

Summary
Chimeric antigen receptor (CAR) T cell therapy has achieved remarkable success in hematological malignancies but remains ineffective in solid tumors, due in part to CAR T cell exhaustion in the solid tumor microenvironment. To study dysfunction of mesothelin-redirected CAR T cells in pancreatic cancer, we establish a robust model of continuous antigen exposure that recapitulates hallmark features of T cell exhaustion and discover, both in vitro and in CAR T cell patients, that CAR-dysregulation is associated with a CD8+ T-to-NK-like-T cell transition. Furthermore, we identify a gene signature defining CAR and TCR dysregulation and transcription factors, including SOX4 and ID3 as key regulators of CAR T cell exhaustion. Our findings shed light on the plasticity of human CAR T cells and demonstrate that genetic downmodulation of ID3 and SOX4 expression can improve the efficacy of CAR T cell therapy in solid tumors by preventing or delaying CAR T cell dysfunction.
Keywords: CAR T cell, pancreatic cancer, T cell dysfunction, immunotherapy, T cell exhaustion, single-cell RNA-seq, immunology, NK-like T cell, cancer, cell transfer therapy, ID3, SOX4
Graphical Abstract
Introduction
T cell exhaustion is a differentiation state acquired when T cells are exposed to persistent antigen stimulation in the setting of chronic viral infection or in response to tumors (Blank et al., 2019). Failure to eliminate antigen results in a progressive loss of effector functions or dysregulation (Pauken and Wherry, 2015). Hallmarks of T cell exhaustion include reduced effector function, distinct epigenetic and transcriptional gene signatures, sustained expression of multiple inhibitory receptors, defective cytokine production, increased chemokine expression, and limited proliferative capacity (Blank et al., 2019; Pauken and Wherry, 2015; Thommen and Schumacher, 2018). Examination of genes upregulated in exhausted CD8+ tumor-infiltrating lymphocytes (TILs) from patients (Guo et al., 2018; Li et al., 2019; Zhang et al., 2018; Zheng et al., 2017) and TILs from mouse models (Khan et al., 2019; Singer et al., 2016) has led to the identification of genes that restrain tumor immunity, including LAYN, Tox, and Gata3. Furthermore, genome-wide CRISPR Cas9 knock-out and knock-in screens in mouse and human CD8+ T cells revealed additional targets such as Mapk14, Dhx37, NR4A, ZC3H12A, Ptpn2, SOSCS1, and TGFBR2 that modulate T cell function (Dong et al., 2019; Guo and Xu, 2020; Gurusamy et al., 2020; Manguso et al., 2017; Roth et al., 2020; Shifrut et al., 2018; Wei et al., 2019). Importantly, engineered CAR and TCR T cells also acquire an exhausted phenotype when they enter the tumor microenvironment (TME) in in vivo models (Chen et al., 2019; Moon et al., 2014; Stromnes et al., 2015), leading to the hypothesis that CAR T cell exhaustion/dysfunction is a major hurdle for CAR T cell therapy (Fraietta et al., 2018a; Fraietta et al., 2018b; Long et al., 2015; Lynn et al., 2019).
We hypothesized that the development of an in vitro CAR T cell model that employs prolonged continuous antigen exposure to drive CAR T cell exhaustion or dysfunction would uncover new perspectives of CAR T cell dysfunction. As opposed to in vivo models, an in vitro model allows for scalability, ease of manipulation, and the ability to study dynamic changes across multiple time points of T cell dysfunction. Despite the tremendous success of CAR T cells in hematological malignancies, patient responses to CAR T cell therapy in solid tumors are not curative. We therefore focused on solid tumors, in particular, pancreatic cancer using pancreatic cancer cells to stimulate mesothelin-directed CAR (M5CAR) T cells. Currently, phase 1 studies are underway evaluating the safety and feasibility of intravenous administration of M5CAR T cells in patients with mesothelin-positive tumors, including mesothelioma, lung, ovarian, and pancreatic cancers (NCT03054298, NCT03323944). Given that CRISPR Cas9 technology now permits safe multiplex gene-editing of human T cells (Stadtmauer et al., 2020), finding inducers of exhaustion in CAR T cells could permit in principle—via inactivation of the inducers— the development of synthetically enhanced CAR T cell therapies designed to treat solid tumors.
Here, we developed and validated an in vitro model of CAR T cell dysfunction that not only recapitulates defined characteristics of T cell exhaustion, but also identifies previously unknown hallmarks of CAR T cell dysfunction: expression of transcription factors and the transition of conventional CD8+ T-to-NK-like T cells. The relevance of these hallmarks of T cell dysfunction is further highlighted by the demonstration of loss of surface CAR and the presence of NK-like CAR T cells in patient samples from CAR T clinical trials. We also employed our in vitro model to identify a gene signature of dysfunction and to reveal that ID3 and SOX4 transcription factors potentiate this dysfunctional gene signature and the associated reduction in CAR T cytotoxicity. Importantly, such reduction in cytotoxicity can be attenuated by disruption of ID3 or SOX4, revealing a potential strategy to enhance the efficacy of CAR T cell therapy in solid tumors.
Results
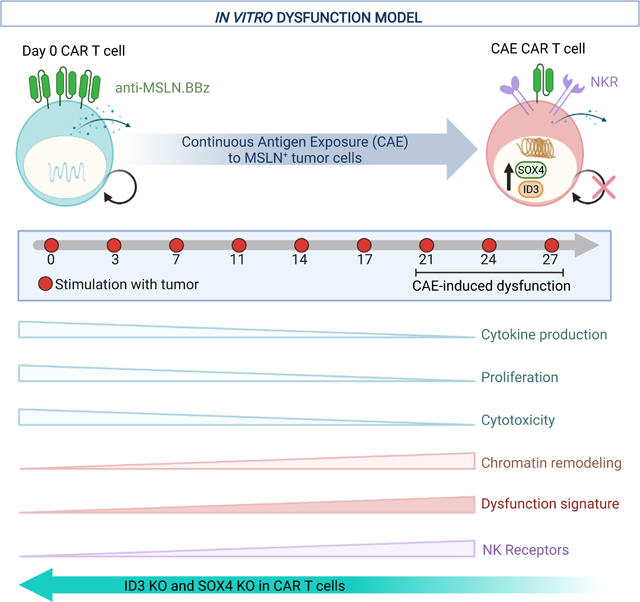
Establishment and validation of an in vitro model of CAR T dysfunction induced by prolonged and continuous antigen exposure (CAE)
To gain a deeper understanding of CAR T cell exhaustion, we developed an in vitro model in which anti-mesothelin CAR (M5CAR) T cells were driven to a dysfunctional state through continuous antigen exposure (CAE). M5CAR contains a human MSLN-binding scFv and CD8α hinge and transmembrane domains fused to 4–1BB and CD3-ζ cytoplasmic signaling domains. To achieve CAE, M5CAR T cells were manufactured from normal donor (ND) peripheral blood mononuclear cells (PBMCs) and repeatedly stimulated with a mesothelin-expressing pancreatic cancer cell line (AsPC-1) such that tumor cells were never cleared by the CAR T cells (Figure 1A and Figure S1A). AsPC-1 express low levels of mesothelin (Figure S1B). After prolonged stimulation (20–35 days), M5CAR T cells lost or decreased doubling capacity-although the time to onset of this dysfunction varied between donors (Figure 1B). Furthermore, although the viability of CAR T cells remained stable at 70 to 80%, the phenotype of apoptotic CAR T cells shifted from early apoptotic to late apoptotic after 18 days of CAE (Figure S1C). We directly measured changes in the number of CD8+ M5CAR T cells by staining for CAR expression on the T cell surface (surCARpos) and observed increasingly reduced levels of surCARpos T cells undergoing prolonged CAE in most donors, similar to (Li et al., 2020) (Figure 1C).
Figure 1: CAR T cell dysfunction develops during chronic antigenic stimulation with reversible loss of cell surface expression of the CAR in vitro and in patients.

(A) Experimental design of CAR T cell dysfunction in vitro model. (B) Population doubling level of M5CAR transduced T cells during CAE, measured by changes in absolute Epcam-CD45+ counts. Five normal donors (ND) were tested. (C) Time-related changes in surface expression of M5CAR on CD8+ T cells. Data from six donors is shown. (D) Percent of sorted CD8+ CAR+ T cells expressing PD-1 and CTLA-4 during CAE. Two donors are shown. (E) M5CAR T cell lysis of AsPC-1 pancreatic tumor cell line before and after CAE measured by xCelligence as real-time impedance (4:1 E:T ratio). Media and non-specific CD19BBz T cells are used as controls. Data are representative of 4 donors (see Figure S1C). (F) Cytokine profile of CD8+ surCAR pos T cells (day 28 CAE, day 0 product and control CD19BBz) co-cultured with AsPC-1 cells. Significance by two-way ANOVA with Tukey’s post hoc test. Data is shown as mean ± SEM. Two additional donors were tested (see Figure S1I). (G) M5CAR genomic DNA detection in CD8+ surface CAR-positive and -negative T cells (right) during CAE. Data from ND150 is shown. (H) Surface CAR expression on CAE CD8+ CAR T cells before and after rest with IL-15. Data from ND150 is shown. (I) Cell killing capacity of CD8+ M5CAR transduced T cells against AsPC-1 cells after 26 days of CAE before and after 24 hrs of rest with IL-15 (7:1 E:T ratio). Data representative of 2 donors is shown as mean ± SEM (see Figure S2C). Significance by Student’s t test. (J) Surface (top) and intracellular (bottom) M5CAR expression on CD8+ T cells from pleural fluid 36 days post-M5CAR T cell infusion (patient #02916–06). M5CAR FMO is shown as negative control (left). See also Figures S1 and S2.
At baseline, CD8+ M5CAR T cells did not express immune checkpoint inhibitors PD-1 or CTLA-4; however, this population exhibited high levels after initial stimulation (day 3), and, as expected, remained elevated above baseline in dysfunctional T cells (Figure 1D). In addition, CAR T cells upregulated the exhaustion marker TIM3 upon prolonged antigen stimulation (Figure S1D). Moreover, we examined tumor cytotoxicity of CAR T cells following CAE (Figure 1E and S1E–F). While day 0 (unstimulated) CD8+ surCARpos M5CAR T cells eliminated tumor cells, day 28 CD8+ surCARpos T cells and non-specific control CD8+ CD19CAR (BBz)-positive T cells did not control tumor growth, revealing that surCARpos T cells become dysfunctional after tumor recognition and CAE. Loss of effector function was not specific to co-culture with the AsPC-1 tumor cell line; similar results were observed when CD8+ M5CAR T cells were continuously stimulated with K562-meso tumor cells, a human myelogenous leukemia cell line engineered to express mesothelin (Figure S1G and S1H). Further, while day 0 CD8+ M5CAR T cells produced high levels of TNF-α and IL-2, CAE CD8+ M5CAR T cells and day 0 CD19BBz antigen control CAR T cells lacked cytokine production (Figure 1F and S1I). Together these data demonstrate that our in vitro model induces progressive CAR T cell dysfunction that is dependent on antigen recognition.
Next, we examined whether this dysfunctional phenotype of CAR T cells in our model is specific to CAR signaling. We collected CD8+ M5CAR T cells following 24 days of CAE, then stimulated with PMA + ionomycin or AsPC-1 cells to measure cytokine production capacity. Both CAE and day 0 cells produced large amounts of IL-2 and IFN-γ after being stimulated with PMA + ionomycin. However, when stimulated with AsPC-1 cells, cytokine production by the CAE cells was significantly reduced (Figure S1J). CAE M5CAR T cells failed to secrete cytokines after prolonged CAR engagement, but still retained the ability to produce cytokines through pharmacologic stimulation by a CAR bypass mechanism, suggesting that downstream signaling remains intact.
Rest restores surface CAR expression and improves cytotoxicity
To further explore the decline in surface CAR expression with CAE, we sorted surface CAR positive (surCARpos) and surface CAR negative (surCARneg) M5CAR T cell populations at 4, 7, and 17 days of CAE. Importantly, by day 17 of CAE, these two populations demonstrated equivalent amounts of genomic CAR DNA by qPCR, indicating that most surface CAR negative cells are transduced CAR T cells with the CAR ligand internalized (Figure 1G, left). To test whether CAE-induced loss of surface CAR is reversible in our model, transduced M5CAR T cells were cultured under CAE (Figure S2A), sorted for surCARneg cells (Figure 1H, left), and then rested with fresh media plus IL-15 for a day. 38% of surCARneg CD8+ T cells regained surface CAR expression (Figure 1H). We next investigated the impact of CAE-induced surface CAR loss on M5CAR T cell effector function by measuring cell killing capacity. Bulk CD8+ T cells collected after CAE could not control tumor growth; however, 24-hour rest with IL-15 dramatically rescued their cytotoxic ability (Figure 1I and Figure S2B and C). Taken together, these results suggest that although loss of surface CAR expression is observed after several weeks of CAE, M5CAR T cells can recover effector function and surface CAR expression with rest and IL-15 supplement.
Having demonstrated reduced surface expression of the M5CAR in vitro under CAE, we examined the clinical relevance of this phenomenon in the human TME. We obtained peritoneal/pleural fluid samples collected after M5CAR T cell infusion from two ovarian cancer patients enrolled on a M5CAR T cell trial (NCT03054298). We identified tumor cells (Figure S2D) and M5CAR CD8+ T cells post-CAR intravenous infusion (Figure 1J and S2E). Although the levels of M5CAR T cells were low as determined by qPCR (not shown), we were able to detect CAR T cells by flow cytometry. Notably, the frequency of intracellular CAR-positive T cells (Figure 1J, bottom right), which represents both surCARpos and surCARneg T cells, was higher than surCARpos T cells alone (Figure 1J, top right), confirming that M5CAR T cells exhibit reduced expression of CAR on the cell surface after infusion in the human TME (Figure 1J, Figure S2E).
Transcriptional dynamics of dysfunctional CAR T cells
To better understand the mechanisms driving loss of CAR T effector function, we performed bulk RNA-seq on CD8+ day 0 product and day 28 CAE surCARpos cells (Figure 2A, Table S1). In parallel, we performed RNA-seq on day 0 and day 28 CAE surCARneg CD8+ T cells (comprising both untransduced T cells and internalized CAR T cells). There was strong correlation of the gene expression signatures for surCARpos and surCARneg populations (Figures 2B and S3A,B), suggesting that CAR T cells acquire the dysregulation signature before developing impaired expression of surface CAR. Since our phenotypic studies were performed in surCARpos cells (see Figure 1) and the mechanisms of dysfunction in this population are unexplored, we decided to focus on this population for the remainder of the bulk RNA-seq analyses.
Figure 2: Transcriptional dynamics of dysfunctional CAR T cells.

(A) Differentially expressed genes between day 0 and 28 CAE surCARpos cells. Genes on the right are upregulated at day 28 (N=521) and genes on the left are downregulated (N=517). Red dots indicate significant genes with adjusted p values <0.05 and fold change >2. Analysis includes four biological replicates. (B) Average gene expression values (TPMs) for day 28 surCARpos compared to day 28 surCARneg for differentially expressed genes defined in Figure 2A (top) and all genes (bottom). (C) Ingenuity Pathway Analysis (IPA) of significant genes from 2A. Red denotes NK and blue denotes exhaustion pathways. (D) Normalized RNA-seq counts of representative NK-related genes. Average of four biological replicates with standard deviation depicted. Statistics by Mann-Whitney U test. (E) Heatmap of genes differentially expressed between day 0, 16, and 28 CAE surCARpos cells (N=762 genes). Average of two biological replicates. (F) IPA upstream regulator analysis of transcription factors predicted to regulate the differentially expressed genes between day 0 and 28, ranked by -log(p value). Gene expression log2 FC (day 28/day 0) is shown on the right. Only transcription factors dysregulated upon CAE are shown. (G, H) Representative ATAC-seq tracks (top) and pooled RNA-seq tracks (bottom) from day 0 and 28 samples at ID3 (G) and KLF2 (H) regulatory regions. Analysis includes four biological replicates. See also Figures S3 and S4, Tables S1–S3.
Next, we investigated how well our model correlates with established in vivo models of T cell exhaustion. 27% of genes upregulated in CAE CD8+ surCARpos T cells overlapped with genes upregulated in exhausted T cells from the chronic lymphocytic choriomeningitis virus (LCMV) mouse model (Pauken et al., 2016), including genes implicated in T cell exhaustion [CTLA4, TOX, TIGIT, NR4A2, NR4A3, HAVCR2 (TIM3), ENTPD1 (CD39), TNFRSF9 (4–1BB)] (Figure S3C). There was also significant overlap between genes downregulated in CAE and exhausted T cells, which included genes known to be expressed in naïve or memory CD8+ T cells (IL7R, LEF1, SELL, Figure S3D). Further, GSEA analysis of our data with the 4 transient states of T cell exhaustion identified in the LCMV mouse model (Beltra et al., 2020) revealed significant enrichment with the intermediate and terminally exhausted T cell populations (Figure S3E), indicating our model recapitulates features of the later stages of T cell exhaustion in mouse T cells.
We also compared our model to tumor-infiltrating lymphocytes (TILs). The single-cell RNA-seq (scRNA-seq) gene signatures of dysfunctional human CD8+ TILs isolated from patients with melanoma (Li et al., 2019), hepatocellular carcinoma (Zheng et al., 2017), colorectal (Zhang et al., 2018), and non-small cell lung cancer (Guo et al., 2018) significantly overlapped with genes upregulated in CAE surCARpos T cells (Figure S3F–S3I). We overlapped datasets from the four cancer types and found a common group of 18 TIL marker genes (Figure S3J), and most of these genes were upregulated in CAE surCARpos T cells (Figure S3K). To determine how applicable our signature is to other CARs, we performed GSEA analysis of the exhaustion signature curated in GD2-directed CARs (Lynn et al., 2019). Genes upregulated in the exhausted CD8+ GD2 CAR T cells were significantly enriched with genes up in day 28 CAE M5CAR T cells, suggesting that at least some of the signaling observed in the 4–1BB mesothelin-directed dysfunctional CAR T cells is conserved in the exhausted GD2–28z CAR T cells (Figure S3L). Taken together, these analyses provide further evidence that our in vitro model of CAR T cell dysfunction aligns with many features of in vivo human and mouse models of T cell exhaustion and dysfunction.
To further illuminate the biological functions of the entire dysregulated gene expression signature identified in Figure 2A, we performed Ingenuity Pathway Analysis (IPA). As expected, T cell exhaustion, PD-1/PD-L1 cancer immunotherapy, and CTLA4 signaling pathways were enriched (Figure 2C; blue, Table S2). Interestingly, several pathways related to natural killer cells (NK cells) were also enriched in the gene expression signature of CAE CD8+ surCARpos T cells (Figure 2C; red). In fact, we noted that multiple NK receptors were upregulated, including KLRC1, KLRC2, KLRC3, KLRB1, KLRD1, and KIR2DL4 (Figure 2D). αβ T cells often upregulate receptors constitutively expressed by NK cells, potentially due to chronic activation by antigens and cytokines (Balin et al., 2018; McMahon et al., 2002; Meresse et al., 2004). To identify whether CAE drives a similar gene expression program in CD4+ T cells, we performed RNA-seq on day 0 and day 28 CAE surCARpos CD4+ T cells and found significant overlap between the CD4+ and CD8+ T cell signatures following CAE, including the upregulation of NK receptors (KLRB1, KLRC1, KLRC2, KLRC3, KLRD1) and other genes in our signature including GNLY, LAYN, CD9, PHLDA1, SOX4, and TNFRSF9, among others (Figures S3M and S3N).
To better understand how gene expression changes over time in our model, we performed RNA-seq on CAE surCARpos CD8+ T cells at day 16 (a middle time point). We identified genes that showed temporal changes in expression between day 0, 16, and 28, (Figure 2E). For example, many NK receptors and exhaustion markers gradually turned on, with moderate expression by day 16 and highest expression by day 28 (cluster 5: KLRD1, KLRC1, KLRC2, KLRC3, TOX, HAVCR2, TIGIT), while other markers remained off or lowly expressed until dramatic upregulation at day 28 (cluster 4: KLRB1, KLRK1). Cluster 6 genes displayed robust activation on day 16 with slight downregulation by day 28, and included inhibitory molecules (CTLA4, LAG3), genes encoding chemokines (CCL3, CCL4, CXCL8), cytotoxic molecules (PRF1, GZMB, NKG7), and T cell activation genes (Boroughs et al., 2020).
Next, we identified potential transcription factors that control the dysregulated gene expression signature in CAE surCARpos T cells. This list included genes that were upregulated (EGR1, ID3, SOX4, RBPJ), as well as downregulated (KLF2, BCL6, LEF1) in CAE surCARpos cells (Figure 2F, Table S3).
We performed ATAC-seq (assay for transposase-accessible chromatin with sequencing) to explore CAE specific regulatory changes in surCARpos cells. Overall, there was a closing of chromatin upon CAE (Figure S4A). Of the sites that opened in CAE, most were in introns, intergenic, and promoter regions consistent with a regulatory role (Figure S4B). We integrated our RNA-seq and ATAC-seq datasets and found that genes upregulated in CAE displayed an opening of chromatin, while genes downregulated in CAE displayed a closing of chromatin (Figure S4C). For example, the upregulated gene ID3 and the downregulated gene KLF2 displayed opening and closing of chromatin at nearby regulatory regions, respectively (Figure 2G and H).
To determine if the epigenetic landscape of the dysfunctional CAR T cells is similar to TCR-mediated exhaustion, we queried ATAC-seq datasets from exhausted human PD1-high TILs (Philip et al., 2017) and found that chromatin sites opening in day 28 CAE cells are also open in exhausted TILs (Figures S4D and S4E). We also observed closing of chromatin in day 28 CAE cells at CD5, CD28, and TCF7, similar to PD1-high human TILs or dysfunctional mouse T cells, as previously reported (Philip et al., 2017) (Figure S4F).
Single-cell analysis of CAE CD8+ T cells reveals co-expression of dysfunction signature genes
We performed scRNA-seq for day 0 and day 20 CAE cells. Of note, this experiment was performed in CAR-transduced CD8+ T cells and thus includes a mixed population of surCARpos, surCARneg, and untransduced CD8+ T cells. We first identified differentially expressed genes (DEGs) between day 0 and 20 CAE cells using “cellfishing” (Sato et al., 2019) and found a strong correlation with our findings using bulk RNA-seq (Figure S4G). Next, we performed a nonlinear dimensionality-reduction technique (uniform manifold approximation and projection, UMAP) followed by unsupervised clustering on cells from day 0 (Figure 3A) and 20 (Figure 3B). The program identified three distinct clusters on day 0 (D0–1, D0–2, D0–3) and four clusters on day 20 (D20–1, D20–2, D20–3, D20–4). Top marker genes were identified for day 20 CAE (Figure 3C) and day 0 cell clusters (Figure S4H). Interestingly, a group of genes upregulated in surCARpos CAE cells identified via bulk genomics (Figure 2) (KLRC1, SOX4, TNFRSF18, RBPJ, RGS16, CCL3) were found to be top marker genes for single-cell clusters D20–1 and D20–4. Furthermore, gene pathway analysis using all DEGs for each cluster revealed enrichment of the term “natural killer signaling” in day 20 CAE cell clusters D20–1 and D20–4, but not D20–2 and D20–3 clusters or day 0 clusters (Figure 3D, Figure S4I). Overlap of the top marker genes for each single cell cluster revealed that genes defining clusters D20–1 and D20–4 significantly overlapped with genes upregulated in day 28 CAE cells via bulk genomics (Figure S4J). Thus, D20–1 and D20–4 clusters likely represent a subpopulation of CAE cells consisting of dysfunctional CD8+ T cells that express NK-associated genes. Genes that were highly expressed in day 0 cells (IL7R, LTB, CD48, HLA-DRB1) were top marker genes for clusters D20–2 and D20–3, suggesting that the cells in these clusters have attributes similar to day 0 cells. Of note, clusters D20–1 and D20–3 were highly enriched for cell-cycle regulated pathways (see Figure 3D).
Figure 3: Single-cell analysis of CAE CD8+ T cells reveals co-expression of dysfunction signature genes.

UMAP projection of sc-RNA seq data from day 0 product (A) and day 20 CAE cells (B) for donor ND388. (C) Heatmap of top 10 marker genes for each cluster defined in B. (D) Gene ontology determined by metascape pathway analysis for each single-cell cluster from the day 20 CAE sample. Columns are cell clusters (from B) and rows are enriched pathways color coded by level of significance. (E) Volcano plot depicting differentially expressed genes between day 20 CAE clusters 1 and 4 (dysfunctional) and clusters 2 and 3 (non-dysfunctional). Genes upregulated in the dysfunctional clusters are on the right side. Red dots indicate significant genes with p<0.05 and log2FC >0.2. (F) Dot plot illustrating the expression level of dysfunction signature, naïve/memory, cell cycle and control genes in day 0 (left) and day 20 CAE (right), donor ND388. Each column represents one cluster as depicted in A and B. (G) Gene regulatory network analysis (PIDC) for day 20 CAE cells. Columns and rows are the top 500 most variable genes determined by Seurat. Depicted on the right are select genes found within the same community, boxed in red. (H) Normalized counts of CAR transcripts from sc-RNA-seq data for day 20 and 28 CAE cells. Pooled cells from dysfunctional and non-dysfunctional clusters from three CAR T donors. Data shown as mean with standard deviation. Significance by Mann-Whitney U test. (I) Percent of cells that express the CAR transcript in dysfunctional and non-dysfunctional clusters. Average of three CAR T donors. Data shown as mean ±SEM. See also Figures S4, S5, and Tables S4 and S5.
We identified all genes specifically expressed in the presumptive dysfunctional clusters (D20–1 and D20–4), compared to clusters D20–2 and D20–3 (Figure 3E, Table S4). Genes with known links to exhaustion, including HAVCR2, ENTPD1, LAYN, CTLA4, PHLDA1, TNFRSF9, NR4A1, PRDM1, and LAG3 were upregulated in the dysfunctional clusters (Figure 3E volcano plot, right side). We then curated an unbiased dysfunction gene signature consisting of the top 30 genes most highly upregulated in day 20 dysfunctional clusters (Figure 3F) of which 24/30 genes were also upregulated in bulk CAE surCARpos T cells (Figure 2A). Genes identified exclusively in scRNA-seq included SRGAP3, DUSP4, and CSF1-genes not currently linked to T cell exhaustion (Figure S4K). Clusters that emerged that were not dysfunctional (D20–2 and D20–3) highly expressed HLA molecules (HLA-DRB1, HLA-DQB1, HLA-DRA, HLA-DPB1) and IL7R, TC2N and FYB1 (see Figure 3E, left side).
We generated dot plots containing the 30 signature genes, as well as naïve/memory markers, cell cycle genes, and control genes (Figure 3F). Of note, many of the dysfunction signature genes were also present in the gene expression signature described for other models of T cell dysfunction (Table S5). As expected, day 20 CAE cells (Figure 3F, right) had two cell clusters that highly expressed the dysfunction signature (clusters D20–1, D20–4), while clusters D20–2 and D20–3 and day 0 cell clusters (Figure 3F, left) did not express this signature. Although not part of our 30 gene signature, CTLA4 was upregulated in D20–1 and D20–4 clusters (Figure S4L). Select T cell activation genes identified in CD19 CAR T cells (CCL3, CCL4, GZMB, and TNFRSF9) (Boroughs et al., 2020) are in our 30 gene signature; however, many inhibitory receptors are also T cell activation genes, and their sustained expression is a hallmark feature of T cell exhaustion (Wherry and Kurachi, 2015).
We investigated whether the dysfunction signature genes were co-expressed within the same single cell using an unbiased gene regulatory network analysis (PIDC) (Chan et al., 2017). One community in day 20 CAE cells included 34 genes that were co-expressed (Figure 3G; boxed in red). Strikingly, 27/30 of our defined dysfunction signature genes (Figure 3F) were contained within this community, confirming that these genes were co-expressed in the same subset of cells and that they had a common regulatory network (Figure 3G).
Importantly, to confirm our single-cell findings, we performed scRNA-seq in two additional donors (ND538 and ND150) for day 0 and 28 CAE cells and found remarkably similar gene expression signatures, despite these cells being collected at later timepoints of CAE (Figure S5A–J). Human donors have variability in the number of days required to reach a dysfunctional state; however, most CAR T donors are dysfunctional by 20 days of CAE.
Given that CAE results in dysfunctional CAR T cells with reduced effector function, we next asked whether we could detect CAR transcripts in our single-cell datasets, and if so, whether cells that express the CAR are preferentially expressed in the dysfunctional cell clusters. We found that the dysfunctional cell clusters expressed significantly more CAR (Figure 3H) and had a higher percentage of cells overall that expressed the CAR (Figure 3I).
Mass and flow cytometry profiling reveals NK-like phenotype of CD8+ CAR T cells under CAE
Next, we examined expression of NK-associated proteins by flow cytometry on surCARpos and surCARneg CD8+ T cells throughout CAE. CD8+ CAR T cells did not express high levels of NK-associated molecules and exhaustion markers before CAE, but exhibited increased expression after CAE with concurrent loss of CD28 (Figure 4A). While most NK receptors increased over time, NKG2C was expressed early, followed by a rapid decline in expression during CAE. Importantly, we could not identify invariant NKT cells (Figure 4A), suggesting NK-like T cells identified in this model need to be separately classified from iNKT cells (Godfrey et al., 2004).
Figure 4: In vivo relevance of CAR and TCR T cell dysfunction signature and the NK-like phenotype.

(A) Time-related changes in NK-associated molecules and PD-1 and CD28 on surCARpos and surCARneg CD8+ T cells during CAE. iNKT are defined as cells with Vα24-Jα18 specific TCRs. Data from ND150 is shown. (B) Experimental design of the recurrent AsPC-1 mouse model. (C) AsPC-1 tumor growth volumes in M5CAR T-treated mice. Red arrows indicate tumors analyzed after recurrence. (D) NK-associated molecules expression in CD8 day 0 product (top) and TILs from a representative AsPC-1 recurrent tumor (bottom). (E) Average expression of NK-associated molecules on CD8 T cells in day 0 product and in three recurrent tumors. Each datapoint represents a single mouse for recurrent tumor data and a single technical replicate staining for day 0 product. Color code for mice data is matched with Figure 4C. (F) PD-1, LAG3, and TIM3 expression in CD8 day 0 product (top) and TILs from a representative AsPC-1 recurrent tumor (bottom). (G) Average expression of checkpoint receptors PD-1, LAG3, and TIM3 in CD8 T cells. Each datapoint represents a single mouse for recurrent tumor data and a single technical replicate staining for day 0 product. Color code for mice data is matched with Figure 4C. (H) CD56 expression in CD8+ surCARpos T cells isolated from DLBCL patients at the peak of CTL019 expansion. (I) Expression of NK-associated molecules and PD-1 on CD8+ surCARpos T cells in day 0 product and day 27 peripheral blood T cells from a patient with DLBCL (#13413–39). (J) Timeline showing the experimental design of NY-ESO-1 TIL mouse model. (K) Heatmap of dysfunction signature genes in NY-ESO-1 reactive CD8+ TILs along with blood (CD8+CD45RO+ T cells) and day 0 infused product. See also Figure S6. Data from (E) and (G) is shown as mean ± SEM and significance were assessed by two-way ANOVA plus Sidak test.
We performed an NK focused cytometry by time-of-flight (CyTOF or mass cytometry) to explore how the dysfunction signature identified by scRNA-seq relates to protein expression levels on CAR T cells. t-distributed stochastic neighbor embedding (t-SNE) plots revealed twenty subpopulations of CD8+ T cells, where CAE CAR T cells had markedly different clusters compared to day 0 product (Figure S6A, red circle denotes cell populations more abundant in CAE T cells). Notably many NK receptors and NK-related proteins were increased in the CAE specific clusters, including the inhibitory receptors (KLRB1, TIGIT, NKG2A, PD-1) and NK-related proteins CD56 and granulysin (Figure S6B). The mass cytometry data closely aligned with our flow cytometry profiling of CD8+ CAR T cells under CAE as shown in Figure 4A. The various subpopulations identified in the CAE cells revealed the NK-like phenotype was heterogeneous. There were two distinct subpopulations of cells that expressed CD56, one group that was KLRB1+ and another group that was KLRB1-. In agreement with our genomics data (see Figure 2B), NK-like phenotypes emerged in both surCARpos and surCARneg cells (Figures 4A and S6B). Overall, these data suggest that a subset of day 0 CD8+ T cells dynamically evolve into NK-like T cells with a distinct phenotype marked by KLRB1 and/or CD56 expression.
In vivo NK receptor upregulation and dysfunction signature gene expression in CAR T cells and TILs
Our observations above of upregulation of NK molecules on CD8+ CAR T cells in vitro during CAE prompted us to test whether this expansion occurs in vivo. AsPC-1 tumors were established in mice and M5CAR T cells were able to eliminate large mesothelin-expressing flank tumors within two weeks after CAR T injection (Figure 4B and C). However, two to four months after initial injection of the CAR T cells, several of the mice relapsed. We analyzed the recurrent tumors and found that the mechanism of tumor relapse was not due to loss of the mesothelin target antigen (Figure S6C). Therefore, we analyzed the infiltrating human T cells in the relapsed tumors and found that nearly all the infiltrating T cells were CD8+ CAR T cells (Figure S6D and E). Intriguingly, the CAR T cells from the recurrent tumors expressed our dysfunction signature with high levels of NK receptors (Figures 4D and E) and checkpoint receptors (Figures 4F and G), unlike the day 0 CAR T product. Further, since the tumors were progressing without losing mesothelin expression, we can be confident that the T cells had lost the ability to control the tumor and are thus dysfunctional.
This finding prompted us to test whether this expansion occurs in patients undergoing CAR T therapy. Diffuse large B-cell lymphoma (DLBCL) patients treated with CD19-directed CAR T cells (CTL019) were retrospectively assessed in a clinical trial (NCT02030834) to determine if any of their circulating CAR T cells exhibited NK-like features. Three of seventeen analyzed DLBCL patients exhibited greater than 5% expansion of the CAR+ NK-like T cell population as early as 10 days post-CAR T infusion of a CD19-directed CAR, and other patients showed detectable expansion (Figure 4H). Notably, the patient with the highest level of NK-like CAR T cells (13413–39) had progressive tumor and failed to respond to the therapy (Schuster et al., 2017). There was sufficient material from patient 13413–39 to analyze additional NK markers in CAR+ T cells. The percentage of NK-like T cells in the day 0 CAR T product was low, but the NK-like CD8+ T cell phenotype was upregulated at day 27 post-CAR T infusion as determined by increased levels of NKG2A, CD94, and CD56 (Figure 4I). We did not detect increased KLRB1 levels; however, this could be explained by the late expression of this marker upon CAE (Figure 4A). In conclusion, these data provide evidence for the acquisition of an NK-like CAR T cell phenotype in some CAR T cell patients.
To determine whether the CAR T dysfunction signature is CAR-specific or more broadly applicable to T cells chronically exposed to antigen, we generated lung tumors that expressed the antigen NY-ESO-1 in a xenograft mouse model, and then injected human T cells specifically engineered to express NY-ESO-1-reactive Ly95 TCR into the tumor (Figure 4J). This generates hypofunctional Ly95 TILs that are unable to eradicate tumor (Moon et al., 2016). Our dysfunction gene signature was expressed at a low level in the infused product and blood CD8+ T cells but, strikingly, 28/30 of the exhaustion and NK signature genes were upregulated in the NY-ESO-1-reactive TCR TILs, including the transcription factors ID3 and SOX4 (Figure 4K).
Transition of CD8+ T cells to NK-like T cells upon continuous antigen stimulation
NK-like T cells have been shown to express both T cell and NK cell markers and are frequently defined as CD3+CD56+ or CD3+KLRB1+ and they often express KLRC1 (Barbarin et al., 2017; Kurioka et al., 2018). UMAP plots of scRNA-seq day 0 versus day 20 CAE cells showed enrichment of cells that co-express CD3, KLRB1, and KLRC1 (Figure 5A, related to UMAPs in Figure 3A and 3B). In addition, flow cytometry analysis using two separate markers for NK-like T cells (CD3+CD56+ and CD3+KLRB1+) revealed a robust expansion of this NK-like T cell population during CAE (Figure 5B).
Figure 5: Transition of CD8+ T cells to NK-like T cells upon continuous antigen stimulation.

(A) NK-like T cell population (CD3+, KLRB1+, and KLRC1) at day 0 (left) and day 20 CAE (right) overlayed on UMAP graphs from Figure 3A and B. (B) Identification of NK-like T cell populations (CD56+ CD3+ and CD3+ KLRB1) during CAE. (C) On left, NK-like T cell frequency (CD3+CD56+) at day 0 and following CD56 depletion. On right, NK-like T cell frequency (CD3+CD56+) with or without CD56 depletion during CAE. Data representative of two donors is shown as mean ± SEM. (D) Single-cell TCR fingerprinting + gene expression analysis in ND150 (left) and ND538 (right). Results are filtered for CD8+ T cells that have the same CDR3 TCR sequence at day 0 and at day 28. Cells were classified as either KLRB1-negative or -positive at day 0 and at day 28 and total number of cells in each category is depicted. (E) Monocle trajectory analysis of ND388 day 20 CAE cells, with single-cell clusters labeled according to their defined clusters in Figure 3B (left). On right, same monocle trajectory but with cells labeled according to expression of the dysfunction gene signature (N= 30 genes, see Figure 3F). (F) Monocle trajectory analysis of ND150 and ND538 day 0 and day 28 CAE cells combined, corresponding to supplemental Figures S5. Cells are labeled according to sample ID (left) or by how highly each cell expresses the dysfunction signature genes (right). See also Figure S6.
Our findings overall demonstrate expansion of an NK-like T cell population upon CAE; however, it is unclear whether these are clonally expanded cells from an NK-like T population existing at day 0, or, in contrast, whether CD8+ T cells acquire NK receptors via plasticity during prolonged antigen exposure. To test this in our in vitro model of CAR T cell dysfunction, we depleted the CD56+ cells from the input day 0 population and repeated the CAE experiment. We note that CD56 is the most frequently used marker to identify human NK and NK-like T cells and hence CD56 depletion is expected to remove both populations from the day 0 product (Barbarin et al., 2017; Seyda et al., 2016). At day 0, the percentage of NK-like T cells was very low (0.69–2.23%, Figure 5A and 5B left). Strikingly, CD56+ depletion had no effect on the percent of NK-like T cells that emerged upon CAE (Figure 5C, right, also see theoretical model, Figure S6F), consistent with transition of CD8+ T cells to NK-like T cells rather than expansion.
To confirm the T cell to NK-like CAR T cell transition, we performed scRNA-seq alongside lineage tracing using T cell receptor (TCR) sequencing at day 0 and 28 CAE (Figure 5D), reasoning that the specific TCR allele would be the same after transition. We filtered for CD8+ cells with TCRs in common between day 0 and 28 (Figure 5D, left). Of these, 36 were KLRB1- at day 0 and by day 28, 17/36 (47%) transitioned to KLRB1+. This was validated independently using another CAR T donor (Figure 5D, right). These results confirm that the NK-like T cells are undergoing transition, and not simply expanding. We note that 96–99% of the TCRs were unique in each sample, providing additional evidence against clonal expansion in our in vitro model (Figure S6G).
To model the changes in transcription that occur as CD8+ T cells transition to NK-like T cells, we performed pseudotime analysis which showed that day 20 CAE clusters (D20–2, D20–3) separated from dysfunctional clusters (D20–1, D20–4), with transcriptional progression from D20–3, D20–2, D20–4 to the D20–1 cluster (Figure 5E, left). Consistent with this progression, cells expressing the dysfunction signature (see Figure 3F, N=30 genes) prominently occupied the end of the trajectory (Figure 5E, right). We used two additional donors to validate these findings and importantly, combined day 0 and 28 CAE samples from both donors together for pseudotime analysis. As expected, day 0 samples clustered together (red and blue cells) on the right side of the trajectory, while day 28 samples (green and purple) clustered together on the left (Figure 5F, left). Furthermore, cells expressing the highest level of dysfunction signature genes (green) clustered on the left side of the trajectory with day 28 CAE cells (Figure 5F, right). Taken together, our dysfunction signature genes associate with transitioned NK-like T cells.
ID3 and SOX4 are potential regulators of the dysfunction signature
Identification of a common transcription factor(s) that controls this CAR T dysfunction signature and NK-like T cell transition could provide an approach to prevent and/or reverse loss of effector function. DEGs identified in our scRNA-seq datasets between day 0 and 20 CAE cells were analyzed by IPA to identify potential transcription factors that regulate the signature. All transcription factors highlighted in the bulk RNA-seq experiment (Figure 2F) were also regulators of the single-cell signature and some, but not all, were themselves differentially expressed in the single-cell dataset (FC indicated to the right, Figure 6A, Table S6). Importantly, ID3 and SOX4 were specifically expressed in the dysfunction clusters (Figure 6B, C, related to Figure 3B), while other transcription factors with the possible exception of TWIST1 that was expressed at low levels, lacked specificity or had less dramatic changes between dysfunctional and non-dysfunctional clusters (Figure 6D and S6H). Consistently, ID3 and SOX4 were co-expressed with the other dysfunction signature genes in CAE T cells (see Figure 3G), suggesting these transcription factors may help to orchestrate the dysregulated gene expression signature.
Figure 6: ID3 and SOX4 are potential regulators of the dysfunction signature.

(A) Select transcription factors predicted to regulate differentially expressed genes between day 0 and day 20 CAE cells in single-cell sequencing datasets, identified using IPA upstream regulator analysis. Depicted are transcription factors that overlap with factors from Figure 2F. On right, gene expression log2 FC (day 20 CAE/day 0) for each transcription factor. NA depicts genes that are not differentially expressed between day 0 and day 20 cells. (B) UMAP plots from Figure 3B showing single-cell transcript levels of ID3 and SOX4 on day 20 CAE cells. Top two clusters are dysfunctional. (C) Violin plots depicting gene expression levels for ID3 and SOX4 for each cluster from day 20 CAE cells (see Figure 3B). (D) Single-cell transcript levels of CDKN2A, BCL6, RBPJ, ID2, and KLF2 illustrated by UMAP plots, corresponding to clusters from Figure 3B (day 20 CAE cells). (E) HOMER motif analysis depicting top 10 enriched transcription factor motifs in bulk ATAC-seq dataset for day 0 samples (left) and day 28 samples (right). Analysis includes four biological replicates. (F) Box plots illustrating the ATAC-seq signal at unchanged peaks (left) and peaks that change between day 0 and day 28 (right). Data are further subdivided depending on whether a SOX4 motif is present. Statistics assessed by Mann-Whitney U test. (G-I) ATAC-seq tracks in regulatory regions at SOX4 motifs from day 0 and 28 CAE samples at dysfunction genes AFAP1L2 (G), CDK6 (H) and CSF1 (I). SOX4 motifs labeled with red bars above tracks. Analysis includes four biological replicates. See also Figures S6 and S7, Table S6.
ID3 is a member of a family of helix-loop-helix transcription factors that do not bind DNA directly, but rather inhibit other transcription factors from binding DNA (Benezra et al., 1990), and thus, ID3 lacks a specific DNA-binding motif. However, SOX4, a member of the SRY-related HMG-box family, has a known DNA motif (Fornes et al., 2020; The UniProt Consortium., 2019). We identified top transcription factor motifs enriched in day 0 samples (left) and day 28 samples (right) using our bulk ATAC-seq datasets (Figure 6E). Day 28-specific peaks were enriched for the SOX17 motif, which is identical to the SOX4 motif (Figure S6I), whereas day 0 peaks displayed no SOX enrichment. Day 28-specific ATAC-seq peaks with a SOX4 motif displayed increased ATAC-seq signal (p=7.9e-07) compared to ATAC-seq peaks that lacked a SOX4 motif, while day 0 samples showed no significant difference (p=.09) (Figure 6F, right). We note that ATAC-seq peaks that did not change between day 0 and 28 (Figure 6F, left, unchanged peaks) showed no specific enrichment for SOX4 motifs. Further, 18/30 of our dysfunction signature genes had chromatin opening at SOX4 motifs in day 28 CAE cells-including AFAP1L2, CDK6, and CSF1 (Figure 6G–I) and NK receptor genes KLRC1 and KLRB1 (Figure S7A and S7B). Our results indicate that CAR T cells develop an opening of chromatin at SOX4 sites upon CAE.
Disruption of ID3 and SOX4 improves CAR T effector function
To investigate whether ID3 and SOX4 regulate the dysfunction signature, T-to-NK-like T transition and drive CAR T dysfunction, we generated ID3 and SOX4 KO CAR T cells using CRISPR-Cas9 (Figure 7A and Figure S7C). We validated the efficiency of KO cells in the day 0 product (Figure S7C). No differences in cytotoxicity (Figure S7D) or T cell subset distribution (naïve, effector, and memory populations) were observed at baseline between WT and KO day 0 CAR T cells (Figure S7E); however, as expected, there were minor differences in T cell subsets between the CAR T donors.
Figure 7: Disruption of ID3 and SOX4 improves CAR T effector function.

(A) Schematic representation of the CRISPR strategy to generate ID3 and SOX4 KO M5CAR T cells. (B) Experimental design for WT, ID3 KO, and SOX4 KO analyses for donors ND566 and ND539. (C) Agarose gel showing ID3 and SOX4 KO detection on cDNA from CD8 sorted populations after CAE for donor ND566. ID3: ID3 PCR, SOX4: SOX4 PCR, Positive Control: histone H3.3, WT: Mock M5CAR, W: water negative control, KO: ID3 KO (in ID3 PCR) and SOX4 KO (in SOX4 PCR). (D) KO quantification of ID3 (ND566 and ND539) and SOX4 (ND566) by cDNA sequencing. Percent indels and fragment deletions upon CAE are shown as mean with standard deviation. (E) UMAP projection of sc-RNA seq data from sorted CD8+ WT, ID3 KO, or SOX4 KO day 24 CAE cells for donor ND566-cells are color coded by KO status. (F) NK-like T cell population at day 24 CAE for donor ND566, depicted by co-expression of CD3, KLRB1, and KLRC1, overlayed on UMAP graphs from Figure 7E. (G) Percentage of NK-like T cells in WT, ID3 KO and SOX4 KO cells, relative to WT (donor ND566). Significance by Fisher’s exact test. (H) UMAP graph from Figure 7E with cells labeled according to expression of the dysfunction gene signature for donor ND566. Dysfunction score for WT, ID3 KO, and SOX4 KO cells for donor ND566 (I) and WT and ID3 KO cells for donor ND539 (J). Significance measured by Mann-Whitney U test. (K) Dot plot illustrating the expression level of dysfunction signature genes in WT, ID3 KO, and SOX4 KO day 24 CAE cells, donor ND566. (L-T) Violin plots depicting gene expression levels from WT, ID3 KO, and SOX4 KO day 24 CAE cells for SOX4 (L), AFAP1L2 (M), CSF1 (N), ID3 (O), LAYN (P), CD9 (Q), TNFRSF18 (R), GNLY (S) and KLRC1 (T) for donor ND566. (U) Cell killing capacity of WT, ID3 KO, and SOX4 KO M5CAR T CAE cells, with controls media alone and day 0 CAR T product. Cells were collected and seeded at 1:8 E:T ratio with AsPC-1 on day 18 (ND539) and day 21 (ND566). Data is presented as mean ± SEM. Significance by two-way ANOVA with Geisser-Greenhouse correction and Dunnet’s post hoc test. See also Figure S7.
To study the role of the transcription factors in driving CAR T dysfunction, we challenged WT, ID3 KO and SOX4 KO CAR T cells with CAE for 20–28 days and analyzed their transcriptional profile and cytotoxic capacity (Figure 7B). Of note, day 0 and CAE conditions showed a similar KO efficiency, suggesting there was no enrichment or depletion of SOX4 or ID3 KO cells during CAE (Figure 7C–D and Figure S7C). To identify if the transcription factors regulate the NK phenotype and/or the dysfunction signature genes, we performed scRNA-seq. WT cells clustered predominantly on the right side, while ID3 and SOX4 KO cells clustered largely on the left (Figure 7E). Interestingly, the KO cluster on the left was depleted of NK-like T cells (Figure 7F) and overall, KO cells showed a significant reduction in the frequency of NK-like T cells compared to WT cells at day 24 (Figure 7G). This finding was validated in an independent CAR T donor for ID3 KO cells at day 20 CAE (Figure S7F).
We calculated a “dysfunction score” for each cell by taking the average expression level of the 30 genes in our signature. Importantly, we found cells that expressed the highest dysfunction score (in red) were clustered to the right (Figure 7H), coincident with the cluster of NK-like T cells (Figure 7F); overall, the KO conditions displayed a significant decrease in the dysfunction score per cell (Figure 7I). This finding was reproduced in an independent CAR T donor for WT and ID3 KO conditions at day 20 CAE (Figure 7J). A dot plot also revealed downregulation of the dysfunction signature in ID3 and SOX4 KO cells (Figure 7K). Interestingly, we detected significant loss of SOX4 expression in the ID3 KO cells, suggesting that SOX4 is a putative ID3 target (Figure 7L). Hence, the ID3 KO cells resembled a double KO as they lacked both ID3 and SOX4 expression. AFAP1L2 and CSF1 (genes upregulated in CAE) displayed chromatin opening in day 28 CAE cells at SOX4 motifs (see Figure 6G and 6I), and these genes were significantly downregulated in KO cells and are thus putative SOX4 target genes (Figure 7M and 7N). Of note, ID3 was significantly downregulated in SOX4 KO cells (Figure 7O), although expression was not abrogated, suggesting ID3 may have additional transcriptional regulators. Select genes significantly downregulated in both KO conditions include LAYN, CD9, TNFRSF18, GNLY, and KLRC1 (Figure 7P–T).
To determine whether KO of ID3 or SOX4 associated with increased effector function, we performed cytotoxicity assays following CAE with WT, ID3 KO, and SOX4 KO cells. Importantly, ID3 and SOX4 KO cells showed enhanced CAR T killing of tumor cells after CAE compared to WT cells (Figure 7U and S7G–I).
Discussion
Several recent studies have suggested that T cell dysfunction is a major contributor to ineffective CAR T cell therapy in solid tumors (Poorebrahim et al., 2021). However, little is known about the mechanisms mediating loss of CAR T cell function. Here we examine how prolonged exposure to tumor antigen (CAE) in an in vitro model, as similarly encountered by CAR T cells in the TME, impacts the efficacy, surface expression, and phenotype of CAR T cells. We show the acquisition of a CAR T dysfunction or exhaustion gene signature and the transcription factors that regulate this transition. Moreover, we identify multiple mechanisms of CAR T dysfunction and demonstrate their relevance to patients treated with CAR T cell therapy.
Indeed, we identified a mechanism of CAR T cell dysfunction whereby cells undergo a transition from T cells to NK-like T cells. Our findings are supported by reports that CD8+ T cells acquire innate like characteristics by expressing NK receptors during chronic antigen exposure (Balin et al., 2018; Seyda et al., 2016; Wencker et al., 2014), and by observations of increased expression of NK receptors on tumor-infiltrating CD8+ T cells isolated from patients with hematological malignancy and solid tumors (Barbarin et al., 2017; Mathewson et al., 2021). Several studies have shown that NKG2A and KLRB1 act as immune checkpoints and that blocking these receptors improves the efficacy of immunotherapies (Abd Hamid et al., 2019; Andre et al., 2018; Mathewson et al., 2021; van Montfoort et al., 2018). Further, CD8+ cytotoxic T lymphocytes (CTLs) expressing cytotoxic granule proteins perforin, granzyme B, granulysin, and NK receptor NKG2C mediate TCR-dependent and independent anti-microbial activity (Balin et al., 2018). Interestingly, in addition to NK receptors, CAR T cells in our in vitro model express all three cytotoxic granule protein genes. Furthermore, plasticity of CTLs to NK-like cells has been observed in celiac disease (Meresse et al., 2004). Together, these data support that NK-like T cells have an important role in immunity and that T cells can undergo a transition to NK-like T cells. Under prolonged CAE, CAR T cells both fail to re-express surface CAR and exhibit a significant decrease in the expression of genes involved in the antigen presentation pathway (see Figure 2C), leading us to speculate that these conditions may select for T cells that transition to NK-like T cells because NK receptors provide needed signals required for T cell survival. Expression of the inhibitory NK receptors, such as CD94-NKG2A, KLRB1 (CD161), TIGIT, and inhibitory KIR may initially serve as a feedback mechanism to dampen excessive stimulatory signaling to avoid activation-induced cell death induced by TCR or CAR.
Single-cell gene expression data from CAE CAR transduced CD8+ T cells uncovers both non-dysfunctional and dysfunctional clusters. The dysfunctional T cell clusters are defined by a robust gene expression signature that includes genes implicated in T cell exhaustion such as HAVCR2 (Sakuishi et al., 2010; Singer et al., 2016), LAYN (Zheng et al., 2017), PHLDA1 (Li et al., 2019), and TNFRSF9 (Mognol et al., 2017) and genes with no known connection to dysfunction including RGS16, SRGAP3, DUSP4, NDFIP2, and CD9. CAR expression was predominately detected in the dysfunctional clusters, with minimal expression in the non-dysfunctional clusters, indicating that chronic stimulation of CAR T cells is driving the dysfunction phenotype. Strikingly, we observed robust alignment of the dysfunction gene signature identified in our in vitro CAR T CAE model with gene expression changes in hypofunctional NY-ESO-1 TILs isolated from in vivo tumors; this important correlation suggests that our dysfunction signature is relevant to gene engineered cell therapy, independent of whether CAR- or TCR-mediated. Further, hypofunctional TILs isolated from mice with relapsing mesothelin positive AsPC-1 tumors following M5CAR T cell injection also expressed NK receptors and exhaustion markers, similar to our in vitro data in Figure 4A. This data, in conjunction with NY-ESO-1 TIL data, provides in vivo demonstration that cells exhibiting the exhaustion signatures are dysfunctional in vivo.
We further investigated the regulatory mechanisms driving CAR T cell dysfunction. We find the transcription factors SOX4 and ID3 regulate genes in the dysfunction signature. Notably, our finding of improved CAR T cell killing in ID3 and SOX4 KO human CAR T cells demonstrates a role for these transcription factors in the dysfunction of CAR T cells. ID3 is important for promoting the thymic development of bipotential NK/T progenitors to an NK cell fate (Leong et al., 2017) and forced expression of ID3 blocks T cell and promotes NK cell development in a fetal thymic organ culture system (Heemskerk et al., 1997). SOX4 has been shown to control thymic production of iNKT cells by inducing microRNA-181 (Mir181) to enhance TCR signaling (Malhotra et al., 2018). ID3 and SOX4 are also key transcription factors in memory CD8+ T cell development (Hu and Chen, 2013; Ji et al., 2011; Yang et al., 2011) and Prdm1 and Id3 expression distinguish distinct CD8+ T cell subsets in acute viral and bacterial infections and tumors (Milner et al., 2020). Our observation that ID3 plays a role in T cell dysfunction is supported by Li et.al., who identify ID3 as one of nineteen transcription factors computationally predicted to regulate dysfunctional melanoma TILs isolated from human patients (Li et al., 2019). Further, Id3hi/Prdm1lo mouse TILs show enrichment of gene-expression signatures associated with progenitor exhausted T cells (Milner et al., 2020) and Id3 expression delineates progenitor exhausted T cells in a LCMV model of chronic viral infection (Utzschneider et al., 2020). However, GSEA analysis of our CAE CAR T cells, which express both ID3 and PRDM1, reveals significant enrichment with genes upregulated in intermediate and terminally exhausted T cells, but not progenitor populations (Beltra et al., 2020). In addition, compared to exhausted WT cells, chronically infected Tox-deficient T cells are negatively enriched for the SOX4 transcription factor network, indicating that Sox4 may collaborate with Tox and other transcription factors in the development of exhaustion upon chronic infection (Khan et al., 2019). Moreover, SOX4 is downregulated in two persistent clonotypes of a mutated KRAS (G12D)-reactive TIL infusion product from a patient with metastatic colorectal cancer (Lu et al., 2019), suggesting that its downregulation may contribute to persistence in adoptive cell therapy. Importantly, our finding of improved CAR T cell killing in ID3 and SOX4 KO human CAR T cells demonstrates a role for these transcription factors in the dysfunction of CAR T cells.
In summary, our robust in vitro model of dysfunction in pancreatic cancer reveals multiple mechanisms of CAR and TCR T cell dysfunction, including features of exhaustion and transition of CD8+ T cells to an NK-like T cell state. Importantly, we demonstrate that these in vitro observations are relevant in vivo both in mouse models of CAR T and TCR dysfunction and in patients after CAR T cell infusion. We further confirm the predictive value of the model whereby disruption of the transcription factors ID3 and SOX4 in CAR T cells diminishes the dysfunctional gene expression signature and, importantly, enhances tumor killing. In conclusion, our in vitro model of human T cell dysfunction provides a validated platform that can lead to the development of new strategies to improve the efficacy of CAR and TCR T cell therapy in solid tumors.
Limitations of the Study
While gene expression signatures obtained from dysfunctional CAR T cells in vitro significantly overlaps with in vivo models of T cell dysfunction, our model does not recapitulate other facets of T cell exhaustion, including the immunosuppressive effects of the TME or tumor cell line specific effects contributing to exhaustion. Further, while our data show that KO of ID3 and SOX4 improves effector function in vitro, this study does not test KO CAR T cells using in vivo models and thus follow-up studies are needed to determine whether these transcription factors function similarly in vivo.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Carl June (cjune@upenn.edu).
Material Availability
CAR construct used in this study will be provided under a material transfer agreement. sgRNAs and primer sequences generated in this study are provided in the Key resources table. Anti M5 idiotype antibody was provided by Novartis.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-human CD45 | Biolegend | Cat# 304032 |
| anti-human CD45 | Biolegend | Cat# 304017 |
| anti-human CD45 | Biolegend | Cat# 304028 |
| anti-human CD3 | Biolegend | Cat# 317322 |
| anti-human CD8 | Biolegend | Cat# 344748 |
| anti-human CD4 | Biolegend | Cat# 357412 |
| anti-human CD4 | Biolegend | Cat# 317440 |
| anti-human CD4 | Biolegend | Cat# 317428 |
| anti-human CD56 | Biolegend | Cat# 304608 |
| anti-human EpCAM | Biolegend | Cat# 324226 |
| anti-human EpCAM | Biolegend | Cat# 324238 |
| anti-human anti-human CD94 | Biolegend | Cat# 305520 |
| anti-human KLRB1 | Biolegend | Cat# 339918 |
| anti-human TIGIT | Biolegend | Cat# 372716 |
| anti-human TCR Va24-Ja18 | Biolegend | Cat# 342922 |
| anti-human PD-1 | Biolegend | Cat# 329928 |
| anti-human TIM3 | Biolegend | Cat# 345014 |
| anti-human LAG3 | Biolegend | Cat# 369315 |
| anti-human Mesothelin | Biolegend | Cat# 530203 |
| anti-human CD45RO | Biolegend | Cat# 304244 |
| anti-human CD8 | BD Pharmingen | Cat# 560179 |
| anti-human CCR7 | BD Pharmingen | Cat# 561271 |
| anti-human NKG2A | R&D Systems | Cat# FAB1059P |
| anti-human Mesothelin | R&D Systems | Cat# FAB32652P |
| anti-human CTLA-4 | eBioscience | Cat# 12–1529-42 |
| anti-human NKG2C (130–103-636, REA205 | Miltenyi Biotec | Cat# 130–103-636 |
| anti-human IgG F(ab’)2 | Jackson ImmunoResearch | Cat# 109–066-006 |
| mouse IgG2a, κ Isotype control Antibody | Biolegend | Cat# 400269 |
| mouse IgG1, κ Isotype control Antibody | Biolegend | Cat# 400126 |
| mouse IgG1, κ Isotype control Antibody | Biolegend | Cat# 400168 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Invitrogen | C7373–03 |
| Biological samples | ||
| T lymphocytes from human healthy donors | UPenn Human Immunology Core | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Live/Dead Aqua | ThermoFisher | Cat# L34957 |
| Zombie NIR (Biolegend) Fixable Viability Kits | Biolegend | Cat# 423106 |
| Apotracker™ Green | Biolegend | Cat# 427403 |
| Alt-R® Cas9 Electroporation Enhancer, 10 nmol | Integrated DNA Technologies | Cat# 1075916 |
| SpyFi Cas9 | Aldeveron | Cat# 9214 |
| P3 Primary cell 4D-nucleofactor X Kit L | Lonza | Cat# V4XP-3024 |
| OpTmizer T Cell Expansion SFM | Gibco | Cat# A1048501 |
| Human AB Serum | GeminiBio | Cat#100–512 |
| Recombinant Human IL-7 | Peprotech | Cat#200–07 |
| Recombinant Human IL-15 | Peprotech | Cat#200–15 |
| DNAse I roche | Sigma | Cat#10104159001 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#1166801 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat#L3000015 |
| Matrigel Membrane Matrix | Corning | Cat#356234 |
| Collagenase D | Sigma | Cat# 11088866001 |
| DNase I from bovine pancreas | Sigma | Cat# 11284932001 |
| EDTA (0.5 M), pH 8.0, RNase-free | Thermo Fisher | Cat# AM9261 |
| FITC Streptavidin | Biolegend | Cat# 405202 |
| Alexa Fluor® 488 Streptavidin | Biolegend | Cat# 405235 |
| APC Streptavidin | Biolegend | Cat# 405235 |
| Critical commercial assays | ||
| RNA Clean & Concentrator™−5 | ZYMO | R1016 |
| EZ-Tn5™ Transposase | Lucigen | TNP92110 |
| SMARTScribe™ Reverse Transcriptase | Takara | 639536 |
| AGENCOURT® AMPURE® XP | beckmancoulter | A63881 |
| DNA Clean & Concentrator™−5 | ZYMO | D4014 |
| TAGMENT DNA BUFFER | Illumina | 15027866 |
| TDE1,TAGMENT DNA ENZYME | Illumina | 15027865 |
| NEBNext® Library Quant Kit for Illumina® | New England Biolabs | E7630L |
| NextSeq 500/550 High Output Kit (75 cycles) v2.5 kit | Illumina | 20024906 |
| NextSeq 500/550 High Output Kit v2.5 (150 Cycles) | Illumina | 20024907 |
| Chromium Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.1 | 10X Genomics | 1000128 |
| Single Index Kit T Set A | 10X Genomics | 1000213 |
| Chromium Single Cell 5’ Library & Gel Bead Kit | 10X Genomics | 1000014 |
| Chromium Single Cell V(D)J Enrichment Kit, Human T Cell | 10X Genomics | 1000005 |
| Chromium Single Cell 3’ Library & Gel Bead Kit v3 | 10X Genomics | 1000092 |
| DynaBeads CD3×28 (Human) | ThermoFisher | Cat# 11131D |
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | Cat# 74609.50 |
| QIAGEN Plasmid Plus Maxi Kit | QIAGEN | Cat# 12963 |
| True-Nuclear™ Transcription Factor Buffer Set | Biolegend | Cat# 424401 |
| Foxp3 / Transcription Factor Staining Buffer Set | Life Technologies | Cat# 00–5523-00 |
| CD56 Microbeads | Miltenyi Biotec | Cat# 130–050-401 |
| CountBright Absolute Counting Beads, (ThermoFisher) | Thermo Fisher | Cat# C36950 |
| LongAmp™ Taq 2X Master Mix | New England Biolabs | Cat# M0287S |
| Vacuum Filter/Storage Systems | Corning | Cat# 430770 |
| Dead cell removal kit | Miltenyi Biotec | Cat# 130–090-101 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE160174 |
| LCMV mouse model naïve and exhausted T cell RNA-seq datasets | (Pauken et al., 2016) | GEO: GSE86881 |
| Human PD1 high CD8 T cell ATAC-seq datasets | (Philip et al., 2017) | GEO: GSE89308 |
| Experimental models: Cell lines | ||
| Human (female) HEK293T | ATCC | CRL-11268 |
| Human (female) K562 | This paper | N/A |
| Human (female) ASPC-1 | ATCC | CRL-1682 |
| Experimental models: Organisms/strains | ||
| NOD/scid/IL2rγ−/− (NSG) | Jackson Laboratory | Cat# 5557 |
| Oligonucleotides | ||
| TSO (SMARTseq2): AAG CAG TGG TAT CAA CGC AGA GTA CAT rGrGrG | (Picelli et al., 2014) | N/A |
| Oligo-dT30VN (SMARTseq2): AAG CAG TGG TAT CAA CGC AGA GTA CTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TVN | (Picelli et al., 2014) | N/A |
| ISPCR (SMARTseq2): AAG CAG TGG TAT CAA CGC AGA GT | (Picelli et al., 2014) | N/A |
| Tn5MErev (SMARTseq2): /5Phos/CT GTC TCT TAT ACA CAT CT | (Picelli et al., 2014) | N/A |
| Tn5ME-A (SMARTseq2): TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG | (Picelli et al., 2014) | N/A |
| Tn5ME-B (SMARTseq2): GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA G | (Picelli et al., 2014) | N/A |
| Ad1_noMX (ATAC-seq): AATGATACGGCGACCACCGAGATCTACACTCGTCGGCAGCGTCAGATGTG | (Corces et al., 2017) | N/A |
| ID3sgRNA#2: 5’-TGGCTAAGCTGAGTGCCTCT-3’ | Integrated DNA Technologies | Hs.Cas9.ID3.1.AA |
| ID3sgRNA#2: 5’-TGGCCAGACTGCGTTCCGAC-3’ | Integrated DNA Technologies | N/A |
| SOX4 sgRNA #1: 5’-GCTGGTGCAAGACCCCGAGT-3’ | Integrated DNA Technologies | Hs.Cas9.SOX4.1.AL |
| SOX4 sgRNA #2: 5’-AGGAGGCGATTCCCAGCTCG-3’ | Integrated DNA Technologies | N/A |
| ID3.PCR F(genomic DNA): 5’-ATAAAGAGGCGTGCCTTCCA-3’ | Genewiz | N/A |
| ID3.PCR.R(gDNA): R 5’- CATCCTTGCCTGGGTGTTCA-3’ | Genewiz | N/A |
| ID3.Seq.F (gDNA): 5’-TTCTCTTTGGGGCACCTCTG-3’ | Genewiz | N/A |
| ID3.Seq.R (gDNA): 5’-GAAGGTGGGGGCCATCAG-3’ | Genewiz | N/A |
| SOX4.PCR.F (gDNA and cDNA): 5’-CGGAGAACTCCTTCCCCAAATC-3’ | Genewiz | N/A |
| SOX4.PCR.R (gDNA and cDNA): 5’-CTCTTTTTCTGCGCCGGTTTG-3’ | Genewiz | N/A |
| SOX4.Seq.F (gDNA and cDNA): 5’-CCGCGAGGGTGTGAGC-3’ | Genewiz | N/A |
| SOX4.Seq.R (gDNA and cDNA): 5’-TGTAGTCGGGGTAGTCAGCC-3’. | Genewiz | N/A |
| ID3.cDNAPCR.F (gDNA): 5’- TTGCAGGTCACTGTAGCGG-3’ | Genewiz | N/A |
| ID3.cDNAPCR.R (gDNA): 5’- AGGCCACAAGTTCACAGTCC-3’ | Genewiz | N/A |
| ID3.cDNASeq.F (gDNA): 5’- TCTTTCTCTTTGGGGCACCTC-3’ | Genewiz | N/A |
| ID3.cDNASeq.R (gDNA): 5’- TGGTGAAGTCAAGTGGGCAG-3’ | Genewiz | N/A |
| H3Histone Poscntrl Human F 5’-AAAGCCGCTCGCAAGAGTGCG-3’ | Genewiz | N/A |
| H3Histone Poscntrl Human R 5’-ACTTGCCTCCTGCAAAGCAC-3’ | Genewiz | N/A |
| Recombinant DNA | ||
| pTRPE M5BBz | This paper | N/A |
| Software and algorithms | ||
| Unique code | This paper | https://github.com/bergerlabupenn/InVitroCARTexh_code_2020 |
| R version 3.6.2 | CRAN | https://cran.r-project.org/ |
| Seurat_3.2.3 | (Butler et al., 2018; Stuart et al., 2019) | https://satijalab.org/seurat/ |
| Cell Ranger v3.1.0 | 10X Genomics | https://www.10xgenomics.com/ |
| sctransform_0.3.2 | (Hafemeister and Satija, 2019) | https://cran.r-project.org/web/packages/sctransform/index.html |
| Metascape | (Zhou et al., 2019) | https://metascape.org/gp/index.html#/main/step1 |
| Monocle 3 | (Qiu et al., 2017; Trapnell et al., 2014) | https://www.bioconductor.org/packages/release/bioc/html/monocle.html |
| Cellfishing.jl | (Sato et al., 2019) | https://github.com/bicycle1885/CellFishing.jl |
| samtools v1.1 | (Li et al., 2009) | http://www.htslib.org/download/ |
| STAR v2.5.2a | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| HTSeq v0.6.1 | (Anders et al., 2015) | https://htseq.readthedocs.io/en/master/install.html |
| HOMER v4.6 | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/introduction/install.html |
| bowtie2 v2.3.4.1 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml |
| FlowJo™ v10.8 Software | BD Life Sciences |
https://www.flowjo.com |
| Ingenuity Pathway Analysis Software | QIAGEN | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| Other | ||
| CRISPick sgRNA designer ( [2020]) | The Broad Institute | https://portals.broadinstitute.org/gppx/crispick/public |
| Benchling sgRNA designer tool (https://www.benchling.com, [2020]) | Benchling | https://benchling.com/ |
| Synthego’s Performance Analysis ICE (short for Inference of CRISPR Edits) tool | Synthego | https://ice.synthego.com/ [2021]). |
| BioRender illustration design tool | BioRender | https://biorender.com/ |
Data and Code Availability
All genomics data have been submitted to the gene expression omnibus database and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
Unique computer code used in this manuscript has been submitted to GitHub and can be accessed using the following link https://github.com/bergerlabupenn/InVitroCARTexh_code_2020.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
AsPC-1, K562, and HEK293T cells were obtained from American Type Culture Collection (ATCC). AsPC-1 cells were grown in D20 media consisting of DMEM/F12 (1:1) (Gibco, Life Technologies), 20% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Gibco, Life Technologies) and K562 and HEK293T cells were cultured in R10 media consisting of RPMI-1640 (Gibco, Life Technologies) with 10% FBS, 2% HEPES (Gibco), 1% of GlutaMAX™ (Gibco), and 1% of penicillin/streptomycin. GFP-expressing cell lines were generated by lentiviral transduction for cell killing assays. All cell lines were routinely authenticated by the University of Arizona Genetics Core and tested for mycoplasma contamination (MycoAlert Mycoplasma Detection Kit, Lonza).
Mice
Animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Six- to eight week-old male NOD/scid/IL2rγ−/− (NSG) were procured from Jackson Laboratories and bred in the vivarium at the University of Pennsylvania in pathogen-free conditions. Mice were maintained under pathogen free conditions.
Human samples
Healthy donor primary T lymphocytes were provided by the University of Pennsylvania Human Immunology Core. Samples are deidentified for compliance with HIPAA rules. Donor sex and age is shown below: ND516 (female, age 37), ND538 (female, 48), ND388 (male, 53), ND534 (male, 28), ND150 (male, 40), ND552 (female, 26), ND539 (male, 39), ND566 (female, 26).
Post-CAR19 infusion PBMCs samples were collected from DLBCL patients who were enrolled in CTL019 clinical trial NCT02030834. Patients enrolled in this trial had received previous primary and salvage therapies, relapsed/residual disease after autologous stem-cell transplantation, or were not eligible for autologous or allogeneic stem-cell transplantation. Post-M5CAR infusion peritoneal/pleural fluid samples were collected from ovarian cancer patients (02916–01 and 02916–06) enrolled on a M5CAR T cell trial (NCT03054298). Patients enrolled in this trial had recurrent disease after at least one prior standard of care chemotherapy for advanced stage disease.
METHOD DETAILS
General cell culture
AsPC-1, K562, and HEK293T cells were obtained from American Type Culture Collection (ATCC). AsPC-1 cells were grown in D20 media consisting of DMEM/F12 (1:1) (Gibco, Life Technologies), 20% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Gibco, Life Technologies) and K562 and HEK293T cells were cultured in R10 media consisting of RPMI-1640 (Gibco, Life Technologies) with 10% FBS, 2% HEPES (Gibco), 1% of GlutaMAX™ (Gibco), and 1% of penicillin/streptomycin. GFP-expressing cell lines were generated by lentiviral transduction for cell killing assays. All cell lines were routinely authenticated by the University of Arizona Genetics Core and tested for mycoplasma contamination (MycoAlert Mycoplasma Detection Kit, Lonza).
Lentiviral vector production
Lentiviral vector production was performed as previously described (Kutner et al., 2009). Briefly, HEK293T cells were transfected with lentiviral CAR and packaging plasmids using Lipofectamine 2000 or Lipofectamine 3000 (Invitrogen) following the manufacturer’s protocol. Lentiviral supernatants were collected at 24- and 48-hours post-transfection and concentrated using high-speed ultracentrifugation. To generate the lentiviral stocks, the resulting concentrated lentivirus batches were resuspended in cold R10 media and stored at −80°C.
Transduction of CAR-redirected human T cells
The M5CAR is a second-generation CAR containing a human MSLN-binding scFv and CD8α hinge and transmembrane domains fused to 4–1BB and CD3-ζ cytoplasmic signaling domains. Primary human CD4+ T and CD8+ T cells from normal donors were provided by University of Pennsylvania Human Immunology Core. CAR T cells were generated as previously described (Carpenito et al., 2009). Briefly, CD4+ and CD8+ T at 1 : 1 ratio at 1 × 10e6 cells/ml were activated with Dynabeads® CD3/CD28 CTS™ (Thermofisher) at a 3 : 1 bead-to-cell ratio. Approximately after 24 hours, T cells were transduced at a multiplicity of infection (MOI) of 3 to 5. At day 5 beads were removed from cultures. T cell cultures were maintained at 8 × 10e5 cells/ml. Cell number and volume were monitored daily using Multisizer 4 Coulter Counter (Beckman). Transduced T cells were cryopreserved when reached the resting state, as determined by cell size.
CAR T cell in vitro dysfunction model
AsPC-1 cells were routinely seeded in 6-well plates at 1 × 10e6 cells/well the day preceding T cell seeding. M5CAR T cells (30 – 50% of transduction efficiency) were thawed and rested at 1 × 10e6 cells/ml in T75 flasks with R10 media. After 24 hours, the T cell number (CD45+EpCAM-) was calculated and 2.5 × 10e5 T cells/well were transferred to the AsPC-1 plates. After 3 – 4 days, the cocultures were thoroughly suspended by frequent pipetting and 300 – 400μl of the cell suspension was used for T cell counting assessment and flow cytometry staining. The remaining cell suspension was spun down and the supernatant (conditioned media) was collected and filtered with a 0.45 μm filter (Corning). The cells were resuspended in media containing equal amounts of conditioned and fresh R10. The resulting T cell suspension was transferred into AsPC-1-coated plates cells (2.5 × 10e5 T cells/well) for continuous co-culture. This process was repeated for 20–35 days.
Flow cytometry and sorting
For flow cytometry and sorting assays of CAE, cell suspensions from M5CAR T cell expansions, in vitro cocultures and recurrent AsPC-1 tumors were stained in fluorescence-activated cell sorting (FACS) buffer consisting of PBS (Gibco), 0.5% bovine serum albumin (BSA) (GEMINI), 2 mM EDTA (Invitrogen), and 100 μg/ml DNase (Roche). CountBright™ Absolute Counting Beads, (ThermoFisher) were used as an internal standard according to the manufacturer’s instructions to calculate absolute cell counts in cell suspensions. Antibodies used for surface and intracellular stainings are detailed in the Key resources table. M5CAR expression was assessed using biotinylated goat anti-human IgG F(ab’)2 (Jackson ImmunoResearch, 109–066-006) followed by streptavidin (FITC-, AF488- or APC-conjugated, see the Key resources table) or using an anti-idiotype antibody provided by Novartis Pharmaceuticals. Live/dead staining was performed using a Live/Dead Aqua (Life Technologies) or Zombie NIR (Biolegend) Fixable Viability Kits following manufacturer’s protocol followed by cell surface staining for 15 min at 4°C in the dark. Apoptosis was assessed using Live/Dead Aqua and Apotracker™ Green according to manufacturer’s instructions Intracellular staining was performed with the True Nuclear and Foxp3/Transcription Factor Staining Buffer set (Thermo Fisher) according to the manufacturer’s instructions. Samples were acquired on an LSRII Fortessa Cytometer (BD Bioscience) and analyzed with FlowJo v10 software (FlowJo, LLC). Sorting assays were performed using a FACS Aria Cytometer (BD Bioscience).
CD56+ cell depletion
MACS Dead cell removal kit and CD56 MicroBeads (Miltenyi Biotec) were used for CD56-positive cell depletion on day 0 CAR T cell products. The CD56-depleted CAR T cell product was subjected to CAE protocol as described above and the frequency of CD56+ T cells was assessed by flow cytometry.
The out-competition model assumes that initial depletion of the NK-like-T cell population would result in altered kinetics of NK-like-T cell abundance over time compared to a non-depleted control group, whereas transitioning assumes similar kinetics between the control and depleted groups. As shown in Figure S6F, in case of out-competition by the CD56-positive cell subset (left panel), the frequency of CD56 in the CD56-depleted cultures increase at a lower rate than in the controls. This growth can be expressed by the formula PT = (P0 − d) × kt. On the other hand, if T cell are transitioning into NK-like-T cells, (S6F, right panel), the frequency of CD56 in the cocultures would increase at the same rate over the time, independently of the initial depletion of the CD56 at the start of the coculture, which can be expressed as PT = (P0 − d) + k × t. PT: percentage CD56-positive cells at time “T”. P0: Percentage CD56-positive cells at time zero. t: time of in vitro stimulation [Days]. k: transition constant. D: percentage CD56-positive cells depleted.
Clinical trial design and research participants
Single-institution pilot safety and feasibility trial was conducted at University of Pennsylvania. This study is registered at www.clinicaltrial.gov as #NCT03054298. 1 – 3 × 10e7 M5CAR T cells/m2 were intravenously infused into patients who were diagnosed with ovarian cancer. Pleural fluid (patient 06) or peritoneal fluid (patient 01) were collected (06: day 36, 01: day 21) and surface and intracellular CAR expression was analyzed by flow cytometry. PBMCs collected from patients who received CD19CAR (CTL019) T cells to treat DLBCL (www.clinicaltrial.gov number, NCT02030834) and CTL019 T cell products were used for identifying NK-like CAR T cells in human. Fifty-two DLBCL patients were enrolled and 35 patients were excluded as CD56 expression was not examined. CTL019 T cell expansion in the patient’s blood was analyzed by qPCR and the peak time point of expansion was selected to examine the frequency of NK-like CAR T cells. To investigate the expression of NK-related molecules on CAR T cells, cryopreserved materials from patient 13413–39 (CTL019 T cell product and PBMCs collected 27 days after CAR T infusion) were thawed and analyzed by flow cytometry.
Production of Human CRISPR-Engineered CAR-T Cells.
Single guide RNA (sgRNA) sequences targeting ID3 and SOX4 were designed using CRISPick sgRNA designer (the Broad Institute [2020]) and Benchling online software (https://www.benchling.com, [2020]) and were synthesized by Integrated DNA Technologies (IDT). Two of five sgRNAs targeting each gene were selected for further experiments after pre-validation. Gene disruption, T cell activation, transduction, expansion, and knockout validation of ID3KO and SOX4KO M5 CAR T cells were performed following an optimized protocol previously described (Agarwal et al., 2021). Briefly, CD4+ and CD8+ T at 1 : 1 ratio were incubated in OpTmizer T Cell expansion media (Gibco) supplemented with 5 ng/mL of huIL-7 and huIL-15 each(Preprotech) (OPT 7/15 media). After 24h, cells were collected and resuspended at 1× 10e8 cells/mL in P3 nucleofection solution (Lonza). The ribonucleoprotein (RNP) complexes were generated by incubating each sgRNA (5 μg per 10 × 10e6 cells) individually with the Cas9 nuclease (Aldevron, 10 μg per 10 × 10e6 cells) for 10 min at room temperature. Cells were electroporated in batches of 10 × 10e6 cells (100 μL) with a mixture of RNP complex plus 16.8 pmol of electroporation enhancer (IDT) into electroporation cuvettes (electroporation code EH111) in a 4D-Nucleofactor X-Unit (Lonza). After electroporation cells were grown in OPT 7/15 media at 5 × 10e6cells/mL at 37°C and activated 4 to 6h later with anti-CD3/anti-CD28 monoclonal antibody-coated magnetic beads. After 24 h, T-cell were lentivirally transduced and expanded as described above.
Since each target locus was defined by two sgRNA cut sites (spanned 100 and 130 bp for SOX4 and ID3, respectively), PCR primers and sequencing primers were designed to detect each target locus. LongAmp™ Taq 2X Master Mix (NEB) was used for target sequence amplification and used following manufacturer’s protocol and NucleoSpin Gel and PCR Clean-up (Macherey-Nagel) was used for DNA purification. Analysis of gene editing efficiency was assessed by Sanger sequencing. We obtained two sets of KO T cells per group: one bearing small insertions and deletions due to a single sgRNA hit, and a second population of CAR T cells bearing a large fragment deletion as a result of a double sgRNA hit. Synthego’s Performance Analysis ICE (short for Inference of CRISPR Edits) tool, was used to calculate the editing efficiency (https://ice.synthego.com/ [2021]). The sequences used for KO generation and editing efficiency validation are listed in the Key resources table. The schematic representation of the in vivo experiments of figure 4 and the knockout strategy in figure panel 7A were created using BioRender.com.
Cytotoxicity assays
Cytotoxic killing of target cells was assessed using a real-time, impedance-based assay with xCELLigence Real-Time Cell Analyzer System (ACEA Biosciences). Briefly, 1 × 10e4 AsPC-1 cells were seeded to the 96-well E-plate. After 24 hours, sorted CD8+ CAE surCARpos T cells (day 28 CAE, day 0 product and CD19BBz) were added to the wells in 4 : 1 E:T ratio. Tumor killing was monitored every 20 min over 4 days. To evaluate the cell killing capacity of WT, ID3 KO and SOX4 KO M5CAR T cells upon CAE, cells were collected on day 18 for ND539 and day 21 and day 28 for ND566 and seeded at 1:8 E:T ratio with AsPC-1. Tumor killing was monitored every 20 min over 8 days. Significant differences between groups were assessed by two-way ANOVA and Dunnet’s post hoc test.
High-throughput cytotoxicity assay using Celigo Image Cytometer (Nexcelom Bioscience) was used to investigate the effects of the resting with cytokine supplement on cytotoxicity of CAR T cells. CD8+ M5CAR T cells were sorted after CAE, counted and the viability assessed using Moxi Flow System (Orflo Technologies). Part of the cell suspension was cocultured with 1.5 × 10e3-2 × 10e3 AsPC-1-GFP cells immediately after sorting in a 7 : 1 E:T ratio and the rest was left resting at 1.0× 10e6 cells/ml in fresh R10 media with IL-15 supplement (20 ng/ml). After 24 hr, cell viability was examined and rested T cells were cocultured with AsPC-1-GFP cells in identical conditions as the non-rested counterparts. The % lysis was calculated by direct cell counting of live fluorescent target cells. % Lysis = (1 – count # of live target cells (GFP) in wells with effector cells / count # of live target cells (GFP) in wells without effector cells) × 100.
Cytokine production
Fifty thousand CD8+ surCARpos T cells (day 28 CAE, day 0 product and CD19BBz) were cocultured with 5 × 10e4 AsPC-1 cells or left in R10 media in 48 well plate. After 48 hr, supernatant was collected and analyzed by high-sensitivity LUMINEX assay according to manufacturer’s instructions (Merck Millipore).
Quantitative real-time PCR (qPCR)
Surface CAR-positive and -negative CD8+ T cells were sorted on days 4, 7, and 17 after CAE and genomic DNA was isolated from sorted cell pellets using an ArcturusTM PicoPureTM DNA Extraction Kit (Applied Biosystems). qPCR was performed in triplicate with TaqMan Gene Expression Master Mix on a 7500Fast Real-Time PCR System (Applied Biosystems) per the manufacturer’s instructions. The validated primers specific to the 4–1BB and CD3ζ fusion gene and probes specific for the fusion fragment and labeled with compatible reporter dyes (FAM or VIC) were used to detect the CAR. The average plasmid copy per cell was calculated based on the factor 0.0063 ng /cell. Nine μL DNA was loaded directly for quantitation by p21 qPCR. A correction factor (CF) was not used for calculating the average % marking and copies/μg DNA as the amount of actual DNA loaded was accurately quantified by p21.
CAR re-expression assay
SurCARneg CD8+ T cells were sorted after 23 days of CAE, rested in fresh R10 media with IL-15 supplement (20 ng/ml) for 24 hrs and examined for surface CAR expression by flow cytometry.
CyTOF
Mass cytometry antibodies were obtained as pre-conjugated metal-tagged antibodies from Fluidigm or prepared using the Maxpar antibody conjugation kit (Fluidigm) according to the manufacturer’s protocol. Following labeling, antibodies were diluted in Candor PBS Antibody Stabilization solution (Candor Bioscience GmbH, Wangen, Germany) supplemented with 0.02% NaN3 to 0.25 mg/mL and stored long-term at 4º C. Each antibody was titrated to optimal staining concentrations using primary human PBMCs.
CAE CD8+ CART cells and CD8+ CART product were washed and resuspended 1:1 with PBS containing EDTA and 20 μM cisplatin for 2 min before quenching 1:1 with CSM (cell staining medium: PBS with 0.5% BSA and 0.02% NaN3) for dead cell discrimination. After washed, the cells were fixed for 10 min at RT using 1.6% paraformaldehyde (PFA) in PBS and frozen in CSM with 10% DMSO at −80ºC. CAE CD8+ CART cells and CD8+ CART product were barcoded with distinct combinations of stable Pd isotypes in Barcode Perm Buffer (Fluidigm). Cells were washed twice with CSM, and once with PBS, and pooled into a single tube. Cells were blocked with human FcR blocking reagent (BD Bioscience) for 10 min at RT. Cells were then incubated with all antibodies targeting cell surface markers for 30 min at RT. After washed, cells were fixed with 1.6% PFA and permeabilized with Perm-S buffer (Fluidigm). Fixed/permeabilized cells were incubated with all antibodies targeting intracellular antigens for 30 min at room temperature. After washed with CSM, cells were incubated in 4% PFA in PBS with 191/193-iridium intercalator (Fluidigm) for 48 hrs. Cells were washed in CSM, PBS, and then deionized H2O. Cells were resuspended in deionized H2O containing EQ four-element beads (Fluidigm) to approximately 1 × 106 cells and then analyzed on Helios CyTOF system (Fluidigm) at Flow Cytometry Core, University of California, San Francisco. The acquisition data were normalized with premessa package and analyzed with cytofkit package (27662185) in R software 3.6.1 (The R foundation for Statistical Computing, Vienna, Austria).
Mouse experiments
NOD/scid/IL2rγ−/− (NSG) mice were purchased from The Jackson Laboratory and bred and housed in the vivarium at the University of Pennsylvania in pathogen-free conditions. Animal studies were approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
For the NY-ESO-1 TIL mouse model, five million A549-A2-ESO tumor cells in 150 μl of Matrigel:PBS (1:1) solution were subcutaneously injected in the flanks of NSG mice. 2 × 10e7 human T cells were activated with anti-CD3 + anti-CD28 microbeads 3:1 and subsequently transduced with 3rd generation high titer lentivirus encoding for the Ly95 TCR. Transduced cells (50% of which were positive for Ly95 TCR) were intravenously injected when tumors reached a mean volume of 150 mm3. Thirty days after T cell injection, mice were sacrificed, tumors were harvested, digested, and processed. The single-cell suspension obtained was then treated with Dead Cell Removal Kit (Miltenyi Biotec) following manufacturer’s protocol, and CD3+ cells were positively selected by using an EasySep cell isolation kit (Stem Cell Technologies). The non-transduced CD8+ T cells from the same donor and the transduced NY-ESO-1 redirected infusion product were also subjected to the same digestion and processing protocols. T cells from the tumor cell suspension were stained with anti-human CD8 and anti-TCRVβ13.1 The donor’s CD8+ T cells were stained with anti-CD8 and anti-CD45RO. NY-ESO-1 T cell infusion product was stained with anti-CD8 and anti-TCRVβ13.1. All three specimens were flow sorted on the BD FACS Aria on the same day for the following populations: CD45 cells isolated from tumor digest – CD8+/ TCRVβ13.1+, donor’s untransduced CD8+ T cells – CD8+/CD45RO+, NY-ESO-1 T cell infusion product – CD8+/ TCRVβ13.1+. Sorted samples were snap frozen, subjected to RNA extraction with Qiazol (Qiagen) and gene expression microarray analyses. For genes with multiple probes, average expression values were used to make the heatmap in R (pheatmap).
For the AsPC-1 recurrence model, NSG mice were subcutaneously injected with 2 × 10e6 AsPC-1 cells suspended in 200 μl Matrigel:PBS (1:1) into the right flank. When the mean of tumor volumes reached 300 mm3, mice were treated with 1 × 10e6 ND552 M5CAR+ T cells. Tumor volumes were calculated as length × with2÷ 2. Tumor growth was weekly assessed by caliper measurement. After primary antitumor response mice were monitored for recurrence. Mice bearing recurrent tumors were sacrificed when reached the maximum size or showed evident signs of disease, and tumors were collected. Fresh tumors were excised and digested in RPMI containing collagenase D (400 Mandl units/mL, Sigma) and DNase I (50 mg/mL, Sigma) for 15 minutes at 37ºC. Enzymatic digestion was stopped with 12 μL/mL EDTA d 0.5 M, pH 8. Tumors were mechanically disrupted and filtered through a 0.7 μm cell strainer (Corning). For flow cytometry stainings, single-cell suspensions were stained with Fixable Dead Cell Dyes followed by FcR-Block treatment (Fc Receptor Blocking Solution, Biolegend) following manufacturer’s recommendations. Positive NK receptor cell subsets in D0 and recurrent samples were determined in sample-matched tumor and Day 0 FMO controls. Positive checkpoint receptor subsets were determined sample-matched tumor and Day 0 isotype controls. for checkpoint receptors. All the isotype controls were incubated at the same final concentration as their corresponding test antibody.
Single-cell RNA-seq and TCR-seq
ScRNA-seq libraries were generated using a Chromium Single-Cell 3’ Library and Gel Bead Kit (10x Genomics) using v3 for CAR T donor ND388 and v3.1 for donors ND539 and ND566 following the manufacturer’s protocol. Briefly, 16,000 CD8+ T cells were sorted by flow cytometry and washed with ice cold PBS + 0.04% BSA. After washing, cells were used to generate single-cell gel beads in emulsion. Following reverse transcription, gel beads in emulsion were disrupted and barcoded complementary DNA was isolated and amplified by PCR for 12 cycles. After fragmentation, end repair, and poly A tailing, samples indexes were added and amplified following the manufacturer’s protocol. The final libraries were quality control checked and sequenced on an Illumina NextSeq 500 with a 150-cycle kit with parameters Read 1: 28, Read 2: 130, Index 1: 8, Index 2: 0. One sample was sequenced per flow cell. For CAR T donors ND150 and ND538, scRNA-seq libraries were generated using Chromium Single-Cell 5’ Library and Gel Bead Kit and TCR libraries were generated using Chromium Single-Cell V(D)J Reagent Kits (10x Genomics) according to the manufacturer’s protocol. Followed same brief protocol as above except amplified cDNA by PCR for 13 cycles. Two uL of post amplified cDNA was used to generate TCR libraries and 50ng of cDNA was used to generate 5’ gene expression libraries. After fragmentation, end repair and poly A tailing, sample indexes were added and amplified following manufacturer’s protocol. The libraries were sequenced on an Illumina NextSeq 500 with a 150-cycle kit with parameters Read 1: 26, Read 2: 130, Index 1: 8, Index 2: 0. One RNA library and one TCR library (8:1 ratio) were pooled and sequenced on one flow cell.
Single-cell RNA-seq analysis
Sequencing data were aligned to the GRCh38 genome, filtered, and then barcodes and unique molecular identifiers were counted using the Cell Ranger v3.1.0 command cellranger count. Data were further analyzed in R using Seurat version 3.1.2 (Butler et al., 2018; Stuart et al., 2019). Briefly, genes that were not detected in at least 3 cells and cells with >5% mitochondrial reads were excluded, as well as cells that express <200 genes or >5000 genes. Data were normalized using sctransform (Hafemeister and Satija, 2019). PCA was performed on the most variable genes which were found based on average expression and variance. Clusters and UMAP were generated from the first 10 PCA dimensions using the default parameter settings in Seurat. Marker genes were determined using the FindAllMarkers function in Seurat where at least 25% of the cells must be expressing the gene. Sctransform normalized expression was used for the heatmap of marker genes, UMAP feature plots, and dot plots. Metascape was used with cluster marker genes for gene ontology analysis (Zhou et al., 2019). Monocle 3 was used for trajectory analysis with the default parameter setting and 100 PCs (Qiu et al., 2017; Trapnell et al., 2014). AddModuleScore was used to project expression of the dysfunction signature genes (N=30) onto the Monocle trajectory. Gene regulatory network inference was performed using the partial information decomposition algorithm, PIDC, on the top 500 variable genes (identified via Seurat) with a threshold for edge inclusion of 15% (Chan et al., 2017). Cellfishing.jl, a software that builds a database from single-cell data to then be queried, was used for differential expression analysis between single cell data sets (day 0 product versus day 20 CAE cells) with the default of 10 k-nearest neighbors (Sato et al., 2019). 1,834 genes were found to be differentially expressed. Data were analyzed using IPA (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis). For IPA analysis, mitochondrial genes were filtered out and only genes with fold change > 2 (N=1,442 genes) were included. Fold change was calculated as the number of cells at day 20 that upregulate the gene divided by the number of cells at day 20 that downregulate the gene compared to day 0 cells. NK-like T cells were identified using raw_counts [“KLRC1”,]>0 & raw_counts[“KLRB1”,]>0 & raw_counts[“CD3E”,]>0. Significant differences in changes in the NK-like T cell populations between WT and KO conditions were measured by Fisher’s exact test.
To identify our 30 gene dysfunction signature, we identified all genes differentially expressed between dysfunctional and non-dysfunctional clusters using Seurat’s FindMarkers function. For donor ND388, differentially expressed genes were identified between dysfunctional clusters D20–1 and D20–4 versus non-dysfunctional clusters D20–2 and D20–3. This list was further filtered by log2FC>0.64 and padj<1.0e22 (padj with Bonferroni correction using all genes in the dataset).
WT, SOX4 KO, and ID3 KO Seurat objects were combined for analysis using the merge function (for donor ND566) and WT and ID3 KO samples were combined for donor ND539. Genes that were not detected in at least 3 cells and cells with >5% mitochondrial reads were excluded, as well as cells that express < 200 genes or >5000 genes. EPCAM expression (tumor marker) was used to identify a contaminating tumor cell cluster which was subsequently removed using seurat’s subset function. CellCycleScoring was used to regress out cell cycle specific clustering using SCTransform vars.to.regress (S.Score, G2M.Score) function. SCT counts of the dysfunction signature genes (N=30) were averaged per cell to create the dysfunction score. Mann-Whitney U test was used to test significance of dysfunction score between WT and KO conditions.
To assess the expression of M5CAR in the scRNA-seq data, the cellranger reference was reindexed (mkref) by adding a single contig for the 627 bp WPRE sequence (a unique sequence in the CAR plasmid) to assembly GRCh38 of the human genome (the gene annotation GTF file was appended with CDS and exon entries spanning the entire sequence and gene_id “Ligand”). To analyze expression of CAR and to determine the percent of cells expressing the CAR (related to Figure 3H and I), we pooled data from three scRNA-seq experiments (ND388 day 20 CAE cells, ND538 and ND150 day 28 CAE cells). Cells belonging to the dysfunctional clusters and non-dysfunctional clusters were defined for each donor separately, see Figures 3 and S5.
Single-cell TCR-seq analysis
Sequencing data were aligned to the vdj-GRCh38-alts-ensembl-3.1.0 genome and processed using the cellranger vdj command in Cell Ranger v3.1.0. To assess receptor persistence, a map of full-length receptor peptide sequences to cell barcodes was loaded at both time points from filtered coverage annotation (FCA) files. Cell barcodes associated with peptide sequences in common to both time points were screened against lists of cell barcodes that express CD8A at both time points; cells without persistent CD8A expression were removed. Remaining cells were screened against barcodes of cells that express KLRB1 at either day 0 or day 28, or not at all. Sankey plots of this distribution were created using the plotly library in R. Maps were also analyzed for the number of cell barcodes associated to each full-length peptide sequence to insure that the data largely obey a one peptide : one barcode rule.
Bulk RNA-seq
RNA-seq libraries were made following the previously established SMARTseq2 protocol (Picelli et al., 2014). Briefly, total RNA was extracted using Qiazol (Qiagen) from 300 cells for day 0, day 16, and day 28 for CD8+ T cells continuously stimulated with antigen (two sorted populations including surface CAR-positive and surface CAR-negative cells). From the same experiment, CD4+ T cells were sorted, and RNA extracted for surface CAR positive cells at day 0 and day 28 CAE. Cells were recovered by RNA Clean and Concentrator spin columns (Zymo), followed by incubation with oligo-dT. The transcription reaction was carried out on 100 pg of cDNA for 1min at 55° C. Libraries were uniquely barcoded (Buenrostro et al., 2013) and amplified for 14 cycles. Fragment size distribution was verified and paired-end sequencing (2 × 75 bp reads) was carried out on an Illumina NextSeq 500.
Bulk RNA-seq analysis
Paired-end data were aligned to human genome assembly GRCh37/hg19 using STAR v2.5.2a
(Dobin et al., 2013) with command-line parameters --outFilterType BySJout
--outFilterMultimapNmax 20 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1
--outFilterMismatchNmax 999 --alignIntronMin 20 --alignIntronMax 1000000
--alignMatesGapMax 1000000. Resulting SAM files were converted to BAM format using samtools v1.1 (samtools view -bS) and BAM files were sorted by position using samtools sort. For CD8+ T cell replicate R1, several libraries were pooled after alignment to enhance coverage using samtools merge as below:
R1 Control Day 0 CAR+ T1: 4-Day0-CD8-CARpos_S7, 16-Day0-CD8-CARpos_S5
R1 Control Day 0 CAR- T1: 3-Day0-CD8-CARneg_S2, 15-Day0-CD8-CARneg_S6
R1 other CAR+ T1: 21-other-CD8-CARpos-10–24-2018_S11, 12-other-CD8-CARpos_S12, 24-other-CD8-CARpos_S10
R1 CAE CAR+ T1: 8-CAE-CD8-CARpos_S9, 20-CAE-CD8-CARpos_S12
R1 CAE CAR- T1: 7-CAE-CD8-CARneg_S6, 19-CAE-CD8-CARneg_S3
HTSeq v0.6.1 (Anders et al., 2015) was used to count aligned tags over gene features with command-line python -m HTSeq.scripts.count -f bam -r pos -s no -t exon -i gene_id BAM_FILE GTF. The GTF was constructed from RefSeq transcripts and UCSC Genome Browser’s annotation of RefSeq transcript IDs to gene symbols. For the antigen exposure and time series analysis, DESeq2 was used to adjust library size and estimate significant differences at an FDR of 0.05. The Wald test was used to assess differences between control day 0 and CAE. Other samples were included to adjust dispersions and library sizes but were not used for the contrast. LRT was used to assess differences along the time course (day 0, day 16, and day 28), with a full model of ~Replicate+Time and a reduced model of ~Replicate. For this analysis other exposure samples were not included. For the antigen exposure analysis (day 0 versus day 28 CAE, see Figure 2A), some genes were filtered out which register as significantly different, but which may be artifacts of the SMART-seq library construction; these fall along the arcs of a parabola in a volcano plot of the data. Lines were interpolated on the plot using genes along the arcs: between IL22 and WDR63 (negative) and between ALK and INBHE (positive). Genes with a perpendicular distance <1 to the lines were removed. For IPA pathway analysis and to overlap DEGs with other datasets, genes were further filtered for padj <0.05 and fold-change >2, (N=1,038 DEGs for CD8+ T cells and N=1,477 DEGs for CD4+ T cells). DESeq2 adjusted counts were used to generate gene expression plots of NK-associated molecules and DEGs (see Figure 2D and S3N). Statistics assessed by Mann-Whitney test (****P <0.0001, ***P <0.001, **P <0.01, *P <0.05). Statistics to compute significance of overlaps were assessed by hypergeometric test.
For IPA upstream regulator analysis, only transcription factors differentially expressed in CAE are shown (p value <2e-5 and log2 fold change >1). However, a full list of upstream regulators can be found in Table S3. For GSEA analyses, DESeq2 adjusted counts for all genes from CD8+ T cell bulk RNA-seq datasets for day 0 and day 28 CAE samples were uploaded. To perform GSEA with the four transient states of exhausted T cells (Figure S3E) we downloaded data from supplemental table 1 (Beltra et al., 2020) to identify genes upregulated compared to at least one of the other 3 transient exhausted T cell states. Only genes with mouse to human orthologues were used for the analysis. GSEA max size parameter of 550 was used. For GSEA analysis of HA GD2 exhausted CAR T cell signature, gene lists related to supplementary table 1 was downloaded from (Lynn et al., 2019). We took the average expression of CD19 or HA samples for CD8+ T cell populations and filtered for genes that are upregulated >2 fold in HA samples compared to CD19 to generate the CD8+ HA GD2 exhausted CAR T signature (N=91 genes) used for GSEA analysis.
To contrast CAR+ and CAR-samples (see Figure 2B and S3A–B), transcripts per kilobase million (TPMs) were calculated for each gene using the bioinfokit.analys module in python. Gene lengths were calculated from the gene models used to run HTSeq, taking the maximum of all summed exon lengths across multiple isoforms as the length of the gene. For illustration purposes, we removed outlying genes with high expression (>15,000) in surCARpos versus surCARneg plots to more easily see where >99% of the genes fall on the correlation plot. However, all genes were included to make calculations, including spearman r.
Tracks were created for RNA-seq by pooling CAR+ samples across all replicates for control day 0 or CAE samples. BED files were filtered to remove alignments extending over 100bp, primarily removing intron-spanning alignments. Coverage maps were created using BEDtools genomeCoverageBed -bg and these were adjusted by multiplying by the RPM coefficient. Resulting bedGraphs were converted to bigWigs using UCSC Genome Browser Tools’ bedGraphToBigWig.
Bulk RNA-seq was compared to single-cell RNA-seq by taking all genes with significant differences in the single-cell data (between day 0 and day 20 CAE, identified using cellfishing.jl software, see description above) and rank-ordering them into ten deciles by log2(day 20 CAE/day 0 control), then representing the bulk RNA-seq log2(day 28 CAE/day 0 control) for each decile by box and whisker. Boxes are heated by the median value. DEGs identified by cellfishing.jl were filtered for genes with low signal-genes had to be expressed in at least 100 cells to be considered for analysis (see Figure S4G).
IPA analysis
Full list of enriched pathways and transcription factors can be found in tables S2, S3, and S6.
ATAC-seq
Omni ATAC-seq libraries were made as previously described (Corces et al., 2017). Briefly, nuclei were isolated from 30,000 sorted CD8+ surface CAR+ T cells, followed by the transposition reaction using Tn5 transposase (Illumina) for 30 min at 37° C with 1000 rpm mixing. Purification of transposed DNA was completed with DNA Clean and Concentrator (Zymo) and fragments were barcoded with ATAC-seq indices (Buenrostro et al., 2013). Final libraries were double size selected using AMPure beads prior to sequencing. Paired-end sequencing (2 × 75 bp reads) was carried out on an Illumina NextSeq 500 platform.
ATAC-seq analysis
Paired-end data were aligned to human genome assembly GRCh37/hg19 using bowtie2 v2.3.4.1 (Langmead and Salzberg, 2012) with parameters --local -X 1000. Resulting SAM files were converted to BAM and filtered for match quality using samtools view -q 5 -bS (samtools v1.1). BAM files across four NextSeq lanes were merged and sorted by read name using samtools merge -n, then PCR de-duplicated with PICARD MarkDuplicates REMOVE_DUPLICATES=True ASSUME_SORT_ORDER=queryname. BAM files were converted to BEDs using BEDtools bamToBed and processed to remove all alignments on chrM. Alignments with a mate distance under 100 bp were kept as sub-nucleosome fragment size signal and others were discarded.
For replicates R1 and R4, re-sequenced libraries were pooled using UNIX cat as follows:
R1 Day 0: 4-Day0-CD8-CARpos-R_S13, 4-Day0-CD8-CARpos-10–24-2018_S6
R1 CAE: 8-CAE-CD8-CARpos-R_S7, 8-CAE-CD8-CARpos-10–24-2018_S5)
R4 Day 0: 2-Day0-CD8-CARpos-REP4-ATAC-re_S17, 2-Day0-CD8-CARpos-REP4-ATAC_S17
R4 CAE: 4-CAE-CD8-CARpos-REP4-ATAC-re_S15, 4-CAE-CD8-CARpos-REP4-ATAC_S18
Peaks were called in the sub-nucleosome fragment fraction of alignments using MACS2 callpeak with parameters -s 42 -q 0.01 and no explicit background control sample. The FDR was subsequently controlled at 0.001.
Robust peak sets for control and CAE were identified in the following way. Peaks in either condition were combined across replicates, merging overlapping loci. Merged peaks without representation (BEDtools intersect) in all four replicates were removed.
To make track visualizations of the ATAC-seq data, an appropriate library size adjustment is necessary. DESeq2 was used to calculate size factors (coefficients for library size adjustment for each sample) from a set of pan-conditional peaks. The robust peak sets for control and CAE were combined, merging overlapping loci. Tag counts were calculated for all pan-conditional peaks across all samples and the resulting table was input to DESeq2 to estimate size factors and get adjusted tag counts at each peak. For each sample, sub-nucleosome sized fragment alignments were converted into a coverage map using BEDtools genomeCoverageBed -bg. Resulting bedGraph files were adjusted for library size by dividing coverage tallies by the DESeq2 size factors. Files were then sorted using UCSC Genome Browser Tools’ bedSort and converted to bigwig format using bedGraphToBigWig.
To compare ATAC-seq to bulk RNA-seq, pan-conditional peaks were filtered to remove peaks overlapping ENCODE blacklisted regions. Remaining peaks were mapped to the nearest RefSeq transcript by TSS. The set of genes up- or down-regulated at FDR 0.05 in the antigen exposure contrast was used to identify mapped peaks, and their DESeq2-adjusted counts were plotted by box-and-whisker. Statistics assessed by Mann-Whitney.
Enriched motifs were identified in peaks specific to control day 0 or CAE using HOMER v4.6 findMotifsGenome.pl with command-line parameters -size 200 -mask (Heinz et al., 2010). Robust peak sets were filtered for any overlap with ENCODE blacklisted regions or with peaks from the other condition (e.g., control day 0 peaks without overlap to CAE peaks) using BEDtools intersect, and these specific peak sets were input to HOMER. The HOMER background (-bg) was set as robust peaks specific to the other condition.
To analyze the enrichment of SOX4 at ATAC-seq peaks, the SOX4 position weight matrix was downloaded from JASPAR (MA0867.2) and scanned against robust CAE-specific peaks (those without overlaps to ENCODE blacklisted regions or control day 0 peaks) or peaks common to control day 0 and CAE stimulation using PWMSCAN, with the FDR controlled at 1E-8. Peaks were divided into those with or without the motif and DESeq2-adjusted values are shown for these peak sets in box-and-whisker (Figure 6F). Statistics assessed by Mann-Whitney. Day 28 CAE peaks present in at least two biological replicates were associated to nearby genes using HOMER v4.6 annotatePeaks.pl. Peaks were examined for the presence of the human SOX4 motif (JASPAR MA0867.2) using PWM_SCAN, with p-value cutoff 1E-6. Our CAR T dysfunction signature genes (N=30) were then queried to determine how many had associated peaks with a SOX4 motif hit.
ATAC-seq analysis of exhausted human TILs
Published data from (Philip et al., 2017)(TIL model) were downloaded from NCBI GEO. FASTQs were trimmed using TrimGalore! v0.6.6 (relying on FASTQC v0.11.2 and cutadapt v2.10) with command-line arguments --paired --fastqc, then aligned to the hg19 assembly of the human genome using bowtie2 v2.3.4.1 with command-line parameters --local -X 1000. Data were filtered for poor alignments using samtools view -q 5 (samtools v1.1, (Li et al., 2009)), then sorted with samtools sort -n and filtered for PCR duplicates using PICARD MarkDuplicates REMOVE_DUPLICATES=True ASSUME_SORT_ORDER=queryname (PICARD v2.21.3-SNAPSHOT). Data were rendered as BED files using bamToBed v2.27.1–65-gc2af1e7-dirty, then processed using python to exclude chrM alignments and filtered to find paired-end alignments smaller than 100 bp (“sub-nucleosome fragments”). Remaining fragments from three biological replicates were pooled. Day 28 CAE-specific peaks present in at least two biological replicates (N = 4,766) were scanned for ATAC-seq enrichment in both our data and in the Philip et al Nature 2017 TIL model (naïve and PD1hi cells) in a 5kb window around the peak center using 50bp increments. Peaks were sorted vertically by the summed Day 28 CAE ATAC-seq signal in descending order and all ATAC-seq traces were rendered as a heatmap using python and the PIL imaging library. To assess the dynamics of the TIL model at day 28 CAE-specific peaks, a background peak set was randomly sampled from all ATAC-seq OCRs from day 0 or day 28 CAE (size = 4,766 with random seed 3399039292705153955). Peak enrichment for our data and the Philip et al Nature 2017 TIL model (naïve and PD1hi cells) was measured over day 28 CAE-specific peaks and background peaks, and then the difference PD1hi-naïve was rendered as a box-and-whisker for the two peak groups (Figure S4E). Statistical significance of the difference between day 28 CAE-specific peaks and background peaks was assessed using a permutation test (coin library in R).
LCMV chronic viral infection data analysis
RNA-seq FASTQ files were downloaded from GEO submission GSE86881 for naïve mouse CD8+ T cells (GSM2309810, GSM2309811) and exhausted CD8+ T cells (GSM2309812, GSM2309813, GSM2309814). FASTQ files were aligned to the mm10 reference genome using STAR and differentially expressed genes between naïve CD8+ T cells and exhausted CD8+ T cells were identified using DESeq2. Only genes with mouse to human homologs were overlapped with CAR T dysfunction gene signature. Homologs were obtained from the Mouse Genome Informatics (MGI) database.
Human cancer TIL overlap analysis
The following published single-cell datasets were overlapped with the CAR T cell dysfunction gene signature. Colorectal cancer exhausted CD8 TIL associated genes were downloaded from supplementary table 5 for the CD8_C07-LAYN specific genes (N=714 genes, including LAYN) (Zhang et al., 2018). Non-small-cell lung cancer exhausted CD8 TIL-associated genes were downloaded from supplementary table 3 for the CD8-C6-LAYN specific genes (N=399 genes) (Guo et al., 2018). Hepatocellular carcinoma exhausted CD8 TIL associated genes were downloaded from supplementary table 4 (N=82 genes) (Zheng et al., 2017). Melanoma exhausted CD8 TIL associated genes were obtained from Figure 2B (genes most correlated with LAG3) and supplemental Figure S2E (genes most correlated with HAVCR2) (N=34) (Li et al., 2019).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism software or R and are represented by the following ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. Statistical significance between two groups was determined using two-tailed unpaired Student’s t test (parametric) or Mann-Whitney (non-parametric). The Benjamini–Hochberg procedure (FDR) was used to adjust p values for multiple testing, unless otherwise noted. Statistical significance between multiple groups of two variables was assessed by two-way ANOVA with post hoc tests. Statistical significance of Venn diagram overlaps between two groups was calculated using hypergeometric tests. The specific tests used for the analyses shown in the supplementary figure are indicated in supplementary figure legends. The specifical tests used for analyses in the main figure legends are detailed in the main figure legends and summarized below. Cytokine profile analysis of CD8+ surCARpos T cells (day 28 CAE, day 0 product and CD19BBz) of Figure 1F was assessed by two-way ANOVA with Tukey’s post hoc test. Cytokine secretion of day 26 CAR T cells before and after 24 hrs of rest in presence of IL-15 (Figure 1H) was calculated with two-tailed unpaired Student’s t test. Differences between day 0 and Day 28 normalized RNA-seq counts of representative NK receptor/marker genes (Figure 2D) was analyzed with Mann-Whitney U test. Significance of the differences on normalized counts of CAR transcripts from sc-RNA-seq data for between non dysfunctional and dysfunctional clusters (Figure 3H) was calculated with Mann-Whitney U test. Statistical differences in protein levels of NK-associated molecules and checkpoint receptors in CD8 T infiltrating recurrent tumors versus day 0 product (Figure 4E and 4G) were assessed by two-way ANOVA with Sidak test for multiple comparisons.
Differences in ATAC-seq signal at peaks specific to day 28 (Figure 6F) were assessed by Mann-Whitney. Differences on the percentage of NK-like T cells between WT, ID3 KO, and SOX4 KO M5CAR T cells (Figure 7G) was measured by Fisher’s exact test. Statistical differences in the dysfunctional score of WT, ID3 KO, and SOX4 KO M5CAR T cells for donor ND566 (Figure 7I) and WT and ID3 KO M5CAR T cells for donor ND539 (Figure 7J) were measured by Mann-Whitney U test. Cell killing statistical differences in WT versus ID3 KO, and SOX4 KO M5CAR T cells was assessed by two-way ANOVA with Geisser-Greenhouse correction and Dunnet’s post hoc test. Data were visualized using the following R packages and functions ggplot2, EnhancedVolcano, pheatmap, RColorBrewer, gplots, dplyr, plotly. See Key Resource Table for versions of R packages utilized in this study.
ADDITIONAL RESOURCES
Clinical samples analyzed in this study were obtained from clinical trials conducted at University of Pennsylvania and registered at www.clinicaltrial.gov as NCT03054298 and NCT02030834.
Supplementary Material
(A) Detailed experimental design of CAR T cell dysfunction in vitro model. (B) Mesothelin expression on AsPC-1 cells, measured by flow cytometry. (C) Apoptosis characterization by Annexin V binding assessment and loss of plasma membrane integrity (L/D Aqua staining), showing live, early apoptotic and late apoptotic M5CAR transduced T cells during CAE. A single normal donor was tested. Data shown as mean with standard deviation (D) Frequency of TIM3+ CD8+ M5CAR T cells on day 0 product and after CAE. Data is representative of two different donors. **P <0.01 by Mann-Whitney test. (E) Tumor cytotoxicity of CD8+ surCARpos T cells (CAE and day 0 product) against AsPC-1 cells using 3 different donor T cells (ND388, ND534, and ND516) measured by xCelligence. (F) Cell killing capacity of CD8+ M5CAR transduced T cells (CAE and day 0 product) against AsPC-1 cells using 2 different donor T cells (ND150 8:1 E:T ratio, ND538 7:1 E:T ratio) measured by Celigo. **** P <0.0001, *** P <0.001, ** P <0.01 by Student’s t test. (G) Mesothelin expression on mesothelin transduced K562 cells (K562-meso), measured by flow cytometry. (H) Kinetics of K562-meso cell lysis incubated with CD8+ M5CAR transduced T cells (day 26 CAE and day 0 product) using 2 different donor T cells (ND150 8:1 E:T ratio, ND538 7:1 E:T ratio) measured by Celigo. *** P <0.001, ** P <0.01, * P < 0.05 by Student’s t test. (I) Cytokine secretion of CD8+ surCARpos T cells (day 28 CAE and day 0 product) stimulated with AsPC-1 for 24 hrs using 2 different donor T cells (ND388 and ND534). CD19BBz CAR T cells were tested as a control for allogeneic recognition of AsPC-1. **** P <0.0001, * P < 0.05 by two-way ANOVA with Tukey’s post hoc test. (J) Cytokine profile of CD8+ surCARpos T cells (day 24 CAE and day 0 product). Sorted CD8+ surCARpos T cells were stimulated with PMA + ionomycin or AsPC-1, and media was used as a control. Two donors were analyzed (ND150 and ND534). **** P <0.0001, *** P <0.001, ** P <0.01, by two-way ANOVA with Tukey’s post hoc test.
(A) Surface CAR expression on day 23 CAE CD8+ M5CAR-transduced T cells before sorting, associated with Figure 1H. (B) Frequency of surCARpos CD8+ T cells used for cell killing assay (day 26), associated with Figure 1I. (C) Cell killing capacity of donor ND538 CD8+ M5CAR-transduced T cells against AsPC-1 cells after 26 days of CAE before and after 24 hrs of rest with IL-15 (7:1 E:T ratio). **P <0.01, *P <0.05 by Student’s t test, data is shown as mean ± SEM. (D) Tumor cells (mesothelin+ CD45-) in human pleural fluid on day 36 after CAR T cell infusion (patient #02916–06) and in peritoneal fluid on day 21 after CAR T cell infusion (patient #02916–01). (E) Surface CAR expression (top) and intracellular M5CAR expression (bottom) on human CD8+ T cells in peritoneal fluid (patient #02916–01) after 21 days of M5CAR T cell infusion. CAR staining (right) and M5CAR FMO control (left).
Average gene expression values (TPMs) for day 0 surCARpos compared to day 0 surCARneg cells for differentially expressed genes defined in Figure 2A (A) and for all genes (B). (C) Venn diagram displaying overlap between genes upregulated in day 28 CAE surCARpos CD8+ T cells (see Figure 2A) and genes upregulated in exhausted CD8+ T cells from the LCMV clone-13 mouse model of chronic viral infection. Only genes with mouse to human orthologs were included. (D) Overlap of genes downregulated in day 28 CAE surCARpos cells (see Figure 2A) and genes downregulated in exhausted T cells from the LCMV clone-13 mouse model of chronic viral infection. (E) GSEA analysis of day 0 and 28 CAE surCARpos RNA-seq samples with exhausted T cell populations from (Beltra et al., 2020) for TEX-progenitor 1, TEX-progenitor 2, TEX-intermediate, and TEX-terminal from the LCMV mouse model of chronic viral infection. Overlap of genes upregulated in day 28 CAE surCARpos cells (see Figure 2A) and genes that define dysfunctional CD8+ TILs from hepatocellular carcinoma patients [HCC] (F), melanoma patients (G), non-small-cell lung cancer patients [NSCLC] (H), and colorectal cancer patients [CRC] (I). (J) Overlap of HCC, melanoma, NSCLC, and CRC dysfunctional TIL signature genes. (K) Heatmap of day 0 and 28 CAE surCARpos RNA-seq samples on CD8+ TIL dysfunction gene signature common between hepatocellular carcinoma, non-small-cell lung cancer, colorectal cancer, and melanoma (18 genes in common from Figure S3J). (L) GSEA analysis of day 0 and 28 CAE surCARpos RNA-seq samples with genes upregulated in CD8+ HA GD2 CAR T cells from (Lynn et al., 2019). (M) Overlap of genes upregulated in CD4+ T cells and CD8+ T cells in day 28 CAE surCARpos cells (compared to day 0 cells) (left) and for genes downregulated (right). (N) Normalized RNA-seq counts of representative dysfunction signature genes in CD4+ T cells at day 0 and day 28 CAE. Average of two biological replicates and data is shown as mean ± SEM. Statistical significance of overlap between two groups (in panels C-D, F-I, M) was calculated using hypergeometric test.
(A) ATAC-seq open chromatin regions specific to day 0 (left) or day 28 CAE (right) surCARpos cells. ATAC-seq analyses include 4 biological replicates. (B) Genomic location of open chromatin regions for day 0 (blue) and day 28 CAE (red) surCARpos cells. (C) Relation of gene expression and chromatin changes during CAE. Average ATAC-seq signal of genes upregulated at day 28 (left) and genes downregulated (right) in day 0 and day 28 CAE surCARpos cells. Average of 4 biological replicates represented with box plots. Statistics assessed by Mann-Whitney. (D) Heatmap of sub-nucleosome ATAC-seq tags for naïve T cells and PD1-high TILs from (Philip et al., 2017) at sites opening in day 28 CAE surCARpos cells compared to day 0 cells. (E) Box plot of PD1-high sub-nucleosome ATAC-seq tags from (Philip et al., 2017) normalized to naïve T cells at day 28 CAE specific sites versus an equal number of randomly sampled other open chromatin regions (OCRs). Difference assessed by permutation test. (F) ATAC-seq tracks from day 0 and day 28 CAE cells at CD5, CD28, and TCF7 regulatory regions. Analysis includes four biological replicates. (G) Decile plot showing correlation between bulk RNA-seq and single-cell RNA-seq datasets. Genes differentially expressed in single-cell data between day 20 CAE and day 0, defined by cellfishing.jl, were divided into 10 groups and sorted by single-cell fold-change, going from lowest to highest (x axis). Y-axis plots the fold change (day 28 CAE/day 0) in the bulk RNA-seq for genes in each group. (H) Heatmap of top 10 marker genes for each day 0 single-cell cluster as defined in Figure 3A, donor ND388. Columns correspond to cells and rows correspond to gene names. (I) Gene ontology determined by metascape pathway analysis for each single-cell day 0 cluster, donor ND388. Columns are cell clusters (defined in Figure 3A) and rows are enriched pathways color coded by level of significance. (J) Venn diagram displaying overlap between genes upregulated in day 28 CAE surCARpos CD8+ T cells from bulk RNA-seq analyses (see Figure 2A) and genes that define single cell clusters D20–1, D20–2, D20–3, and D20–4 from day 20 CAE cells (see Figure 3B). Significance calculated using hypergeometric test. (K) Violin plots depicting gene expression levels from day 20 CAE cells (donor ND388) for SRGAP3, DUSP4, and CSF1. X axis is cell clusters defined in Figure 3B. (L) Violin plot of day 20 CAE cells for CTLA4, donor ND388 (padj 1.2e-11, log2FC 0.20).
Single-cell analysis of day 0 product and day 28 CAE cells for donor ND538 (A-E) and donor ND150 (F-J). (A) UMAP projection of single-cell gene expression data from day 0 cells, made using Seurat. Each dot corresponds to one cell and cell clusters are color coded. (B) UMAP projection of single-cell gene expression data from day 28 CAE T cells, made using Seurat. Each dot corresponds to one cell and cell clusters are color-coded. (C) Heatmap of top 10 marker genes for each day 28 CAE cluster as defined in (B). Columns correspond to cells and rows correspond to gene names. (D) Volcano plot depicting differentially expressed genes between day 28 CAE cluster 1 (dysfunctional) and clusters 2 and 3 (non-dysfunctional), also see (E). Genes upregulated in the dysfunctional cluster are on the right side and genes downregulated are on the left. The x-axis is log2(fold change) and y-axis is -log10(p value). Red dots indicate significant genes with p<0.05 and log2FC >0.5. (E) Dot plot illustrating the expression level of genes in day 0 (left) and day 28 CAE cell clusters (right). Genes included are dysfunction signature genes (N=30), naïve/memory, cell cycle, and control genes. Each column represents one cluster. The size of the circle represents the percent of cells expressing the gene in each cluster and the color depicts how highly expressed the gene is within that cluster. The number of cells in each cluster is written beneath cluster identity. Donor ND150 (F) UMAP projection of single-cell gene expression data from day 0 cells. (G) UMAP projection of single-cell gene expression data from day 28 CAE cells. (H) Heatmap of top 10 marker genes for each day 28 CAE cluster as defined in (G). Columns correspond to cells and rows correspond to gene names. (I) Volcano plot depicting differentially expressed genes between day 28 cluster 3 (non-dysfunctional) and clusters 1,2,4, and 5 (dysfunctional), also see (J). Genes upregulated in the dysfunctional clusters are on the right side and genes downregulated are on the left. The x axis is log2 (fold change) and y axis is -log10(p value). Red dots indicate significant genes with p<0.05 and log2FC >0.5. (J) Dot plot illustrating the expression level of genes in day 0 (left) and day 28 CAE (right). Genes included are dysfunction signature genes (N=30), naïve/memory, cell cycle and control genes. Each column represents one cluster. The number of cells in each cluster is written beneath cluster identity.
(A) Phenotypical change between day 0 products (left) and day 29 CAE samples (right) profiled by mass cytometry using a NK flow panel. Data from 2 donors (ND150 and ND538) are shown. (B) Expression of surface M5CAR and NK-associated molecules (CD161, TIGIT, CD56, NKG2A, NKG2C) and granulysin on day 0 product (top) and day 29 CAE CD8+ T cells (bottom). Red circles highlight subpopulations of CD8+ T cells more abundant under CAE. (C) Representative plot showing mesothelin expression from an AsPC-1 recurrent tumor associated with Figure 4B, and the FMO control (right). (D) Frequency of CD8+ T cells infiltrating recurrent tumors (n=3 mice). Data is shown as mean ± SEM. (E) Representative dot plot of surface CAR (sCAR) and intracellular (iCAR) staining (left) and FMO controls (middle). Percentage of CD8 T cells expressing surface and intracellular CAR isolated from recurrent tumors from Figure 4B (n=3), (right). (F) Model for CD56 depletion assay. Expected percentage of NK-like T cells (CD56+, y-axis) and time (x-axis) during continuous antigen stimulation for out competition model (left) and transition model (right). Control cells start with regular CAR T cell population at day 0 and CD56-depleted starts with day 0 CAR T cells depleted of CD56. (G) TCR single-cell sequencing data for day 0 and day 28 CAE for donors ND150 and ND538. Y axis is the percentage of CDR3 sequences, and x-axis is the number of cells that have that CDR3 sequence. Illustrates that between 96–99% of CDR3 sequences are unique. (H) Single-cell transcript levels of FOS, EGR1, FOXP3, MYB, LEF1, CREM, TWIST1 illustrated by UMAP plots, corresponding to clusters from Figure 3B (day 20 CAE cells). Top two clusters are dysfunctional. (I) Transcription factor motif for SOX4 from Jaspar database (top) and SOX4 and SOX17 motifs from UniProt database (bottom).
(A-B) Representative ATAC-seq tracks in regulatory regions at SOX4 motifs from day 0 and day 28 CAE samples at NK receptor and dysfunction genes KLRC1 (A) and KLRB1 (B). SOX4 motifs labeled with red bars above tracks. Analysis includes four biological replicates. (C) Agarose gel (top) and KO efficiency by genomic DNA sequencing (bottom) showing CRISPR edits on KO T cells. ID3-specific and SOX4-specific PCR targeting the edited region of each transcription factor shows the appearance of two bands in KO UTD and KO M5CAR T cells, corresponding to two edited populations derived from different sgRNA hits as depicted in Figure 7A. The KO efficiency quantification from two donors (ND566 and ND539). WT: wildtype, UTD: non-electroporated un-transduced T cells (no M5CAR), M5: T cells electroporated with M5CAR. Data is shown as mean ± SEM from two CAR T donors. (D) Cytotoxicity assessment of day 0 products at 1:1 (left) and 1:10 (right) E:T ratio on ND539 (top) and ND566 (bottom) in WT, ID3 KO, and SOX4 KO M5CAR T cells. Media used as a control. Data is shown as mean ± SEM. (E) Flow cytometry characterization of naïve, central memory, effector memory and effector subsets on day 0 product with CD45RO and CCR7 expression for WT, ID3 KO, and SOX4 KO M5CAR T cells for donor ND566 (left) and ND539 (right). (F) Percentage of NK-like T cells in WT and ID3 KO cells, relative to WT (donor ND539). Significance measured by Fisher’s exact test. (G) In vitro killing assay of ND539 WT, ID3 KO, and SOX4 KO M5CAR T cells. Cells were collected on day 18 of CAE and seeded at 1:8 E:T ratio with AsPC-1 on day 18. WT day 0 cells are used as a positive control and media is used as a negative control. Data is shown as mean ± SEM. (H) In vitro killing assay of ND566 WT, ID3 KO, and SOX4 KO M5CAR T cells. Cells were collected on day 21 of CAE and seeded at 1:8 E:T ratio. Data is shown as mean ± SEM. (I) In vitro killing assay of ND566 WT, ID3 KO, and SOX4 KO M5CAR T cells. Cells were collected on day 28 of CAE and seeded at 1:8 E:T ratio. Data is shown as mean ± SEM. Supplemental excel tables
Table S1: Differentially expressed genes between day 0 and day 28 CAE CAR T cells from bulk RNA-seq experiments. Related to Figure 2A.
Table S2: Full list of Ingenuity Pathway Analysis pathways enriched in differentially expressed genes between day 0 and day 28 CAE CAR T cells from bulk RNA-seq experiments. Related to Figure 2C.
Table S3: All upstream regulators (transcription factors) predicted to regulate differentially expressed genes between day 0 and day 28 CAE CAR T cells from bulk RNA-seq experiments. Related to Figure 2F.
Table S4: Differentially expressed genes between dysfunctional clusters (D20–1, D20–4) and non-dysfunctional clusters (D20–2, D20–3) from scRNA-seq experiment in donor ND388. Related to Figure 3E.
Table S5: Table of dysfunction signature genes (N=30). X marks genes that are upregulated in LCMV exhausted T cells or defines dysfunctional TILs in scRNA-seq experiments. HCC (hepatocellular carcinoma), NSCLC (non-small-cell lung cancer), CRC (colorectal carcinoma), LCMV (lymphocytic choriomeningitis virus). Related to Figures 3 and S3.
Table S6: All upstream regulators (transcription factors) predicted to regulate differentially expressed genes between day 0 and day 20 CAE CAR T cells from scRNA-seq experiment in donor ND388. Related to Figure 6A.
Highlights.
CAR T cells under chronic antigen stimulation show hallmarks of T cell exhaustion
CAR dysregulation is associated with a CD8+ T to NK-like T cell transition
CAR T cells with NK-like transition were identified in patients after treatment
Unlike WT CAR T cells, ID3 and SOX4 knockout CAR T cells retain anti-tumor immunity
Acknowledgements
Research supported by a SU2C-Lustgarten Foundation Translational Cancer Research Team Grant, grant Numbers SU2-CAACR-DT21-17 and SU2C-RT6162. S.L.B. is supported by NIH grant CA078831. C.R.G is supported by NIH grant CA232466 and the American Cancer Society - Rob Kugler - Postdoctoral Fellowship. R.M.Y and C.H.J are supported by NIH grant P01CA214278. C.H.J. and L.L.L. are supported by the Parker Institute for Cancer Immunotherapy. P.C.R. is supported by the National Institutes of Health/National Cancer Institute grant 5T32CA009140. We acknowledge the Parnassus Flow Cytometry Core, in part supported by the NIH DRC Center Grant P30 DK063720 and S10 1S10OD018040-01 for use of the CyTOF instrument. The authors wish to thank E. John Wherry and Austin L. Good for their valuable feedback on this manuscript.
Footnotes
Declaration of Interests
R.M.Y., S.G., S.F.L., S.M.A., M.R., and C.H.J. are inventors on patents and/or patent applications licensed to Novartis Institutes of Biomedical Research and receive license revenue from such licenses. R.M.Y. is an inventor on patents and/or patent applications licensed to Tmunity Therapeutics and receives license revenue from such licenses. C.H.J is a scientific founder of Tmunity Therapeutics and DeCART Therapeutics, and is a member of the scientific advisory boards of AC Immune, BluesphereBio, Cabaletta, Carisma, Cartography, Cellares, Celldex, Decheng, Poseida, Verismo, WIRB-Copernicus and Ziopharm. S.J.S. is a consultant, on the scientific advisory board and receives research support from Genentech/Roche, Novartis and Juno Therapeutics. S.J.S. is a consultant for AlloGene, AstraZeneca, BeiGene, Regeneron and Tesa Therapeutics. S.J.S. is a consultant and on the scientific advisory board for Loxo Oncology. S.J.S. is on the scientific advisory board for Nordic Nanovector. S.J.S. is a consultant and receives research support from Celgene. S.F.L receives research funding from Tmunity Therapeutics and Cabaletta. M.R. is on the scientific advisory board of AbClon Inc and consulted for BMS, nanoString, GSK, and Bayer. M.R. is the scientific founder of viTToria biotherapeutics. L.L.L. is on the scientific advisory boards for Alector, Atreca, Dragonfly, DrenBio, Morphosys, Nkarta, Obsidian Therapeutics, Rubius, SBI, and Innovent. SMA receives research funding from Tmunity Therapeutics, RAPT, and Incyte Corporation and is scientific advisor for Trizell, BioArdis and Verismo.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abd Hamid M, Wang RZ, Yao X, Fan P, Li X, Chang XM, Feng Y, Jones S, Maldonado-Perez D, Waugh C, et al. (2019). Enriched HLA-E and CD94/NKG2A Interaction Limits Antitumor CD8(+) Tumor-Infiltrating T Lymphocyte Responses. Cancer Immunol Res 7, 1293–1306. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Wellhausen N, Levine BL, and June CH (2021). Production of Human CRISPR-Engineered CAR-T Cells. J Vis Exp. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W. (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, Blery M, Bonnafous C, Gauthier L, Morel A, et al. (2018). Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 175, 1731–1743.e1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balin SJ, Pellegrini M, Klechevsky E, Won ST, Weiss DI, Choi AW, Hakimian J, Lu J, Ochoa MT, Bloom BR, et al. (2018). Human antimicrobial cytotoxic T lymphocytes, defined by NK receptors and antimicrobial proteins, kill intracellular bacteria. Sci Immunol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbarin A, Cayssials E, Jacomet F, Nunez NG, Basbous S, Lefèvre L, Abdallah M, Piccirilli N, Morin B, Lavoue V, et al. (2017). Phenotype of NK-Like CD8(+) T Cells with Innate Features in Humans and Their Relevance in Cancer Diseases. Front Immunol 8, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, et al. (2020). Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52, 825–841 e828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benezra R, Davis RL, Lockshon D, Turner DL, and Weintraub H. (1990). The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell 61, 49–59. [DOI] [PubMed] [Google Scholar]
- Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, et al. (2019). Defining ‘T cell exhaustion’. Nat Rev Immunol 19, 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroughs AC, Larson RC, Marjanovic ND, Gosik K, Castano AP, Porter CBM, Lorrey SJ, Ashenberg O, Jerby L, Hofree M, et al. (2020). A Distinct Transcriptional Program in Human CAR T Cells Bearing the 4–1BB Signaling Domain Revealed by scRNA-Seq. Mol Ther 28, 2577–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, et al. (2009). Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A 106, 3360–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TE, Stumpf MPH, and Babtie AC (2017). Gene Regulatory Network Inference from Single-Cell Data Using Multivariate Information Measures. Cell Syst 5, 251–267.e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP, and Rao A. (2019). NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, Park JJ, Kim HR, Errami Y, Guzman CD, et al. (2019). Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 178, 1189–1204.e1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranašić D, et al. (2020). JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48, D87–d92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O’Connor RS, Hwang WT, et al. (2018a). Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 24, 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, Cogdill AP, Morrissette JJD, DeNizio JE, Reddy S, et al. (2018b). Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558, 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, and Van Kaer L. (2004). NKT cells: what’s in a name? Nat Rev Immunol 4, 231–237. [DOI] [PubMed] [Google Scholar]
- Guo J, and Xu C. (2020). Screening for the Next-Generation T Cell Therapies. Cancer Cell 37, 627–629. [DOI] [PubMed] [Google Scholar]
- Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 24, 978–985. [DOI] [PubMed] [Google Scholar]
- Gurusamy D, Henning AN, Yamamoto TN, Yu Z, Zacharakis N, Krishna S, Kishton RJ, Vodnala SK, Eidizadeh A, Jia L, et al. (2020). Multi-phenotype CRISPR-Cas9 Screen Identifies p38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell 37, 818–833.e819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R. (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk MH, Blom B, Nolan G, Stegmann AP, Bakker AQ, Weijer K, Res PC, and Spits H. (1997). Inhibition of T cell and promotion of natural killer cell development by the dominant negative helix loop helix factor Id3. J Exp Med 186, 1597–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, and Chen J. (2013). A genome-wide regulatory network identifies key transcription factors for memory CD8(+) T-cell development. Nat Commun 4, 2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Pos Z, Rao M, Klebanoff CA, Yu Z, Sukumar M, Reger RN, Palmer DC, Borman ZA, Muranski P, et al. (2011). Repression of the DNA-binding inhibitor Id3 by Blimp-1 limits the formation of memory CD8+ T cells. Nat Immunol 12, 1230–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, et al. (2019). TOX transcriptionally and epigenetically programs CD8. Nature 571, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurioka A, Klenerman P, and Willberg CB (2018). Innate-like CD8+ T-cells and NK cells: converging functions and phenotypes. Immunology 154, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutner RH, Zhang XY, and Reiser J. (2009). Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 4, 495–505. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong JW, Wagner JA, Ireland AR, and Fehniger TA (2017). Transcriptional and post-transcriptional regulation of NK cell development and function. Clin Immunol 177, 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen J, Blank CU, et al. (2019). Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 176, 775–789 e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Qiu S, Chen J, Jiang S, Chen W, Jiang J, Wang F, Si W, Shu Y, Wei P, et al. (2020). Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 53, 456–470 e456. [DOI] [PubMed] [Google Scholar]
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. (2015). 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 21, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Jia L, Zheng Z, Tran E, Robbins PF, and Rosenberg SA (2019). Single-Cell Transcriptome Analysis Reveals Gene Signatures Associated with T-cell Persistence Following Adoptive Cell Therapy. Cancer Immunol Res 7, 1824–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, et al. (2019). c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra N, Qi Y, Spidale NA, Frascoli M, Miu B, Cho O, Sylvia K, and Kang J. (2018). SOX4 controls invariant NKT cell differentiation by tuning TCR signaling. J Exp Med 215, 2887–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathewson ND, Ashenberg O, Tirosh I, Gritsch S, Perez EM, Marx S, Jerby-Arnon L, Chanoch-Myers R, Hara T, Richman AR, et al. (2021). Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 184, 1281–1298 e1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon CW, Zajac AJ, Jamieson AM, Corral L, Hammer GE, Ahmed R, and Raulet DH (2002). Viral and bacterial infections induce expression of multiple NK cell receptors in responding CD8(+) T cells. J Immunol 169, 1444–1452. [DOI] [PubMed] [Google Scholar]
- Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, Raulet DH, Lanier LL, Groh V, Spies T, et al. (2004). Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 21, 357–366. [DOI] [PubMed] [Google Scholar]
- Milner JJ, Toma C, He Z, Kurd NS, Nguyen QP, McDonald B, Quezada L, Widjaja CE, Witherden DA, Crowl JT, et al. (2020). Heterogenous Populations of Tissue-Resident CD8. Immunity 52, 808–824.e807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, Hoffmann A, Hogan PG, Rao A, and Trifari S. (2017). Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc Natl Acad Sci U S A 114, E2776–e2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon EK, Ranganathan R, Eruslanov E, Kim S, Newick K, O’Brien S, Lo A, Liu X, Zhao Y, and Albelda SM (2016). Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin Cancer Res 22, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Pure E, Milone MC, et al. (2014). Multifactorial T-cell Hypofunction That Is Reversible Can Limit the Efficacy of Chimeric Antigen Receptor-Transduced Human T cells in Solid Tumors. Clin Cancer Res 20, 4262–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, and Wherry EJ (2015). Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, and Sandberg R. (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Poorebrahim M, Melief J, Pico de Coana Y, W SL, Cid-Arregui A, and Kiessling R. (2021). Counteracting CAR T cell dysfunction. Oncogene 40, 421–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma YA, and Trapnell C. (2017). Single-cell mRNA quantification and differential analysis with Census. Nat Methods 14, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Li PJ, Blaeschke F, Nies JF, Apathy R, Mowery C, Yu R, Nguyen MLT, Lee Y, Truong A, et al. (2020). Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell 181, 728–744.e721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, and Anderson AC (2010). Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 207, 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Tsuyuzaki K, Shimizu K, and Nikaido I. (2019). CellFishing.jl: an ultrafast and scalable cell search method for single-cell RNA sequencing. Genome Biol 20, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, et al. (2017). Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med 377, 2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyda M, Elkhal A, Quante M, Falk CS, and Tullius SG (2016). T Cells Going Innate. Trends Immunol 37, 546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, and Marson A. (2018). Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971.e1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, et al. (2016). A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 166, 1500–1511.e1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, et al. (2020). CRISPR-engineered T cells in patients with refractory cancer. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen H, Cuevas C, Dotson AM, Tan X, Hotes JL, Greenberg PD, et al. (2015). T Cells Engineered against a Native Antigen Can Surmount Immunologic and Physical Barriers to Treat Pancreatic Ductal Adenocarcinoma. Cancer Cell 28, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R. (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–d515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommen DS, and Schumacher TN (2018). T Cell Dysfunction in Cancer. Cancer Cell 33, 547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, Shi W, and Kallies A. (2020). Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat Immunol 21, 1256–1266. [DOI] [PubMed] [Google Scholar]
- van Montfoort N, Borst L, Korrer MJ, Sluijter M, Marijt KA, Santegoets SJ, van Ham VJ, Ehsan I, Charoentong P, André P, et al. (2018). NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 175, 1744–1755.e1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, Wang Y, Wang YD, Qian C, Xu B, et al. (2019). Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wencker M, Turchinovich G, Di Marco Barros R, Deban L, Jandke A, Cope A, and Hayday AC (2014). Innate-like T cells straddle innate and adaptive immunity by altering antigen-receptor responsiveness. Nat Immunol 15, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, and Kurachi M. (2015). Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, Li HS, Rivera RR, Lind KC, D’Cruz LM, et al. (2011). The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol 12, 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, Gao R, Kang B, Zhang Q, Huang JY, et al. (2018). Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272. [DOI] [PubMed] [Google Scholar]
- Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. (2017). Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169, 1342–1356 e1316. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Detailed experimental design of CAR T cell dysfunction in vitro model. (B) Mesothelin expression on AsPC-1 cells, measured by flow cytometry. (C) Apoptosis characterization by Annexin V binding assessment and loss of plasma membrane integrity (L/D Aqua staining), showing live, early apoptotic and late apoptotic M5CAR transduced T cells during CAE. A single normal donor was tested. Data shown as mean with standard deviation (D) Frequency of TIM3+ CD8+ M5CAR T cells on day 0 product and after CAE. Data is representative of two different donors. **P <0.01 by Mann-Whitney test. (E) Tumor cytotoxicity of CD8+ surCARpos T cells (CAE and day 0 product) against AsPC-1 cells using 3 different donor T cells (ND388, ND534, and ND516) measured by xCelligence. (F) Cell killing capacity of CD8+ M5CAR transduced T cells (CAE and day 0 product) against AsPC-1 cells using 2 different donor T cells (ND150 8:1 E:T ratio, ND538 7:1 E:T ratio) measured by Celigo. **** P <0.0001, *** P <0.001, ** P <0.01 by Student’s t test. (G) Mesothelin expression on mesothelin transduced K562 cells (K562-meso), measured by flow cytometry. (H) Kinetics of K562-meso cell lysis incubated with CD8+ M5CAR transduced T cells (day 26 CAE and day 0 product) using 2 different donor T cells (ND150 8:1 E:T ratio, ND538 7:1 E:T ratio) measured by Celigo. *** P <0.001, ** P <0.01, * P < 0.05 by Student’s t test. (I) Cytokine secretion of CD8+ surCARpos T cells (day 28 CAE and day 0 product) stimulated with AsPC-1 for 24 hrs using 2 different donor T cells (ND388 and ND534). CD19BBz CAR T cells were tested as a control for allogeneic recognition of AsPC-1. **** P <0.0001, * P < 0.05 by two-way ANOVA with Tukey’s post hoc test. (J) Cytokine profile of CD8+ surCARpos T cells (day 24 CAE and day 0 product). Sorted CD8+ surCARpos T cells were stimulated with PMA + ionomycin or AsPC-1, and media was used as a control. Two donors were analyzed (ND150 and ND534). **** P <0.0001, *** P <0.001, ** P <0.01, by two-way ANOVA with Tukey’s post hoc test.
(A) Surface CAR expression on day 23 CAE CD8+ M5CAR-transduced T cells before sorting, associated with Figure 1H. (B) Frequency of surCARpos CD8+ T cells used for cell killing assay (day 26), associated with Figure 1I. (C) Cell killing capacity of donor ND538 CD8+ M5CAR-transduced T cells against AsPC-1 cells after 26 days of CAE before and after 24 hrs of rest with IL-15 (7:1 E:T ratio). **P <0.01, *P <0.05 by Student’s t test, data is shown as mean ± SEM. (D) Tumor cells (mesothelin+ CD45-) in human pleural fluid on day 36 after CAR T cell infusion (patient #02916–06) and in peritoneal fluid on day 21 after CAR T cell infusion (patient #02916–01). (E) Surface CAR expression (top) and intracellular M5CAR expression (bottom) on human CD8+ T cells in peritoneal fluid (patient #02916–01) after 21 days of M5CAR T cell infusion. CAR staining (right) and M5CAR FMO control (left).
Average gene expression values (TPMs) for day 0 surCARpos compared to day 0 surCARneg cells for differentially expressed genes defined in Figure 2A (A) and for all genes (B). (C) Venn diagram displaying overlap between genes upregulated in day 28 CAE surCARpos CD8+ T cells (see Figure 2A) and genes upregulated in exhausted CD8+ T cells from the LCMV clone-13 mouse model of chronic viral infection. Only genes with mouse to human orthologs were included. (D) Overlap of genes downregulated in day 28 CAE surCARpos cells (see Figure 2A) and genes downregulated in exhausted T cells from the LCMV clone-13 mouse model of chronic viral infection. (E) GSEA analysis of day 0 and 28 CAE surCARpos RNA-seq samples with exhausted T cell populations from (Beltra et al., 2020) for TEX-progenitor 1, TEX-progenitor 2, TEX-intermediate, and TEX-terminal from the LCMV mouse model of chronic viral infection. Overlap of genes upregulated in day 28 CAE surCARpos cells (see Figure 2A) and genes that define dysfunctional CD8+ TILs from hepatocellular carcinoma patients [HCC] (F), melanoma patients (G), non-small-cell lung cancer patients [NSCLC] (H), and colorectal cancer patients [CRC] (I). (J) Overlap of HCC, melanoma, NSCLC, and CRC dysfunctional TIL signature genes. (K) Heatmap of day 0 and 28 CAE surCARpos RNA-seq samples on CD8+ TIL dysfunction gene signature common between hepatocellular carcinoma, non-small-cell lung cancer, colorectal cancer, and melanoma (18 genes in common from Figure S3J). (L) GSEA analysis of day 0 and 28 CAE surCARpos RNA-seq samples with genes upregulated in CD8+ HA GD2 CAR T cells from (Lynn et al., 2019). (M) Overlap of genes upregulated in CD4+ T cells and CD8+ T cells in day 28 CAE surCARpos cells (compared to day 0 cells) (left) and for genes downregulated (right). (N) Normalized RNA-seq counts of representative dysfunction signature genes in CD4+ T cells at day 0 and day 28 CAE. Average of two biological replicates and data is shown as mean ± SEM. Statistical significance of overlap between two groups (in panels C-D, F-I, M) was calculated using hypergeometric test.
(A) ATAC-seq open chromatin regions specific to day 0 (left) or day 28 CAE (right) surCARpos cells. ATAC-seq analyses include 4 biological replicates. (B) Genomic location of open chromatin regions for day 0 (blue) and day 28 CAE (red) surCARpos cells. (C) Relation of gene expression and chromatin changes during CAE. Average ATAC-seq signal of genes upregulated at day 28 (left) and genes downregulated (right) in day 0 and day 28 CAE surCARpos cells. Average of 4 biological replicates represented with box plots. Statistics assessed by Mann-Whitney. (D) Heatmap of sub-nucleosome ATAC-seq tags for naïve T cells and PD1-high TILs from (Philip et al., 2017) at sites opening in day 28 CAE surCARpos cells compared to day 0 cells. (E) Box plot of PD1-high sub-nucleosome ATAC-seq tags from (Philip et al., 2017) normalized to naïve T cells at day 28 CAE specific sites versus an equal number of randomly sampled other open chromatin regions (OCRs). Difference assessed by permutation test. (F) ATAC-seq tracks from day 0 and day 28 CAE cells at CD5, CD28, and TCF7 regulatory regions. Analysis includes four biological replicates. (G) Decile plot showing correlation between bulk RNA-seq and single-cell RNA-seq datasets. Genes differentially expressed in single-cell data between day 20 CAE and day 0, defined by cellfishing.jl, were divided into 10 groups and sorted by single-cell fold-change, going from lowest to highest (x axis). Y-axis plots the fold change (day 28 CAE/day 0) in the bulk RNA-seq for genes in each group. (H) Heatmap of top 10 marker genes for each day 0 single-cell cluster as defined in Figure 3A, donor ND388. Columns correspond to cells and rows correspond to gene names. (I) Gene ontology determined by metascape pathway analysis for each single-cell day 0 cluster, donor ND388. Columns are cell clusters (defined in Figure 3A) and rows are enriched pathways color coded by level of significance. (J) Venn diagram displaying overlap between genes upregulated in day 28 CAE surCARpos CD8+ T cells from bulk RNA-seq analyses (see Figure 2A) and genes that define single cell clusters D20–1, D20–2, D20–3, and D20–4 from day 20 CAE cells (see Figure 3B). Significance calculated using hypergeometric test. (K) Violin plots depicting gene expression levels from day 20 CAE cells (donor ND388) for SRGAP3, DUSP4, and CSF1. X axis is cell clusters defined in Figure 3B. (L) Violin plot of day 20 CAE cells for CTLA4, donor ND388 (padj 1.2e-11, log2FC 0.20).
Single-cell analysis of day 0 product and day 28 CAE cells for donor ND538 (A-E) and donor ND150 (F-J). (A) UMAP projection of single-cell gene expression data from day 0 cells, made using Seurat. Each dot corresponds to one cell and cell clusters are color coded. (B) UMAP projection of single-cell gene expression data from day 28 CAE T cells, made using Seurat. Each dot corresponds to one cell and cell clusters are color-coded. (C) Heatmap of top 10 marker genes for each day 28 CAE cluster as defined in (B). Columns correspond to cells and rows correspond to gene names. (D) Volcano plot depicting differentially expressed genes between day 28 CAE cluster 1 (dysfunctional) and clusters 2 and 3 (non-dysfunctional), also see (E). Genes upregulated in the dysfunctional cluster are on the right side and genes downregulated are on the left. The x-axis is log2(fold change) and y-axis is -log10(p value). Red dots indicate significant genes with p<0.05 and log2FC >0.5. (E) Dot plot illustrating the expression level of genes in day 0 (left) and day 28 CAE cell clusters (right). Genes included are dysfunction signature genes (N=30), naïve/memory, cell cycle, and control genes. Each column represents one cluster. The size of the circle represents the percent of cells expressing the gene in each cluster and the color depicts how highly expressed the gene is within that cluster. The number of cells in each cluster is written beneath cluster identity. Donor ND150 (F) UMAP projection of single-cell gene expression data from day 0 cells. (G) UMAP projection of single-cell gene expression data from day 28 CAE cells. (H) Heatmap of top 10 marker genes for each day 28 CAE cluster as defined in (G). Columns correspond to cells and rows correspond to gene names. (I) Volcano plot depicting differentially expressed genes between day 28 cluster 3 (non-dysfunctional) and clusters 1,2,4, and 5 (dysfunctional), also see (J). Genes upregulated in the dysfunctional clusters are on the right side and genes downregulated are on the left. The x axis is log2 (fold change) and y axis is -log10(p value). Red dots indicate significant genes with p<0.05 and log2FC >0.5. (J) Dot plot illustrating the expression level of genes in day 0 (left) and day 28 CAE (right). Genes included are dysfunction signature genes (N=30), naïve/memory, cell cycle and control genes. Each column represents one cluster. The number of cells in each cluster is written beneath cluster identity.
(A) Phenotypical change between day 0 products (left) and day 29 CAE samples (right) profiled by mass cytometry using a NK flow panel. Data from 2 donors (ND150 and ND538) are shown. (B) Expression of surface M5CAR and NK-associated molecules (CD161, TIGIT, CD56, NKG2A, NKG2C) and granulysin on day 0 product (top) and day 29 CAE CD8+ T cells (bottom). Red circles highlight subpopulations of CD8+ T cells more abundant under CAE. (C) Representative plot showing mesothelin expression from an AsPC-1 recurrent tumor associated with Figure 4B, and the FMO control (right). (D) Frequency of CD8+ T cells infiltrating recurrent tumors (n=3 mice). Data is shown as mean ± SEM. (E) Representative dot plot of surface CAR (sCAR) and intracellular (iCAR) staining (left) and FMO controls (middle). Percentage of CD8 T cells expressing surface and intracellular CAR isolated from recurrent tumors from Figure 4B (n=3), (right). (F) Model for CD56 depletion assay. Expected percentage of NK-like T cells (CD56+, y-axis) and time (x-axis) during continuous antigen stimulation for out competition model (left) and transition model (right). Control cells start with regular CAR T cell population at day 0 and CD56-depleted starts with day 0 CAR T cells depleted of CD56. (G) TCR single-cell sequencing data for day 0 and day 28 CAE for donors ND150 and ND538. Y axis is the percentage of CDR3 sequences, and x-axis is the number of cells that have that CDR3 sequence. Illustrates that between 96–99% of CDR3 sequences are unique. (H) Single-cell transcript levels of FOS, EGR1, FOXP3, MYB, LEF1, CREM, TWIST1 illustrated by UMAP plots, corresponding to clusters from Figure 3B (day 20 CAE cells). Top two clusters are dysfunctional. (I) Transcription factor motif for SOX4 from Jaspar database (top) and SOX4 and SOX17 motifs from UniProt database (bottom).
(A-B) Representative ATAC-seq tracks in regulatory regions at SOX4 motifs from day 0 and day 28 CAE samples at NK receptor and dysfunction genes KLRC1 (A) and KLRB1 (B). SOX4 motifs labeled with red bars above tracks. Analysis includes four biological replicates. (C) Agarose gel (top) and KO efficiency by genomic DNA sequencing (bottom) showing CRISPR edits on KO T cells. ID3-specific and SOX4-specific PCR targeting the edited region of each transcription factor shows the appearance of two bands in KO UTD and KO M5CAR T cells, corresponding to two edited populations derived from different sgRNA hits as depicted in Figure 7A. The KO efficiency quantification from two donors (ND566 and ND539). WT: wildtype, UTD: non-electroporated un-transduced T cells (no M5CAR), M5: T cells electroporated with M5CAR. Data is shown as mean ± SEM from two CAR T donors. (D) Cytotoxicity assessment of day 0 products at 1:1 (left) and 1:10 (right) E:T ratio on ND539 (top) and ND566 (bottom) in WT, ID3 KO, and SOX4 KO M5CAR T cells. Media used as a control. Data is shown as mean ± SEM. (E) Flow cytometry characterization of naïve, central memory, effector memory and effector subsets on day 0 product with CD45RO and CCR7 expression for WT, ID3 KO, and SOX4 KO M5CAR T cells for donor ND566 (left) and ND539 (right). (F) Percentage of NK-like T cells in WT and ID3 KO cells, relative to WT (donor ND539). Significance measured by Fisher’s exact test. (G) In vitro killing assay of ND539 WT, ID3 KO, and SOX4 KO M5CAR T cells. Cells were collected on day 18 of CAE and seeded at 1:8 E:T ratio with AsPC-1 on day 18. WT day 0 cells are used as a positive control and media is used as a negative control. Data is shown as mean ± SEM. (H) In vitro killing assay of ND566 WT, ID3 KO, and SOX4 KO M5CAR T cells. Cells were collected on day 21 of CAE and seeded at 1:8 E:T ratio. Data is shown as mean ± SEM. (I) In vitro killing assay of ND566 WT, ID3 KO, and SOX4 KO M5CAR T cells. Cells were collected on day 28 of CAE and seeded at 1:8 E:T ratio. Data is shown as mean ± SEM. Supplemental excel tables
Table S1: Differentially expressed genes between day 0 and day 28 CAE CAR T cells from bulk RNA-seq experiments. Related to Figure 2A.
Table S2: Full list of Ingenuity Pathway Analysis pathways enriched in differentially expressed genes between day 0 and day 28 CAE CAR T cells from bulk RNA-seq experiments. Related to Figure 2C.
Table S3: All upstream regulators (transcription factors) predicted to regulate differentially expressed genes between day 0 and day 28 CAE CAR T cells from bulk RNA-seq experiments. Related to Figure 2F.
Table S4: Differentially expressed genes between dysfunctional clusters (D20–1, D20–4) and non-dysfunctional clusters (D20–2, D20–3) from scRNA-seq experiment in donor ND388. Related to Figure 3E.
Table S5: Table of dysfunction signature genes (N=30). X marks genes that are upregulated in LCMV exhausted T cells or defines dysfunctional TILs in scRNA-seq experiments. HCC (hepatocellular carcinoma), NSCLC (non-small-cell lung cancer), CRC (colorectal carcinoma), LCMV (lymphocytic choriomeningitis virus). Related to Figures 3 and S3.
Table S6: All upstream regulators (transcription factors) predicted to regulate differentially expressed genes between day 0 and day 20 CAE CAR T cells from scRNA-seq experiment in donor ND388. Related to Figure 6A.
Data Availability Statement
All genomics data have been submitted to the gene expression omnibus database and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
Unique computer code used in this manuscript has been submitted to GitHub and can be accessed using the following link https://github.com/bergerlabupenn/InVitroCARTexh_code_2020.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.