

Abstract
ROS1 fusion proteins resulting from chromosomal rearrangements of the ROS1 gene are targetable oncogenic drivers in diverse cancers. Acquired resistance to targeted inhibitors curtails clinical benefit and response durability. Entrectinib, a NTRK/ROS1/ALK targeted tyrosine kinase inhibitor (TKI), was approved for the treatment of ROS1 fusion-positive NSCLC in 2019. In addition, lorlatinib and repotrectinib are actively being explored in the setting of treatment naïve or crizotinib-resistant ROS1 fusion driven NSCLC. Here, we employed an unbiased forward mutagenesis screen in Ba/F3 CD74-ROS1 and EZR-ROS1 cells to identify resistance liabilities to entrectinib, lorlatinib, and repotrectinib. ROS1F2004C emerged as a recurrent entrectinib resistant mutation and ROS1G2032R was discovered in entrectinib and lorlatinib-resistant clones. Cell-based and modeling data show that entrectinib is a dual type I/II mode inhibitor, and thus liable to both types of resistant mutations. Comprehensive profiling of all clinically relevant kinase domain mutations showed that ROS1 L2086F is broadly resistant to all type I inhibitors, but remains sensitive to type II inhibitors. ROS1F2004C/I/V are resistant to type I inhibitors, entrectinib and crizotinib, and type II inhibitor, cabozantinib, but retain sensitivity to the type I macrocyclic inhibitors. Development of new, more selective type II ROS1 inhibitor(s) or potentially cycling type I and type II inhibitors may be one way to expand durability of ROS1 targeted agents.
Introduction
Chromosomal rearrangements that generate oncogenic ROS1 fusion proteins are oncogenic drivers in a diverse subset of cancers (1). ROS1-positive cancers are targetable with kinase inhibitors that have offered enormous clinical benefit to patients (1–6). Crizotinib was FDA approved for treating advanced-stage ROS1-rearranged NSCLC (2016), followed by entrectinib (2019) (1). Entrectinib is now experiencing broad clinical adoption for the treatment of ROS1-positive cancer (2). Several additional kinase inhibitors with potent ROS1 inhibitory activity (ROS1i) are under development, including lorlatinib, repotrectinib, ensartinib, and taletrectinib (7–11).
Despite robust benefit of targeted TKI, therapeutic resistance eventually emerges as a clinical challenge in the majority of patients (1,12–19). Thus, knowledge of the spectrum of resistance is necessary to facilitate development of second line treatments (12,20,21). Known mechanisms of resistance include mutation of the ROS1 kinase domain that hinder drug binding (kinase-intrinsic), and bypass pathway alteration wherein tumor cells develop ROS1-independence by activating parallel or downstream signaling pathways (1). Previous studies reported kinase-intrinsic resistance in 8% (18), 38% (15) or 52% (14) of crizotinib resistant patients, and 43% of lorlatinib-resistant patients (15). ROS1G2032R represents a historically recurrent crizotinib-resistant mutation; however, the spectrum of mutations is expanding as new inhibitors are utilized in the clinical setting (14,15,20,22,23). ROS1L2086F was recently reported in lorlatinib-resistant patients (7,15) and a taletrectinib-resistant patient who responded to cabozantinib with this mutation (10). There are no published patient cases of repotrectinib resistance to date. In preclinical studies, ROS1G2032R confers entrectinib-resistance (15,24,25). One entrectinib-resistant patient had a ROS1F2004V mutation, and transition to lorlatinib achieved clinical response (21). Outside this data, no systematic preclinical studies on the resistance liabilities for entrectinib in ROS1-driven cancer models have been reported.
Kinase inhibitors are classified as type I, I½, II, III, IV, V, and VI, based on their binding preferences for distinct kinase conformations (26–29). Type I, I ½ and II inhibitors are more frequently encountered in the pharmacological landscape of cancer therapeutics. Type I inhibitors interact with the ATP binding pocket of the DFG-in kinase conformation while type II inhibitors bind the allosteric pocket of the DFG-out conformation (29,30). Of the currently clinically available inhibitors, our work previously characterized cabozantinib as the only type II ROS1 inhibitor (20).
Diverse 5’ fusion partner genes have been identified in ROS1 rearrangements associated with tumors. The role of 5’ ROS1 fusion partner on subcellular localization on activation of downstream signaling was recently explored (31). Here, we performed unbiased forward mutagenesis screening assays to identify mechanisms of resistance to entrectinib, lorlatinib and repotrectinib using CD74-ROS1 and EZR-ROS1-dependent Ba/F3 cells. To complement cell-based studies, we performed molecular dynamics simulations and molecular docking of inhibitors in wildtype and mutant ROS1 kinases. Cumulatively, our results herein provide further insight into ROS1 next-generation TKI resistance mechanisms, and demonstrate the activity of a broad panel of first and next-generation ROS1 inhibitors against ROS1 kinase domain mutations.
Materials and Methods
Inhibitors
Brigatinib (AP26113), cabozantinib (XL184), ceritinib (LDK378), crizotinib (PF-02341066), entrectinib (RXDX-101), foretinib (GSK1363089), and cycloheximide were purchased from Selleckchem. Lorlatinib (PF-6463922) and repotrectinib (TPX-0005) were purchased from MedChem Express.
Cell Culture
Ba/F3, U20S, and HEK293T cells were purchased from American Type Culture Collection. Parental Ba/F3 cells were cultured in complete medium [RPMI medium 1640 with 10% FBS, 2mmol/L L-glutamine, penicillin, streptomycin and 2ng/mL recombinant murine IL-3]. HEK293T cells were cultured in complete medium [DMEM with 10% (v/v) BGS, 2mmol//L L-glutamine, penicillin, streptomycin]. All cells were tested for mycoplasma every 6 months using the Lonza MycoAlert™ PLUS Kit. The latest date of mycoplasma testing was July 22nd 2021.
DNA transfections
HEK293T cells were transiently transfected using TransIT-LT1 transfection reagent (Mirus Bio) according to the manufacturer’s specifications.
Microscopy
U2OS cells were seeded at a density of 50,000 cells in 35 mm glass bottom dishes. 150–500 ng pCDNA monomeric YFP (mYFP)-tagged ROS1 fusion proteins (CD74-ROS1, EZR-ROS1, and GOPC-ROS1) were transfected using XtremeGene (Roche) per manufacturers protocol. Twenty hours after transfection, cells were mounted on heated stage of Nikon/Yokogawa CSU-W1 Spinning Disk Confocal Microscope. Z-stacks of live cells were acquired at the lowest exposure needed to visualize fusion proteins using a 100× 1.49 Apo TIRF objective. Time lapse imaging was performed with capture rate of 1 frame every second. Image processing was done with Fiji (Image J) software. Time-lapse movies are shown at a rate of 5 frames per second.
Accelerated Mutagenesis Resistance Screen
Ba/F3 cells expressing CD74-ROS1 or EZR-ROS1 were treated overnight with N-ethyl-N-nitrosourea (ENU) (50μg/mL), pelleted, washed, and resuspended in complete RPMI-10% FBS medium. After 48 hours, cells were plated in 96-well plates (2×104 cells per well) in 200 μL complete medium supplemented with the varying concentrations of cabozantinib, crizotinib, entrectinib, lorlatinib, or repotrectinib as indicated. The plates were monitored for outgrowth every 2–3 days for four weeks. Well outgrowth was determined using an inverted microscope and media color. Wells exhibiting outgrowth were transferred and expanded in 24-well plates with 1 mL fresh complete medium supplemented with the same concentration of inhibitor. At confluency, cells were harvested, pelleted and DNA was extracted using QuickExtract™ DNA Extraction Solution (Lucigen). In situations of 100% or near 100% resistant cell outgrowth, DNA was extracted from a randomly selected subset of wells. The CD74-ROS1 or EZR-ROS1 kinase and C-terminal domains were PCR amplified using the primers ROS1 5707F (5’-GACAAAGAGTTGGCTGAGCTG-3’) and ROS1 REV_14 (5’-TCAGACCCATCTCCATATCCA-3’) and bidirectionally sequenced using ROS1 6198F (5’-CTGTGTCTACTTGGAACGGATG-3’) and ROS1 6304R (5’-TCTCTGGCGAGTCCAAAGTC-3’). Chromatographs were aligned to identify mutations using Benchling software
Cell Line Generation
The CD74-ROS1 and EZR-ROS1 mutants were made using site-directed mutagenesis following manufacturer’s protocol (Agilent) using primers listed in Supplementary Table 1. Platinum-E cells (Cell Biolabs, Inc) were transfected with pBABE CD74-ROS1 or pCX4 EZR-ROS1 wildtype and mutant constructs using Biotool DNA transfection reagent to generate replication incompetent, ecotropic retrovirus. Ba/F3 cells were maintained at densities between 0.5×106 and 1.5×106 cells per mL and infected with retrovirus. Cells stably expressing the respective fusions were selected with puromycin treatment (2 μg/ml) before IL-3 withdrawal. To generate IL-3 independent, stable Ba/F3 cell lines, the cells were washed three times with complete medium and seeded at 0.5×106 cells per mL. Cells were counted every 2 days and were expanded as needed to maintain a density of <1.5×106 cells per mL. Cells that grew out after IL-3 withdrawal were maintained in IL-3 free complete medium and used for in vitro assays. For experimental rigor, all transformed (post-IL3 withdrawal) cell lines were sequenced to verify the presence of the desired mutation. Cells were harvested, pelleted, and DNA was extracted using QuickExtract™ DNA Extraction Solution (Lucigen). The ROS1 kinase and C-terminal domains were PCR amplified using the primers ROS1 5707F (5’-GACAAAGAGTTGGCTGAGCTG-3’) and ROS1 REV_14 (5’-TCAGACCCATCTCCATATCCA-3’) and bidirectionally sequenced using ROS1 6198F (5’-CTGTGTCTACTTGGAACGGATG-3’) and ROS1 6304R (5’-TCTCTGGCGAGTCCAAAGTC-3’). Chromatographs were aligned using Benchling software to confirm that only the desired mutations were present and that no other mutations were introduced during viral transduction.
Cell Viability Assays
All inhibitors were prepared as 1 mmol/L stocks in DMSO. Inhibitors were distributed at 2x indicated final concentrations into 384-well plates pre-seeded with 25 μl per well of complete medium using a D300 Digital Dispenser (Hewlett-Packard). Ba/F3 cells expressing wildtype or mutant CD74-ROS1 or wildtype or mutant EZR-ROS1 constructs were seeded at 1000 cells per well in a volume of 25 μl using a Multidrop Combi Reagent Dispenser (Thermo Scientific). Plates were incubated for 72 hours at 37°C, 5% CO2. Viability was measured using a methanethiosulfate (MTS)-based assay (BioVision) and read on a Biotek Synergy 2 plate reader. Each condition was assayed in triplicate. Data were normalized using Microsoft Excel, and IC50 values were calculated using a nonlinear regression analysis in GraphPad Prism.
Immunoblot Analysis
Ba/F3 CD74-ROS1, CD74-ROS1F2004C, CD74-ROS1I2239*, EZR-ROS1, and EZR-ROS1F2004C were treated with the indicated concentrations of inhibitors for 4–6 hours prior to harvest. Ba/F3 cell lines and transfected HEK293T cells were pelleted, washed with ice-cold PBS, and lysed in cell lysis buffer supplemented with 0.25% deoxycholate, 0.05% SDS, and protease and phosphatase inhibitors. Protein concentration was determined using the Pierce™ BCA Protein Assay kit (ThermoFisher Scientific). After protein quantification, lysates were either used for immunoprecipitation with anti-Flag affinity gel (Bimake) or extracted with Laemelli sample buffer for 10 min at 75°C and lysates were run on 4–12% Bis-Tris or 4–20% Tris-glycine precast gradient gels (Invitrogen; ThermoFisher Scientific). Proteins were transferred to nitrocellulose membranes (Prometheus) and probed with phospho-ROS1 Y2274 [3078; 1:1000; Cell Signaling Technology], phospho-ROS1Y2114 [PA5-37350;1:1000; Invitrogen], ROS1 [D4D6; 1:1000; Cell Signaling Technology], phospho-p44/42 MAPK [9101; 1:1000, Cell Signaling Technology], ERK2 [sc-1647; 1:1000; Santa Cruz], p44/42 MAPK (ERK1/2) [4696, Cell Signaling Technology], phospho-S6 [4858; 1:1000, Cell Signaling Technology], S6 [2216; 1:1000, Cell Signaling Technology], GAPDH [OTI2D9;1:5000; Origene], phospho-SHP2 [A5278;1:1000; Bimake], phospho-STAT3 [9145; 1:1000; Cell Signaling Technology], STAT3 [9139; 1:1000; Cell Signaling Technology], Actin [JLA20; 1:5000; Developmental Studies Hybridoma Bank], phospho-Akt [4060; 1:1000; Cell Signaling Technology], Akt [9272; 1:1000; Cell Signaling Technology], phospho-mTOR [5536;1:2000; Cell Signaling Technology], mTOR [2983; 1:2000; Cell Signaling Technology], phospho-4EBP1 [; 1:2000; Cell Signaling Technology], DYKDDDDK (Flag) [8H8L17;1:1000; Invitrogen], phospho-MEK1 [A5191;1:1000;Bimake], GFP [TA150032;1:5000; Origene], and phospho-p70S6K [A5033;1:1000; Bimake]. Signal was detected using a BioRad ChemiDoc imaging station or a LI-COR Odyssey imaging system with use of HRP-conjugated or IR dye secondary antibodies, respectively.
Cycloheximide treatment
Ba/F3 CD74-ROS1 and CD74-ROS1I2239* were seeded at 1×106 cells per mL and treated with 50μg/mL cycloheximide (CHX). At 2, 4, 6, and 8 hours post-CHX addition, 2×106 cells were pelleted, washed with ice-cold PBS and lysed in cell lysis buffer supplemented with 0.25% deoxycholate, 0.05% SDS, and protease and phosphatase inhibitors.
Molecular Dynamic Simulations
The ROS1 kinase domain structure was modeled in both the active and inactive forms using the homology modeling module within YASARA version 17.12.2 as described earlier (32). Mutations of interest were introduced into the kinase domain for active and inactive conformations, and the structures were energy minimized using an AMBER14 forcefield. Point mutations can affect the function of the ROS1 active and inactive kinase domains by altering the structure, stability, dynamics, and/or interactions with target substrates and drugs. Therefore, molecular dynamics simulations were performed on the ROS1 WT and mutant kinases to sample the local conformational space using periodic boundary conditions under an NPT ensemble. The kinase domains were solvated using 16068 TIP3 water molecules in 0.9% sodium chloride, pH 7.4, 298 K, at atmospheric pressure, using a simulation cell with a distance minimum of 8 angstrom in all directions from the protein. Electrostatic interactions were computed using the particle mesh Ewald (PME) simulation method with a 10 angstrom short-range electrostatic cutoff. The short-range cutoff used for Van der Waals interactions during the simulation was also 10 angstrom. First, each system was energy-minimized for 5000 steps each using steepest-decent and conjugate gradient algorithms; subsequently, the solvent and ions were equilibrated for 100 ps in constant pressure (NPT) ensembles, while the substrates of the protein were restrained harmonically using a force constant of 1000 kJ mol−1 nm−2. Finally, the proteins and protein-ligand complexes were simulated via MD for 150 ns and snapshots of all trajectories were recorded every 250 ps for further analysis. The RMSD for the C-alpha atoms between the initial minimized structure and individual poses during the simulation were calculated for the A-loop (residues 1950–1960), P-loop (residues 2101–2126), the C-helix (residues 1986–2004) and the entire kinase model (RMSD total). The data were plotted using Prism GraphPad software.
Principal Component Analysis (PCA)
PCA, a statistical technique that can identify independent motions in proteins, was conducted on the molecular dynamics trajectories for the various active and inactive conformations of WT and mutant ROS1 kinase domains. Each simulation snapshot was plotted on the subspace spanned by the first three principal components. Cluster analysis through Wolfram Mathematica was performed to identify conformational clusters, and the “average” conformational pose representing the center of each cluster was computed (33). The poses were energy minimized (32,33) and the hydrogen bonding was optimized. Each of these poses were used in subsequent docking experiments at a pH 7.4.
Molecular Docking
Molecular docking studies were performed using VINA Autodock version 4.2 program (The Scripps Research Institute, La Jolla, CA, USA). Each pose, corresponding to the center of a cluster identified from the PCA analysis, was used for the docking experiments. Before docking any drug into the active site, the kinase poses were examined for incorrect or missing sidechains. Substrate cofactor and water molecules were deleted from the protein, and a simulation cell of 30 × 30 × 30 Å was centered on the drug-binding active site. Five rotameric structural ensembles corresponding to each pose were calculated, and the drugs were then docked with VINA AutoDock 32 times for each rotamer, producing a total of 160 docks per drug for each cluster center. Ligand conformation and position was randomized before the docking routine initiated its search for a local minimum. The binding energies are plotted as a histogram for all kinase conformations for a given drug.
Results
CD74-ROS1 and EZR-ROS1 exhibit distinct subcellular localization but similar signaling pathway activation and inhibitor sensitivity
ROS1 fusion-positive NSCLC patients represent the largest clinical cohort treated with ROS1 targeted inhibitors (ROS1i). CD74-ROS1 and EZR-ROS1 are recurrent fusions, present in ~44% and ~16% of NSCLC patients, respectively. GOPC-ROS1 is expressed in several cancer subtypes with prevalence in glioblastoma (1). Domain organization of these fusions in reference to ROS1 receptor is illustrated in Fig. 1A. To examine their subcellular localization, we expressed fluorescently tagged CD74-ROS1, EZR-ROS1, SLC34A2-ROS1, and GOPC-ROS1 in U2OS cells and live imaging revealed that these fusions reside in distinct intracellular compartments (Fig. 1B, Supplementary Movies 1–8). CD74-ROS1 is in the endoplasmic reticulum (ER) where it is in structures resembling stacked ER cisternae. CD74-ROS1 has two transmembrane domains; however, in our experiments the protein was not localized to the plasma membrane in any cells. EZR-ROS1 containing an amino-terminal ‘Four-point-one, Ezrin, Radixin, Moesin’ (FERM) domain exhibits punctate localization in the juxta-plasma membrane region (Fig. 1B, Supplementary Movies); this is consistent with the distribution of normal ezrin (34,35). GOPC-ROS1 is a cytoplasmic protein, and SLC34A2-ROS1 distributes to the ER and in puncta that bear strong resemblance to membraneless cytoplasmic granules as recently reported for ALK and RET fusions (36) (Fig. 1B, Supplementary Movies). Representative supplementary movies depict Z-stack animation and time-lapse imaging of CD74-ROS1, EZR-ROS1, GOPC-ROS1 and SLC34A2-ROS1. Taken together, these data show that the 5’ fusion partners target ROS1 kinase to discrete subcellular locations.
Figure 1. Characterization of subcellular localization, activity, and inhibitor sensitivity of ROS1 fusions.
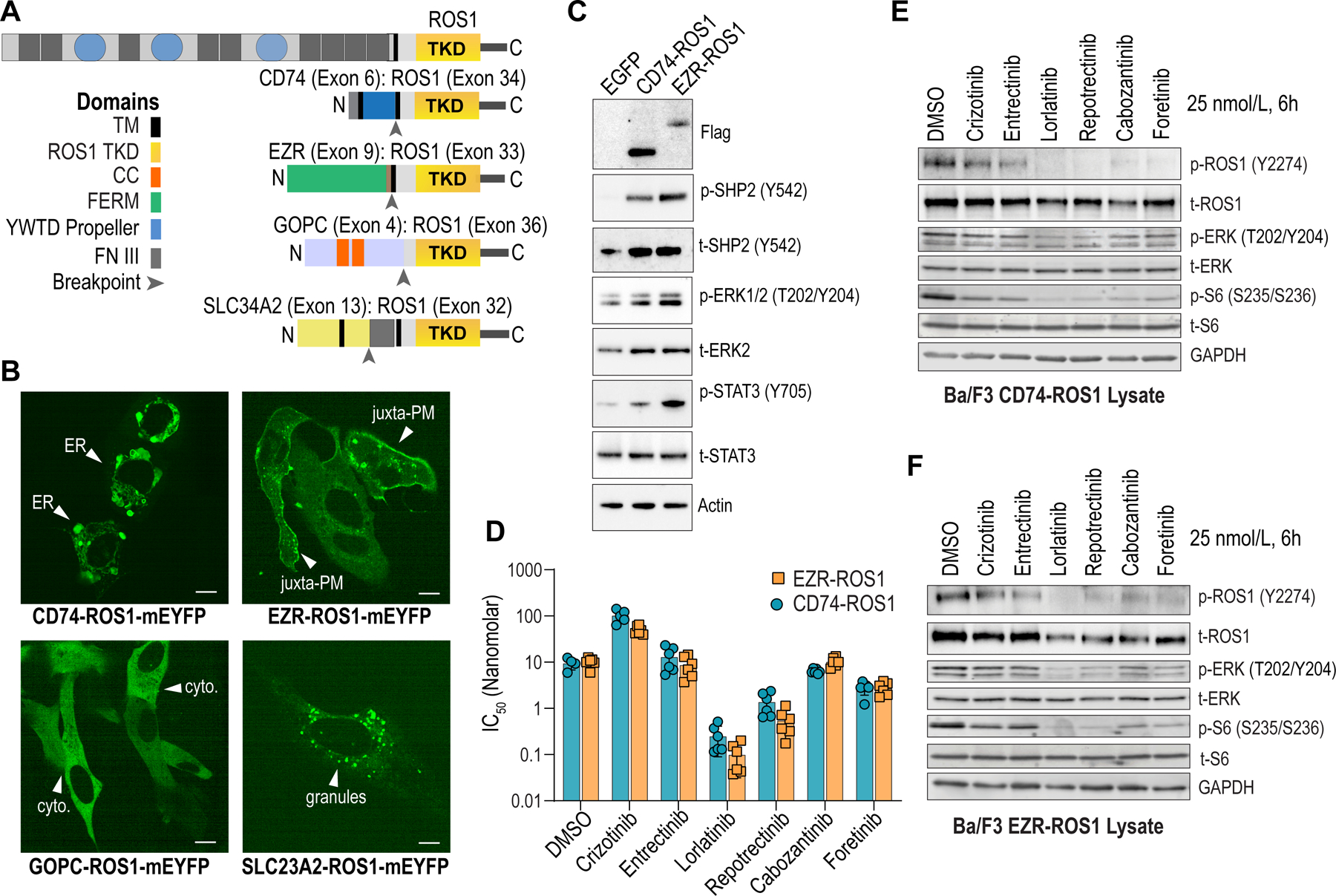
A, Domain organization of ROS1 fusions. TKD - ROS1 tyrosine kinase domain; TM – Transmembrane domain; FERM – Protein 4.1 Ezrin Radixin, Moesin domain; CC – coiled-coiled domain; YWTD Propeller - β Propeller domain; FN III – fibronectin type III repeat. B, Live cell imaging (spinning disk confocal microscopy) of monomeric yellow fluorescent protein (mYFP)-tagged ROS1 fusion proteins. White arrowheads indicate endoplasmic reticulum (ER), juxta-plasma membrane (juxta-PM), cytoplasm (cyto), and membraneless cytoplasmic granules (granules). C, Immunoblot analysis of the phosphorylated and total proteins shown in transfected HEK293T cell lysates. Anti-Flag antibody was used to detect Flag-tagged ROS1 fusion. D, IC50 values indicate relative potency (nanomolar) of the indicated ROS1i for Ba/F3 CD74-ROS1 or EZR-ROS1; six replicate data points are shown. E & F, Immunoblot analysis of the phosphorylated and total proteins shown DMSO or ROS1i-treated (25 nmol/L, 6 hours) in cell lysates generated from Ba/F3 CD74-ROS1 and EZR-ROS1, respectively. Representative immunoblots from two independent experiments are shown.
To test if CD74-ROS1 and EZR-ROS1 exhibit differences in catalytic activity and/or stimulation of downstream signaling pathways, we transiently transfected HEK293T cells with Flag-tagged ROS1 fusions, and assessed relative ROS1 auto-phosphorylation. Immunoprecipitation of Flag-CD74-ROS1 and Flag-EZR-ROS1 showed equivalent ROS1 phosphorylation (pROS1 relative to tROS1) (Supplementary Fig. 1A). Immunoblotting for signaling effectors in cell lysates shows that both fusion proteins drive robust activation of SHP2, ERK1/2, and STAT3, modest activation of mTOR and 4EBP1 but no measurable activation of AKT or p70S6K in this model system (Fig. 1C, Supplementary Fig. S1 B–D).
To compare relative TKI sensitivity of CD74-ROS1 and EZR-ROS1, we generated transformed Ba/F3 cell lines. Ba/F3 is a well-established model system to interrogate drug efficacy and mechanisms of resistance (20,22,23,25,37). No substantive difference in the transformative capacity of CD74-ROS1 versus EZR-ROS1 expressing cells was noted (Supplementary Fig. 1E). Dose-response cell viability data show that Ba/F3 CD74-ROS1 and EZR-ROS1 cells exhibit comparable sensitivity to crizotinib, ceritinib entrectinib, and foretinib (Fig. 1D, Supplementary Fig. 1F - H). While EZR-ROS1 cells are modestly more sensitive to lorlatinib and repotrectinib than CD74-ROS1, these differences are unlikely to be biologically meaningful given sub-nanomolar IC50. Immunoblotting shows that 6-hour treatment with 25 nmol/L inhibitor (crizotinib, entrectinib, lorlatinib, repotrectinib, cabozantinib and foretinib) reduces ROS1, ERK1/2 and S6 phosphorylation that correlates to observed potency in Ba/F3 cell viability assays (Fig. 1E–F). Overall, CD74-ROS1 and EZR-ROS1 exhibit similar catalytic activity and inhibitor sensitivity despite exhibiting differences in subcellular localization.
Accelerated mutagenesis screening to discover entrectinib-resistant ROS1 mutations
Entrectinib is rapidly becoming the front-line therapy for ROS1-positive cancer patients (2). The spectrum of crizotinib-resistant ROS1 mutations in preclinical (22,23) and clinical settings is known (5,12–14,18,38). In contrast, resistance profiling for entrectinib in ROS1-driven cancer models has not been fully explored. Here we conducted a saturated mutagenesis screen using Ba/F3 CD74-ROS1 or EZR-ROS1 cells. We observed a concentration-dependent reduction in the percentage of wells and spectrum of mutations that exhibited outgrowth in the case of Ba/F3 CD74-ROS1 cells (Supplementary Data & Supplementary Figure S2). Specifically, 10.6%, 1.5%, and 0.26% of wells with CD74-ROS1 cells exhibited resistant populations when challenged with 25, 50 or 100 nmol/L entrectinib, respectively. We did not recover any resistant CD74-ROS1 clones with 1000 nmol/L entrectinib challenge. In the case of EZR-ROS1, 100% of the wells grew out when challenged with 50 and 100 nmol/L entrectinib and 22.9% of wells grew out with 250 nmol/L entrectinib. Using Sanger sequencing, we profiled recovered clones for acquired ROS1 intracellular domain mutations, including the juxtamembrane (JM) domain, tyrosine kinase domain (TKD), and carboxy-terminal domain mutations (C-term). We found the following ROS1 TKD mutations in CD74-ROS1 cells that grew out in entrectinib challenged wells: F2004C, Q2012K, D2033N, M2073T and K2117Q. Three C-term truncating mutations (stop gained) were also found: Y2227*, I2239*, E2280*. Additional kinase domain mutations, V1979A, L1982F, S1986T, G2032R and D2113G co-occurred with I2239*. Given the limitations of Sanger sequencing, we were unable to definitively conclude whether the C-term truncation mutants that co-occurred with the ROS1 TKD mutation were on the same strand, in the same cell (as a compound mutation), or whether this represents two subclonal cell populations – one harboring the C-term truncation and the second with the TKD mutations. For testing the impact of C-term truncation, we engineered I2239* alone or as a compound mutation with S1986F and F2004C as described below.
In entrectinib resistant EZR-ROS1 cells, a juxtamembrane deletion mutant, A1924_I1934del was recovered in a subset of clones, and the following ROS1 TKD mutations were found: F2004C, D2113N, and G2032R/G2331C (Supplementary Fig. S2, Supplementary Data). A subset of EZR-ROS1 resistant cells also harbored ROS1R2116K concurrent with other kinase domain mutation. We retrospectively discovered that ROS1R2116K was present a priori in ≤ 10% of parental cells, and was likely introduced during viral transduction since retroviral replication is error-prone. We tested the impact of ROS1R2116K and found that it has no influence on catalytic activity, transformative capacity, and importantly ROS1i sensitivity as compared to wildtype EZR-ROS1 cells (Supplementary Fig. 3).
G2032R and F2004C are recurrent resistant liabilities to entrectinib treatment.
To evaluate the sensitivity of these mutations to entrectinib as well as other ROS1i, we re-engineered CD74-ROS1 and EZR-ROS1 mutant cell lines. Engineered Ba/F3 CD74-ROS1 V1979A, L1982F, S1986T, F2004C, Q2012K, G2032R, D2033N, M2073T, and D2113G retained transformative capacity (Supplementary Fig. 4A). All cell lines were validated with Sanger sequencing after IL-3 withdrawal to ensure that no additional mutations were gained during the retroviral transduction or withdrawal steps. Surprisingly, with the exception of CD74-ROS1F2004C and CD74-ROS1G2032R, the other TKD mutants recovered from clones that grew out in ENU assay (V1979A, L1982F, S1986T, Q2012K, D2033N, M2073T, D2113G) were sensitive to entrectinib in re-engineered cells (Table 1), suggesting that these mutations likely do not affect entrectinib binding. CD74-ROS1G2032R is an established multi-ROS1 TKI resistant clinical liability (14,38) and thus validates the methodology.
Table 1.
Entrectinib sensitivity in re-engineered Ba/F3 cell lines
| Fusion | Mutation | ROS1 Domain | Entrectinib IC50 (nmol/L) |
|---|---|---|---|
| CD74-ROS1 | - | 19.4 ± 2.3 | |
| CD74-ROS1 | V1979A | Kinase | 7.3 ± 1.1 |
| CD74-ROS1 | L1982F | Kinase | 5.2 ± 0.3 |
| CD74-ROS1 | S1986T | Kinase | 5.3 ± 0.9 |
| CD74-ROS1 | F2004C | Kinase | 177 ± 9.8 |
| CD74-ROS1 | Q2012K | Kinase | 19.7 ± 3.8 |
| CD74-ROS1 | G2032R | Kinase | > 2000 |
| CD74-ROS1 | D2033N | Kinase | 21.4 ± 4.2 |
| CD74-ROS1 | M2073T | Kinase | 11.4 ± 1.0 |
| CD74-ROS1 | D2113G | Kinase | 21.4 ± 2.9 |
| CD74-ROS1 | I2239* | C-term truncation | 10.6 ± 0.6 |
| EZR-ROS1 | - | 5.2 ± 0.9 | |
| EZR-ROS1 | A1924_I1934del | Juxtamembrane | 3.4 ± 0.2 |
| EZR-ROS1 | F2004C | Kinase | 79.2 ± 7.9 |
| EZR-ROS1 | G2032R | Kinase | 767.0 ± 26.3 |
| EZR-ROS1 | D2113N | Kinase | 15.9 ± 2.1 |
While ROS1G2032R was characterized extensively in the past, less is known about the F2004 substitutions. Here we further explored the impact of ROS1F2004C substitution on ROS1i-sensitivity. Immunoblotting of Ba/F3 CD74-ROS1F2004C cells treated with first and next generation ROS1i showed that CD74-ROS1F2004C exhibits resistance to entrectinib, crizotinib, cabozantinib and taletrectinib (at 25 nmol/L) (Fig. 2A). A higher concentration (150 nmol/L) of entrectinib partially inhibits ROS1 autophosphorylation but the effector pathways (SHP2 and ERK1/2) are not sufficiently suppressed (Fig. 2A). In contrast, next-generation macrocyclic inhibitors, lorlatinib and repotrectinib, retain potency for CD74-ROS1F2004C, including downstream attenuation of SHP2 and ERK1/2 phosphorylation.
Figure 2. ROS1F2004C induces resistance to entrectinib.
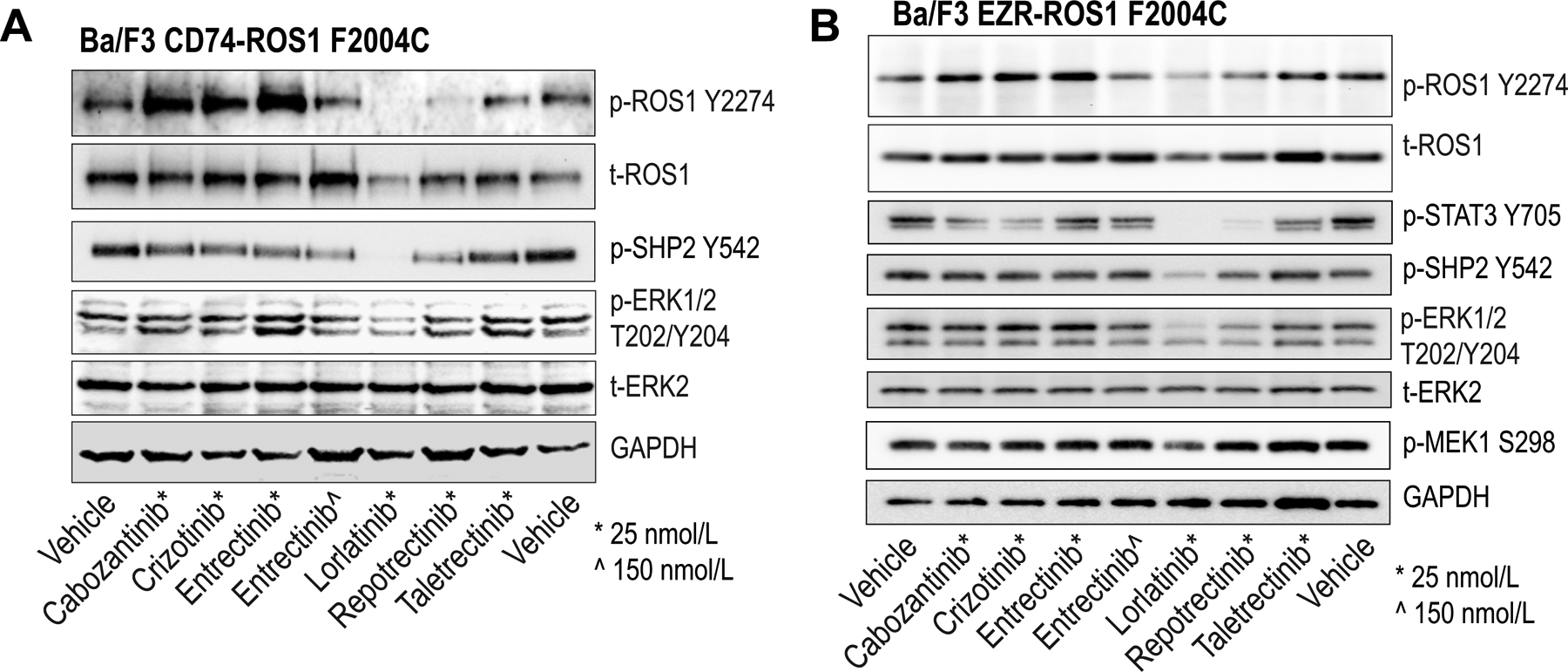
A & B, Immunoblot analysis of the phosphorylated and total proteins shown from Ba/F3 CD74-ROS1F2004C (A) and Ba/F3 EZR-ROS1F2004C (B) lysates treated with the indicated inhibitors for 4 hours. Vehicle indicates DMSO treatment.
C-term truncating CD74-ROS1L2223* was previously observed in mutagenesis screenings with ceritinib (23) and cabozantinib (20), however, its functional impact is still unknown. Thus, here we engineered Ba/F3 CD74-ROS1I2239* and confirmed that CD74-ROS1I2239* retains catalytic activity, however effector pathway activation (phosphorylation of SHP2, STAT3, ERK1/2, p70S6K and S6) was attenuated in CD74-ROS1I2239* cells as compared to CD74-ROS1WT cells (Supplementary Fig. S5A). We consistently observed ~3–7 fold higher total protein levels of CD74-ROS1I2239* compared to CD74-ROS1WT and in reference to GFP, a surrogate marker for viral copy number integration (Supplementary Fig. S5B). Thus, we hypothesized that C-term truncation stabilizes ROS1 resulting in a longer-half life. To test this, we treated cells with the protein synthesis inhibitor, cycloheximide, and observed that the half-life of CD74-ROS1I2239* is ≥ 2-fold longer than CD74-ROS1WT (Supplementary Fig. S5C, D). Ba/F3 CD74-ROS1I2239* cells have 3.8-fold lower sensitivity to crizotinib (IC50 = 35.2 versus 9.2 nmol/L for CD74-ROS1WT cells); however we noted no meaningful differences in effectiveness of ROS1i inhibitors (Supplementary Fig. 5E, F). Immunoblotting confirms on-target inhibition of CD74-ROS1I2239* (p-ROS1 Y2214 and Flag), and of downstream effectors (Supplementary Fig. 5E, G).
As with CD74-ROS1, we re-engineered the following Ba/F3 EZR-ROS1 cell lines in order to test the potential contribution of mutations recovered in ENU assays: A1924_I1934del, F2004C, G2032R, G2032R/G2331C, and D2113N. All mutants retained transformative capacity (Supplementary Fig. 4B). EZR-ROS1F2004C and EZR-ROS1G2032R are 13.4- and 76-fold less sensitive to entrectinib, respectively, compared to EZR-ROS1WT. EZR-ROS1D2113N exhibits only a 3-fold decrease in sensitivity to entrectinib (Table 1). The JM mutant, EZR-ROS1A1924_I1934del modestly sensitizes to entrectinib, and thus likely does not contribute to cellular resistance. Immunoblotting of ROS1i-treated Ba/F3 EZR-ROS1F2004C cell lysates confirms resistance of this mutant to cabozantinib, crizotinib, entrectinib and taletrectinib (25 nmol/L); while 150 nmol/L entrectinib treatment partially inhibits ROS1, it is insufficient to block effector pathway phosphorylation (Fig 2B).
Molecular dynamic simulations reveal entrectinib is a type I/II inhibitor and its binding is impacted by F2004C mutation, particularly in the DFG-out conformation
Kinase domains are dynamic structures that adopt numerous conformations, which affect inhibitor binding. These conformations are broadly categorized as DFG-in (active, Type I) or DFG-out (inactive, Type II) with the DFG motif referring to the consequential amino acids, aspartic acid (D), phenylalanine (F), glycine (G) at the start of the structurally-flexible activation loop (A-loop). There is no crystal structure of the ROS1 DFG-out conformation; however, we previously generated a ROS1 DFG-out homology model that we validated via experimental mutagenesis studies (20). The superimposed models of wildtype ROS1 DFG-in and DFG-out highlight the structural differences between these conformations (Fig. 3A); the P-loop, αC-helix, and A-loop; the hinge region and ATP-binding pocket differences are illustrated. The majority of ROS1i (crizotinib, entrectinib, lorlatinib, repotrectinib and brigatinib) are classified as ATP-competitive type I inhibitors that bind to ROS1 DFG-in. We previously established that cabozantinib and foretinib are type II inhibitors that preferentially bind to ROS1 DFG-out and partially occupy the hydrophobic pocket near the ATP-binding site (20). Notably, in this previous study, we identified the F2004 substitutions as a resistant liability for these type II inhibitors.
Figure 3. Structural assessment of ROS1F2004C with molecular dynamic simulation and inhibitor docking studies reveal entrectinib binds both DFG-in and DFG-out ROS1 kinase domain.
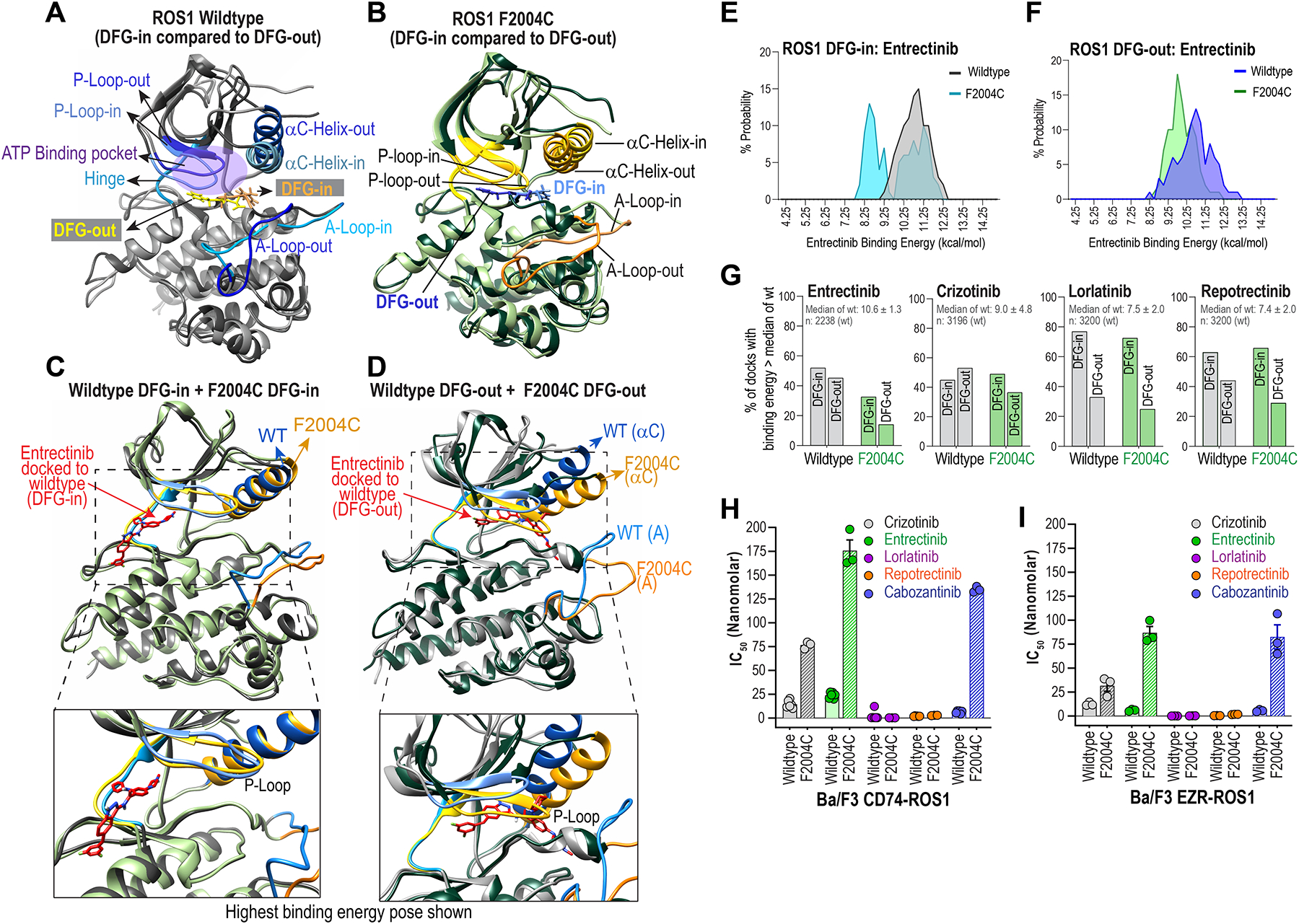
A & B, Superimposed wildtype ROS1 (A) and ROS1F2004C mutant crystal structure models in DFG-in and DFG-out conformations are shown to highlight distinctions in structural features: Activation loop (A-Loop), αC Helix, P-loop, ATP-binding pocket, and the positioning of the aspartic acid (D), phenyalanine (F) and glycine (G), i.e., DFG motif in the two conformations. C & D, Superimposed crystal structures show conformational differences between wildtype ROS1 and ROS1F2004C in their DFG-in (C) and DFG-out (D) states. E & F, Histograms show binned probability distribution of binding energy (kcal/mol) for ROS1WT and ROS1F2004C in the DFG-in (E) and DFG-out (F) kinase conformation. Binding energies were determined with Yasara; in this case the higher binding energies reflect more favorable binding. G, Percentage of docks whose binding energy was greater than the median binding energy of indicated inhibitor docking to ROS1WT DFG-in and ROS1WT DFG-out is plotted. The median binding energy ± standard deviation and the total number of docks for the ROS1WT DFG-in & ROS1WT DFG-out conformations is indicated inset within graph and panel below graph. H & I, IC50s of crizotinib, entrectinib, lorlatinib, repotrectinib and cabozantinib as derived from dose-response cell viability assay with Ba/F3 CD74-ROS1 (H) and Ba/F3 EZR-ROS1 (I), wildtype or F2004C mutant cells.
In this study, we uncovered ROS1F2004C as recurrent entrectinib-resistant mutation in the unbiased ENU accelerated mutagenesis experiments described above. To interrogate the structural basis of resistance and differences between ROS1WT and ROS1F2004C in their DFG-in and DFG-out states, we developed models of ROS1F2004C DFG-in and DFG-out (Figure 3A–D). Superimposed wildtype ROS1 and ROS1F2004C DFG-in and DFG-out are shown in Fig. 3A and 3B, respectively. Mutations can affect inhibitor interactions both directly by altering molecular contacts, and indirectly through altering kinase structure, stability, and dynamics. To investigate these differences, we performed molecular dynamics (MD) simulations of ROS1WT and ROS1F2004C DFG-in and DFG-out conformations. ROS1WT and ROS1F2004C were stable during the course of the simulation, and no large-scale conformational change was observed (Supplementary Fig. 6). Root mean square deviation (RMSD) of the protein was calculated from the simulations (Supplementary Fig. S6). The A-loop of ROS1F2004C-DFG-in exhibits substantially higher flexibility than A-loop of ROS1WT-DFG-in during simulation (Supplementary Fig. S6A). ROS1F2004CDFG-out A-loop is marginally more dynamic than ROS1WT-DFG-out A-loop (Supplementary Fig. S6B). ROS1F2004C P-loop is slightly more rigid ROS1WT P-loop in the DFG-in conformation, whereas both wildtype and mutant are relatively stable in the DFG-out conformation (Supplementary Fig. S6C and D). The αC-helix is relatively dynamic in both ROS1F2004C and ROS1WT, however the overall change in angle of rotation appears slightly reduced in ROS1F2004C compared to ROS1WT in the DFG-out conformation (Supplementary Fig. S6E and F). These data suggest two, not mutually exclusive possibilities: the enhanced dynamics of ROS1F2004C destabilizes entrectinib binding, or the alterations in P loop and α-C helix alter the binding pocket.
Since kinase domains adopt a dynamic ensemble of poses, we employed Principal Component Analysis (PCA) and a clustering algorithm to identify putative conformational clusters, and computed an average pose for each group of poses. Since PCA is a statistical technique used to identify the most essential independent correlations, or components, within a data set, here, it helped reveal relevant global structural changes by plotting them in a conformational space. PCA analyses reveal that ROS1WT & ROS1F2004C sample multiple poses in DFG-in and DFG-out conformations and that the four kinases, ROS1WT and ROS1F2004C in DFG-in or DFG-out, adopt distinct conformational clusters (Supplementary Fig. S7A, B). Comparison of six different poses for each of these four kinase subtypes is shown in Supplementary Figure S7C and D. The observed increase in dynamicity of the DFG-out conformation, especially for ROS1F2004C, is apparent (Supplementary Figure S7C, D).
To estimate inhibitor-binding affinities for ROS1WT and ROS1F2004C, we performed molecular docking studies for entrectinib, crizotinib, lorlatinib and repotrectinib. The average poses for the WT and variant kinase in the DFG-in and DFG-out states were docked with each inhibitor one hundred and sixty times. Although high-resolution crystal structure(s) of entrectinib bound ROS1 are not available, it is presumed that entrectinib is a canonical type I inhibitor. However, our docking studies indicate that entrectinib exhibits high affinity to specific poses of both ROS1 DFG-in (type I) and DFG-out (type II) kinases, involving unique binding pockets. In ROS1WT DFG-in, the inhibitor occludes the ATP-binding pocket through contacts with residues in the hinge region and solvent front (Supplementary Fig. 8A); in the DFG-out conformation, the highest affinity binding involves the back hydrophobic pocket and residues F2004, F2075 and R2078. (Supplementary Fig. S8B). As shown in Fig. 3C, the superimposed DFG-in conformations of ROS1F2004C and ROS1WT adopt relatively similar conformations. In contrast, the DFG-out conformations of ROS1F2004C show enhanced dynamics within the P-loop, α-C helix, and the A-loop relative to ROS1WT (Fig. 3D), and likely adversely affect entrectinib-binding in this kinase conformation.
To quantitatively assessthe impact of ROS1F2004C on entrectinib binding, we binned binding energies from the molecular docking studies for ROS1F2004C and ROS1WT in DFG-in and DFG-out states as depicted in the histograms in Fig. 3E and F, respectively. Here, higher binding energies indicate more favorable interactions. In both DFG-in and DFG-out, entrectinib exhibits weaker binding to ROS1F2004C, as indicated by the leftward shift towards lower binding energy states. The entrectinib binding energy histogram for ROS1F2004C DFG-in is bimodal; examination of the raw data indicates this is due to a generally lowered entrectinib binding affinity in one of the six poses adopted by ROS1F2004C DFG-in. However, understanding this conformer’s effect on the overall ensemble affinity is difficult from this data alone since the protein’s residence time in each pose in the cellular context is not known. We also performed docking studies with crizotinib, lorlatinib and repotrectinib using DFG-in and DFG-out poses of ROS1F2004C and ROS1WT (Supplementary Fig. S9). Crizotinib binding to both ROS1F2004C DFG-in and DFG-out is weakened, albeit not as severely as for entrectinib in ROS1F2004C DFG-out. Lorlatinib and repotrectinib exhibit no reductions in binding to ROS1F2004C DFG-in or DFG-out conformations. Next, we computed the percentage of docks to ROS1F2004C that had binding energy greater than the median for the corresponding drug:wild-type kinase dock (Fig. 3G, Table 2). These data reveal new observations regarding the nuanced binding preferences of these four ROS1 inhibitors for the DFG-in or DFG-out conformations, and the corresponding impact of the F2004C mutation on these interactions. Entrectinib binds equivalently to both ROS1 DFG-in and ROS1 DFG-out, however its binding is most strongly weakened for ROS1F2004C DFG-out. To our surprise, in silico docking data suggest that crizotinib also binds both ROS1 DFG-in and DFG-out (Fig. 3G, Supplementary Figure S9), and its interaction with ROS1F2004C is disrupted only in the DFG-out conformation (Fig. 3G). Lorlatinib is a preferential type I inhibitor with best binding to DFG-in, and ROS1F2004C has no effect on lorlatinib binding in either conformation. Finally, repotrectinib may bind some DFG-out poses of ROS1, and slight disruption is observed only in the case of ROS1F2004C DFG-out. Taken together, these MD simulation and docking studies suggest that entrectinib, and potentially crizotinib, are not ‘true’ type I (DFG-in) or type II (DFG-out) inhibitors, but may operate as type I/II dual mode inhibitors depending on resident kinase pose and conformation contexts. Ba/F3 cell viability assays were employed to confirm inhibitor preferences predicted through computational modeling. Cell-based IC50 data indicate that CD74-ROS1F2004C (Fig. 3H) and EZR-ROS1F2004C (Fig. 3I) cells display high resistance to entrectinib and cabozantinib, partial resistance to crizotinib, and no resistance to lorlatinib and repotrectinib.
Table 2.
Number of docks with binding energy greater than the median binding energy for wildtype kinase per inhibitor.
| Entrectinib | Crizotinib | Lorlatinib | Repotrectinib | |
|---|---|---|---|---|
| Wildtype DFG-in | 589 of 1120 | 484 of 1118 | 864 of 1120 | 700 of 1120 |
| Wildtype DFG-out | 532 of 1188 | 1116 of 2078 | 716 of 2080 | 900 of 2080 |
| F2004C DFG-in | 209 of 638 | 292 of 638 | 459 of 640 | 426 of 640 |
| F2004C DFG-out | 90 of 640 | 180 of 478 | 122 of 480 | 138 of 480 |
Lorlatinib and repotrectinib are potent ROS1 inhibitors with narrow kinase-intrinsic resistance liabilities
To compare the spectrum of resistance liabilities of entrectinib to those of the macrocyclic inhibitors, lorlatinib and repotrectinib, we performed accelerated mutagenesis followed by lorlatinib or repotrectinib challenge. In both CD74-ROS1 and EZR-ROS1 cells, the overall incidence of lorlatinib and repotrectinib-resistance was substantially lower compared to entrectinib (Supplementary Data). Sanger sequencing revealed ROS1 S1986F, L2026M, G2032R, M2073T and I2239* in lorlatinib-resistant CD74-ROS1 cells (Supplementary Figure 10A), and the A1924_I1934del, K1980E, F2004C, G2032R+G2331C and D2113N in lorlatinib-resistant EZR-ROS1 cells (Supplementary Fig. S10B). Repotrectinib-resistance in CD74-ROS1 was rare and all four resistant wells had ROS1WT kinase domain. Repotrectinib-resistant EZR-ROS1 cells had similar spectrum of mutations as lorlatinib-resistant EZR-ROS1 cells: A1924_I1934del, F2004C, G2032R/G2331C and D2113N (Supplementary Fig. S10C). For testing lorlatinib-sensitivity, we re-engineered CD74-ROS1S1986F and CD74-ROS1L2026M; all other mutant cell lines were already generated for testing entrectinib-resistance (Fig. 2). Dose-response data show that, as expected, the ROS1G2032R is the only mutation that measurably affects lorlatinib-sensitivity in both fusion proteins (Supplementary Fig. S10D, E). The F2004C and D2113N mutations increase the repotrectinib IC50 of EZR-ROS1 cells by 4.8 and 2.4-fold respectively, however the absolute IC50 remains below 10 nmol/L (Supplementary Fig. S10F).
A limited ENU assay with crizotinib and cabozantinib challenge was performed using Ba/F3 EZR-ROS1 cells in order to assess if different fusion partner would influence resistant mutation spectrum as compared to previous data for CD74-ROS1 for these inhibitors (20,22,23) (Supplementary Fig. S11). ROS1G2101A (xDFG) was the only new mutation recovered with crizotinib challenge in Ba/F3 EZR-ROS1 cells (Supplementary Fig. 11A, B). ROS1F2004C emerges as a frequent cabozantinib-resistant substitution (Supplementary Fig.11C and D); these data are consistent with F2004C as a resistance liability to inhibitors that bind the type II or DFG-out conformation that we previously reported (20).
Spectrum of resistance to first and next-generation ROS1 inhibitors.
In addition to the ROS1 mutations recovered via Sanger Sequencing from ENU assay as described above, we investigated a panel of clinically relevant mutations for their sensitivity to first and next-generation Type I or type II ROS1 inhibitors. For this, we generated ROS1 S1986F, F2004V (for comparison to F2004C), L2026M, D2033N and L2086F in CD74-ROS1 (Fig. 4A) and EZR-ROS1 (Supplementary Figures S12). ROS1 F2004I was also engineered in both fusions (Supplementary Figures S13 and S14). We tested the sensitivity of these mutants to the following ROS1i: (a) FDA approved inhibitors that we propose are functioning as Type I/II or Type I1/2 dual mode binding – crizotinib and entrectinib; (b) type II mode inhibitors – cabozantinib and foretinib (tool compound only); (c) next-generation macrocyclic inhibitors developed to overcome crizotinib resistance – lorlatinib and repotrectinib. Ba/F3 CD74-ROS1 dose-response cell viability data are presented in Fig. 4A. Corresponding data for these mutants in Ba/F3 EZR-ROS1 are in Supplementary Figure S12. These data show that ROS1G2032R and ROS1L2086F are highly resistant to crizotinib, entrectinib and lorlatinib. Repotrectinib is also unable inhibit ROS1L2086F; however it retains activity for G2032R albeit with reduced sensitivity compared to wildtype. In contrast, both these mutations retain sensitivity for the type II inhibitors, cabozantinib and foretinib. Consistent with our previous findings (20), ROS1F2004C hinders binding of type II inhibitors (cabozantinib and foretinib), and as we defined via structural modeling here, the type I/II inhibitors, particularly entrectinib. ROS1L2026M exhibits reduced inhibitor sensitivity only to crizotinib, and may be a unique resistant mutation for it. ROS1D2033N mutation increases crizotinib IC50 in CD74-ROS1 and EZR-ROS1 by ~3- and ~14-fold respectively, and similarly entrectinib IC50 in CD74-ROS1 and EZR-ROS1 cells by 2-fold and ~17-fold, respectively. ROS1S1986F, in our hands, did not impose meaningful resistance to any inhibitors. Immunoblotting confirms that crizotinib, entrectinib, taletrectinib (a new Type I inhibitor), lorlatinib or repotrectinib are unable to engage ROS1L2086F whereas cabozantinib and foretinib are highly effective, especially at the 100 nM concentration tested herein (Fig. 4B, C). A summary of cell-based IC50 of all mutants tested for CD74-ROS1 and EZR-ROS1 (in comparison with wildtype) is presented in Fig. 4D and 4E, respectively. Supplementary Figure S13 and 14 show dose-response curves for other mutants not depicted in Figure 4A.
Figure 4. Activity of first and next-generation ROS1 inhibitors against spectrum of ROS1 intracellular mutations in CD74-ROS1 and EZR-ROS1.
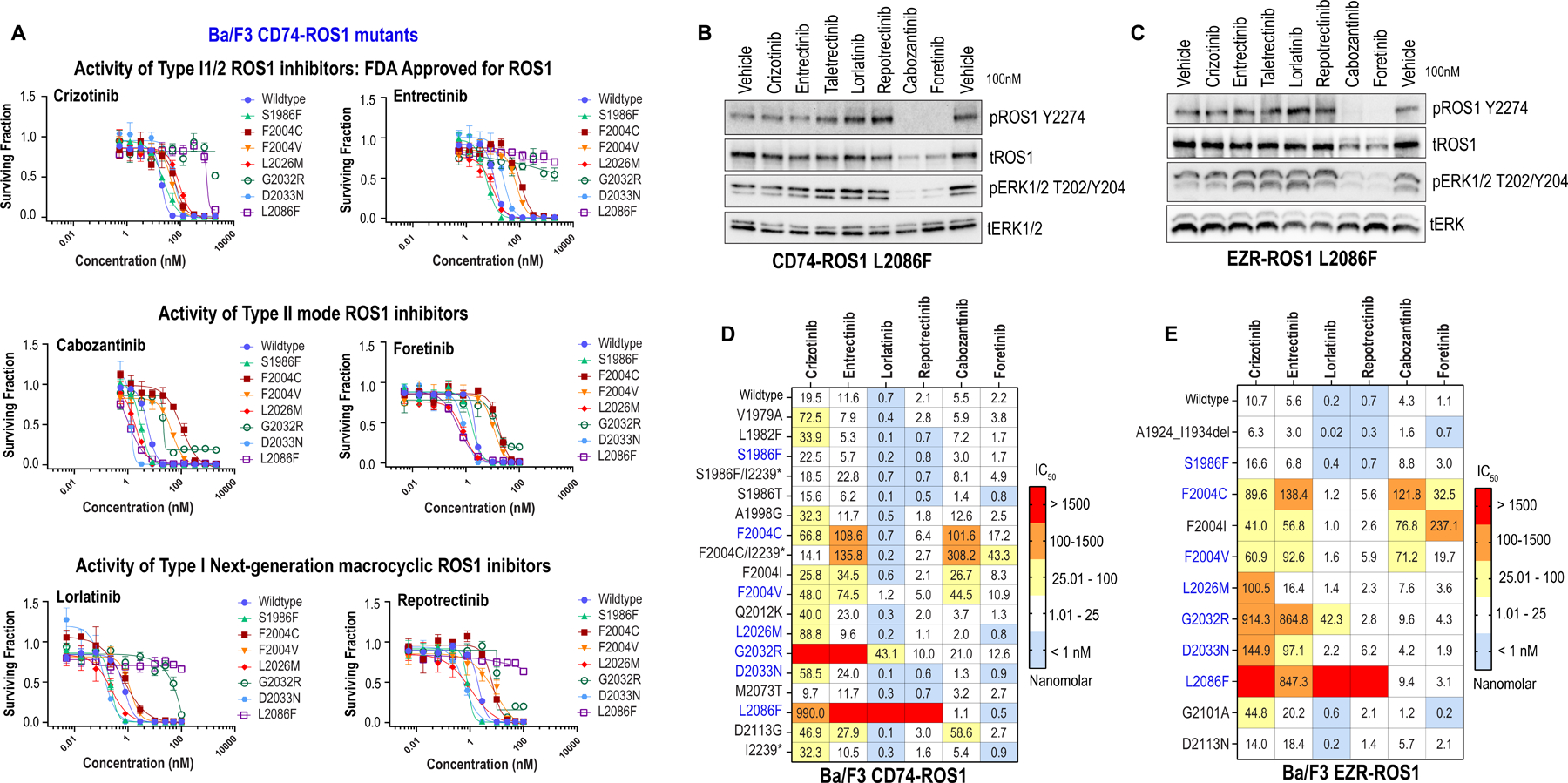
A, Dose response cell viability assays of clinically relevant (S1986F, F2004C, F2004V, L2026M, G2032R, D2033N and L2086F compared to wildtype) ROS1 kinase domain mutations in Ba/F3 CD74-ROS1 cells. Average ± SEM shown for all data. B & C, Immunoblotting of Ba/F3 CD74-ROS1L2086F and EZR-ROS1L2086F cells treated for 6 h with 100 nM of indicated inhibitors. Phosphorylated and total ROS1 and ERK1/2 are shown. D & E, Heat map of IC50 values (nanomolar) of crizotinib, entrectinib, lorlatinib, repotrectinib, cabozantinib and foretinib for Ba/F3 CD74-ROS1 (D) and EZR-ROS1 (E) mutant cell lines. IC50 are average of two to four replicates for each mutant and each inhibitor. Color scale for heatmap in indicated in the figure.
Discussion
Recent integrated analysis of entrectinib efficacy in ROS1-positive NSCLC showed that 77% of patients had an objective response (2). These data establish entrectinib as a promising new therapeutic option for ROS1 fusion-positive patients, and expansion of its benefits are being explored in non-NSCLC indications, including CNS tumors in adults and children (STARTRK-2 & STARTRK-NG clinical trials). Entrectinib resistance was explored in the clinical setting via circulating tumor DNA sequencing and revealed ROS1G2032R and ROS1F2004C/I mutations in 28% of patients (39). While ROS1G2032R is a known multi-ROS1 TKI resistant mutation, and has been previously characterized as an entrectinib-resistant mutation (40,41), the role of ROS1 F2004 amino acid substitutions on ROS1i resistance was not studied. Entrectinib resistance was not previously profiled in preclinical studies. Thus, here we performed accelerated mutagenesis screening with entrectinib, lorlatinib and repotrectinib with the goal of generating a broad pre-clinical profile of the spectrum of resistance to first- and next-generation inhibitors
While there has only been one report of a ROS1 patient with a mutation at the F2004 position, mutations at the homologous ALK F1174 position have been identified in multiple cancers including neuroblastoma and non-small cell lung cancer (NSCLC). These mutations are known to be resistant to type I inhibitors crizotinib and ceritinib, but remain sensitive to lorlatinib (42–46). No data has been published on entrectinib sensitivity of ALKF1174 mutations in an ALK fusion context, but one patient harboring non-fusion ALKF1174L showed a complete response when treated with entrectinib (47). A key observation from our accelerated mutagenesis studies herein is that substitutions of the ROS1 F2004 position may emerge as recurrent entrectinib-resistant liabilities. We previously established that ROS1F2004C is a particular problem for type II or DFG-out binding mode inhibitors. However, entrectinib is presumed to be a type I (DFG-in) binding mode inhibitor. Here, our experiments establish ROS1 F2004 substitutions as a recurrent entrectinib-resistance liability. These data suggest that entrectinib may also bind the type II or DFG-out ROS1 kinase conformations. To address this, we explored the structural basis for this reduced binding using computational chemistry approaches. Data from rigorous molecular dynamic simulation and molecular docking studies of entrectinib bound to ROS1WT and ROS1F2004C leads us to conclude that entrectinib, and surprisingly to some extent also crizotinib, are not strictly type I mode inhibitors. Docking studies show that entrectinib binds with high affinity to specific kinase poses of both the ROS1 DFG-in (type I) and ROS1 DFG-out (type II) conformations. Thus, entrectinib may be liable to a subset of both type I and type II binding mode resistance mutations.
Lorlatinib and repotrectinib are potent, brain penetrating, next generation macrocyclic ROS1 inhibitors that are currently being evaluated in clinical trials for ROS1 (4,7,8). Cell-based data presented herein shows that these macrocyclic ROS1 inhibitors retain sensitivity for ROS1 F2004 substitutions, and thus may represent true type I inhibitors. To generate a comprehensive reference map of the functional impact of ROS1 kinase domain mutations, we tested clinically relevant ROS1 mutations, L2026M, F2004V, G2032R, and L2086F, alongside those discovered via ENU assays, for their sensitivity to first and second generation ROS1 inhibitors. Lorlatinib and repotrectinib have higher intrinsic potency for wildtype ROS1 kinase domain, however as has been previously reported, solvent front mutation G2032R as well as L2086F, reduce or abrogate the functional effectiveness of these agents, respectively. These cell-based data validate that cabozantinib and the tool compound foretinib are likely true type II inhibitors, binding the back hydrophobic pocket within the kinase domain, preferentially, since they retain nanomolar inhibitory potency for ROS1L2086F while developing significant resistance to ROS1F2004C/V.
ROS1 C-term truncations were previously observed in resistance screening via ENU accelerated mutagenesis (20,23), however their functional impact was unexplored until now. Here we showed that removal of the ROS1 C-term increases the total level of truncated CD74-ROS1 fusion by 2 to 7-fold at steady state, and this is due to longer half-life. From a structural perspective, the ROS1 C-term is a highly disordered, and possesses multiple phosphorylation, adaptor protein binding, and putative ubiquitination sites. We propose that the ROS1 C-term may be a sensor for ROS1 catalytic activity, and govern protein stability via ubiquitination. Future studies are needed to discern the mechanistic basis of ROS1 fusion turnover and the role of the C-term in its regulation. In addition to exhibiting a longer half-life, CD74-ROS1I2239X has modestly reduced crizotinib-sensitivity. Intriguingly, the compound mutants, S1986F+I2239* and F2004C+I2239*, where the kinase domain mutation is introduced in the CD74-ROS1I2239* context, exhibit reversal of crizotinib-resistance compared to S1986F and F2004C mutants, alone. These data offer a hypothesis that the C-term may play a role in regulating ROS1 kinase conformations that are favorable for crizotinib-binding.
The distinct subcellular localizations of CD74-ROS1 and EZR-ROS1 observed in this study, and of other ROS1-fusions shown previously by Neel et al. (31), may influence relative activation of effector pathways. For example, EZR-ROS1 activated SHP2 and STAT3 phosphorylation to a greater extent than CD74-ROS1. It is unclear how the amplitude or duration of activation of downstream pathways contributes to drug resistance; in our studies EZR-ROS1 cells displayed a greater propensity for developing entrectinib resistance than CD74-ROS1 cells. However, there are multiple caveats to consider. First, ENU accelerated mutagenesis approaches are susceptible to inter- and intra-experimental variations, and thus, the higher occurrence of entrectinib resistant clones, especially ROS1 F2004C mutated cells, may be a consequence of stochastic probability. Second, while both the EZR-ROS1 and CD74-ROS1 cDNAs are in murine retroviral vector backbones, they are in different versions of the vector. Thus, unknown factors relating to viral titer and mutagenic propensity may result in these differences. Finally, since these studies were conducted exclusively in the Ba/F3 or HEK293 cell models, the influence or impact of cancer cell lineage cannot be ascertained. Indeed, one can hypothesize that incidence of resistance and the mutagenic pathways that are involved in acquisition of kinase domain independent resistance will vary in cancers of different lineages. Future studies aimed at developing paired, isogenic ROS1 TKI resistance cell models starting with endogenous ROS1 fusions and lineage authentic human cell backgrounds will be necessary, particularly to map the spectrum of bypass or kinase-extrinsic resistance pathways.
Taken together, our data demonstrate that entrectinib is potentially liable to both type I (e.g., ROS1G2032R and ROS1L2086F) and type II (e.g., ROS1F2004C/I/V) inhibitor binding mode resistance mutations. Meanwhile effectiveness of the promising next-generation macrocyclic inhibitors, lorlatinib and repotrecintib will be hampered by ROS1G2032R (for lorlatinib) and particularly by ROS1L2086F. We propose that development of new, more selective type II ROS1 inhibitors may be essential for long-term disease control. A compelling treatment plan may involve metronomic dosing, wherein next generation type I and type II inhibitors are cycled, in order to quench subclonal emergence of distinct kinase-intrinsic resistance liabilities to these agents. We hope that our data inform strategies for early detection of resistance, and development of appropriate second-line treatment strategies for extending the window of therapeutic durability for ROS1 fusion positive patients.
Supplementary Material
Acknowledgements:
We would like to thank Dr. Nicolle Hofmann for assistance with cloning the mYFP tagged CD74-, EZR-, SLC34A2-, and GOPC-ROS1 constructs, and Sudarshan Iyer for assisting with generation of pENTR-EZR-ROS1 construct. This work was partially funded by R01 CA233495-01A1 and an American Cancer Society (ACS) grant (RSG-19-082-01-TBG) to M.A.D.
Conflicts of Interest:
Romel Somwar has received research grants from Merus, Helsinn Healthcare, Elevation Oncology Inc. and LOXO Oncology. These funding are not related to the current study. All other authors have no potential conflict of interest to disclose.
References
- 1.Drilon A, Jenkins C, Iyer S, Schoenfeld A, Keddy C, Davare MA. ROS1-dependent cancers - biology, diagnostics and therapeutics. Nat Rev Clin Oncol 2021;18(1):35–55 doi 10.1038/s41571-020-0408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drilon A, Siena S, Dziadziuszko R, Barlesi F, Krebs MG, Shaw AT, et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1–2 trials. Lancet Oncol 2020;21(2):261–70 doi 10.1016/S1470-2045(19)30690-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, Lee J, et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372–001 and STARTRK-1). Cancer Discov 2017;7(4):400–9 doi 10.1158/2159-8290.CD-16-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw AT, Felip E, Bauer TM, Besse B, Navarro A, Postel-Vinay S, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol 2017. doi 10.1016/S1470-2045(17)30680-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371(21):1963–71 doi 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw AT, Solomon BJ. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2015;372(7):683–4 doi 10.1056/NEJMc1415359. [DOI] [PubMed] [Google Scholar]
- 7.Shaw AT, Solomon BJ, Chiari R, Riely GJ, Besse B, Soo RA, et al. Lorlatinib in advanced ROS1-positive non-small-cell lung cancer: a multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol 2019;20(12):1691–701 doi 10.1016/S1470-2045(19)30655-2. [DOI] [PubMed] [Google Scholar]
- 8.Drilon A, Ou SI, Cho BC, Kim DW, Lee J, Lin JJ, et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations. Cancer Discov 2018;8(10):1227–36 doi 10.1158/2159-8290.CD-18-0484. [DOI] [PubMed] [Google Scholar]
- 9.Yun MR, Kim DH, Kim S-Y, Joo H-S, Lee YW, Choi HM, et al. Repotrectinib Exhibits Potent Antitumor Activity in Treatment-Naïve and Solvent-Front–Mutant ROS1-Rearranged Non–Small Cell Lung Cancer. Clinical Cancer Research 2020;26(13):3287–95 doi 10.1158/1078-0432.ccr-19-2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papadopoulos KP, Borazanci E, Shaw AT, Katayama R, Shimizu Y, Zhu VW, et al. U.S. Phase I First-in-human Study of Taletrectinib (DS-6051b/AB-106), a ROS1/TRK Inhibitor, in Patients with Advanced Solid Tumors. Clinical Cancer Research 2020;26(18):4785–94 doi 10.1158/1078-0432.ccr-20-1630. [DOI] [PubMed] [Google Scholar]
- 11.Lovly CM, Heuckmann JM, de Stanchina E, Chen H, Thomas RK, Liang C, et al. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res 2011;71(14):4920–31 doi 10.1158/0008-5472.CAN-10-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drilon A, Somwar R, Wagner JP, Vellore NA, Eide CA, Zabriskie MS, et al. A Novel Crizotinib-Resistant Solvent-Front Mutation Responsive to Cabozantinib Therapy in a Patient with ROS1-Rearranged Lung Cancer. Clin Cancer Res 2016;22(10):2351–8 doi 10.1158/1078-0432.ccr-15-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Facchinetti F, Loriot Y, Kuo MS, Mahjoubi L, Lacroix L, Planchard D, et al. Crizotinib-Resistant ROS1 Mutations Reveal a Predictive Kinase Inhibitor Sensitivity Model for ROS1- and ALK-Rearranged Lung Cancers. Clin Cancer Res 2016;22(24):5983–91 doi 10.1158/1078-0432.ccr-16-0917. [DOI] [PubMed] [Google Scholar]
- 14.Gainor JF, Tseng D, Yoda S, Dagogo-Jack I, Friboulet L, Lin JJ, et al. Patterns of Metastatic Spread and Mechanisms of Resistance to Crizotinib in ROS1-Positive Non-Small-Cell Lung Cancer. JCO Precis Oncol 2017;2017 doi 10.1200/PO.17.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin JJ, Choudhury NJ, Yoda S, Zhu VW, Johnson TW, Sakhtemani R, et al. Spectrum of Mechanisms of Resistance to Crizotinib and Lorlatinib in ROS1 Fusion-Positive Lung Cancer. Clinical Cancer Research 2021:clincanres.0032 doi 10.1158/1078-0432.ccr-21-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin JJ, Johnson T, Lennerz JK, Lee C, Hubbeling HG, Yeap BY, et al. Resistance to lorlatinib in ROS1 fusion-positive non-small cell lung cancer. Journal of Clinical Oncology 2020;38(15_suppl):9611- doi 10.1200/JCO.2020.38.15_suppl.9611. [DOI] [Google Scholar]
- 17.Lin JJ, Shaw AT. Recent Advances in Targeting ROS1 in Lung Cancer. J Thorac Oncol 2017;12(11):1611–25 doi 10.1016/j.jtho.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCoach CE, Le AT, Gowan K, Jones K, Schubert L, Doak A, et al. Resistance Mechanisms to Targeted Therapies in ROS1(+) and ALK(+) Non-small Cell Lung Cancer. Clin Cancer Res 2018;24(14):3334–47 doi 10.1158/1078-0432.CCR-17-2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato H, Schoenfeld AJ, Siau E, Lu YC, Tai H, Suzawa K, et al. MAPK Pathway Alterations Correlate with Poor Survival and Drive Resistance to Therapy in Patients with Lung Cancers Driven by ROS1 Fusions. Clin Cancer Res 2020;26(12):2932–45 doi 10.1158/1078-0432.CCR-19-3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davare MA, Vellore NA, Wagner JP, Eide CA, Goodman JR, Drilon A, et al. Structural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitors. Proc Natl Acad Sci U S A 2015;112(39):E5381–90 doi 10.1073/pnas.1515281112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dimou A, Ou SI, Doebele RC. Dramatic Response to Lorlatinib in a Patient With CD74-ROS1-Positive Lung Adenocarcinoma With Acquired F2004V Mutation. JCO Precis Oncol 2019;3 doi 10.1200/PO.19.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davare MA, Saborowski A, Eide CA, Tognon C, Smith RL, Elferich J, et al. Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proc Natl Acad Sci U S A 2013;110(48):19519–24 doi 10.1073/pnas.1319583110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katayama R, Kobayashi Y, Friboulet L, Lockerman EL, Koike S, Shaw AT, et al. Cabozantinib overcomes crizotinib resistance in ROS1 fusion-positive cancer. Clin Cancer Res 2015;21(1):166–74 doi 10.1158/1078-0432.CCR-14-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ardini E, Menichincheri M, Banfi P, Bosotti R, De Ponti C, Pulci R, et al. Entrectinib, a Pan–TRK, ROS1, and ALK Inhibitor with Activity in Multiple Molecularly Defined Cancer Indications. Molecular Cancer Therapeutics 2016;15(4):628–39 doi 10.1158/1535-7163.mct-15-0758. [DOI] [PubMed] [Google Scholar]
- 25.Katayama R, Gong B, Togashi N, Miyamoto M, Kiga M, Iwasaki S, et al. The new-generation selective ROS1/NTRK inhibitor DS-6051b overcomes crizotinib resistant ROS1-G2032R mutation in preclinical models. Nature Communications 2019;10(1) doi 10.1038/s41467-019-11496-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dar AC, Shokat KM. The Evolution of Protein Kinase Inhibitors from Antagonists to Agonists of Cellular Signaling. Annual Review of Biochemistry 2011;80(1):769–95 doi 10.1146/annurev-biochem-090308-173656. [DOI] [PubMed] [Google Scholar]
- 27.Modi V, Dunbrack RL. Defining a new nomenclature for the structures of active and inactive kinases. Proceedings of the National Academy of Sciences 2019;116(14):6818–27 doi 10.1073/pnas.1814279116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zuccotto F, Ardini E, Casale E, Angiolini M. Through the “Gatekeeper Door”: Exploiting the Active Kinase Conformation. Journal of Medicinal Chemistry 2010;53(7):2681–94 doi 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]
- 29.Roskoski R Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacological Research 2021:105463 doi 10.1016/j.phrs.2021.105463. [DOI] [PubMed] [Google Scholar]
- 30.Roskoski R Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacological Research 2016;103:26–48 doi 10.1016/j.phrs.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 31.Neel DS, Allegakoen DV, Olivas V, Mayekar MK, Hemmati G, Chatterjee N, et al. Differential Subcellular Localization Regulates Oncogenic Signaling by ROS1 Kinase Fusion Proteins. Cancer Research 2019;79(3):546–56 doi 10.1158/0008-5472.can-18-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Somwar R, Hofmann NE, Smith B, Odintsov I, Vojnic M, Linkov I, et al. NTRK kinase domain mutations in cancer variably impact sensitivity to type I and type II inhibitors. Commun Biol 2020;3(1):776 doi 10.1038/s42003-020-01508-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Savage JC, Shinde P, Yao Y, Davare MA, Shinde U. A Broccoli aptamer chimera yields a fluorescent K(+) sensor spanning physiological concentrations. Chemical communications (Cambridge, England) 2021;57(11):1344–7 doi 10.1039/d0cc07042d. [DOI] [PubMed] [Google Scholar]
- 34.Neisch AL, Fehon RG. Ezrin, Radixin and Moesin: key regulators of membrane-cortex interactions and signaling. Curr Opin Cell Biol 2011;23(4):377–82 doi 10.1016/j.ceb.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bosanquet DC, Ye L, Harding KG, Jiang WG. FERM family proteins and their importance in cellular movements and wound healing (review). Int J Mol Med 2014;34(1):3–12 doi 10.3892/ijmm.2014.1775. [DOI] [PubMed] [Google Scholar]
- 36.Tulpule A, Guan J, Neel DS, Allegakoen HR, Lin YP, Brown D, et al. Kinase-mediated RAS signaling via membraneless cytoplasmic protein granules. Cell 2021. doi 10.1016/j.cell.2021.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warmuth M, Kim S, Gu XJ, Xia G, Adrian F. Ba/F3 cells and their use in kinase drug discovery. Curr Opin Oncol 2007;19(1):55–60 doi 10.1097/CCO.0b013e328011a25f. [DOI] [PubMed] [Google Scholar]
- 38.Awad MM, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med 2013;368(25):2395–401 doi 10.1056/NEJMoa1215530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doebele RD RC, Drilon A, Shaw A, Wolf J, Farago AF, Dennis L, Riehl T, Simmons B, Wu C, Chang C, Choeurng V, Wilson TR. Genomic landscape of entrectinib resistance from ctDNA analysis in STARTRK-2. Annals of Oncology Volume 30 (suppl_5): v851–v934. 10.1093/annonc/mdz3942019. [DOI] [Google Scholar]
- 40.Katayama R, Gong B, Togashi N, Miyamoto M, Kiga M, Iwasaki S, et al. The new-generation selective ROS1/NTRK inhibitor DS-6051b overcomes crizotinib resistant ROS1-G2032R mutation in preclinical models. Nat Commun 2019;10(1):3604 doi 10.1038/s41467-019-11496-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin JJ, Choudhury NJ, Yoda S, Zhu VW, Johnson TW, Sakhtemani R, et al. Spectrum of Mechanisms of Resistance to Crizotinib and Lorlatinib in ROS1 Fusion-Positive Lung Cancer. Clin Cancer Res 2021. doi 10.1158/1078-0432.CCR-21-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, et al. The Neuroblastoma-Associated F1174L ALK Mutation Causes Resistance to an ALK Kinase Inhibitor in ALK-Translocated Cancers. Cancer Research 2010;70(24):10038–43 doi 10.1158/0008-5472.can-10-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK Inhibitor Ceritinib Overcomes Crizotinib Resistance in Non–Small Cell Lung Cancer. Cancer Discovery 2014;4(6):662–73 doi 10.1158/2159-8290.cd-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, et al. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov 2016;6(1):96–107 doi 10.1158/2159-8290.CD-15-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaw AT, Solomon BJ, Besse B, Bauer TM, Lin C-C, Soo RA, et al. ALK Resistance Mutations and Efficacy of Lorlatinib in Advanced Anaplastic Lymphoma Kinase-Positive Non–Small-Cell Lung Cancer. Journal of Clinical Oncology 2019;37(16):1370–9 doi 10.1200/jco.18.02236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discovery 2016;6(10):1118–33 doi 10.1158/2159-8290.cd-16-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Desai AV, Robinson GW, Basu EM, Foster J, Gauvain K, Sabnis A, et al. Updated entrectinib data in children and adolescents with recurrent or refractory solid tumors, including primary CNS tumors. Journal of Clinical Oncology 2020;38(15_suppl):107- doi 10.1200/JCO.2020.38.15_suppl.107.31774706 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.