

Summary
Elucidating regulatory relationships between transcription factors (TFs) and target genes is fundamental to understanding how cells control their identity and behavior. Unfortunately, existing computational gene regulatory network (GRN) reconstruction methods are imprecise, computationally burdensome, and fail to reveal dynamic regulatory topologies. Here, we present Epoch, a reconstruction tool that uses single-cell transcriptomics to accurately infer dynamic networks. We apply Epoch to identify the dynamic networks underpinning directed differentiation of mouse embryonic stem cells (ESCs) guided by multiple signaling pathways, and we demonstrate that modulating these pathways drives topological changes that bias cell fate potential. We also find that Peg3 rewires the pluripotency network to favor mesoderm specification. By integrating signaling pathways with GRNs, we trace how Wnt activation and PI3K suppression govern mesoderm and endoderm specification, respectively. Finally, we identify regulatory circuits of patterning and axis formation that distinguish in vitro and in vivo mesoderm specification.
Keywords: single-cell RNA-seq, gene regulatory network, network inference, directed differentiation, gastrulation, embryoid body, Peg3, Wnt
Graphical abstract
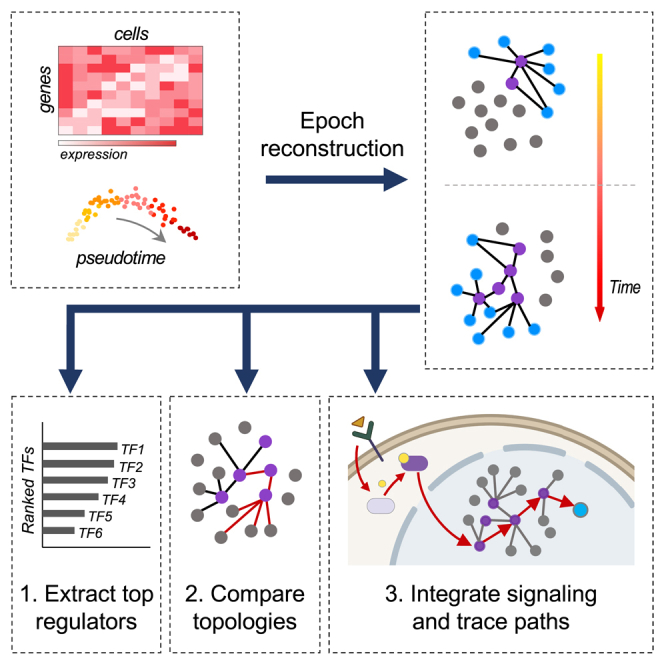
Highlights
-
•
Epoch is a single-cell GRN reconstruction tool that infers dynamic GRNs
-
•
Integrating signaling pathways with GRNs enables tracing activation of genetic programs
-
•
Peg3 rewires the pluripotency GRN to specify mesoderm fate
-
•
Signaling pathways alter GRN topology to bias lineage potential
Cahan and colleagues developed a gene regulatory network reconstruction tool, Epoch, which leverages single-cell transcriptomics to infer and analyze dynamic networks. The authors applied Epoch to uncover the networks underlying in vitro mESC-directed differentiation and compared these to their in vivo counterparts. This revealed signaling-induced network topology shifts that biased lineage potential.
Introduction
Gene regulatory networks (GRNs) model the regulatory relationship between a set of regulators, or transcription factors (TFs), and their target genes. The topology of these networks, defined by edges that map regulatory interactions between TFs and targets, offers a molecular-level view of a controlled system in which genes work together as part of a framework to accomplish specific cell functions (Karlebach and Shamir, 2008; Le Novère, 2015). Uncovering the topology of these GRNs is fundamental in answering a number of questions, including understanding how cellular identity is maintained and established (Davidson and Erwin, 2006), elucidating mechanisms of disease caused by dysfunctional gene regulation (Morgan et al., 2020; Qin et al., 2019), and finding novel drug targets among others (Carro et al., 2010). In the context of cell fate engineering, the mapping of GRNs would enable the identification of TFs required to activate the expression of target genes so as to control cell fate transitions or cell behavior (Cahan et al., 2021; Rackham et al., 2016). Unfortunately, how best to map these relationships remains both an experimental and a computational challenge.
Experimental approaches, including chromatin immunoprecipitation sequencing (ChIP-seq), have identified regulatory targets and TF binding site motifs in certain cell lines and cell types. Similarly, chromatin accessibility assays that detect TF-binding site (TFBS) footprints, such as assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) (Buenrostro et al., 2013), enable the inference of regulatory networks. However, these approaches are limited in scope (e.g., ATAC-defined networks are limited to TFs with known motifs) and scalability (e.g., it is infeasible to perform ChIP-seq for all TFs in all cell types). Therefore, computational methods to infer GRNs are needed. Methods that leverage advances in data collection and machine learning to enable the statistical inference of GRNs via gene expression data include those based on information theory (Faith et al., 2007; Margolin et al., 2006; Meyer et al., 2007), ensemble learning (Huynh-Thu et al., 2010), Bayesian theory (Hartemink, 2005; Yu et al., 2004), and ordinary differential equations (ODEs) (di Bernardo et al., 2005). Unfortunately, the tools developed to date suffer from several drawbacks, including low precision and sensitivity. The leading contributors to poor performance include difficulty in distinguishing direct from indirect interactions (Marbach et al., 2010), the confounding effects of Simpson’s paradox (Trapnell, 2015), and the fact that bulk derived data does not offer perturbation sufficient to detect regulatory relationships (Stark et al., 2003). Attempts to ameliorate these by aggregation across methods have achieved modest success (Marbach et al., 2012).
With the advent of single-cell RNA-seq (scRNA-seq) (Klein et al., 2015; Macosko et al., 2015; Zheng et al., 2017), computational techniques have emerged that try to take advantage of the resolution offered by single-cell transcriptomics to infer GRN structures (Aibar et al., 2017; Matsumoto et al., 2017; Qiu et al., 2020). To date, these methods suffer from limitations similar to those of their bulk counterparts, including low precision and sensitivity (Chen and Mar, 2018) and high computational burden, which limits users’ ability to hone analysis through iterative application (Bonnaffoux et al., 2019). In addition to higher resolution and access to more perturbations, single-cell data allow for computational modeling of dynamic processes, such as differentiation, by ordering cells along a trajectory following linear or more complex graph structures (Haghverdi et al., 2016; Qiu et al., 2011; Street et al., 2018; Trapnell et al., 2014). While many new scRNA-seq GRN methods use pseudotemporal analysis to aid in reconstruction, they are limited in their ability to explain how network topology evolves over time. We define a dynamic topology as one that changes the edge-level regulatory relationships between TFs and targets over time, which thereby increases the number of reachable cell states (Figure 1). This implies that for a given TF-target relationship, the modulation of the TF in a particular context or time point would lead to changes in target expression, but modulation of the same TF in another context or time point would not lead to changes in target expression. The dynamic and noncommutative nature of regulatory networks permits independent control of genetic programs that would otherwise be simultaneously activated with non-sequential, or combinatorial, logic (Letsou and Cai, 2016). Such shifts in network topology can arise for several reasons, including the presence or absence of co-factors, epigenomic modifications, or changes in chromatin accessibility. Thus, for GRNs to more accurately model the emergence of distinct cell fates, they must encapsulate this dynamic behavior of changing GRN topology. Moreover, because GRN topology dictates how a cell responds to perturbation, uncovering the dynamic GRN aids in understanding how the landscape of reachable cell states changes over time, which has implications in the quest to engineer cell fate.
Figure 1.

Effector activity on static and dynamic networks
Given a static GRN, manipulating signaling activity may result in an effector guiding cell state via two possibilities: directly, the effector may regulate a set of fate-specific genes, or indirectly, the effector may regulate a set of TFs that further regulate the same fate-specific genes. Both cases result in the activation of the same program. Alternatively, given a dynamic network, the state space of possible cell fates increases since the result of effector activity is dependent on a changing network topology. As a result, multiple sets of genetic programs may be regulated through the same signaling mechanism. Furthermore, these shifts in GRN topology maybe be induced by the signaling activity itself.
For those reasons, we developed a computational GRN reconstruction tool called Epoch, which uses single-cell transcriptomics to efficiently reconstruct dynamic networks. There are several features that distinguish Epoch from other methods: first, reconstruction is limited to dynamically expressed genes. Second, Epoch uses an optional “cross-weighting” strategy to reduce false positive interactions. Third, Epoch divides pseudotime into epochs, or discrete time periods, extracts a dynamic network, and predicts the most influential regulators in driving topology changes. Finally, Epoch includes a number of functionalities to aid in network analysis and comparison, including the integration of GRNs with major signaling pathways and subsequent tracing through the GRN of the shortest paths from signaling effectors to selected target genes. We compared the performance of Epoch to commonly used computational GRN reconstruction tools using synthetically generated data, in vivo mouse muscle development data, and in vitro directed differentiation data. To demonstrate the utility of Epoch, we applied it to mouse embryonic stem cells (ESCs) undergoing directed differentiation to measure the extent to which signaling pathways influence cell fate by altering GRN topology rather than by direct regulation of pathway effectors (Figure 1).
Results
Epoch relies on single-cell analysis techniques to infer dynamic network structures
Epoch takes as minimum input processed single-cell transcriptomic data and pseudotime or equivalent annotation from any trajectory inference method (Figure 2A). Its first step is to limit reconstruction to dynamically expressed genes to focus on genes playing an active role in cell state changes. To accomplish this, Epoch models gene expression across pseudotime using a generalized additive model. Upon filtering the data for dynamically expressed genes, Epoch reconstructs an initial static network via a CLR-like (Context Likelihood of Relatedness) method, using either Pearson correlation or mutual information (MI) to infer interactions (Faith et al., 2007).
Figure 2.

Epoch workflow and benchmarking
(A) Epoch relies on a 3-step process to reconstruct dynamic network structures: (1) extraction of dynamic genes and CLR, (2) application of cross-weighting, and (3) extraction of the dynamic network.
(B) BEELINE benchmarking on synthetic data. Epoch using mutual information (MI) and Pearson, denoted by “MI” and “P,” respectively. “X” indicates the use of cross-weighting. The red line indicates the median AUPR of the best-performing method, Epoch using MI + cross-weighting on size 5,000 cell datasets. Kruskal-Wallis p < 2.2 × 10−16. See also Figures S2 and S3.
In an optional step, which we have called “cross-weighting,” Epoch next refines the network via a cross-correlation-based weighting scheme. This helps to reduce false positives that may result from indirect interactions or that represent non-logical interactions, ultimately improving precision (Figure S1A). In this step, the expression profiles over pseudotime for each TF-target pair are aligned and progressively shifted to determine an average offset value at which maximum correlation is achieved between the two profiles. A graded-decline weighting factor is computed based on this offset, and it is used to negatively weight interactions that are less likely to be true positives.
Once a static network has been inferred, Epoch extracts a dynamic network. This begins with Epoch breaking down pseudotime into epochs, based on pseudotime, cell ordering, k-means or hierarchical clustering, sliding window similarity, or user-defined assignment. With the exception of user-defined assignment, all methods of defining epochs are, to some extent, automated, with epoch definitions learned directly from the data. Genes are assigned to epochs based on their activity along pseudotime. Epoch then fractures the static network into a dynamic one, composed of “epoch networks,” representing active interactions within a particular epoch, and “transition networks,” describing how an epoch network transitions into a subsequent epoch network.
As Epoch was initially built with the improvement of cell fate engineering protocols in mind, we included in the framework network analysis and comparison capabilities. For example, Epoch will identify influential TFs within a given static or dynamic network by extracting top regulators that appear to have the most influence on network state. Specifically, Epoch ranks TFs by PageRank (Brin and Page, 1998) in the context of their epoch networks, and will also look for TFs that simultaneously score high in betweenness and degree centralities as compared to all of the other TFs. In addition, Epoch can integrate major signal transduction pathways with reconstructed networks. This can be used to determine the shortest paths through the networks from pathway effectors to groups of specified target genes, allowing users to identify topologies capable of activating or repressing specific groups of genes.
Finally, Epoch is modularly designed such that it can be broken into individual steps and flexibly merged and used with any trajectory inference, network reconstruction, and network refinement tools.
To assess the performance of Epoch in reconstructing static networks, we benchmarked it against multiple variations of CLR (Faith et al., 2007) and GENIE3 (Huynh-Thu et al., 2010) (including the original methods themselves and variations in which the methods were embedded within the Epoch framework) on in vivo, in vitro, and in silico datasets (Figure S2; Note S1). Our results indicated that key steps in the Epoch framework improved overall static network reconstruction, as limiting reconstruction to dynamically expressed genes and applying the cross-weighting scheme both led to increased fold improvement in area under the precision-recall curve (AUPR) over random. We further compared Epoch to recently developed GRN reconstruction methods designed for single-cell data. We used the benchmarking platform BEELINE (Pratapa et al., 2020) to assess the performance of Epoch against 11 other single-cell methods across synthetic and curated datasets (Figures 2B and S3). Our results demonstrated that Epoch with cross-weighting outperformed all other methods based on AUPR and execution time.
scRNA-seq of early in vitro mouse ESC-directed differentiation
With the goal of exploring the dynamic GRN topology underlying lineage specification in gastrulation, we collected scRNA-seq data from days (d) 0 through 4 of in vitro mESC-directed differentiation guided by four separate treatments encouraging primitive streak formation (Figure 3A). Briefly, cells were allowed to differentiate in serum-free differentiation media before being treated with 1 of 4 treatments on d2: Wnt3a and activin A alone (WA), or with one of Bmp4 (WAB), GSK inhibitor (WAG), or Noggin (WAN). Multiple rounds of differentiation were staggered such that we could harvest samples representative of d0–d4 at one time for sequencing using the MULTI-seq protocol (McGinnis et al., 2019). After barcode classification, samples were preprocessed using the SCANPY pipeline (Wolf et al., 2018), and RNA velocity analysis was performed (Bergen et al., 2020; La Manno et al., 2018) (Figures 3B and 3C).
Figure 3.

mESC in vitro-directed differentiation
(A) Directed differentiation protocol. Cells were harvested from d0 through d4 samples for MULTI-seq.
(B and C) Clustering (B) and (C) RNA velocity for MULTI-seq data.
(D) Select marker gene expression. See also Figure S4C.
(E) Cell populations by treatment.
(F) Quantification of cell fate distribution based on treatment comparing mesendoderm and neuroectoderm, endoderm and mesoderm, cluster ‘7,0′ and ‘7,1′ along endoderm fate, and cluster distribution along neuroectoderm fate.
We detected three distinct lineages, neuroectoderm (based on Sox1 expression), mesoderm (Mesp1), and endoderm (Foxa2), a result confirmed by comparison to gastrulation data with SingleCellNet (SCN) (Tan and Cahan, 2019) (Figures 3D and S4; Table S1; Note S2). RNA velocity additionally supported our cluster annotations. We found that the majority of the differentiating cells transitioned toward the neuroectoderm (clusters ‘0,2′, ‘0,3′, 4, 2, 5, 9), with smaller populations transitioning toward mesoderm (cluster 8) and endoderm (clusters ‘7,1′ and ‘7,0′).
We observed differences between induction treatments in terms of the distribution of cells among the different populations (Figures 3E and 3F). Notably, cells treated with WAG exhibited a stunted trajectory toward neuroectoderm but a fuller trajectory toward mesoderm. This was in contrast to cells treated with the remaining three treatments (WAB, WAN, and WA), in which very few cells differentiated toward mesodermal fate, but instead exhibited strong neuroectodermal or endodermal commitment. Furthermore, while WAB and WA cells tended to transition into cluster ‘7,0′ (furthest along endodermal development), WAN and WAG cells were more likely to stay in cluster ‘7,1′.
We applied Epoch to reconstruct the networks underlying the data, using latent time (Bergen et al., 2020) to order cells. Latent time was divided into three epochs, a dynamic network was extracted for each lineage, and top regulators were extracted via PageRank and Betweenness-Degree (Figure S5; Table S2; Note S3).
Peg3 is a central regulator in mesodermal WAG networks
We next asked why WAG-treated cells had the greatest propensity for mesoderm fate. We hypothesized that treatment-specific GRNs underpinned differences in fate potential among treatments. To explore this hypothesis, we reconstructed and compared dynamic networks for each treatment along the mesodermal path (which we refer to as the treatment-specific networks: WAG, WAB, WAN, and WA networks), and we compared these against the full reconstructed mesoderm network, which is reconstructed from all cells regardless of treatment, and which we refer to as the mesoderm network.
We sought to identify regulators that drive differences in mesoderm reachability between the treatments. We performed community detection on epoch 2, epoch 3, and their transition of the mesoderm network and identified distinct TF communities, or modules (Figures 4A and S6A). We assessed the activity of each module by looking at the average expression of member genes across latent time for each treatment (members predicted to be repressed were not included in the average so as not to improperly depress this measure of activity) (Figures 4B and S6B). At least three modules exhibited strong activation in the WAG treatment, but were inactive or weakly activated in the remaining three treatments. The TFs in these modules were Peg3, Lhx1, Hoxb2, Foxc1, Tshz1, Meis2, Mesp1, Tbx6, Foxc2, Prrx2, Meox1, and Notch1. Upon extracting the differential network, defined as the network containing the interactions that are more specific to a treatment network by edge weight (see supplemental experimental procedures), between WAG and WA, we identified the top differential regulators of these TFs themselves (Figures 4C and S6C). Interestingly, Peg3 was an upstream regulator of the majority of these TFs in the WAG treatment, but had a negligible role in the WA treatment.
Figure 4.

Mesodermal network analysis
(A) The epoch 3 subnetwork of the mesodermal dynamic GRN. TFs are colored by community and faded by betweenness. Blue and red edges represent activating and repressive edges, respectively.
(B) Average community expression over time by treatment along the mesodermal path. Communities shown (each row) are from the epoch 2 subnetwork (yellow), transition (green), and epoch 3 subnetwork (aqua).
(C) The epoch 3 differential network between WAG and WA. Interactions in this network represent edges present in the WAG mesodermal network that are not present in the WA mesodermal network. TFs are colored by community and faded by betweenness. Blue and red edges represent activating and repressive edges, respectively.
(D) Peg3 and Dnmt3a expression in a sampled portion of relevant cell types in gastrulation data adapted from Grosswendt et al. (2020). Peg3 ANOVA p < 2 × 10−16, Dnmt3a ANOVA p < 2 × 10−16.
The central influence of Peg3 in the WAG network implied a pathway linking glycogen synthase kinase 3 (GSK3) inhibition, Peg3 expression, and mesodermal gene expression. GSK3 contributes to DNA methylation at imprinted loci, including Peg3, in mESCs via Dnmt3a (Meredith et al., 2015), potentially mediated by N-Myc (Popkie et al., 2010). Consistent with a model whereby GSK3 maintains repressive methylation of Peg3 via Dnmt3a activity, we found that Dnmt3a and Peg3 expression were largely mutually exclusive, and there was low-to-no Dnmt3a expression in the WAG-treated cells (Figure S6D). This phenomenon occurs in vivo, too, as we found a relatively high expression of Dnmt3a in epiblast cells, which was tapered in primitive streak and mesodermal cells (Figure 4D) from a single-cell mouse gastrulation dataset (Grosswendt et al., 2020). In contrast, Peg3 expression was low in epiblast and increased in the primitive streak, mirroring our in vitro data, and suggesting that Peg3 may have a role in orchestrating the exit of pluripotency and specification of mesodermal fate in vivo.
Tracing signaling cascades to germ layer transcriptional programs
We next sought to trace pathways from signaling effectors to the activation of mesodermal fate. To determine which signal transduction pathways were active and when along the mesodermal trajectory, we computed the average expression of targets of 18 signaling effector TFs (available as part of the Epoch framework) across latent time broken down by treatment. As expected, targets of the Wnt effector Lef1 were activated early in WAG, whereas targets of the transforming growth factor-β (TGF-β)/bone morphogenetic protein (BMP) effector Smad4 were activated to the highest extent and longest duration in WAB (Figure 5A). Notch targets were activated in WAB, WAN, and WA, but not in WAG, along the mesodermal lineage (and weakly activated along the neuroectodermal lineage), consistent with the role of Notch in specifying neuroectodermal fate in the neuroectoderm-mesendoderm fate decision (Androutsellis-Theotokis et al., 2006; Aubert et al., 2002).
Figure 5.

Integrating signaling effectors and tracing paths to target genes
(A) Average expression of targets of signaling effectors by treatment over time. Pathway and effector are labeled on each row.
(B) Shortest path analysis showing length of shortest paths from Wnt effector targets (each row) to mesodermal target genes (each column) within treatment-specific networks. Path lengths are normalized against average path length within a network. Blue and red indicate paths that activate and repress the target gene, respectively. If such a path does not exist, then it has length of infinity and is therefore white.
(C) The epoch 3 subnetwork of the endodermal dynamic network. TFs are colored by community and faded by betweenness. Blue and red edges represent activating and repressive edges, respectively.
(D) Shortest path analysis showing length of shortest paths from PI3K suppression (targets of Foxo1 activation) (each row) to endodermal genes (each column) within treatment-specific networks. As before, path lengths are normalized against average path length within a network. Blue and red indicate activation and repression of the target gene, respectively. If a path does not exist, it has length of infinity and is therefore white.
Because Wnt signaling is essential for mesodermal fate, we looked for paths connecting Wnt effectors to the TFs in the WAG-specific modules defined previously. We computed the shortest paths from targets of Wnt effectors to the module TFs in each treatment-specific dynamic network (using edge lengths inversely proportional to the cross-weighted score) (Figure 5B), finding that no paths from Wnt to many of the module TFs existed in WAB, WAN, and WA.
Our signaling effector target analysis also revealed increased Foxo1 activity in WAB-, WAN-, and WA-treated cells as compared to WAG-treated cells, indicating the suppression of phosphatidylinositol 3-kinase (PI3K) signaling along the WAB, WAN, and WA trajectories (Figure 5A). The establishment of definitive endoderm (DE) requires the suppression of PI3K signaling (McLean et al., 2007; Yu et al., 2015), and in induced pluripotent stem cell (iPSC)-DE differentiation Foxo1 binds to DE-formation-related genes and its inhibition impedes DE establishment (Nord et al., 2020). We therefore hypothesized that a second fate choice between mesodermal and endodermal fates further exacerbated the uneven mesodermal fate preference between the treatments. Our analysis indicates that in the mesodermal-endodermal fate choice, the majority of WAG-treated cells differentiate toward the mesoderm in contrast to WAN and WA in which cells preferentially differentiate toward DE. WAB-treated cells exhibited a mixed potential split roughly equally along the two paths.
We sought to elucidate a possible explanation for the role of Foxo1 in this fate choice by searching for the shortest paths toward a set of endodermal genes. Specifically, similar to the previous mesodermal analysis, we applied community detection to the full endodermal network and identified the cluster containing Sox17 and Foxa2, known master regulators of endodermal fate (Figure 5C). This cluster included the TFs Sox17, Foxa2, Bmp2, Cited1, Foxa1, Gata6, Hhex, Lhx1, and Tcf7l2. For each treatment-specific network, we computed the shortest paths from Foxo1 targets to these genes (Figure 5D). We found that many of these regulators, including both Sox17 and Foxa2, were not reachable from Foxo1 in the WAG network, consistent with the observation that WAG-treated cells preferentially differentiated toward mesodermal fate over endodermal fate. Interestingly, Foxa2 was not reachable in the WAN network, although Sox17 was. We believe this may explain the differences in the distribution of cells between the endodermal clusters ‘7,1′ and ‘7,0′. Of the cells that specify endodermal fate, WA- and WAB-treated cells more fully transitioned into cluster ‘7,0′. In contrast, WAN-treated cells remained mostly in cluster ‘7,1′ and were less likely to differentiate further into ‘7,0′ . Previous studies in directed differentiation of human ESCs toward DE have implicated a regulatory role of Sox17 in establishing DE to be upstream of Foxa2, the loss of which impairs foregut and subsequent hepatic endoderm differentiation (Genga et al., 2019). This is consistent with our cell-type annotations of cluster ‘7,1′ (definitive endoderm) and ‘7,0′ (gut endoderm), and offers a possible mechanism of the discrepancy in cell fate preferences between treatments. These results demonstrate that WAB, WAN, and WA networks do not allow for the activation of mesoderm programs from their cognate effectors, but instead assume topologies that are preferential for endodermal fate, implying profound structural changes in the networks that underlie the treatment-specific differences we see in cell fate. Furthermore, this analysis provides a basis for identifying which signaling pathways must be targeted for directing certain fate transitions—for example, through an exhaustive search of effector targets.
TF-target gene differences among treatments illuminate rewiring of network topology by signaling pathways
Given the apparent restructuring of GRN topology by signaling activity, we aimed to understand the extent to which network-wide topology changes were responsible for differences in cell fate potential between treatments. To answer this, we performed two analyses focusing on the differences in targets of TFs across the four treatment networks.
First, we asked whether TFs actively expressed in all four treatments regulated the same set of target genes. To this end, we focused on the treatment-specific networks along the mesodermal path, and narrowed our analysis to the 72 TFs that were active in all 4 treatments during d3 or d4. We quantified the overlap between the targets of each TF in a pairwise comparison of the treatments using the Jaccard similarity (Figure 6A). As a baseline, we implemented a bootstrapping method in which we reconstructed 10 networks for each treatment (using 400 sampled cells each); for each TF, we calculated the average Jaccard similarity of its targets among pairwise comparisons of the reconstructed networks within a treatment. This gave us a baseline, or expected, Jaccard similarity for each TF in each treatment. Overall, we found that overwhelmingly, target differences between treatments were significantly greater than the baselines, indicating vast network topology differences among the treatment networks.
Figure 6.

Similarity of targets of TFs in different treatments
(A) Jaccard similarity of targets of 72 TFs. The targets of each TF in a treatment are compared to those in the other 3 treatments (light blue). As a baseline (turquoise), targets of each TF in bootstrapped reconstructed networks within a treatment are pairwise compared. All pairs p < 2 × 10−16.
(B) The 13 TFs selected for GSEA and their average pairwise Jaccard similarity among the treatment-specific networks.
(C) Summarized results of performing GSEA on targets of TFs in (B). Heatmap shows rankings (top 10 shown) based on the frequency a term is considered enriched among the 13 TFs (1 = most frequently enriched term).
Second, we asked whether these differences in predicted targets had functional consequences in affecting fate potential. To this end, we focused on the 13 TFs among the 72 mentioned above that exhibited the largest differences in predicted targets between treatments (Figure 6B). For each treatment, we performed gene set enrichment analysis (GSEA) on the predicted targets of these TFs (Figure 6C). In line with our observations, we found enrichment for heart development and mesoderm formation in only the WAG treatment, despite the fact that the networks we analyzed were reconstructed along the mesodermal path. Our results are consistent with previous literature that hinted at environment-based differences in the fate potential of mESCs in that Esrrb/Nanog double knockdown impedes self-renewal in 2i alone but not in the presence of 2i and leukemia inhibitory factor (LIF) (Dunn et al., 2014). These results support our hypothesis that manipulating signaling activity results in a topological restructuring of the GRN, ultimately guiding cell fate potential.
Patterning and neuroectoderm programs drive differences between in vivo and in vitro gastrulation and mesoderm specification
Finally, we aimed to understand the extent to which our in vitro mESC-derived mesodermal cells established a GRN resembling that of their in vivo counterparts. To this end, we sampled 250 cells from previously published gastrulation stage embryos for each of 4 annotated populations that corresponded to our in vitro mesoderm lineage populations: Epiblast, Primitive streak early, Primitive streak late, and Mesoderm presomitic. We then used Epoch to reconstruct an in vivo dynamic network from these sampled data. After reconstruction, we extracted the top regulators in each epoch (Figure 7A). Of the 22 unique top regulators in epochs 2 and 3, at least 17 have known roles in guiding exit from pluripotency, mesoderm specification, or somitogenesis (Note S4), corroborating the usefulness of Epoch in identifying important TFs driving dynamic processes. Of note, Peg3 ranked highly in epoch 2 of the in vivo network, bolstering our earlier prediction of Peg3 as an orchestrator of the pluripotent-to-mesodermal fate transition in vitro and in vivo.
Figure 7.

Comparison to in vivo gastrulation and mesoderm specification
(A) Top regulators of each epoch in the in vivo mesoderm network as predicted by PageRank and Betweenness-Degree.
(B) Shortest path analysis showing length of shortest paths from Wnt effector targets (each row) to mesodermal target genes (each column) in the in vivo network. Path lengths are normalized against average path length in the network. Blue and red indicate paths that activate and repress the target gene, respectively. If such a path does not exist, then it has length of infinity and is white.
(C) Top 10 enriched terms for in vivo- and in vitro-specific modules based on the Combined Score from Enrichr GSEA analysis. See also Figure S7.
Substantial differences between top regulators of the in vivo and in vitro networks suggested broader topology differences. Thus, we further applied the signaling pathway tracing analysis to the in vivo network. Analogous to the in vitro networks, we looked for paths connecting Wnt effectors to the same TFs in the in vitro WAG-specific modules (Figure 7B). As expected, all of the TFs were reachable. Furthermore, Wnt activation and these target TFs were highly connected, with paths existing from almost every effector target to every TF, implying the existence of robust and coordinated control over this mesoderm module.
Finally, we sought to directly compare the topological and functional differences between the in vivo and in vitro mesoderm networks. To directly compare topological differences, we applied a threshold to both networks, keeping the top 2% of non-zero-weighted edges in each. We then extracted the differential network corresponding to the in vivo-specific interactions and the differential network corresponding to the in vitro-specific interactions (data not shown). We performed community detection on both and measured the activity of each resulting module by assessing the average expression of member genes across time in the in vivo and in vitro data (Figures S7A and S7B).
Of the in vivo-specific modules, three had low or insignificant activation in the in vitro data, despite being strongly activated in the in vivo data. To understand the functional consequences of this, we applied GSEA to each of these modules (Figures 7C and S7C). Two modules showed enrichment for multiple pathways related to patterning and axis specification. Thus, a large but not unexpected difference between the in vivo and in vitro mesoderms is the lack of activation of patterning programs in the in vitro ESC-derived mesodermal cells.
Conversely, of the in vitro-specific modules, we isolated those that had low or insignificant activation in the in vivo data but were strongly activated in the in vitro data. Within the first epoch, three communities satisfied this criterion. We hypothesized that differences in this early epoch could drive fate differences between the two networks at a later time. GSEA on these modules revealed that one was enriched for the positive regulation of stem cell proliferation and the regulation of mRNA splicing, while another was enriched for terms related to fluid shear stress, DNA methylation, and male gonad development (Table S3). Meanwhile, GSEA on in vitro-specific modules from the second and third epochs revealed the underlying activation of neural-related programs in the in vitro ESC-derived mesoderm (Figures 7C and S7D). Most of the modules were enriched for neuroectoderm processes, implying that the in vitro differentiated cells failed to completely inhibit the default neuroectoderm lineage and instead retained a network topology capable of activating at least portions of neuroectoderm programs. This aligns with our observation that the majority of the in vitro-differentiated cells tended toward the neuroectoderm lineage. Our results suggest that to more efficiently produce mesodermal cells, emphasis should be placed on disrupting network module topologies responsible for neural programs.
Discussion
Here, we presented a GRN reconstruction tool, Epoch, which leverages single-cell transcriptomics and efficiently infers dynamic network structures. We show that it outperforms methods designed for bulk and single-cell GRN reconstruction in both synthetically generated and real-world datasets. It is computationally efficient, facilitating the optimization of network topology or iterative simulations of changes in network structure. Finally, the utility of Epoch is enhanced by its flexibility; there are no strict requirements on the flavor of pseudotemporal input, and its workflow is structured in discrete steps, allowing users to pick and choose or substitute portions of the workflow, making it straightforward to incorporate emerging analysis techniques into the framework of Epoch.
To demonstrate the practical utility of Epoch, we applied it to scRNA-seq data from d0 through d4 in in vitro mESC-directed differentiation undergoing 4 treatments (WAG, WAB, WAN, and WA). This analysis revealed several topological features that are likely to play important roles in specifying germ layer fate during directed differentiation.
First, we identified a set of Peg3-controlled TF modules that promote mesoderm emergence, which were preferentially activated in WAG. This, coupled with prior data linking GSK3 to the regulation of imprinted genes (Meredith et al., 2015), preferential expression of Peg3 in mesoderm and somites (Kuroiwa et al., 1996), and the inhibitory influence of Peg3 on pluripotency and reprogramming (Theka et al., 2017), implicate Peg3 as a candidate to improve the efficiency of directed differentiation toward mesodermal fates.
Second, by integrating Epoch-reconstructed dynamic GRNs with signaling pathways, we were able to trace paths from signaling effectors through GRNs to the regulation of germ-layer-specific gene batteries. This revealed not only direct targets of signal pathways but also broad network remodeling that altered accessibility to targets, thereby honing the capacity and potential for distinct fates. Specifically, we found that paths from Wnt signaling effectors to mesodermal programs were confined to the WAG network. Similarly, we found condition-specific paths between the suppression of PI3K signaling, subsequent Foxo1 activation, and endoderm specification. Collectively, these analyses supported our hypothesis that signaling-induced topological differences altered the cell fate landscape, resulting in distinct propensities for each germ layer. The nature of these topological differences remains unclear. One possibility is direct alteration of the epigenomic state, and thus GRN, by signaling pathways. We point to the reported effect of GSK over Peg3 methylation as an example of such a phenomenon. Further studies to elucidate differences in epigenomic states between mESCs undergoing directed differentiation by modulation of distinct signaling pathways will aid in clarifying whether such a mechanism is in play.
Finally, we explored how in vivo versus in vitro mesoderm specification GRNs compare, revealing two broad GRN differences. First, the in vivo GRN established patterning and axis-specification programs that were lacking in the in vitro network, consistent with incomplete recapitulation of the self-organization present in in vivo gastrulation (ten Berge et al., 2008; Glykofrydis et al., 2021). Second, the in vitro GRN retained a topology partially favorable for neuroectoderm development, suggesting that the cells undergoing in vitro-directed differentiation incompletely suppressed the neuroectoderm fate. Overall, this analysis suggests that guiding cells toward a more faithful mesodermal fate at a higher rate will likely require steps to disrupt the retention of neuroectoderm-favoring networks while promoting the establishment of appropriate patterning-related programs.
Our results ultimately suggest a model of differentiation that is driven by the activation of genetic programs and honed by network topology changes. In other words, signaling-induced restructuring of GRNs alters the fate potential landscape, which allows for independent control of multiple genetic programs and thereby increases the diversity of reachable cell states. In the case of mESC-directed differentiation, GRN topology clearly restricted the potential of cells toward specific fates, resulting in different propensities for mesoderm, endoderm, and neuroectoderm depending on treatment. Finally, while it may be the case that both signaling activities described (i.e., the effector regulation of distinct targets and the restructuring of network topology) occur concurrently, it is ultimately difficult to resolve their relative timing.
The methodology presented here, available in the R package Epoch, is broadly applicable to any biological question that benefits from uncovering dynamic regulatory networks, their comparisons, and their interfacing with signaling pathways. In particular, Epoch can be used to understand cell fate transitions at branch points in lineage trajectories, to uncover key regulators driving these decisions, and to trace paths from signal transduction events to the activation or repression of transcriptional programs. Such an approach provides a powerful strategy to not only elucidate dynamic, multiscale processes in development but also to identify signaling pathways to modulate for the purposes of directing cell identity transitions in vitro.
Experimental procedures
Epoch workflow
Epoch takes as input processed (normalized and log-transformed) single-cell transcriptomic data and accompanying pseudotime (or equivalent) annotation. The Epoch workflow is based on three strategies: (1) the extraction of dynamically expressed genes and subsequent static reconstruction with CLR, (2) network refinement using a cross-correlation based strategy called cross-weighting, and (3) the extraction of a dynamic network and the subsequent identification of top regulators.
Identification of dynamically expressed genes
Limiting network reconstruction to dynamically expressed genes serves two purposes. First, it focuses the network on interactions that more likely play a role in the observed biological process. Second, it reduces the instances of false positive interactions and improves precision by limiting possible edges between temporally variable genes. To select for such genes, Epoch models individual genes across the annotated pseudotime using a generalized additive model (GAM). Specifically, Epoch uses the ‘gam’ package (Gaussian family, LOESS smooth pseudotime) to fit a model for each gene using the backfitting algorithm. Genes are considered dynamic based on the significance of the smooth term (pseudotime). Alternatively, users can specify finding dynamic genes via TradeSeq (Van den Berge et al., 2020), a recently developed tool that identifies dynamic changes in gene expression via a GAM based on the negative binomial distribution.
Cross-weighting
After inferring an initial network structure, Epoch can apply cross-weighting. The objective of this step is to negatively weight edges in the initial network that are unlikely to be true interactions, which may, for example, be representative of indirect interactions. To this end, for every TF-target pair in the initial network, Epoch computes cross-correlation across a given lag time (defaults to one-fifth of total pseudotime). After ordering the lag times by decreasing correlation, Epoch computes an offset value in which maximum correlation is achieved, defined by default as the top one-third of the ranked lag times. Finally, Epoch scores the offset values of each interaction:
where the offset, x, is computed as described, and maximum and minimum windows can be altered by the user if desired. Z-scores from the initial network are weighted accordingly, ultimately filtering out false positives. At this step, Epoch will return an appended GRN table, including the offset value and new weighted score for each interaction.
Default parameters for cross-weighting are specified within Epoch, and were chosen based on empirical improvements in performance across synthetic and real datasets (ranging in number of genes, number of cells, and simple trajectory types). For example, we found that optimal lag time varied to some extent with dataset size, with larger datasets requiring a larger lag time to catch target response to regulator expression changes (Figure S1E). Smaller lag times tended to correlate with decreased AUPR, likely because the lag time was not sufficient to catch the point of maximum cross-correlation. AUPR also begins to gradually decrease at larger lag times, although this effect is much less pronounced. We found that our default lag (set to one-fifth of the dataset size) resulted in high AUPR across various datasets. Similarly, we also examined various minimum and maximum windows and found that our default values resulted in optimal AUPR (Figure S1F) across datasets of varying sizes. However, users may want to modify these parameters to increase or decrease the leniency of the weighting for a number of reasons, such as when applying Epoch to more complex trajectories with large numbers or more complex state changes (and, correspondingly, a large number of epochs). In this case, we would recommend finding optimal lag and window in an analogous method. Specifically, this would entail designing a modular network (and corresponding GRN) representative of the more complex trajectory, using this network to simulate synthetic datasets, and performing a parameter sweep to optimize reconstruction AUPR. Synthetic network design and simulation for both simple and complex trajectories can be done via platforms such as Dyngen (Cannoodt et al., 2021).
Dynamic network extraction
Epoch will extract a dynamic network from the reconstructed static network. Specifically, the process begins by breaking pseudotime into epochs. A number of options are available to users to accomplish this. Briefly, Epoch can define the epochs based on pseudotime, equal cell ordering (resulting in equal number of cells per epoch), k-means or hierarchical clustering, sliding window similarity, or user-defined manual assignment. With the exception of user-defined assignment and sliding window similarity, the number of epochs is specified by the user, and can be determined by examining the heatmap of gene expression across pseudotime and estimating the rough number of expression states represented in the data. We recommend smoothing the data to aid in heatmap visualization, which can be done through Epoch. Alternatively, it is unnecessary to supply the number of epochs if using the similarity method, which will automatically detect epochs based on correlation between groups of cells along a sliding window across pseudotime. After epochs are defined, Epoch will assign genes to epochs, based on their activity along pseudotime. In brief, this is either based on activity (i.e., genes active in any epochs will be assigned to those epochs) or on differential expression (i.e., genes are assigned based on whether they are differentially expressed in an epoch). Specifically, if genes are assigned by activity, Epoch will compute, for each gene, a threshold against which average expression of the gene in an epoch is compared. This threshold can be modified by user input. If, instead, genes are assigned by differential expression, a p-value threshold is used to determine assignments. Finally, “orphan genes,” which we define as dynamically expressed genes that are not assigned to any epoch, are assigned to the epoch in which their average expression is maximum.
Based on these assignments, Epoch will fracture the static network into a dynamic one composed of epoch networks and transition networks. Specifically, an edge between regulator and target gene appears in an epoch network if the regulator is assigned to that epoch. Furthermore, an edge will appear in a transition subnetwork under two conditions: (1) For an activating edge, the target is not active in the source epoch, the target is active in the subsequent epoch, and the regulator is active, or (2) for a repressive edge, the target is active in the source epoch, is not active in the subsequent epoch, and the regulator is active. Following this step, Epoch will return a dynamic network represented by a list of individual GRN tables. This includes the epoch networks or essentially the dynamic network, as well as transition networks that describe how an epoch network may transition into a subsequent epoch network.
Top regulator prediction
Epoch uses two graph theoretic methods to predict the “top regulators,” the TFs that appear to have the most influence in driving changes in or maintaining topology. First, Epoch will rank regulators by weighted PageRank. In brief, the PageRank centrality measures the importance of nodes in a network based on the number and quality of links of which a node is a part. In essence, the most influential nodes are likely to interact with many influential nodes. Second, Epoch will rank regulators by the product of normalized betweenness and normalized degree. Here, the assumption is that the most influential nodes are likely to be traversed by many shortest paths (and thus have high betweenness) and interact with many other nodes (and thus have high degree). In Epoch, PageRank, betweenness, and degree centralities are implemented through the ‘igraph’ package. By default, cross-weighted edge weights are used, but can be further specified by the user. In both top regulator prediction methods, Epoch will return ranked lists of nodes and further specify their corresponding PageRank or normalized betweenness, normalized degree, and betweenness-degree product.
Data and code availability
Epoch is available as a package in R, and code and tutorials can be found at https://github.com/pcahan1/epoch. Data are available at GEO under accession number GEO: GSE177051.
Author contributions
E.Y.S. and P.C. developed Epoch with assistance from J.Y.K., performed analysis, and wrote the manuscript. A.S. performed in vitro differentiation experiments with the assistance of E.Y.S. Q.B. performed the mouse muscle development experiments. P.C. conceptualized and supervised the project.
Conflicts of interest
The authors declare no competing interests.
Acknowledgments
We would like to thank Yuqi Tan, Ray Cheng, Eric Kernfeld, and David Johanson. We also thank Chris McGinnis for help with the MULTI-seq protocol and associated reagents. This work was supported by the NIH under grant R35GM124725 to P.C. and by the NSF Graduate Research Fellowship under grant no. DGE-1746891 to E.Y.S.
Published: January 27, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2021.12.018.
Supplemental information
References
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androutsellis-Theotokis A., Leker R.R., Soldner F., Hoeppner D.J., Ravin R., Poser S.W., Rueger M.A., Bae S.-K., Kittappa R., McKay R.D.G. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823–826. doi: 10.1038/nature04940. [DOI] [PubMed] [Google Scholar]
- Aubert J., Dunstan H., Chambers I., Smith A. Functional gene screening in embryonic stem cells implicates Wnt antagonism in neural differentiation. Nat. Biotechnol. 2002;20:1240–1245. doi: 10.1038/nbt763. [DOI] [PubMed] [Google Scholar]
- ten Berge D., Koole W., Fuerer C., Fish M., Eroglu E., Nusse R. Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell. 2008;3:508–518. doi: 10.1016/j.stem.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berge K., Roux de Bézieux H., Street K., Saelens W., Cannoodt R., Saeys Y., Dudoit S., Clement L. Trajectory-based differential expression analysis for single-cell sequencing data. Nat. Commun. 2020;11:1201. doi: 10.1038/s41467-020-14766-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen V., Lange M., Peidli S., Wolf F.A., Theis F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020;38:1408–1414. doi: 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- di Bernardo D., Thompson M.J., Gardner T.S., Chobot S.E., Eastwood E.L., Wojtovich A.P., Elliott S.J., Schaus S.E., Collins J.J. Chemogenomic profiling on a genome-wide scale using reverse-engineered gene networks. Nat. Biotechnol. 2005;23:377–383. doi: 10.1038/nbt1075. [DOI] [PubMed] [Google Scholar]
- Bonnaffoux A., Herbach U., Richard A., Guillemin A., Gonin-Giraud S., Gros P.-A., Gandrillon O. WASABI: a dynamic iterative framework for gene regulatory network inference. BMC Bioinformatics. 2019;20:220. doi: 10.1186/s12859-019-2798-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brin S., Page L. The anatomy of a large-scale hypertextual web search engine. Computer Networks ISDN Syst. 1998;30:107–117. [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahan P., Cacchiarelli D., Dunn S.-J., Hemberg M., de Sousa Lopes S.M.C., Morris S.A., Rackham O.J.L., Del Sol A., Wells C.A. Computational stem cell biology: open questions and guiding principles. Cell Stem Cell. 2021;28:20–32. doi: 10.1016/j.stem.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannoodt R., Saelens W., Deconinck L., Saeys Y. Spearheading future omics analyses using dyngen, a multi-modal simulator of single cells. Nat. Commun. 2021;12:3942. doi: 10.1038/s41467-021-24152-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro M.S., Lim W.K., Alvarez M.J., Bollo R.J., Zhao X., Snyder E.Y., Sulman E.P., Anne S.L., Doetsch F., Colman H., et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Mar J.C. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinformatics. 2018;19:232. doi: 10.1186/s12859-018-2217-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson E.H., Erwin D.H. Gene regulatory networks and the evolution of animal body plans. Science. 2006;311:796–800. doi: 10.1126/science.1113832. [DOI] [PubMed] [Google Scholar]
- Dunn S.J., Martello G., Yordanov B., Emmott S., Smith A.G. Defining an essential transcription factor program for naïve pluripotency. Science. 2014;344:1156–1160. doi: 10.1126/science.1248882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith J.J., Hayete B., Thaden J.T., Mogno I., Wierzbowski J., Cottarel G., Kasif S., Collins J.J., Gardner T.S. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genga R.M.J., Kernfeld E.M., Parsi K.M., Parsons T.J., Ziller M.J., Maehr R. Single-cell RNA-sequencing-based CRISPRi screening resolves molecular drivers of early human endoderm development. Cell Rep. 2019;27:708–718.e10. doi: 10.1016/j.celrep.2019.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykofrydis F., Cachat E., Berzanskyte I., Dzierzak E., Davies J.A. Bioengineering self-organizing signaling centers to control embryoid body pattern elaboration. ACS Synth. Biol. 2021;10:1465–1480. doi: 10.1021/acssynbio.1c00060. [DOI] [PubMed] [Google Scholar]
- Grosswendt S., Kretzmer H., Smith Z.D., Kumar A.S., Hetzel S., Wittler L., Klages S., Timmermann B., Mukherji S., Meissner A. Epigenetic regulator function through mouse gastrulation. Nature. 2020;584:102–108. doi: 10.1038/s41586-020-2552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L., Büttner M., Wolf F.A., Buettner F., Theis F.J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods. 2016;13:845–848. doi: 10.1038/nmeth.3971. [DOI] [PubMed] [Google Scholar]
- Hartemink A.J. Reverse engineering gene regulatory networks. Nat. Biotechnol. 2005;23:554–555. doi: 10.1038/nbt0505-554. [DOI] [PubMed] [Google Scholar]
- Huynh-Thu V.A., Irrthum A., Wehenkel L., Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS One. 2010;5:e12776. doi: 10.1371/journal.pone.0012776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlebach G., Shamir R. Modelling and analysis of gene regulatory networks. Nat. Rev. Mol. Cell Biol. 2008;9:770–780. doi: 10.1038/nrm2503. [DOI] [PubMed] [Google Scholar]
- Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D.A., Kirschner M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroiwa Y., Kaneko-Ishino T., Kagitani F., Kohda T., Li L.L., Tada M., Suzuki R., Yokoyama M., Shiroishi T., Wakana S., et al. Peg3 imprinted gene on proximal chromosome 7 encodes for a zinc finger protein. Nat. Genet. 1996;12:186–190. doi: 10.1038/ng0296-186. [DOI] [PubMed] [Google Scholar]
- Letsou W., Cai L. Noncommutative biology: sequential regulation of complex networks. PLoS Comput. Biol. 2016;12:e1005089. doi: 10.1371/journal.pcbi.1005089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A., et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D., Prill R.J., Schaffter T., Mattiussi C., Floreano D., Stolovitzky G. Revealing strengths and weaknesses of methods for gene network inference. Proc. Natl. Acad. Sci. U S A. 2010;107:6286–6291. doi: 10.1073/pnas.0913357107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D., Costello J.C., Küffner R., Vega N.M., Prill R.J., Camacho D.M., Allison K.R., DREAM5 Consortium. Kellis M., Collins J.J., et al. Wisdom of crowds for robust gene network inference. Nat. Methods. 2012;9:796–804. doi: 10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin A.A., Nemenman I., Basso K., Wiggins C., Stolovitzky G., Dalla Favera R., Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006;7(Suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto H., Kiryu H., Furusawa C., Ko M.S.H., Ko S.B.H., Gouda N., Hayashi T., Nikaido I. SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics. 2017;33:2314–2321. doi: 10.1093/bioinformatics/btx194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis C.S., Patterson D.M., Winkler J., Conrad D.N., Hein M.Y., Srivastava V., Hu J.L., Murrow L.M., Weissman J.S., Werb Z., et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods. 2019;16:619–626. doi: 10.1038/s41592-019-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean A.B., D’Amour K.A., Jones K.L., Krishnamoorthy M., Kulik M.J., Reynolds D.M., Sheppard A.M., Liu H., Xu Y., Baetge E.E., et al. Activin a efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3-kinase signaling is suppressed. Stem Cells. 2007;25:29–38. doi: 10.1634/stemcells.2006-0219. [DOI] [PubMed] [Google Scholar]
- Meredith G.D., D’Ippolito A., Dudas M., Zeidner L.C., Hostetter L., Faulds K., Arnold T.H., Popkie A.P., Doble B.W., Marnellos G., et al. Glycogen synthase kinase-3 (Gsk-3) plays a fundamental role in maintaining DNA methylation at imprinted loci in mouse embryonic stem cells. Mol. Biol. Cell. 2015;26:2139–2150. doi: 10.1091/mbc.E15-01-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer P.E., Kontos K., Lafitte F., Bontempi G. Information-theoretic inference of large transcriptional regulatory networks. EURASIP J. Bioinform. Syst. Biol. 2007;2007:79879. doi: 10.1155/2007/79879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D., Studham M., Tjärnberg A., Weishaupt H., Swartling F.J., Nordling T.E.M., Sonnhammer E.L.L. Perturbation-based gene regulatory network inference to unravel oncogenic mechanisms. Sci. Rep. 2020;10:14149. doi: 10.1038/s41598-020-70941-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord J., Schill D., Pulakanti K., Rao S., Cirillo L.A. The transcription factor FoxO1 is required for the establishment of the human definitive endoderm. BioRxiv. 2020 doi: 10.1101/2020.12.22.423976. [DOI] [Google Scholar]
- Le Novère N. Quantitative and logic modelling of molecular and gene networks. Nat. Rev. Genet. 2015;16:146–158. doi: 10.1038/nrg3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popkie A.P., Zeidner L.C., Albrecht A.M., D’Ippolito A., Eckardt S., Newsom D.E., Groden J., Doble B.W., Aronow B., McLaughlin K.J., et al. Phosphatidylinositol 3-kinase (PI3K) signaling via glycogen synthase kinase-3 (Gsk-3) regulates DNA methylation of imprinted loci. J. Biol. Chem. 2010;285:41337–41347. doi: 10.1074/jbc.M110.170704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratapa A., Jalihal A.P., Law J.N., Bharadwaj A., Murali T.M. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods. 2020;17:147–154. doi: 10.1038/s41592-019-0690-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin G., Yang L., Ma Y., Liu J., Huo Q. The exploration of disease-specific gene regulatory networks in esophageal carcinoma and stomach adenocarcinoma. BMC Bioinformatics. 2019;20:717. doi: 10.1186/s12859-019-3230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P., Simonds E.F., Bendall S.C., Gibbs K.D., Bruggner R.V., Linderman M.D., Sachs K., Nolan G.P., Plevritis S.K. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X., Rahimzamani A., Wang L., Ren B., Mao Q., Durham T., McFaline-Figueroa J.L., Saunders L., Trapnell C., Kannan S. Inferring causal gene regulatory networks from coupled single-cell expression dynamics using scribe. Cell Syst. 2020;10:265–274.e11. doi: 10.1016/j.cels.2020.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham O.J.L., Firas J., Fang H., Oates M.E., Holmes M.L., Knaupp A.S., FANTOM Consortium. Suzuki H., Nefzger C.M., Daub C.O., et al. A predictive computational framework for direct reprogramming between human cell types. Nat. Genet. 2016;48:331–335. doi: 10.1038/ng.3487. [DOI] [PubMed] [Google Scholar]
- Stark J., Brewer D., Barenco M., Tomescu D., Callard R., Hubank M. Reconstructing gene networks: what are the limits? Biochem. Soc. Trans. 2003;31:1519–1525. doi: 10.1042/bst0311519. [DOI] [PubMed] [Google Scholar]
- Street K., Risso D., Fletcher R.B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics. 2018;19:477. doi: 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y., Cahan P. SingleCellNet: a computational tool to classify single cell RNA-seq data across platforms and across species. Cell Syst. 2019;9:207–213.e2. doi: 10.1016/j.cels.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theka I., Sottile F., Aulicino F., Garcia A.C., Cosma M.P. Reduced expression of Paternally Expressed Gene-3 enhances somatic cell reprogramming through mitochondrial activity perturbation. Sci. Rep. 2017;7:9705. doi: 10.1038/s41598-017-10016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. Defining cell types and states with single-cell genomics. Genome Res. 2015;25:1491–1498. doi: 10.1101/gr.190595.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Smith V.A., Wang P.P., Hartemink A.J., Jarvis E.D. Advances to Bayesian network inference for generating causal networks from observational biological data. Bioinformatics. 2004;20:3594–3603. doi: 10.1093/bioinformatics/bth448. [DOI] [PubMed] [Google Scholar]
- Yu J.S.L., Ramasamy T.S., Murphy N., Holt M.K., Czapiewski R., Wei S.-K., Cui W. PI3K/mTORC2 regulates TGF-β/Activin signalling by modulating Smad2/3 activity via linker phosphorylation. Nat. Commun. 2015;6:7212. doi: 10.1038/ncomms8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Epoch is available as a package in R, and code and tutorials can be found at https://github.com/pcahan1/epoch. Data are available at GEO under accession number GEO: GSE177051.