

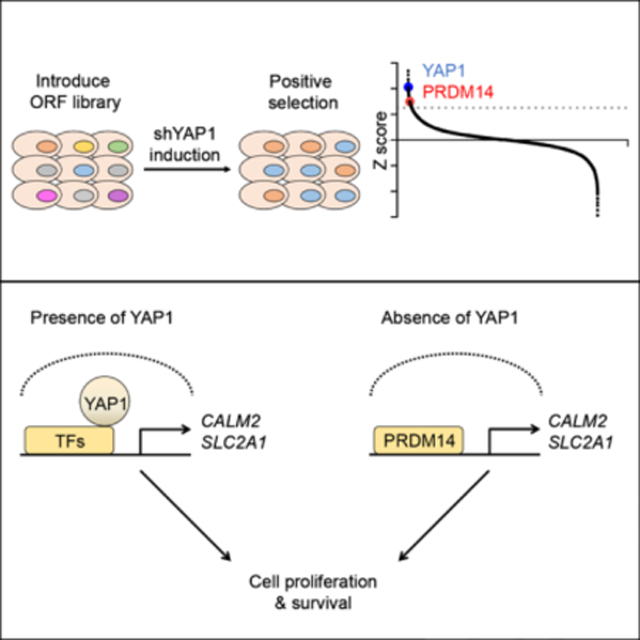
SUMMARY
The transcriptional co-activator YAP1 oncogene is the downstream effector of the Hippo pathway, which regulates tissue homeostasis, organ size, regeneration and tumorigenesis. Multiple cancers are dependent on sustained expression of YAP1 for cell proliferation, survival and tumorigenesis, but the molecular basis of this oncogene dependency is not well understood. To identify genes that can functionally substitute for YAP1, we performed a genome-scale genetic rescue screen in YAP1-dependent colon cancer cells expressing an inducible YAP1-specific shRNA. We found that the transcription factor PRDM14 rescued cell proliferation and tumorigenesis upon YAP1 suppression in YAP1-dependent cells, xenografts, and colon cancer organoids. YAP1 and PRDM14 individually activated the transcription of calmodulin 2 (CALM2) and a glucose transporter SLC2A1 upon YAP1 suppression; and CALM2 or SLC2A1 expression was required for the rescue of YAP1 suppression. Together, these findings implicate PRDM14-mediated transcriptional upregulation of CALM2 and SLC2A1 as key components of oncogenic YAP1 signaling and dependency.
eTOC Blurb
Multiple cancers are dependent on sustained expression of YAP1 for cell proliferation, survival, and tumorigenesis, but whether other genes can functionally substitute for YAP1 is not clear. Here, Kim et al. show that PRDM14-mediated transcriptional upregulation of CALM2 and SLC2A1 allow YAP1-dependent cancers to survive in the absence of YAP1.
Graphical Abstract
INTRODUCTION
The Hippo pathway was originally identified as a key signaling pathway that controls organ size in Drosophila melanogaster. In mammals, the Hippo pathway plays critical regulatory functions in tissue homeostasis, organ size control, regeneration and tumorigenesis (Meng et al., 2016; Mo et al., 2014; Moroishi et al., 2015; Zanconato et al., 2016). In the mammalian Hippo pathway, mammalian STE20-like protein (MST) and the large tumor suppressor (LATS) kinases negatively regulate yes-associated protein (YAP1) and transcriptional co-activator with PDZ-binding motif (TAZ), which are closely related paralogues and the major downstream effectors of this pathway. YAP1 and TAZ function as transcriptional co-activators that shuttle between the cytoplasm and the nucleus, where they induce the expression of genes typically associated with cell proliferation and survival through interactions with transcriptional factors such as TEA domain family members (TEAD). As a result, unrestricted YAP1 and TAZ activation leads to increased organ size during development, and in adult tissues, such as liver and colon, promotes tumorigenesis (Meng et al., 2016; Moroishi et al., 2015; Nishio et al., 2012; Zhou et al., 2011; Zhou et al., 2009).
Previous studies have used the intestinal epithelium as an experimental model to study the complex roles of Hippo pathway and YAP1 oncogene (Avruch et al., 2012; Ou et al., 2017). Loss of function studies in vivo have shown that Yap1 is dispensable for normal intestinal development and homeostasis, but following chemical injury or gamma irradiation, Yap1 is required for the intestinal stem cell pool and crypt regeneration (Cai et al., 2010; Gregorieff et al., 2015). Genetic studies have shown that Yap1 is required for adenoma formation in the Apcmin mouse model of colon cancer (Cai et al., 2010; Gregorieff et al., 2015). Similarly, studies in human colon cancer cell lines have shown that YAP1 is required for cell proliferation, survival and tumorigenesis (Rosenbluh et al., 2012; Zhou et al., 2011), and a number of clinical reports have linked high levels of YAP1 expression and activity to colon cancer progression and overall poor prognosis (Wierzbicki and Rybarczyk, 2015). In summary, YAP1 is dispensable for normal intestinal development and homeostasis but required for intestinal tumorigenesis.
PRDM14 belongs to the PRDI-BF1 and RIZ homology domain-containing (PRDM) transcriptional regulators. Among the PRDM family members, PRDM14 is specifically expressed in preimplantation embryos, primordial germ cells, and embryonic stem cells in vitro, and accordingly plays a key role in the regulation of pluripotency and epigenetic reprogramming (Nakaki and Saitou, 2014). Gene amplification and/or overexpression of PRDM14 has been reported in breast cancer, leukemia and testicular/intracranial germ cell tumors (Dettman et al., 2011; Moelans et al., 2010; Nishikawa et al., 2007; Ruark et al., 2013). More recently, PRDM14 expression has been shown to promote a malignant phenotype and correlate with poor prognosis in colorectal cancers (Igarashi et al., 2020). Although PRDM14 has been well studied in embryonic stem cells, its role in cancer is not well understood.
Based on previous observations that YAP1 is required for cell proliferation, survival and tumorigenesis of colon cancers, we were particularly interested in signaling mechanisms activated by this oncogene. To this end, we performed a genome-scale genetic rescue screen to identify open reading frames (ORFs) that sustain cell proliferation, survival and tumorigenesis phenotypes of YAP1-dependent colon cancer cell lines in the setting of YAP1 suppression.
RESULTS
Genome-scale ORF screen identifies genes that rescue cell proliferation upon YAP1 suppression in YAP1-dependent cells
We performed a genome-scale ORF screen to identify genes that support the proliferation of YAP1-dependent colon cancer cells upon suppression of YAP1 (Figure 1A). We tested a panel of colon cancer cell lines to identify those that required YAP1 expression for cell proliferation. We found that the HT29, SKCO1, SW48 and COLO320 colon cancer cell lines depend on YAP1 expression for proliferation and selected these cell lines for further study (Figure S1A). In parallel, we tested several doxycycline-inducible short hairpin RNAs (shRNA) targeting the 3’ untranslated region (UTR) of YAP1 and selected one that induced the most efficient depletion (pLKO-Tet-On shYAP1–9) to use in our experiments (Figure S1B). We then generated single-cell clones of HT29, SKCO1, SW48 and COLO320 cells stably expressing this selected doxycycline-inducible shRNA (HT29, SKCO1, SW48 and COLO320 shYAP1, Figure 1B) and confirmed that suppression of YAP1 by doxycycline treatment inhibited cell proliferation of these shYAP1-expressing single-cell clones. Expression of a YAP1 ORF, which lacks the YAP1 3’UTR and thus cannot be suppressed by this YAP1-specific shRNA, rescued cell proliferation upon suppression of YAP1 (Figure 1B), confirming that the observed decrease in proliferation was due to suppressing YAP1.
Figure 1. Genome-scale ORF screen identifies genes that rescue cell proliferation upon YAP1 suppression in YAP1-dependent cells.

(A) Schematic of the genome-scale ORF screens to identify ORFs that rescue cell proliferation upon suppression of YAP1 in YAP1-dependent cells.
(B) Immunoblots and clonogenic proliferation assays of HT29, SKCO1, SW48 and COLO320 shYAP1 cells expressing GFP or YAP1 with or without YAP1 suppression. Data represent mean ± standard error of the mean (SEM) normalized to cell proliferation in water treated conditions. P values were calculated using a paired Student’s two-tailed t test; n = 3, **, p<0.01, *, p<0.05. Water, without YAP1 suppression; Dox, with YAP1 suppression.
(C) Distribution of Z scores for all screened ORFs. The average of two replicates is shown. The gray line indicates 2.5 standard deviations above the mean. Venn diagram illustrates the overlap in ORFs with a Z score > 2.5 across all screened cell lines.
Using these shYAP1 expressing single-cell clones, we introduced 17,255 uniquely barcoded ORFs from the human ORFeome library collection 8.1 (Yang et al., 2011) into these cells in a pooled format. We then treated cells with doxycycline to induce YAP1 suppression and cultured these cells for 14 days. Following cell harvest, genomic DNA purification and sequencing, we identified enriched ORFs in doxycycline treated samples, compared to control samples, as candidate genes that functionally substitute for YAP1 (Figure 1A). We ranked the relative enrichment of each ORF and defined hits as ORFs with a Z score greater than 2.5. We identified 232, 182, 329 and 186 hits in HT29, SKCO1, SW48 and COLO320 shYAP1 cells, respectively and found that PRDM14 was the only gene that scored in all four cell lines (Figure 1C, S1C and Table S1). When we used a Z score greater than 2, we found YAP1 among 8 genes scoring in all four cell lines (Figure 1C and S1D).
To identify common pathways and interactions among the candidate genes from ORF screens, we performed a meta-analysis of pathway and protein-protein interaction (PPI) enrichment using the Metascape suite of tools (http://metascape.org) (Tripathi et al., 2015) (Figure S2A and S2B). From the pathway enrichment analysis within the Molecular Signatures Database (MSigDB) (Subramanian et al., 2005), we found that the candidate genes from our ORF screens were enriched in pathways and processes related to cell cycle, growth, EGFR, MAPK and PI3K signaling, etc. (Figure S2A). In addition, to understand the potential PPI complexes among the candidate genes from ORF screens, we performed a PPI meta-analysis (Figure S2B). We merged candidate gene lists from all four ORF screens into one list and generated a single PPI network representing the full interactome (Figure S2B top network). We applied the Molecular Complex Detection (MCODE) algorithm to identify densely connected network components. From this PPI meta-analysis, we found PPI clusters with most notable enrichment of interactions within components of RTK/PI3K signaling, GPCR signaling and cell cycle (Figure S2B).
PRDM14 rescues YAP1 suppression in YAP1-dependent cells
We focused our attention on PRDM14 since it was the only gene that scored in all four colon cancer cell lines at the higher cutoff value (Z score > 2.5) (Figure 1C, S1C and Table S1). We observed by immunoblotting that endogenous protein expression levels of YAP1 and PRDM14 in all four colon cancer cell lines screened were similar (Figure 2A control lanes and S3A). To validate that PRDM14 promoted cell proliferation upon YAP1 suppression, we performed a rescue experiment in the screened doxycycline-inducible shYAP1 cell lines. We expressed GFP as a negative control, YAP1 as a positive control and PRDM14 in HT29, SKCO1, SW48 and COLO320 shYAP1 cells in parallel and performed clonogenic proliferation assays using crystal violet staining following YAP1 suppression. We then determined the rescue level of each gene by normalizing the cell proliferation of YAP1-suppressed cells to that of YAP1-intact cells. We found that suppression of YAP1 eliminated 70% of cells expressing the negative control GFP (Figure 2A and S3B). Exogenous YAP1 expression rescued YAP1 suppression at approximately 80% relative to YAP1-intact cells. In this setting, we found that PRDM14 expression rescued the proliferation effect of YAP1 suppression to a similar level as exogenous YAP1 expression (Figure 2A, S3B and S3C).
Figure 2. PRDM14 rescues YAP1 suppression in YAP1-dependent cells and xenograft models.

(A) Immunoblots and clonogenic proliferation assays of HT29, SKCO1, SW48 and COLO320 shYAP1 cells expressing GFP, YAP1 or PRDM14 with or without YAP1 suppression. Data represent mean ± SEM normalized to cell proliferation in water treated conditions. P values were calculated using a paired Student’s two-tailed t test; n = 3, **, p<0.01, *, p<0.05. Water, without YAP1 suppression; Dox, with YAP1 suppression.
(B) Immunodeficient mice were fed with doxycycline diet 2 days before injection of HT29 shYAP1 cells expressing indicated ORFs. Data represent tumor volume on day 28. P values were calculated using an unpaired Student’s two-tailed t test; n = 3, ****, p<0.0001.
(C) HT29 shYAP1 cells expressing indicated ORFs were injected in immunodeficient mice. YAP1 suppression was initiated with doxycycline diet on day 14 when tumor volume reached 100 mm3. Y-axis indicates percentage change in tumor volume on day 28 relative to day 14. Each bar represents one tumor. Data are representative of three independent experiments.
See also Figure S3.
To better understand the functional relationship between YAP1 and PRDM14, we investigated whether depletion of PRDM14 affects cell proliferation in YAP1-dependent cells and whether YAP1 expression rescues this effect. We found that YAP1-dependent HT29, SKCO1 and SW48 cells are also dependent on PRDM14 for cell proliferation (Figure S3D). We then performed clonogenic proliferation assays in HT29, SKCO1 and SW48 cells expressing either GFP or YAP1 in the setting of PRDM14 depletion. We found that expression of YAP1 was not able to rescue PRDM14 depletion in these cell lines (Figure S3E). To interrogate the functional relationship between YAP1 and PRDM14 further, we also tested whether PRDM14 is required for YAP1-induced cell transformation. We expressed YAP1 in immortalized HA1E cells (Hahn et al., 1999) and confirmed that YAP1 induced anchorage-independent colony formation (Figure S3F). We found that depletion of PRDM14 abrogated YAP1-driven anchorage-independent colony formation (Figure S3F). Together, these observations suggest that PRDM14 acts downstream of YAP1.
PRDM14 rescues YAP1 suppression in xenograft models
To investigate whether PRDM14 also rescued YAP1 suppression during tumor formation, we performed an in vivo rescue experiment using the HT29 shYAP1 cell line. This cell line was selected as it has been previously established as a mouse xenograft model (Radulovic et al., 1991; Teng et al., 2016). We expressed GFP, YAP1 or PRDM14 in HT29 shYAP1 cells and injected these cells subcutaneously into immunodeficient mice fed with a doxycycline-containing diet 2 days prior to cell injection. We observed that GFP expressing cells failed to form tumors while cells expressing exogenous YAP1 formed tumors. Notably, PRDM14 expressing cells also formed tumors (Figure 2B), indicating that PRDM14 expression rescued YAP1 suppression to promote tumor formation. In addition to tumor formation, to assess whether PRDM14 rescued YAP1-driven tumor growth, we injected HT29 shYAP1 cells expressing GFP, YAP1 or PRDM14 subcutaneously into immunodeficient mice. Once tumors reached 100 mm3, we induced YAP1 suppression by the treatment of doxycycline-containing diet into mice. Following YAP1 suppression, GFP expressing tumors regressed while exogenous YAP1 expressing tumors continued to grow. We found that PRDM14 also allowed YAP1-dependent tumors to grow upon suppression of YAP1 (Figure 2C). We also observed this PRDM14-mediated rescue of YAP1 suppression for tumorigenic growth in in vivo xenograft experiments using SW48 cell line (Figure S3G and S3H).
Transcriptional activity of PRDM14 is required to rescue YAP1 suppression
PRDM14 is a member of the PRDM family of transcriptional regulators and composed of a single PR/SET domain and six tandemly repeated zinc fingers (Figure 3A). The PR/SET domain defines a large group of histone methyl-transferases and PRDM14 regulates transcription of target genes by directly binding to regulatory regions through zinc finger domains (Nakaki and Saitou, 2014). To identify regions of PRDM14 necessary to promote cell proliferation upon YAP1 suppression, we expressed PRDM14 domain-specific mutants in HT29 and SW48 shYAP1 cells (Figure 3A and 3B). Expression of a PRDM14 mutant harboring deletion of the PR/SET domain (PRDM14 ΔSET domain) rescued cell proliferation upon YAP1 suppression, equivalent to what we observed with wild-type PRDM14 (PRDM14 WT) expression in these cells. Thus, histone methyl-transferase activity is not required for PRDM14 activity in the context of YAP1 suppression. However, expression of both PRDM14 truncation mutants lacking zinc finger domains (PRDM14 ΔZinc finger and PRDM14 ΔSET+ΔZinc finger) disrupted the ability of PRDM14 to rescue cells from YAP1 suppression (Figure 3B lower panels). These observations suggest that PRDM14 mediates cell proliferation upon suppression of YAP1 through direct DNA interactions through zinc finger domains, which, in turn, affects transcriptional activity.
Figure 3. PRDM14 domain structure and function analysis.

(A) PRDM14 domain structure and PRDM14 mutants.
(B) Immunoblots and clonogenic proliferation assays of HT29 and SW48 shYAP1 cells expressing empty vector, PRDM14 wild-type or mutants with or without YAP1 suppression. * in the immunoblot represents the exogenously expressing PRDM14-specific band. Data represent mean ± SEM normalized to cell proliferation in water treated conditions. P values were calculated using a paired Student’s two-tailed t test; n = 3, ***, p<0.001, **, p<0.01.
PRDM14 and YAP1 regulate overlapping transcriptional programs
Because transcriptional activity was essential for PRDM14 to rescue YAP1 suppression, we hypothesized that PRDM14 induces changes in gene expression allowing cells to proliferate in the setting of YAP1 suppression. To investigate transcriptional programs regulated by PRDM14 and YAP1, we performed RNA-sequencing in HT29, SKCO1 and SW48 cells, in the setting of independent depletion of YAP1 or PRDM14 (shYAP1 or shP14), or independent overexpression of YAP1 or PRDM14 (YAP1 OE or P14 OE) (Figure S4A, S4B and Table S2). We then analyzed and compared differential gene expression profiles and identified common target genes of YAP1 and PRDM14 that were both up-regulated and down-regulated by modulation of YAP1 and PRDM14 expression (Figure S4A and Table S2). We then performed pathway enrichment analysis within the MSigDB (Subramanian et al., 2005) and found that YAP1 and PRDM14 regulate overlapping pathways including cell cycle and growth factor signaling (Figure S4B).
To identify direct transcriptional target genes of YAP1 and PRDM14 that are necessary and sufficient to rescue YAP1 dependence, we performed chromatin immunoprecipitation followed by sequencing (ChIP-sequencing) in HT29, SKCO1 and SW48 shYAP1 cells expressing V5-tagged YAP1 or V5-tagged PRDM14 upon suppression of YAP1 (Figure 4A). We identified 3387, 5371 and 2457 genomic loci that were both bound by YAP1 and PRDM14 in HT29, SKCO1 and SW48 shYAP1 cells, respectively (Figure 4B). In parallel, we performed RNA-sequencing in HT29, SKCO1 and SW48 shYAP1 cells expressing GFP, YAP1 or PRDM14 upon suppression of YAP1, to identify genes upregulated by the expression of YAP1 or PRDM14 compared to GFP upon suppression of YAP1 (Figure 4A). We then integrated two datasets using Binding and Expression Target Analysis (BETA) (Wang et al., 2013): the genomic loci bound by YAP1 or PRDM14 with genes that were upregulated by expression of YAP1 or PRDM14 when YAP1 was suppressed.
Figure 4. PRDM14 and YAP1 regulate overlapping transcriptional programs.

(A) Schematics of ChIP-sequencing and RNA-sequencing experiments.
(B) Venn diagrams illustrate the overlaps in YAP1 and PRDM14 bound peaks upon YAP1 suppression in each cell line. Heatmaps represent YAP1 and PRDM14 common bound peaks in each cell line identified by ChIP-sequencing. YAP1 and PRDM14 common bound peaks are ranked from the strongest to the weakest signal in YAP1 ChIP in each cell line.
(C) Venn diagrams illustrate the overlaps in YAP1 and PRDM14 target genes upon YAP1 suppression in each cell line or across cell lines.
(D) YAP1 and PRDM14 binding status at genomic loci of CALM2 promoter and SLC2A1 enhancer. H3K27Ac status at these loci is used as a marker for active regions for transcription factor binding.
We identified 747, 1045 and 401 genes that were both bound and upregulated by YAP1 and PRDM14 in HT29, SKCO1 and SW48 shYAP1 cells, respectively. Among these genes, we focused on 12 genes that were targets of YAP1 and PRDM14 in all of the cell lines (Figure 4C and Table S3). Among these 12 genes, we found that gene expression of CALM2 and SLC2A1 was not only upregulated by expression of exogenous YAP1 and PRDM14 upon suppression of YAP1 but also downregulated by suppression of YAP1 compared to YAP1-intact cells. CALM2 is a member of the calmodulin family that plays a role in signaling pathways, cell cycle progression and proliferation (Berchtold and Villalobo, 2014), while SLC2A1 is a major glucose transporter that is associated with tumor progression, metastasis, and poor prognosis in cancers (de Wit et al., 2012; Wang et al., 2017; Younes et al., 1996). Upregulation of SLC2A1 by YAP1 was previously shown to dictate the oncogenic phenotypes of breast cancer cells and enable organ growth by modulating glycolysis (Cox et al., 2018; Lin and Xu, 2017). Therefore, in the following experiments, we focused on CALM2 and SLC2A1 as direct target genes of PRDM14 and YAP1 that are important for YAP1 dependency.
PRDM14 and YAP1 regulate expression of CALM2 and SLC2A1
To directly confirm that PRDM14 and YAP1 bind to CALM2 and SLC2A1 promoters or enhancers, we compared our PRDM14 and YAP1 ChIP-sequencing data with H3K27Ac ChIP-sequencing data as a marker for active regions for transcription factor binding. We noted an enrichment of PRDM14 and YAP1 at the CALM2 promoter and SLC2A1 enhancer (Figure 4D). To further confirm that PRDM14 and YAP1 bind to these regions, we performed ChIP-qPCR with primers flanking the CALM2 promoter and SLC2A1 enhancer and observed a 5- to 10-fold enrichment of PRDM14 and YAP1 binding to these regions compared to control regions (Figure 5A). We then assessed whether the expression of CALM2 and SLC2A1 was regulated by PRDM14 and YAP1. Consistent with our integrated analysis of RNA-sequencing and ChIP-sequencing data, we observed that CALM2 and SLC2A1 mRNA transcripts decreased upon YAP1 suppression in GFP expressing cells. This decrease was reversed by expression of exogenous YAP1 or PRDM14 (Figure 5B). Confirming these findings, we observed that protein levels of CALM2 and SLC2A1 corresponded to these changes in CALM2 and SLC2A1 mRNA levels (Figure 5C). Furthermore, we found that expression of CALM2 and SLC2A1 was decreased upon suppression of PRDM14 in YAP1-dependent HT29, SKCO1 and SW48 cells (Figure S3D and S5A) whose proliferation is also dependent on the expression of PRDM14, CALM2 and SLC2A1 (Figure S3D and S5B). Together, these observations indicate that PRDM14 and YAP1 binding to the CALM2 promoter or SLC2A1 enhancer restores CALM2 and SLC2A1 expression that was decreased upon YAP1 suppression and that the regulation of these genes is essential for cell survival upon suppression of YAP1.
Figure 5. PRDM14 and YAP1 regulate expression of CALM2 and SLC2A1.

(A) ChIP-qPCR of HT29, SKCO1 and SW48 shYAP1 cells expressing V5-YAP1 or V5-PRDM14 upon YAP1 suppression to assess YAP1 or PRDM14 DNA binding at CALM2 promoter and SLC2A1 enhancer regions shown in Figure 4D. Data represent mean ± SEM normalized to DNA binding in negative control regions. P values were calculated using a paired Student’s two-tailed t test; n = 3, ****, p<0.0001, ***, p<0.001, **, p<0.01, *, p<0.05.
(B) mRNA expression of CALM2 and SLC2A1 in HT29, SKCO1 and SW48 shYAP1 cells expressing GFP, YAP1 or PRDM14 with or without YAP1 suppression. Data represent mean ± SEM normalized to mRNA expression in GFP expressing cells without YAP1 suppression. P values were calculated using a paired Student’s two-tailed t test; n = 3, ****, p<0.0001, ***, p<0.001, **, p<0.01.
(C) Immunoblots of HT29, SKCO1 and SW48 shYAP1 cells expressing GFP, YAP1 or PRDM14 with or without YAP1 suppression. Water, without YAP1 suppression; Dox, with YAP1 suppression. Cytoplasmic, cytoplasmic fraction; Plasma membrane, plasma membrane fraction.
CALM2 and SLC2A1 expression rescues YAP1 suppression in YAP1-dependent cells and xenograft models
From our genome-scale screening data, we noted that CALM2 and SLC2A1 scored just below the cutoff we used to define hits (Figure S5C and Table S1). Therefore, to investigate whether CALM2 and SLC2A1 promoted cell proliferation upon YAP1 suppression, we performed a rescue experiment in YAP1-dependent cells. We expressed GFP, YAP1, PRDM14, CALM2 or SLC2A1 in HT29, SKCO1 and SW48 cells expressing shYAP1 and performed clonogenic proliferation assays following YAP1 suppression. We found that CALM2 or SLC2A1 expression indeed rescued the proliferation effect of YAP1 suppression, similar to what we observed when we expressed exogenous YAP1 or PRDM14 (Figure 6A). In addition, we performed clonogenic proliferation assays in HT29, SKCO1 and SW48 cells expressing either GFP, CALM2 or SLC2A1 in the setting of PRDM14 depletion (Figure S3E). We found that CALM2 or SLC2A1 expression rescued cell proliferation following PRDM14 depletion (Figure S3E). We then tested whether CALM2 and SLC2A are synergistic in their ability to rescue the YAP1 or PRDM14 depletion phenotype by performing clonogenic proliferation assays in HT29, SKCO1 and SW48 cells expressing either GFP, CALM2, SLC2A1 or both CALM2 and SLC2A1 in the setting of YAP1 or PRDM14 depletion (Figure S3E and S5D). We observed that expression of both CALM2 and SLC2A1 showed stronger rescue effects on the YAP1 or PRDM14 depletion phenotype than expression of either CALM2 or SLC2A1 alone. However, they were not synergistic in their ability to rescue the YAP1 or PRDM14 depletion phenotype (Figure S3E and S5D).
Figure 6. CALM2 and SLC2A1 are required for PRDM14 to rescue YAP1 suppression.

(A) Clonogenic proliferation assays of HT29, SKCO1 and SW48 shYAP1 cells expressing indicated ORFs with or without YAP1 suppression.
(B) Immunodeficient mice were fed with doxycycline diet 2 days before injection of HT29 shYAP1 cells expressing indicated ORFs. Data represent tumor volume on day 28. P values were calculated using an unpaired Student’s two-tailed t test; n = 3, ****, p<0.0001.
(C) HT29 shYAP1 cells expressing indicated ORFs were injected in immunodeficient mice. YAP1 suppression was initiated with doxycycline diet on day 14 when tumor volume reached 100 mm3. Y-axis indicates percentage change in tumor volume on day 28 relative to day 14. Each bar represents one tumor. Data are representative of three independent experiments.
(D) Clonogenic proliferation assays of HT29, SKCO1 and SW48 shYAP1 cells expressing indicated ORFs and shRNAs with or without YAP1 suppression.
(E) Clonogenic proliferation assays of HT29, SKCO1 and SW48 shYAP1 cells expressing indicated ORFs with or without YAP1 suppression and a CALM2 inhibitor, W-7 or a SLC2A1 inhibitor, BAY-876 treatment.
(F) Proposed mechanism of PRDM14-mediated rescue of cell proliferation and survival upon YAP1 suppression.
All data represent mean ± SEM normalized to cell proliferation in water treated conditions (A), in shGFP expressing cells without YAP1 suppression (D), or in DMSO treated cells without YAP1 suppression (E). P values were calculated using a paired Student’s two-tailed t test; n = 3, ****, p<0.0001, ***, p<0.001, **, p<0.01, *, p<0.05, ns, not significant.
To further probe CALM2 and SLC2A1 rescue of YAP1 suppression during tumor formation and maintenance, we adapted our previous in vivo rescue experiment using the HT29 shYAP1 cells expressing GFP, YAP1, PRDM14, CALM2 or SLC2A1 placed subcutaneously into immunodeficient mice. We found that CALM2 or SLC2A1 expression rescued YAP1 suppression both to promote tumor formation (Figure 6B) and maintain YAP1-driven tumor growth (Figure 6C).
CALM2 and SLC2A1 are required for PRDM14 rescue of YAP1 suppression
To investigate whether CALM2 and SLC2A1 are required for PRDM14 to rescue YAP1 suppression, we performed a rescue experiment in HT29, SKCO1 and SW48 shYAP1 cells expressing GFP, YAP1 or PRDM14 in which we suppressed expression of CALM2 or SLC2A1 by CALM2 or SLC2A1-specific shRNAs. We found that suppression of CALM2 or SLC2A1 abrogated the ability of PRDM14 to rescue YAP1 suppression (Figure 6D). Consistent with this observation, we also found that suppression of CALM2 or SLC2A1 with a CALM2 inhibitor, W-7 or a SLC2A1 inhibitor, BAY-876 treatment decreased the ability of PRDM14 to rescue YAP1 suppression (Figure 6E). Together, these findings suggest that CALM2 and SLC2A1 are significant downstream targets of PRDM14 contributing to the rescue of cell proliferation upon suppression of YAP1 (Figure 6F).
Interaction of PRDM14 and YAP1 with TEAD transcription factors
Because SLC2A1 has been previously reported as part of a YAP1/TEAD signaling axis (Cox et al., 2018; Lin and Xu, 2017), we investigated whether PRDM14 rescues cell proliferation and SLC2A1 expression upon YAP1 suppression by interacting with TEAD. First, we tested whether depletion of TEAD affects the fitness of YAP1-dependent cells (Figure S6A). We found that YAP1-dependent cells are also dependent on TEAD transcription factors for proliferation and survival (Figure S6A). Next, we performed clonogenic proliferation assays to test whether TEAD is involved in the PRDM14-mediated rescue of YAP1 suppression. We found that depletion of TEAD abrogated the rescue effect of PRDM14 upon YAP1 suppression, suggesting that PRDM14 genetically interacts with TEAD to rescue YAP1 suppression (Figure S6B). In addition, we confirmed that depletion of TEAD indeed downregulates the expression of SLC2A1 (Figure S6C). As we found in clonogenic proliferation assays (Figure S6B), we also found that PRDM14 rescues SLC2A1 expression upon YAP1 suppression in a TEAD dependent manner (Figure S6C). However, we did not detect a physical interaction between PRDM14 and TEAD by performing co-immunoprecipitation experiments (Figure S6D). In addition, overexpression of TEAD2 did not rescue YAP1 suppression in YAP1-dependent cells (Figure S6E). Together, these findings suggest the existence of a genetic but indirect interaction between PRDM14 and TEAD and that TEAD is required for PRDM14-mediated rescue of YAP1 suppression. However, TEAD by itself is not sufficient for rescue of YAP1 suppression. These observations reinforce the concept that YAP1 and PRDM14 promote cell survival by regulating overlapping portions of transcriptional programs necessary for cell survival but do not activate a single transcriptional program.
Interaction of PRDM14 and YAP1 with Ras effector pathways
Prior work indicates that YAP1-regulated expression of the EGFR ligand amphiregulin leads to activation of ERK and AKT (Overholtzer et al., 2006; Zhang et al., 2009), and PRDM14 activates AKT-mTORC1 signaling by regulating transcription of proto-oncogene TCL1, AKT kinase coactivator. (Yamaji et al., 2013). When we searched for transcriptional programs regulated by both PRDM14 and YAP1 in our RNA-sequencing and ChIP-sequencing data (Figure 4C) using MSigDB (Subramanian et al., 2005), we found upregulation of MEK and AKT signaling pathways (Figure S4C) among the oncogenic signatures (Liberzon et al., 2015). To investigate whether expression of PRDM14 or YAP1 reactivated the MAPK and PI3K signaling pathways, we expressed GFP, YAP1 or PRDM14 in HT29, SKCO1 and SW48 shYAP1 cells and evaluated the phosphorylation status of ERK1/2, AKT and S6 upon suppression of YAP1. We found that PRDM14 or YAP1 expression restored the phosphorylation of ERK1/2, AKT and S6 that was decreased upon suppression of YAP1 (Figure S6F). To investigate whether reactivation of MAPK and PI3K pathways was necessary for the ability of PRDM14 and YAP1 to rescue YAP1 suppression, we treated HT29, SKCO1 and SW48 shYAP1 cells expressing PRDM14 or YAP1 with the AKT inhibitor, MK-2206 or GDC-0068 or MEK inhibitor, trametinib or selumetinib. Treatment with either AKT or MEK inhibitor decreased the ability of PRDM14 and YAP1 to rescue YAP1 suppression compared to cells expressing GFP (Figure S6G and S6H). Thus, PRDM14 may activate the MAPK and PI3K pathways to rescue YAP1 suppression.
CALM2 is a member of the calmodulin family that plays a role in signaling pathways, cell cycle progression and proliferation (Berchtold and Villalobo, 2014). Previous studies show that calmodulin binds to several tyrosine kinase receptors including the EGFR and ErbB2 and modulates their tyrosine kinase activities and signaling (Li et al., 2004; Martin-Nieto and Villalobo, 1998; Sanchez-Gonzalez et al., 2010; White et al., 2011). In addition, calmodulin-mediated activation of AKT was reported to regulate survival of c-Myc-overexpressing mouse mammary carcinoma cells (Deb et al., 2004). Thus, we hypothesized that restoration of CALM2 expression by PRDM14 upon suppression of YAP1 might be involved in the reactivation of MAPK and PI3K pathways. To test this hypothesis, we assessed the effect of CALM2 inhibitor, W-7 treatment on PRDM14-mediated restoration of phosphorylation status of ERK1/2, AKT and S6 upon suppression of YAP1. We found that suppression of CALM2 decreased the ability of PRDM14 to restore the activities of MAPK and PI3K pathways (Figure S6F). Therefore, these findings suggest that PRDM14 rescues YAP1 suppression in part through CALM2-mediated reactivation of MAPK and PI3K signaling pathways.
PRDM14 rescues YAP1 suppression in both mouse and human colon cancer organoid models
To investigate whether PRDM14 rescues YAP1 depletion in other biological and colon cancer models, we used colon organoid models derived from Apc wild-type (Apc WT, Apcflox/flox; tdRFPLSL or Lgr5-eGFP-CreER; Apcflox/flox; tdRFPLSL; td−), Apc knockout (Apc KO, Lgr5-eGFP-CreER; Apcflox/flox; tdRFPLSL; td+) and Apc knockout Kras mutant (Apc KO;Kras G12D, Adeno-Cre treatment of Apcflox/flox; LSL-KrasG12D) mice as well as APC and KRAS mutant colorectal cancer patient tumors. We used these models because Yap1 is known to be activated in Apcmin mouse colon cancer and required for intestinal tumorigenesis in this model (Cai et al., 2010; Gregorieff et al., 2015). In addition, Apc KO mouse organoids are reported to be more sensitive to the treatment of dasatinib, the inhibitor of YES1, which phosphorylates and activates YAP1, compared to Apc WT mouse organoids (Rosenbluh et al., 2012). Using the Apc WT and KO mouse colon organoid models, we tested whether Yap1 is required for the viability of Apc KO organoids and whether PRDM14 expression rescues the viability of Apc KO organoids following Yap1 suppression. First, we confirmed that Yap1 expression is increased in Apc KO organoids compared to WT organoids (Figure S7A and S7B). In addition, we found that expression levels of transcriptional target genes, Calm2 and Slc2a1 are also increased in Apc KO organoids compared to WT organoids (Figure S7A).
We generated Apc WT, Apc KO or Apc KO;Kras G12D organoids stably expressing doxycycline-inducible shRNAs targeting Yap1 and confirmed that doxycycline treatment induced Yap1 knockdown in these organoid lines (Figure S7C). In this setting, we also observed that expression of Calm2 and Slc2a1 was downregulated after suppressing endogenous Yap1 (Figure S7D). We then performed CellTiter-Glo 3D cell viability assays and found that Apc KO and Apc KO;Kras G12D organoids are more sensitive to Yap1 depletion than Apc WT organoids for their viability (Figure 7A). To investigate whether PRDM14 expression rescues Yap1 suppression in this context, we performed a rescue experiment in these dox-inducible shYap1-expressing Apc KO and Apc KO;Kras G12D organoids (Figure 7B). We expressed either Luciferase, YAP1 or PRDM14 in these lines and performed viability assays following Yap1 suppression. We found that PRDM14 expression as well as exogenous YAP1 expression rescued the viability effect induced by Yap1 depletion (Figure 7B).
Figure 7. PRDM14 rescues YAP1 suppression in both mouse and human colon cancer organoid models.

(A) CellTiter-Glo 3D cell viability assays of Apc WT, Apc KO and Apc KO;Kras G12D mouse colon organoids expressing dox-inducible shYap1 with or without YAP1 suppression.
(B) Immunoblots and CellTiter-Glo 3D cell viability assays of Apc KO and Apc KO;Kras G12D shYap1 organoids expressing LUC, YAP1 or PRDM14 with or without YAP1 suppression.
(C) Representative images, YAP1 mRNA expression, distribution and quantification of APC and KRAS mutant patient-derived colon organoids expressing dox-inducible shYAP1 (0544 + shYAP1–3) with or without YAP1 suppression. Scale bar, 20 μm.
(D) Immunoblots and CellTiter-Glo 3D cell viability assays of 0544 shYAP1–3 organoids expressing LUC, YAP1 or PRDM14 with or without YAP1 suppression.
(E) mRNA expression of CALM2 and SLC2A1 in 0544 shYAP1–3 organoids expressing LUC, YAP1 or PRDM14 with or without YAP1 suppression.
All data represent mean ± SEM normalized to cell viability in water treated conditions (A, B, D) or to mRNA expression in LUC expressing cells without YAP1 suppression (E). P values were calculated using a paired Student’s two-tailed t test; n = 3, ****, p<0.0001, ***, p<0.001, **, p<0.01, *, p<0.05. Water, without YAP1 suppression; Dox, with YAP1 suppression.
In addition to these mouse colon organoids, we also used a patient-derived colon organoid model (0544) harboring both APC and KRAS mutations (Table S4). To test whether YAP1 is required for the viability of this patient-derived colon organoid, we generated organoid line stably expressing doxycycline-inducible shYAP1 (0544 + shYAP1–3) (Figure 7C) and assessed viability with or without doxycycline treatment. We found that these organoids are dependent on YAP1 for their propagation (Figure 7C). We then tested whether PRDM14 expression rescues viability of these dox-inducible shYAP1-expressing organoids upon YAP1 suppression (Figure 7D). Specifically, we expressed either Luciferase, YAP1 or PRDM14 in this organoid line and assessed viability with or without doxycycline treatment. We found that either PRDM14 or YAP1 expression was able to rescue the viability of these organoids following YAP1 suppression (Figure 7D). We also confirmed that expression levels of CALM2 and SLC2A1, the common target genes of YAP1 and PRDM14 were rescued by either YAP1 or PRDM14 expression following YAP1 depletion (Figure 7E). Together, these observations demonstrate that relationship among YAP1, PRDM14 and KRAS are generalizable in both murine and human colon cancer models.
Amplification or overexpression of PRDM14 and YAP1 in colorectal adenocarcinoma
We then interrogated The Cancer Genome Atlas (TCGA) and Pan-Cancer Atlas mRNA expression data derived from the genomic characterization of colorectal adenocarcinoma tissue samples available on cBioPortal (Gao et al., 2013). Specifically, we compared mRNA transcript levels of YAP1 and PRDM14 in both normal and colorectal cancer tissues (Figure S7E). We found that YAP1 is uniformly expressed in normal colon tissues and overexpressed in some of the colorectal cancer tissues (Figure S7E). On the other hand, PRDM14 expression level is generally low in normal colon tissues and elevated in a subset of colorectal cancer tissues (Figure S7E). We also found that gene amplification or overexpression of PRDM14 and YAP1 are mutually exclusive in this set of colorectal adenocarcinoma samples (Figure S7F). These observations reinforce the conclusion that PRDM14 substitutes for YAP1 in colon cancers and that tumors appear to overexpress either YAP1 or PRDM14, not both.
DISCUSSION
Using a systematic functional approach, we identified PRDM14 as a gene whose expression rescued cell proliferation upon suppression of YAP1 in YAP1-dependent colon cancer cell lines. In mouse xenograft models, we found that PRDM14 rescued tumorigenesis of YAP1-driven tumors upon suppression of YAP1. Through integration of transcriptional factor-binding data and differential gene expression data, we identified a PRDM14-driven transcriptional program, involving CALM2 and SLC2A1, necessary for the survival of YAP1-dependent cancers. Notably, PRDM14-induced upregulation of CALM2 and SLC2A1 was required to rescue YAP1 suppression, indicating CALM2 and SLC2A1 as downstream targets of PRDM14 essential for YAP1 signaling and dependency (Figure 6F). We also confirmed this PRDM14-mediated rescue of YAP1 suppression in both mouse and human colon cancer organoid models. Furthermore, we found that gene amplification or overexpression of PRDM14 and YAP1 are mutually exclusive in colorectal adenocarcinoma, suggesting that some tumors require PRDM14 expression for survival and maintenance. Therefore, this study provides further insights into genes and pathways regulated by YAP1 in a cancer context.
Prior work showed that Hippo signaling dysfunction induces cancer cell addiction to YAP1 (Han et al., 2018), indicating YAP1 as the major downstream effector of the Hippo pathway in cancers. However, whereas amplifications of YAP1 have been observed in liver, breast, and esophageal cancers (Muramatsu et al., 2011; Overholtzer et al., 2006; Zender et al., 2006), and loss-of-function mutations of SAV1, MOB1 and NF2, the upstream regulators of YAP1 in the Hippo pathway, have been described in some cancers (Evans, 2009; Jiang et al., 2019; Kosaka et al., 2007; Rouleau et al., 1993; Trofatter et al., 1993), these genetic alterations are relatively uncommon compared with the observed prevalence of YAP1 overexpression across many cancer types (Steinhardt et al., 2008). These observations suggest that YAP1 may contribute to cancer development and maintenance in both Hippo pathway regulation-dependent and independent mechanisms. Indeed, in some contexts, YAP1 appears to act as a tumor suppressor (Barry et al., 2013; Cheung et al., 2020), highlighting the context dependent function of YAP1. The finding that PRDM14 and YAP1 regulate an overlapping set of genes required for YAP1-induced tumor phenotypes, suggests that these targets are likely to be both dependent and independent of Hippo signaling involving TEAD family transcription factors.
Disruption of normal cell differentiation occurs in many cancers and is thought to be a key step in cell transformation. Both YAP1 and PRDM14 are involved in the maintenance of the self-renewal of stem cells in part through the suppression of genes involved in differentiation (Cai et al., 2010; Chia et al., 2010; Gregorieff et al., 2015; Tsuneyoshi et al., 2008), suggesting that aberrant expression of YAP1 or PRDM14 contributes to transformation by affecting cell fate decisions.
Moreover, overexpression or amplification of YAP1 has been implicated in resistance to several types of targeted therapies (Huang et al., 2013; Kapoor et al., 2014; Kim et al., 2016; Lee et al., 2015; Lin et al., 2015a; Lin et al., 2015b; Shao et al., 2014; Song et al., 2015; Touil et al., 2014; Xiao et al., 2016; Yoshikawa et al., 2015; Zhao et al., 2014). We and others have also reported that YAP1 signaling functionally replaces KRAS in KRAS-dependent cancer cells and Yap1 amplifications are observed in tumors that escape suppression of Kras in Kras-driven murine pancreatic ductal adenocarcinomas (Kapoor et al., 2014; Shao et al., 2014). These resistant tumors often exhibit altered differentiation states, such as the epithelial to mesenchymal transition or transition from one differentiation state to another (Niederst et al., 2015; Oser et al., 2015), which may play a key role in their resistance to targeted therapies. In such tumors, the expression of YAP1 or PRDM14 may alter differentiation programs and facilitate resistance.
Multiple cancers are dependent on sustained YAP1 transcriptional activity for both tumor initiation and maintenance pointing to YAP1 and the pathways regulating YAP1 as a relevant therapeutic target. Since YAP1 is dispensable for normal development and homeostasis of the colonic epithelium (Cai et al., 2010), targeting YAP1 is a possible therapeutic strategy. In this study, our finding that PRDM14-induced upregulation of CALM2 and SLC2A1 bypasses YAP1 suppression predicts a potential resistance mechanism of YAP1-targeted therapy and implicates PRDM14 as a therapeutic target in YAP1-driven cancers.
Limitations of the Study
Here, we focused on investigating how PRDM14 functionally substitutes for YAP1 in YAP1-dependent colon cancers. This question should be extended to other types of cancers and stem cells. In addition, although we showed that PRDM14-CALM2-mediated reactivation of MAPK and PI3K pathways rescues YAP1 suppression (Figure S6F), other pathways enriched by YAP1 and PRDM14 expression such as the TGFB/SMAD pathway (Figure S4C) may also be involved in the rescue of YAP1 suppression. Moreover, since upregulation of SLC2A1 by YAP1 has been reported to modulate glycolysis (Cox et al., 2018; Lin and Xu, 2017), regulation of glucose metabolism by PRDM14 through SLC2A1 may also be involved in cell survival and tumorigenesis induced by YAP1.
STAR METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, William C. Hahn (william_hahn@dfci.harvard.edu).
MATERIALS AVAILABILITY
Plasmids generated in this study are available by request to the lead contact, William C. Hahn (william_hahn@dfci.harvard.edu).
DATA AND CODE AVAILABILITY
The datasets generated in this study are publicly available at NCBI GEO (GSE182432) and SRA (PRJNA756046) as of the date of publication.
No codes were generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293T and colon cancer cell lines were obtained from ATCC. Cells were maintained in DMEM (293T; Gibco #11995–065), McCoy’s 5A (HT29; Gibco #16600–082), EMEM (SKCO1; Corning #10–009-CV), RPMI 1640 (SW48, COLO320; Corning #10–040-CV) or MEM Alpha (HA1E; Gibco #12571–063) supplemented with 100 units/mL of penicillin, 100 μg/mL of streptomycin (Gibco #15140–122) and 10% fetal bovine serum (MilliporeSigma), and incubated at 37°C in 5% CO2.
Primary cell cultures
Apc WT and Apc KO mouse colon organoids were created from an Apcflox/flox; tdRFPLSL mouse and Lgr5-eGFP-CreER; Apcflox/flox; tdRFPLSL mouse flank injected with tamoxifen in vivo. Apc KO; Kras G12D mouse colon organoids were a gift from Kevin Haigis lab at Dana-Farber Cancer Institute and were generated through adeno-Cre treatment of organoids derived from an Apcflox/flox; LSL-KrasG12D mouse. Intestinal cells harvested from the Lgr5-eGFP-CreER; Apcflox/flox; tdRFPLSL mice were dissociated and sorted for td+ and td− populations, and collected in 50% DMEM 50% FBS recovery media with 1ug/mL Rock inhibitor added. Then, the cells were spun down and plated in Matrigel (Corning #354234). Wnt/R-spondin-deprived medium, DMEM/F12 with HEPES (MilliporeSigma) containing 20% FBS, 1% penicillin/streptomycin and 50 ng/mL recombinant mouse EGF (Life Technologies #PMG8041), was used for culturing mouse colon organoids. For the first 2–3 days after seeding, the media was also supplemented with 10 μM ROCK inhibitor Y-27632 (MilliporeSigma #Y0503) and 10 μM SB431542 (Sigma Aldrich #616461), an inhibitor for the transforming growth factor (TGF)-β type I receptor to avoid anoikis. For passage, mouse colon organoids were dispersed by TrypLE Express and transferred to fresh Matrigel. Passage was performed every 3–4 days with a 1:3–1:5 split ratio.
Patient-derived colon organoids were established from colorectal cancer patient samples obtained after participant consent and approval from the Gastrointestinal Cancer Center at the Dana-Farber Cancer Institute (protocol numbers 03–189, 17–000, and 18–060). Model 0544 was derived from a tumor sample of a colon cancer patient who was a 52-year-old female at the time of the biopsy. To establish organoids, tumor samples were minced and incubated with 5 mg/mL collagenase XI (MilliporeSigma #C9407), 10 μg/mL DNase I (StemCell #07900) and 10 μM Y-27632 (MilliporeSigma #Y0503). The digested tumors were embedded in domes of growth factor reduced Matrigel (Corning #356231), overlaid with intestinal organoid growth media (IOGM) containing Advanced DMEM/F12 (Gibco), 1X glutamax, 10 mmol/L HEPES, 100 U/mL penicillin-streptomycin (Life technologies), 1X B-27 supplement (Thermo Fisher Scientific #17504044), 100 ng/mL Noggin (Thermo Fisher Scientific #PHC1506), 1.25 mM N-acetylcysteine (MilliporeSigma #A9165), 10 mM Nicotinamide (MilliporeSigma #N3376), 10 uM SB202190 (MilliporeSigma #S7067), 50 ng/mL EGF (Life Technologies #PMG8041), 0.5 uM A 83–01 (Tocris #2939), 1 uM PGE2 (Tocris #2296). IOGM was changed every 2–3 days and organoids were passaged every 7–10 days via enzymatic digestion in TrypLE Express (Thermo Fisher Scientific) for 10 min at 37°C. Y-27632 was included in IOGM for 2 days following enzymatic digestion (Rendo et al., 2020; van de Wetering et al., 2015).
Mouse models
5-week-old female NCR-nude (CrTac:NCr-Foxn1nu) mice were obtained from Taconic Biosciences. Maximum of 5 mice were housed per individual ventilated cage and were maintained under optimum hygienic conditions, air-conditioned with 10 air changes per hour. The environment was continuously monitored with temperature target ranges of 22 ± 2°C, relative humidity of 45–65% and 12-hour light/12-hour dark cycle. All procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee of the Dana-Farber Cancer Institute (protocol number 04–101).
Human subjects
Patient data from previous studies were used in this study. Cancer patient whole exomes and transcripts levels were analyzed based on datasets in cBioPortal (Gao et al., 2013).
METHOD DETAILS
Lentiviral infection
ORFs in pLX317 or pLX304 backbones and shRNAs in pLKO.1 backbone were obtained from the Genetic Perturbation Platform at the Broad Institute (Kim et al., 2014). Target sequences of shRNAs are provided in Table S5. Lentiviruses were produced in 293T cells by transfecting with VSVG, delta8.9 and ORF or shRNA plasmids using TransIT (Mirus Bio). Virus was harvested 72 hours after transfection (Moffat et al., 2006). Cells were infected with lentivirus with 10 μg/mL of polybrene (MilliporeSigma #TR-1003-G). Infected cells were selected with 2 μg/mL of puromycin dihydrochloride (Thermo Fisher Scientific #Gibco A1113803) for 2 days or 10 μg/mL of blasticidin S HCl (Thermo Fisher Scientific #Gibco A1113903) for 5 days.
Generation of doxycycline-inducible shYAP1 cells
HT29, SKCO1, SW48 and COLO320 cells were seeded at 250,000, 500,000, 1,000,000 and 1,000,000 cells per well, respectively, in 6-well plates. Cells were infected with lentiviruses with 10 μg/mL of polybrene to integrate doxycycline-inducible YAP1 shRNAs using the pLKO-Tet-On backbone (Novartis). Target sequences of shRNAs are provided in Table S5. Plates were spun for 30 minutes at 2250 rpm at 30°C. 24 hours after infection, cells were selected with 1 mg/mL of G418 sulfate (Thermo Fisher Scientific #10131–035) for 7 days. Subsequently, cells were seeded at 0.3 cells per well in 96-well plates to allow selection of single-cell clones. Thirty clones per cell line were assessed, and HT29, SKCO1, SW48 and COLO320 shYAP1 single-cell clones were selected based on effectiveness of YAP1 suppression upon doxycycline (Clontech #631311) treatment.
YAP1 rescue screen
For the YAP1 rescue screen in HT29, SKCO1, SW48 and COLO320 doxycycline-inducible shYAP1 cells, 80×106 cells were infected per replicate with 30–40% infection efficiency to obtain at least 1,000 cells per ORF after selection (20×106 surviving cells containing 17,255 ORFs from the human ORFeome library collection 8.1 (Yang et al., 2011)). Two biological replicates were performed. Cells were seeded at 3×106 cells per well in 12-well plates and infected with the amount of virus determined during optimization using the Genetic Perturbation Platform virus titering protocol (https://portals.broadinstitute.org/gpp/public/resources/protocols), with 10 μg/mL of polybrene. Plates were spun for 2 hours at 2000 rpm at 30°C. Approximately 6 hours after infection, all wells within a replicate were pooled and split into 15 cm dishes. 24 hours after infection, cells were selected with 2 μg/mL of puromycin and expanded for 6 days. 60×106 cells were seeded in 15 cm dishes and allowed to adhere for 24 hours. On day 0, 1 mg/ml of doxycycline was treated to the cells to induce YAP1 suppression. Cells were passaged in doxycycline or fresh media containing doxycycline was added every 3–4 days. Cells were harvested 14 days after initiation of doxycycline treatment. Genomic DNA was purified using the Qiagen DNA Blood Maxi Kit (#51194) according to the manufacturer’s protocol. The barcodes corresponding to each ORF were amplified using PCR and analyzed by next generation sequencing.
Enriched barcodes were analyzed as follows: Each sample was normalized to a total of 1 million barcode reads. The number of each barcode after normalization was calculated to its log2 value. The log2 value of each barcode in the water treated cells was subtracted from the doxycycline treated cells to obtain the log2 fold-change value of each barcode. The averages and standard deviations (SD) of the log2 fold-change values in all samples were determined, and Z scores for each barcode were calculated as follows: Z Barcode X = (log2 fold-change value Barcode X - average) / SD. The Z score was used to evaluate the enrichment of a certain ORF in the doxycycline treated cells compared to the water treated cells.
Meta-analysis of ORF screening data
Meta-analysis of pathway and protein-protein interaction (PPI) enrichment was performed with candidate genes from ORF screens using the Metascape suite of tools (http://metascape.org) (Tripathi et al., 2015). Pathway enrichment analysis was performed within the Molecular Signatures Database (MSigDB) (Subramanian et al., 2005). PPI meta-analysis was performed with the merged candidate gene lists from all four ORF screens based on the BioGrid, InWeb_IM and OmniPath databases per the standard metascape algorithm. The Molecular Complex Detection (MCODE) algorithm was applied to identify densely connected network components. The network displays and integrated data representations for each figure were derived from standard plots generated by the Metascape software.
Immunoblots and antibodies
Cell lysate was purified using RIPA buffer (MilliporeSigma #R0278) containing protease inhibitor cocktail (Cell Signaling #5871S) and phosphatase inhibitors, sodium fluoride and sodium orthovanadate (NEB #P0759, #0758). Immunoblots were performed by separating 20–50 μg of cell lysate per sample on 4%−12% Bis-Tris gel (Invitrogen NuPAGE) and transferring the gel to the nitrocellulose membrane using iBlot gel transfer device. Membranes were blocked for 1 hour at room temperature and incubated with the primary antibodies overnight at 4°C, and then incubated with the secondary antibodies (LiCor Biosciences #926–32210, #926–32211, #926–68020, #926–68021) for 2 hours at room temperature. Immunoblots were visualized by infrared imaging (LI-COR).
For SLC2A1 immunoblots, cytoplasmic and plasma membrane fractions were purified using Plasma Membrane Protein Extraction Kit (Thermo Fisher Scientific #NC1053482) according to the manufacturer’s protocol. Because boiling caused aggregation of membrane proteins and poor resolution by SDS-PAGE, cell lysate was denatured for 30 minutes at room temperature following the addition of NuPAGE LDS sample buffer (#NP0007) as this prevented the aggregation of membrane proteins. 40 μg of cytoplasmic or plasma membrane fraction per sample was used for immunoblots in the same way.
Primary antibodies were obtained from Cell Signaling (YAP1 #4912S, GAPDH #5174S, Na,K-ATPase #3010S, p-ERK1/2 (T202/Y204) #4370S, ERK1/2 #9107S, p-AKT (S473) #4060S, AKT #2920S, p-S6 (S235/236) #4858S, S6 #2317S, TEAD1 #12292), MilliporeSigma (PRDM14 #ABD121, β-Actin #A5316), Novus (CALM2 #NBP2–14871), Abcam (SLC2A1 #ab115730, TEAD4 #ab97460), Invitrogen (V5 #R96025) and Bethyl Laboratories (CBFA2T2 #A303–593A).
Co-immunoprecipitation
HT29 shYAP1 cells stably expressing empty vector or Flag-tagged PRDM14 were harvested and lysed with 0.5% Triton X-100 buffer [50 mM Tris–Cl (pH 7.5), 150 mM NaCl, 1 mM EDTA and 0.5% Triton X-100]. Cell lysate (1 mg in 1 mL) was incubated with 40 μL of the 50% anti-Flag M2 magnetic bead (MilliporeSigma #M8823) suspension per reaction overnight at 4°C. The beads were washed three times with TBS (50 mM Tris HCl,150 mM NaCl, pH 7.4), eluted with Flag peptides (MilliporeSigma #F3290) and boiled with NuPAGE LDS sample buffer (#NP0007). CBFA2T2 is a known binding partner of PRDM14 (Tu et al., 2016), thus, its immunoblot was included as a positive control.
Clonogenic proliferation assays
Cells were seeded at 2,500–10,000 cells per well in 24-well plates and allowed to adhere for 24 hours. On day 0, cells were treated with water as a control or with 1 mg/ml of doxycycline to induce YAP1 suppression. Media was changed every 3–4 days with or without doxycycline treatment. On day 7, cells were fixed with 10% formalin and stained with 0.5% crystal violet in 10% ethanol for 20 minutes. Crystal violet uptake was extracted with 10% acetic acid and quantified by measuring absorbance at 565 nm using a SpectraMax M5 microplate reader (Molecular Devices).
Anchorage-independent colony formation assays
A total of 4 mL of 0.6% bottom agar in MEM Alpha supplemented with 100 units/mL of penicillin, 100 μg/mL of streptomycin and 20% fetal bovine serum was solidified in 6-well plates. 10,000 HA1E cells expressing GFP or YAP1 with shGFP or shPRDM14 were added to a total of 5 mL of 0.4% top agar in HA1E growth medium and layered onto bottom agar. After 21 days, colonies were stained with 0.1% crystal violet followed by washing with distilled water. Stained colonies were examined under a dissecting microscope and counted.
In vivo xenografts
For tumor formation experiment, 5-week-old female Taconic NCR-nude (CrTac:NCr-Foxn1nu) mice were fed with doxycycline-containing diet 2 days before cell injection. 106 HT29 or SW48 shYAP1 cells expressing GFP, YAP1, PRDM14, CALM2 or SLC2A1 were subcutaneously injected into the top, left and right flanks of each immunodeficient mouse. Tumor size was measured with caliper every 3–4 days. For tumor maintenance experiment, HT29 or SW48 shYAP1 cells expressing GFP, YAP1, PRDM14, CALM2 or SLC2A1 were subcutaneously injected into the immunodeficient mice. When tumor volume reached 100 mm3 on day 14, YAP1 suppression was initiated with doxycycline-containing diet. Tumor size was measured with caliper every 3–4 days. Tumor volume was calculated by the formula: volume = length × width2 × 0.5. All procedures were performed according to protocols approved by the Institutional Animal Care and Use Committees of the Dana-Farber Cancer Institute (protocol number 04–101).
Organoid transduction and CellTiter-Glo 3D cell viability assays
For generation of doxycycline-inducible shYAP1 organoids (0544 + shYAP1–3), organoids were digested into a single cell suspension in TrypLE Express before infection with lentivirus (pLKO-Tet-On shYAP1–3). Single cells were centrifuged with virus and 10 μg/mL polybrene at 2000 rpm for 1 hour. Following centrifugation, cells were incubated at 37°C for 6 hours before being washed and plated in Matrigel domes. After 24 hours in intestinal organoid growth media (IOGM), organoids were selected in 500 μg/mL of G418 (Thermo Fisher Scientific #10131–035) for 10 days. Following expansion, organoids were passaged and treated with doxycycline for 7 days. Doxycycline containing IOGM was refreshed every three days.
For generation of doxycycline-inducible shYap1 mouse colon organoids and expression of ORFs in all organoid lines, organoids were dissociated into a single cell suspension and infected with lentivirus in organoid growth media with 10 μg/mL of polybrene and 10 μM of Y-27632 followed by centrifugation at 2000 rpm for 2 hours at 32°C. After overnight incubation at 37°C, the cells were detached from the plate, collected and re-plated in Matrigel domes. After 24 hours in organoid growth media, organoids were selected with 1 mg/mL of G418 or 2 μg/mL of puromycin for 7 days.
For CellTiter-Glo 3D cell viability assays (Promega #G9683), 2,000 cells were seeded in a single 5 μL Matrigel aliquot into each well of 96-well plate containing 100 μL of organoid growth media. 1 mg/mL of doxycycline was added to the media at 24 hours after organoid plating. Doxycycline containing organoid growth media was changed every 2–3 days. After 7 days, 100 μL of CellTiter-Glo 3D reagent was added into each well of 96-well plate and the plate was shaken for 30 min at room temperature to allow the Matrigel to dissolve. Plate was read on a SpectraMax M5 microplate reader (Molecular Devices).
Whole exome sequencing
For whole exome sequencing in the patient-derived organoid model (0544), total cellular DNA was isolated using the DNeasy Blood & Tissue Mini Kit (Qiagen #69504). Quality of isolated genomic DNA was verified by following methods: 1) DNA degradation and contamination were monitored on 1% agarose gels. 2) DNA concentration was measured using Qubit® DNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies). 100X whole exome sequencing was performed by Novogene Corporation Inc Service using Illumina NovaSeq 6000 with an average read depth of at least 20 million reads. The genomic DNA sample was randomly fragmented by sonication (Covaris) to the size of 180–280 bp fragments. Burrows-Wheeler Aligner (BWA) was utilized to map the paired-end clean reads to the human reference genome (hg38, http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/analysisSet/hg38.analysisSet.2bit). SAMtools (Li et al., 2009) was used for sorting the BAM files, and Picard was utilized to mark duplicate reads. Following genomic variant detection, annotation of variants with the tool ANNOVAR (Wang et al., 2010) in multiple aspects are performed, including protein coding changes, genomic regions affected by the variants, allele frequency, deleteriousness prediction according to Novogene internal analysis pipeline.
Quantitative PCR
RNA was purified using an RNeasy Plus Mini Kit (Qiagen #74134). cDNA was synthesized using High Capacity RNA-to-cDNA Kit (Invitrogen #4387406) and analyzed by quantitative PCR using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific #4367659) on a QuantStudio 6 Flex PCR system (Applied Biosystems) according to the manufacturer’s recommendations. Primer sequences used for qRT-PCR are provided in Table S6.
RNA-sequencing
For RNA-sequencing in HT29, SKCO1 and SW48 shYAP1 cells expressing GFP, YAP1 or PRDM14, 1,000,000–4,000,000 cells were seeded in 10 cm dishes and allowed to adhere for 24 hours. Subsequently, cells were treated with water as a control or with 1 mg/ml of doxycycline for 2 days. Total RNA was extracted using an RNeasy Plus Mini Kit (Qiagen #74134). RNA sequencing libraries were prepared using a NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB #E7420). The concentration of each cDNA library was quantified with the KAPA Complete Kit (ABI Prism) (Kapa Biosystems #KK4835). Libraries were pooled for sequencing using the HiSeq 2500. Reads were mapped to the reference human genome (hg19) using Tophat 2.0.2 (Trapnell et al., 2010) and genome annotation from the UCSC genome browser (Kent et al., 2002). Transcripts were assembled and tested for abundance and differential expression using Cufflinks 2.0.2 module (Trapnell et al., 2010) with default setting on GenePattern (Reich et al., 2006).
ChIP-sequencing
For ChIP-sequencing in HT29, SKCO1 and SW48 shYAP1 cells expressing YAP1 or PRDM14, 10–20×106 cells were seeded in 15 cm dishes and allowed to adhere for 24 hours. Cells were then treated with water as a control or with 1 mg/ml of doxycycline for 2 days. Cells were fixed with serum-free media containing 1% formaldehyde (Thermo Fisher Scientific #F79–500) for 10 minutes at 37°C. Crosslinking was stopped by adding 0.125M of glycine for 5 minutes at 37°C. Cells were collected and lysed with Sarkosyl lysis buffer (0.25% Sarkosyl, 1mM DTT, protease inhibitors in 0.3M RIPA buffer). Samples were sonicated using Bioruptor at high setting for a total time of 30 minutes and centrifuged to remove insoluble materials. Lysate was then added with A/G Dyna magnetic beads (Thermo Fisher Scientific #10001D, #10003D) that were pre-bound to 5 μg of V5 antibody (Cell Signaling #13202) or H3K27Ac antibody (Abcam #ab4729), and incubated overnight at 4°C. The beads were washed with 6 times with 0.3M RIPA buffer (0.1% SDS, 1% Triton X-100, 10 mM Tris-HCl, 1 mM EDTA, 0.1% NaDOC, 0.3M NaCl) followed by the last wash with TE buffer. DNA was reverse-crosslinked by adding 100 μl of buffer (1% SDS, 0.1M NaHCO3) and incubating for 16 hours at 65°C at 600 rpm. Supernatant containing DNA was collected. Sample DNA as well as its input was purified using QIAquick PCR Purification Kit (Qiagen #28104) and quantified using Quant-iT PicoGreen dsDNA (Thermo Fisher Scientific #P7589). Equal amount of each DNA and input of each sample was used for quantitative PCR. Primer sequences used for ChIP-qPCR are provided in Table S6. Inputs and sample DNAs were prepared as the sequencing libraries using the ThruPLEX DNA-seq Kit (Rubicon Genomics #R400427). Libraries were sequenced using 75-bp single reads on Illumina platform at the Dana-Farber Cancer Institute core facility. All samples were processed using the ChiLin pipeline (Qin et al., 2016) for reads mapping and peak calling. Heatmaps were generated using deepTools (Ramirez et al., 2016) and peaks at the specific genomic loci were shown using IGV viewer. Integration of RNA-sequencing and ChIP-sequencing datasets was performed using the BETA-plus package (Wang et al., 2013).
Gene expression analysis
To identify enriched gene sets, gene set enrichment analysis was performed with genes that are bound and upregulated by YAP1 and PRDM14 using Molecular Signatures Database (MSigDB) (Subramanian et al., 2005), C6 collection of oncogenic signatures (Liberzon et al., 2015).
cBioPortal analysis
TCGA and Pan-Cancer Atlas data available on cBioPortal (Gao et al., 2013) was queried for PRDM14 and YAP1 genetic alteration rates in colorectal adenocarcinoma samples. Differential gene expression of PRDM14 and YAP1 between normal and tumor tissues was analyzed using TIMER (http://timer.cistrome.org/) (Li et al., 2020).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was determined using a paired Student’s two-tailed t test and shown in the figure legends. For in vivo experiments, an unpaired Student’s two-tailed t test was used to assess differences in tumor volumes between groups. All graphs represent the mean ± SEM of at least three independent experiments, unless indicated otherwise.
Supplementary Material
Table S1. Results of genome scale ORF screens. Related to Figure 1.
Z scores in genome scale ORF screens ordered by Gene ID.
Table S2. Gene expression profiling in the context of depletion or overexpression of YAP1 or PRDM14. Related to Figure 3 and 4.
Statistically significantly (q < 0.05) differentially expressed genes by independent depletion or overexpression of YAP1 or PRDM14 ordered by Gene Symbol.
Table S3. YAP1 and PRDM14 target genes. Related to Figure 4.
Genes bound by and upregulated by YAP1 or PRDM14 upon YAP1 suppression based on BETA ordered by Gene Symbol.
Table S4. Whole exome sequencing data for patient-derived colon organoid model. Related to Figure 7.
APC and KRAS variants in 0544 organoids identified by whole exome sequencing.
Table S5. Target sequences of shRNAs. Related to STAR methods.
Table S6. Primer sequences used for qRT-PCR and ChIP-qPCR. Related to STAR methods.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-YAP | Cell Signaling Technolody | Cat# 4912, RRID:AB_2218911 |
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technolody | Cat# 5174, RRID:AB_10622025 |
| Rabbit polyclonal anti-Na,K-ATPase | Cell Signaling Technolody | Cat# 3010, RRID:AB_2060983 |
| Rabbit monoclonal anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling Technolody | Cat# 4370, RRID:AB_2315112 |
| Mouse monoclonal anti-p44/42 MAPK (Erk1/2) | Cell Signaling Technolody | Cat# 9107, RRID:AB_10695739 |
| Rabbit monoclonal anti-phospho-Akt (Ser473) | Cell Signaling Technolody | Cat# 4060, RRID:AB_2315049 |
| Mouse monoclonal anti-Akt (pan) | Cell Signaling Technolody | Cat# 2920, RRID:AB_1147620 |
| Rabbit monoclonal anti-phospho-S6 Ribosomal Protein (Ser235/236) | Cell Signaling Technolody | Cat# 4858, RRID:AB_916156 |
| Mouse monoclonal S6 Ribosomal Protein | Cell Signaling Technolody | Cat# 2317, RRID:AB_2238583 |
| Rabbit monoclonal anti-TEAD1 | Cell Signaling Technolody | Cat# 12292, RRID:AB_2797873 |
| Rabbit polyclonal anti-PRDM14 | MilliporeSigma | Cat# ABD121 |
| Mouse monoclonal anti-beta-Actin | MilliporeSigma | Cat# A5316, RRID:AB_476743 |
| Rabbit polyclonal anti-Calmodulin 2 | Novus | Cat# NBP2-14871 |
| Rabbit monoclonal anti-Glucose Transporter GLUT1 (SLC2A1) | Abcam | Cat# ab115730, RRID:AB_10903230 |
| Rabbit polyclonal anti-TEAD4 | Abcam | Cat# ab97460, RRID:AB_10680436 |
| Mouse monoclonal anti-V5 Epitope Tag | Invitrogen | Cat# R96025, RRID:AB_159313 |
| Rabbit polyclonal anti-CBFA2T2 | Bethyl Laboratories | Cat# A303-593A, RRID:AB_11125147 |
| Rabbit monoclonal anti-V5-Tag | Cell Signaling Technolody | Cat# 13202, RRID:AB_2687461 |
| Rabbit polyclonal anti-Histone H3 (acetyl K27) | Abcam | Cat# ab4729, RRID:AB_2118291 |
| IRDye 800CW Goat anti-Mouse IgG | LI-COR Biosciences | Cat# 926-32210, RRID:AB_621842 |
| IRDye 800CW Goat anti-Rabbit IgG | LI-COR Biosciences | Cat# 926-32211, RRID:AB_621843 |
| IRDye 680LT Goat anti-Mouse IgG | LI-COR Biosciences | Cat# 926-68020, RRID:AB_10706161 |
| IRDye 680LT Goat anti-Rabbit IgG | LI-COR Biosciences | Cat# 926-68021, RRID:AB_10706309 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| G418 sulfate | Thermo Fisher Scientific | Cat# 10131-035 |
| Puromycin dihydrochloride | Thermo Fisher Scientific | Cat# A1113803 |
| Blasticidin S HCl | Thermo Fisher Scientific | Cat# A1113903 |
| Doxycycline | Clontech | Cat# 631311 |
| MK-2206 2HCl | Selleck Chemicals | Cat# 1078 |
| Ipatasertib (GGC-0068) | Selleck Chemicals | Cat# 2808 |
| Trametinib | Selleck Chemicals | Cat# 2673 |
| Selumetinib | Selleck Chemicals | Cat# 1008 |
| W-7, hydrochloride | MilliporeSigma | Cat# 681629 |
| BAY-876 | Tocris Bioscience | Cat# 6199 |
| Polybrene | MilliporeSigma | Cat# TR-1003-G |
| anti-Flag M2 magnetic bead | MilliporeSigma | Cat# M8823 |
| Flag peptide | MilliporeSigma | Cat# F3290 |
| Matrigel | Corning | Cat# 354234 |
| EGF | Life Technologies | Cat# PMG8041 |
| Y-27632 | MilliporeSigma | Cat# Y0503 |
| SB431542 | MilliporeSigma | Cat# 616461 |
| Collagenase XI | MilliporeSigma | Cat# C9407 |
| DNase I | StemCell | Cat# 07900 |
| Growth factor reduced Matrigel | Corning | Cat# 356231 |
| B-27 supplement | Thermo Fisher Scientific | Cat# 17504044 |
| Noggin | Thermo Fisher Scientific | Cat# PHC1506 |
| N-acetylcysteine | MilliporeSigma | Cat# A9165 |
| Nicotinamide | MilliporeSigma | Cat# N3376 |
| SB202190 | MilliporeSigma | Cat# S7067 |
| A 83-01 | Tocris | Cat# 2939 |
| PGE2 | Tocris | Cat# 2296 |
| Critical Commercial Assays | ||
| DNA Blood Maxi Kit | Qiagen | Cat# 51194 |
| Plasma Membrane Protein Extraction Kit | Thermo Fisher Scientific | Cat# NC1053482 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74134 |
| NEBNext Ultra Directional RNA Library Prep Kit for Illumina | NEB | Cat# E7420 |
| KAPA Complete Kit (ABI Prism) | Kapa Biosystems | Cat# KK4835 |
| QIAquick PCR Purification Kit | Qiagen | Cat# 28104 |
| Quant-iT PicoGreen dsDNA | Thermo Fisher Scientific | Cat# P7589 |
| ThruPLEX DNA-seq Kit | Rubicon Genomics | Cat# R400427 |
| DNeasy Blood & Tissue Mini Kit | Qiagen | Cat# 69504 |
| CellTiter-Glo 3D Cell Viability Assay | Promega | Cat# G9683 |
| Deposited Data | ||
| ORF screen data | This study | Table S1 |
| RNA-seq and ChIP-seq data | This study | GSE182432 |
| Whole exome sequencing data | This study | PRJNA756046 |
| Analysis of genetic alteration in cancer | cBioPortal (Gao et al., 2013) | https://www.cbioportal.org/ |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL-3216, RRID:CVCL_0063 |
| HT-29 | ATCC | Cat# HTB-38, RRID:CVCL_0320 |
| SK-CO-1 | ATCC | Cat# HTB-39, RRID:CVCL_0626 |
| SW48 | ATCC | Cat# CCL-231, RRID:CVCL_1724 |
| COLO 320 | ATCC | Cat# CCL-220, RRID:CVCL_0219 |
| HA1E | Previous study (Hahn et al., 1999) | N/A |
| Experimental Models: Organisms/Strains | ||
| NCR-nude mouse (CrTac:NCr-Foxn1nu) | Taconic Biosciences | NCRNU-F |
| Apc WT and KO mouse colon organ oid models (Apcflox/flox; tdRFPLSL and Lgr5-eGFP-CreER; Apcflox/flox; tdRFPLSL) | This study | N/A |
| Apc KO;Kras G12D mouse colon or ganoid model (Adeno-Cre treatment of Apcflox/flox; LSL-KrasG12D) | Kevin Haigis lab at Dana-Farber Cancer Institute | N/A |
| Patient-derived colon organoid model (0544) | This study | N/A |
| Oligonucleotides | ||
| Table S6 | This study | N/A |
| Recombinant DNA | ||
| pLKO-Tet-On | Novartis | N/A |
| Human ORFeome library collection 8.1 | Broad Institute | N/A |
| pLX317 GFP | Broad Institute | BRDN0000559466 |
| pLX317 ANPEP | Broad Institute | TRCN0000476147 |
| pLX317 GRAMD1B | Broad Institute | TRCN0000481176 |
| pLX317 HDAC6 | Broad Institute | TRCN0000471558 |
| pLX317 HSP90AA1 | Broad Institute | TRCN0000491541 |
| pLX317 MAP3K5 | Broad Institute | TRCN0000488584 |
| pLX317 PIK3CB | Broad Institute | TRCN0000467602 |
| pLX317 PRDM14 | Broad Institute | TRCN0000471194 |
| pLX317 YAP1 | Broad Institute | TRCN0000477457 |
| pLX317 WWTR1 | Broad Institute | TRCN0000472318 |
| pLX317 CALM2 | Broad Institute | TRCN0000479230 |
| pLX317 SLC2A1 | Broad Institute | TRCN0000476750 |
| pLX317 TEAD2 | Broad Institute | TRCN0000478936 |
| pLX304 GFP | Broad Institute | ccsbBroad304_99986 |
| pLX304 PRDM14 | Broad Institute | ccsbBroad304_08822 |
| pLX304 YAP1 | Broad Institute | ccsbBroad304_07601 |
| pLX304 CALM2 | Broad Institute | ccsbBroad304_00207 |
| pLX304 SLC2A1 | Broad Institute | ccsbBroad304_01540 |
| pLKO.1 puro shGFP | Previous Study (Kim et al., 2014) | N/A |
| pLKO.1 puro shYAP1-1 | Broad Institute | TRCN0000107265 |
| pLKO.1 puro shYAP1-2 | Broad Institute | TRCN0000107266 |
| pLKO.1 puro shPRDM14-1 | Broad Institute | TRCN0000018523 |
| pLKO.1 puro shPRDM14-2 | Broad Institute | TRCN0000018525 |
| pLKO.1 puro shPRDM14-3 | Broad Institute | TRCN0000018524 |
| pLKO.1 puro shPRDM14-4 | Broad Institute | TRCN0000018527 |
| pLKO.1 puro shPRDM14-5 | Broad Institute | TRCN0000018526 |
| pLKO.1 puro shCALM2-1 | Broad Institute | TRCN0000195431 |
| pLKO.1 puro shCALM2-2 | Broad Institute | TRCN0000196508 |
| pLKO.1 puro shCALM2-3 | Broad Institute | TRCN0000052580 |
| pLKO.1 puro shSLC2A1-1 | Broad Institute | TRCN0000043583 |
| pLKO.1 puro shSLC2A1-2 | Broad Institute | TRCN0000043587 |
| pLKO.1 puro shTEAD1/3/4-1 | This study | N/A |
| pLKO.1 puro shTEAD1/3/4-2 | This study | N/A |
| pLX307 P2A GFP Luciferase | This study | N/A |
| pLX307 P2A GFP YAP1 | This study | N/A |
| pLX307 P2A GFP PRDM14 | This study | N/A |
| Software and Algorithms | ||
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Metascape | Previous study (Tripathi et al., 2015) | http://metascape.org |
| ChiLin | Previous study (Qin et al., 2016) | http://cistrome.org/chilin/ |
| deepTools | Previous study (Ramirez et al., 2016) | https://deeptools.readthedocs.io/en/develop/ |
| Tuxedo Suite RNA-seq analysis package on GenePattern | Previous study (Trapnell et al., 2010) | https://www.genepattern.org/ |
| BETA-plus package on Cistrome | Previous study (Wang et al., 2013) | http://cistrome.org/ |
| Gene Set Enrichment Analysis | Previous study (Subramanian et al., 2005) | https://www.gsea-msigdb.org/gsea/index.jsp |
| SAMtools | Previous study (Li et al., 2009) | http://www.htslib.org/ |
| ANNOVAR | Previous study (Wang et al., 2010) | https://annovar.openbioinformatics.org/ |
| TIMER | Previous study (Li et al., 2020) | http://timer.cistrome.org/ |
Highlights.
PRDM14 substitutes for oncogenic YAP1 in YAP1-dependent colon cancers.
PRDM14 activates the transcription of CALM2 and SLC2A1 to rescue YAP1 suppression.
PRDM14 is essential for survival of YAP1-dependent colon cancer organoid models.
PRDM14 is amplified or overexpressed in a subset of colorectal adenocarcinomas.
ACKNOWLEDGEMENTS
This work was supported by the National Cancer Institute’s Office of Cancer Genomics Cancer Target Discovery and Development (CTD2) initiative as well as grants from the U.S. National Institutes of Health/National Cancer Institute: U01 CA176058 (to W.C.H.), U01 CA199253 (to W.C.H.), P01 CA203655 (to W.C.H.), K00 CA212221 (to J.P.R.) and research funding from Bristol-Myers-Squibb, Merck, Servier and Janssen (to M.G.).
DECLARATION OF INTERESTS
W.C.H. is a consultant for ThermoFisher, Solasta Ventures, MPM Capital, KSQ Therapeutics, iTeos, Tyra Biosciences, Frontier Medicine, Jubilant Therapeutics, RAPPTA Therapeutics, Function Oncology and Calyx. M.G. receives research funding from Bristol-Myers-Squibb, Merck, Servier and Janssen. J.H.H. is a consultant for Astrin Biosciences, Principal Investigator for Caris Life Sciences Genitourinary disease working group.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Avruch J, Zhou D, and Bardeesy N (2012). YAP oncogene overexpression supercharges colon cancer proliferation. Cell Cycle 11, 1090–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, Fuchs CS, Magness ST, Smits R, Ogino S, et al. (2013). Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 493, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold MW, and Villalobo A (2014). The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim Biophys Acta 1843, 398–435. [DOI] [PubMed] [Google Scholar]
- Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, and Pan D (2010). The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev 24, 2383–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung P, Xiol J, Dill MT, Yuan WC, Panero R, Roper J, Osorio FG, Maglic D, Li Q, Gurung B, et al. (2020). Regenerative Reprogramming of the Intestinal Stem Cell State via Hippo Signaling Suppresses Metastatic Colorectal Cancer. Cell Stem Cell 27, 590–604 e599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia NY, Chan YS, Feng B, Lu X, Orlov YL, Moreau D, Kumar P, Yang L, Jiang J, Lau MS, et al. (2010). A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 468, 316–320. [DOI] [PubMed] [Google Scholar]
- Cox AG, Tsomides A, Yimlamai D, Hwang KL, Miesfeld J, Galli GG, Fowl BH, Fort M, Ma KY, Sullivan MR, et al. (2018). Yap regulates glucose utilization and sustains nucleotide synthesis to enable organ growth. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit M, Jimenez CR, Carvalho B, Belien JA, Delis-van Diemen PM, Mongera S, Piersma SR, Vikas M, Navani S, Ponten F, et al. (2012). Cell surface proteomics identifies glucose transporter type 1 and prion protein as candidate biomarkers for colorectal adenoma-to-carcinoma progression. Gut 61, 855–864. [DOI] [PubMed] [Google Scholar]
- Deb TB, Coticchia CM, and Dickson RB (2004). Calmodulin-mediated activation of Akt regulates survival of c-Myc-overexpressing mouse mammary carcinoma cells. J Biol Chem 279, 38903–38911. [DOI] [PubMed] [Google Scholar]
- Dettman EJ, Simko SJ, Ayanga B, Carofino BL, Margolin JF, Morse HC 3rd, and Justice MJ (2011). Prdm14 initiates lymphoblastic leukemia after expanding a population of cells resembling common lymphoid progenitors. Oncogene 30, 2859–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DG (2009). Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis 4, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, and Wrana JL (2015). Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 526, 715–718. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, and Weinberg RA (1999). Creation of human tumour cells with defined genetic elements. Nature 400, 464–468. [DOI] [PubMed] [Google Scholar]
- Han H, Yang B, Nakaoka HJ, Yang J, Zhao Y, Le Nguyen K, Bishara AT, Mandalia TK, and Wang W (2018). Hippo signaling dysfunction induces cancer cell addiction to YAP. Oncogene 37, 6414–6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi H, Taniguchi H, Nosho K, Ishigami K, Koide H, Mitsuhashi K, Okita K, Takemasa I, Imai K, and Nakase H (2020). PRDM14 promotes malignant phenotype and correlates with poor prognosis in colorectal cancer. Clin Transl Oncol 22, 1126–1137. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Zhang Y, Leung JY, Fan C, Popov KI, Su S, Qian J, Wang X, Holtzhausen A, Ubil E, et al. (2019). MERTK mediated novel site Akt phosphorylation alleviates SAV1 suppression. Nat Commun 10, 1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, et al. (2014). Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Kim M, Lee MS, Kim CH, and Lim DS (2014). The MST1/2-SAV1 complex of the Hippo pathway promotes ciliogenesis. Nat Commun 5, 5370. [DOI] [PubMed] [Google Scholar]
- Kosaka Y, Mimori K, Tanaka F, Inoue H, Watanabe M, and Mori M (2007). Clinical significance of the loss of MATS1 mRNA expression in colorectal cancer. Int J Oncol 31, 333–338. [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Sanchez-Torres J, Del Carpio A., Salas V, and Villalobo A (2004). The ErbB2/Neu/HER2 receptor is a new calmodulin-binding protein. Biochem J 381, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, and Liu XS (2020). TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res 48, W509–W514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, and Xu X (2017). YAP1-TEAD1-Glut1 axis dictates the oncogenic phenotypes of breast cancer cells by modulating glycolysis. Biomed Pharmacother 95, 789–794. [DOI] [PubMed] [Google Scholar]
- Martin-Nieto J, and Villalobo A (1998). The human epidermal growth factor receptor contains a juxtamembrane calmodulin-binding site. Biochemistry 37, 227–236. [DOI] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, and Guan KL (2016). Mechanisms of Hippo pathway regulation. Genes Dev 30, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo JS, Park HW, and Guan KL (2014). The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep 15, 642–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moelans CB, de Weger RA, Monsuur HN, Vijzelaar R, and van Diest PJ (2010). Molecular profiling of invasive breast cancer by multiplex ligation-dependent probe amplification-based copy number analysis of tumor suppressor and oncogenes. Mod Pathol 23, 1029–1039. [DOI] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. [DOI] [PubMed] [Google Scholar]
- Moroishi T, Hansen CG, and Guan KL (2015). The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu T, Imoto I, Matsui T, Kozaki K, Haruki S, Sudol M, Shimada Y, Tsuda H, Kawano T, and Inazawa J (2011). YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis 32, 389–398. [DOI] [PubMed] [Google Scholar]
- Nakaki F, and Saitou M (2014). PRDM14: a unique regulator for pluripotency and epigenetic reprogramming. Trends Biochem Sci 39, 289–298. [DOI] [PubMed] [Google Scholar]
- Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, et al. (2015). RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 6, 6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa N, Toyota M, Suzuki H, Honma T, Fujikane T, Ohmura T, Nishidate T, Ohe-Toyota M, Maruyama R, Sonoda T, et al. (2007). Gene amplification and overexpression of PRDM14 in breast cancers. Cancer Res 67, 9649–9657. [DOI] [PubMed] [Google Scholar]
- Nishio M, Hamada K, Kawahara K, Sasaki M, Noguchi F, Chiba S, Mizuno K, Suzuki SO, Dong YY, Tokuda M, et al. (2012). Cancer susceptibility and embryonic lethality in Mob1a/1b double-mutant mice. J Clin Invest 122, 4505–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oser MG, Niederst MJ, Sequist LV, and Engelman JA (2015). Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol 16, e165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou C, Sun Z, Li S, Li G, Li X, and Ma J (2017). Dual roles of yes-associated protein (YAP) in colorectal cancer. Oncotarget 8, 75727–75741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, and Haber DA (2006). Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A 103, 12405–12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Mei S, Wu Q, Sun H, Li L, Taing L, Chen S, Li F, Liu T, Zang C, et al. (2016). ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC Bioinformatics 17, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic S, Miller G, and Schally AV (1991). Inhibition of growth of HT-29 human colon cancer xenografts in nude mice by treatment with bombesin/gastrin releasing peptide antagonist (RC-3095). Cancer Res 51, 6006–6009. [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, and Mesirov JP (2006). GenePattern 2.0. Nat Genet 38, 500–501. [DOI] [PubMed] [Google Scholar]
- Rendo V, Stoimenov I, Mateus A, Sjoberg E, Svensson R, Gustavsson AL, Johansson L, Ng A, O’Brien C, Giannakis M, et al. (2020). Exploiting loss of heterozygosity for allele-selective colorectal cancer chemotherapy. Nat Commun 11, 1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, et al. (2012). beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151, 1457–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, Hoang-Xuan K, Demczuk S, Desmaze C, Plougastel B, et al. (1993). Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 363, 515–521. [DOI] [PubMed] [Google Scholar]
- Ruark E, Seal S, McDonald H, Zhang F, Elliot A, Lau K, Perdeaux E, Rapley E, Eeles R, Peto J, et al. (2013). Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat Genet 45, 686–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Gonzalez P, Jellali K, and Villalobo A (2010). Calmodulin-mediated regulation of the epidermal growth factor receptor. FEBS J 277, 327–342. [DOI] [PubMed] [Google Scholar]
- Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, et al. (2014). KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhardt AA, Gayyed MF, Klein AP, Dong J, Maitra A, Pan D, Montgomery EA, and Anders RA (2008). Expression of Yes-associated protein in common solid tumors. Hum Pathol 39, 1582–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng HW, Hung MH, Chen LJ, Chang MJ, Hsieh FS, Tsai MH, Huang JW, Lin CL, Tseng HW, Kuo ZK, et al. (2016). Protein tyrosine phosphatase 1B targets PITX1/p120RasGAP thus showing therapeutic potential in colorectal carcinoma. Sci Rep-Uk 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. (2015). Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, Eldridge R, Kley N, Menon AG, Pulaski K, et al. (1993). A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 72, 791–800. [DOI] [PubMed] [Google Scholar]
- Tsuneyoshi N, Sumi T, Onda H, Nojima H, Nakatsuji N, and Suemori H (2008). PRDM14 suppresses expression of differentiation marker genes in human embryonic stem cells. Biochem Biophys Res Commun 367, 899–905. [DOI] [PubMed] [Google Scholar]
- Tu S, Narendra V, Yamaji M, Vidal SE, Rojas LA, Wang X, Kim SY, Garcia BA, Tuschl T, Stadtfeld M, et al. (2016). Co-repressor CBFA2T2 regulates pluripotency and germline development. Nature 534, 387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, et al. (2015). Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ye C, Chen C, Xiong H, Xie B, Zhou J, Chen Y, Zheng S, and Wang L (2017). Glucose transporter GLUT1 expression and clinical outcome in solid tumors: a systematic review and meta-analysis. Oncotarget 8, 16875–16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, and Liu XS (2013). Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 8, 2502–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CD, Li Z, and Sacks DB (2011). Calmodulin binds HER2 and modulates HER2 signaling. Biochim Biophys Acta 1813, 1074–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki PM, and Rybarczyk A (2015). The Hippo pathway in colorectal cancer. Folia Histochem Cytobiol 53, 105–119. [DOI] [PubMed] [Google Scholar]
- Yamaji M, Ueda J, Hayashi K, Ohta H, Yabuta Y, Kurimoto K, Nakato R, Yamada Y, Shirahige K, and Saitou M (2013). PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell 12, 368–382. [DOI] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. (2011). A public genome-scale lentiviral expression library of human ORFs. Nat Methods 8, 659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes M, Lechago LV, and Lechago J (1996). Overexpression of the human erythrocyte glucose transporter occurs as a late event in human colorectal carcinogenesis and is associated with an increased incidence of lymph node metastases. Clin Cancer Res 2, 1151–1154. [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, and Piccolo S (2016). YAP/TAZ at the Roots of Cancer. Cancer Cell 29, 783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, et al. (2006). Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125, 1253–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ji JY, Yu M, Overholtzer M, Smolen GA, Wang R, Brugge JS, Dyson NJ, and Haber DA (2009). YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat Cell Biol 11, 1444–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Zhang Y, Wu H, Barry E, Yin Y, Lawrence E, Dawson D, Willis JE, Markowitz SD, Camargo FD, et al. (2011). Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc Natl Acad Sci U S A 108, E1312–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou DW, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, et al. (2009). Mst1 and Mst2 Maintain Hepatocyte Quiescence and Suppress Hepatocellular Carcinoma Development through Inactivation of the Yap1 Oncogene. Cancer Cell 16, 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Results of genome scale ORF screens. Related to Figure 1.
Z scores in genome scale ORF screens ordered by Gene ID.
Table S2. Gene expression profiling in the context of depletion or overexpression of YAP1 or PRDM14. Related to Figure 3 and 4.
Statistically significantly (q < 0.05) differentially expressed genes by independent depletion or overexpression of YAP1 or PRDM14 ordered by Gene Symbol.
Table S3. YAP1 and PRDM14 target genes. Related to Figure 4.
Genes bound by and upregulated by YAP1 or PRDM14 upon YAP1 suppression based on BETA ordered by Gene Symbol.
Table S4. Whole exome sequencing data for patient-derived colon organoid model. Related to Figure 7.
APC and KRAS variants in 0544 organoids identified by whole exome sequencing.
Table S5. Target sequences of shRNAs. Related to STAR methods.
Table S6. Primer sequences used for qRT-PCR and ChIP-qPCR. Related to STAR methods.
Data Availability Statement
The datasets generated in this study are publicly available at NCBI GEO (GSE182432) and SRA (PRJNA756046) as of the date of publication.
No codes were generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.