

Abstract
Advances in population scale genomic sequencing have greatly expanded the understanding of the inherited basis of cardiovascular disease (CVD). Reanalysis of these genomic datasets identified an unexpected risk factor for CVD, somatically acquired DNA mutations. In this review, we provide an overview of somatic mutations and their contributions to CVD. We focus on the most common and well described manifestation, clonal hematopoiesis of indeterminate potential (CHIP). We also review the currently available data regarding how somatic mutations lead to tissue mosaicism in various forms of CVD, including atrial fibrillation and aortic aneurism associated with Marfan Syndrome. Finally, we highlight future research directions given current knowledge gaps and consider how technological advances will enhance the discovery of somatic mutations in CVD and management of patients with somatic mutations.
Cardiovascular disease (CVD) is the leading cause of mortality, accounting for 32% of all deaths worldwide1. A mainstay of the management of CVD is risk factor modification2. Well established risk factors include modifiable lifestyle factors such as diet, alcohol consumption, tobacco use, and exercise while more recently, environmental as well as social determinants of health are partially modifiable risk factors contributing to the development of CVD3–5. There is also strong evidence supporting non-modifiable risk factors such as age, biological sex, and inherited genetics in the development of CVD6–8. Increasingly, somatic or acquired mutations in a variety of tissues have been identified as risk factors for the development of CVD, some with a substantial impact on the development and severity of CVD.
Most investigations into cardiovascular disease genetics focus on inherited genetic mutations; however, individuals acquire mutations throughout their lifespan. Although acquired mutations have historically been a focus of cancer genomics, recent technological advances in genome sequencing have enabled a new ability to catalog this axis of genetic diversity. These technological advances have resulted in an emerging appreciation for how acquired mutations contribute to diseases beyond cancer. These new technologies include error-corrected deep sequencing and single cell simultaneous multi-omics; both of which have scaled to higher throughput while at historically low costs. Simultaneous with the development of large-scale biobanks and tissue repositories, these technologies and methods have allowed for the discovery of mutations with low variant allele frequencies (VAF) in patients with CVD and led to a greater understanding of the somatic determinants of CVD.
In this review, we introduce the concept of somatic mutations leading to tissue mosaicism and subsequent disease, while providing an overview of the current understanding of the origin of this phenomenon. The most prominent example of somatic mosaicism is clonal hematopoiesis of indeterminate potential (CHIP) which has broad ranging effects across the CVD spectrum exerted primarily through an inflammatory mechanism. We then examine other somatic mutations in CVD, providing a framework for the consideration of somatic mutations in CVD more broadly. Lastly, we consider how new longitudinal cohorts with deep phenotyping and precision medicine derived clinical trials in combination with novel methods, such as deep error-corrected sequencing and single cell sequencing will enable re-appraisal of prior work and new discoveries.
Somatic Mutations and Mosaicism
Mutations occur throughout the life of an individual due to a variety of biological mechanisms9. Developmental timing determines the anatomical distribution, allele frequency, and clinical manifestations of these somatic mutations10. Widely known through our understanding of cancer development and progression, somatic mutations and subsequent tissue mosaicism result in a spectrum of clinical manifestations with malignancy at one extreme and quiescent mutations producing no clinically relevant disease at the other. Technological advancements over the past decade, including enhanced somatic variant calling, microdissection, and spatial genetics, have revealed mosaicism to be common both in health and disease11–13.
Somatic mutations robustly occur as early as the first cellular division of embryogenesis, continuing throughout embryogenesis and into adulthood via several mechanisms12,14. (Figure 1, left panel) The most common and reliable mechanism for somatic mutation formation is spontaneous deamination of 5-methylcytosine to thymine, primarily at methylated CpG dinucleotides15. Left unidentified for repair, this alteration is passed to progeny cells throughout the lifespan of the organism in a linear fashion and can serve as a marker of aging16. Small insertions and deletions (indels) may also commonly arise during non-homologous joining of DNA double-strand breaks and replication error by DNA polymerase, though at considerably lower rates17,18. Larger structural variations, such as kilobase-sized insertions, deletions, loss-of-heterozygosity, and rearrangements are even more rare19.
Figure 1. Mechanisms of somatic mutations giving rise to tissue mosaicism.
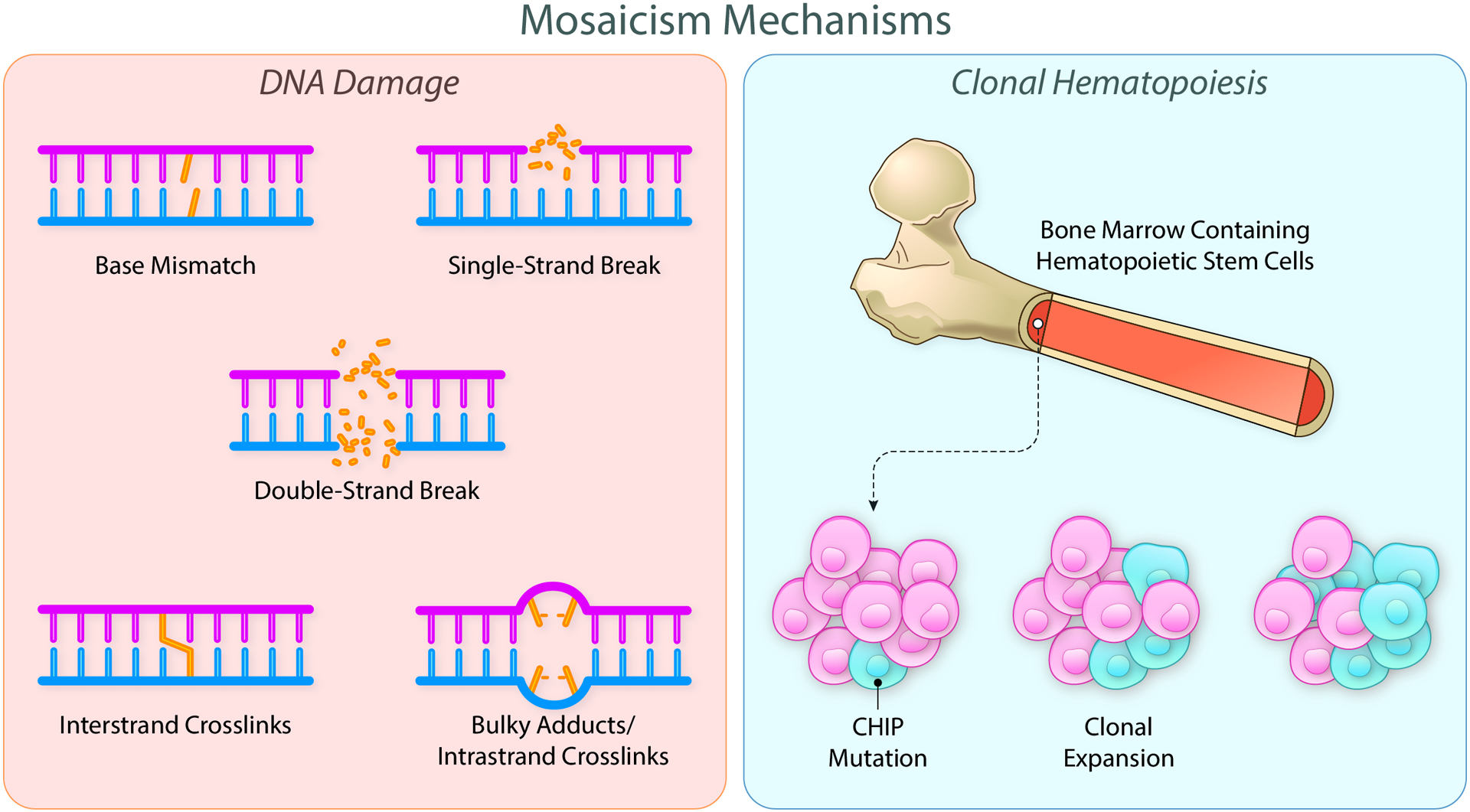
Mosaicism results from somatic DNA mutations obtained throughout the lifespan of the individual. Mutations arise from a variety of mechanisms including base mismatches, single and double strand breaks, and various crosslinks (left panel). When these mutations occur in driver genes within hematopoietic stem cells (blue cells), a survival advantage can be conferred, leading to the enhanced proliferation of the mutated cells (right panel). Clonal hematopoiesis of indeterminate potential results from this selective advantage. Created with BioRender.com. Illustration Credit: Ben Smith.
Within cardiomyocytes, a cell that rarely undergoes division, somatic mutations were thought to be uncommon, reinforced by several studies using next generation sequencing (NGS) to evaluate primary cardiac tissue13,20,21. However, single-cell whole genome sequencing (WGS) was recently used to identify single nucleotide variants (SNVs) from 48 single cardiomyocytes in 10 healthy individuals. Remarkably, this revealed human cardiomyocytes to have as many as 4,000 to 30,000 somatic SNVs per cell22. However, given the non-dividing nature of cardiomyocytes, somatic mutations derived after organogenesis would not have the ability pass these mutations to progeny. This contrasts with somatic mutations within stem cell progenitors which can subsequently pass mutations to daughter cells, creating tissue mosaics.
Clonal Hematopoiesis of Indeterminate Potential
Unlike cardiomyocytes, hematopoietic stem cells (HSPCs) are constantly dividing. Somatic mutations in select genes confer a competitive advantage, leading to a clonal proliferation termed CHIP. HSPCs harboring CHIP driver mutations give rise to a similarly mutated population of peripherally circulating blood cells, collectively called a clone23,24 (Figure 1, right panel). While patients harboring driver mutations are at higher risk for myeloproliferative neoplasms, CHIP nomenclature is used to distinguish from cancer-related clonality as relatively few individuals with CHIP go on to develop blood cancers. HSPCs with CHIP driver mutations harbor the potential for malignant conversion with the acquisition of additional somatic mutations that would enable unchecked growth and organ dysfunction25. An important aspect of CHIP diagnosis and prognosis is clone size. Currently, CHIP is considered present if the VAF reaches 2%, corresponding to 4% of circulating cells, presuming heterozygosity26,27.
CHIP is strongly linked to aging and is estimated to affect greater than 10% of individuals older than 70 years of age28–30. Patients with CHIP have a tenfold increased risk of developing blood cancer; however, this risk does not fully account for the 30–40% increased risk of mortality associated with this condition. Rather, CHIP patients have higher rates of ischemic stroke and cardiovascular disease which accounts for the increased mortality (figure 2). In fact, the risk of developing coronary disease is twice as high in patients with the three most common CHIP mutations, DNMT3A, TET2, and ASXL1 while having a JAK2 mutation conferred a 12-fold relative risk of incident coronary artery disease (CAD)31.
Figure 2. Somatic mutations in cardiovascular disease.
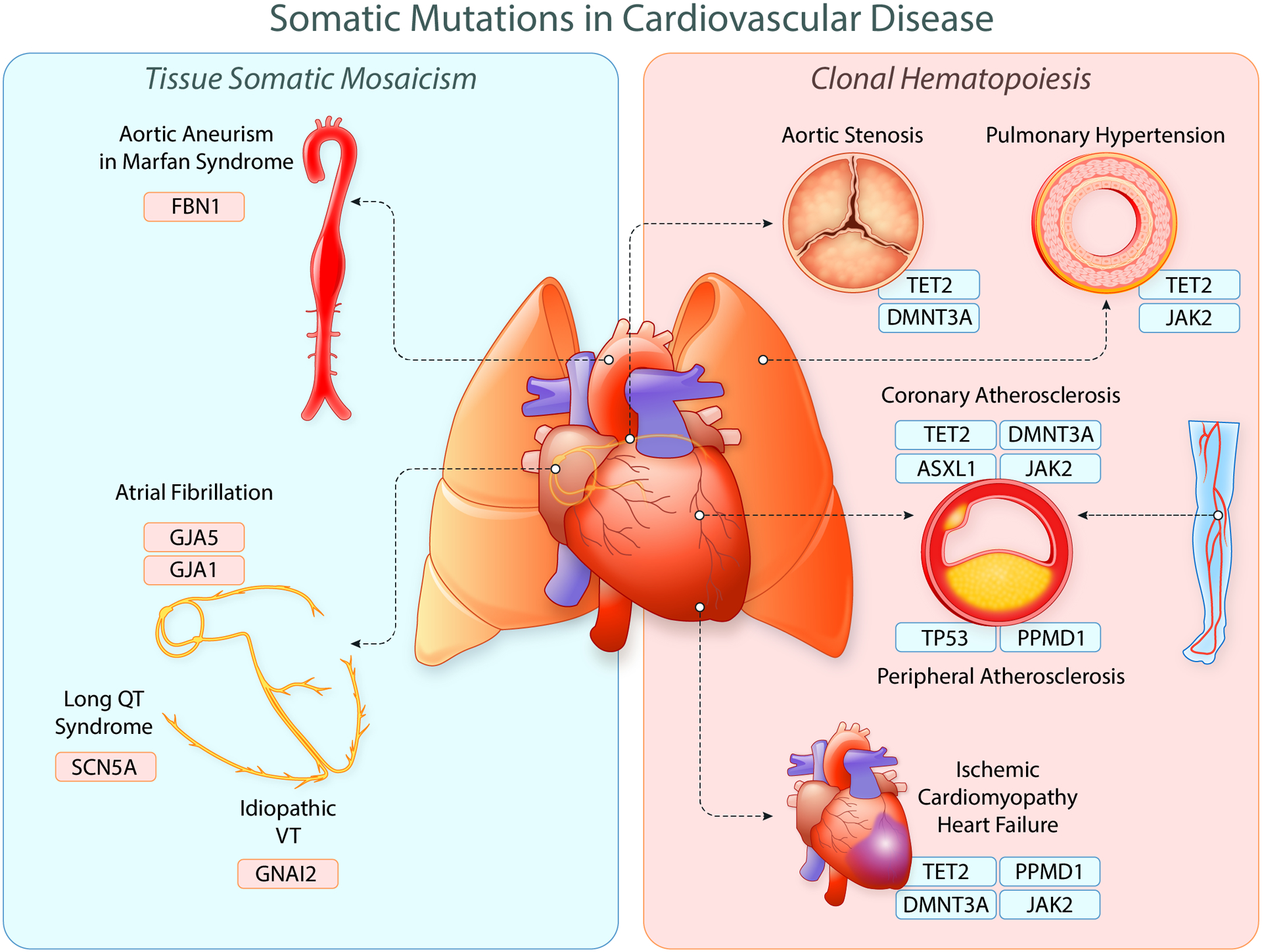
Somatic mutations can lead to a variety of manifestations in cardiovascular disease ranging from conduction system alterations to the development of atherosclerotic coronary disease. The most commonly identified somatic mutations are CHIP mutations within hematopoietic stem cells which lead to deleterious downstream effects across the cardiovascular system. Due to technological advancements, somatic mutations are increasingly being identified and characterized across cardiovascular tissues. Created with BioRender.com. Illustration Credit: Ben Smith.
Although there are ~75 CHIP driver mutations, the most common, DNMT3A, TET2, and ASXL1, account for two-thirds of patients with CHIP26,30,31. DNMT3A and TET2 are involved in DNA methylation while ASXL1 is a chromatin regulator. Loss of function mutations in both DNMT3A or TET2 show enhanced renewal capability in mice in vitro leading to the development of clonal populations32. ASXL1 mediated histone modification is essential in normal hematopoiesis among HSPCs; however, a clear mechanism for clonal expansion has not yet been elucidated in the setting of CHIP33. JAK2, PPM1D, and TP53 are also notable CHIP mutations due to their influence on CVD. TP53 is involved with DNA damage response (DDR) and HSPCs harboring mutations in this gene have a competitive advantage over neighbors within the same compartment34. PPM1D is also part of the DDR pathway and mutations confer a proliferative advantage through resistance to p53 mediated apoptosis35. JAK2 is part of the cellular JAK/STAT signaling pathway and activating mutations allow for unopposed cellular proliferation, supporting clonal expansion36.
While CHIP mutations occur in somatic cells, mounting evidence suggests the presence of a germline predilection towards the development of CHIP mutations in HPSCs. In 2009, a GWAS-based analysis identified an allele in the JAK2 locus that predisposes to the development of JAK2p.Val617Phe (JAK2V617F) derived MPNs, a finding that has been affirmed by several other studies and lends credence to the notion of inherited risk of clonality37–41. Later, telomerase reverse transcriptase (TERT), a key enzyme in the maintenance of telomeres, was found to be associated with incident CHIP in a whole genome-based GWAS among a large Icelandic cohort30. The same germline TERT loci was again identified in a large study of nearly 100,000 genomes29. It is notable that TERT is constitutively expressed in HPSCs whereas most cells within the body lack expression of this gene42. TERT itself has been associated with CVD in several GWAS, but the role of telomeres and their regulatory environment as it pertains to HPSC clonality and downstream effects is not fully known and remains a focus for future research43,44. Several novel SNVs were identified in the same large study of 100,000 genomes, furthering the hypothesis of germline risk of CHIP29. This group identified an intergenic region near TET2 (rs144418061) in patients with African ancestry which confers a 2.4-fold increased risk of CHIP, an association that was equal among the three most common CHIP driver mutations (DNMT3A, TET2, and ASXL1). Further work from the same paper showed a single locus within the intron of the T-cell leukemia/lymphoma 1A gene (TCL1A, rs2887399) leading to a 1.23-fold risk of acquiring DNMT3A-specific CHIP. There is partial overlap between the germline loci that give rise to CHIP mutations and germline loci associating with CVD that suggests a potential common genetic source for the development of CVD. The degree to which germline variants determine CHIP risk continue to be refined through larger studies, but the impact can be extrapolated from data from the above studies. For example, since the TET2 SNV rs144418061 confers a 2.4 fold increased risk of CHIP development for each copy of the risk allele, a 50 year old individual with one copy of this allele is at a similar risk to a 65 year old individual without this allele45,46. Future studies will further refine our understanding of the germline determinants of CHIP development and progression.
CHIP in Cardiovascular Disease
Atherosclerotic Coronary Artery Disease
Somatic mutations in HSPCs leading to CHIP are strongly associated with the development of coronary artery disease29,31,47. This association was confirmed across several of the CHIP driver mutations including DNMT3A, TET2, ASXL1 and JAK2. Causality in humans is further supported by data showing that there is a dose-response relationship with VAF/clone size and atherosclerosis severity as assessed by coronary artery calcification scoring31. Studies in animal and cell models have revealed further mutation-specific mechanistic insights into the development of coronary disease.
TET2 is the second most common CHIP mutation and has been the subject of multiple mechanistic studies by independent research groups evaluating atherosclerosis and coronary disease. Competitive bone marrow transplantation of TET2−/− HSPCs in a mouse model of hypercholesterolemia-driven atherosclerosis recapitulated the clonal expansion seen in TET2 CHIP patients without alterations in blood counts48. Post bone marrow transplant, the TET2-deficient cells expanded markedly in the marrow and led to a 60% increase in atherosclerotic plaque size48. A contemporaneous study evaluated the effects of TET2 inactivation within all hematopoietic cells on the background of a diet-driven atherosclerotic mouse model to find similarly, that TET2 inactivation led to 2.7 fold larger atherosclerotic lesion area31. Pursuing this mechanism further, myeloid lineage-specific TET2-knockout mice and macrophages showed accelerated atherosclerosis development dependent on an enhanced CXC chemokine expression and subsequent secretion of IL-1β and IL-631,48. These findings are supported by prior studies in primary cells where TET2 was found to participate in the active suppression of IL-6 transcription during inflammation in innate myeloid cells, including dendritic cells and macrophages. Furthermore, loss of TET2 resulted in the upregulation of several inflammatory mediators, including IL-649. These fundamental studies have translational relevance as clinical data has pointed to a putative inflammatory mechanism implicating IL-6 in the development of coronary disease. One study evaluated the effects of a genetically mediated reduction in IL-6 signaling on CVD event rates. Utilizing a common variant in the IL-6 receptor gene that disrupts IL-6 signaling, whole exomes were evaluated revealing that those with the mutated IL-6 signaling had significantly lower CVD event rates50. Furthering this notion, serum IL-6 levels are highly predictive of coronary artery disease as measured in patients of intermediate risk referred for coronary angiography51. In a sub-study of the CANTOS clinical trial, inhibition of IL-1β, an upstream mediator of IL-6, reduced major adverse cardiovascular events among high-risk atherosclerosis patients with CKD52. Building on this finding, the initial results of the IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE) trial, found that IL-6 inhibition was effective at reducing C-Reactive protein (CRP) levels as well as fibrinogen, and lipoprotein(a), all biomarkers relevant to the development of atherosclerosis53.
Similar to TET2, clonal hematopoiesis associated with JAK2V617F mutations have significant effects on coronary disease. JAK2V617F mutations lead to cytokine-independent activation of the JAK–STAT pathway, resulting in proliferation of mature myeloid cells, mechanistically distinct from DNMT3A and TET2 driver mutations54. Mice with myeloid-specific JAK2V617F mutations develop accelerated atherosclerosis in concert with cellular proliferation underpinned by AIM2 inflammasome activation and IL-1β production55. This is similar to what was observed in mice transplanted with JAK2V617F positive bone marrow after irradiation56. These events led to an overall increase in the burden of inflammatory macrophages within atherosclerotic lesions but also to increased necrotic core formation and putative plaque instability. These effects were prevented with the use of an IL-1β inhibitor55,56.
Ischemic Cardiomyopathy and Heart Failure
After initial associations with CVD and CAD, CHIP was next mechanistically linked to congestive heart failure (CHF) in animal models and human epidemiological cohorts57,58. Initial investigation began in murine models of heart failure. One group evaluated TET2 inactivation in two separate models of murine heart failure. TET2 inactivation led to cardiac dysfunction in both models. They posited this was through an IL-1β mediated mechanism as IL-1β was upregulated in their experimental models. Furthermore, inactivation of the upstream mediator, NOD-, LRR- and pyrin domain-containing protein 3 inflammasome complex (NLRP3), protected against the development of heart failure in both models and prevented several markers of cardiac dysfunction59. Clustered regularly interspaced short palindromic repeats (CRISPR) technology was later used by this same group to introduce inactivating mutations in TET2 and DNMT3A in bone marrow cells which were subsequently engrafted into lethally irradiated mice. When challenged with an infusion of angiotensin II, mice with inactivating mutations in TET2 or DNMT3A displayed greater cardiac hypertrophy, decreased cardiac function, and higher levels of fibrosis when compared to wild type controls60. TET2 inactivation promoted the expression of IL-1β, IL-6 and CCL5 whereas DNMT3A inactivation promoted the expression of CXCL1, CXCL2, IL-6 and CCL5 when stimulated with lipopolysaccharide in a macrophage cell line. These results are supportive of an inflammatory mechanism for the enhanced effects of CHIP in these two specific mutations60.
Beyond the CHIP genes that modulate epigenetics, JAK2, PPM1D, and TP53 also have notable cardiovascular consequences across several models of murine heart failure. Mice that underwent transduction of myeloid-restricted JAK2V617F cells had larger infarct size in a LAD-ligation model of myocardial injury and subsequently were found to have a greater reduction in cardiac function when compared to controls as a result of this insult61. Consistent with prior studies, myeloid-restricted JAK2V617F mice had greater expression of IL-6 and IL-1β but had no alterations in overall cell counts. Using the same transduction model, this group sought to evaluate pressure-overload heart failure through transverse aortic constriction surgery. In this setting, mice with myeloid-restricted JAK2V617F had greater cardiac hypertrophy and fibrosis along with decreased cardiac function61. Furthermore, heart tissue from these mice displays greater macrophage infiltration and IL-6 transcript expression61. In a separate line of work by another group, mice transplanted with bone marrow containing PPM1D-mutant cells were more susceptible to stress-induced cardiac remodeling and dysfunction. PPM1D-mutant macrophages displayed DDR pathway suppression and greater cytokine production in response to cardiotoxic stress via chemotherapy. Notably, NLRP3 inflammasome inhibition reversed the mouse phenotype that was conferred by the transplantation of the PPM1D-mutant cells. These data suggest gain-of-function mutations in PPM1D can contribute to chemotherapy induced cardiomyopathy through an inflammatory mechanism and inhibition of upstream mediators provide potential therapeutic targets for the treatment of this condition62. Adding to this, Sano et al. showed doxorubicin treatment can lead to the rapid expansion of a pre-existing TP53 clone, producing enhanced cardiotoxicity through a neutrophil mediated mechanism which further supports the important role of DDR CHIP mutations in the development of CVD63.
Human data on CHIP in heart failure appear concordant with animal and cellular models. At the single cell level, Abplanalp et al. focused on DNMT3A mutations within monocytes and T cells and showed monocytes from heart failure patients with DNMT3A mutations have an increased proinflammatory signature compared to heart failure controls58. This included increased expression of IL-6, CXCL2, IL-1β, tumor necrosis factor (TNF), and NLRP3. These findings were experimentally corroborated using a DNMT3A-silenced human monocyte cell line. Interestingly, widespread genetic changes were found within both monocytes and T cells regardless of mutational status, suggesting a pleiotropic effect of DNMT3A mutations.
In a clinical study of 200 older and mostly male patients with CHF, the participants underwent bone marrow biopsy and their samples were subsequently sequenced for CHIP mutations. Worse long-term clinical heart failure outcomes, as measured by death or death combined with heart failure hospitalization, were found in patients with either DNMT3A or TET2 mutations compared with non-CHIP CHF patients. There was a statistically significant dose-response association between clone size and clinical outcome, suggestive of a causal relationship64. Subsequently, a large prospective cohort study of 50,000 patients showed there was an association between overall CHIP presence and incident HF. Gene-based analyses in this study demonstrated significant associations specifically for TET2, JAK2, and ASXL1, but interestingly, not DNMT3A57.
Aortic Stenosis
CHIP has been linked to poor outcomes after transcatheter aortic valve implantation (TAVI) in calcific aortic stenosis (AS). In one study, eight patients with severe degenerative AS and three controls underwent single-cell RNA sequencing analyses of circulating peripheral monocytes. AS patients who carried DNMT3A or TET2 CHIP-driver mutations had increased monocyte expression of IL-1β, IL-6R, and NLRP3, all potent mediators of inflammation. Importantly, there were no significant differences in circulating levels of IL-6 or high-sensitivity C-reactive protein in this cohort of patients, leading the authors to speculate that patients with DNMT3A or TET2 driver mutations may be primed for excessive inflammatory responses65. Other studies have noted increased circulating levels of IL-6 in patients with AS and have correlated this with disease progression. Therefore, increased inflammatory potential via IL-6 is a possible explanatory mechanism for poor outcomes in the time period after TAVI66.
Another study evaluated 279 sequentially enrolled older patients undergoing TAVI for critical AS in Germany67. Enrolled patients were sequenced for the presence of either TET2 or DNMT3A CHIP driver mutations and then followed with the primary endpoint of all-cause mortality. Notably, the evaluated CHIP mutations were enriched in patients with severe calcified AV stenosis undergoing TAVI. The primary finding of the study shows patients carrying either a DNMT3A or TET2 CHIP driver mutations experienced significantly worse clinical outcome for death during the first 8 months after TAVI with a hazard ratio of 3.1 when compared to non-CHIP controls. The authors subsequently performed FACS on a subset of the patients revealing a skew towards pro-inflammatory t-cell polarization in DNMT3A-CHIP patients and increased levels of circulating non-classical monocytes in TET2-CHIP patients. Inflammatory infiltrate in surgically removed mineralized aortic valves is composed of macrophages, mast cells, CD4+ T cells and CD8+ T cells68 and as such, the cellular alterations seen in these CHIP mutations add to the notion that an inflammatory mechanism might be critical to poor outcomes in AS.
Peripheral Artery Disease and Venous Thromboembolism
Observations linking CHIP to atherosclerosis beyond the coronary vasculature are emerging. One group has identified TP53 as a potential driver of atherosclerosis in vascular beds across the body69. This group leveraged whole exome sequences and tested whether CHIP was associated with increased risk of PAD and atherosclerosis within multiple arterial beds. Their preprinted work revealed the novel finding that DDR TP53 and PPM1D CHIP associates with incident PAD. Specifically, there were significant associations between TP53 and CAD, aortic and peripheral aneurysms, and chronic mesenteric ischemia. A mouse model of atherosclerosis transplanted with 20% Trp53−/− bone marrow cells proved sufficient to accelerate atherosclerosis development through a macrophage-dependent process69. This stands in contrast to previous work on DNMT3A and TET2 CHIP, where elevated expression of pro-inflammatory cytokines IL-6 and IL-1β in the aortic wall promote atherosclerosis suggesting a distinct mechanism for injury in this CHIP subtype.
To date there have been no studies on the effects of CHIP on VTE in the absence of MPN. There is one study to report negative results, where the presence of non-JAK2 CHIP mutations seem to have no impact on VTE formation in the setting of MPNs70 For JAK2, patients with MPN found to have JAK2V617F mutations had significantly elevated rates of venous thrombosis compared to controls in a large-scale clinical cohort study71. They postulated this effect was through an enhanced neutrophil extracellular trap (NET) formation, an important factor in thrombosis. Treatment with ruxolitinib, a JAK1/2 inhibitor, decreased NET formation in vitro and decreased thrombosis in JAK2V617F mice in vivo71.
Pulmonary Hypertension
Evaluating a large PAH cohort revealed mutations in TET2 were associated with PAH independent of previously established PAH genes. Interestingly, a significant proportion of these mutations were predicted to be germline rather than somatic (75% vs 25%, respectively). The identification of somatic variants, perhaps limited by read depth of whole exome sequencing, may have underestimated the prevalence of somatic mutations. Furthermore, primary lung tissue was not used to corroborate their findings. To investigate the underlying mechanisms of their clinical findings, a mouse model of conditional hematopoietic TET2 knockout was sufficient to induce PH typified by marked vascular remodeling and profound microvascular loss secondary to increased inflammation72. This model had increased levels of IL-1B and the phenotype was recovered when treated with antibody-mediated IL-1B blockade. A more recent study found that JAK2 CHIP was associated with the development of PAH in transgenic mice as well as mice transplanted with JAK2V617F bone marrow cells73. These effects were mediated through the enhanced differentiation of neutrophils in pulmonary arterial regions leading to upregulation of chemokine activity and subsequent arterial remodeling. However, unlike similar models of clonal hematopoiesis, this model harbored baseline hematological differences, such as elevated white blood cell count which could confound their results. An analysis of a 70 person PAH cohort identified an increase JAK2V617F mutations in PAH cases (7%) compared to aged matched controls (0%)73.
Primary Tissue Somatic Mosaicism in CVD
Marfan Syndrome
Marfan Syndrome (MFS) is a hereditary connective tissue disorder which typically results from heterozygous pathogenic variants in the FBN1 gene, encoding fibrillin-1. Cardiovascular manifestations include thoracic and abdominal aneurysms and dissections. About 75% of cases have a positive family history whereas as many as 25% arise sporadically. Using clinically diagnosed probands, one group found 5 individuals with somatically-derived mutations in FBN1 in blood samples74. Pathogenic mosaics had been described only rarely before with two prior case reports, and therefore were thought to be primarily clinically asymptomatic as is commonly observed in parents of patients with spontaneous MFS75,76. Clinically, these five patients were heterogeneous in terms of typical MFS manifestations. Though still rare, this study and others illustrate somatically acquired mutations in the FBN1 gene are an important phenomenon and should be part of regular screening77. This information is especially important regarding genetic counseling and family planning in the proband.
Atrial Fibrillation
Atrial fibrillation is the most common arrhythmia, affecting ~1% of the population. There has been significant effort to define the genetic architecture of this disease82,83. Several pathogenic familial mutations have been identified and more recently GWAS-derived SNVs have been added together to produce clinically usable polygenic risk scores84,85. Somatic mutations have been described in a minority of cases with several studies reporting that there were no somatic variants within their respective populations. A 2015 study evaluating paired DNA from lymphocytes and left atrial appendages of 25 atrial fibrillation patients using high-depth NGS could not reveal any significant somatic mutations and subsequently concluded that atrial-specific tissue mutations are rare and that somatic mosaicism within the atria is unlikely to significantly contribute to AF pathogenesis21. Similarly, a later paired-tissue study evaluated blood and left atrial tissue DNA (harvested from posterior left atrial wall, between the pulmonary veins) from 44 AF patients also revealed no somatic variants within the studied cardiac tissue86. However, both studies were designed to evaluate valvular AF and were limited by sample size. Conversely, other reports suggest somatic mutations could contribute to the development of AF. Gollob et al showed 4 out of 15 patients with early onset idiopathic AF had heterozygous mutations in the GJA5 (connexin 40) gene in surgically harvested left atrial tissue but not in peripheral lymphocytes87. Likewise, in a selected group of 10 patients undergoing surgical PVI, 1 was found to have a mutation in the coding region of the connexin 43 gene in atrial tissue only in a study from another group88. Studies using paired cardiac tissue and DNA from blood samples combined with high-quality deep sequencing technology represent the gold standard for the determination of somatic variants and additional studies with more patients are needed to fully evaluate this phenomenon.
Long QT Syndrome
Somatic mosaicism has also been described as a rare cause of Long QT syndrome (LQTS). One group characterized an index patient with LQTs revealing the mosaic presence of a mutated SCN5A gene89. Unsure if this could be causative of the patient’s presentation, the researchers created a model simulation which suggested somatic mosaicism within the Purkinje system can lead to abnormal electrophysiological propagation consistent with LQTS, offering a potential explanation for the development of an arrhythmia-prone substrate. Furthermore, somatic mosaicism appeared to account for 0.17% of undiagnosed cases of LQTS in a large cohort of patients being evaluated for genetically derived arrhythmias, supporting the notion that LQTS is rarely derived from somatic mutations but could be considered in sporadic cases89.
Idiopathic VT
Idiopathic ventricular tachycardia occurs in patients without clear structural heart disease and in the absence of other arrhythmia syndromes such as LQTS. Notably, these arrhythmias primarily arise from the right ventricular outflow tract (RVOT), a defining characteristic of idiopathic VT. One group identified a focal somatic myocardial mutation in GNIA2 present in the RVOT, the site of the arrhythmogenic substrate, but nowhere else in the sampled myocardium90. GNAI2 codes for the alpha subunit of guanine nucleotide binding protein, part of a larger family of G proteins which are involved in inhibition of adenylyl cyclase, activation of PI-3 kinase, and modulation of K+ and Ca2+ channels91. The absence of these inhibitor proteins were shown to predispose transgenic mice to ventricular arrhythmias92. Taken together, focal somatic mutations present in the RVOT could be contributory in the development of idiopathic VT. However, there have been no follow up studies and no human studies have recapitulated these findings to date.
Congenital Heart Disease
Congenital heart disease (CHD) represents a broad spectrum of cardiovascular dysfunction that is present at birth and has historically been associated with inherited genetics. However, there exists a significant proportion of CHD cases that occur in families without a history of CHD, suggesting a mutational acquisition early in embryological development. Due to enhanced analytical methods, somatic mutations have been identified as contributing to CHD in recent years78–80. These findings have been very recently reviewed by Morton et al. and can provide the interested reader with detail beyond the scope of the current work81.
Heteroplasmy
Another facet of somatic mosaicism within the broader context of CVD is heteroplasmy. Heteroplasmy describes the presence of different mitochondrial DNA within the same organism resulting in a mosaic pattern within a particular tissue. This is thought to be an age-related phenomenon resulting from large deletions of mitochondrial DNA (mtDNA) leading to alterations in the generation of ATP or Ca2+ handling, which is especially important within the cardiac tissues93,94. Heteroplasmy has been identified as potentially contributory in arrhythmia, heart failure, and atherosclerosis while decreased mtDNA copy number, a specific type of acquired heteroplasmy is associated with incident CVD across several large, well characterized cardiovascular cohorts95.
In an animal model of accelerated accumulation of mtDNA deletions within cardiomyocytes, aged animals had significantly higher rates of arrhythmia than their control counterparts96. Similarly, a 2017 study using transgenic mice observed higher rates of spontaneous and inducible cardiac arrhythmias after experimental myocardial infarction among mice with elevated mtDNA mutations97. In humans, a prospective study evaluating high-risk patients undergoing CABG were more likely to have postoperative AF if higher levels of mitochondrial dysfunction was present in right atrial tissue98. Another study appeared to confirm these results; this group evaluated 88 paired atria-blood tissue samples for specific mtDNA deletions. mtDNA deletions were closely associated with age and were present in significantly higher quantities in patients with AF99. A 2006 study revealed mtDNA mutations present in atrial tissue but not in mtDNA from peripheral blood cells of patients with chronic AF, leading the researchers to conclude that oxidative injury and large mtDNA deletions in cardiac muscle are increased in patients with chronic AF, which may lead to the pathogenesis of AF100. More recently, mtDNA copy number was inversely associated with the risk of incident AF in several large population-based prospective cohort studies (Atherosclerosis Risk in Communities (ARIC) study, the Multi-Ethnic Study of Atherosclerosis (MESA), and the Cardiovascular Health Study (CHS)), independent of traditional risk factors for the development of AF. The investigators found mitochondrial DNA copy number is proportional to the transcription of mitochondrial genes and is a marker of mitochondrial dysfunction101,102. This study however did not directly assess mtDNA copy number in atrial tissue, rather from peripheral blood where it is presumed to be a surrogate indicator for mtDNA in heart tissue. Additional studies are needed to confirm the role of heteroplasmy in the development of arrhythmia, especially atrial fibrillation.
Mitochondrial function is essential to cardiac physiology which is especially relevant in cardiac aging and heart failure. Mitochondrial dysfunction has been highly correlated with declining cardiac function and extensively reviewed elsewhere103–105. Alterations in mtDNA are one mechanism for mitochondrial dysfunction and have been associated with heart failure. In an animal model of ischemic cardiomyopathy, mtDNA copy number is decreased in the post MI failing myocardium which correlated with increased cardiac remodeling and systolic dysfunction106. In humans, there is reduced mtDNA replication and depletion of mtDNA in heart failure while up to 22% of idiopathic dilated cardiomyopathy cases can be attributed to mtDNA mutations107,108.
Regarding atherosclerotic coronary disease, there have been multiple studies to correlate mtDNA content with the presence of coronary disease109,110. One single-center study was able to identify mtDNA content in peripheral blood mononuclear cells as a predictor for CHD and further was able to correlate mtDNA content with severity of coronary atherosclerosis110. Higher rates of heteroplasmy have also been identified in several studies of post-mortem aorta samples with increased levels of atherosclerosis111–113. There does indeed appear to be a link between the presence of mtDNA mutations or changes in copy number to atherogenesis however, the mechanisms leading to this are incompletely understood. It is possible that the mitochondrial damage leads to increased LDL oxidation and subsequent formation of atherogenic LDL species or activation of the NLRP3 inflammasome modulating the inflammation axis to promote plaque formation however additional studies are needed to confirm these theories114–116.
Outlook for Somatic Mosaicism and CVD
The evaluation of somatic mosaicism and its most commonly identified form, CHIP, in CVD is a new and growing field based on advances in sequencing technology and is primed to expand considerably in the upcoming years. In the CHIP space, there is a lack of data regarding phenotype over time. There are no published studies following CHIP clone size over time and therefore we do not know the complete natural history of this nascent condition, particularly as it relates to CVD. At least part of the difficulty with establishing this data lies with the current diagnostic tools available. With the development of a clinical CHIP bioassay, and deep error-corrected sequencing, patients could be more easily identified, and clone size followed in a longitudinal manner.
Similarly, we also have limited data as to the specific CHIP mutations and their effects on downstream pathologies. For example, are DNMT3A R882H hotspot mutations equally pathogenic as DNMT3A loss of function mutations? The distinction of clone type is an important one since CHIP represents a heterogenous set of mutations and subsequent clinical impacts. Identification of the natural history for a specific mutation will be critical to individual risk estimation and implementation of prevention strategies. Larger prospective studies are needed to assist patients and clinicians to risk stratify patients in this manner. With the growth of mega-biobanks, such as NIH All of Us, UK Biobank and others, we expect that additional data will be available to answer these questions in the next few years.
Excitingly, based on the findings reviewed here, there are putative therapeutic targets for CHIP patients on the horizon. Given the findings of several large-scale trials testing anti-inflammatory approaches such as the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS), Low Dose Colchicine 1 and 2 (LoDoCo), Colchicine Cardiovascular Outcomes Trial (COLCOT) there is strengthening evidence to suggest that inflammation plays a causal role in the pathogenesis of CVD. Indeed, a subgroup analysis of the CANTOS data revealed that patients who experienced the most inflammation reduction as measured by high-sensitivity CRP or IL-6 levels while on treatment with canakinumab were also the patients who derived the most benefit through decreased event rates117. Building on this, one team has sequenced a large proportion of the CANTOS participants to then sub-select for TET2 CHIP patients within the study and found treatment with the IL-1B inhibitor decreased relative risk of major CV events by 64% in TET2 CHIP patients compared to 15% overall118. In sum, the current available research supports a role of inflammation in the most common CHIP mutations, likely mediated via an IL-6 mechanism which makes the upcoming trial of Ziltivekimab (a selective IL-6 antagonist) compared to placebo in people with CVD (ZEUS) of particular interest; however, there are no current clinical trials specifically evaluating treatments for patients with CHIP. Together, these developments lend support for the notion that a precision medicine approach to address the unique pathophysiology of CHIP is increasingly feasible.
New technologies will catalyze ongoing efforts to identify and detect somatic mutations in all cardiovascular tissues and determine their clinical consequences. The development of deep error-corrected sequencing has led to an enhanced ability to identify somatic variants and we anticipate this trend to continue in the identification of somatic mosaicism within cardiovascular diseases. Revisitation of prior work may indeed yield divergent results, especially in the setting of evaluating somatic mutations within primary cardiovascular tissues. Combined with cutting edge bulk sequencing, emerging but low-throughput methods such as single cell DNA sequencing and multi-omics will shed light on the accumulation of mutations throughout the life of a particular cell and reveal critical aspects of pathophysiology as it pertains to cellular function over time, opening the door to new therapeutic modalities and the treatment of CVD.
Sources of Funding
This work was supported by NIH grant DP5 OD029586, a Burroughs Wellcome Fund Career Award for Medical Scientists, the E.P. Evans Foundation, RUNX1 Research Program and the Vanderbilt University Medical Center Brock Family Endowment to Dr. Bick.
Non-Standard Abbreviations
- AS
Aortic stenosis
- CAD
Coronary artery disease
- CANTOS
Canakinumab Anti-inflammatory Thrombosis Outcome Study
- CH
Clonal hematopoiesis
- CHIP
Clonal hematopoiesis of indeterminate potential
- COLCOT
Colchicine Cardiovascular Outcomes Trial
- CRP
C-Reactive protein
- HPSC
Hematopoietic stem cell
- Indel
Insertion and deletion
- LoDoCo
Low Dose Colchicine Trial
- NGS
Next generation sequencing
- NLRP3
NOD-, LRR- and pyrin domain-containing protein 3 inflammasome complex
- SNVs
Single nucleotide variants
- TERT
Telomerase reverse transcriptase
- VAF
Variant allele fraction
- WGS
Whole genome sequencing
Footnotes
Disclosures
AGB is on the scientific advisory board of TenSixteen Bio.
References
- 1.Cardiovascular diseases (CVDs) [Internet]. [cited 2021 Oct 4];Available from: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)
- 2.Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, Himmelfarb CD, Khera A, Lloyd-Jones D, McEvoy JW, et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140:e563–e595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kivimäki M, Steptoe A. Effects of stress on the development and progression of cardiovascular disease. Nat Rev Cardiol. 2018;15:215–229. [DOI] [PubMed] [Google Scholar]
- 4.Havranek EP, Mujahid MS, Barr DA, Blair IV, Cohen MS, Cruz-Flores S, Davey-Smith G, Dennison-Himmelfarb CR, Lauer MS, Lockwood DW, et al. Social Determinants of Risk and Outcomes for Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation. 2015;132:873–898. [DOI] [PubMed] [Google Scholar]
- 5.Bowen KJ, Sullivan VK, Kris-Etherton PM, Petersen KS. Nutrition and Cardiovascular Disease—an Update. Curr Atheroscler Rep. 2018;20:8. [DOI] [PubMed] [Google Scholar]
- 6.Sniderman AD, Furberg CD. Age as a modifiable risk factor for cardiovascular disease. The Lancet. 2008;371:1547–1549. [DOI] [PubMed] [Google Scholar]
- 7.Schunkert H Expanding the spectrum of CVD genetics. Nat Rev Cardiol. 2018;15:77–78. [DOI] [PubMed] [Google Scholar]
- 8.O’Neil A, Scovelle AJ, Milner AJ, Kavanagh A. Gender/Sex as a Social Determinant of Cardiovascular Risk. Circulation. 2018;137:854–864. [DOI] [PubMed] [Google Scholar]
- 9.Franco I, Helgadottir HT, Moggio A, Larsson M, Vrtačnik P, Johansson A, Norgren N, Lundin P, Mas-Ponte D, Nordström J, et al. Whole genome DNA sequencing provides an atlas of somatic mutagenesis in healthy human cells and identifies a tumor-prone cell type. Genome Biol. 2019;20:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet. 2013;14:307–320. [DOI] [PubMed] [Google Scholar]
- 11.Coorens THH, Moore L, Robinson PS, Sanghvi R, Christopher J, Hewinson J, Przybilla MJ, Lawson ARJ, Spencer Chapman M, Cagan A, et al. Extensive phylogenies of human development inferred from somatic mutations. Nature. 2021;597:387–392. [DOI] [PubMed] [Google Scholar]
- 12.Park S, Mali NM, Kim R, Choi J-W, Lee J, Lim J, Park JM, Park JW, Kim D, Kim T, et al. Clonal dynamics in early human embryogenesis inferred from somatic mutation. Nature. 2021;1–5. [DOI] [PubMed] [Google Scholar]
- 13.Moore L, Cagan A, Coorens THH, Neville MDC, Sanghvi R, Sanders MA, Oliver TRW, Leongamornlert D, Ellis P, Noorani A, et al. The mutational landscape of human somatic and germline cells. Nature. 2021;597:381–386. [DOI] [PubMed] [Google Scholar]
- 14.Abascal F, Harvey LMR, Mitchell E, Lawson ARJ, Lensing SV, Ellis P, Russell AJC, Alcantara RE, Baez-Ortega A, Wang Y, et al. Somatic mutation landscapes at single-molecule resolution. Nature. 2021;593:405–410. [DOI] [PubMed] [Google Scholar]
- 15.Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287:560–561. [DOI] [PubMed] [Google Scholar]
- 16.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. Quiescent Hematopoietic Stem Cells Accumulate DNA Damage during Aging that Is Repaired upon Entry into Cell Cycle. Cell Stem Cell. 2014;15:37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomasetti C, Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, Ling H, Hetrick KN, Pugh EW, Amos C, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44:642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, et al. Evidence for Cardiomyocyte Renewal in Humans. Science. 2009;324:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts JD, Longoria J, Poon A, Gollob MH, Dewland TA, Kwok P-Y, Olgin JE, Deo RC, Marcus GM. Targeted deep sequencing reveals no definitive evidence for somatic mosaicism in atrial fibrillation. Circ Cardiovasc Genet. 2015;8:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walsh C, Choudhury S, Huang A, Kim JJ, Morillo K, Maury E, Zhou ZZ, Kenny C, Lee E, Chen M. Somatic mutations in single human cardiomyocytes demonstrate accelerated age-related DNA damage and cell fusion [Internet]. 2021. [cited 2021 Oct 4];Available from: https://www.researchsquare.com/article/rs-84503/v1
- 23.Turhan AG, Humphries RK, Phillips GL, Eaves AC, Eaves CJ. Clonal Hematopoiesis Demonstrated by X-Linked DNA Polymorphisms after Allogeneic Bone Marrow Transplantation. N Engl J Med. 1989;320:1655–1661. [DOI] [PubMed] [Google Scholar]
- 24.Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, Karchin R, Kinzler KW, Vogelstein B, Nowak MA. Accumulation of driver and passenger mutations during tumor progression. Proceedings of the National Academy of Sciences. 2010;107:18545–18550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valent P, Kern W, Hoermann G, Milosevic Feenstra J, Sotlar K, Pfeilstöcker M, Germing U, Sperr W, Reiter A, Wolf D, et al. Clonal Hematopoiesis with Oncogenic Potential (CHOP): Separation from CHIP and Roads to AML. IJMS. 2019;20:789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N Engl J Med. 2014;371:2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N Engl J Med. 2014;371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, Szeto MD, Liao X, Leventhal MJ, Nasser J, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020;586:763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, Thorgeirsson TE, Sigurdsson A, Gudjonsson SA, Gudmundsson J, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130:742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell. 2011;20:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamamoto K, Goyama S, Asada S, Fujino T, Yonezawa T, Sato N, Takeda R, Tsuchiya A, Fukuyama T, Tanaka Y, et al. A histone modifier, ASXL1, interacts with NONO and is involved in paraspeckle formation in hematopoietic cells. Cell Reports. 2021;36:109576. [DOI] [PubMed] [Google Scholar]
- 34.Bondar T, Medzhitov R. p53-Mediated Hematopoietic Stem and Progenitor Cell Competition. Cell Stem Cell. 2010;6:309–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dudgeon C, Shreeram S, Tanoue K, Mazur SJ, Sayadi A, Robinson RC, Appella E, Bulavin DV. Genetic variants and mutations of PPM1D control the response to DNA damage. Cell Cycle. 2013;12:2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N Engl J Med. 2005;352:1779–1790. [DOI] [PubMed] [Google Scholar]
- 37.Kilpivaara O, Mukherjee S, Schram AM, Wadleigh M, Mullally A, Ebert BL, Bass A, Marubayashi S, Heguy A, Garcia-Manero G, et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2V617F-positive myeloproliferative neoplasms. Nat Genet. 2009;41:455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trifa AP, Cucuianu A, Petrov L, Urian L, Militaru MS, Dima D, Pop IV, Popp RA. The G allele of the JAK2 rs10974944 SNP, part of JAK2 46/1 haplotype, is strongly associated with JAK2 V617F-positive myeloproliferative neoplasms. Ann Hematol. 2010;89:979–983. [DOI] [PubMed] [Google Scholar]
- 39.Trifa AP, Bănescu C, Bojan AS, Voina CM, Popa Ștefana, Vișan S, Ciubean AD, Tripon F, Dima D, Popov VM, et al. MECOM, HBS1L-MYB, THRB-RARB, JAK2, and TERT polymorphisms defining the genetic predisposition to myeloproliferative neoplasms: A study on 939 patients. Am J Hematol. 2018;93:100–106. [DOI] [PubMed] [Google Scholar]
- 40.Olcaydu D, Harutyunyan A, Jäger R, Berg T, Gisslinger B, Pabinger I, Gisslinger H, Kralovics R. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet. 2009;41:450–454. [DOI] [PubMed] [Google Scholar]
- 41.Jones AV, Chase A, Silver RT, Oscier D, Zoi K, Wang YL, Cario H, Pahl HL, Collins A, Reiter A, et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet. 2009;41:446–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zimmermann S, Martens UM. Telomeres, senescence, and hematopoietic stem cells. Cell Tissue Res. 2008;331:79–90. [DOI] [PubMed] [Google Scholar]
- 43.Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AFR, Barbalic M, Gieger C, et al. Large-scale association analyses identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, Jan Hottenga J, Fischer K, Esko T, Surakka I, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45:422–427e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science. 2019;366:eaan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silver AJ, Bick AG, Savona MR. Germline risk of clonal haematopoiesis. Nat Rev Genet. 2021;22:603–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Honigberg MC, Zekavat SM, Niroula A, Griffin GK, Bick AG, Pirruccello JP, Nakao T, Whitsel EA, Farland LV, Laurie C, et al. Premature Menopause, Clonal Hematopoiesis, and Coronary Artery Disease in Postmenopausal Women. Circulation. 2021;143:410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu C-L, Sano S, Muralidharan S, Rius C, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, Zhao D, Liu Y, Wang C, Zhang X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, Libby P, Kathiresan S, Natarajan P. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation. 2020;141:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wainstein MV, Mossmann M, Araujo GN, Gonçalves SC, Gravina GL, Sangalli M, Veadrigo F, Matte R, Reich R, Costa FG, et al. Elevated serum interleukin-6 is predictive of coronary artery disease in intermediate risk overweight patients referred for coronary angiography. Diabetol Metab Syndr. 2017;9:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ridker PM, MacFadyen JG, Glynn RJ, Koenig W, Libby P, Everett BM, Lefkowitz M, Thuren T, Cornel JH. Inhibition of Interleukin-1β by Canakinumab and Cardiovascular Outcomes in Patients With Chronic Kidney Disease. Journal of the American College of Cardiology. 2018;71:2405–2414. [DOI] [PubMed] [Google Scholar]
- 53.Ridker PM, Devalaraja M, Baeres FMM, Engelmann MDM, Hovingh GK, Ivkovic M, Lo L, Kling D, Pergola P, Raj D, et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. The Lancet. 2021;397:2060–2069. [DOI] [PubMed] [Google Scholar]
- 54.James C, Ugo V, Le Couédic J-P, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. [DOI] [PubMed] [Google Scholar]
- 55.Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, Liu W, Thomas DG, Hajebrahimi MA, Pircher J, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592:296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang W, Liu W, Fidler T, Wang Y, Tang Y, Woods B, Welch C, Cai B, Silvestre-Roig C, Ai D, et al. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2 V617F Mice. Circ Res [Internet]. 2018. [cited 2021 Oct 6];123. Available from: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.118.313283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu B, Roberts MB, Raffield LM, Zekavat SM, Nguyen NQH, Biggs ML, Brown MR, Griffin G, Desai P, Correa A, et al. Association of Clonal Hematopoiesis With Incident Heart Failure. Journal of the American College of Cardiology. 2021;78:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abplanalp WT, Cremer S, John D, Hoffmann J, Schuhmacher B, Merten M, Rieger MA, Vasa-Nicotera M, Zeiher AM, Dimmeler S. Clonal Hematopoiesis–Driver DNMT3A Mutations Alter Immune Cells in Heart Failure. Circ Res. 2021;128:216–228. [DOI] [PubMed] [Google Scholar]
- 59.Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. Journal of the American College of Cardiology. 2018;71:875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ Res. 2018;123:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sano S, Wang Y, Yura Y, Sano M, Oshima K, Yang Y, Katanasaka Y, Min K-D, Matsuura S, Ravid K, et al. JAK2-Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC: Basic to Translational Science. 2019;4:684–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yura Y, Miura-Yura E, Katanasaka Y, Min K-D, Chavkin N, Polizio AH, Ogawa H, Horitani K, Doviak H, Evans MA, et al. The Cancer Therapy-Related Clonal Hematopoiesis Driver Gene Ppm1d Promotes Inflammation and Non-Ischemic Heart Failure in Mice. Circ Res. 2021;129:684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sano S, Wang Y, Ogawa H, Horitani K, Sano M, Polizio AH, Kour A, Yura Y, Doviak H, Walsh K. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI Insight. 2021;6:e146076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, Schmid T, Brüne B, Wagner S, Serve H, et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019;4:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abplanalp WT, Mas-Peiro S, Cremer S, John D, Dimmeler S, Zeiher AM. Association of Clonal Hematopoiesis of Indeterminate Potential With Inflammatory Gene Expression in Patients With Severe Degenerative Aortic Valve Stenosis or Chronic Postischemic Heart Failure. JAMA Cardiol. 2020;5:1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.El Husseini D, Boulanger M-C, Mahmut A, Bouchareb R, Laflamme M-H, Fournier D, Pibarot P, Bossé Y, Mathieu P. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: Implication for calcific aortic valve disease. Journal of Molecular and Cellular Cardiology. 2014;72:146–156. [DOI] [PubMed] [Google Scholar]
- 67.Mas-Peiro S, Hoffmann J, Fichtlscherer S, Dorsheimer L, Rieger MA, Dimmeler S, Vasa-Nicotera M, Zeiher AM. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. European Heart Journal. 2020;41:933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mathieu P, Bouchareb R, Boulanger M-C. Innate and Adaptive Immunity in Calcific Aortic Valve Disease. Journal of Immunology Research. 2015;2015:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zekavat SM, Viana-Huete V, Zuriaga MA, Uddin MM, Trinder M, Paruchuri K, Matesanz N, Zorita V, Ferrer-Pérez A, Amorós-Pérez M, et al. TP53-mediated clonal hematopoiesis confers increased risk for incident peripheral artery disease [Internet]. 2021. [cited 2021 Aug 26];2021.08.22.21262430. Available from: https://www.medrxiv.org/content/10.1101/2021.08.22.21262430v1 [DOI] [PMC free article] [PubMed]
- 70.Dunbar A, Bolton KL, Devlin SM, Sanchez-Vega F, Gao J, Mones JV, Wills J, Kelly D, Farina M, Cordner KB, et al. Genomic profiling identifies somatic mutations predicting thromboembolic risk in patients with solid tumors. Blood. 2021;137:2103–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, Silver AJ, Adams D, Castellano CA, Schneider RK, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med 2018;10:eaan8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Potus F, Pauciulo MW, Cook EK, Zhu N, Hsieh A, Welch CL, Shen Y, Tian L, Lima P, Mewburn J, et al. Novel Mutations and Decreased Expression of the Epigenetic Regulator TET2 in Pulmonary Arterial Hypertension. Circulation. 2020;141:1986–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kimishima Y, Misaka T, Yokokawa T, Wada K, Ueda K, Sugimoto K, Minakawa K, Nakazato K, Ishida T, Oshima M, et al. Clonal hematopoiesis with JAK2V617F promotes pulmonary hypertension with ALK1 upregulation in lung neutrophils. Nat Commun. 2021;12:6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arnaud P, Morel H, Milleron O, Gouya L, Francannet C, Da Costa A, Le Goff C, Jondeau G, Boileau C, Hanna N. Unsuspected somatic mosaicism for FBN1 gene contributes to Marfan syndrome. Genet Med. 2021;23:865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rekondo J, Robledo-Inarritu M, Vado Y, Pérez de Nanclares G, Arós F. Marfan Syndrome Caused by Somatic Mosaicism in an FBN1 Splicing Mutation. Revista Española de Cardiología (English Edition). 2016;69:520–521. [DOI] [PubMed] [Google Scholar]
- 76.Blyth M, Foulds N, Turner C, Bunyan D. Severe Marfan syndrome due to FBN1 exon deletions. Am. J. Med. Genet 2008;146A:1320–1324. [DOI] [PubMed] [Google Scholar]
- 77.Fernández-Álvarez P, Codina-Sola M, Valenzuela I, Teixidó-Turá G, Cueto-González A, Paramonov I, Antolín M, López-Grondona F, Vendrell T, Evangelista A, et al. A systematic study and literature review of parental somatic mosaicism of FBN1 pathogenic variants in Marfan syndrome. J Med Genet. 2021;jmedgenet-2020–107604. [DOI] [PubMed] [Google Scholar]
- 78.Manheimer KB, Richter F, Edelmann LJ, D’Souza SL, Shi L, Shen Y, Homsy J, Boskovski MT, Tai AC, Gorham J, et al. Robust identification of mosaic variants in congenital heart disease. Hum Genet. 2018;137:183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hsieh A, Morton SU, Willcox JAL, Gorham JM, Tai AC, Qi H, DePalma S, McKean D, Griffin E, Manheimer KB, et al. EM-mosaic detects mosaic point mutations that contribute to congenital heart disease. Genome Medicine. 2020;12:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Morton SU, Quiat D, Seidman JG, Seidman CE. Genomic frontiers in congenital heart disease. Nat Rev Cardiol. 2021;1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roselli C, Rienstra M, Ellinor PT. Genetics of Atrial Fibrillation in 2020: GWAS, Genome Sequencing, Polygenic Risk, and Beyond. Circ Res. 2020;127:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shoemaker MB, Roden DM. Atrial Fibrillation Is a Complex Trait: Very Complex. Circ Res. 2020;127:244–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kertai MD, Mosley JD, He J, Ramakrishnan A, Abdelmalak MJ, Hong Y, Shoemaker MB, Roden DM, Bastarache L. Predictive Accuracy of a Polygenic Risk Score for Postoperative Atrial Fibrillation After Cardiac Surgery. Circ: Genomic and Precision Medicine [Internet]. 2021. [cited 2021 Nov 28];14. Available from: https://www.ahajournals.org/doi/10.1161/CIRCGEN.120.003269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Khurshid S, Mars N, Haggerty CM, Huang Q, Weng L-C, Hartzel DN, Regeneron Genetics Center, Lunetta KL, Ashburner JM, Anderson CD, et al. Predictive Accuracy of a Clinical and Genetic Risk Model for Atrial Fibrillation. Circ Genom Precis Med. 2021;14:e003355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gregers E, Ahlberg G, Christensen T, Jabbari J, Larsen KO, Herfelt CB, Henningsen KM, Andreasen L, Thiis JJ, Lund J, et al. Deep sequencing of atrial fibrillation patients with mitral valve regurgitation shows no evidence of mosaicism but reveals novel rare germline variants. Heart Rhythm. 2017;14:1531–1538. [DOI] [PubMed] [Google Scholar]
- 87.Gollob MH, Jones DL, Krahn AD, Danis L, Gong X-Q, Shao Q, Liu X, Veinot JP, Tang ASL, Stewart AFR, et al. Somatic Mutations in the Connexin 40 Gene ( GJA5 ) in Atrial Fibrillation. N Engl J Med. 2006;354:2677–2688. [DOI] [PubMed] [Google Scholar]
- 88.Thibodeau IL, Xu J, Li Q, Liu G, Lam K, Veinot JP, Birnie DH, Jones DL, Krahn AD, Lemery R, et al. Paradigm of Genetic Mosaicism and Lone Atrial Fibrillation: Physiological Characterization of a Connexin 43–Deletion Mutant Identified From Atrial Tissue. Circulation. 2010;122:236–244. [DOI] [PubMed] [Google Scholar]
- 89.Priest JR, Gawad C, Kahlig KM, Yu JK, O’Hara T, Boyle PM, Rajamani S, Clark MJ, Garcia STK, Ceresnak S, et al. Early somatic mosaicism is a rare cause of long-QT syndrome. PNAS. 2016;113:11555–11560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lerman BB, Dong B, Stein KM, Markowitz SM, Linden J, Catanzaro DF. Right ventricular outflow tract tachycardia due to a somatic cell mutation in G protein subunitalphai2. J. Clin. Invest 1998;101:2862–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Beals CR, Wilson CB, Perlmutter RM. A small multigene family encodes Gi signal-transduction proteins. Proceedings of the National Academy of Sciences. 1987;84:7886–7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zuberi Z, Nobles M, Sebastian S, Dyson A, Lim SY, Breckenridge R, Birnbaumer L, Tinker A. Absence of the Inhibitory G-Protein Gα i2 Predisposes to Ventricular Cardiac Arrhythmia. Circ: Arrhythmia and Electrophysiology. 2010;3:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Larsson N-G. Somatic Mitochondrial DNA Mutations in Mammalian Aging. Annu. Rev. Biochem 2010;79:683–706. [DOI] [PubMed] [Google Scholar]
- 94.von Kleist-Retzow J-C, Hornig-Do H-T, Schauen M, Eckertz S, Dinh TAD, Stassen F, Lottmann N, Bust M, Galunska B, Wielckens K, et al. Impaired mitochondrial Ca2+ homeostasis in respiratory chain-deficient cells but efficient compensation of energetic disadvantage by enhanced anaerobic glycolysis due to low ATP steady state levels. Experimental Cell Research. 2007;313:3076–3089. [DOI] [PubMed] [Google Scholar]
- 95.Ashar FN, Zhang Y, Longchamps RJ, Lane J, Moes A, Grove ML, Mychaleckyj JC, Taylor KD, Coresh J, Rotter JI, et al. Association of Mitochondrial DNA Copy Number With Cardiovascular Disease. JAMA Cardiol. 2017;2:1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baris OR, Ederer S, Neuhaus JFG, von Kleist-Retzow J-C, Wunderlich CM, Pal M, Wunderlich FT, Peeva V, Zsurka G, Kunz WS, et al. Mosaic Deficiency in Mitochondrial Oxidative Metabolism Promotes Cardiac Arrhythmia during Aging. Cell Metabolism. 2015;21:667–677. [DOI] [PubMed] [Google Scholar]
- 97.Stöckigt F, Beiert T, Knappe V, Baris OR, Wiesner RJ, Clemen CS, Nickenig G, Andrié RP, Schrickel JW. Aging-related mitochondrial dysfunction facilitates the occurrence of serious arrhythmia after myocardial infarction. Biochemical and Biophysical Research Communications. 2017;493:604–610. [DOI] [PubMed] [Google Scholar]
- 98.Greenberg JW, Lancaster TS, Schuessler RB, Melby SJ. Postoperative atrial fibrillation following cardiac surgery: a persistent complication. European Journal of Cardio-Thoracic Surgery. 2017;52:665–672. [DOI] [PubMed] [Google Scholar]
- 99.Lai L-P, Tsai C-C, Su M-J, Lin J-L, Chen Y-S, Tseng Y-Z, Huang SKS. Atrial Fibrillation Is Associated With Accumulation of Aging-Related Common Type Mitochondrial DNA Deletion Mutation in Human Atrial Tissue*. Chest. 2003;123:539–544. [DOI] [PubMed] [Google Scholar]
- 100.Park H-W, Ahn Y, Jeong M-H, Cho J-G, Park J-C, Kang J-C, Shin M-G, Shin J-H, Suh S-P, Ryang D-W, et al. Chronic atrial fibrillation associated with somatic mitochondrial DNA mutations in human atrial tissue. Journal of Clinical Pathology. 2006;60:948–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Malik AN, Czajka A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion. 2013;13:481–492. [DOI] [PubMed] [Google Scholar]
- 102.Zhao D, Bartz TM, Sotoodehnia N, Post WS, Heckbert SR, Alonso A, Longchamps RJ, Castellani CA, Hong YS, Rotter JI, et al. Mitochondrial DNA copy number and incident atrial fibrillation. BMC Med. 2020;18:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marín-García J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev. 2008;13:137–150. [DOI] [PubMed] [Google Scholar]
- 104.Akhmedov AT, Rybin V, Marín-García J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Fail Rev. 2015;20:227–249. [DOI] [PubMed] [Google Scholar]
- 105.Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. 2018;128:3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. [DOI] [PubMed] [Google Scholar]
- 107.Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA Replication Impairs Mitochondrial Biogenesis In Human Failing Hearts Karamanlidis: Mitochondrial biogenesis in human heart failure. Circ Res. 2010;106:1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Arbustini E, Diegoli M, Fasani R, Grasso M, Morbini P, Banchieri N, Bellini O, Dal Bello B, Pilotto A, Magrini G, et al. Mitochondrial DNA Mutations and Mitochondrial Abnormalities in Dilated Cardiomyopathy. The American Journal of Pathology. 1998;153:1501–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen S, Xie X, Wang Y, Gao Y, Xie X, Yang J, Ye J. Association between leukocyte mitochondrial DNA content and risk of coronary heart disease: A case-control study. Atherosclerosis. 2014;237:220–226. [DOI] [PubMed] [Google Scholar]
- 110.Liu L-P, Cheng K, Ning M-A, Li H-H, Wang H-C, Li F, Chen S-Y, Qu F-L, Guo W-Y. Association between peripheral blood cells mitochondrial DNA content and severity of coronary heart disease. Atherosclerosis. 2017;261:105–110. [DOI] [PubMed] [Google Scholar]
- 111.Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Mitochondrial Mutations are Associated with Atherosclerotic Lesions in the Human Aorta. Clinical and Developmental Immunology. 2012;2012:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Changes of mitochondria in atherosclerosis: Possible determinant in the pathogenesis of the disease. Atherosclerosis. 2013;227:283–288. [DOI] [PubMed] [Google Scholar]
- 113.Sazonova MA, Sinyov VV, Barinova VA, Ryzhkova AI, Zhelankin AV, Postnov AY, Sobenin IA, Bobryshev YV, Orekhov AN. Mosaicism of Mitochondrial Genetic Variation in Atherosclerotic Lesions of the Human Aorta. BioMed Research International. 2015;2015:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yu E, Calvert PA, Mercer JR, Harrison J, Baker L, Figg NL, Kumar S, Wang JC, Hurst LA, Obaid DR, et al. Mitochondrial DNA Damage Can Promote Atherosclerosis Independently of Reactive Oxygen Species Through Effects on Smooth Muscle Cells and Monocytes and Correlates With Higher-Risk Plaques in Humans. Circulation. 2013;128:702–712. [DOI] [PubMed] [Google Scholar]
- 115.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. [DOI] [PubMed] [Google Scholar]
- 116.Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim Y-N, Kim SS, Kim DH, Hur KY, Kim HK, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83–92. [DOI] [PubMed] [Google Scholar]
- 117.Ridker PM, Libby P, MacFadyen JG, Thuren T, Ballantyne C, Fonseca F, Koenig W, Shimokawa H, Everett BM, Glynn RJ. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). European Heart Journal. 2018;39:3499–3507. [DOI] [PubMed] [Google Scholar]
- 118.Svensson EC, Madar A, Campbell CD, He Y, Sultan M, Healey ML, D’Aco K, Fernandez A, Wache-Mainier C, Ridker PM, et al. Abstract 15111: TET2-Driven Clonal Hematopoiesis Predicts Enhanced Response to Canakinumab in the CANTOS Trial: An Exploratory Analysis. Circulation. 2018;138:A15111–A15111. [Google Scholar]