

Summary
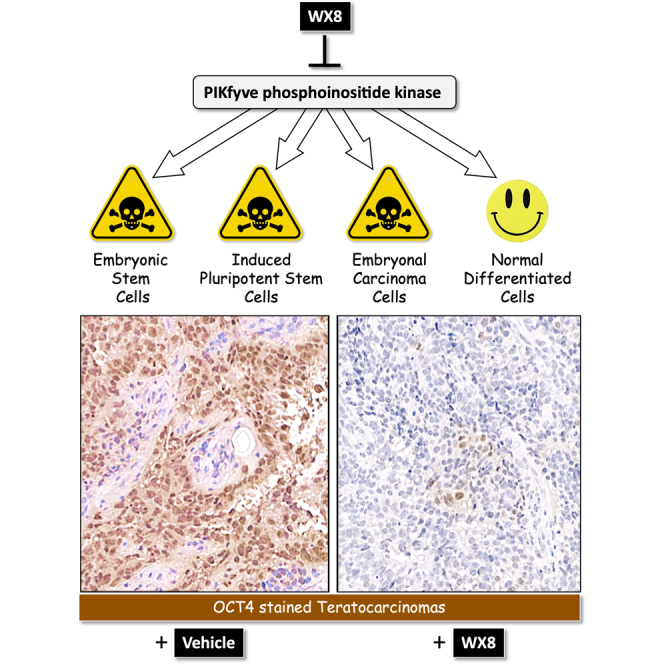
Inhibition of PIKfyve phosphoinositide kinase selectively kills autophagy-dependent cancer cells by disrupting lysosome homeostasis. Here, we show that PIKfyve inhibitors can also selectively eliminate pluripotent embryonal carcinoma cells (ECCs), embryonic stem cells, and induced pluripotent stem cells under conditions where differentiated cells remain viable. PIKfyve inhibitors prevented lysosome fission, induced autophagosome accumulation, and reduced cell proliferation in both pluripotent and differentiated cells, but they induced death only in pluripotent cells. The ability of PIKfyve inhibitors to distinguish between pluripotent and differentiated cells was confirmed with xenografts derived from ECCs. Pretreatment of ECCs with the PIKfyve specific inhibitor WX8 suppressed their ability to form teratocarcinomas in mice, and intraperitoneal injections of WX8 into mice harboring teratocarcinoma xenografts selectively eliminated pluripotent cells. Differentiated cells continued to proliferate, but at a reduced rate. These results provide a proof of principle that PIKfyve specific inhibitors can selectively eliminate pluripotent stem cells in vivo as well as in vitro.
Keywords: embryonic stem cells, induced pluripotent stem cells, embryonal carcinoma stem cells, cancer stem cells, autophagy, autophagosome, lysosome
Graphical abstract
Highlights
-
•
PIKfyve inhibitors selectively kill pluripotent stem cells
-
•
Differentiated cells remain viable
-
•
Cell death occurs by non-canonical apoptosis
-
•
PIKfyve inhibition selectively eliminates stem cells from teratocarcinomas
In this article, DePamphilis and colleagues show that PIKfyve inhibitors can selectively eliminate human pluripotent embryonal carcinoma cells, embryonic stem cells, and induced pluripotent stem cells under conditions where differentiated cells remain viable. This study provides a proof of principle that PIKfyve specific inhibitors can selectively eliminate pluripotent stem cells in vivo as well as in vitro.
Introduction
Pluripotent embryonic stem cells (ESCs) are essential for normal mammalian development (Martello and Smith, 2014; Smith, 2017). Remarkably, a wide variety of somatic cells can be reprogrammed into an embryonic-like pluripotent state, termed induced pluripotent stem cells (iPSCs), that provides a powerful tool for regenerative medicine (Takahashi and Yamanaka, 2013). Unfortunately, ESCs and iPSCs also give rise to germ cell tumors from which pluripotent cancer stem cells, termed embryonal carcinoma cells (ECCs), are derived (Oosterhuis and Looijenga, 2019). Genes commonly required in both pluripotent cells and cancer cells include the core pluripotency genes NANOG, POU5F1 (OCT4), and SOX2, together with the MYC transcription factor (Lee et al., 2013). In addition, both ESCs and cancer cells require the Geminin/GMNN gene to prevent aberrant DNA replication during cell proliferation (Adler-Wailes et al., 2017; Huang et al., 2015; Zhu and Depamphilis, 2009), whereas neither ESCs nor cancer cells require the transcription factor and tumor suppressor gene p53 for either cell-cycle arrest or programmed cell death (Jaiswal et al., 2020). Thus, a major concern in the application of pluripotent stem cells to regenerative medicine is their ability to cause cancer (Gorecka et al., 2019; Martin, 2017).
Previous studies have shown that autophagy-dependent cancer cells can be terminated by inhibition of the PIKFYVE phosphatidylinositol kinase that is essential for lysosome homeostasis (Ikonomov et al., 2019). PIKfyve activity is inhibited with remarkable specificity by the WX8 family of seven small molecule analogs whose affinity for the PIKfyve protein is 90 to 15,000 times greater than for their secondary target PIP4K2C (Sharma et al., 2019). These inhibitors mimic the effects of PIKfyve small interfering RNA (siRNA), and either dominant negative mutations in the PIKFYVE gene or disruption of the PIKFYVE gene nullify the effects of these molecules on cell metabolism, thereby confirming that PIKfyve is the critical target (Gayle et al., 2017; Ikonomov et al., 2019; Lu et al., 2014; O'Connell and Vassilev, 2021; Sharma et al., 2019).
PIKfyve specific inhibitors prevent lysosome fission without preventing homotypic lysosome fusion (Sharma et al., 2019), inhibit trafficking of molecules into lysosomes (Sharma et al., 2019), and inhibit maturation of cathepsins (Gayle et al., 2017; Sharma et al., 2019). They also disrupt endosome trafficking (de Lartigue et al., 2009; Ikonomov et al., 2001) and prevent heterotypic fusion between lysosomes and autophagosomes (Sharma et al., 2019). Consequently, cells that depend on autophagy under standard culture conditions, such as autophagy-dependent cancer cells, are sensitive to inhibition of PIKfyve under conditions where non-malignant cells remain viable (Gayle et al., 2017; Ikonomov et al., 2019; Sharma et al., 2019). The fact that ablation of the PIKfyve gene prevents mouse development beyond the blastocyst stage (Ikonomov et al., 2011) suggests that ESCs are autophagy dependent and therefore would be sensitive to PIKfyve inhibitors.
Here, we report that PIKfyve specific inhibitors WX8 and Apilimod can induce non-canonical apoptosis in ESCs, iPSCs, and ECCs in vitro under conditions that have little, if any, effect on the viability of differentiated cells. Moreover, WX8 can selectively eliminate ECCs from teratocarcinomas, thereby converting a rapidly growing malignant tumor into a slow-growing benign tumor. Thus, PIKfyve inhibitors have therapeutic potential in treating germ cell neoplasia and in preventing spontaneous tumor formation during targeted gene therapy and cell replacement therapy.
Results
WX8-induced cell death in mouse embryonic stem cells
The WX8 family of PIKfyve inhibitors was discovered in a high-throughput screen for compounds that inhibit Geminin, a protein essential to prevent DNA re-replication-dependent apoptosis in cells derived from human cancers, but not in cells derived from normal tissues (Lee et al., 2012; Zhu et al., 2011). As Geminin is also essential in mouse ESCs to prevent DNA re-replication-dependent apoptosis during self-renewal and teratoma formation (Adler-Wailes et al., 2017; Hosogane et al., 2017; Huang et al., 2015), we reasoned that PIKfyve might also be essential for mouse ESC viability.
To test this hypothesis, the same mouse ESCs used to evaluate the role of Geminin were cultured under self-renewal conditions in the presence of 3 μM WX8, the most potent of five PIKfyve inhibitors analyzed together (Sharma et al., 2019). Within 8 h, cells were densely packed with cytoplasmic vacuoles (Figure 1A), confirming that WX8 had inhibited lysosome fission, but not lysosome fusion (Sharma et al., 2019). Fluorescence-activated cell sorting (FACS) (Figure 1B) revealed that this phenomenon was accompanied by a transient accumulation of cells with >4N DNA content (Figure 1C), consistent with excess DNA replication. Within 32 h, 50% of the cells contained <2N DNA, a hallmark of cell death (Elmore, 2007). Cell death was confirmed by the permeability of cells to trypan blue (Figure 1D). Within 72 h, >90% of the cells contained <2N DNA (Table 1). The half maximal inhibitory concentration (IC50) was 1 μM WX8.
Figure 1.

WX8 induced loss of viability and DNA degradation in mouse ESCs
(A) Mouse Gmnn(fl/fl);ER-Cre/+ ESCs were cultured in the presence of 3 μM WX8. WX8 was dissolved in dimethyl sulfoxide (DMSO) at 20 mM concentrations and stored at −20°C. By 8 h, cells were densely populated with cytoplasmic vacuoles (enlarged lysosomes) compared with the vehicle (DMSO). Magnification was 20×.
(B) Cells were cultured in the presence of 3 μM WX8 for the indicated times. Attached and unattached cells were combined, permeabilized, and stained with PI, and their relative DNA content was quantified by FACS using a FACSCalibur flow cytometer (BD Biosciences) according to the manufacturer’s instructions. Raw data were analyzed using FlowJo software. Cells with <2N DNA content are indicated.
(C) The fraction of cells with <2N DNA and the fraction with >4N DNA were quantified as in (B) and displayed as a function of time in 3 μM WX8.
(D) Cells were cultured for 72 h in the indicated concentration of WX8 and then stained with trypan blue to distinguish viable cells from dead cells. The total fraction (attached plus unattached) of unstained (viable) ESCs was counted using a hemocytometer. The WX8 concentration that resulted in 50% reduction in viability (IC50) is indicated. SEM for 3 independent repetitions is indicated.
Table 1.
Sensitivity of mammalian cells to WX8
| Cells (origin) | Cell line | WX8 IC50 (μM) |
||
|---|---|---|---|---|
| ATPa | Live Cellsb | DNAc | ||
| Mouse stem cells | ||||
| ESC (blastocyst) | Gmnn(fl/fl); ER-Cre/+ | 1.0 | Yes | |
| Human stem cells | ||||
| ESC (blastocyst) | WA01/H1 | 1.6 | 1.9 | 2 |
| ESC (blastocyst) | WA09/H9 | 0.26 | 2.5 | 5 |
| iPSC (H108 skin fibroblast) | SCUi24 | 0.4 | 2.5 | 1 |
| ECC (lung epithelial) | TERA-2 | 3 | 5 | 5 |
| ECC (testis epithelial) | NTERA-2 | 0.48 | 1.3/0.2 | 3.5/0.4 |
| ECC (testis epithelial) | GCT27D.C1 | 0.28 | 0.2 | |
| Differentiated cell lines | ||||
| Normal skin fibroblast | HFF-1 | 28 | 75/>10 | 50/>10 |
| Normal skin fibroblast | H108 | 15 | 50 | 140 |
| Normal lung epithelial | HBEC | 6 | 140 | 20 |
| Normal kidney epithelial | VeroE6 | 14 | 60 | 60 |
| Differentiated stem cells | ||||
| Differentiated ESCs | H1-diff | >10 | ||
| Differentiated iPSCs | SCUi24-diff | >10 | ||
| Teratocarcinoma xenografts | ||||
| ECC (testis epithelial) | NTERA-2 | Eliminated stem cells and inhibited tumor growth | ||
| ECC (testis epithelial) | GCT27D.C1 | |||
Average of cellular ATP levels of three independent assays were quantified using the Promega CellTiter-Glo Luminescent Cell Viability Assay kit.
The fraction of live cells was determined by FACS of nonpermeabilized cells stained with annexin-V and PI.
The fraction of cells with less <2N DNA was determined by FACS of permeabilized cells stained with PI. H1 ESCs and SCUi24 iPSCs differentiated spontaneously when cultured in standard conditions for fibroblast and epithelial cell lines (Fang et al., 2005). Teratocarcinoma xenografts were produced from ECCs inoculated subcutaneously into immunocompromised mice and treated by intraperitoneal injections of WX8. IC50 values for Apilimod (Figure S4) are indicated for NTERA-2 and HFF-1 cells as WX8/Apilimod.
WX8 reduced viability in human pluripotent stem cells
To determine whether or not the results with mouse ESCs applied to human pluripotent stem cells, ESC (H1, H9), iPSC (SCUi24), and ECC (TERA-2, NTERA-2, GCT27D.C1) lines were challenged with WX8. H1 and H9 were derived from human blastocysts. SCUi24 was derived from human skin fibroblasts. TERA-2 was derived from a lung teratocarcinoma (Fogh et al., 1977). NTERA-2 was derived from a xenograft tumor of TERA-2 (Andrews et al., 1984, 1985). GCT27D.C1 was isolated from a patient with testicular teratocarcinoma (Pera et al., 1987).
Human ESCs and iPSCs contained normal karyotypes (Mallon et al., 2013, 2014; Su et al., 2016), produced teratomas when inoculated subcutaneously into immunocompromised mice, and expressed the human pluripotent cell biomarker SSEA4 (Figures S1A and S1B). Human ECCs also expressed OCT4, SOX2, and NANOG under self-renewal conditions (Figure S1C), and they produced teratocarcinomas enriched with pluripotent cells when inoculated subcutaneously into immunocompromised mice (Figure S1D) (described below). Similar to mouse ESCs, WX8 induced cytoplasmic vacuolation in all human pluripotent and differentiated cells (for example, Figure S2A), as previously reported for all PIKfyve inhibitors (Sharma et al., 2019). WX8 also induced loss of viability in ESCs, iPSCs, and ECCs, as quantified by loss of ATP (Figures S2B–S2D), with IC50 values from 0.26 to 3 μM WX8 (Table 1, ATP).
PIKfyve inhibitors induced DNA loss in human pluripotent stem cells
Cell death is commonly accompanied by DNA loss (Elmore, 2007). Therefore, to determine whether or not the ATP loss induced by WX8 was accompanied by cell death, the accumulation of stem cells with <2N DNA content was quantified by FACS. The results revealed that the fraction of stem cells with <2N DNA increased with time in WX8 with a concomitant decrease in cells containing 2N DNA (G1 phase) or 4N DNA (G2 and M phases) (Figure 2A). Within 4 days, >80% of the cells contained <2N DNA. Similarly, WX8 induced DNA loss in H1 and H9 ESCs, SCUi24 iPSCs, as well as in TERA-2 and GCT27D.C1 ECCs (Figures 2B–2D). The IC50 values for pluripotent stem cells varied from 0.2 to 5 μM WX8 (Table 1 DNA). The effects of WX8 on human pluripotent cells were confirmed using Apilimod (Figures S4D and S4E), another potent PIKfyve specific inhibitor (Gayle et al., 2017).
Figure 2.

WX8 induced DNA loss selectively in human pluripotent stem cells
(A) NTERA-2 (shown) and GCT27D.C1 ECCs were cultured with 5 μM WX8 for the times indicated, then permeabilized, stained with propidium iodide, and subjected to FACS. The fraction of cells with <2N DNA is indicated in each FACS profile.
(B) Time courses for accumulation of NTERA-2 (●) and GCT27D.C1 (◆) ECCs with <2N DNA content. Data were normalized to cells cultured with vehicle at each time point.
(C and D) The concentration of WX8 required for 50% of the cells to contain <2N DNA (IC50 values) were determined for H1 (▪), H9 (●), SCUi24 (△), GCT27D.C1 (◆), NTERA-2 (●) and TERA-2 (◯).
(E–G) IC50 values for HFF-1, Vero6, H108, and HBEC were determined (vertical dotted line). Error bars indicate SEM for 3 independent repetitions. To generate a logarithmic scale, time 0 h was plotted as 0.1 h. IC50 values are in Table 1.
DNA loss resulted from PCD
To determine whether WX8 induced programmed cell death (PCD) or necrosis in pluripotent cells, human ECCs, ESCs, and iPSCs were cultured in the presence of WX8 and then stained with both annexin-V and propidium iodide (PI). Membrane lipid asymmetry is lost during PCD, and phosphatidylserine becomes exposed on the outer leaflet of the plasma membrane, which allows annexin-V binding (Crowley et al., 2016). When the membranes of cells become sufficiently permeabilized so that PI can enter and bind to DNA, the cells have undergone PCD. Cells that stain with PI, but not with annexin V, died from necrosis. Cell death was quantified by the depletion of live cells with concomitant accumulation of cells stained with annexin-V and cells stained with both annexin-V and PI (Figures 3A and S6A). The fraction of live cells (Q3) and cells undergoing PCD (Q1+Q2) were plotted against WX8 concentration (Figures 3B and S5B).
Figure 3.

WX8 induced programmed cell death selectively only in human pluripotent stem cells
(A) Fibroblasts and ECCs were seeded at densities such that cells cultured in the absence of WX8 were not confluent at the end of the experiment. Cells were treated with various concentrations of WX8 for 4 days, stained with annexin-V and propidium iodide (PI) without prior permeabilization using an Annexin V-FITC Apoptosis Detection Kit, and then analyzed in a Becton Dickinson LSR-II using FACS Diva software.
(B) SEM is indicated for average of three independent assays. Cells cultured in the presence of vehicle (0 μM WX8) were defined as 100% viable cells (Q3 • ▪) and 0% dead cells (Q1+Q2 ◯ □) so that only changes due to WX8 were represented. Time 0 h was plotted as 0.1 h to generate a logarithmic scale. Similar results were obtained with ESCs and iPSCs (Figure S5).
WX8 induced PCD in all three types of pluripotent cells. The IC50 values for ESC and iPSC lines ranged from 1.9 to 2.5 μM WX8 (Table 1 Live Cells). The IC50 values for ECC lines ranged from 0.28 to 5 μM WX8. The IC50 for NTERA-2 was four times less than its parent TERA-2, and patient-derived GCT27D.C1 was five times less than NTERA-2 ECCs (Table 1, Live Cells), suggesting that culturing pluripotent cell lines selects for autophagy independence. Apilimod was about 10 times more effective than WX8 on NTERA-2 cells. After 4 days in culture with 5 μM WX8, necrotic cells accounted for <1% to 18% of total cells. Thus, PIKfyve inhibitors induced primarily PCD.
Differentiated cells were insensitive to PIKfyve inhibitors
To determine whether or not pluripotent stem cells were significantly more sensitive to PIKfyve inhibitors than differentiated cells, four established nonmalignant differentiated cell lines were cultured in the presence of WX8 under conditions of exponential cell proliferation, equivalent to self-renewal of pluripotent cells. HFF-1 foreskin fibroblasts are normal newborn mesenchymal cells from the dermis that produce collagen and other extracellular matrix proteins. H108 skin fibroblasts are the cells from which SCUi24 iPSCs were derived. HBEC lung epithelial cells are frequently used to derive iPSCs, and VeroE6 kidney epithelial cells are commonly used to propagate human viruses.
As previously reported (Sharma et al., 2019), WX8 rapidly induced cytoplasmic vacuolization in differentiated human cells (Figure S2A). However, in contrast to stem cells, differentiated cells remained viable under conditions wherein stem cells did not. ATP loss occurred, on average, at an IC50 of 1.2 μM WX8 in human pluripotent cells (Figures S2B and S2C) and at an IC50 of 15.8 μM WX8 in differentiated cells (Figure S2D). This 13-fold difference in ATP loss reflected the ability of WX8 to inhibit cell proliferation, not cell death.
Cell death was confirmed by the accumulation of cells with less than G1 phase DNA content (<2N DNA) during proliferation of cells in the presence of WX8 (Figure 2). Comparable results were obtained with Apilimod (Figure S5A). DNA loss occurred, on average, at an IC50 of 2.8 μM WX8 in human pluripotent cells and at an IC50 of 67.5 μM WX8 in differentiated cells (Table 1 DNA). This 24-fold difference in DNA loss reflected the ability to induce death in pluripotent cells under conditions in which differentiated cells remained viable.
PCD was confirmed by staining live cells with annexin-V and PI (Figures 2 and S5). Comparable results were obtained with Apilimod (Figure S4E). Human pluripotent cells initiated PCD, on average, at an IC50 of 2.3 μM WX8, whereas differentiated cells initiated PCD at an IC50 of 81.3 μM WX8 (Table 1 live cells). This 35-fold difference in the initiation of PCD was comparable to the difference in DNA loss, thereby confirming that PIKfyve inhibitors can selectively kill pluripotent cells under conditions where differentiated cells remain viable.
Differences between the sensitivity to PIKfyve inhibitors of differentiated cells compared with pluripotent cells were cell line dependent. Thus, the range of sensitivity to WX8-induced cell death (DNA loss) between pluripotent cells and differentiated cells varied from 5-fold (HBEC/H9) to 700-fold (H108/GCT27DC1). Similarly, the range of sensitivity to WX8-induced inhibition of proliferation in differentiated cells varied from insensitive (IC50 >100 μM WX8) to sensitive. For example, WX8 inhibited proliferation of VeroE6 cells (IC50 0.45 μM WX8) under conditions that did not restrict viability (ATP loss, IC50 14 μM WX8) or induced PCD (IC50 60 μM WX8); the cells remained attached to the dish until PCD was evident (Figure S3). Thus, PIKfyve inhibitors can inhibit proliferation of some differentiated cells without inducing PCD.
Differentiation was accompanied by PIKfyve insensitivity
To determine whether or not differentiation of pluripotent cells was accompanied by insensitivity to PIKfyve inhibitors, ESCs were allowed to differentiate spontaneously by culturing them to 80% confluency and then gradually switching to differentiation medium for 4 weeks to allow morphological transformation (Fang et al., 2005). Differentiation was confirmed by the loss of OCT4 mRNA and protein (Figures 4B and 4C). Spontaneous differentiation of H1 ESCs suppressed induction of PCD by WX8 (Figure 4A). Their IC50 increased from 1.9 to >10 μM WX8 (Table 1 Live Cells). Similarly, spontaneous differentiation of SCUi24 iPSCs suppressed the accumulation of cells with <2N DNA (Figure 4E) almost to the level in HFF-1 fibroblasts (Figure 4D). These results confirmed that the ability of WX8 to induce cell death in pluripotent stem cells was lost when they differentiated.
Figure 4.

Resistance to WX8 accompanied differentiation of pluripotent stem cells
(A) Spontaneously differentiated H1 ESCs (H1-diff) were cultured with WX8 for 4 days, stained with annexin-V and PI, and then analyzed by FACS. The fraction of cells undergoing programmed cell death (Q1+Q2) is given.
(B) Oct4 RNA in H1 ESCs and H1-diff ESCs was quantified by real-time PCR. Error bars indicate SEM for three independent repetitions.
(C) OCT4 protein in H1 ESCs and H1-diff ESCs was detected by immunoblotting.
(D) HFF-1 fibroblasts were cultured with the indicated concentration of WX8 for 4 days. Cells were permeabilized and then stained with PI, and cellular DNA content was determined by FACS.
(E) Pluripotent SCUi24 iPSCs and spontaneously differentiated SCUi24 iPSCs were cultured as in (D).
Cell death followed disruption of autophagy
To determine whether or not the effects of WX8 on pluripotent stem cells were linked to autophagy, the time course for WX8-induced cell death was compared with changes in autophagosome associated proteins. LC3-II is a phosphatidylethanolamine conjugate of MAP1LC3A/LC3-I that is recruited to autophagosome membranes. SQSTM1/p62 is an autophagy receptor that interacts directly with LC3-II as well as with ubiquitinated proteins. WX8 has been shown to prevent fusion of autophagosomes with lysosomes, thereby disrupting autophagic flux with a concomitant increase in LC3-II and p62 proteins (Sharma et al., 2019).
Two WX8-sensitive pluripotent cell lines (NTERA-2 ECCs and H1 ESCs) were compared to WX8-insensitive H108 fibroblasts. Cells were cultured with WX8 for up to 96 h. U2OS cells were included in each analysis to provide a common standard by which one blot could be compared with another. Attached and unattached cells were combined, and total protein extracts were subjected to immunoblotting (Figures 5A–5C).
Figure 5.

Non-apoptotic cell death followed disruption of autophagy
(A–D) (A) NTERA-2 ECCs, (B) H108, (C) H1, and (D) H1 differentiated cells were cultured in the presence of 2.5 μM WX8. At the indicated times, total cell extracts were subjected to immunoblotting to detect PARP and cleaved PARP (cPARP), caspase-3 (CASP3) and cleaved caspase-3 (cCASP3), p62, LC3-I, LC3-II, and GAPDH. Histones were detected with Ponceau-S. In addition, NTERA-2 cells were cultured with 1 μM Adriamycin (+Adr lane) and H108 cells with 2 μM Adriamycin for 24 h to induce double-stranded DNA breaks. A standard sample of untreated U2OS cells was included to allow direct comparison of immunoblots. Immunoblots were selected that detected the indicated proteins in U2OS. As LC3 levels in WX8-treated cells were much greater than LC3 levels in untreated U2OS cells, lighter exposures were included to distinguish LC3-II more clearly from LC3-1. Lighter exposures were used to quantify changes in protein levels (Figure S6).
(E) The ratio of p62 in H108 (•), NTERA-2 (▪), and H1 (▴) cells to p62 in untreated U2OS cells is compared with the accumulation of NTERA-2 cells with<2N DNA (□).
(F) The fractions of cleaved PARP and CASP3 proteins quantified in H108, NTERA-2, and H1 cells.
Visual inspection revealed that WX8 induced accumulation of LC3 and p62 protein in all three cell lines. Quantifying these proteins in shorter exposure times (Figure S6A) and normalizing them to the histones revealed that WX8 stimulated accumulation of p62 protein 13-fold in H108, 9-fold in NTERA-2, and <2-fold in H1 cells (Figure 5E). Similarly, WX8 stimulated accumulation of total LC3 protein 10-fold in H108, 4-fold in NTERA-2, and 2-fold in H1 cells (Figure S6A). WX8 induction of p62 and LC3 protein in H108 fibroblasts (Figure 5B) and differentiated H1 ESCs (Figure 5D) were comparable.
These results revealed that WX8 induction of PCD followed disruption of autophagy. The ratio of LC3-II to LC3-I began accumulating rapidly in each cell line after 1 to 2 h of exposure to WX8 and reached a plateau between 10 and 48 h (Figure S6A). This event was followed by p62 protein accumulation from 10 to 48 h (Figure 5E). The accumulation of cells with <2N DNA content began after 10 h with WX8 and continued unabated (Figure 5E), suggesting that induction of PCD by PIKfyve inhibitors in pluripotent cells was a consequence of disrupting autophagic flux. Cell death resulted in significantly reduced accumulation of LC3 and p62 in WX8-sensitive cells compared with insensitive cells.
Cell death occurred via non-canonical apoptosis
WX8 induced ATP loss, DNA loss, binding of annexin-V, and membrane permeabilization in ESCs, iPSCs, and ECCs, but not in differentiated cells, thereby revealing that WX8 induced PCD in pluripotent cells, but not in differentiated cells. To determine whether or not cell death resulted from apoptosis, the temporal relationship between cleavage of poly(ADP-ribose) polymerase (PARP), caspase-3 (CASP3), and loss of cellular DNA was analyzed. PARP binds to single-strand disruptions in DNA, thereby triggering DNA repair or cell death with concomitant cleavage of PARP protein (Soldani and Scovassi, 2002). Canonical apoptosis involves cleavage of CASP3 (Nikoletopoulou et al., 2013).
WX8 induced 41% PARP cleavage in NTERA-2 and 20% in H1, but <2% CASP3 cleavage (Figures 5A, 5C, and 5F) or CASP12 cleavage (Figures S6C–S6E); cleaved CASP7 was not detected. These events coincided with accumulation of LC3 and p62 (Figures 5A–5D). In contrast, >90% of both PARP and CASP3 was cleaved in response to doxorubicin (Adriamycin) (Figures 5A, 5C, and 5F), an anti-cancer drug that triggers apoptosis by inducing double-stranded DNA breaks in proliferating cells (Tewey et al., 1984). Thus, WX8 induced cell death in ECCs and ESCs via a non-canonical form of apoptosis that produces primarily single-stranded rather than double-stranded DNA breaks. Neither WX8 nor Adriamycin induced cleavage of either PARP or CASP3 in H108 fibroblasts (Figures 5B and 5F) because normal differentiated cells respond to DNA damage by cell-cycle arrest or senescence (Campisi and d'Adda di Fagagna, 2007).
WX8 inhibited teratocarcinoma formation
TERA-2 was derived from a lung teratocarcinoma (Fogh et al., 1977), and NTERA-2 was derived from a xenograft tumor of TERA-2 (Andrews et al., 1984, 1985). To determine whether or not WX8 affected the ability of ECCs to form tumors, NTERA-2 cells were cultured overnight with either 10 μM WX8 or its vehicle (DMSO). WX8 rapidly induced cytoplasmic vacuolation, thereby demonstrating its efficacy in disrupting lysosome homeostasis, but the cells remained attached to the plate, and their viability was confirmed by their inability to be stained with trypan blue. The cells were collected by trypsinization, suspended in complete culture medium supplemented with Matrigel Matrix (Corning Life Sciences) and either vehicle or 10 μM WX8. Cells were inoculated subcutaneously into immunocompromised mice. Palpable tumors appeared within 3 weeks, and their sizes were monitored thereafter (Figure 6A). At 7 weeks, the tumors were excised, photographed, and stained for OCT4 protein (Figure 6B). The rate of tumor growth inhibition (TGI) was 65% in ECCs pretreated briefly with WX8, thereby providing proof of principle that cells exposed to WX8 were less capable of tumor formation. Both vehicle and WX8-pretreated NTERA-2 cells gave rise to tumors with indistinguishable histology and large clusters of OCT4-expressing cells. Therefore, pretreatment of ECCs reduced, but did not eliminate, their ability to form a potentially malignant tumor.
Figure 6.

WX8 suppressed initiation and expansion of ECC-derived tumors
(A) NTERA-2 ECCs were cultured overnight with either 10 μM WX8 or vehicle (DMSO). Both vehicle- and WX8-treated cells remained attached to the plate, but the WX8-treated cells were densely vacuolated. The attached cells were isolated and suspended in culture medium containing 50% Matrigel Matrix and either 10 μM WX8 or vehicle. Immunocompromised mice were inoculated with 200 μL containing 2 million WX8-treated cells (●) on one flank and with 2 million vehicle-treated cells (◯) on the opposite flank. Tumor volumes from three mice were averaged together and plotted as a function of time post-treatment.
(B) Tumors were excised after 7 weeks, and slices were stained for OCT4 protein to identify pluripotent cells.
(C) Eight mice were inoculated with untreated NTERA-2 ECCs. When tumors were palpable, mice received daily intraperitoneal injections of either vehicle (◯) or 40 mg WX8/kg (●) mouse.
(D) Eight mice were inoculated with untreated GCT27D.C1 ECCs and treated as in (C).
(E) Tumor slices from (D) were immune-stained for OCT4, SOX2, and Ki67 proteins.
(F) The average number of OCT4, SOX2, and Ki67-stained cells were quantified by counting the number of Ki67-positive cells in three different sections of each tissue section. All error bars indicate ± SEM.
WX8 depleted teratocarcinomas of pluripotent stem cells
To determine whether or not WX8 could selectively kill pluripotent cells in vivo as well as in vitro, subcutaneous tumor xenografts were produced from ECCs. Once the tumors were palpable, the mice received daily intraperitoneal injections of either vehicle or WX8. Tumors produced from either NTERA-2 cells (Figure 6C) or GCT27D.C1 cells (Figure 6D) were markedly reduced by WX8. TGI was 71% for NTERA.2 tumors and 78% for GCT27D.C1 tumors, and tumors from WX8-treated mice were visibly smaller than tumors from vehicle-treated mice (Figures S6F and S6G). Mouse body weights were maintained, and their dispositions remained normal throughout the experiment (Figures S6F and S6G).
Tumors from vehicle-treated mice were enriched with densely packed clusters of OCT4- and SOX2-expressing cells (Figure 6E), characteristic of cancer stem cells in patient-derived germ cell tumors (Liu et al., 2010). Examination of larger sections through these tumors revealed puddles of OCT4- and SOX2-expressing cells scattered throughout the tumor (Figure S7), characteristic of human teratocarcinomas (Jones et al., 2004a, 2004b). Proliferating cells were identified by staining for Ki67, an antigen unique to proliferating cells and expressed throughout the mitotic cell cycle (Inwald et al., 2013).
Comparison of randomly selected images provided by the NIH pathology laboratory that contained an image scale revealed that WX8 had selectively eliminated cells expressing OCT4 or SOX2 protein with no detectable effect on proliferating cells (Figures 6E and 6F). Total cell extracts of tumor tissue confirmed a marked reduction of OCT4 and SOX2 protein in WX8-treated mice compared with the vehicle control (Figure S7). WX8 also inhibited expression of OCT4, SOX2, NANOG, and STAT3 proteins relative to GAPDH protein during self-renewal of ECCs in vitro (Figure S1C). STAT3 is a transcription factor involved in maintaining pluripotency in ESCs (Hall et al., 2009). These results revealed that WX8 can selectively eliminate pluripotent stem cells with little effect on proliferating cells. Most of the differentiated cells within these teratocarcinomas continued to proliferate during treatment with WX8, although at a slower rate (Figure 6D), as observed with differentiated cells in culture (Sharma et al., 2019).
Discussion
Despite their different origins, pluripotent stem cells share a variety of biological and genetic characteristics, including tumorigenesis. The PIKfyve specific inhibitors, such as WX8 and Apilimod, exhibit therapeutic potential in selectively eliminating pluripotent stem cells from cancers, thereby converting a malignant tumor into a benign tumor. Thus, they could also be used to eliminate the residual pluripotent cells in genetic engineering and cell replacement therapies to prevent them from producing tumors.
Previous studies have shown that the PIKFYVE gene is essential for preimplantation mouse development and that ablation of PIKFYVE reduced DNA synthesis in mouse embryonic fibroblasts (Ikonomov et al., 2011). The results presented here show that PIKfyve specific inhibitors, represented by WX8 (Sharma et al., 2019) and Apilimod (Gayle et al., 2017), can induce cell death selectively in both mouse and human ESCs, human iPSCs, and human ECCs under conditions where spontaneously differentiated ESCs and iPSCs as well as differentiated cells remained viable. Proliferating differentiated cells were up to 70 times more resistant to ATP loss than proliferating pluripotent stem cells, 470 times more resistant to live cell loss (PCD), and 700 times more resistant to DNA loss. Experiments in progress will determine whether or not the level of PIKfyve protein in PIKfyve-sensitive cells differs significantly from PIKfyve-insensitive cells.
Variation in the sensitivity of different pluripotent cell lines reflects the fact that pluripotency exists in multiple forms, as evidenced by the transition of ESCs from a naive state to a primed state to a lineage committed state (Irie et al., 2018; Smith, 2017) and by the transition of unipotent primordial germ cells through a pluripotent state during post-implantation development (Magnusdottir and Surani, 2014). In addition, cellular stress during passage of human pluripotent cells promotes spontaneous differentiation (Chen et al., 2014), as reflected in a reduced sensitivity of pluripotent cells to WX8 when they were passaged in vitro.
The problems inherent with analysis of cultured cells were eliminated by analysis of ECC-induced teratocarcinomas in mouse xenografts. Whereas cell culture inevitably selects for cells that proliferate well in vitro, formation of teratomas or teratocarcinomas in vivo selects only for pluripotent stem cells. Thus, proof of principle that PIKfyve specific inhibitors can selectively eliminate pluripotent stem cells was confirmed by the ability of WX8 to selectively eliminate the pluripotent cells from these tumors. The remaining cells continued to proliferate, but at a reduced rate relative to teratocarcinomas in untreated mice. This was consistent with reports that PIKfyve is required for proliferation to varying extents in various cell types (Gayle et al., 2017; Ikonomov et al., 2019; Sharma et al., 2019) and confirmed by WX8 treatment of VeroE6 cells.
WX8 induced a non-canonical form of apoptosis, as demonstrated by concomitant ATP loss, binding of annexin-V, membrane permeabilization, and DNA loss that did not involve caspase-3 cleavage. The canonical form of apoptosis is the only PCD that employs CASP3, CASP6, and CASP7 (Nikoletopoulou et al., 2013). It is activated by DNA damage, unfolded proteins, reactive oxygen species, or disruption of cell division. These events activate initiator CASP2, CASP8, CASP9, or CASP10, which then activate effectors CASP3, CASP6, and CASP7, which then degrade cellular proteins. Apoptosis is commonly recognized by DNA fragmentation, DNA loss, binding of annexin-V to the cell membrane, cell permeabilization, and cleavage of caspase-3. Thus, double-stranded DNA damage caused by Adriamycin induced apoptosis in pluripotent mouse (Jaiswal et al., 2020) and human cells (Figure 5). However, WX8 did not induce cleavage of CASP3, CASP7, or CASP12 under conditions where cleaved PARP was readily evident and where CASP3 cleavage was induced by Adriamycin. Similar results have been reported for Apilimod induction of PCD in cancer cells (Gayle et al., 2017). Thus, PIKfyve inhibition induces a non-canonical form of apoptosis.
Previous studies have shown that the WX8 family of PIKfyve inhibitors can selectively kill autophagy-dependent forms of melanoma and B-cell non-Hodgkin’s lymphoma with little or no harm to non-malignant cells (Gayle et al., 2017; Sharma et al., 2019). The results presented here show that PIKfyve inhibitors can also target pluripotent cancer stem cells. Cancers such as osteosarcoma, acute lymphoblastic leukemia, and germ cell neoplasia are confined to young adults, primarily between the ages of 10 and 35 years. The origins of all three of these cancers can be traced to progenitor cells that express pluripotent stem cell biomarkers (Abarrategi et al., 2016; Bonnet and Dick, 1997; Hersmus et al., 2017), frequently referred to as cancer stem cells, and ECCs are pluripotent cancer stem cells that give rise to germ cell neoplasia (Hersmus et al., 2017; Oosterhuis and Looijenga, 2019; Sekita et al., 2016). Teratocarcinomas (malignant) and teratomas (benign) account for 95% of testicular cancers and 70% of ovarian tumors that occur principally during adolescence and early adulthood (Bustamante-Marin et al., 2013; Kline and Bazzett-Matabele, 2010). In addition, teratomas are the most common tumor in newborns, appearing in about 1/35,000 live births (Lakhoo, 2010).
Current chemotherapy for germ cell tumors consists of DNA-damaging agents bleomycin, etoposide, and cisplatin (Pierpont et al., 2017), which are highly toxic to all proliferating cells. In contrast, the WX8 family of PIKfyve inhibitors are reversible, specific for PIKfyve, about 1000 times less toxic to normal cells than to autophagy-dependent cancer cells, and their potency is both concentration and time dependent (Gayle et al., 2017; Sharma et al., 2019). Moreover, they have the potential for converting a malignant teratocarcinoma into a benign teratoma as well as to reduce the growth of the teratoma. POU5F1 (OCT4) and NANOG expression predicts aggressive tumor behavior and worse clinical outcome in several types of cancer, and forced expression of these genes increases tumorigenesis, whereas depletion lowers tumorigenic potential (Filipponi et al., 2019).
Human iPSCs are currently used in the development of targeted gene therapy (Li et al., 2020) and regenerative medicine (Kawamura et al., 2016; Martin, 2017). However, the remarkable similarities between cancer cells and pluripotent cells raises the risk that reprogrammed stem cells or their differentiated progeny will develop into tumors (Gorecka et al., 2019; Lee et al., 2013). PIKfyve inhibitors also selectively killed ESCs, iPSCs, and ECCs in vitro. Moreover, pretreatment of ECCs until cytoplasmic vacuolation was evident and the cells remained attached to the dish significantly diminished their subsequent ability to produce a teratocarcinoma. Remarkably, intraperitoneal injections of WX8 into mice bearing teratocarcinomas not only inhibited tumor growth but also eliminated the pluripotent stem cells. The remaining teratoma cells continued to proliferate, but at a reduced rate. Therefore, PIKfyve specific inhibitors also offer a means to prevent or eliminate tumors that arise when pluripotent cells are used therapeutically.
Experimental procedures
Cell culture
Mouse Gmnn(fl/fl); ER-Cre/+ ESCs (Huang et al., 2015), human ESCs WA01/H1 and WA09/H9 (WiCell, Madison, WI), and human iPSCs SCUi24 (Stem Cell Unit/NIH, Bethesda, MD) (Mallon et al., 2013, 2014; Su et al., 2016) have been described. HBEC lung cells were from David Schrump (Shukla et al., 2017). Human skin fibroblasts H108 were from Pamela Robey (NIDCR/NIH). Human foreskin fibroblasts HFF-1 (SCRC-1041), osteosarcoma epithelial U2OS (HTB-96), TERA-2 (HTB-106), and NTERA-2 cl.D1 (CRL-1973) ECCs and monkey kidney VeroE6 (CRL-1586) were from the American Type Culture Collection. Human patient derived pluripotent GCT27D.C1 ECCs were provided by Martin F. Pera.
Mouse ESCs were cultured as described (Jaiswal et al., 2020). Human ESCs and iPSCs were maintained feeder-free in mTeSR1 medium (STEMCELL Technologies, Vancouver, BC, Canada) on tissue culture plates coated with ESC-qualified Matrix (Corning Life Sciences, San Jose, CA) at 2.5% for ESCs and 1.25% for iPSCs. TERA-2 was cultured in McCoy’s 5a Medium (ATCC) with 15% fetal bovine serum (FBS; MilliporeSigma). NTERA-2 cl.D1 was cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific) with 10% FBS. H108 was maintained in alpha-MEM with 20% FBS and GlutaMAX (Thermo Fisher Scientific). VeroE6 and U2OS were cultured in DMEM with 10% FBS. HFF-1 (Sharma et al., 2019), HBEC, HSAEC (Shukla et al., 2017), and GCT27D.C1 (Pera et al., 1987) were cultured as described. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2. No antibiotics or antimycotics/antifungal were used.
WX8 (Specs, ChemDiv), Apilimod (Selleck Chemicals), and Adriamycin (MilliporeSigma) were purchased.
Immunoblot analysis
Whole-cell lysates were prepared using RIPA buffer (50 mM Tris-HCl [pH 7.4], 1% Triton X-100, 10% glycerol, 0.1% SDS, 2 mM EDTA, and 0.5% deoxycholic, 50 mM NaCl, and 50 mM NaF) with protease inhibitor and phosphatase inhibitor cocktails (MilliporeSigma). Total protein extracts were fractionated in 4–12% Bis-Tris gel with MOPS-SDS running buffer (Thermo Fisher Scientific), transferred to nitrocellulose (Invitrogen, Carlsbad, CA) and incubated in 5% non-fat milk/Tris-buffered saline (TBS). Membranes were incubated overnight at 4°C with primary antibody, washed with 0.1% Tween 20 in TBS, incubated with Li-Cor secondary antibody conjugate, and the signal quantitated using the Odyssey infrared imager (Li-Cor, Lincoln, NE). Proteins were identified by antibody specificity and molecular weights using Mark12 protein standards (Thermo Fisher Scientific).
Antibodies
Antibodies against OCT4 (2750S), NANOG (4903), SOX2 (3579), p62 (7114), LC3 I/II (12,741), STAT3 (9139T), CASP3 (9662S), cleaved-CASP3 (9664), PARP (9542S), cleaved-PARP (5625S), and Ki67 (9449S) were from Cell Signaling Technology (Beverly, MA). GAPDH antibody (AC002) was from American Research Products (Belmont, MA).
Flow cytometry analysis
Cell death was measured using the Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, San Diego, CA). Fluorescein isothiocyanate (FITC) and propidium iodide (PI) fluorescence were detected with BD LSR-II using FACS Diva software (Becton Dickinson). Cell surface staining was done with either PE-conjugated anti-SSEA-4 (sc-21704 PE) antibody or isotype-matched normal antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Cells with <2N DNA content were detected using FACSCalibur flow cytometer (BD Biosciences) according to the manufacturer instructions. Raw data were analyzed using FlowJo software.
Real-time PCR
Total RNA was extracted from cells using RNeasy Mini Kit (Qiagen, Germantown, MD), and 300 ng RNA was reverse transcribed using a cDNA synthesis kit (Thermo Fisher Scientific). Expression of OCT4(POU5F1) gene was determined by real-time PCR using Taqman assay probe Hs04260367_gH (Applied Biosystems). Amplification of GAPDH (Hs02786624_g1, Applied Biosystems) served as control. Relative quantification of genes was performed using the ΔΔ Ct method (Chakraborty et al., 2013).
Xenografts
Xenografts were prepared, treated, and characterized as described (Adler-Wailes et al., 2017). Pretreated NTERA-2 cells were cultured overnight with either 10 μM WX8 or the same amount of vehicle (DMSO). NTERA-2 cells remained attached to the plate, although they were highly vacuolated as a result of lysosome fusion in the absence of lysosome fission. Attached cells were collected by trypsinization and counted using a hemocytometer. Equal numbers of cells were suspended in complete culture medium plus 50% Matrigel Matrix and either vehicle or 10 μM WX8. Volumes of 200 μL containing 2 million cells were inoculated subcutaneously into lower right flanks of 6-week-old outbred female BALB/c nude mice (Jackson Laboratories, #007850). Each mouse received WX8-treated cells in one flank and vehicle-treated cells in the opposite flank. Palpable tumors appeared after 6 weeks. Tumor volumes were monitored thereafter. The length and width of each tumor was measured with calipers. Tumor volume was calculated as (length) × (width)2 × 0.5 (Rago et al., 2007), and the percentage tumor growth inhibition (TGI) was calculated (Zou et al., 2018).
In post-treatment studies, volumes of 200 μL containing either 2 million NTERA2 cells or 5 million GCT27D.C1 cells in 50% Matrigel Matrix were inoculated subcutaneously into lower right flanks of 6-week-old outbred female BALB/c nude mice. Palpable tumors appeared within 8 weeks. Tumor volumes were monitored thereafter. WX8 diluted in sunflower oil was administered daily by intraperitoneal (i.p.) injection of 40 mg WX8/kg mouse. Maximum solubility of WX8 was about 30 mg WX8/350μL DMSO. Based on the body weight of the mouse, 12 to 15 μL of WX8 solution was mixed with sunflower seed oil for a total volume of 200 μL. This solution was transferred by pipette into an insulin syringe (28G needle) after removing the piston from the syringe.
Toxicity studies were carried out using doses from 20 to 100 mg/kg body weight. The maximum dose at which no indication of mouse impairment was observed was 40 mg/kg. The control group was injected with the same volume of vehicle (DMSO). Mice were euthanized 15 days later, and tumors were harvested for analysis.
Teratomas were generated by subcutaneous inoculation of 5 million iPSCs in 50% Matrigel Matrix. Palpable tumors appeared within 8 weeks. Mice were euthanized 4 weeks later, and tumors were harvested for histology. The research protocol for this study was approved by the research animal medicine branch (RAMB) of NICHD's Division of Intramural Research.
Histology and immunohistochemistry
Tissues were fixed overnight either at 4°C in Bouin’s fixative, or at room temperature in fresh 4% paraformaldehyde in phosphate-buffered saline, embedded in paraffin, and sectioned at 5 μm. Immunohistochemical staining for OCT4, SOX2, and Ki67 was done using heat-mediated antigen retrieval in EDTA, pH 8.0. Primary antibodies were incubated overnight at room temperature followed by biotinylated secondary antibody and then horseradish peroxidase conjugate avidin-biotin complex (ABC Elite kit; Vector Laboratories). Positive cells were visualized with DAB (3,3′-diaminobenzidine tetrahydrochloride; Invitrogen). Slides were counterstained with hematoxylin. Histology images were from a Nanozoomer histology slide scanner (Hamamatsu Photonics). Positively stained nuclei were quantified using ImageJ IHC Toolbox software.
Author contributions
Conceptualization, M.L.D., A.R.C.; Methodology, A.R.C., M.L.D.; Investigation, A.R.C., S.K.J., C.E.O., A.V., J.F.A.; Writing-Original draft, M.L.D.; Writing- Review and editing, A.R.C., S.K.J., C.E.O., A.V., J.F.A., B.S.M., M.F.P., M.L.D.; Resources, B.S.M., M.F.P.; Funding Acquisition, M.L.D.; Supervision, M.L.D.
Conflicts of interest
The authors declare no competing interests.
Acknowledgments
Flow cytometry was performed by the NHLBI and NICHD Flow Cytometry core facility. Histology and immunohistochemistry were done by the NHLBI Pathology Core Facility. Pamela Robey (NIDCR) and David Schrump (NCI) provided H108 and HBEC cells, respectively. This work was supported by the National Institute of Child Health and Human Development intramural research program (ZIA HD000506, ZIA HD000507).
Published: January 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2021.12.013.
Supplemental information
References
- Abarrategi A., Tornin J., Martinez-Cruzado L., Hamilton A., Martinez-Campos E., Rodrigo J.P., Gonzalez M.V., Baldini N., Garcia-Castro J., Rodriguez R. Osteosarcoma: cells-of-origin, cancer stem cells, and targeted therapies. Stem Cells Int. 2016;2016:3631764. doi: 10.1155/2016/3631764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler-Wailes D.C., Kramer J.A., DePamphilis M.L. Geminin is essential for pluripotent cell viability during teratoma formation, but not for differentiated cell viability during teratoma expansion. Stem Cell Dev. 2017;26:285–302. doi: 10.1089/scd.2016.0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews P.W., Damjanov I., Simon D., Banting G.S., Carlin C., Dracopoli N.C., Fogh J. Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2. Differentiation in vivo and in vitro. Lab. Invest. 1984;50:147–162. [PubMed] [Google Scholar]
- Andrews P.W., Damjanov I., Simon D., Dignazio M. A pluripotent human stem-cell clone isolated from the TERA-2 teratocarcinoma line lacks antigens SSEA-3 and SSEA-4 in vitro, but expresses these antigens when grown as a xenograft tumor. Differentiation. 1985;29:127–135. doi: 10.1111/j.1432-0436.1985.tb00305.x. [DOI] [PubMed] [Google Scholar]
- Bonnet D., Dick J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Bustamante-Marin X., Garness J.A., Capel B. Testicular teratomas: an intersection of pluripotency, differentiation and cancer biology. Int. J. Dev. Biol. 2013;57:201–210. doi: 10.1387/ijdb.130136bc. [DOI] [PubMed] [Google Scholar]
- Campisi J., d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Chakraborty A.R., Robey R.W., Luchenko V.L., Zhan Z., Piekarz R.L., Gillet J.P., Kossenkov A.V., Wilkerson J., Showe L.C., Gottesman M.M., et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: rationale to combine romidepsin with an MEK inhibitor. Blood. 2013;121:4115–4125. doi: 10.1182/blood-2012-08-449140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.G., Mallon B.S., McKay R.D., Robey P.G. Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell. 2014;14:13–26. doi: 10.1016/j.stem.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley L.C., Marfell B.J., Scott A.P., Waterhouse N.J. Quantitation of apoptosis and necrosis by annexin V binding, propidium iodide uptake, and flow cytometry. Cold Spring Harb. Protoc. 2016;2016:953–957. doi: 10.1101/pdb.prot087288. [DOI] [PubMed] [Google Scholar]
- de Lartigue J., Polson H., Feldman M., Shokat K., Tooze S.A., Urbe S., Clague M.J. PIKfyve regulation of endosome-linked pathways. Traffic. 2009;10:883–893. doi: 10.1111/j.1600-0854.2009.00915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z.F., Jin F., Gai H., Chen Y., Wu L., Liu A.L., Chen B., Sheng H.Z. Human embryonic stem cell lines derived from the Chinese population. Cell Res. 2005;15:394–400. doi: 10.1038/sj.cr.7290307. [DOI] [PubMed] [Google Scholar]
- Filipponi D., Emelyanov A., Muller J., Molina C., Nichols J., Bulavin D.V. DNA damage signaling-induced cancer cell reprogramming as a driver of tumor relapse. Mol. Cell. 2019;74:651–663.e8. doi: 10.1016/j.molcel.2019.03.002. [DOI] [PubMed] [Google Scholar]
- Fogh J., Wright W.C., Loveless J.D. Absence of HeLa cell contamination in 169 cell lines derived from human tumors. J. Natl. Cancer Inst. 1977;58:209–214. doi: 10.1093/jnci/58.2.209. [DOI] [PubMed] [Google Scholar]
- Gayle S., Landrette S., Beeharry N., Conrad C., Hernandez M., Beckett P., Ferguson S.M., Mandelkern T., Zheng M., Xu T., et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood. 2017;129:1768–1778. doi: 10.1182/blood-2016-09-736892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorecka J., Kostiuk V., Fereydooni A., Gonzalez L., Luo J., Dash B., Isaji T., Ono S., Liu S., Lee S.R., et al. The potential and limitations of induced pluripotent stem cells to achieve wound healing. Stem Cell Res. Ther. 2019;10:87. doi: 10.1186/s13287-019-1185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J., Guo G., Wray J., Eyres I., Nichols J., Grotewold L., Morfopoulou S., Humphreys P., Mansfield W., Walker R., et al. Oct4 and LIF/Stat3 additively induce Kruppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell. 2009;5:597–609. doi: 10.1016/j.stem.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Hersmus R., van Bever Y., Wolffenbuttel K.P., Biermann K., Cools M., Looijenga L.H. The biology of germ cell tumors in disorders of sex development. Clin. Genet. 2017;91:292–301. doi: 10.1111/cge.12882. [DOI] [PubMed] [Google Scholar]
- Hosogane M., Bosu L., Fukumoto E., Yamada H., Sato S., Nakayama K. Geminin is an indispensable inhibitor of Cdt1 in mouse embryonic stem cells. Genes Cells. 2017;22:360–375. doi: 10.1111/gtc.12482. [DOI] [PubMed] [Google Scholar]
- Huang Y.Y., Kaneko K.J., Pan H., DePamphilis M.L. Geminin is essential to prevent DNA re-replication-dependent apoptosis in pluripotent cells, but not in differentiated cells. Stem Cells. 2015;33:3239–3253. doi: 10.1002/stem.2092. [DOI] [PubMed] [Google Scholar]
- Ikonomov O.C., Sbrissa D., Delvecchio K., Xie Y., Jin J.P., Rappolee D., Shisheva A. The phosphoinositide kinase PIKfyve is vital in early embryonic development: preimplantation lethality of PIKfyve-/- embryos but normality of PIKfyve+/- mice. J. Biol. Chem. 2011;286:13404–13413. doi: 10.1074/jbc.M111.222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomov O.C., Sbrissa D., Shisheva A. Mammalian cell morphology and endocytic membrane homeostasis require enzymatically active phosphoinositide 5-kinase PIKfyve. J. Biol. Chem. 2001;276:26141–26147. doi: 10.1074/jbc.M101722200. [DOI] [PubMed] [Google Scholar]
- Ikonomov O.C., Sbrissa D., Shisheva A. Small molecule PIKfyve inhibitors as cancer therapeutics: translational promises and limitations. Toxicol. Appl. Pharmacol. 2019;383:114771. doi: 10.1016/j.taap.2019.114771. [DOI] [PubMed] [Google Scholar]
- Inwald E.C., Klinkhammer-Schalke M., Hofstadter F., Zeman F., Koller M., Gerstenhauer M., Ortmann O. Ki-67 is a prognostic parameter in breast cancer patients: results of a large population-based cohort of a cancer registry. Breast Cancer Res. Treat. 2013;139:539–552. doi: 10.1007/s10549-013-2560-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie N., Sybirna A., Surani M.A. What can stem cell models tell us about human germ cell biology? Curr. Top Dev. Biol. 2018;129:25–65. doi: 10.1016/bs.ctdb.2018.02.010. [DOI] [PubMed] [Google Scholar]
- Jaiswal S.K., Oh J.J., DePamphilis M.L. Cell cycle arrest and apoptosis are not dependent on p53 prior to p53-dependent embryonic stem cell differentiation. Stem Cells. 2020:1091–1106. doi: 10.1002/stem.3199. [DOI] [PubMed] [Google Scholar]
- Jones T.D., Ulbright T.M., Eble J.N., Baldridge L.A., Cheng L. OCT4 staining in testicular tumors: a sensitive and specific marker for seminoma and embryonal carcinoma. Am. J. Surg. Pathol. 2004;28:935–940. doi: 10.1097/00000478-200407000-00014. [DOI] [PubMed] [Google Scholar]
- Jones T.D., Ulbright T.M., Eble J.N., Cheng L. OCT4: a sensitive and specific biomarker for intratubular germ cell neoplasia of the testis. Clin. Cancer Res. 2004;10:8544–8547. doi: 10.1158/1078-0432.CCR-04-0688. [DOI] [PubMed] [Google Scholar]
- Kawamura A., Miyagawa S., Fukushima S., Kawamura T., Kashiyama N., Ito E., Watabe T., Masuda S., Toda K., Hatazawa J., et al. Teratocarcinomas arising from allogeneic induced pluripotent stem cell-derived cardiac tissue constructs provoked host immune rejection in mice. Sci. Rep. 2016;6:19464. doi: 10.1038/srep19464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline R.C., Bazzett-Matabele L.B. Adnexal masses and malignancies of importance to the colorectal surgeon. Clin. Colon Rectal Surg. 2010;23:63–71. doi: 10.1055/s-0030-1254292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhoo K. Neonatal teratomas. Early Hum. Dev. 2010;86:643–647. doi: 10.1016/j.earlhumdev.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Lee A.S., Tang C., Rao M.S., Weissman I.L., Wu J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013;19:998–1004. doi: 10.1038/nm.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.Y., Johnson R.L., Wichterman-Kouznetsova J., Guha R., Ferrer M., Tuzmen P., Martin S.E., Zhu W., DePamphilis M.L. High-throughput screening for genes that prevent excess DNA replication in human cells and for molecules that inhibit them. Methods. 2012;57:234–248. doi: 10.1016/j.ymeth.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Yang Y., Hong W., Huang M., Wu M., Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct. Target Ther. 2020;5:1. doi: 10.1038/s41392-019-0089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A., Cheng L., Du J., Peng Y., Allan R.W., Wei L., Li J., Cao D. Diagnostic utility of novel stem cell markers SALL4, OCT4, NANOG, SOX2, UTF1, and TCL1 in primary mediastinal germ cell tumors. Am. J. Surg. Pathol. 2010;34:697–706. doi: 10.1097/PAS.0b013e3181db84aa. [DOI] [PubMed] [Google Scholar]
- Lu Y., Dong S., Hao B., Li C., Zhu K., Guo W., Wang Q., Cheung K.H., Wong C.W., Wu W.T., et al. Vacuolin-1 potently and reversibly inhibits autophagosome-lysosome fusion by activating RAB5A. Autophagy. 2014;10:1895–1905. doi: 10.4161/auto.32200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusdottir E., Surani M.A. How to make a primordial germ cell. Development. 2014;141:245–252. doi: 10.1242/dev.098269. [DOI] [PubMed] [Google Scholar]
- Mallon B.S., Chenoweth J.G., Johnson K.R., Hamilton R.S., Tesar P.J., Yavatkar A.S., Tyson L.J., Park K., Chen K.G., Fann Y.C., McKay R.D. StemCellDB: the human pluripotent stem cell database at the National Institutes of Health. Stem Cell Res. 2013;10:57–66. doi: 10.1016/j.scr.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallon B.S., Hamilton R.S., Kozhich O.A., Johnson K.R., Fann Y.C., Rao M.S., Robey P.G. Comparison of the molecular profiles of human embryonic and induced pluripotent stem cells of isogenic origin. Stem Cell Res. 2014;12:376–386. doi: 10.1016/j.scr.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martello G., Smith A. The nature of embryonic stem cells. Annu. Rev. Cell Dev. Biol. 2014;30:647–675. doi: 10.1146/annurev-cellbio-100913-013116. [DOI] [PubMed] [Google Scholar]
- Martin U. Therapeutic application of pluripotent stem cells: challenges and risks. Front Med. (Lausanne) 2017;4:229. doi: 10.3389/fmed.2017.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoletopoulou V., Markaki M., Palikaras K., Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta. 2013;1833:3448–3459. doi: 10.1016/j.bbamcr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- O'Connell C.E., Vassilev A. Combined inhibition of p38MAPK and PIKfyve synergistically disrupts autophagy to selectively target cancer cells. Cancer Res. 2021;81:2903–2917. doi: 10.1158/0008-5472.CAN-20-3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterhuis J.W., Looijenga L.H.J. Human germ cell tumours from a developmental perspective. Nat. Rev. Cancer. 2019;19:522–537. doi: 10.1038/s41568-019-0178-9. [DOI] [PubMed] [Google Scholar]
- Pera M.F., Blasco Lafita M.J., Mills J. Cultured stem-cells from human testicular teratomas: the nature of human embryonal carcinoma, and its comparison with two types of yolk-sac carcinoma. Int. J. Cancer. 1987;40:334–343. doi: 10.1002/ijc.2910400309. [DOI] [PubMed] [Google Scholar]
- Pierpont T.M., Lyndaker A.M., Anderson C.M., Jin Q., Moore E.S., Roden J.L., Braxton A., Bagepalli L., Kataria N., Hu H.Z., et al. Chemotherapy-induced depletion of OCT4-positive cancer stem cells in a mouse Model of malignant testicular cancer. Cell Rep. 2017;21:1896–1909. doi: 10.1016/j.celrep.2017.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rago C., Huso D.L., Diehl F., Karim B., Liu G., Papadopoulos N., Samuels Y., Velculescu V.E., Vogelstein B., Kinzler K.W., Diaz L.A., Jr. Serial assessment of human tumor burdens in mice by the analysis of circulating DNA. Cancer Res. 2007;67:9364–9370. doi: 10.1158/0008-5472.CAN-07-0605. [DOI] [PubMed] [Google Scholar]
- Sekita Y., Nakamura T., Kimura T. Reprogramming of germ cells into pluripotency. World J. Stem Cell. 2016;8:251–259. doi: 10.4252/wjsc.v8.i8.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G., Guardia C.M., Roy A., Vassilev A., Saric A., Griner L.N., Marugan J., Ferrer M., Bonifacino J.S., DePamphilis M.L. A family of PIKFYVE inhibitors with therapeutic potential against autophagy-dependent cancer cells disrupt multiple events in lysosome homeostasis. Autophagy. 2019;15:1694–1718. doi: 10.1080/15548627.2019.1586257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla V., Rao M., Zhang H., Beers J., Wangsa D., Wangsa D., Buishand F.O., Wang Y., Yu Z., Stevenson H.S., et al. ASXL3 is a novel pluripotency factor in human respiratory epithelial cells and a potential therapeutic target in small cell lung cancer. Cancer Res. 2017;77:6267–6281. doi: 10.1158/0008-5472.CAN-17-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. Formative pluripotency: the executive phase in a developmental continuum. Development. 2017;144:365–373. doi: 10.1242/dev.142679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldani C., Scovassi A.I. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis. 2002;7:321–328. doi: 10.1023/a:1016119328968. [DOI] [PubMed] [Google Scholar]
- Su W.X., Li Q.Z., Zhang L.Q., Fan G.L., Wu C.Y., Yan Z.H., Zuo Y.C. Gene expression classification using epigenetic features and DNA sequence composition in the human embryonic stem cell line H1. Gene. 2016;592:227–234. doi: 10.1016/j.gene.2016.07.059. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induced pluripotent stem cells in medicine and biology. Development. 2013;140:2457–2461. doi: 10.1242/dev.092551. [DOI] [PubMed] [Google Scholar]
- Tewey K.M., Rowe T.C., Yang L., Halligan B.D., Liu L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226:466–468. doi: 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- Zhu W., Depamphilis M.L. Selective killing of cancer cells by suppression of geminin activity. Cancer Res. 2009;69:4870–4877. doi: 10.1158/0008-5472.CAN-08-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W., Lee C.Y., Johnson R.L., Wichterman J., Huang R., DePamphilis M.L. An image-based, high-throughput screening assay for molecules that induce excess DNA replication in human cancer cells. Mol. Cancer Res. 2011;9:294–310. doi: 10.1158/1541-7786.MCR-10-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J., Liu Y., Wang J., Liu Z., Lu Z., Chen Z., Li Z., Dong B., Huang W., Li Y., et al. Establishment and genomic characterizations of patient-derived esophageal squamous cell carcinoma xenograft models using biopsies for treatment optimization. J. Transl. Med. 2018;16:15. doi: 10.1186/s12967-018-1379-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.