

Abstract
Sphingolipids are essential lipid components in the intestinal epithelial cells (IEC) along the intestinal tract. They play crucial roles in maintaining barrier integrity, regulating nutrient absorption, and acting as signaling molecules to regulate regeneration and differentiation of intestinal mucosa [1]. Ceramide is the central sphingolipid species and the precursor of all complex sphingolipids and other downstream simple intermediates like sphingosine (SPH), ceramide-1-phosphate (C-1-P), and sphingosine-1-phosphate (S-1-P). It is also a critical signaling molecule regulating numerous physiologic and pathologic processes. This review will summarize the metabolism of ceramides in the gut and their regulation in inflammatory bowel diseases and colorectal cancer.
Keywords: Ceramide, Intestine, Inflammatory bowel disease, Colon cancer, NAFLD
1. Introduction
Sphingolipids are abundant lipid species in the gut, and knowledge about their bioactivity is expanding as more studies demonstrate their involvement in several major physiologic and pathologic intestinal processes. Sphingolipids constitute a class of thousands of lipids defined by their carbon amino-alcohol backbones, with ceramide as the central sphingolipid species and the precursor of all complex sphingolipids. Modifications to this ceramides backbone generate the vast and diverse family of sphingolipids, which conduct distinct cellular functions. These lipids are expressed in the small intestine and colon mucosal cells, although the level in the small intestine is over twofold higher than in colonic mucosa [2]. These differences result from excessive and rapid differentiation and exfoliation of mucosal cells in the upper gastrointestinal tract. Even though ceramides have significant clinical implications in cell growth, survival, apoptosis, and inflammation with very well-defined mechanisms in other tissues, their role in the gut has been relatively underexplored. This review will discuss the physiological and pathological role of ceramides in the intestine and assess the diagnostic and therapeutic implications in inflammatory bowel diseases (IBD) and colon cancer.
2. Ceramides synthesis and metabolism
2.1. De Novo Synthesis pathway (Figure 1).
Figure. 1.
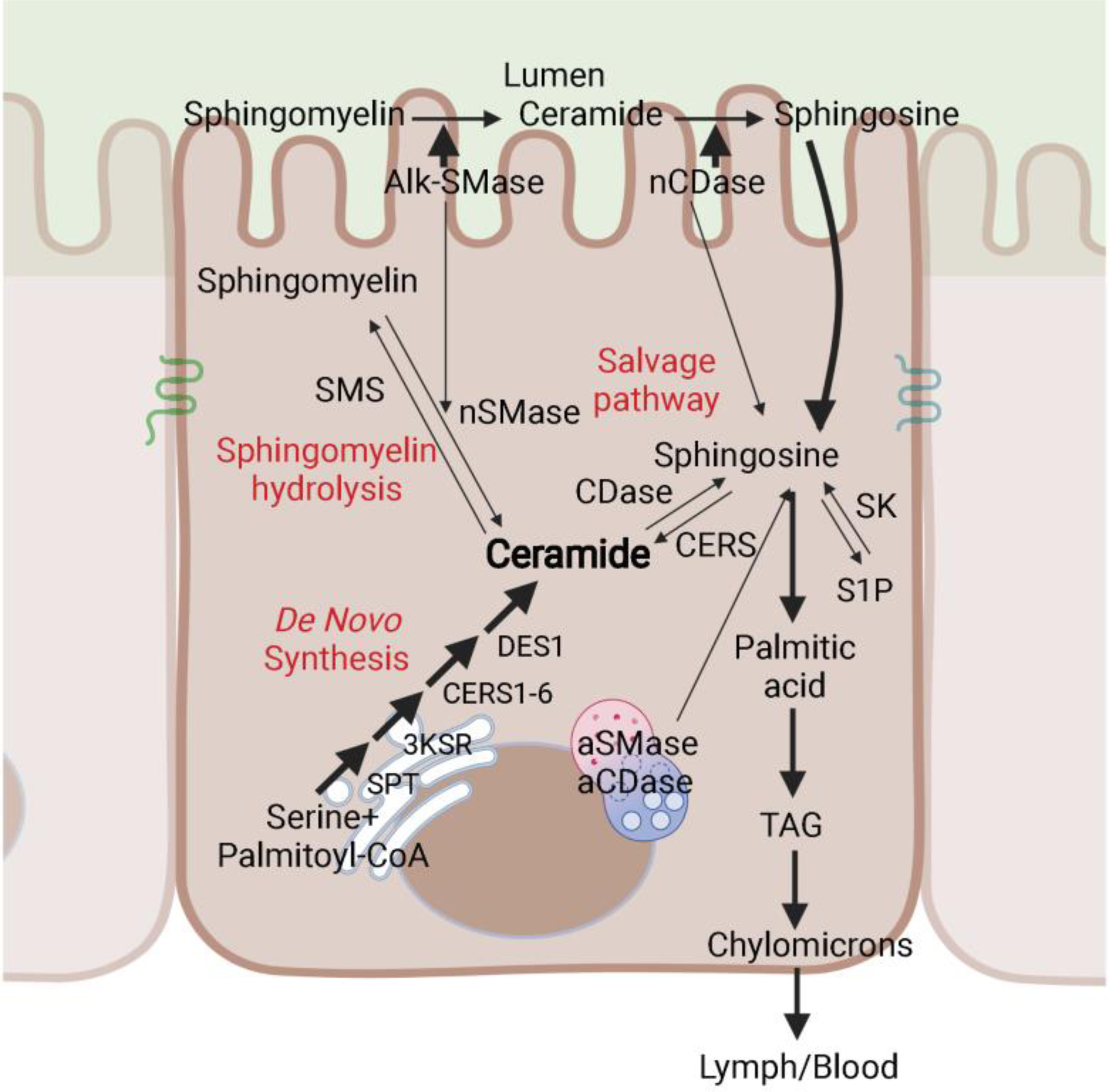
Ceramide production pathways in the intestinal epithelium. Schematic depiction of the major pathways controlling ceramide levels in the intestinal epithelial cells: de novo synthesis, hydrolysis of complex sphingolipids (for example, sphingomyelin hydrolysis) and the salvage pathway. SPT, serine palmitoyltransferase; 3KSR, 3-ketosphinganine reductase; CERS, ceramide synthase; DES1, dihydroceramide desaturase 1; aCDase, acid ceramidase; nCDase, neutral ceramidase; aSMase, acid sphingomyelinase; nSMase, neutral sphingomyelinase; alk-SMase, alkaline sphingomyelinase; SMS, sphingomyelin synthase; TAG, triacylglyceride.
Ceramides in the gastrointestinal tract are synthesized mainly through the de novo synthesis pathway. This anabolic reaction begins in the endoplasmic reticulum with the condensation of palmitoyl-CoA and serine, catalyzed by the enzyme serine palmitoyltransferase (SPT), to produce 3-ketosphinganine [3]. The following three reactions are conducted by 3-ketosphinganine reductase (3KSN), six (dihydro) ceramide synthase (CERS 1–6), and two dihydroceramide desaturases (DES1 and 2) to generate sphinganine, dihydroceramide, ceramide, and phytoceramide respectively. Excepting CERS3, five of the six CERS enzymes are expressed in the intestinal mucosal cells [4], leading to the generation of a diverse ceramide pool with variable acyl chain lengths ranging from 14- to 34-carbon atoms [5]. DES1 is a ubiquitous enzyme that is present in every tissue, while DES2 is restricted largely to epithelial tissues such as the gut, kidney and skin. DES2 has the ability to make phytoceramides, as well as ceramides, in the mouse small intestine [6]. Once synthesized, ceramides are transported to the Golgi apparatus, where sphingomyelin (SM) and glucosylceramide (GlcCer) synthesis occur.
2.2. Salvage pathway (Figure 1).
The salvage pathway—which involves the reacylation of ceramide from sphingosine—also exists in the intestine, but probably on a lesser scale compared to that in other cell types, where salvage can account for 50–90% of sphingolipid biosynthesis [7]. Dietary SM is mainly digested in the jejunum into sphingosine and sphinganine by abundant alkaline sphingomyelinase (alk-SMase) and neutral ceramidase (nCDase) released from both the intestinal epithelium and the liver. Free sphingosine and sphinganine are rapidly absorbed and metabolized by intestinal mucosal cells. The majority of them are further metabolized into palmitic acid and utilized for chylomicron assembly [8]; only a small percentage of sphingoid bases are reincorporated into mucosal ceramide or complex sphingolipids [8; 9].
Although the primary function of alk-SMase and nCDase is dietary sphingolipid digestion, some evidence suggests that they could have a modest effect on IEC ceramides by hydrolyzing sphingolipids at the apical membrane. Nilsson’s group overexpressed alk-SMase in Cos-7 cells, which caused a 30% reduction of SM [10]. Hannun’s group found that knocking down nCDase in HT-29 and HCT116 cells increased total ceramide to 170% and 160% of the control cell, respectively [11]. Acid sphingomyelinase (aSMase) [12] and acid ceramidase (aCDase) present in the lysosomes and alkaline ceramidase (alk-CDase) located in the endoplasmic reticulum and Golgi apparatus are also expressed in the intestinal epithelial cells [13; 14] and likely contribute to the salvage pathway.
2.3. Sphigomyelin hydrolysis pathway (Figure 1).
Sphingomyelin hydrolysis is catalyzed by neutral SMase. Neutral SMase is expressed in mucosal cells of the small intestine, although at lower levels than alk-SMase [12; 15]. Although nothing has been reported about the association between neutral SMase and intestinal disorders, this enzyme may be responsible for transforming SM into ceramide under various stress conditions.
2.4. Ceramide lowering mechanisms (Figure 1).
Ceramides produced in the endoplasmic reticulum can be transported to the Golgi apparatus, where they are converted into complex sphingolipids including sphingomyelins, glucosylceramides and gangliosides. Ceramides can also be deacylated by a family of ceramidases, which liberate a fatty acid and sphingosine. Sphingosine can be phosphorylated by sphingosine kinases 1&2, which are two major regulators of inflammation and tumorigenesis in the intestine to produce S-1-P [16; 17]. Sphingosine can be converted back into ceramide by the CERS enzymes through the salvage pathway mentioned above. The complex sphingolipids (such as sphingomyelin) can be catabolized to regenerate ceramides during stress by sphingomyelinases through the sphingomyelin hydrolysis pathway.
2.5. Bacteria-derived sphingolipids (Figure 1).
Recent studies demonstrate that commensal bacteria-derived sphingolipids can change the host ceramide level and modulate intestinal homeostasis [18; 19]. Bacteroidetes, the most abundant bacterial species in the mammalian intestine, is the only gut commensal that produces sphingolipids [20]. Brown et al. generated a sphingolipid-deficient Bacteroides strain and demonstrated that mono-inoculation of this strain into germ-free mice significantly changed the composition of ceramide in the cecum and worsened inflammation in the intestine [18]. Ley’s group did a series of delicate studies showing that bacterial-derived sphingolipids can be absorbed by intestinal epithelial cells, processed, and transported to the liver through the portal vein. Moreover, bacterial-derived sphingolipids significantly inhibit the de novo sphingolipid synthesis pathway, decreasing certain ceramide species in both the intestine and liver [19].
2.6. Regulation of ceramide metabolism in the gut.
Previous studies have shown that TNF-α, IL-1, and other cytokines increase ceramides levels in different tissues and cell lines by activating SMase [21]. These mechanisms are mirrored in the intestine and intestine-derived cell lines [22; 23]. Moreover, several bacterial toxins such as lipopolysaccharide (LPS), p-fimbriae, and the B-subunit of Shiga toxin increase ceramide levels through a TLR4-dependent response [24]. Ceramide levels in the intestine can also be regulated pharmaceutically. For example, ursodeoxycholic acid (secondary bile acid), 5-ASA (anti-inflammatory substance), and psyllium (dietary fiber supplement) induce intestinal ceramide accumulation by activating alk-SMase and simultaneously reducing CDase activities [15; 25].
3. Ceramide in the inflammatory bowel diseases
Inflammatory bowel diseases (IBD) are prevalent globally with an incidence increasing in both developed and developing countries [26]. The common forms of IBD are Crohn’s disease (CD) and ulcerative colitis (UC). Genetic predisposition and adverse environmental risk factors, mainly related to patients’ diet and microbiome, result in a weakened intestinal barrier and contribute to the etiology of IBD [27; 28; 29].
3.1. Ceramides dysregulation in IBD patients.
Several groups have reported that intestinal sphingolipids are altered in conditions of IBD, but the consensus is lacking. For example, Bazarganipour et al. found that reductions in de novo sphingolipid synthesis in colon tissue are associated with the etiology of UC [30]. However, Diab and colleagues found that UC patients had increased very long-chain ceramides in the IEC from an active inflammation group (treatment-naïve UC patients) compared with that from the remission patient group [31]. The contrasting results from these two studies could be related to the different experimental design: Bazarganipour used control samples from adjacent tissues collected from the same patient, who is experiencing a heightened immune response; Diab and colleagues used control tissues from the rectum of healthy individuals, who are not experiencing the disease. Nonetheless, while these studies show potential dysregulation of sphingolipid metabolism in affected tissues, the directionality is controversial. Notably, a genome-wide association (GWAS) study identified multiple new susceptible loci for UC, including upregulated ORMDL3, a significant negative regulator of SPT activity [32].
3.2. Ceramides and the intestinal epithelial barrier function.
The intestinal physical barrier function relies mainly on intact epithelial tight junctions to prevent penetration of macromolecules and bacteria infiltration [33; 34; 35; 36]. Treatment with myriocin, a potent inhibitor of SPT [37], or fumonisin B1, an inhibitor of CERS [38], induces severe gut toxicity, partially due to barrier function disruption. Knocking out Sptlc2, the first and rate-limiting enzyme in the ceramide de novo synthesis pathway, in the intestinal epithelium caused acute gut leakage and mouse death [39; 40; 41]. Immunostaining of the gut mucosa showed a dramatic reduction of mucin 2, the major protein in the mucus layer secreted by goblet cells, and E-cadherin, an essential plasma membrane tight junction protein, in the knockout mice. Two other groups found that knocking out Cers2, the gene encoding the CERS2 enzyme that generates very long-chain ceramides, disrupts barrier function and aggravates dextran sulfate sodium (DSS)-induced colitis [42; 43]. Using the bone marrow chimeric mice model, the Park group confirmed that the development of colitis in the Cers2 null mice is mainly a gut tissue autonomous phenotype rather than a consequence of dysregulated systemic immune response [43]. Although these two groups found similar phenotypes, including weight loss and intestinal permeability and inflammation, they identified different target molecules accounting for the disrupted barrier function. One group reported decreased expression of JAM-A [43], while the other group attributed their findings to loss of zonula occludins-1 (ZO-1) and occludin. It is worth noting that SPT deletion results in more severe pathologic damages than those from the Cers2 null mice, indicating that other sphingolipids may compensate for the lack of very long-chain ceramides. Both studies in Cers2 null mice found a compensatory increase of long-chain C16 ceramide and normalizing C16 ceramide levels by myriocin treatment worsened intestinal permeability [44]. However, another group reported that mice lacking CERS6, which primarily synthesizes C16 ceramide, didn’t develop leaky gut—with or without DSS treatment [45].
Although the studies mentioned above showed that inhibiting de novo ceramide synthesis disrupts intestinal barrier function, other reports indicate that ceramide accumulation can damage barrier function. Rogler’s group found that adding exogenous SMase to Caco-2 cells efficiently decreased sphingomyelins and increased ceramides in the membrane raft, where tight junctions are located. Meanwhile, epithelial permeability was increased unrelated to apoptosis, as assessed by measuring the transepithelial flux of fluorescein-sulfonic [46].
3.3. Ceramides and the intestinal inflammatory response.
The inflammation network in the intestine is complicated by the exceptional active tissue turnover rate and the heavy burden of commensal flora. A slight imbalance in the extent and intensity can turn inflammation, which helps repair damaged tissue and fight infection under physiologic conditions, into uncontrolled detrimental acute or chronic inflammation, increasing the risk of IBD. As mentioned above, ceramides are essential for maintaining an intact nonspecific barrier in intestinal mucosa to protect enterocytes against digestive enzymes, bile salts, acidic gastric juice, or bacteria insults. Nevertheless, ceramides are involved in IBD etiology by regulating the immune response in the intestinal epithelium and systemically. Numerous studies showed that ceramides are essential lipid mediators regulating inflammation in different organs [2; 47; 48; 49; 50]. In the small intestinal epithelial cells, ceramides were reported to increase NF-kB activity, a key molecule with pro-inflammatory properties, by reducing IkB-a and IkB-b protein levels [22; 23].
Besides increased barrier permeability, Grosch’s group also found activated immune response in the naïve Cers2 null mice, manifested by increased blood monocytes and higher IL-10 levels. DSS treatment worsened this phenotype and increased regulatory T cells (Tregs) and IL-17+ T-cells in the circulation and colon, which was not observed in CerS2 +/+ mice [42]. Deleting Cers6 increases the disease activity index in DSS-induced acute colitis. Instead of a change in intestinal barrier function, this group revealed that the pathologic change is associated with an increased S-1-P level and neutrophil infiltration in the colon tissue [45]. Thus, whether these effects are a direct result of decreased C16 ceramide or secondary to the increase of S-1-P requires further study.
Interestingly, Matthew et al. did an adoptive transfer of wild type or Cers6 deficient splenocytes to the RAG1-deficient mice. Wild-type splenocyte transfer caused significant weight loss and colitis in the recipients, while transplant of Cers6 deficient splenocytes didn’t. These studies indicate that lowering ceramides upregulate inflammation in the intestinal epithelium but represses it in the splenocytes. Most of the small-molecule drugs developed for IBD target the S-1-P signaling pathway, noting that S-1-P is a critical systemic immunoregulator [51]. The findings of IEC or intestine autonomous effects of de novo synthesized ceramides emphasize that regulation of this pathway is crucial, given that none of the therapeutic strategies for IBD are satisfactorily reduced rates of remission and relapse.
As the most abundant sphingolipid in mammalian cells, SM produces a significant amount of ceramides involved in numerous inflammatory processes triggered by pro-inflammatory agonists. Cytokines, such as TNF-α, interferon-γ (IFN- γ), interleukins [52], or platelet-activating factor (PAF) [53] are potent stimulators of SMase activity and have been shown to induce inflammation in different cell types. Systemic inhibition of aSMase, an essential sphingomyelin hydrolase ubiquitously expressed in most cell types, leads to amelioration of DSS-induced colitis, possibly by inhibiting the immune cell response in this mouse model [54; 55]. Alk-SMase and nCDase are the major enzymes that are responsible for SM degradation in the intestinal lumen and mucosa [56]. The unbalanced expression of these two enzymes changed the ceramide level in the gut [15]. Hannun and Obeid found that nCDase expression increased in the epithelium of UC patients and that nCDase deletion increases ceramides in the epithelium and worsens inflammation in DSS-induced colitis [57]. Surprisingly, they found a paradoxical increase of S-1-P and a subsequent Cox-2 upregulation in the nCDase−/− mice, which they assume to be due to the reverse function of this enzyme [58]. Another study by Wang et al. showed that alkaline ceramidase 3 (ACER3) mediates the immune response by upregulating levels of C18:1-ceramide in cells of the innate immune system and colonic epithelial cells. Acer3 deficiency increases several inflammatory cytokines locally and systemically and aggravates colitis in the DSS-induced murine colitis model [14; 59].
4. Ceramide in colon cancer
Ceramides regulate fundamental processes in cancer cells, including cell death, proliferation, autophagy, and drug resistance [60]. Evidence suggests that extracellular and intracellular signals alter ceramide homeostasis by depositing dietary sphingolipids or modifying endogenous enzymatic activity, which may play a role in colon cancer development. Although this review is focused on ceramides and their related enzymes, it is essential to note that their downstream metabolites, such as C-1-P and S-1-P, and the complex SM and glycosphingolipids also play a significant role in colon carcinogenesis [61; 62]. Thus, regulation of ceramide can be pro-death or pro-survival depending on the balance between ceramide species, the rate of their synthesis and degradation, and the cancer type-specific downstream effects.
4.1. Ceramide metabolism dysregulation in human colon cancer tissue.
A bevy of recent studies has reported dysregulated ceramides and their related enzymes in colon cancer patients [63; 64]. Table 1 summarizes the genes that have been tested in human tumors.
Table 1.
Ceramide metabolism in human colon cancer tissue compared to adjacent normal mucosa
| Gene | Analysis type | Ceramide level | References |
|---|---|---|---|
| SPTLC1 | In silico analysis ↑ | N.I. | [40; 65] |
| mRNA ↑ | C16 dihydroceramide ↑ C16 ceramide ↑ sphingosine ↑ |
[40] | |
| SPTLC2 | In sillico analysis ↑ | N.I. | [40] |
| mRNA ↑ | C16 dihydroceramide ↑ C16 ceramide ↑ Sphingosine ↑ |
[40] | |
| CERS1 | mRNA ↑ | C18 ceramide ↓ | [64] |
| CERS2 | mRNA ↑ | C24, C24:1 ceramide ↑ | [64] |
| In silico analysis ↑ | N.I. | [65] | |
| CERS5 | mRNA ↑ | C16 ceramide ↑ | [64] |
| In silico analysis ↑ | N.I. | [65] | |
| mRNA ↑ | N.I. | [66] | |
| CERS6 | mRNA ↑ | C16 ↑ | [40; 64] |
| In silico analysis ↑ | N.I. | [40; 65] | |
| mRNA ↓ | N.I. | [63] | |
| SMPD1 | Activity (35%)* ↓ | N.I. | [68] |
| mRNA ↓ | N.I. | [72] | |
| SMPD2 | Activity (50%)* ↓ | N.I. | [68] |
| NPP7 | Activity (75%)* ↓ | N.I. | [67; 68; 69; 70] |
Enzyme activity of the colorectal carcinoma tissue compared with the adjacent normal mucosa.
De novo sphingolipid synthesis enzymes, SPT and CERS, were dysregulated in human colorectal cancer tissue in multiple human studies. Notably, CERS enzymes are essential for both de novo synthesis and salvage production of ceramides. Among the six isoforms, CERS1, 2, 5, and 6 are dysregulated in colon cancer tissues [64; 65; 66]. In most of these studies, CERS gene expression and corresponding ceramide levels are upregulated, indicating a role for ceramides or other sphingolipids in the activation of tumorigenesis.
Alternative findings were obtained following the manipulation of sphingomyelinases, which can regenerate ceramides from sphingomyelin. Hertervig et al. compared the enzyme activity of three sphingomyelinases (aSMase, nSMase, alk-SMase) from colorectal carcinoma and normal mucosa of 18 patients. The enzymatic activity of all three SMases decreased, with alk-SMase the most downregulated, in the cancerous tissues [67]. Alk-SMase—the enzyme that hydrolyzes most dietary sphingomyelin in the gut—was also downregulated in adenoma and carcinoma tissue [67; 68; 69; 70]. In mice, knockout of alk-SMase enhanced colonic tumorigenesis following treatment with azoxymethane and dextran sulfate sodium (AOM/DSS), suggesting that the enzyme may have an anti-cancer role [71]. Similarly, the acidic sphingomyelinase aSMase was downregulated in lesions from several patient cohorts compared with normal colon mucosa tissue from unrelated individuals [72]. In cultured colon cancer cell lines, sphingomyelinases have been shown to have a panoply of actions on cell death, in both positive and negative directions [72; 73; 74; 75].
The role of nSMase and the three ceramidases in human colon cancer are not well characterized; however, numerous studies in colon cancer cell lines and genetically modified mouse models indicate that they may be involved in the colon tumorigenesis processes.
Collectively, these studies suggest that ceramides produced by de novo synthesis are pro-tumorigenic, while ceramides derived from sphingomyelinases have a more ambiguous role.
4.2. Modulation of ceramides production in murine models.
Under naïve conditions, ablation of Cers2 null mice and decrease in long-chain ceramides didn’t elicit an observable intestinal pathology. However, when challenged with DSS, the knockout mice displayed increased intestinal permeability and exacerbated colitis [42; 43]. Additionally, Grosch’s group found an enhanced tumor burden in the large intestine of Cers2 null mice compared with control mice after 12 weeks of AOM/DSS, indicating that loss of very long-chain ceramides plays a determinant role in chronic inflammation-related colonic tumorigenesis [42]. On the contrary, Acer3 deficiency increased long-chain ceramide C18:1 in colonic epithelial cells and induced low-grade dysplasia in a DSS-induced murine colitis model [13; 58]. Interestingly, C18:1 ceramide but not C18:0 ceramide potentiated LPS induced expression of pro-inflammatory cytokines. These studies suggest that acyl chain length or degree of unsaturation influences the biological function of individual ceramide species. The very long-chain ceramides seem to be protective in the DSS-induced colitis, while the long-chain ceramides may contribute to tissue dysfunction.
Like Cers2 null mice, alk-SMase null mice didn’t develop any spontaneous tumors in the absence of carcinogenic factors [71]; however, knockout mice treated with the procarcinogen AOM developed more aberrant crypt foci (ACF) than wild-type mice. Neither group developed tumors. Interestingly, co-treatment of AOM/DSS resulted in increased tumorigenesis in knockout mice. 32% of alk-SMase null tumors were adenocarcinomas, while all tumors in the wild-type mice were adenomas. These results indicate that loss of alk-SMase function facilitates tumorigenesis and malignant transformation. Mechanistic studies revealed that tumorigeneses was related to decreased ceramides and increased platelet-activating factor (PAF) levels. Furthermore, S-1-P unexpectedly increased in the colon of alk-CDase null mice, again demonstrating the complexity of sphingolipid modulation by stress stimuli.
A recent study from the Hannun group identified nCDase as a regulator of cell survival in colon cancer cells with minimal effects on non-cancerous cells. Knocking out the ASAH2 gene in colon cancer cell lines or mice resulted in the loss of β-catenin and inhibition of ERK, significant components of pathways relevant for colon cancer development. This study also demonstrated that inhibition of nCDase in an HT-29 cell xenograft model increased ceramide and delayed tumor growth. Most importantly, mice lacking nCDase treated with AOM were protected from tumor formation, indicating that nCDase may emerge as a therapeutic target in colon cancer [11].
4.3. Modulation of ceramides production in colon cancer cell lines (Table 2).
Table 2.
Ceramide metabolism in colon cancer cell lines
| Cell line | Intervention | Enzymes (mRNA) and Ceramides | Cellular consequences |
|---|---|---|---|
| SW403 | C2&C6 ceramide | N.I. | Apoptosis[76] |
| Ceramidase inhibitors | Total ceramide ↑ | Apoptosis [76] | |
| Carmofur | aCDase activity ↓ Total ceramide ↑ |
Anti-proliferation [85] | |
| HCT-116 p53−/− | Oxaliplatin & 5-FU |
CerS2 ↑ C24, C24:1 ↑ |
Cell growth arrest [80] |
| HCT-116 | C2 ceramide | - | Apoptosis[77] |
| Oxaliplatin & 5-FU |
CerS5, CerS6 ↑ C14, C16 ↑ |
Cell growth arrest, autophagy [80] | |
| Staurosporine | De novo synthesis ↑ | Apoptosis [87] | |
| Celecoxib |
CerS6, Sptlc2 ↑ Total ceramides and sphingosine ↑ |
Apoptosis, antiproliferation[80; 88] | |
| Curcumin | De novo synthesis ↑ | Apoptosis [86] | |
| Carboxychromanols | Total ceramide and sphingomyelin ↑ | Anti-inflammation Apoptosis [89] | |
| Caco-2 | 5FU | CerS4, CerS5 ↑ | Apoptosis [82] |
| Mevalonate | Smpd1 ↓ | Anti-proliferation[75] | |
| Curcumin | Smpd1 ↓ | Apoptosis [74] | |
| Ursodeoxycholic acid | (monolayer) Smpd1 & Smpd2 ↓ (polarized) Npp7 ↑ |
Anti-proliferation Apoptosis [15] |
|
| HT-29 | C2 ceramide | N.I. | Apoptosis [77] |
| Fenretinide | Total dihydroceramides ↑ | Apoptosis and Necrosis [90] | |
| 5FU | CerS4, CerS5 ↓ | Apoptosis [82] | |
| Psyllium | Npp7 ↑ | Anti-proliferation[83] | |
| Ursolic acid | Npp7 ↑ | Anti-proliferation[84] | |
| Butyric acid |
Smpd1 ↑ Total ceramide ↑ |
N.I.[73] | |
| Curcumin | De novo synthesis ↑ | Apoptosis [86] | |
| SW480(TRAIL sensitive) |
TRAIL |
CesS6 ↑ C16 ↑ |
Apoptosis[91] |
| DLD-1 | 5FU |
Smpd1 ↑ Total ceramide ↑ |
Apoptosis [72] |
| Curcumin | De novo synthesis ↑ | Apoptosis [86] |
Ceramides were initially regarded as pro-apoptotic signaling molecules based on many early studies carried out in colon cancer cells and many other cancer cell lines [63]. However, the number of reports implicating elevated rates of ceramide de novo synthesis in many tumors is growing. Exogenous ceramides induce mitochondria cytochrome c release and apoptotic cell death in colon cancer cell lines SW403, HCT-116 and HT-29 [76; 77; 78]. Moreover, many studies have shown that chemotherapeutic drugs and ingredients from natural plants modulate cell proliferation and apoptosis through the regulation of ceramide levels and the enzymes required for their generation (Table 2).
Adenomas progress into carcinomas and malignancy coincides with the inactivation of the p53 gene in 50% of the tumors. The reciprocal regulation of p53 and ceramides in the context of tumorigenesis was recently reviewed [79]. Sebastian et al. reported differential regulation of ceramide metabolism in the HCT-116 cell line with (wild-type) or without p53 expression [80]. Two chemotherapeutics, oxaliplatin and 5-fluorouracil (5-FU), lead to a substantial increase in CERS5 and CERS6 expression and C16:0-ceramide levels in the wide type HCT-116; conversely, these drugs increase CERS2 expression and C24 and C24:1 ceramides in p53 deficient cells. Surprisingly, knockdown of CERS5 expression leads to an increased sensitivity of wild-type cells, but not p53 deficient cells, to chemotherapeutic treatment. This mechanistic study revealed that the protective effect of CERS5 expression might be attributed to the downregulation of chemotherapeutic-induced autophagy. Accordingly, treatment with the CERS inhibitor fumonisin B1 significantly sensitizes cells to chemotherapeutics.
Ruijuan et al. identified another p53 target, ACER2, in the HCT-116 cell line [81]. They found two p53 response elements in the first intron of the ACER2 gene. Ionizing radiation (IR) increased ACER2 mRNA and protein levels in a p53-dependent manner. Interestingly, low-dose IR-induced ACER2 expression was protective, due to downstream S-1-P generation. High-dose IR induces ACER2 expression to a greater extent and causes DNA fragmentation and caspase 3/7 activation through sphingosine accumulation. These results emphasize the need for attention regarding modulation of the whole enzyme network when therapeutically targeting sphingolipids in colon cancer.
In Caco-2 and HT-29 cells, 5-FU induces apoptosis and a concomitant downregulation of CERS4 and CERS5 [82]. Alternatively, 5-FU-induced apoptosis is related to the upregulation of aSMase and its degradation product ceramide in DLD-1 cells [72]. These findings imply that ceramides generated from the de novo synthesis pathway are pro-survival, while those from the sphingomyelinase hydrolyzation are pro-apoptotic. This assumption is supported by findings that psyllium and ursolic acid induce HT-29 cell death through activation of alk-SMase [83; 84] and that carmofur inhibits SW403 cell proliferation through inhibition of nCDase [85].
Another example is curcumin, a potent natural chemopreventive agent for colon cancer, which conducts its pro-apoptotic effect in a cell-type-specific way. Curcumin induces cell death in HCT-116, HT-29, and DLD-1 cancer cells through upregulation of de novo ceramide synthesis, which is attenuated by myriocin treatment [86]. In Caco-2 cells, curcumin decreases the aSMase protein level, inhibits DNA synthesis, and causes cell death [74].
Another striking finding from del Solar et al. showed that staurosporine induces apoptosis in HCT-116 colon cancer cells and the CCD-112 non-cancerous colon-derived cell line, although upregulation of de novo ceramide synthesis was only observed in HCT-116 cells [87]. This differential regulation of ceramide synthesis holds for other cancer cell lines – Hela S3 and MCF-7 cells versus non-cancerous RPE-1 cells – indicating that accumulation of specific ceramides and dihydroceramides in HCT-116 cells during apoptosis is a cancer-specific phenomenon. In support of this finding, Garcia et al. also found that the nCDase inhibitor C6 urea-ceramide caused cell death in a panel of colon cancer cell lines but not in non-cancerous intestinal epithelial RIE-1 cells. Moreover, nCDase knockout mice didn’t show any significant intestinal pathology but were protected from AOM-induced tumor formation [11].
The research findings from the cancer cell lines have given us important clues to understand the intricate mechanisms of ceramides’ involvement in gut tumorigenesis. Surveillance of the changes in specific ceramides and the expression of ceramide-metabolizing enzymes could profoundly impact therapeutic effects.
5. Ceramide, microbiome, and the gut-liver axis (Figure 2)
Figure 2. Ceramide in microbiome-gut-liver axis.
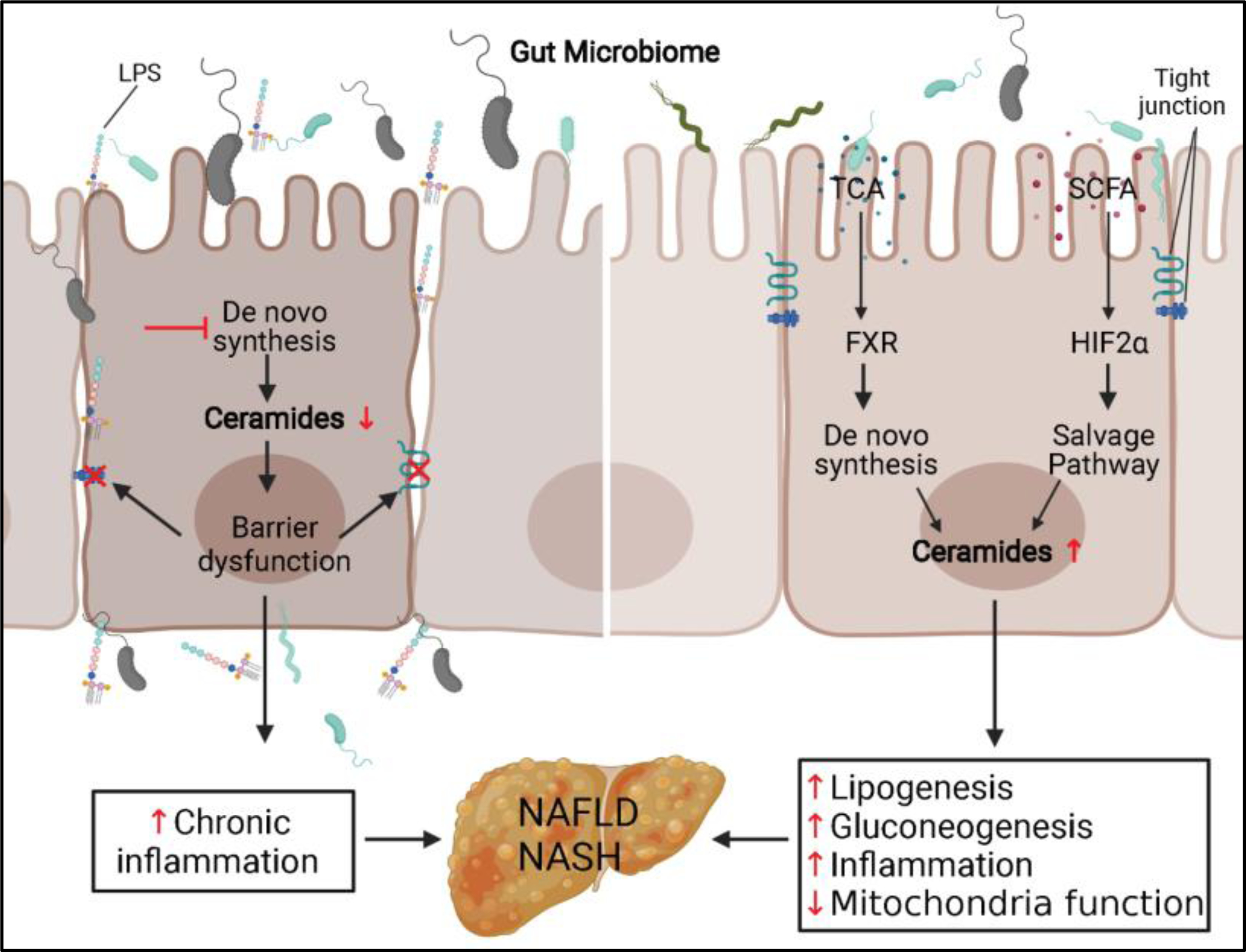
(Left) Inhibition of sphingolipid de novo synthesis pathway decreases ceramides level and leads to barrier dysfunction in the IEC. Bacteria and their metabolites infiltration in the IEC cause chronic inflammation and increase the risk of NAFLD and NASH. (Right) Schematic depiction of the role ceramides played in two pathways that microbiome regulates the metabolites in the gut lumen and causes liver steatosis and NASH. LPS, lipopolysaccharide; TCA, taurocholic acid; SCFA, short-chain fatty acid; FXR, farnesoid X receptor; HIF2α, hypoxia-inducible factor 2α; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis.
Communication within the gut–liver axis regulates systemic metabolic homeostasis, and dysregulation of this process potentiates metabolic disorders including obesity, insulin resistance, non-alcoholic fatty liver disease (NAFLD), and non-alcoholic steatohepatitis (NASH). The bidirectional interchange of information through the portal vein and bile acid secretion integrates the genetic background of the organism with environmental factors such as dietary and hygiene habits.
An intact intestinal barrier is essential for healthy gut-liver communication. Under physiologic conditions, the gut microbiome is excluded from portal circulation and liver by an intact intestinal barrier. In some pathologic conditions, like IBD, increased permeability of the intestinal barrier leads to the invasion of microbes and their metabolites into the liver, causing chronic inflammation. Ceramide plays a significant role in maintaining barrier function, as mentioned above. Ceramides are also involved in gut microbiota-liver communication through intestinal nuclear receptor farnesoid X receptor (FXR). FXR is expressed in both the liver and intestine, where it regulates the synthesis and transport of bile acids and is a crucial modulator of bile acid homeostasis and enterohepatic circulation [92; 93]. FXR in the intestine controls bile acid uptake in the ileum and bile acid synthesis in the liver through FGF15/19 expression in the enterocytes[94]. Using a series of elegantly designed experiments, including intestinal genetic or agonist/antagonist modification of FXR function, the Gonzalez group showed that inhibiting intestinal FXR activity in models of diet-induced or genetic obesity decreases ceramide levels in the intestine and circulation, which resolved obesity, insulin resistance, and hepatic steatosis and suppressed hepatic gluconeogenesis. Mechanistically, bacteria-modified bile acid could inhibit FRX activity, which decreases numerous genes involved in ceramide metabolism in the intestine. Lower systemic ceramide levels restored browning in adipose tissue [94; 95]. Decreases in portal vein ceramide levels act as a signaling molecule to decrease lipogenesis and promote lipid oxidation in the liver, thus mitigating liver steatosis [96]. Furthermore, ceramide depletion restored the mitochondrial TCA cycle function and inhibited hepatic gluconeogenesis in the high fat diet (HFD) fed animals [97]. Thus, an intestinal FXR-ceramide signaling axis mediates the influence of gut microbiota on metabolic disorders.
Hypoxia-inducible factor 2α (HIF2α) also mediates ceramide production. The same group led by Gonzalez generated an intestinal-specific Hif2α knockout mouse, which was protected from HFD-induced obesity and hepatic steatosis. Interestingly, these metabolic improvements were correlated with decreased ceramides in the intestine and systemic circulation. The investigators determined that the ceramide-lowering effect of Hif2α ablation is mediated by inhibition of an important enzyme, Neu3, in the salvage pathway of ceramide production. These studies imply that diet, microbiota, and other stress factors might converge on ceramides to regulate liver and systemic metabolic homeostasis.
Future perspectives
Considering the multiple roles of ceramides as intestinal barrier components, precursors of complex and simple sphingolipids, and signaling molecules for cell survival, proliferation, differentiation, and inflammation, further study is warranted to delineate physiologic and pathologic processes of ceramide in the gut. Variable stress stimuli act on multiple pathways of ceramide generation and regulate levels of individual ceramide molecular species in distinct subcellular compartments to execute specific functions [98]. Ideally, a comparison of all the enzymes involved in ceramide generation at transcriptional and protein levels, as well as lipidomic analysis of human cancer tissue, should be conducted to decipher the dysregulation of the sphingolipid network and identify targetable enzymes involved in pathologic processes of IBD and intestinal cancers. The generation of more targeted genetic animal models is warranted to understand the function of various enzymes in the heterogeneous cell populations of the intestinal epithelium. Most of the data implicating ceramides within intestinal pathologies have relied on loss-of-function interventions that deplete ceramides. However, few studies have evaluated gain-of-function models allowing for selective induction of endogenous ceramide in vivo. Moreover, although ceramides are well accepted as mediators of cellular stress signaling, their specific protein partners are not very clear. Early studies showed that ceramide transport protein CERT [99], the inhibitor 2 of protein phosphatase 2A (I2PP2A) [100], cathepsin D [101] could be the immediate downstream effectors of ceramides. But, lots of ceramides-controlled cellular processes cannot be explained by these pathways. New techniques using bifunctional ceramide or sphingosine analog, pacCer or pacSph, that can detect ceramide-protein interaction identified direct ceramide targets such as steroidogenic acute regulatory protein D7 (StarD7) and mitochondrial fission factor (MFF), which unravels new potential for therapeutic development [102; 103; 104].
Acknowledgements
Funding: This work was supported by the National Institutes of Health (DK115824, DK116888, and DK116450 to SAS); the Juvenile Diabetes Research Foundation (JDRF 3-SRA-2019-768-A-B to SAS); the American Diabetes Association (to SAS); the American Heart Association (to SAS); the Margolis Foundation (to SAS); and the United States Department of Agriculture (2019-67018-29250 to Y.L.).
Footnotes
Declaration of competing interest
SAS is a founder and shareholder with Centaurus Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kurek K, Piotrowska DM, Wiesiolek P, Chabowski A, and Zendzian-Piotrowska M, [Role of sphingolipids in digestive system]. Postepy Hig Med Dosw (Online) 66 (2012) 868–75. [DOI] [PubMed] [Google Scholar]
- [2].Duan RD, and Nilsson A, Metabolism of sphingolipids in the gut and its relation to inflammation and cancer development. Prog Lipid Res 48 (2009) 62–72. [DOI] [PubMed] [Google Scholar]
- [3].Merrill AH Jr., De novo sphingolipid biosynthesis: a necessary, but dangerous, pathway. J Biol Chem 277 (2002) 25843–6. [DOI] [PubMed] [Google Scholar]
- [4].Futerman AH, and Riezman H, The ins and outs of sphingolipid synthesis. Trends Cell Biol 15 (2005) 312–8. [DOI] [PubMed] [Google Scholar]
- [5].Park JW, Park WJ, and Futerman AH, Ceramide synthases as potential targets for therapeutic intervention in human diseases. Biochim Biophys Acta 1841 (2014) 671–81. [DOI] [PubMed] [Google Scholar]
- [6].Omae F, Miyazaki M, Enomoto A, Suzuki M, Suzuki Y, and Suzuki A, DES2 protein is responsible for phytoceramide biosynthesis in the mouse small intestine. Biochem J 379 (2004) 687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kitatani K, Idkowiak-Baldys J, and Hannun YA, The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal 20 (2008) 1010–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nilsson A, Metabolism of sphingomyelin in the intestinal tract of the rat. Biochim Biophys Acta 164 (1968) 575–84. [DOI] [PubMed] [Google Scholar]
- [9].Schmelz EM, Crall KJ, Larocque R, Dillehay DL, and Merrill AH Jr., Uptake and metabolism of sphingolipids in isolated intestinal loops of mice. J Nutr 124 (1994) 702–12. [DOI] [PubMed] [Google Scholar]
- [10].Duan RD, Bergman T, Xu N, Wu J, Cheng Y, Duan J, Nelander S, Palmberg C, and Nilsson A, Identification of human intestinal alkaline sphingomyelinase as a novel ecto-enzyme related to the nucleotide phosphodiesterase family. J Biol Chem 278 (2003) 38528–36. [DOI] [PubMed] [Google Scholar]
- [11].Garcia-Barros M, Coant N, Kawamori T, Wada M, Snider AJ, Truman JP, Wu BX, Furuya H, Clarke CJ, Bialkowska AB, Ghaleb A, Yang VW, Obeid LM, and Hannun YA, Role of neutral ceramidase in colon cancer. FASEB J 30 (2016) 4159–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Duan RD, Verkade HJ, Cheng Y, Havinga R, and Nilsson A, Effects of bile diversion in rats on intestinal sphingomyelinases and ceramidase. Biochim Biophys Acta 1771 (2007) 196–201. [DOI] [PubMed] [Google Scholar]
- [13].Duan RD, Hertervig E, Nyberg L, Hauge T, Sternby B, Lillienau J, Farooqi A, and Nilsson A, Distribution of alkaline sphingomyelinase activity in human beings and animals. Tissue and species differences. Dig Dis Sci 41 (1996) 1801–6. [DOI] [PubMed] [Google Scholar]
- [14].Xu R, Antwi Boasiako P, and Mao C, Alkaline ceramidase family: The first two decades. Cell Signal 78 (2021) 109860. [DOI] [PubMed] [Google Scholar]
- [15].Liu F, Cheng Y, Wu J, Tauschel HD, and Duan RD, Ursodeoxycholic acid differentially affects three types of sphingomyelinase in human colon cancer Caco 2 cells. Cancer Lett 235 (2006) 141–6. [DOI] [PubMed] [Google Scholar]
- [16].Nagahashi M, Hait NC, Maceyka M, Avni D, Takabe K, Milstien S, and Spiegel S, Sphingosine-1-phosphate in chronic intestinal inflammation and cancer. Adv Biol Regul 54 (2014) 112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nielsen OH, Li Y, Johansson-Lindbom B, and Coskun M, Sphingosine-1-Phosphate Signaling in Inflammatory Bowel Disease. Trends in molecular medicine 23 (2017) 362–374. [DOI] [PubMed] [Google Scholar]
- [18].Johnson EL, Heaver SL, Waters JL, Kim BI, Bretin A, Goodman AL, Gewirtz AT, Worgall TS, and Ley RE, Sphingolipids produced by gut bacteria enter host metabolic pathways impacting ceramide levels. Nat Commun 11 (2020) 2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brown EM, Ke X, Hitchcock D, Jeanfavre S, Avila-Pacheco J, Nakata T, Arthur TD, Fornelos N, Heim C, Franzosa EA, Watson N, Huttenhower C, Haiser HJ, Dillow G, Graham DB, Finlay BB, Kostic AD, Porter JA, Vlamakis H, Clish CB, and Xavier RJ, Bacteroides-Derived Sphingolipids Are Critical for Maintaining Intestinal Homeostasis and Symbiosis. Cell Host Microbe 25 (2019) 668–680 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Olsen I, and Jantzen E, Sphingolipids in Bacteria and Fungi. Anaerobe 7 (2001) 103–112. [Google Scholar]
- [21].Hannun YA, and Linardic CM, Sphingolipid breakdown products: anti-proliferative and tumor-suppressor lipids. Biochim Biophys Acta 1154 (1993) 223–36. [DOI] [PubMed] [Google Scholar]
- [22].Homaidan FR, Chakroun I, and El-Sabban ME, Regulation of nuclear factor-kappaB in intestinal epithelial cells in a cell model of inflammation. Mediators Inflamm 12 (2003) 277–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Colell A, Coll O, Mari M, Fernandez-Checa JC, and Garcia-Ruiz C, Divergent role of ceramide generated by exogenous sphingomyelinases on NF-kappa B activation and apoptosis in human colon HT-29 cells. FEBS Lett 526 (2002) 15–20. [DOI] [PubMed] [Google Scholar]
- [24].Fischer H, Ellstrom P, Ekstrom K, Gustafsson L, Gustafsson M, and Svanborg C, Ceramide as a TLR4 agonist; a putative signalling intermediate between sphingolipid receptors for microbial ligands and TLR4. Cell Microbiol 9 (2007) 1239–51. [DOI] [PubMed] [Google Scholar]
- [25].Duan RD, Cheng Y, Hansen G, Hertervig E, Liu JJ, Syk I, Sjostrom H, and Nilsson A, Purification, localization, and expression of human intestinal alkaline sphingomyelinase. J Lipid Res 44 (2003) 1241–50. [DOI] [PubMed] [Google Scholar]
- [26].Ye Y, Pang Z, Chen W, Ju S, and Zhou C, The epidemiology and risk factors of inflammatory bowel disease. Int J Clin Exp Med 8 (2015) 22529–42. [PMC free article] [PubMed] [Google Scholar]
- [27].Pierik M, Yang H, Barmada MM, Cavanaugh JA, Annese V, Brant SR, Cho JH, Duerr RH, Hugot JP, McGovern DP, Paavola-Sakki P, Radford-Smith GL, Pavli P, Silverberg MS, Schreiber S, Taylor KD, Vlietinck R, and I.B.D.I.G. Consortium, The IBD international genetics consortium provides further evidence for linkage to IBD4 and shows gene-environment interaction. Inflamm Bowel Dis 11 (2005) 1–7. [DOI] [PubMed] [Google Scholar]
- [28].Ramos GP, and Papadakis KA, Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin Proc 94 (2019) 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xavier RJ, and Podolsky DK, Unravelling the pathogenesis of inflammatory bowel disease. Nature 448 (2007) 427–34. [DOI] [PubMed] [Google Scholar]
- [30].Bazarganipour S, Hausmann J, Oertel S, El-Hindi K, Brachtendorf S, Blumenstein I, Kubesch A, Sprinzl K, Birod K, Hahnefeld L, Trautmann S, Thomas D, Herrmann E, Geisslinger G, Schiffmann S, and Grosch S, The Lipid Status in Patients with Ulcerative Colitis: Sphingolipids are Disease-Dependent Regulated. J Clin Med 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Diab J, Hansen T, Goll R, Stenlund H, Ahnlund M, Jensen E, Moritz T, Florholmen J, and Forsdahl G, Lipidomics in Ulcerative Colitis Reveal Alteration in Mucosal Lipid Composition Associated With the Disease State. Inflamm Bowel Dis 25 (2019) 1780–1787. [DOI] [PubMed] [Google Scholar]
- [32].McGovern DP, Gardet A, Torkvist L, Goyette P, Essers J, Taylor KD, Neale BM, Ong RT, Lagace C, Li C, Green T, Stevens CR, Beauchamp C, Fleshner PR, Carlson M, D’Amato M, Halfvarson J, Hibberd ML, Lordal M, Padyukov L, Andriulli A, Colombo E, Latiano A, Palmieri O, Bernard EJ, Deslandres C, Hommes DW, de Jong DJ, Stokkers PC, Weersma RK, Consortium NIG, Sharma Y, Silverberg MS, Cho JH, Wu J, Roeder K, Brant SR, Schumm LP, Duerr RH, Dubinsky MC, Glazer NL, Haritunians T, Ippoliti A, Melmed GY, Siscovick DS, Vasiliauskas EA, Targan SR, Annese V, Wijmenga C, Pettersson S, Rotter JI, Xavier RJ, Daly MJ, Rioux JD, and Seielstad M, Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet 42 (2010) 332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D, Vermeire S, Dewit O, de Vos M, Dixon A, Demarche B, Gut I, Heath S, Foglio M, Liang L, Laukens D, Mni M, Zelenika D, Van Gossum A, Rutgeerts P, Belaiche J, Lathrop M, and Georges M, Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet 3 (2007) e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gassler N, Rohr C, Schneider A, Kartenbeck J, Bach A, Obermuller N, Otto HF, and Autschbach F, Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol 281 (2001) G216–28. [DOI] [PubMed] [Google Scholar]
- [35].Buhner S, Buning C, Genschel J, Kling K, Herrmann D, Dignass A, Kuechler I, Krueger S, Schmidt HH, and Lochs H, Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation? Gut 55 (2006) 342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Soderholm JD, Olaison G, Peterson KH, Franzen LE, Lindmark T, Wiren M, Tagesson C, and Sjodahl R, Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn’s disease. Gut 50 (2002) 307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Genin MJ, Gonzalez Valcarcel IC, Holloway WG, Lamar J, Mosior M, Hawkins E, Estridge T, Weidner J, Seng T, Yurek D, Adams LA, Weller J, Reynolds VL, and Brozinick JT, Imidazopyridine and Pyrazolopiperidine Derivatives as Novel Inhibitors of Serine Palmitoyl Transferase. J Med Chem 59 (2016) 5904–10. [DOI] [PubMed] [Google Scholar]
- [38].Bouhet S, Hourcade E, Loiseau N, Fikry A, Martinez S, Roselli M, Galtier P, Mengheri E, and Oswald IP, The mycotoxin fumonisin B1 alters the proliferation and the barrier function of porcine intestinal epithelial cells. Toxicol Sci 77 (2004) 165–71. [DOI] [PubMed] [Google Scholar]
- [39].Li Z, Kabir I, Tietelman G, Huan C, Fan J, Worgall T, and Jiang XC, Sphingolipid de novo biosynthesis is essential for intestine cell survival and barrier function. Cell Death Dis 9 (2018) 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li Y, Chaurasia B, Kaddai V, Wilkerson JL, Maschek JA, Cox J, Wei P, Bensard C, Meikle PJ, Clevers H, Shayman JA, Hirabayashi Y, Holland WL, Rutter J, and Summers SA, Serine palmitoyltransferase controls stemness of intestinal progenitors. bioRxiv (2020). [Google Scholar]
- [41].Ohta E, Ohira T, Matsue K, Ikeda Y, Fujii K, Ohwaki K, Osuka S, Hirabayashi Y, and Sasaki M, Analysis of development of lesions in mice with serine palmitoyltransferase (SPT) deficiency - Sptlc2 conditional knockout mice. Exp Anim 58 (2009) 515–24. [DOI] [PubMed] [Google Scholar]
- [42].Oertel S, Scholich K, Weigert A, Thomas D, Schmetzer J, Trautmann S, Wegner MS, Radeke HH, Filmann N, Brune B, Geisslinger G, Tegeder I, and Grosch S, Ceramide synthase 2 deficiency aggravates AOM-DSS-induced colitis in mice: role of colon barrier integrity. Cell Mol Life Sci 74 (2017) 3039–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kim YR, Volpert G, Shin KO, Kim SY, Shin SH, Lee Y, Sung SH, Lee YM, Ahn JH, Pewzner-Jung Y, Park WJ, Futerman AH, and Park JW, Ablation of ceramide synthase 2 exacerbates dextran sodium sulphate-induced colitis in mice due to increased intestinal permeability. J Cell Mol Med 21 (2017) 3565–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Siddique MM, Li Y, Wang L, Ching J, Mal M, Ilkayeva O, Wu YJ, Bay BH, and Summers SA, Ablation of dihydroceramide desaturase 1, a therapeutic target for the treatment of metabolic diseases, simultaneously stimulates anabolic and catabolic signaling. Mol Cell Biol 33 (2013) 2353–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Helke K, Angel P, Lu P, Garrett-Mayer E, Ogretmen B, Drake R, and Voelkel-Johnson C, Ceramide Synthase 6 Deficiency Enhances Inflammation in the DSS model of Colitis. Sci Rep 8 (2018) 1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bock J, Liebisch G, Schweimer J, Schmitz G, and Rogler G, Exogenous sphingomyelinase causes impaired intestinal epithelial barrier function. World J Gastroenterol 13 (2007) 5217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Claycombe KJ, Wu D, Nikolova-Karakashian M, Palmer H, Beharka A, Paulson KE, and Meydani SN, Ceramide mediates age-associated increase in macrophage cyclooxygenase-2 expression. J Biol Chem 277 (2002) 30784–91. [DOI] [PubMed] [Google Scholar]
- [48].Teichgraber V, Ulrich M, Endlich N, Riethmuller J, Wilker B, De Oliveira-Munding CC, van Heeckeren AM, Barr ML, von Kurthy G, Schmid KW, Weller M, Tummler B, Lang F, Grassme H, Doring G, and Gulbins E, Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med 14 (2008) 382–91. [DOI] [PubMed] [Google Scholar]
- [49].Wu J, Nilsson A, Jonsson BA, Stenstad H, Agace W, Cheng Y, and Duan RD, Intestinal alkaline sphingomyelinase hydrolyses and inactivates platelet-activating factor by a phospholipase C activity. Biochem J 394 (2006) 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Li Y, Talbot CL, and Chaurasia B, Ceramides in Adipose Tissue. Front Endocrinol (Lausanne) 11 (2020) 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Parigi TL, Roda G, Argollo M, Gilardi D, and Danese S, Is there a role for therapeutic sphingolipids in inflammatory bowel disease? Expert Rev Gastroenterol Hepatol 14 (2020) 47–54. [DOI] [PubMed] [Google Scholar]
- [52].Jenkins RW, Canals D, Idkowiak-Baldys J, Simbari F, Roddy P, Perry DM, Kitatani K, Luberto C, and Hannun YA, Regulated secretion of acid sphingomyelinase: implications for selectivity of ceramide formation. J Biol Chem 285 (2010) 35706–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Goggel R, Winoto-Morbach S, Vielhaber G, Imai Y, Lindner K, Brade L, Brade H, Ehlers S, Slutsky AS, Schutze S, Gulbins E, and Uhlig S, PAF-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nat Med 10 (2004) 155–60. [DOI] [PubMed] [Google Scholar]
- [54].Xiong Y, Zhu XD, Wan P, Ren YP, Wang C, Yan RW, Guo Y, and Bai AP, Inhibition of ASM activity ameliorates DSS-induced colitis in mice. Prostaglandins Other Lipid Mediat 140 (2019) 26–30. [DOI] [PubMed] [Google Scholar]
- [55].Sakata A, Ochiai T, Shimeno H, Hikishima S, Yokomatsu T, Shibuya S, Toda A, Eyanagi R, and Soeda S, Acid sphingomyelinase inhibition suppresses lipopolysaccharide-mediated release of inflammatory cytokines from macrophages and protects against disease pathology in dextran sulphate sodium-induced colitis in mice. Immunology 122 (2007) 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nilsson A, and Duan RD, Alkaline sphingomyelinases and ceramidases of the gastrointestinal tract. Chem Phys Lipids 102 (1999) 97–105. [DOI] [PubMed] [Google Scholar]
- [57].Snider AJ, Wu BX, Jenkins RW, Sticca JA, Kawamori T, Hannun YA, and Obeid LM, Loss of neutral ceramidase increases inflammation in a mouse model of inflammatory bowel disease. Prostaglandins Other Lipid Mediat 99 (2012) 124–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].El Bawab S, Birbes H, Roddy P, Szulc ZM, Bielawska A, and Hannun YA, Biochemical characterization of the reverse activity of rat brain ceramidase. A CoA-independent and fumonisin B1-insensitive ceramide synthase. J Biol Chem 276 (2001) 16758–66. [DOI] [PubMed] [Google Scholar]
- [59].Wang K, Xu R, Snider AJ, Schrandt J, Li Y, Bialkowska AB, Li M, Zhou J, Hannun YA, Obeid LM, Yang VW, and Mao C, Alkaline ceramidase 3 deficiency aggravates colitis and colitis-associated tumorigenesis in mice by hyperactivating the innate immune system. Cell Death Dis 7 (2016) e2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ogretmen B, and Hannun YA, Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 4 (2004) 604–16. [DOI] [PubMed] [Google Scholar]
- [61].Ogretmen B, Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer 18 (2018) 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Nganga R, Oleinik N, and Ogretmen B, Mechanisms of Ceramide-Dependent Cancer Cell Death. Adv Cancer Res 140 (2018) 1–25. [DOI] [PubMed] [Google Scholar]
- [63].Abdul Aziz NA, Mokhtar NM, Harun R, Mollah MM, Mohamed Rose I, Sagap I, Mohd Tamil A, Wan Ngah WZ, and Jamal R, A 19-Gene expression signature as a predictor of survival in colorectal cancer. BMC Med Genomics 9 (2016) 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chen L, Chen H, Li Y, Li L, Qiu Y, and Ren J, Endocannabinoid and ceramide levels are altered in patients with colorectal cancer. Oncol Rep 34 (2015) 447–54. [DOI] [PubMed] [Google Scholar]
- [65].Jang SW, Park WJ, Min H, Kwon TK, Baek SK, Hwang I, Kim S, and Park JW, Altered mRNA expression levels of the major components of sphingolipid metabolism, ceramide synthases and their clinical implication in colorectal cancer. Oncol Rep 40 (2018) 3489–3500. [DOI] [PubMed] [Google Scholar]
- [66].Kijanka G, Hector S, Kay EW, Murray F, Cummins R, Murphy D, MacCraith BD, Prehn JH, and Kenny D, Human IgG antibody profiles differentiate between symptomatic patients with and without colorectal cancer. Gut 59 (2010) 69–78. [DOI] [PubMed] [Google Scholar]
- [67].Hertervig E, Nilsson A, Björk J, Hultkrantz R, and Duan RD, Familial adenomatous polyposis is associated with a marked decrease in alkaline sphingomyelinase activity: a key factor to the unrestrained cell proliferation? British journal of cancer 81 (1999) 232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Hertervig E, Nilsson A, Nyberg L, and Duan RD, Alkaline sphingomyelinase activity is decreased in human colorectal carcinoma. Cancer 79 (1997) 448–53. [PubMed] [Google Scholar]
- [69].Hertervig E, Nilsson A, Nilbert M, and Duan RD, Reduction in alkaline sphingomyelinase in colorectal tumorigenesis is not related to the APC gene mutation. Int J Colorectal Dis 18 (2003) 309–13. [DOI] [PubMed] [Google Scholar]
- [70].Sjöqvist U, Hertervig E, Nilsson A, Duan RD, Ost A, Tribukait B, and Löfberg R, Chronic colitis is associated with a reduction of mucosal alkaline sphingomyelinase activity. Inflamm Bowel Dis 8 (2002) 258–63. [DOI] [PubMed] [Google Scholar]
- [71].Chen Y, Zhang P, Xu SC, Yang L, Voss U, Ekblad E, Wu Y, Min Y, Hertervig E, Nilsson A, and Duan RD, Enhanced colonic tumorigenesis in alkaline sphingomyelinase (NPP7) knockout mice. Mol Cancer Ther 14 (2015) 259–67. [DOI] [PubMed] [Google Scholar]
- [72].Jung JH, Taniguchi K, Lee HM, Lee MY, Bandu R, Komura K, Lee KY, Akao Y, and Kim KP, Comparative lipidomics of 5-Fluorouracil-sensitive and -resistant colorectal cancer cells reveals altered sphingomyelin and ceramide controlled by acid sphingomyelinase (SMPD1). Sci Rep 10 (2020) 6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wu J, Cheng Y, Jönsson BA, Nilsson A, and Duan RD, Acid sphingomyelinase is induced by butyrate but does not initiate the anticancer effect of butyrate in HT29 and HepG2 cells. J Lipid Res 46 (2005) 1944–52. [DOI] [PubMed] [Google Scholar]
- [74].Cheng Y, Kozubek A, Ohlsson L, Sternby B, and Duan RD, Curcumin decreases acid sphingomyelinase activity in colon cancer Caco-2 cells. Planta medica 73 (2007) 725–30. [DOI] [PubMed] [Google Scholar]
- [75].Chen Y, Xu SC, and Duan RD, Mevalonate inhibits acid sphingomyelinase activity, increases sphingomyelin levels and inhibits cell proliferation of HepG2 and Caco-2 cells. Lipids Health Dis 14 (2015) 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Selzner M, Bielawska A, Morse MA, Rudiger HA, Sindram D, Hannun YA, and Clavien PA, Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer. Cancer Res 61 (2001) 1233–40. [PubMed] [Google Scholar]
- [77].Ahn EH, and Schroeder JJ, Sphingoid bases and ceramide induce apoptosis in HT-29 and HCT-116 human colon cancer cells. Exp Biol Med (Maywood) 227 (2002) 345–53. [DOI] [PubMed] [Google Scholar]
- [78].Dindo D, Dahm F, Szulc Z, Bielawska A, Obeid LM, Hannun YA, Graf R, and Clavien PA, Cationic long-chain ceramide LCL-30 induces cell death by mitochondrial targeting in SW403 cells. Mol Cancer Ther 5 (2006) 1520–9. [DOI] [PubMed] [Google Scholar]
- [79].Jeffries KA, and Krupenko NI, Ceramide Signaling and p53 Pathways. Adv Cancer Res 140 (2018) 191–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Brachtendorf S, Wanger RA, Birod K, Thomas D, Trautmann S, Wegner MS, Fuhrmann DC, Brune B, Geisslinger G, and Grosch S, Chemosensitivity of human colon cancer cells is influenced by a p53-dependent enhancement of ceramide synthase 5 and induction of autophagy. Biochim Biophys Acta Mol Cell Biol Lipids 1863 (2018) 1214–1227. [DOI] [PubMed] [Google Scholar]
- [81].Xu R, Garcia-Barros M, Wen S, Li F, Lin CL, Hannun YA, Obeid LM, and Mao C, Tumor suppressor p53 links ceramide metabolism to DNA damage response through alkaline ceramidase 2. Cell Death Differ 25 (2018) 841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Mojakgomo R, Mbita Z, and Dlamini Z, Linking the ceramide synthases (CerSs) 4 and 5 with apoptosis, endometrial and colon cancers. Exp Mol Pathol 98 (2015) 585–92. [DOI] [PubMed] [Google Scholar]
- [83].Cheng Y, Ohlsson L, and Duan RD, Psyllium and fat in diets differentially affect the activities and expressions of colonic sphingomyelinases and caspase in mice. The British journal of nutrition 91 (2004) 715–23. [DOI] [PubMed] [Google Scholar]
- [84].Andersson D, Liu JJ, Nilsson A, and Duan RD, Ursolic acid inhibits proliferation and stimulates apoptosis in HT29 cells following activation of alkaline sphingomyelinase. Anticancer research 23 (2003) 3317–22. [PubMed] [Google Scholar]
- [85].Realini N, Solorzano C, Pagliuca C, Pizzirani D, Armirotti A, Luciani R, Costi MP, Bandiera T, and Piomelli D, Discovery of highly potent acid ceramidase inhibitors with in vitro tumor chemosensitizing activity. Sci Rep 3 (2013) 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Moussavi M, Assi K, Gomez-Munoz A, and Salh B, Curcumin mediates ceramide generation via the de novo pathway in colon cancer cells. Carcinogenesis 27 (2006) 1636–44. [DOI] [PubMed] [Google Scholar]
- [87].del Solar V, Lizardo DY, Li N, Hurst JJ, Brais CJ, and Atilla-Gokcumen GE, Differential Regulation of Specific Sphingolipids in Colon Cancer Cells during Staurosporine-Induced Apoptosis. Chem Biol 22 (2015) 1662–70. [DOI] [PubMed] [Google Scholar]
- [88].Schiffmann S, Ziebell S, Sandner J, Birod K, Deckmann K, Hartmann D, Rode S, Schmidt H, Angioni C, Geisslinger G, and Grosch S, Activation of ceramide synthase 6 by celecoxib leads to a selective induction of C16:0-ceramide. Biochem Pharmacol 80 (2010) 1632–40. [DOI] [PubMed] [Google Scholar]
- [89].Jang Y, Park NY, Rostgaard-Hansen AL, Huang J, and Jiang Q, Vitamin E metabolite 13’-carboxychromanols inhibit pro-inflammatory enzymes, induce apoptosis and autophagy in human cancer cells by modulating sphingolipids and suppress colon tumor development in mice. Free Radic Biol Med 95 (2016) 190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Maurer BJ, Melton L, Billups C, Cabot MC, and Reynolds CP, Synergistic cytotoxicity in solid tumor cell lines between N-(4-hydroxyphenyl)retinamide and modulators of ceramide metabolism. J Natl Cancer Inst 92 (2000) 1897–909. [DOI] [PubMed] [Google Scholar]
- [91].White-Gilbertson S, Mullen T, Senkal C, Lu P, Ogretmen B, Obeid L, and Voelkel-Johnson C, Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene 28 (2009) 1132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Gonzalez FJ, Nuclear receptor control of enterohepatic circulation. Compr Physiol 2 (2012) 2811–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Matsubara T, Li F, and Gonzalez FJ, FXR signaling in the enterohepatic system. Mol Cell Endocrinol 368 (2013) 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kliewer SA, and Mangelsdorf DJ, Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig Dis 33 (2015) 327–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, Brocker CN, Desai D, Amin SG, Bisson WH, Liu Y, Gavrilova O, Patterson AD, and Gonzalez FJ, Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun 6 (2015) 10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, Cai J, Qi Y, Fang ZZ, Takahashi S, Tanaka N, Desai D, Amin SG, Albert I, Patterson AD, and Gonzalez FJ, Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest 125 (2015) 386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Xie C, Jiang C, Shi J, Gao X, Sun D, Sun L, Wang T, Takahashi S, Anitha M, Krausz KW, Patterson AD, and Gonzalez FJ, An Intestinal Farnesoid X Receptor-Ceramide Signaling Axis Modulates Hepatic Gluconeogenesis in Mice. Diabetes 66 (2017) 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Hannun YA, and Obeid LM, Many ceramides. J Biol Chem 286 (2011) 27855–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Hanada K, Kumagai K, Yasuda S, Miura Y, Kawano M, Fukasawa M, and Nishijima M, Molecular machinery for non-vesicular trafficking of ceramide. Nature 426 (2003) 803–9. [DOI] [PubMed] [Google Scholar]
- [100].Mukhopadhyay A, Saddoughi SA, Song P, Sultan I, Ponnusamy S, Senkal CE, Snook CF, Arnold HK, Sears RC, Hannun YA, and Ogretmen B, Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J 23 (2009) 751–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R, Weber T, Saftig P, Peters C, Brunner J, Kronke M, and Schutze S, Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J 18 (1999) 5252–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Walls SM, Cammarato A, Chatfield DA, Ocorr K, Harris GL, and Bodmer R, Ceramide-Protein Interactions Modulate Ceramide-Associated Lipotoxic Cardiomyopathy. Cell Rep 22 (2018) 2702–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Bockelmann S, Mina JGM, Korneev S, Hassan DG, Muller D, Hilderink A, Vlieg HC, Raijmakers R, Heck AJR, Haberkant P, and Holthuis JCM, A search for ceramide binding proteins using bifunctional lipid analogs yields CERT-related protein StarD7. J Lipid Res 59 (2018) 515–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Hammerschmidt P, Ostkotte D, Nolte H, Gerl MJ, Jais A, Brunner HL, Sprenger HG, Awazawa M, Nicholls HT, Turpin-Nolan SM, Langer T, Kruger M, Brugger B, and Bruning JC, CerS6-Derived Sphingolipids Interact with Mff and Promote Mitochondrial Fragmentation in Obesity. Cell 177 (2019) 1536–1552 e23. [DOI] [PubMed] [Google Scholar]