins increases the antitumor treatment efficacy [29–32].
In this study, we mimicked the treatment in NPC cells in vitro and developed IR-induced radioresistant cell lines. Then, we investigated the phenotypic and functional characteristics of our models. Furthermore, we reported MCL-1 as a modulator of acquired resistance in NPCs receiving IR treatment.
Materials and methods
Cell culture and reagents
Parental (relatively sensitive to IR, has been previously reported [33]) and radioresistant subpopulations of NPC cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen) at 37 °C and 5% CO2. Cells were plated in 6-well or 12-well plates (Corning Inc., Corning, NY, USA) and the plating efficiencies of cell lines used are higher than 90%. Cells are treated with cycloheximide (CHX; 66-81-9, Abcam, Cambridge, MA, USA), MG132 (S2619, Selleck, Houston, TX, USA), N-acetylcysteine (S0077, Beyotime, Jiangsu, China), or MK2206 (S1078, Selleck) according to studies.
Cell clonogenic survival and cell viability assays
Cells (5 × 103) were suspended and seeded into 6-well plates after receiving the indicated dose of irradiation, which utilizing patented x-ray irradiation technology, cultured for 10–12 days, fixed, and stained with 0.4% methylene blue (Sigma-Aldrich, St Louis, MO, USA). Colonies were defined as >50 cells. For the cell viability assay, cells were seeded into a 96-well plate (Corning) and cultured for 4 days. At various time points after seeding, the cells in each well were stained with MTS (3-(4,5-dimethylthiazol-2-yl)−5-(3-carboxymethoxyphenyl)−2-(4-sulfophenyl)−2H-tetrazolium; G5421, Promega, Madison, WI, USA), and the OD490 was determined using a microplate reader.
Spheroid formation assay
Single-cell suspensions containing 500–800 cells were seeded in 12-well ultra-low-attachment culture plates and cultured in serum-free DMEM/F12 (11320082, Invitrogen) supplemented with 20 ng/mL epidermal growth factor (EGF; PHG0311, Invitrogen) and 10 ng/mL basic fibroblast growth factor (bFGF; PHG0360, Invitrogen) for 10 days. The formed spheroids were counted, and representative images were acquired via microscopy.
Lentiviral transduction studies
An MCL1 expression construct was generated by subcloning the PCR amplified, full-length human MCL1 cDNA into pBABE-puro plasmid; an empty vector (Vec) was used as a control. Cells stably expressing either an MCL1 short hairpin RNA (shRNA) or a scrambled, nontarget shRNA (shLuc) were established using the LV3 plasmid according to the manufacturer’s instructions [34]. The targets of shRNA-1 (sh1) and shRNA-2 (sh2) were 5′-CTTCCATGTAGAGGACCTAGA-3′, and 5′-GCCTAGTTTATCACCAATAAT-3′, respectively. Virus production and infection were performed as previously described [34], and stable cell lines were selected using 2 mg/mL puromycin for 7 days.
Flow cytometry
For side population (SP) analysis, the cells were harvested and incubated with Hoechst 33342 dye (5 mg/mL, Sigma) with or without Fumitremorgin C (FTC, ABCG2 inhibitor, 50 μM, Sigma) and incubated in the dark for 90 min at 37 °C with intermittent mixing. The cells were then subjected to flow cytometry.
ROS analysis
Cells were blocked for 30 min in 5% bovine serum albumin (BSA) and incubated with Dichlorofluorescin-diacetate (DCFH-DA) (S0033S, Beyotime) for 30 min in the dark at room temperature. Images were acquired via a high-throughput confocal microscope (Olympus, Tokyo, Japan).
Western blotting
Immunoblotting was performed according to the standard method as described previously [34]. Primary antibodies were from Cell Signaling Technology (Danvers, MA, USA), including those recognizing cleaved poly(ADP-Ribose) polymerase (PARP; 5625), caspase-3 (9662), β-actin (4970), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 2118), α-tubulin (2144), MCL-1 (D35A5), BCL-2 (2872), BCL-xL (2762), octamer-binding protein 4 (OCT4; 2750), Nanog (4903), SRY box 2 (SOX2; 14962), hypoxia inducible factor 1 alpha (HIF-1α; 3716), γH2AX(5438), protein kinase B (AKT) (4685), phospho-AKT (4060), S6 ribosomal protein (2217), phospho-S6 ribosomal protein (4858), eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1; 9644), and phospho-4E-BP1 (9451).
Real-time reverse transcription-quantitative PCR
The mRNA levels of OCT4, Nanog, SOX2 and MCL-1 were measured by real-time RT-PCR according to the manufacturer’s instructions [35]. Briefly, Total RNA was extracted from cultured cell lines using TRIzol reagent (Invitrogen) and then reverse-transcribed using a cDNA Synthesis Kit (Takara, 6111A). Real-time qPCR was performed using a SYBR PCR Kit (LifeScience, 04707516001). The house keeping gene GAPDH was used as the internal normalization control to calculate the mRNA levels of the different genes.
Tumor xenograft experiments
The protocol for the xenograft experiments comply with the ARRIVE guidelines, and was approved by the Institutional Animal Care and Use Committee of Sun Yat-Sen University Cancer Center. Female BALB/c nude mice (4 weeks old, 16–18 g; Animal Center of Guangdong Province) were housed in barrier facilities. For the tumorigenicity assay, mice were randomly divided into groups. The indicated number tumor cells (2 × 104, 5 × 104 or 15 × 104) were suspended in 50 μL of culture medium containing 50% Matrigel (356243, BD Biosciences, San Jose, CA, USA) and subcutaneously inoculated into the mice (n = 10 per group). The mice were monitored every 3 days to measure tumor formation. All mice were euthanized at 5 weeks after injection. The tumor-initiating cell frequency (TIF) was calculated using extreme limiting dilution analysis (ELDA) software (http://bioinf.wehi.edu.au/software/elda/). For the radiation response in vivo assay, mice were inoculated subcutaneously with S26 and S26-R (radioresistant) cells (5 × 106 in 100 μL of sterile phosphate-buffered saline (PBS), n = 6 in each group). MK2206 or vehicle (control) was administered daily by gavage (120 mg/kg). The tumor volume and body weight were recorded every 3 days. At day 32, the mice were euthanized, the primary tumors were weighed, and tumor samples were collected for western blotting analysis.
Statistics
All statistical analyses were carried out using SPSS 21.0 (IBM Corp., Armonk, NY, USA). The data are presented as the mean ± standard error (SE) of at least three independent experiments. Comparisons between the groups were performed using one-way analysis of variance (ANOVA). A two-tailed Student’s t-test was used to compare the data between two groups. All cell culture experiments were performed independently in triplicate at least three times. p-values < 0.05 were considered statistically significant.
Results
Development of the acquisition of radioresistance in NPC cells after Ionizing radiation
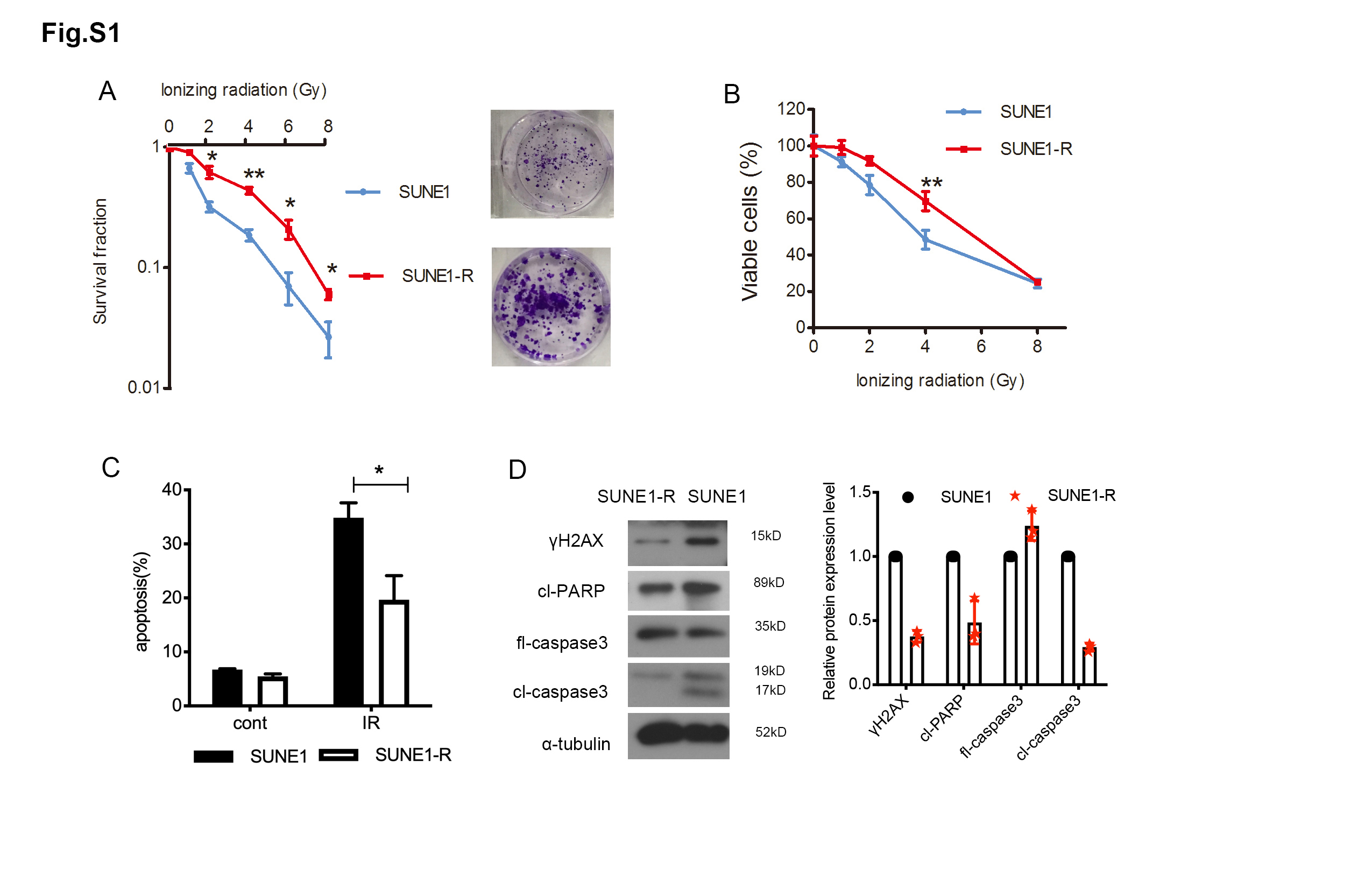
We have reported that S26 cells (a mono-clone derived from CNE-2 cells, a poorly differentiated human NPC cell line) were relatively sensitive to radiation therapy and chemotherapy [33]. To mimic the process of acquired resistance in patients after ionizing radiation (IR) treatment, S26 NPC cells were irradiated by daily exposure (5 days per week, for 6 weeks) to single fractions of 2 Gy X-ray radiation. The acquired radioresistant cell line, S26-R, was developed. Radioresistance was confirmed by growth fraction assays. The survival ability of radioresistant S26-R cells was significantly higher than that of their parental S26 cells when exposed to a gradient dose of radiation (Fig. 1A). Less inhibition of proliferation was also observed in the S26-R cell line under IR (Fig. 1B). To rule out that the effect is cell line specific, we developed radioresistant SUNE1-R cells from another NPC cell line, SUNE1 (Fig. S1A, B). Radiation exerts its cytotoxicity by inducing apoptosis. When exposed to radiation treatment, radioresistant S26-R and SUNE1-R cells showed significantly decreased apoptosis and lower protein levels of H2AX variant histone (γH2AX), cleaved PARP, and cleaved caspase-3, which are all well-known markers of DNA damage and apoptosis, indicating reduced induction of cell death (Fig. 1C, D and Fig. S1C, D).
Fig. 1. Repeated ionizing radiation induces a risk of radioresistance in NPC cells.

A (Left) Cell survival curve for treatment with increased doses of ionizing radiation in S26 and S26-R cells, *p < 0.05, **p < 0.01, Student’s t-test. (Right) representative images of colonies formed after treatment with the indicated dose of ionizing radiation in S26 and S26-R cells. B A cell proliferation curve was constructed from the MTS assay results, data are presented as the mean ± S.D. values, **p < 0.01, Student’s t-test. C, D S26 and S26-R cells were treated with or without 4 Gy of radiation for 48 h, then subjected to flow cytometry analysis of apoptosis *p < 0.05, Student’s t-test (C) and western blotting of the apoptotic markers (D); α-tubulin was used as loading control. (NPC, nasopharyngeal carcinoma; MTS, 3-(4,5-dimethylthiazol-2-yl)−5-(3-carboxymethoxyphenyl)−2-(4-sulfophenyl)−2H-tetrazolium.).
Ionizing radiation treatment enriches the CSC subpopulation
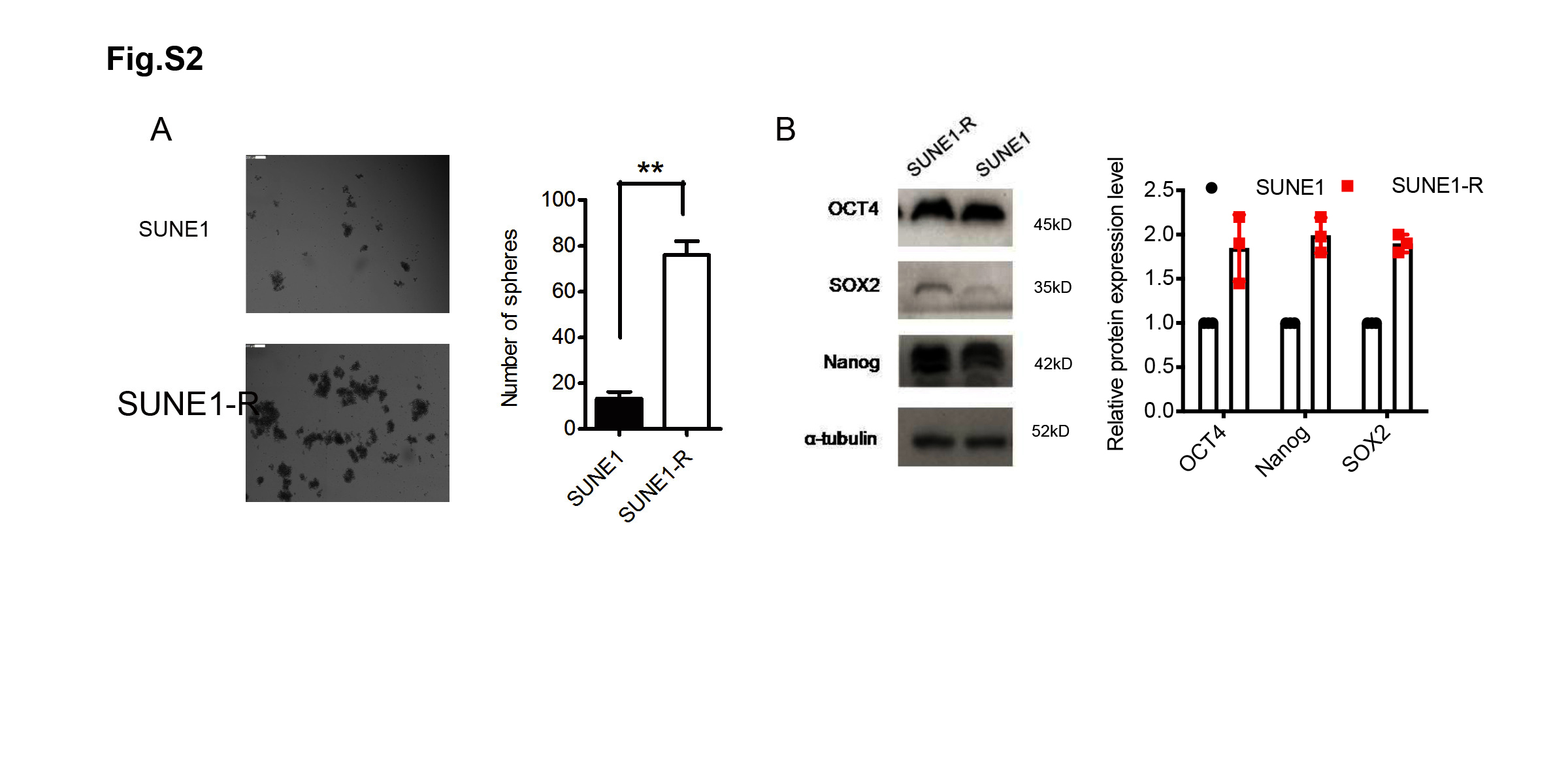
Stem cells have the capability to form spheres under suspension culture conditions. The cells that survived after IR showed a significantly increased sphere formation ability and size (Fig. 2A) and the percentage of subpopulation (SP) cells, a CSC marker for NPC cells (p < 0.001, Student’s t-test; Fig. 2B). Consistently, western blotting and quantitative PCR showed that levels of CSC markers (OCT4, Nanog, and SOX2) were elevated dramatically in radioresistant S26-R and SUNE1-R cells compared with those in their parental cells (Fig. 2C and S2).
Fig. 2. Ionizing radiation enhanced CSC subpopulation enrichment and radioresistance in vitro and in vivo.

A Single-cell suspensions were seeded in ultra-low-attachment culture plates. The formed spheroids were counted via microscopy. Representative images are shown, the number (upper) and the size (lower) of S26 and S26-R cells were compared, **p < 0.01, Student’s t-test, Scale bar: 200 μm. B Percentages of SP cells are shown in the left panel; three independent experiments were performed; The right panels show the upregulation of SP in cells S26-R NPC cells, **p < 0.01, Student’s t-test. C (Left) mRNA levels of stem-cell markers were determined by quantitative PCR analysis, ACTB (encoding β-actin) was used as a control. (Right) protein levels of stem-cell markers as determined using western blotting; β-actin was used as the loading control. D A total of 2 × 104, 5 × 104, and 15 × 104 of S26 and its radio-resistant S26-R NPC cells were subcutaneously injected into NOD/SCID mice (n = 10 mice/group). A summary of tumorigenicity in mice is shown. The TIF and p-value were calculated by using ELDA software. (NPC, nasopharyngeal carcinoma; CSC, cancer stem cell; SP, side population; NOD, non-obese diabetic; SCID, severe combined immunodeficiency; TIF, tumor-initiating cell frequency; ELDA, extreme limiting dilution analysis).
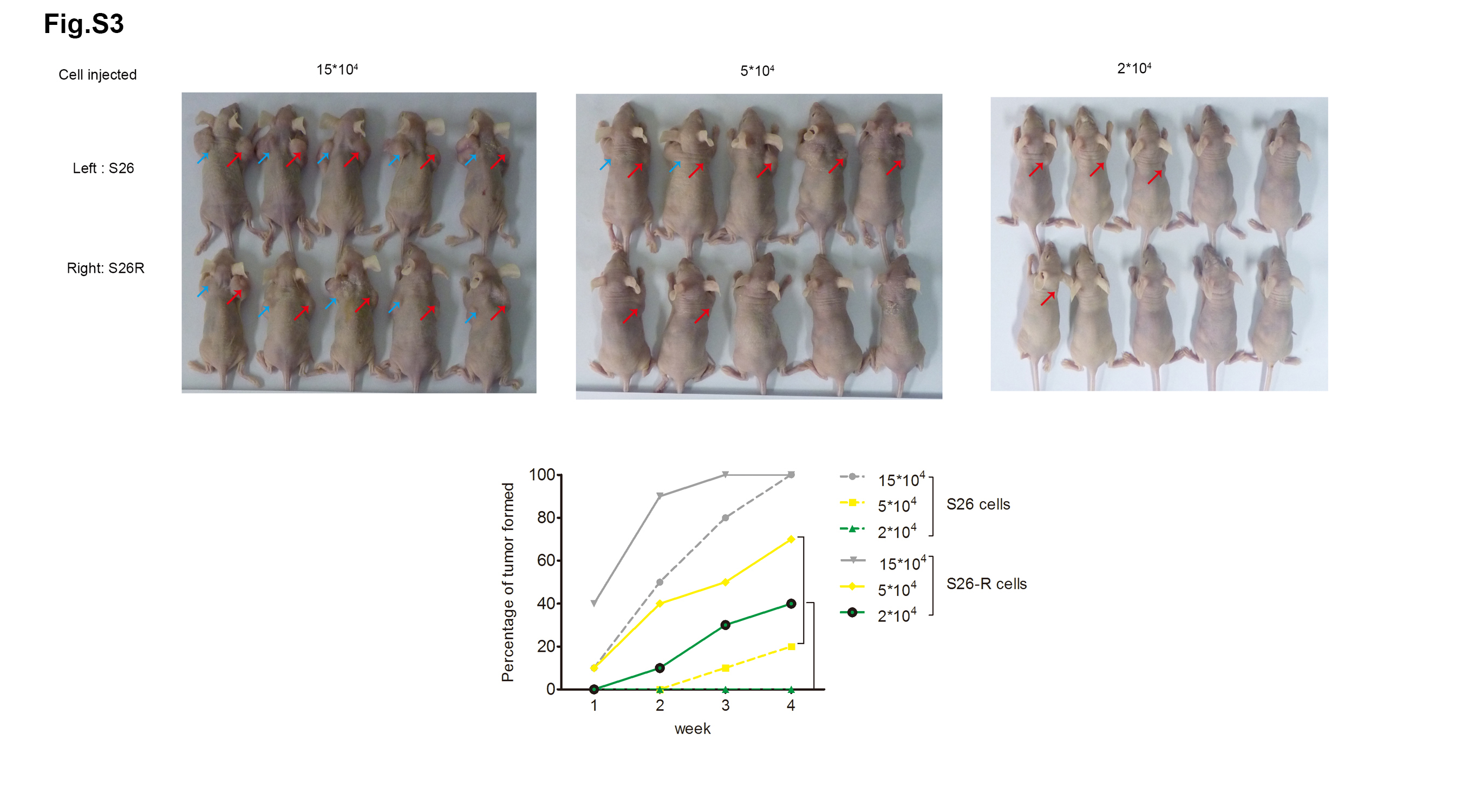
Then, we inoculated radioresistant S26-R cells and their parental S26 cells subcutaneously into nude mice at densities of 2 × 104, 5 × 104, and 15 × 104 cells, respectively. Parental S26 cells injected at 15 × 104 cells formed tumors in all nude mice, similar to S26-R cells, but at a much slower rate. When injected at a lower density (5 × 104 cells), 7 of 10 nude mice formed tumors in the S26-R group, while parental S26 cells formed tumors in 2 of 10 nude mice. When 2 × 104 cells were injected, parental S26 cells failed to form tumors in nude mice, whereas tumors formed in four of 10 mice bearing S26-R cells (Fig. 2D and Fig. S3). The above data confirmed that NPC cells after IR showed stem-cell phenotypes.
The enhanced stem-cell phenotypes induced by IR were MCL-1-dependent
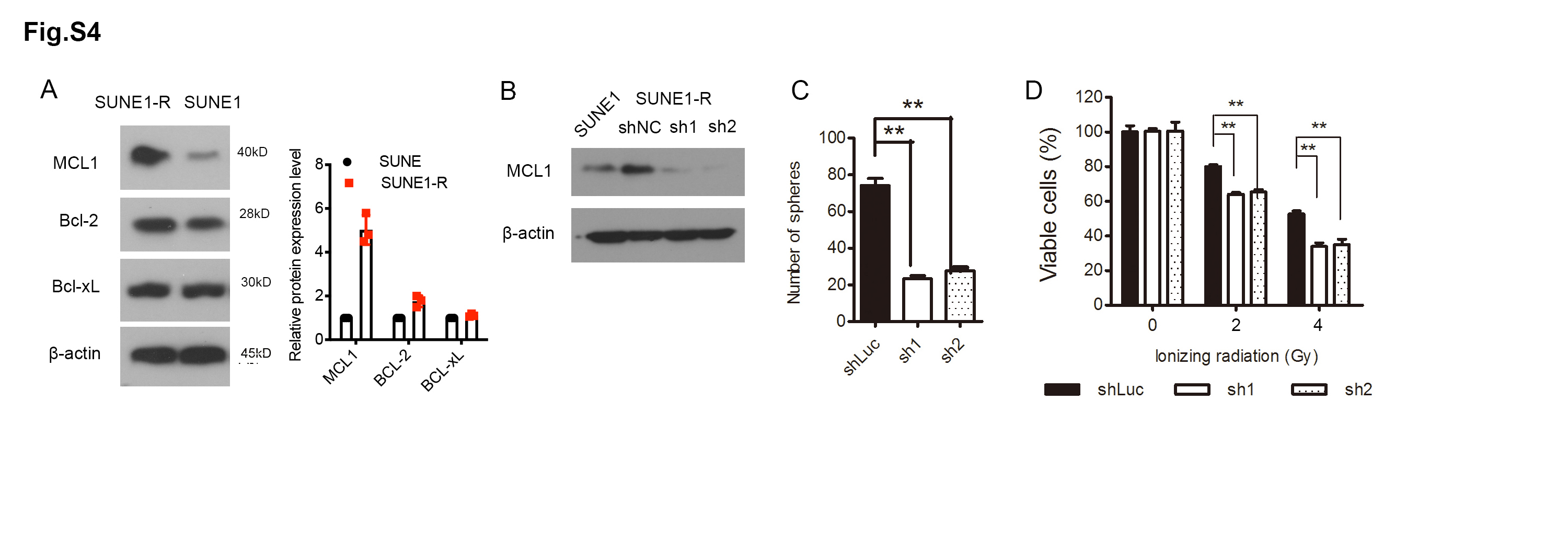
Increased antiapoptotic BCL-2 protein expression has been implicated in the development of resistance to radiation therapies. In our models, higher levels of MCL-1 and BCL-2 were found in S26-R and SUNE1-R cells after exposure to radiation, while nonsignificant changes in BCL-xL were also observed (Fig. 3A and Fig. S4A).
Fig. 3. MCL-1 depletion in radioresistant NPC cells inhibited stem-cell characteristic and restored radiation sensitivity.

S26 NPC cells were irradiated by daily exposure (5 days per week, for 6 weeks) to single fractions of 2 Gy X-ray radiation to acquire radioresistant cell line S26-R. A Western blotting analysis of BCL-2 family members expression in S26 and its radio-resistant S26-R cells. B–F S26-R cells stably transfected with shRNA targeting MCL1 (sh1, sh2) or scrambled shRNA (shNC) were analyzed as follows. (B, left panel) Representative images of single-cell suspensions in ultra-low-attachment culture plates are shown. (Right panel) The formed spheroids were counted via microscopy (upper) and the size of spheroids were compared (lower), **p < 0.01, Student’s t-test, Scale bar: 200 μm. C Percentages of SP cells are shown in the left panel, and the right panels compare the SP formed in S26-R cells transfected with the shRNA targeting MCL-1 or its control, n = 3; *p < 0.05, Student’s t-test. D Protein levels of stem-cell markers were determined by western blotting, tubulin was used as the loading control. E Cell viability of S26-R cells transfected with shRNA targeting MCL-1 or its control after treatment with ionizing radiation determined by the MTS assay, **p < 0.01, Student’s t-test. F Cell survival curve for treatment with increased doses of ionizing radiation in S26-R cells transfected with shRNA targeting MCL-1 or its control, *p < 0.05, **p < 0.01, Student’s t-test. G, H S26-R cells transfected with shRNA targeting MCL1 or its control were treated with indicated dose of radiation, then necrotic cells are revealed by propidium iodide staining (G) and apoptotic cells are revealed by flow cytometry analysis (H). Qualification results are shown, **p < 0.01, Student’s t-test. (NPC, nasopharyngeal carcinoma; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; shRNA, short hairpin RNA; SP, side population).
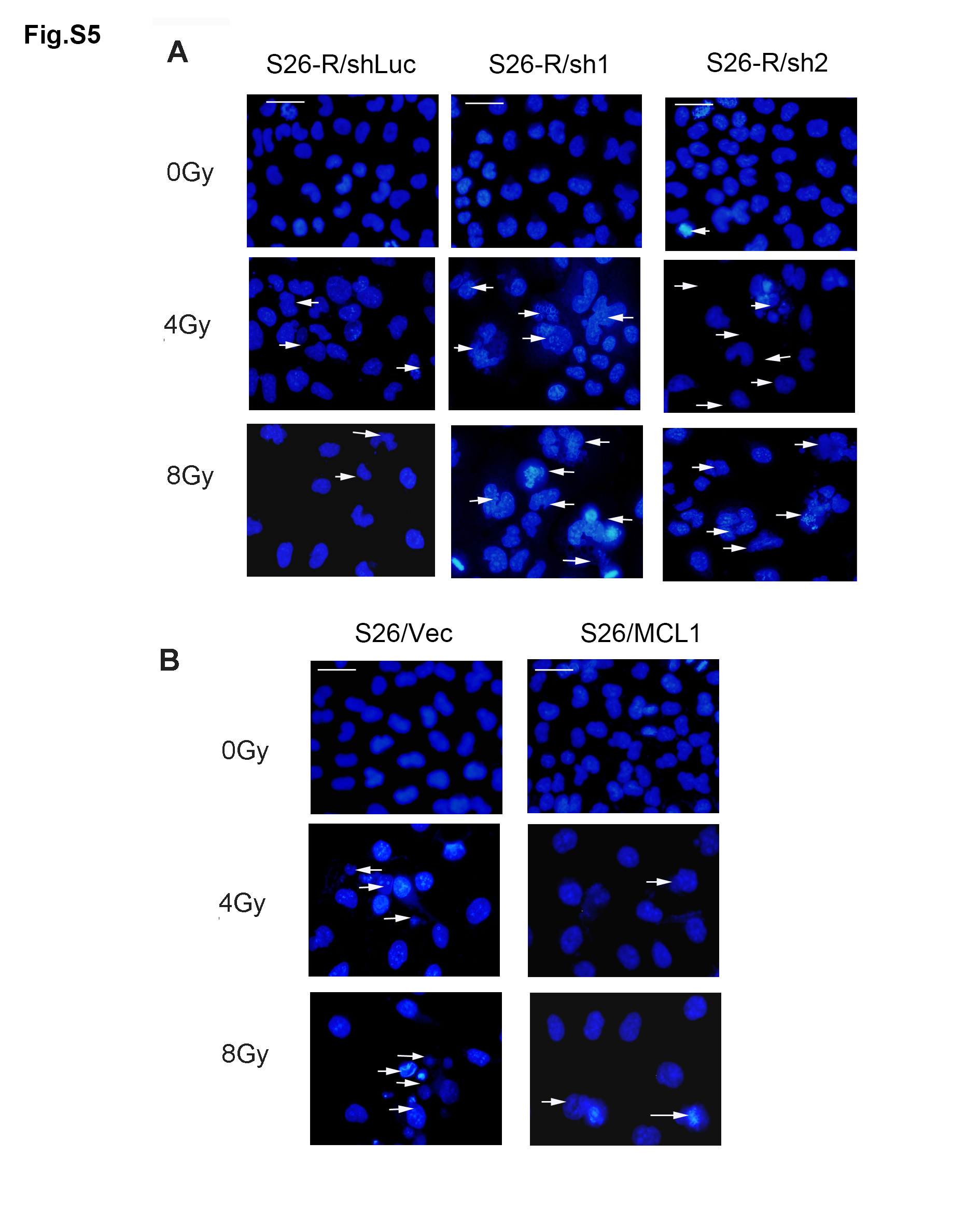
To test the role of MCL-1 in NPC CSC subpopulation enrichment, we knocked down MCL1 and found that its depletion reduced the sphere formation ability/size of radioresistant NPC cells (Fig. 3B). Knockdown of MCL1 reduced the percentage of SP cells compared with that in the parental S26 cells (Fig. 3C, from 43.6% to 25.7% and 26.6%, respectively, p < 0.05, Student’s t-test) and decreased the levels of CSC markers, as indicated by western blotting analysis (Fig. 3D). Moreover, proliferation assays showed that silencing MCL1 expression contributed to inhibition of cell proliferation (Fig. 3E) and clonogenic cell survival (Fig. 3F). Ionizing radiation-induced DNA damage results in an increase in nuclear chromatin condensation [36]. Knockdown of MCL1 increased nuclear condensation and nuclear fragmentation significantly when exposed to a gradient dose of radiation (Fig. 3G and Fig. S5A; p < 0.01 for both comparisons). Radiation induced significantly increased apoptosis in cells with MCL1 silencing (Fig. 3H). Additionally, we observed a significant decreased in sphere formation and vulnerability to IR treatment in SUNE1-R cells when MCL1 was knocked down (Fig. S4B–D).
Consistently, the overexpression of MCL1 increased the sphere formation ability and the size of S26 cells (Fig. 4A). S26 cells with high levels of MCL-1 increased the percentage of the SP cells (Fig. 4B, from 0.07% to 16.8%, p < 0.001, Student’s t-test) and stem-cell marker expression (Fig. 4C). Moreover, increasing MCL-1 levels contributed to radioresistance (Fig. 4D) and decreased nuclear condensation (Fig. 4E and Fig. S5B) upon exposure to radiation.
Fig. 4. Expression of MCL-1 in NPC cells induced CSC characteristic and radiation resistance.

S26 cells stably overexpressing MCL-1 or empty vector (Vec) were analyzed as follows. A The representative images of single-cell suspensions are shown (left panel) and spheroids were counted (right upper panel), and the size were measured (right lower panel), **p < 0.01, Student’s t-test. B Percentages of SP cells are shown in the left panel, and the right panels compare the populations of SP, n = 3, *p < 0.05, Student’s t-test. C Protein levels of stem-cell markers were determined by western blotting, GAPDH was used as the loading control. D Cell viability after treatment with ionizing radiation determined by MTS assay, **p < 0.01, Student’s t-test. E Qualification of nuclear condensation after treatment with different doses of radiation are shown, **p < 0.01, Student’s t-test. (NPC, nasopharyngeal carcinoma; CSC, cancer stem cell; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; SP, side population).
Activation of the ROS-AKT feedback loop regulates the protein synthesis of MCL-1 in vitro
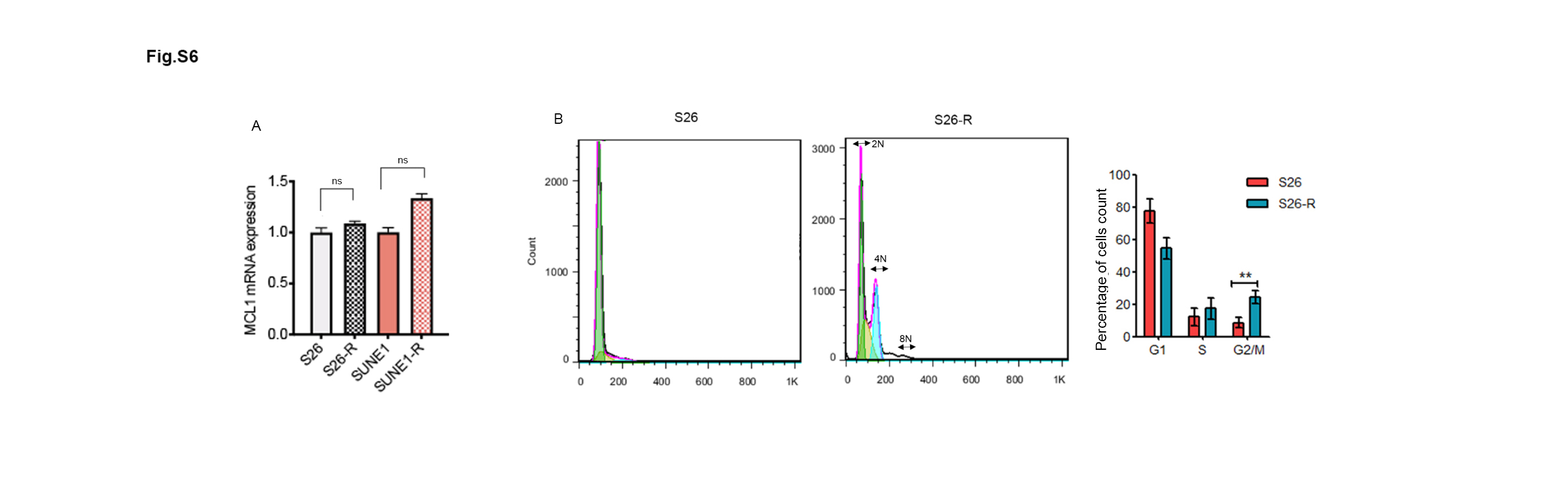
Regulation of MCL-1 expression occurs at the transcriptional, translational, and posttranslational levels. We measured the mRNA levels of MCL1 using quantitative PCR and found that the mRNA level was not altered in radioresistant S26 and SUNE1 NPC cells (Fig. S6A). We treated cells with CHX, a translation inhibitor, and collected protein at indicated time points to test existing MCL-1 protein levels. The results showed that MCL-1 protein stability was increased slightly in S26-R cells when compared with that in S26 cells (Fig. 5A). Then, the MCL-1 synthesis level was compared between S26 and S26-R cells treated with cycloheximide to remove MCL-1 proteins and then treated with MG132 to obtain newly synthesized proteins. Radioresistant S26-R cells exhibited increased synthesis of the MCL-1 protein (Fig. 5B).
Fig. 5. ROS-AKT feedback loop activation regulates the protein synthesis of MCL-1.

A S26 and S26-R cells were treated with CHX (50 μg/mL), lysates were collected at the indicated times. (Upper) Immunoblotting analysis of MCL-1 levels. (Lower) MCL-1 levels were quantified and normalized to the signal of β-actin. B S26-R and S26 cells were treated with CHX (50 μg/mL) or combined with MG132 (5 μM) for 2 h. Cell lysates were subjected to western blotting. β-actin was used as the loading control. C, D S26-R and S26 cells were treated with the AKT inhibitor MK2206 (1 μM) for 2 h, and then cell lysates were subjected to immunoblotting (C), β-actin was used as the loading control, and quantification of ROS levels is shown (D) (n = 3); *p < 0.05, **p < 0.01 compared with the 0 h control. E, F S26 and S26-R cells were treated with N-acetylcysteine (NAC, 5 μM) for 2 h, E Quantification of ROS levels measured by DCFH-DA is shown (n = 3; *p < 0.05, **p < 0.01 compared with the 0 h control); F cell lysates were subjected to immunoblotting, β-actin was used as the loading control. G S26-R cells and s26 cells were treated with the AKT inhibitor or NAC, then the cells were cultured for 10 days, and the formed spheroids were counted and compared, **p < 0.01, *p < 0.05, Student’s t-test. H, I S26-R cells and S26 cells were treated with the AKT inhibitor or NAC for 2 h, and then cells received the indicated dose of irradiation, and cell viability was determined by the MTS assay 2 days post-irradiation, and clony formation was determined by stained with methylene blue and counted 10 days post-irradiation, **p < 0.01, *p < 0.05, Student’s t-test. (CHX, cycloheximide; ROS, reactive oxygen species; DCFH-DA, dichlorofluorescin-diacetate; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium).
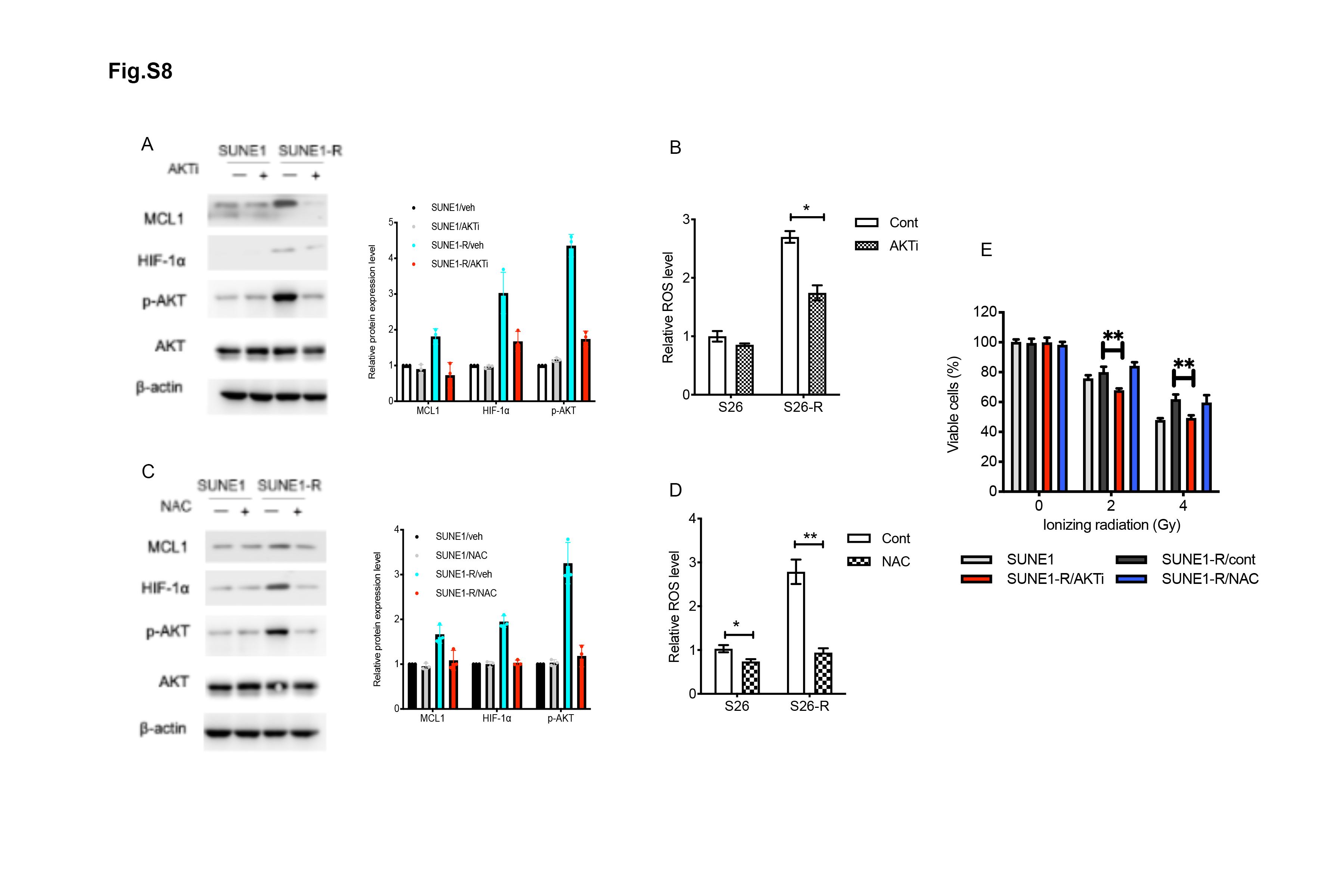
The process of protein synthesis is active during mitosis. DNA content analysis by fluorescence microscopy confirmed that S26-R cells had arrested at the G2/M phase, and showed polyploid multinucleate cell formation (Fig. S6B), suggesting that S26-R cells had advantage for protein synthesis. The AKT-mechanistic target of rapamycin (mTOR) signaling pathway has been reported to be involved in the protein synthesis process [37, 38]. To further investigate the mechanism of increased synthesis of MCL-1 in IR-induced NPC cells, we tested the AKT pathway signal and found it was activated in S26-R cells. The AKT inhibitor MK2206 efficiently blocked the activation of AKT, as detected by a decrease in AKT and its downstream ribosomal S6 kinase or 4EBP1 phosphorylation, which consequently caused downregulation of MCL-1 protein expression (Fig. 5C).
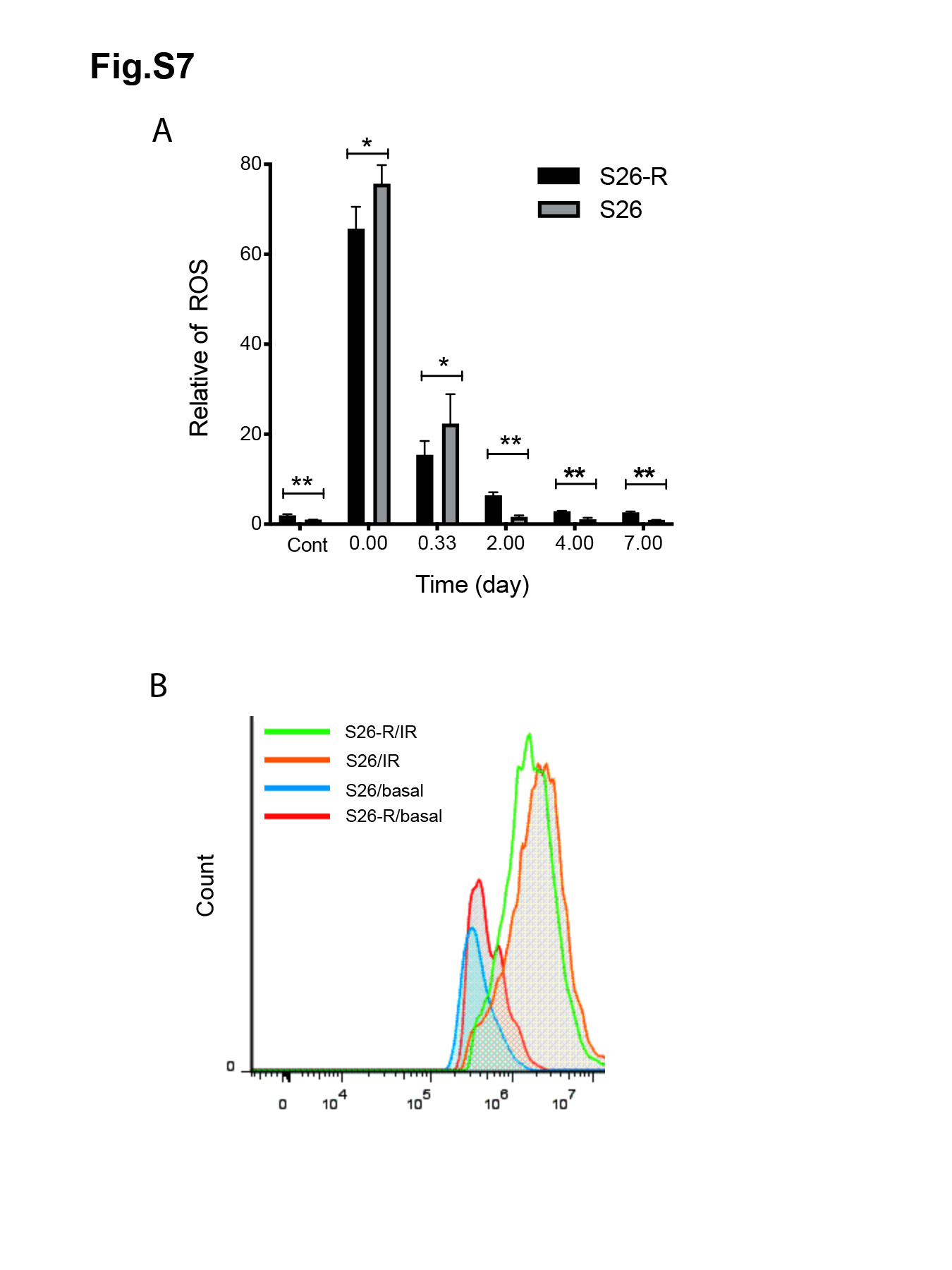
High levels of ROS are the main driver for cell death induced by irradiation. However, persistent ROS could be a key step in the transformation of cancer cells to CSCs [39, 40]. To determine the effects of ROS generation between IR-treated and untreated NPC cells, we performed an DCFH-DA assay. We observed an obvious increase in ROS soon after radiation and a quick decrease after 8 h, and then a slow decrease to the basal level over 1 to 2 weeks in S26 cells (Fig. S7A). S26-R cells have higher basal level of ROS without treatment when compared with S26 (fold- change = 1.95 ± 0.25, Fig. S7B). However, S26-R cells displayed relatively lower level of ROS generation in the first day after IR treatment (fold-change = 0.86 ± 0.05 in 0 h; fold-change = 0.68 ± 0.11 in 8 h, Fig. S7B), but a significant increase after 2 days(fold-change = 3.45 ± 0.36, Fig. S7B) through to 1 week when compared with S26 cells, suggesting a persistently higher basal level of ROS in the process of CSC formation (Fig. S7B). AKT inhibitor treatment cause a slight decrease in HIF-1α levels (a direct target of ROS) and significant downregulation of intracellular ROS levels (Fig. 5C, D). Then, N-acetylcysteine (NAC) was applied to inhibit basal ROS levels, and the efficiency of NAC was confirmed by reduced ROS and HIF-1α levels. Downregulation of AKT and phosphorylation of its downstream ribosomal S6 kinase were consistently observed (Fig. 5E, F). To confirm the impact of ROS-AKT on MCL1 synthesis, we use another NPC cell line, SUNE1, to test MCL1 level under inhibitor and NAC treatment. Again, we observed AKT inhibitor treatment lead to MCL1, HIF-1α downregulation and ROS level (Fig. S8A, B). N-acetylcysteine was also applied to treat SUNE1 cells. The effect of NAC was confirmed by reduced ROS level and HIF-1α expression (Fig. S8C, D). Interestingly, NAC treatment benefited MCL1 inhibition (Fig. S8C).
The above data suggested that the ROS-AKT axis forms a positive feedback loop that regulates MCL-1 protein synthesis, which is closely related to the CSC phenotype induced by irradiation (Fig. 5G, H, I and S8E). Inhibition of AKT increased the sensitivity of radioresistant cells to radiation therapy. Unexpectedly, ROS inhibition failed to reversed resistance to IR (Fig. 5H). We suspect that elevated intracellular ROS causes lethal damage induced by irradiation, allowing ROS inhibition to improve cell survival while receiving irradiation.
Inhibition of AKT-MCL-1 signaling increased the antitumor effect of radiation therapy in vivo
As described above, the AKT inhibitor acted as a tool to reverse radioresistance. To examine the effect of AKT-MCL-1 signaling on radiation sensitivity in vivo, nude mice were injected subcutaneously with S26-R cells transfected with MCL1 shRNA1 or control shRNA. All mice received daily therapy. Mice injected with control S26-R cells were randomly assigned to receive the AKT inhibitor or vehicle (control) treatment. The weights of the mice in each group were comparable (data not shown), suggesting that the treatment was tolerable for the mice. Tumor-bearing S26-R cells with MCL-1 depletion grew slowly compared with S26-R cells with control shRNA treatment. Interestingly, the AKT inhibitor treatment inhibited the growth of the tumors formed by S26-R cells, but not to the same extent as that of MCL-1 depletion (Fig. 6A–C). Immunoblotting of tumor lysates showed an obvious reduction in MCL-1 levels in the MCL-1 knockdown group, but little effect on AKT/ HIF-1α pathway molecules while compared with vehicle treatment group, suggesting that MCL-1 acts as a downstream factor in the AKT/ HIF-1α signaling pathway. AKT inhibitor treatment reduced MCL-1 protein levels, even though the reduction was not comparable to MCL-1 knockdown in the tumors. We also observed the size of tumor in mice was closely correlated with MCL-1 expression. Moreover, AKT inhibitor significantly suppressed HIF-1α and phosphor-AKT expression, indicating HIF-1α, at least partially, was regulated by AKT pathway (Fig. 6D).
Fig. 6. Inhibition of AKT-MCL-1 signaling increased antitumor effect of radiation therapy in vivo.

S26-R cells transfected with MCL-1 shRNA(MCL1/kd) or negative control shRNA were injected subcutaneously into BALB/c nude mice. Then, S26-R cells transfected with the control scrambled shRNA were treated with AKT inhibitor (MK2206, AKTi) or vehicle (veh, used as control) during treatment with radiation. A Tumor volume growth curves; data are shown as the mean ± SD. (**p < 0.01, ***p < 0.001, Student’s t-test). B Images of tumors from all mice in the indicated groups (n = 6/group). C Weight of tumors formed in the indicated groups; data are shown as the mean ± SD. (**p < 0.01, ***p < 0.001, Student’s t-test). D Tumor lysates were subjected to immunoblotting, β-actin was used as the loading control (left panel). Levels of the indicated proteins were quantified and normalized to the signal of β-actin, and phospho-AKT were normalized total AKT and β-actin (right panel).
Discussion
MCL-1-mediated chemotherapy resistance was attributed to resistance to apoptotic cell death or compromised homologous recombination-mediated DNA double-strand break repair [41]. Furthermore, the correlation between MCL-1 overexpression and stem cell-like potential has been reported previously in several cancer types, such as breast cancer [42], leukemia [43, 44], lung cancer [45], and glioblastoma [46]. In our study, we examined an additional role for MCL-1 in irradiation-induced cancer stem-like cells and acquired radioresistance in NPC.
Regulation of MCL-1 expression occurs at the transcriptional, translational, and posttranslational levels. In response to stimuli, such as growth factors or cytotoxic drugs, MCL1 transcription is enhanced by activating transcription factors (ATF5, STAT, and HIF-1α) [47, 48]. Despite the suggestion that HIF-1α activates MCL1 transcription, we found that the MCL1 mRNA level remained relatively constant even though HIF-1α expression was increased significantly. This observation ruled out transcriptional regulation as being responsible for IR-induced MCL-1 upregulation.
MCL-1 differs from other BCL-2 family members by a unique characteristic: It can be modified on sites within PEST (proline, glutamic acid, serine, and threonine) on its N-terminal region, affecting its rate of turnover [27, 49]. The degradation of MCL-1 was mainly attributed to the presence of amino acid residues marked for ubiquitination by USP9X when exposed to irradiation, which further promoted radioresistance [50]. Other posttranslational modifications (phosphorylation, caspase cleavage) are also dispensable for MCL-1 stability, closely correlating with the response to targeted therapy, chemotherapeutics, and oxidative stress [51–57]. Consistent with previous studies, MCL-1 downregulation was slightly delayed after proteasome inhibitor treatment in IR-induced radioresistant NPC cells during the observation period. Moreover, we observed an increase in MCL-1 synthesis in our model. MCL-1 protein synthesis has been reported to be blocked in Hela cells following a single fraction of ultraviolet irradiation [58]. Early-phase repression of protein synthesis, which helps to save energy in times of cellular stress, would not last long, with recovery of protein synthesis delayed until 48 h following high-dose IR treatment [59]. However, in cells that survived under multiple fractionated irradiation, enhanced protein synthesis later becomes the predominant mode [60]. Thus, our results suggested that MCL-1 accumulation is caused by an increase in MCL-1 stability and more importantly, enhanced protein synthesis.
Changes in protein synthesis capacity are required to maintain the demands of proliferating tumor cells [61]. Multiple mechanisms appear to participate in the process of protein synthesis. Upregulated phosphorylation of eIF4A, a key component of the translation and downstream effector of the AKT signaling pathway, plays a vital role [61]. AKT inhibition downregulated the phosphorylation of 4EBP1 and S6 and markedly reduced the levels of MCL-1 [62]. The AKT/ MCL-1 signaling pathway has been reported to play a prominent role in mediating antiapoptotic signals in chronic lymphocytic leukemia B cells [63] or in resistance to BCL-2/PARP inhibitors, or BH3 mimetics [64–66]. Enhanced AKT/mTOR signaling also contributes to X-ray and carbon ion beam irradiation resistance [67]. Our results demonstrated that the AKT signaling pathway controls the protein synthesis of MCL-1 in vitro (Fig. 6). Targeting AKT achieved limited success compared with MCL1 knockdown, and we suspected that the reason for the disappointing activity of the drugs was the induction of multiple feedback loops that causes overactivation of upstream or compensatory pathways, including PI3K and ERK, potentially blocking the antitumor effects of the inhibitors [68, 69]. A combination of bypass inhibitors could be applied to attenuate reactivation of the AKT signaling pathway.
Anti-cancer strategies always include chemotherapeutic drugs or irradiation to mediating ROS enhancement. High concentration of ROS induced by ionizing radiation is a key factor in the damage effect of the body. Reducing ROS with NAC treatment benefited cell survival at short-term post-irradiation (Fig. 5H and S8E). However, inhibition of ROS production significant hinder cell colony formation in long term (Fig. 5I). In fact, growing evidence supports the view that a moderate level of ROS is required for some cellular functions, including cell proliferation and angiogenesis, which are vital for tumorigenesis [70]. Cancer cells normally adapt to persistent oxidative stress by enriching their stem-like potential [71]. Human breast CSCs maintain persistent ROS generation that provide radioprotection, representing a possible explanation for tumor recurrence with therapy [18, 72]. Persistent prolonged ROS-induced oxidative stress expands the clonal selection of cancer cells, gradually making these clonal cells form subsets with new features that surviving from radiation exposure [73].
Small increases in ROS would be expected to activate the PI3K/AKT pathway [74–77]. In contrast, PI3K/AKT signaling contributes to increased ROS levels through direct modulation of mitochondrial bioenergetics, or indirectly through the production of ROS as a metabolic by-product [78]. In the present study, we found inhibition of AKT pathway helps reduction of ROS generation in vivo and in vitro. We also showed that ionizing radiation produced large amounts of ROS rapidly, which then decreased at 8 h post-irradiation, and maintained a consistently higher basal level subsequently, suggesting a relatively increased basal level of ROS is involved in the IR-induced stem-cell phenotype. Inhibition of ROS, however, failed to reverse cellular resistance to irradiation (Fig. 5G), strongly indicating the indispensable role of high concentration of ROS in IR-induced cell death. The synthesis of MCL-1 is controlled by a positive feedback loop of ROS/AKT signaling, which helps to maintain persistent intracellular MCL-1 levels. Although chronic oxidative stress is critical to the CSC phenotype induced by IR, redox cancer drugs have not yet showed convincing antitumor activity. How to maintain the subtle balance of intracellular ROS levels to increase treatment success remains a significant challenge.
In summary, we have shown that ionization radiation induces positive feedback activation of the ROS/AKT axis, which mediates increased synthesis of MCL-1, and contributes to CSC enrichment. In turn, CSC enrichment leads to resistance to radiation therapy in irradiated NPC tumors. Thus, a combinatorial approach of depleting MCL-1 in conjunction with IR might provide an important therapeutic improvement for the IR-induced resistant NPC.
Supplementary information
revided supplementary description-without redmarks
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We would like to thank Elixigen (http://www.elixigen.com/Service.asp) for English language editing. This work was supported by the Natural Science Foundation of Guangdong Province [grant number 2020A1515010208 (to YL)]; and the National Natural Science Functional of China [grant numbers 81773354 (to YY), 81972619 (to LJ), 81672874 (to LJ)].
Author contributions
YY and ZM guided the whole work. YL and YY conceptualized and designed the study. YL, FN, and AX performed most of the experiments. LJ helped with validation and methodology. CL and HL helped to provide research data for review. ZM and XS collected and analyzed clinical data with software. YL and FN wrote the manuscript. YW helped to revise the manuscript. All authors read and approved the final manuscript.
Data availability
The data used to support the findings of this study are included within the article and supplementary. Otherwise, some data are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Sun Yat-Sen University Cancer Center.
Footnotes
Edited by Dr. Shankar Varadarajan
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Ying-Ying Liang, Fei-Yu Niu, An-An Xu.
Contributor Information
Zhi-Wen Mo, Email: mozhiwen308@163.com.
Ya-Wei Yuan, Email: yuanyawei@gzhmu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41419-022-04551-z.
References
- 1.Wang DH, Luo HD, Huo ZJ, Chen MK, Han ZB, Huang MC, et al. Irradiation-induced dynamic changes of gene signatures reveal gain of metastatic ability in nasopharyngeal carcinoma. Am J Cancer Res. 2019;3:479–95. [PMC free article] [PubMed]
- 2.Yang CF, Peng LX, Huang TJ, Yang GD, Chu QQ, Liang YY, et al. Cancer stem-like cell characteristics induced by EB virus-encoded LMP1 contribute to radioresistance in nasopharyngeal carcinoma by suppressing the p53-mediated apoptosis pathway. Cancer Lett. 2014;344:260–71. doi: 10.1016/j.canlet.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Xie T, Zhong X, Jiang HL, Li R, Wang BY, et al. Melatonin reverses nasopharyngeal carcinoma cisplatin chemoresistance by inhibiting the Wnt/beta-catenin signaling pathway. Aging. 2020;12:5423–38. doi: 10.18632/aging.102968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen W, Hu GH. Biomarkers for enhancing the radiosensitivity of nasopharyngeal carcinoma. Cancer Biol Med. 2015;12:23–32. doi: 10.7497/j.issn.2095-3941.2014.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kong F, Zhou J, Du C, He X, Kong L, Hu C, et al. Long-term survival and late complications of intensity-modulated radiotherapy for recurrent nasopharyngeal carcinoma. BMC Cancer. 2018;18:1139. doi: 10.1186/s12885-018-5055-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY, Kim CH, et al. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol Cancer. 2017;16:10. doi: 10.1186/s12943-016-0577-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moncharmont C, Levy A, Guy JB, Falk AT, Guilbert M, Trone JC, et al. Radiation-enhanced cell migration/invasion process: a review. Crit Rev Oncol Hematol. 2014;92:133–42. doi: 10.1016/j.critrevonc.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 8.Chargari C, Goodman KA, Diallo I, Guy JB, Rancoule C, Cosset JM, et al. Risk of second cancers in the era of modern radiation therapy: does the risk/benefit analysis overcome theoretical models? Cancer Metastasis Rev. 2016;35:277–88. doi: 10.1007/s10555-016-9616-2. [DOI] [PubMed] [Google Scholar]
- 9.Gray M, Turnbull AK, Ward C, Meehan J, Martinez-Perez C, Bonello M, et al. Development and characterisation of acquired radioresistant breast cancer cell lines. Radiat Oncol. 2019;14:64. doi: 10.1186/s13014-019-1268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Li X, Zhang N, Yang Q, Moran MS. Low doses ionizing radiation enhances the invasiveness of breast cancer cells by inducing epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2011;412:188–92. doi: 10.1016/j.bbrc.2011.07.074. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen GH, Murph MM, Chang JY. Cancer stem cell radioresistance and enrichment: where frontline radiation therapy may fail in lung and esophageal cancers. Cancers. 2011;3:1232–52. doi: 10.3390/cancers3011232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 13.Ghisolfi L, Keates C.A, Hu XW, Lee DK, Li C.J. Ionizing radiation induces stemness in cancer cells. PLoS One. 2012;8:e43628. [DOI] [PMC free article] [PubMed]
- 14.Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol. 2012;44:2144–51. doi: 10.1016/j.biocel.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei P, Niu M, Pan S, Zhou Y, Shuai C, Wang J, et al. Cancer stem-like cell: a novel target for nasopharyngeal carcinoma therapy. Stem Cell Res Ther. 2014;5:44. doi: 10.1186/scrt433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reid PA, Wilson P, Li Y, Marcu LG, Bezak E. Current understanding of cancer stem cells: review of their radiobiology and role in head and neck cancers. Head Neck. 2017;39:1920–32. doi: 10.1002/hed.24848. [DOI] [PubMed] [Google Scholar]
- 17.Li F, Zhou K, Gao L, Zhang B, Li W, Yan W, et al. Radiation induces the generation of cancer stem cells: a novel mechanism for cancer radioresistance. Oncol Lett. 2016;12:3059–65. doi: 10.3892/ol.2016.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–3. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang CW, Chen YS, Chou SH, Han CL, Chen YJ, Yang CC, et al. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res. 2014;74:6291–305. doi: 10.1158/0008-5472.CAN-14-0626. [DOI] [PubMed] [Google Scholar]
- 20.Pasto A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G, et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget. 2014;5:4305–19. doi: 10.18632/oncotarget.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Francesco EM, Sotgia F, Lisanti MP. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochemical J. 2018;475:1611–34. doi: 10.1042/BCJ20170164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. 2017;26:633–647 e637. doi: 10.1016/j.cmet.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balcer-Kubiczek EK. Apoptosis in radiation therapy: a double-edged sword. Exp Oncol. 2012;34:277–85. [PubMed] [Google Scholar]
- 24.Le Gouill S, Podar K, Harousseau JL, Anderson KC. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle. 2004;3:1259–62. doi: 10.4161/cc.3.10.1196. [DOI] [PubMed] [Google Scholar]
- 25.Adams KW, Cooper GM. Rapid turnover of mcl-1 couples translation to cell survival and apoptosis. J Biol Chem. 2007;282:6192–200. doi: 10.1074/jbc.M610643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warren CFA, Wong-Brown MW, Bowden NA. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019;10:177. doi: 10.1038/s41419-019-1407-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. 2018;9:5341. doi: 10.1038/s41467-018-07551-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang YY, Deng XB, Zeng LS, Lin XT, Shao XF, Wang B, et al. RASSF6-mediated inhibition of Mcl-1 through JNK activation improves the anti-tumor effects of sorafenib in renal cell carcinoma. Cancer Lett. 2018;432:75–83. doi: 10.1016/j.canlet.2018.05.048. [DOI] [PubMed] [Google Scholar]
- 29.Trisciuoglio D, Tupone MG, Desideri M, Di Martile M, Gabellini C, Buglioni S, et al. BCL-XL overexpression promotes tumor progression-associated properties. Cell Death Dis. 2017;8:3216. doi: 10.1038/s41419-017-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cournoyer S, Addioui A, Belounis A, Beaunoyer M, Nyalendo C, Le Gall R, et al. GX15-070 (Obatoclax), a Bcl-2 family proteins inhibitor engenders apoptosis and pro-survival autophagy and increases Chemosensitivity in neuroblastoma. BMC Cancer. 2019;19:1018. doi: 10.1186/s12885-019-6195-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luanpitpong S, Poohadsuan J, Samart P, Kiratipaiboon C, Rojanasakul Y, Issaragrisil S. Reactive oxygen species mediate cancer stem-like cells and determine bortezomib sensitivity via Mcl-1 and Zeb-1 in mantle cell lymphoma. Biochim Biophys Acta Mol Basis Dis. 2018;1864:3739–53. doi: 10.1016/j.bbadis.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 32.Hu Y, Yague E, Zhao J, Wang L, Bai J, Yang Q, et al. Sabutoclax, pan-active BCL-2 protein family antagonist, overcomes drug resistance and eliminates cancer stem cells in breast cancer. Cancer Lett. 2018;423:47–59. [DOI] [PubMed]
- 33.Liang YY, Chen MY, Hua YJ, Chen S, Zheng LS, Cao X, et al. Downregulation of Ras association domain family member 6 (RASSF6) underlies the treatment resistance of highly metastatic nasopharyngeal carcinoma cells. PLoS ONE. 2014;9:e100843. doi: 10.1371/journal.pone.0100843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang YY, Deng XB, Lin XT, Jiang LL, Huang XT, Mo ZW, et al. RASSF1A inhibits PDGFB-driven malignant phenotypes of nasopharyngeal carcinoma cells in a YAP1-dependent manner. Cell Death Dis. 2020;11:855. doi: 10.1038/s41419-020-03054-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang YY, Zheng LS, Wu YZ, Peng LX, Cao Y, Cao X, et al. RASSF6 promotes p21(Cip1/Waf1)-dependent cell cycle arrest and apoptosis through activation of the JNK/SAPK pathway in clear cell renal cell carcinoma. Cell Cycle. 2014;13:1440–9. doi: 10.4161/cc.28416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tawar U, Bansal S, Shrimal S, Singh M, Tandon V. Nuclear condensation and free radical scavenging: a dual mechanism of bisbenzimidazoles to modulate radiation damage to DNA. Mol Cell Biochem. 2007;305:221–33. doi: 10.1007/s11010-007-9546-y. [DOI] [PubMed] [Google Scholar]
- 37.Chu N, Salguero AL, Liu AZ, Chen Z, Dempsey DR, Ficarro SB, et al. Akt Kinase Activation Mechanisms Revealed Using Protein Semisynthesis. Cell. 2018;174:897–907 e814. doi: 10.1016/j.cell.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiol (Bethesda) 2006;21:362–9. doi: 10.1152/physiol.00024.2006. [DOI] [PubMed] [Google Scholar]
- 39.Harada H, Itasaka S, Kizaka-Kondoh S, Shibuya K, Morinibu A, Shinomiya K, et al. The Akt/mTOR pathway assures the synthesis of HIF-1alpha protein in a glucose- and reoxygenation-dependent manner in irradiated tumors. J Biol Chem. 2009;284:5332–42. doi: 10.1074/jbc.M806653200. [DOI] [PubMed] [Google Scholar]
- 40.Ghanbari Movahed Z, Rastegari-Pouyani M, Mohammadi MH, Mansouri K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed Pharmacother. 2019;112:108690. doi: 10.1016/j.biopha.2019.108690. [DOI] [PubMed] [Google Scholar]
- 41.Mattoo AR, Pandita RK, Chakraborty S, Charaka V, Mujoo K, Hunt CR, et al. MCL-1 Depletion impairs DNA double-strand break repair and reinitiation of stalled DNA replication forks. Mol Cell Biol. 2017;37:e00535–16. [DOI] [PMC free article] [PubMed]
- 42.Annunziato S, de Ruiter JR, Henneman L, Brambillasca CS, Lutz C, Vaillant F, et al. Comparative oncogenomics identifies combinations of driver genes and drug targets in BRCA1-mutated breast cancer. Nat Commun. 2019;10:397. doi: 10.1038/s41467-019-08301-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiskus W, Cai T, DiNardo CD, Kornblau SM, Borthakur G, Kadia TM, et al. Superior efficacy of cotreatment with BET protein inhibitor and BCL2 or MCL1 inhibitor against AML blast progenitor cells. Blood Cancer J. 2019;9:4. doi: 10.1038/s41408-018-0165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Youn H, Lee HK, Sohn HR, Park UH, Kim EJ, Youn B, et al. RaRF confers RA resistance by sequestering RAR to the nucleolus and regulating MCL1 in leukemia cells. Oncogene. 2018;37:352–62. doi: 10.1038/onc.2017.329. [DOI] [PubMed] [Google Scholar]
- 45.Weeden CE, Ah-Cann C, Holik AZ, Pasquet J, Garnier JM, Merino D, et al. Dual inhibition of BCL-XL and MCL-1 is required to induce tumour regression in lung squamous cell carcinomas sensitive to FGFR inhibition. Oncogene. 2018;37:4475–88. doi: 10.1038/s41388-018-0268-2. [DOI] [PubMed] [Google Scholar]
- 46.Cameron BD, Traver G, Roland JT, Brockman AA, Dean D, Johnson L, et al. Bcl2-expressing quiescent type B neural stem cells in the ventricular-subventricular zone are resistant to concurrent temozolomide/X-irradiation. Stem Cells. 2019;37:1629–39. doi: 10.1002/stem.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhattacharyya A, Chattopadhyay R, Hall EH, Mebrahtu ST, Ernst PB, Crowe SE. Mechanism of hypoxia-inducible factor 1 alpha-mediated Mcl1 regulation in Helicobacter pylori-infected human gastric epithelium. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1177–1186. doi: 10.1152/ajpgi.00372.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Blaskovich MA, Forinash KD, Sebti SM. Withacnistin inhibits recruitment of STAT3 and STAT5 to growth factor and cytokine receptors and induces regression of breast tumours. Br J Cancer. 2014;111:894–902. doi: 10.1038/bjc.2014.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Blasio A, Vento R, Di Fiore R. Mcl-1 targeting could be an intriguing perspective to cure cancer. J Cell Physiol. 2018;233:8482–98. doi: 10.1002/jcp.26786. [DOI] [PubMed] [Google Scholar]
- 50.Warr MR, Mills JR, Nguyen M, Lemaire-Ewing S, Baardsnes J, Sun KL, et al. Mitochondrion-dependent N-terminal processing of outer membrane Mcl-1 protein removes an essential Mule/Lasu1 protein-binding site. J Biol Chem. 2011;286:25098–107. doi: 10.1074/jbc.M111.218321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Demelash A, Pfannenstiel LW, Tannenbaum CS, Li X, Kalady MF, DeVecchio J, et al. Structure-function analysis of the Mcl-1 protein identifies a novel senescence-regulating domain. J Biol Chem. 2015;290:21962–75. doi: 10.1074/jbc.M115.663898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang S, Zhang M, Jing Y, Yin X, Ma P, Zhang Z, et al. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat Commun. 2018;9:215. doi: 10.1038/s41467-017-02693-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cherok E, Xu S, Li S, Das S, Meltzer WA, Zalzman M, et al. Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5-dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol Biol Cell. 2017;28:396–410. doi: 10.1091/mbc.E16-04-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gasca J, Flores ML, Giraldez S, Ruiz-Borrego M, Tortolero M, Romero F, et al. Loss of FBXW7 and accumulation of MCL1 and PLK1 promote paclitaxel resistance in breast cancer. Oncotarget. 2016;7:52751–65. doi: 10.18632/oncotarget.10481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wakatsuki S, Tokunaga S, Shibata M, Araki T. GSK3B-mediated phosphorylation of MCL1 regulates axonal autophagy to promote Wallerian degeneration. J Cell Biol. 2017;216:477–93. doi: 10.1083/jcb.201606020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene. 2004;23:5301–15. doi: 10.1038/sj.onc.1207692. [DOI] [PubMed] [Google Scholar]
- 57.Hagenbuchner J, Kiechl-Kohlendorfer U, Obexer P, Ausserlechner MJ. A novel Mcl1 variant inhibits apoptosis via increased Bim sequestration. Oncotarget. 2013;4:1241–52. doi: 10.18632/oncotarget.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, et al. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–86. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Braunstein S, Badura ML, Xi Q, Formenti SC, Schneider RJ. Regulation of protein synthesis by ionizing radiation. Mol Cell Biol. 2009;29:5645–56. doi: 10.1128/MCB.00711-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stickel S, Gomes N, Su TT. The role of translational regulation in survival after radiation damage; an opportunity for proteomics analysis. Proteomes. 2014;2:272–90. doi: 10.3390/proteomes2020272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ertel F, Nguyen M, Roulston A, Shore GC. Programming cancer cells for high expression levels of Mcl1. EMBO Rep. 2013;14:328–36. doi: 10.1038/embor.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meja K, Stengel C, Sellar R, Huszar D, Davies BR, Gale RE, et al. PIM and AKT kinase inhibitors show synergistic cytotoxicity in acute myeloid leukaemia that is associated with convergence on mTOR and MCL1 pathways. Br J Haematol. 2014;167:69–79. doi: 10.1111/bjh.13013. [DOI] [PubMed] [Google Scholar]
- 63.Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating antiapoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood. 2008;111:846–55. doi: 10.1182/blood-2007-05-089037. [DOI] [PubMed] [Google Scholar]
- 64.Coloff JL, Macintyre AN, Nichols AG, Liu T, Gallo CA, Plas DR, et al. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 2011;71:5204–13. doi: 10.1158/0008-5472.CAN-10-4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mattoo AR, Joun A, Jessup JM. Repurposing of mTOR complex inhibitors attenuates MCL-1 and sensitizes to PARP inhibition. Mol Cancer Res: MCR. 2019;17:42–53. doi: 10.1158/1541-7786.MCR-18-0650. [DOI] [PubMed] [Google Scholar]
- 66.Lee YC, Wang LJ, Huang CH, Shi YJ, Chang LS. ABT-263-induced MCL1 upregulation depends on autophagy-mediated 4EBP1 downregulation in human leukemia cells. Cancer Lett. 2018;432:191–204. doi: 10.1016/j.canlet.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 67.Sato K, Azuma R, Imai T, Shimokawa T. Enhancement of mTOR signaling contributes to acquired X-ray and C-ion resistance in mouse squamous carcinoma cell line. Cancer Sci. 2017;108:2004–10. doi: 10.1111/cas.13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sathe A, Chalaud G, Oppolzer I, Wong KY, von Busch M, Schmid SC, et al. Parallel PI3K, AKT and mTOR inhibition is required to control feedback loops that limit tumor therapy. PLoS ONE. 2018;13:e0190854. doi: 10.1371/journal.pone.0190854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rozengurt E, Soares HP, Sinnet-Smith J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Therap. 2014;13:2477–88. doi: 10.1158/1535-7163.MCT-14-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52:192–203. doi: 10.1038/s12276-020-0384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dando I, Cordani M, Dalla Pozza E, Biondani G, Donadelli M, Palmieri M. Antioxidant mechanisms and ROS-related microRNAs in cancer stem cells. Oxid Med Cell Longev. 2015;2015:425708. doi: 10.1155/2015/425708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mahalingaiah PK, Singh KP. Chronic oxidative stress increases growth and tumorigenic potential of MCF-7 breast cancer cells. PloS ONE. 2014;9:e87371. doi: 10.1371/journal.pone.0087371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu J, Wang Z. Increased oxidative stress as a selective anticancer therapy. Oxid Med Cell Longev. 2015;2015:294303. doi: 10.1155/2015/294303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamamori T, Yasui H, Yamazumi M, Wada Y, Nakamura Y, Nakamura H, et al. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic Biol Med. 2012;53:260–70. doi: 10.1016/j.freeradbiomed.2012.04.033. [DOI] [PubMed] [Google Scholar]
- 75.Okoh VO, Felty Q, Parkash J, Poppiti R, Roy D. Reactive oxygen species via redox signaling to PI3K/AKT pathway contribute to the malignant growth of 4-hydroxy estradiol-transformed mammary epithelial cells. PLoS ONE. 2013;8:e54206. doi: 10.1371/journal.pone.0054206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Deng S, Dai G, Chen S, Nie ZG, Zhou JL, Fang HS, et al. Dexamethasone induces osteoblast apoptosis through ROS-PI3K/AKT/GSK3β signaling pathway. Biomed Pharmacother. 2019;110:602–8. [DOI] [PubMed]
- 77.Zhao Y, Hu X, Liu Y, Dong S, Wen Z, He W, et al. ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol Cancer. 2017;16:79. doi: 10.1186/s12943-017-0648-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koundouros N, Poulogiannis G. Phosphoinositide 3-kinase/Akt signaling and redox metabolism in cancer. Front Oncol. 2018;8:160. doi: 10.3389/fonc.2018.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
revided supplementary description-without redmarks
Data Availability Statement
The data used to support the findings of this study are included within the article and supplementary. Otherwise, some data are available from the corresponding author on reasonable request.