

Abstract
Wolfram syndrome (WFS) is a rare autosomal recessive disorder characterized by diabetes mellitus and insipidus, progressive optic atrophy and sensorineural deafness. An increased incidence of psychiatric disorders has also been reported in WFS patients. There are two subtypes of WFS. Type 1 (WFS1) is caused by mutations in the WFS1 gene and type 2 (WFS2) results from mutations in the CISD2 gene. Existing Wfs1 knockout mice exhibit many WFS1 cardinal symptoms including diabetic nephropathy, metabolic disruptions and optic atrophy. Far fewer studies have examined loss of Cisd2 function in mice. We identified B6.DDY-Cisd2m1Lmt, a mouse model with a spontaneous mutation in the Cisd2 gene. B6.DDY-Cisd2m1Lmt mice were initially identified based on the presence of audible sonic vocalizations as well as decreased body size and weight compared to unaffected wildtype littermates. Although Wfs1 knockout mice have been characterized for numerous behavioral phenotypes, similar studies have been lacking for Cisd2 mutant mice. We tested B6.DDY-Cisd2m1Lmt mice in a battery of behavioral assays that model phenotypes related to neurological and psychiatric disorders including anxiety, sensorimotor gating, stress response, social interaction and learning and memory. B6.DDY-Cisd2m1Lmt mice displayed hypoactivity across several behavioral tests, exhibited increased stress response and had deficits in spatial learning and memory and sensorimotor gating compared to wildtype littermates. Our data indicate that the B6.DDY-Cisd2m1Lmt mouse strain is a useful model to investigate potential mechanisms underlying the neurological and psychiatric symptoms observed in WFS.
Keywords: Cisd2, Wfs1, Wolfram Syndrome, behavior, mutant
1.0. Introduction
Wolfram syndrome (WFS) is a rare autosomal recessive disorder affecting 1 in 770,000 individuals. Two subtypes, Wolfram Syndrome Type 1 (WFS1) and Type 2 (WFS2), are caused by mutations in the WFS1 and CISD2 genes, respectively. WFS1 is more common, implicated in 90% of patients [1] and characterized by juvenile onset of key clinical features commonly referred to as DIDMOAD: diabetes insipidus, diabetes mellitus, progressive vision loss due to optic atrophy and deafness [2]. WFS2 was more recently characterized [3] and presents with similar clinical features except for diabetes insipidus [4]. WFS patients have a shortened life expectancy of 25-49 years and death is primarily due to respiratory failure resulting from autonomic dysfunction [1, 2].
In addition to the cardinal symptoms described above, neurological dysfunction and psychiatric disorders including anxiety, depression, bipolar disorder, schizophrenia, suicidal behavior and psychosis have been reported in WFS patients [5–7] [6, 8]. Moreover, first degree relatives of WFS patients are 26-fold more likely to require psychiatric hospitalizations [6] suggesting that WFS carrier status increases risk for psychiatric disease. It is important to note that many of the studies reporting psychiatric manifestations in WFS were conducted prior to the discovery of the WFS2 subtype and identification of the CISD2 gene. The incidence of concomitant psychiatric symptoms in WFS2 is less frequently reported, but mood disorders and mild depression in WFS2 patients have been noted [9, 10]. Thus, existing data provide compelling evidence that WFS genes play a role in the development of psychiatric disorders, or are involved in mechanistic pathways that, when perturbed, might increase risk.
Mechanistic studies of psychiatric disorders in human cohorts are challenging due to the heterogeneous nature of these disorders, the impact of unknown or unknowable environmental factors and the inability to access relevant brain tissue. To overcome this, animal models have been used to investigate complex diseases and allow for the interrogation of disease mechanisms at multiple levels under controlled environmental conditions and on a stable genetic background. Furthermore, mice can be genetically manipulated to study the role of a gene or genes in a disease and provide reproducible data, making them extremely powerful model systems.
Wfs1 mouse knockouts have been produced and exhibit cardinal signs of WFS including glucose intolerance and overt diabetes [11–15]. Wfs1 knockout mice have also been assessed in numerous behavioral assays and show deficits in active and passive avoidance behavior and associative and spatial learning, decreased social interaction, increased anxiety-like behavior and higher stress-induced corticosterone (CORT) compared to wildtype mice [12, 16]. Cisd2 knockout mice have also been produced and exhibit a premature aging phenotype and shortened life span that has been attributed to mitochondrial degeneration [17]. Cisd2 knockout mice also successfully recapitulate multiple clinical features of WFS including impaired glucose tolerance and optic atrophy [17]. However, behavioral characterization of Cisd2 knockout mice has been limited primarily to locomotor activity [18]. Assessment of a broader range of assays is critical for determining the function of the Cisd2 gene in behavior.
We identified a spontaneous mutation of the Cisd2 gene that arose in the commercial mouse breeding colony of the DDY/JclSidSeyFrkJ (DDY) inbred mouse strain at the Jackson Laboratory. B6.DDY-Cisd2m1Lmt mice have a single base deletion in the second exon of the gene. The deletion results in a frameshift and insertion of an early stop codon that is presumed to truncate the CISD2 protein. In the present study, we have characterized B6.DDY-Cisd2m1Lmt in a battery of behavioral assays that model phenotypes relevant to neurological and psychiatric disorders. Insight into how WFS1 and CISD2 might increase risk for psychiatric disease can inform our understanding of potential mechanisms that more generally increase risk for psychiatric disorders and ultimately have a major impact on the diagnosis and treatment of mental illness.
2.0. Methods and Materials
2.1. Animals
Several males in a shipment of DDY inbred mice purchased from the Jackson Laboratory were found to exhibit audible chirping noises. Affected DDY males were crossed to C57BL/6J (B6) females and all mice resulting from this cross are referred to as having a B6.DDY background. Female mice from these crosses (N1) were backcrossed to affected DDY males to produce an N2 generation (B6.DDYxDDY; N=40) that was used for initial linkage mapping described below. N2 mice were backcrossed an additional six times to B6 and intercrossed to produce littermates of all three genotypes for behavioral and glucose tolerance studies.
2.2. Mapping and Identification of Causal Polymorphism
Twenty one affected and nineteen unaffected B6.DDYxDDY N2 mice were genotyped using the Mouse Universal Genotyping Array (MUGA; Neogen Corporation, Lincoln, NE) using previously described methods [19]. A panel of simple sequence length polymorphisms (SSLPs) and single nucleotide polymorphisms (SNPs) that differed between B6 and DDY were identified and used to narrow the critical region (Supplemental Table 1). SSLPs were genotyped with microsatellite markers using standard procedures [20]. SNPs were genotyped using the Sequenom MassArray platform (Sequenom, San Diego, CA) following the manufacturers protocols as previously described [21]. Genomic DNA from one affected and one unaffected animal (both on homozygous DDY inbred background) was sent to the Broad Institute Mouse Resequencing Center for exome sequencing targeted to the critical region on Chromosome 3 spanning 135,234,644-136,111,550 base pairs. The Agilent SureSelect Mouse Exome array and Illumina HiSeq were used to generate sequence data to a coverage of at least 20x or higher for 80% of the targets. Sequence was visualized and variants identified using the Integrative Genomics Viewer (IGV, v2.6.3). All genetic marker locations are reported in mouse build GRCm38 (mm10).
2.3. Assessment of Gene Expression by Quantitative PCR
Total RNA was isolated from one brain hemisphere from 3 wildtype and 3 mutant mice using the RNeasy Plus Mini kit (QIAGEN) per the manufacturer’s instructions. The QuantiNOVA Reverse Transcription kit (QIAGEN) was used to reverse transcribe the total RNA to cDNA. Reverse transcription quantitative real time PCR (RT-qPCR) was performed using the QuantiNOVA SYBR PCR kit (QIAGEN) with custom designed Cisd2 primers (Forward Primer- 5’ TGGCCCGCATCGTGAAGGTG 3’, Reverse primer- 5’ GAATGGGCGCACTGCGAGGT 3’) and a commercially available Gapdh primer assay kit (QIAGEN). RT-PCR products for each sample were validated using gel electrophoresis to confirm product size and sequenced to confirm the presence or absence of the Cisd2 deletion in mutant and wildtype samples, respectively. Each sample was run in triplicate and values were averaged and normalized to GAPDH using the delta Ct (∆Ct) formula: ∆Ct= Ct (sample) - Ct (GAPDH). The average of the three wildtype Cisd2 values was used as the control average. Delta delta Ct (∆∆Ct) values for each sample were calculated using the formula: ∆∆Ct = Ct (sample)- Ct (control average). Fold gene expression was then determined using the formula 2−(∆∆Ct ) [22]. Data were log transformed and genotype differences were analyzed by t-test using SPSS for Macintosh (v24).
2.4. Phenotyping Pipeline Design
Separate cohorts of B6.DDY-Cisd2m1Lmt mutant mice including heterozygote and wildtype littermates were tested in two behavioral pipelines, each consisting of three behavioral tests. 21 wildtype (11 male, 10 female), 45 heterozygote (20 male, 25 female) and 17 mutant (8 male, 9 female) mice were tested in Pipeline 1 for open field behavior, prepulse inhibition (PPI) and stress restraint. Average age at the start of Pipeline 1 was 61 days (±2.3 days). 28 wildtype (14 male, 14 female), 28 heterozygote (14 male, 14 female) and 28 mutant (14 male, 14 female) mice were tested in Pipeline 2 in the light/dark assay, social interaction and Morris Water Maze tests. Average age at the start of Pipeline 2 was 64 days (±4.8 days). A separate cohort of 18 wildtype (8 male, 10 female), 37 heterozygote (17 male, 20 female) and 18 mutant (8 male, 10 female) mice were tested for intraperitoneal (IP) glucose tolerance. Average age of mice tested for glucose tolerance was 83 days (±2 days). Data from 3 animals are missing from restraint studies for at least one timepoint due to inability to collect sufficient plasma for assessing corticosterone.
2.4.1. General Behavioral Procedures
All mice were tested during the light part of the light/dark cycle between the hours of 8AM-12PM. On each testing day, mice were transported from the housing colony to the procedure room immediately prior to behavioral assessment. Mice were returned to the housing colony immediately after testing. There was at least one day off with no testing between each behavioral test with the exception of the social interaction and Morris Water Maze tests that were separated by 4 days of no testing. Mice were not handled on days when there were no scheduled behavioral tests. Behavioral chambers were cleaned after each test subject with a light bleach solution (0.25%) with the exception of the Morris Water Maze and tubes used for stress restraint. Stress restraint tubes were not reused during a test session.
2.4.2. Open Field
The open field arena (ENV-515-16, Med Associates, St. Albans, VT, USA), measured 17x17x13cm and consisted of a white Plexiglas floor and four clear Plexiglas walls. The walls are surrounded by infrared detection beams on the X, Y and Z axes used to detect horizontal and vertical activity of the animal throughout the duration of the testing session. The open field (OF) chamber was fitted with two overhead light fixtures containing 28-V lamps all within a sound attenuating box (73.5x59x59 cm). Light levels on the arena floor were 24 lux in the center, 10 lux in the corners and 13 lux along the walls. Each mouse was placed into the OF testing chamber and allowed to freely move about and explore for 30 minutes. Immediately upon completion of testing, mice were placed back into home cages. Distance moved (cm), ambulatory counts, time and episodes, time resting, vertical counts, vertical time, stereotypy time and counts, jump time and counts and average velocity (cm/s) were recorded over the 30-minute test session. Percent time spent and distance moved in the center of the testing chamber were recorded during the first 10 minutes of the assay as a measure of anxiety-like behavior. All behaviors were acquired using the manufacturers data acquisition software (Activity Monitor v5.9.725; Med Associates).
2.4.3. Startle Response and Prepulse Inhibition
Acoustic startle response and PPI were assessed using SR-LAB test stations (San Diego Instruments, San Diego, CA). Each startle chamber consisted of a clear Plexiglas cylinder (16cm long x 8.75cm diameter) mounted to a Plexiglas frame seated upon a piezoelectric transducer. The Plexiglas cylinders were contained within ventilated, sound attenuating, enclosures (with a small fan) and illuminated by a 15 W light bulb stationed in the ceiling. Light levels in the acoustic startle chambers averaged approximately 103 lux. Acoustic stimuli (white noise) were delivered through a small speaker mounted on the enclosure ceiling. A digital sound level meter was used to verify acoustic stimuli at the start of each test session. At the start of each test session, mice were weighed and placed into the startle chambers with the house light and fan turned on and continuous white noise (70dB) was presented through the overhead speaker. After a 5-minute acclimation period, the mice were tested for a total of 42 trials with random inter-trial intervals ranging from 10-15 seconds. There were 7 different trial types - no stimulus, a 40msec, 120-db acoustic stimulus alone, and five prepulse trials with a 20msec acoustic prepulse of 74, 78, 82, 86 and 90dB followed 100ms later by a 120-db acoustic stimulus. Each trial type was repeated in 6 different blocks and the order of trial types was randomized across each block although the same order was maintained across all mice. Startle response amplitudes for each trial were recorded over a 165ms period beginning at the onset of each startle stimulus and defined as the mean startle response across recording window. PPI was calculated as [100 − [response amplitude with prepulse stimulus/response amplitude with startle stimulus alone] * 100].
2.4.4. Stress Restraint
Mice were tested for CORT levels prior to and following a restraint stress under fluorescent laboratory lighting – 180-205 lux. Each mouse was removed from the home cage and whole blood was collected from the retro-orbital sinus using a micro-hematocrit capillary tube. Blood was collected from all mice in a cage within 1 minute to avoid increases in CORT due to anticipatory stress. Mice were immediately placed into a cylindrical Broome-Style restraint tube (Plas-Labs, Inc, Lansing, MI) for 10 minutes. Immediately following restraint, a second capillary tube of whole blood was collected from the alternate retro-orbital sinus and the mouse was returned to its home cage. Whole blood was centrifuged at 11,700 rpm for 13 minutes to isolate serum. Serum CORT levels (in ng/mL) were measured before and after restraint using a radioimmunoassay (RIA; MP Biomedicals, Santa Ana, CA) following the manufacturers standard protocol.
2.4.5. Light/Dark Assay
The light/dark apparatus consisted of a 42x42x30 cm open field arena (Versamax420 Animal Activity Monitoring System, AccuScan Instruments Inc., Columbus, OH, USA) with a white Plexiglas floor and clear Plexiglas walls. The arena was surrounded by 16 photobeams along each side that allowed for tracking of both horizontal and vertical activity. A black Plexiglas box (40x21x13 cm) occupied one-half of the arena and had a 10x3 cm opening to the light side and holes on all four sides that allowed detection of movement by the photobeams. Light levels in the light side of the apparatus were approximately 155 lux at floor level. Mice were tested following the procedures of Crawley and Goodwin [23]. Mice were placed in the light side of the arena facing the dark insert and allowed to freely explore for 10 minutes. The total time spent (s), distance traveled (cm) and entries into each side of the arena (light vs dark) and transitions between the two sides were recorded using the manufacturers data collection software (VersaMap, AccuScan Instruments, Inc). Percent time in the light chamber was calculated post hoc.
2.4.6. Social Interaction
Social interaction was tested in a rectangular, three-chambered apparatus fabricated with clear polycarbonate. The entire apparatus was 62.9cm W x 42.5cm D x 22.2cm H, divided into 3 17.8cm wide compartments separated by 4.4cm thick walls with retractable doorways. The light levels in the social arena were approximately 320-340 lux. Each test session included a 10-min habituation period followed by a 10-min social interaction test. During the habituation phase, mice were placed into the middle chamber with access to all 3 chambers. Time spent in all three chambers and transitions from the center to left and center to right chambers were recorded using EthoVision video tracking software (v7, Noldus). Once the habituation phase was completed, the test mouse was restricted to the central chamber while an unfamiliar, sex-matched C57BL/6J mouse was placed inside a wire containment cage (Galaxy Cup, Spectrum Diversified Designs Inc, Streetsboro, Ohio) in one of the two adjacent chambers, described hereafter as the “social-chamber”. The wire cage allowed for nose-to-nose contact but prevented direct full-body contact. An empty wire cage was placed in the opposing chamber, hereafter referred to as the “non-social chamber”. The doorways were reopened and the subject mouse was allowed to explore all three chambers with the stranger mouse present. The side housing the stranger mouse was counterbalanced across test subjects. The time spent in the social, non-social and central chambers as well as the 1-inch “social zone” surrounding the wire cage containing the stranger mouse was recorded using the EthoVision video tracking software. Entries into each chamber were also recorded as described above. Social interaction behavior was calculated as the percent change in the time spent in the social chamber during the social interaction test as compared to the habituation period. Social approach was calculated as time spent in the social zone as a percentage of total time spent in the social chamber.
2.4.7. Morris Water Maze
The Morris water maze was a 122cm diameter circular pool filled with water to a depth of 45cm (24-28°C). The water was made opaque with white, non-toxic, poster paint. Mice were tested under fluorescent laboratory lighting – 180-205 lux. The pool consisted of four quadrants (Q1-Q4) with visual cues located near each quadrant for visual association (see Supplemental Figure 3a). Mice were tested for their ability to locate an escape platform during two different trial types: 1) platform acquisition (training) trials and 2) a probe trial in the absence of the escape platform. During the platform acquisition testing, mice were trained over 4 consecutive days for 4 trials each day. For each trial, mice were randomly placed into one of the four quadrants. Each mouse was placed in each quadrant on each training day and the order of quadrant placement was balanced across genotypes. During each training trial, mice had to locate a visible platform that was just below the surface of the water in the target quadrant. Mice were given 60 seconds to find the platform. Mice that reached the platform in the 60 seconds were left on the platform for 10 seconds before the trial ended. Animals that did not locate the platform during the trial were placed onto the platform for 30 seconds to identify visual cues before being removed from the pool to begin the next trial. During the training period, the time it took for each mouse to find the platform (s) or escape latency over the course of each trial, distance to the escape platform or path length (cm) and velocity (cm/s) were recorded. For each training day, a mean value for all 4 trials was calculated for each mouse and each measure (Supplemental Table 2). On the fifth day, mice were tested during a 60-second probe trial. In the probe trial, the platform was removed from the pool and each animal was placed into Quadrant 3 and allowed to explore the maze for 60 seconds. During the probe trial, the time spent in each quadrant and velocity (in cm/s) was recorded. All measures were recorded using an automated tracking system (Ethovision v7, Noldus Information Technology, Netherlands).
2.4.8. Glucose Tolerance
The ability to regulate blood glucose was measured using an IP glucose tolerance test. Mice were fasted for at least 3 hours prior to testing and weighed at the start of each test session. At the start of the test session, 2 mm of the tail tip was removed using a scalpel blade. The first drop of blood was discarded and the second drop was placed onto a OneTouch Ultra test strip (LifeScan, Inc.) that was inserted into a OneTouch UltraMini Blood Glucose Monitoring System (LifeScan, Inc.) glucose meter to record baseline glucose levels in mg/dL. Additional blood samples were collected from the tail at 15, 30, 60, 90, and 120 minutes following an IP injection of D-(+)-glucose solution (45%, Sigma Life Science) in a volume of 0.01ml/g of body weight and additional blood glucose levels were obtained as described above.
2.5. Statistical Analyses
Data from behavioral tests from each pipeline was analyzed using the SPSS statistical software for Macintosh (v 24). Data were first assessed for normality using the Shapiro-Wilk test in SPSS. Data that were not normally distributed (p<0.05) were transformed using a rank-based inverse normal transformation using the original group mean and standard deviation. Normally distributed data were analyzed by ANOVA with genotype, sex and test-specific parameters (as described below) as independent variables. Weight was included as a covariate for assessment of acoustic startle response. Significant genotype differences were analyzed post hoc with Tukeys HSD. Repeated measures ANOVA was conducted for PPI, glucose tolerance and the Morris Water Maze where the same measures were collected across several trials for each animal. Behaviors that used repeated measures for analysis were first assessed for sphericity using Mauchly’s Test of Sphericity. If Mauchly’s test yielded a p>0.05, sphericity was assumed. If the p-value for Mauchly’s test was <0.05, the data violated the assumption of sphericity and Greenhouse-Geisser (Greenhouse-Geisser Epsilon <0.75) or Huynh-Feldt (Greenhouse-Geisser Epsilon >0.75) corrections were used. Significant between subject effects were followed up with post hoc Tukeys HSD analysis to identify differences between groups.
3.0. Results
3.1. Identification of causal mutation
Initial analysis of genotype data from 21 affected and 19 unaffected N2 mice from a backcross of affected DDY animals with B6 identified a single, 5.3 Mb region on Chromosome 3 bounded by markers rs46827828 (132,031,621 bp) and rs31236589 (137,317,747 bp). All mice that were DDY homozygotes in this region exhibited the chirping phenotype. Additional genotyping with a panel of 14 SSLP and SNP markers (Supplemental Table 1) narrowed the mapped locus to a 498Kb critical region bounded by markers rs37841890 (135,234,644 bp) and rs30309434 (135,732,112 bp) (Fig 1a). Targeted exome sequencing of this region and sequence analysis identified a deletion of a single guanine nucleotide in exon 2 of the Cisd2 gene at 135,411,194 bp. The deletion was present in all 14 reads from the mutant and 0 out of 31 reads from the inbred DDY reference sequence. No other sequence differences were observed. The B6.DDY-Cisd2m1Lmt deletion is predicted to cause a frameshift and introduce a premature termination codon. Translation of the altered transcript would lead to a truncated CISD2 protein with altered C-terminal protein sequence (Fig 1b). Analysis of the altered C-terminal protein sequence using Protein-BLAST (blast.ncbi.nlm.nih.gov) identified no significant homology with any other Mus musculus protein family.
Figure 1. Identification of causal mutation.

a. Fine mapping with a panel of SNP markers in 63 affected mice identified 7 key recombinants that narrowed the critical region to 498kb bounded by rs37841890 and rs30309434. b. Exome sequencing identified the deletion of a single guanine nucleotide in exon 2 of Cisd2 in B6.DDY-Cisd2m1Lmt mice. The single base deletion is predicted to result in a frameshift that introduces a string of aberrant amino acids and a premature stop codon. c. Cisd2 mRNA levels were lower in brain tissue from mutant (MUT) compared to wildtype (WT) mice but the difference was not significant.
3.2. Assessment of Gene Expression by Quantitative PCR
RT-qPCR was performed to assess Cisd2 gene expression in whole brain tissue from 3 wildtype and 3 mutant mice. Although lower gene expression was observed in the B6.DDY-Cisd2m1Lmt mutants (Fig 1c), the difference was not significant. (t (4) = −2.5; p=0.064).
3.3. Physical characteristics of B6.DDY-Cisd2m1Lmt mice.
Our original observation and identification of the B6.DDY-Cisd2m1Lmt was based on audible “chirping” vocalizations. We also noted that mutant mice appeared smaller than heterozygous and wildtype littermates. Analysis of body weight data at weaning confirmed that B6.DDY-Cisd2m1Lmt weighed significantly less than wildtype mice at weaning (approximately 20 days of age; p = 0.03; Supplemental Table 2). No sex differences in weight were observed at weaning. Mutant mice also weighed significantly less than both wildtype (p = 5.1x10−9) and heterozygous (p = 5.1x10−9) littermates at the start of behavioral testing (61 days of age ± 2.3 days). Male mice, regardless of genotype, had significantly higher body weight compared to female mice (p = 3.9x10−44). We also observed a significant genotype x sex interaction. Mutant male mice weighed significantly less than both heterozygous (p = 5.1x10−9) and wildtype (p = 8.0x10−9) littermates, whereas females mutants weighed significantly less than wildtype (p =0.001) but not heterozygous littermates.
3.4. Behavioral Testing
Mean values and standard deviations for all behavioral variables separated by genotype and sex are shown in Supplemental Table 2.
Behavioral Pipeline 1
3.5. Open Field
Mice were tested in the open field to assess locomotor activity, exploratory and anxiety-related behavior. Behavioral measures collected in the open field were highly correlated, so we employed a data reduction strategy to reduce the number of variables for analysis. A factor analysis using varimax rotation was conducted on all open field measures recorded during the entire 30-minute session. Behaviors loaded onto two components with Eigenvalues >1 that collectively explained 81% of the variance. Component one (Eigenvalue = 7.9) explained 66% of the variance and included locomotor and exploratory behaviors including distance, ambulatory time, counts and episodes, stereotypy time and counts, vertical time and counts and time resting. The second component (Eigenvalue = 1.9) explained 15% of the variance and included average velocity, jump counts and time. Regression scores from the factor analysis were saved and analyzed by ANOVA to examine the effects of genotype and sex. We observed significant genotype (F(2,82) = 5.4;p = 0.006), sex (F(1,82) = 5.0;p = 0.028) and genotype x sex interaction (F(2,82) = 3.4;p = 0.37) effects for the first principal component. Male mutants have significantly lower scores (were less active) than both wildtype and heterozygous littermates (Fig 2a). Females exhibited no significant differences regardless of genotype. For the second principal component, males had significantly lower scores than females (F(1,82) = 55.0;p = 1.3x10−10) but there were no genotype (F(2,82) = 1.0;p = 0.36) or genotype x sex effects (F(1,82) = 1.6; p=0.21) (Fig 2b).
Figure 2. Locomotor activity in the open field.
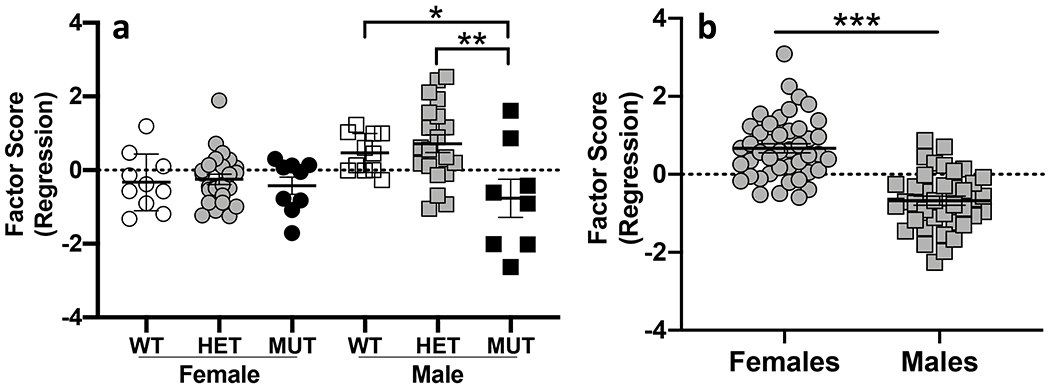
a. B6.DDY-Cisd2m1Lmt (MUT) males were less active in the open field compared to both heterozygous (HET) and wildtype (WT) males. b. Male mice had significantly lower factor scores compared to female mice for the second principal component. Each data point represents an individual mouse. Error bars are standard error of the mean. * p < 0.05, ** p < 0.01, *** p < 0.001.
In order to assess anxiety-related behavior, we also examined time spent and locomotor activity in the center of the arena during the first 10-min of open field testing before mice habituated to the novel environment. We observed no significant genotype or sex differences for either of these behaviors (data not shown).
3.6. Startle Response and Prepulse Inhibition
There were no significant genotype, sex or sex x genotype interaction effects on acoustic startle response. Sensorimotor gating was assessed by examining PPI of the startle response. Repeated measures ANOVA identified a significant effect of prepulse decibel level on prepulse inhibition (F(2.7, 207.6) = 243.1; p = 3.1 x 10−64). The amplitude of the startle response decreased as the prepulse decibel level increased (Fig 3a) but there was no significant genotype, sex or genotype x sex interaction on prepulse inhibition at any prepulse level. There was a significant main effect of genotype (F(2,77) = 7.2; p = 0.001) on prepulse inhibition of the startle response. Wildtype (p = 0.007) and heterozygous (p=0.001) mice exhibited greater PPI than B6.DDY-Cisd2m1Lmt mice across all prepulse levels (Fig 3b).
Figure 3. Startle response and prepulse inhibition.
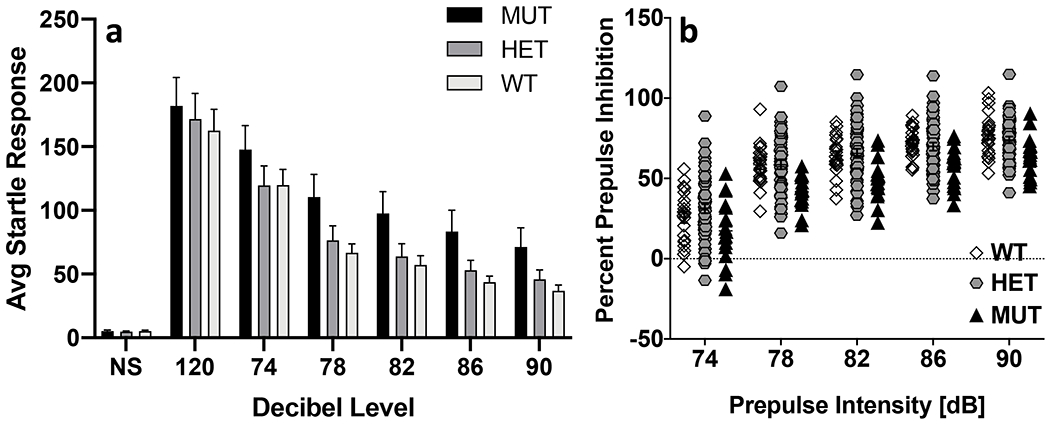
a. Startle response decreased as the prepulse decibel level increased in all groups (NS- no stimulus). b. B6.DDY-Cisd2m1Lmt mutant mice (MUT) exhibited significantly lower PPI compared to both wildtype (WT) and heterozygous (HET) littermates across all prepulse levels. Each data point represents an individual mouse. Error bars are standard error of the mean.
3.7. Stress Restraint
Basal stress and stress responsivity were assessed by measuring serum CORT levels prior to and following a 10-minute restraint. An ANOVA yielded a significant effect of time (F(1,149)=643.2; p=6.3x10−56), genotype (F(2,149)=7.9; p=5.7x10−4) and sex (F(1,149)=12.6; p=5.1x10−4). We also observed a significant time x sex interaction (F(1,149)=7.0; p=0.009) but no genotype x sex interaction (F(2,160)=1.7; p=0.191). CORT levels increased significantly at ten minutes relative to baseline regardless of genotype or sex (Fig 4a). B6.DDY-Cisd2m1Lmt mutants had significantly higher CORT levels than either wildtype (p=1.0x10−4) or heterozygous (p=0.013) littermates (Fig 4a). Post-restraint CORT levels differed significantly between males and females (p=1.1 x 10−5) but basal levels did not differ significantly between sexes (p=0.367) (Fig 4b).
Figure 4. Basal stress and stress reactivity.
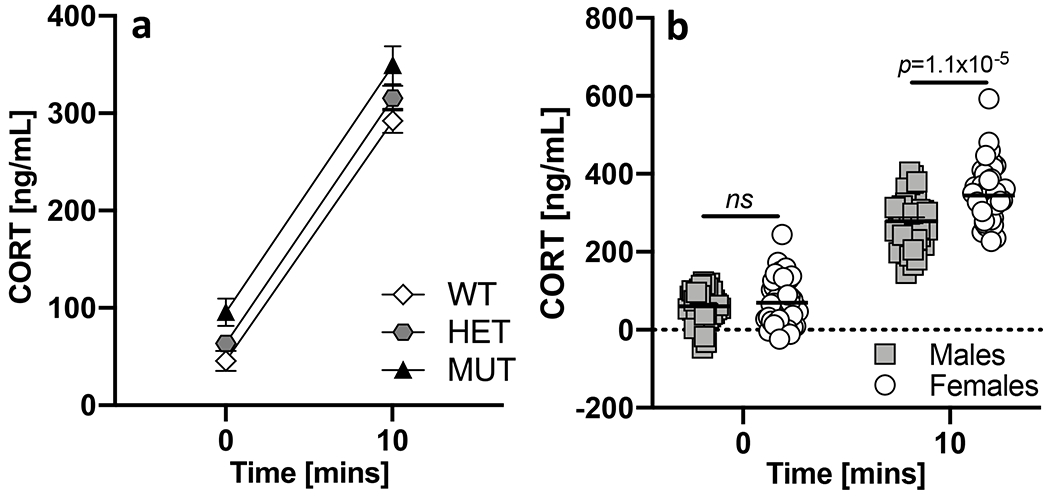
a. CORT levels increased significantly after a 10-minute restraint regardless of sex or genotype. B6.DDY-Cisd2m1Lmt mutant mice (MUT) had significantly higher CORT than either wildtype (WT) or heterozygous (HET) littermates. b. Pre-restraint CORT levels were not significantly different between male and female mice. Female mice had significantly greater post- restraint CORT levels compared to male mice. Each data point represents an individual mouse. Error bars are standard error of the mean.
Behavioral Pipeline 2
3.8. Light/Dark Assay
Entries into, time spent and total transitions between the light and dark chambers and total distance in the light/dark arena were measured to assess anxiety-related behavior and locomotor activity (Supp Fig 1). We observed significant genotype differences for percent time spent in the light chamber (Fig 5a; F(2,78) = 3.8; p = 0.026). Mutant mice spent significantly more time in the light chamber compared to wildtype (p = 0.031) but not heterozygous littermates (p = 0.438). We also observed significant genotype differences for distance moved in the entire L/D arena (F(2,78) = 4.1; p = 0.021) and the dark chamber (F(2,78) = 4.9; p=0.010) and for number of transitions between light and dark chambers (F(2,78) = 5.0; p= 0.009). Mutant B6.DDY-Cisd2m1Lmt mice move significantly less than heterozygous (p=0.034) and wildtype mice (p=0.015) in the dark chamber (Fig 5b) and significantly less than heterozygotes (p=0.021) in the entire arena. Mutant mice also made significantly fewer transitions than heterozygotes (p=0.006). We also observed sex effects for total distance (F(1,78) = 21.0; p = 1.7x10−5;) and distance in both the light (F(1,78) = 4.6; p = 0.035) and dark (F(1,78) = 36.5; p = 4.9x10−8) compartments. Female mice exhibited higher locomotor activity for all three measures compared to males. Female mice also made more transitions between the light and dark compartments (F(1,78) = 13.9; p = 3.6x10−4). We did not observe any significant sex x genotype interactions for any phenotype measured in the light/dark assay.
Figure 5. Locomotor activity in the light/dark assay.
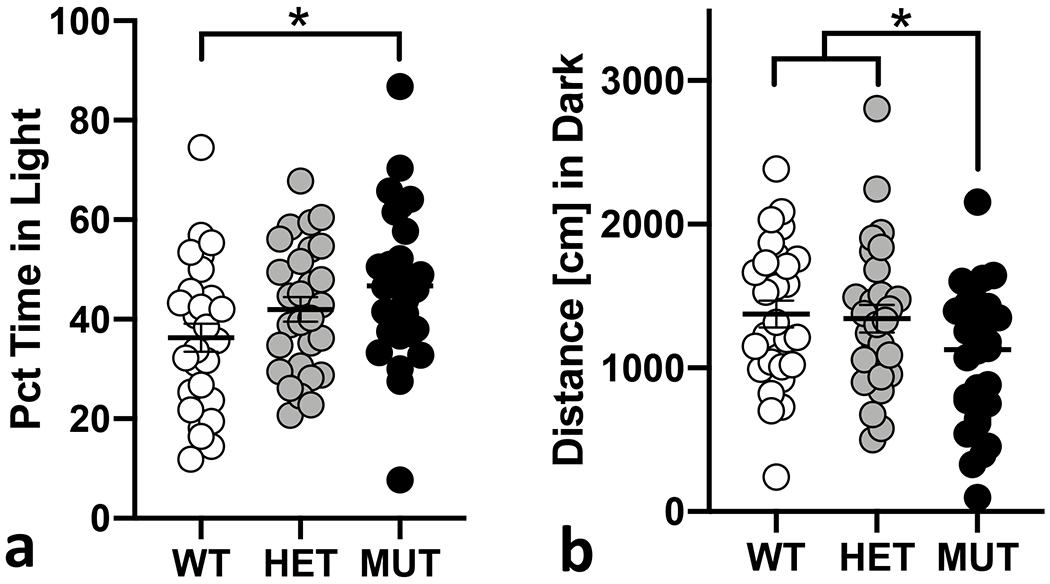
a. Mutant mice (MUT) spent significantly more time in the light chamber compared to wildtype (WT) littermates. b. MUT mice moved significantly less compared to both WT and heterozygous (HET) littermates. Each data point represents an individual mouse. Error bars are standard error of the mean. *p< 0.05, ** p< 0.01, *** p< 0.001.
3.9. Social Interaction
Social interaction behavior was defined as the percent change in the time spent in the social chamber during the social interaction test as compared to the habituation period (Supp Fig 2). We observed no significant effect of genotype on the percent change in time spent in the social chamber. Females spent significantly less time in the social chamber than males (F(1,77) = 5.6; p = 0.021). We observed no significant genotype or sex effect on social approach behavior as measured by percent time spent in the social zone. We did observe a significant effect of both genotype and sex but no genotype x sex interaction on the total number of transitions between the chambers in the social interaction arena during both the habituation (genotype, F(2,77) = 9.0; p = 3.0 x 10−4; sex, F(1,77) = 12.7; p = 6.3 x 10−4) and social interaction sessions (genotype, F(2,82) = 7.3; p = 1.3 x 10−3; sex, F(1,82) = 9.2; p = 0.003). In both cases, mutant mice made significantly fewer transitions than heterozygous or wildtype littermates (all p < 0.01) and males made fewer transitions than females (all p < 0.01). We also assessed transitions to the social chamber vs non-social chamber. Mice made significantly more transitions to the social chamber (F(1,165) = 7.0; p = 0.009) regardless of genotype, but we observed no significant genotype by chamber interaction.
3.10. Morris Water Maze
Spatial learning and memory were measured in the Morris Water Maze (Supp Fig 3). We observed a significant decrease in escape latency (F(2.96,231.2) = 48.1; p=5.66 x 10−24), path length to platform (F(2.99,233.1) = 52.3; p = 8.5 x 10−26) and velocity (F(2.86,222.7) = 8.53; p=3.1 x 10−5) across the 4 training days.
We observed a significant effect of genotype on escape latency (F(2,78) = 13.0; p= 1.4 x 10−5), path length (F(2,78) = 4.8; p = 0.011) and velocity (F (2,78) = 4.8; p = 0.01) during training trials. B6.DDY-Cisd2m1Lmt mutant mice took significantly more time to reach the platform than wildtype (p = 1.9 x 10−5) and heterozygous (p = 0.001) littermates (Fig 6a) and also traveled a greater distance to reach the platform compared to wildtype mice (p = 0.01) and heterozygotes, although that difference was not significant (p = 0.09; Fig 6b). Mutant mice also moved at a slower speed compared to both wildtype (p = 0.017) and heterozygous (p = 0.034) littermates (Fig 6c). Females traveled greater path lengths to reach the platform (F(1,78) = 8.9; p=0.004; Supp Fig 3b) and at significantly higher velocity (F(1,78) = 11.4; p=0.001) than males (Supp. Fig 3c).
Figure 6. Escape latency and path length in the Morris water maze.
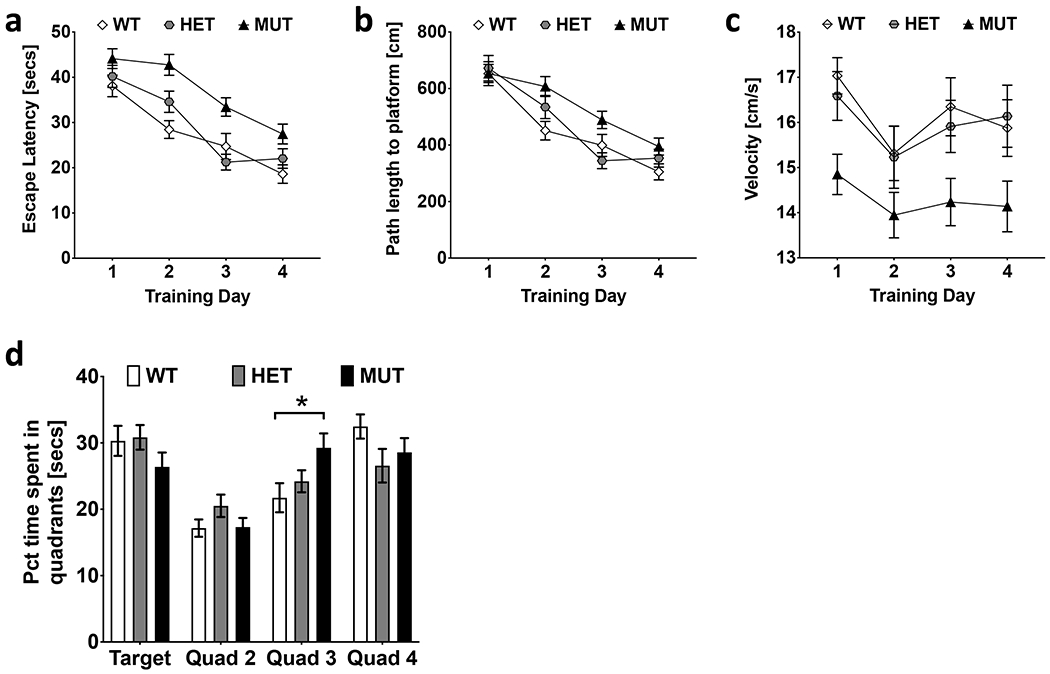
a. B6.DDY-Cisd2m1Lmt mutant mice (MUT) had a greater escape latency compared to both wildtype (WT) and heterozygous (HET) littermates across all training days. b. MUT mice traveled a significantly greater path length to reach the platform compared to WT mice. c. The swim speed for MUT mice was significantly slower compared to both WT and HET mice. d. MUT mice spent significantly more time in quadrant 3 compared to WT mice on probe day. Error bars shown are standard error mean *p < 0.05, **p < 0.01, ***p < 0.001.
Time spent in each quadrant of the Morris Water Maze was measured during the probe trial. We observed a significant difference in percent time spent in each quadrant (F(3,312) = 20.5; p = 3.6 x 10−12) and a significant interaction of percent time in quadrant x genotype (F(6,312) = 2.89; p = 0.009). All mice, regardless of genotype, spent significantly less time in quadrant 2 compared to all other quadrants (all p<0.001). Mutant mice spent significantly more time in quadrant 3 compared to wildtype mice (p=0.030). Average time spent in the target quadrant (quadrant 1) was not significantly higher than time spent in any other quadrant across all genotypes (Fig 6d). No significant sex differences or interactions were observed.
3.11. Glucose Tolerance
The ability to regulate blood glucose was measured using an IP glucose tolerance test. Blood glucose levels were monitored at 5 time points following an IP injection of 45% glucose solution. Repeated measures ANOVA revealed a significant effect of time (F(4.4,295.0) = 248.9; p = 1.3 x 10−97) on blood glucose levels. Blood glucose levels increased in the first 15 minutes following glucose administration and started decreasing after 30 minutes in all mice. We also observed a significant interaction of time x genotype x sex (F(8.8,295.0) = 2.4; p = 0.012). The time course of glucose levels differed across genotype. Female B6.DDY-Cisd2m1Lmt mutants had lower glucose than wildtype females 15- and 30-min post glucose exposure. The same difference was not observed in males (Fig 7a, b).
Figure 7. Blood glucose regulation.
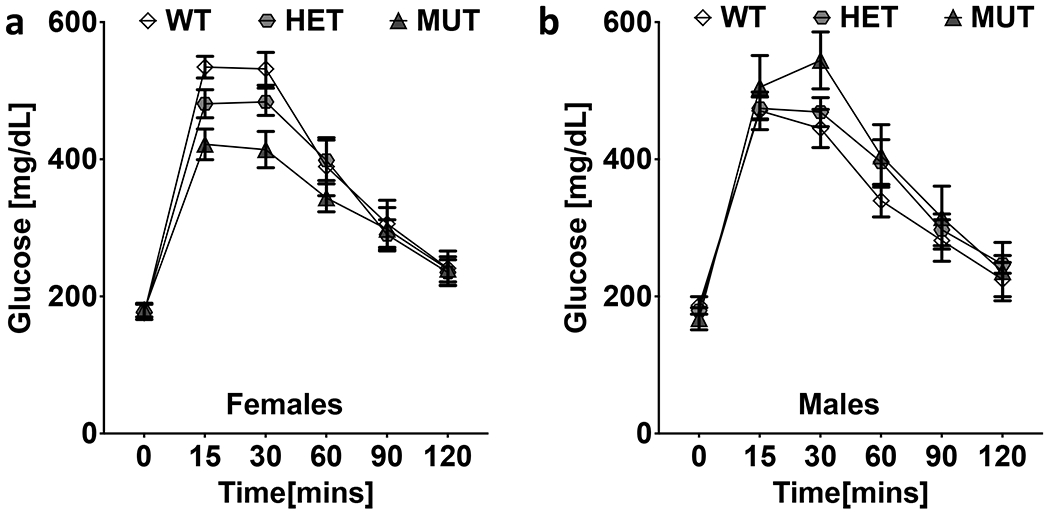
a. Female mutant (MUT) mice had lower glucose levels than wildtype females at 15- and 30-min post glucose exposure. b. Glucose levels in male mice did not differ significantly by genotype.
4.0. Discussion
Wolfram syndrome (WFS) is a progressive, debilitating disease that significantly decreases lifespan and overall quality of life. Along with the primary diagnostic criteria, WFS patients also suffer from significant neurological and psychiatric symptoms, but the underlying causes remain largely unknown. Studying behavioral outcomes of gene dysfunction in animal models can provide insights into the mechanisms that increase risk for neurological symptoms in WFS patients. While WFS1 is more common, understanding the biology underlying both WFS1 and WFS2 will be informative for developing novel treatment and intervention strategies.
We present here a series of behavioral studies conducted in mice carrying a spontaneous mutation in Cisd2, the gene implicated in WFS2. A single base pair deletion in Cisd2 in B6.DDY-Cisd2m1Lmt mice is predicted to result in a frameshift that alters the amino acid sequence at the C-terminus and introduces a premature termination codon. Our quantitative PCR analyses suggest that B6.DDY-Cisd2m1Lmt mutant mice show an overall reduction of Cisd2 expression in the brain although the difference did not reach statistical significance. The basis for reduced Cisd2 expression in B6.DDY-Cisd2m1Lmt mice is unknown. It is possible that the mutant transcript is targeted for nonsense mediated decay (NMD). Although we cannot definitively rule out this possibility, we do not think NMD represents the most likely mechanism. Only mRNAs with premature termination codons located 50-55 nucleotides upstream of the final exon-exon junction are efficiently degraded by NMD [24]. The B6.DDY-Cisd2m1Lmt premature termination codon is located downstream of this boundary. Additional experiments would be necessary to identify the mechanism(s) that alter gene expression in B6.DDY-Cisd2m1Lmt mice. Proteomic studies are also warranted to investigate the effects of the mutation on protein production.
Numerous studies have been published describing physical, physiological, neurological and behavioral phenotypes in Wfs1 knockout mice. Studies examining WFS symptoms in Cisd2 knockout mice have also been published but there are currently no publications describing the effects of loss of Cisd2 function on behavior with the exception of locomotor activity. The present study was designed to perform initial behavioral characterization of B6.DDY-Cisd2m1Lmt mice. We selected a battery of behavioral assays that are relevant to neurological and psychiatric disorders. Specifically, we chose assays that overlap with those that have been assessed in Wfs1 knockout mice since both WFS1 and WFS2 subtypes have a similar clinical profile. Our results indicate that B6.DDY-Cisd2m1Lmt mice are hypoactive, have higher levels of CORT and have deficits in learning and memory but show normal glucose tolerance.
The most consistent phenotype we observed in B6.DDY-Cisd2m1Lmt mutant mice was reduced locomotor activity. Hypoactivity was observed across multiple behavioral assays that depend upon locomotor activity including the open field, light/dark and social interaction tests. In the open field, hypolocomotion was only observed in mutant males (Fig 2a). Male-specific hypoactivity in the open field has also been observed in Cisd2 knockout mice (International Mouse Phenotyping Consortium; J:165965; www.mousephenotype.org). We did not, however, identify sex-specific genotype effects in locomotor behavior in any other behavioral assay. Deficits in mitochondrial integrity may contribute to the locomotor phenotype observed in Cisd2 knockout mice and B6.DDY-Cisd2m1Lmt mice. Cisd2 knockout mice have mitochondrial abnormalities [17, 25] and it has been suggested that mitochondrial dysfunction affects locomotor behavior [26]. Additional studies to examine mitochondrial function in B6.DDY-Cisd2m1Lmt mice are warranted. The hypolocomotion observed in B6.DDY-Cisd2m1Lmt mutant mice might also result from exposure to elevated CORT levels. B6.DDY-Cisd2m1Lmt mice displayed significantly higher CORT levels compared to both wildtype and heterozygous littermates. Previous studies have shown that chronic exposure to CORT decreases locomotor activity in laboratory mice [27, 28]. Several studies have also linked increased CORT with social deficits [29, 30]. Decreased social interaction has been reported in Wfs1 knockouts [16]. Although male B6.DDY-Cisd2m1Lmt mutant and heterozygotes spent less time in the social chamber, the difference was not significant. (Supp Fig 2a).
Although behavioral studies of Cisd2 knockout mice have been limited to locomotor assessment, Wfs1 knockout mice have been characterized for numerous behavioral measures including several that we assessed in B6.DDY-Cisd2m1Lmt mice. In the Morris Water Maze, B6.DDY-Cisd2m1Lmt mice had longer escape latencies and traveled further to reach the platform across training days compared to wildtype and heterozygous littermates. Kato et. al also found that Wfs1 knockout mice had longer escape latencies compared to wildtype controls in the Morris Water Maze, suggesting abnormal spatial learning [16]. However, based on their observation that distance traveled did not differ between KO and wildtype mice, Kato et. al hypothesized that differences in escape latency might be due to reduced locomotor activity in Wfs1 KO mice. Although B6.DDY-Cisd2m1Lmt mice also display general hypoactivity and significantly lower velocity in the Morris Water Maze compared to wildtype control mice, we do not think that hypoactivity is the primary cause of genotype differences in escape latency. We draw this conclusion based on our observation that B6.DDY-Cisd2m1Lmt mutant mice travel a greater distance to reach the platform compared to wildtype littermates. Similar to Kato et. al, control mice in our experiments did not spend significantly more time in the target quadrant compared to the three other quadrants during the probe test so spatial memory was not assessed.
B6.DDY-Cisd2m1Lmt mice exhibited significantly less PPI compared to both wildtype and heterozygote littermates. To our knowledge, this is the first study that identifies PPI deficits in a mouse model of WFS. Deficits in PPI are a hallmark of schizophrenia which is one of several psychiatric disorders observed in the WFS patients [31, 32]. Kato et al. also examined acoustic startle and PPI in Wfs1 knockout mice at 12- and 31- weeks of age and observed no significant effect of genotype. We note here that behavioral differences between Wfs1 knockout and B6.DDY-Cisd2m1Lmt mice could be due to the different genetic backgrounds used to generate the models. The Wfs1 knockout mice used by Kato et al were generated on a 129Sv background while B6.DDY-Cisd2m1Lmt mice are on a B6.DDY background. Further exploration of PPI in WFS models would add considerably to the literature based on the relevance of this phenotype in schizophrenia.
Perhaps the most striking characteristic of B6.DDY-Cisd2m1Lmt mice is their audible “chirping” vocalizations. Similar vocalizations have previously been reported in Wfs1, but not Cisd2 knockouts [12]. In Wfs1 knockout mice, stressful situations increase vocalizations and administration of diazepam blocks these vocalizations suggesting that this behavior may reflect the anxiety-state of the mice. Anecdotally, B6.DDY-Cisd2m1Lmt exhibit these vocalizations in the home cage and we have not observed an increase in vocalizations upon handling or during testing. Another potential explanation is that the use of sonic vocalizations arises due to hearing deficits in the mice. Sensorineural hearing impairment is a cardinal symptom of WFS, although to our knowledge no one has yet reported hearing loss phenotypes in either Wfs1 or Cisd2 knockout mice. If B6.DDY-Cisd2m1Lmt mice do experience hearing loss, they may be unable to develop normal ultrasonic communication in the absence of normal auditory cues. We don’t believe this is likely since others have argued that auditory feedback is not essential for the development of ultrasonic vocalization in mice [33] based on observations that deaf mice develop normal ultrasonic vocalizations [34, 35]. Based on the reduction in startle response observed during PPI testing (Fig 3a), B6.DDY-Cisd2m1Lmt are able to detect white noise bursts as low as 74 dB suggesting that they do not experience complete hearing loss by early adulthood. Future studies that assess hearing loss over a range of frequencies and at different ages are necessary as are precise measurements of both sonic and ultrasonic vocalizations in B6.DDY-Cisd2m1Lmt mice and other WFS mouse models.
B6.DDY-Cisd2m1Lmt mice did not have impaired glucose tolerance, a landmark symptom of WFS that has been observed in both Wfs1 and Cisd2 knockout mice [11] [12–15, 17]. This disparity could be due to the difference in genetic backgrounds on which these models were generated, the age at which the B6.DDY-Cisd2m1Lmt mice were tested or some combination of both. We tested glucose tolerance in B6.DDY-Cisd2m1Lmt mice at 11-12 weeks of age. Previous studies have reported impaired glucose tolerance in Wfs1 and Cisd2 mice at a similar age although one study found that Wfs1 mice exhibited normal glucose tolerance at 8 weeks, mild impairment at 12 weeks and more severe impairment at 16 and 24 weeks [14]. B6.DDY-Cisd2m1Lmt mice used in these studies had been backcrossed to B6 for seven generations and still have residual DDY/JclSidSeyFrkJ background that might delay the development of glucose intolerance. It is also possible that the Cisd2 allele carried by B6.DDY-Cisd2m1Lmt mice generates a truncated version of the protein that retains some functional activity. Testing B6.DDY-Cisd2m1Lmt mice at later ages will be necessary to determine if these mice go on to develop impaired glucose tolerance. Additional studies aimed at understanding the potential role of the strain background on glucose tolerance in these mice can also provide insight into genetic and biological mechanisms that contribute to this phenotype in human WFS2 patients.
Our work primarily addresses behavioral phenotypes in B6.DDY-Cisd2m1Lmt mice, but there remain other interesting avenues for future study. Cisd2 encodes the endoplasmic reticulum intermembrane small protein, ERIS, which localizes to the mitochondrial outer membrane, endoplasmic reticulum and the mitochondria associated endoplasmic reticulum membranes [3, 36]. Cisd2 knockout mice exhibit mitochondrial dysfunction with ongoing mitochondrial breakdown and highlight the interconnectivity between Cisd2 functionality, mitochondrial homeostasis and mitochondrial integrity [37]. The reduction in mitochondrial integrity and function can lead to both nerve and muscle degeneration, which may contribute to the hypoactivity observed in Cisd2 knockout and B6.DDY-Cisd2m1Lmt mice [25]. Numerous studies have also linked mitochondrial dysfunction and the progression of psychiatric pathology including schizophrenia, bipolar disorder, Parkinson’s, Alzheimer’s and major depressive disorder [38]. This leads to the hypothesis that dysfunction caused by mutations in human CISD2 might also contribute to increased risk for psychiatric illness [2, 17]. Assessment of mitochondrial function in B6.DDY-Cisd2m1Lmt will be critical in delineating the role that loss of mitochondrial integrity may play in this new WFS2 animal model.
We have characterized B6.DDY-Cisd2m1Lmt mice in a battery of behavioral tests. These mice displayed sonic vocalizations, hypoactivity, decreased social interactions and deficits in learning and memory and prepulse inhibition. Our B6.DDY-Cisd2m1Lmt mice did not exhibit one of the key features of WFS2, impaired glucose tolerance. As we stated previously, phenotype differences between B6.DDY-Cisd2m1Lmt mice and other Wolfram Syndrome models could be due to background genetic effects [39]. Previous evidence supports an effect of genetic background on the diabetogenic effects of Wfs1 deficiency [11]. Future studies will be pivotal for characterizing behavioral and non-behavioral WFS symptoms in this novel WFS2 model and determining the extent to which the observed phenotypes are influenced by mitochondrial dysfunction, genetic background and age.
It is interesting to note that heterozygote behavior was comparable to the wildtype in most of our assays. Increased incidence of psychiatric illness has been reported among first-degree relatives of WFS patients. Our data in mice indicate that carriers of the B6.DDY-Cisd2m1Lmt mutation do not exhibit altered behavioral profiles. However, our studies are limited to a specific genetic alteration, inbred strain background and behavioral panel. Regardless, B6.DDY-Cisd2m1Lmt mice are a unique model with which to study the genetic and biological mechanisms that contribute to the symptomatology of WFS2. The knowledge gained from these mutants will be useful for developing treatment strategies for improving quality of life and possibly extending the lifespan of WFS patients.
Supplementary Material
Acknowledgements:
This work was supported by funding from the Foundation of Hope (walkforhope.com) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (U54 HD079124).
References
- 1.Khanim F, et al. , WFS1/wolframin mutations, Wolfram syndrome, and associated diseases. Hum Mutat, 2001. 17(5): p. 357–67. [DOI] [PubMed] [Google Scholar]
- 2.Barrett TG and Bundey SE, Wolfram (DIDMOAD) syndrome. J Med Genet, 1997. 34(10): p. 838–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amr S, et al. , A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am J Hum Genet, 2007. 81(4): p. 673–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rigoli L and Di Bella C, Wolfram syndrome 1 and Wolfram syndrome 2. Curr Opin Pediatr, 2012. 24(4): p. 512–7. [DOI] [PubMed] [Google Scholar]
- 5.Swift RG, Sadler DB, and Swift M, Psychiatric findings in Wolfram syndrome homozygotes. Lancet, 1990. 336(8716): p. 667–9. [DOI] [PubMed] [Google Scholar]
- 6.Swift RG, et al. , Predisposition of Wolfram syndrome heterozygotes to psychiatric illness. Mol Psychiatry, 1998. 3(1): p. 86–91. [DOI] [PubMed] [Google Scholar]
- 7.Kinsley BT, et al. , Morbidity and mortality in the Wolfram syndrome. Diabetes Care, 1995. 18(12): p. 1566–70. [DOI] [PubMed] [Google Scholar]
- 8.Chaussenot A, et al. , Neurologic features and genotype-phenotype correlation in Wolfram syndrome. Ann Neurol, 2011. 69(3): p. 501–8. [DOI] [PubMed] [Google Scholar]
- 9.Rondinelli M, et al. , Wolfram syndrome 2: a novel CISD2 mutation identified in Italian siblings. Acta Diabetol, 2015. 52(1): p. 175–8. [DOI] [PubMed] [Google Scholar]
- 10.El-Shanti H, et al. , Homozygosity mapping identifies an additional locus for Wolfram syndrome on chromosome 4q. Am J Hum Genet, 2000. 66(4): p. 1229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishihara H, et al. , Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet, 2004. 13(11): p. 1159–70. [DOI] [PubMed] [Google Scholar]
- 12.Luuk H, et al. , Wfs1-deficient mice display impaired behavioural adaptation in stressful environment. Behav Brain Res, 2009. 198(2): p. 334–45. [DOI] [PubMed] [Google Scholar]
- 13.Koks S, et al. , Wfs1 gene deletion causes growth retardation in mice and interferes with the growth hormone pathway. Physiol Genomics, 2009. 37(3): p. 249–59. [DOI] [PubMed] [Google Scholar]
- 14.Riggs AC, et al. , Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia, 2005. 48(11): p. 2313–21. [DOI] [PubMed] [Google Scholar]
- 15.Noormets K, et al. , Sex differences in the development of diabetes in mice with deleted wolframin (Wfs1) gene. Exp Clin Endocrinol Diabetes, 2011. 119(5): p. 271–5. [DOI] [PubMed] [Google Scholar]
- 16.Kato T, et al. , Behavioral and gene expression analyses of Wfs1 knockout mice as a possible animal model of mood disorder. Neurosci Res, 2008. 61(2): p. 143–58. [DOI] [PubMed] [Google Scholar]
- 17.Chen YF, et al. , Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev, 2009. 23(10): p. 1183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morgan H, et al. , EuroPhenome: a repository for high-throughput mouse phenotyping data. Nucleic Acids Res, 2010. 38(Database issue): p. D577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan AP, et al. , The Mouse Universal Genotyping Array: From Substrains to Subspecies. G3 (Bethesda), 2015. 6(2): p. 263–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tarantino LM, et al. , Confirmation of quantitative trait loci for alcohol preference in mice. Alcohol Clin Exp Res, 1998. 22(5): p. 1099–105. [PubMed] [Google Scholar]
- 21.Pletcher MT, et al. , Use of a dense single nucleotide polymorphism map for in silico mapping in the mouse. PLoS Biol, 2004. 2(12): p. e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ and Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 2001. 25(4): p. 402–8. [DOI] [PubMed] [Google Scholar]
- 23.Crawley J and Goodwin FK, Preliminary report of a simple animal behavior model for the anxiolytic effects of benzodiazepines. Pharmacol Biochem Behav, 1980. 13(2): p. 167–70. [DOI] [PubMed] [Google Scholar]
- 24.Hug N, Longman D, and Caceres JF, Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res, 2016. 44(4): p. 1483–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanki T and Klionsky DJ, Mitochondrial abnormalities drive cell death in Wolfram syndrome 2. Cell Res, 2009. 19(8): p. 922–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Z, et al. , Improvement of cognitive and motor performance with mitotherapy in aged mice. Int J Biol Sci, 2020. 16(5): p. 849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singleton JM and Garland T Jr., Influence of corticosterone on growth, home-cage activity, wheel running, and aerobic capacity in house mice selectively bred for high voluntary wheel-running behavior. Physiol Behav, 2019. 198: p. 27–41. [DOI] [PubMed] [Google Scholar]
- 28.Burke SJ, et al. , One week of continuous corticosterone exposure impairs hepatic metabolic flexibility, promotes islet beta-cell proliferation, and reduces physical activity in male C57BL/6J mice. J Steroid Biochem Mol Biol, 2019. 195: p. 105468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veenit V, et al. , Increased corticosterone in peripubertal rats leads to long-lasting alterations in social exploration and aggression. Front Behav Neurosci, 2013. 7: p. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iniguez SD, et al. , Social defeat stress induces a depression-like phenotype in adolescent male c57BL/6 mice. Stress, 2014. 17(3): p. 247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gowda GS, et al. , Schizophrenia in Wolfram Syndrome (DIDMOAD Syndrome): A case report in support of the mitochondrial dysfunction hypothesis. Schizophr Res, 2018. 195: p. 574–575. [DOI] [PubMed] [Google Scholar]
- 32.Itokawa M, et al. , Identification of a male schizophrenic patient carrying a de novo balanced translocation, t(4; 13)(p16.1; q21.31). Psychiatry Clin Neurosci, 2004. 58(3): p. 333–7. [DOI] [PubMed] [Google Scholar]
- 33.Hammerschmidt K, et al. , Mice lacking the cerebral cortex develop normal song: insights into the foundations of vocal learning. Sci Rep, 2015. 5: p. 8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hammerschmidt K, et al. , Mice do not require auditory input for the normal development of their ultrasonic vocalizations. BMC Neurosci, 2012. 13: p. 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahrt EJ, et al. , Engineered deafness reveals that mouse courtship vocalizations do not require auditory experience. J Neurosci, 2013. 33(13): p. 5573–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiley SE, et al. , Wolfram Syndrome protein, Miner1, regulates sulphydryl redox status, the unfolded protein response, and Ca2+ homeostasis. EMBO Mol Med, 2013. 5(6): p. 904–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen YF, et al. , Cisd2 mediates mitochondrial integrity and life span in mammals. Autophagy, 2009. 5(7): p. 1043–5. [DOI] [PubMed] [Google Scholar]
- 38.Rezin GT, et al. , Mitochondrial dysfunction and psychiatric disorders. Neurochem Res, 2009. 34(6): p. 1021–9. [DOI] [PubMed] [Google Scholar]
- 39.Wolfer DP, Crusio WE, and Lipp HP, Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci, 2002. 25(7): p. 336–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.