

Abstract
Niemann–Pick disease type C1 (NPC1) is a rare, prematurely fatal lysosomal storage disorder which exhibits highly variable severity and disease progression as well as a wide-ranging age of onset, from perinatal stages to adulthood. This heterogeneity has made it difficult to obtain prompt diagnosis and to predict disease course. In addition, small NPC1 patient sample sizes have been a limiting factor in acquiring genome-wide transcriptome data. In this study, primary fibroblasts from an extensive cohort of 41 NPC1 patients were used to validate our previous findings that the lysosomal quantitative probe LysoTracker can be used as a predictor for age of onset and disease severity. We also examined the correlation between these clinical parameters and RNA expression data from primary fibroblasts and identified a set of genes that were significantly associated with lysosomal defects or age of onset, in particular neurological symptom onset. Hierarchical clustering showed that these genes exhibited distinct expression patterns among patient subgroups. This study is the first to collect transcriptomic data on such a large scale in correlation with clinical and cellular phenotypes, providing a rich genomic resource to address NPC1 clinical heterogeneity and discover potential biomarkers, disease modifiers, or therapeutic targets.
Subject terms: Medical genetics, Diseases of the nervous system, Endocrine system and metabolic diseases, Medical research
Introduction
Niemann–Pick disease type C1 (NPC1) is a rare, recessive, lysosomal storage disorder associated with neurodegenerative phenotypes as well as hepatomegaly and splenomegaly (OMIM #257220). Clinical manifestations of NPC1 disease vary and often correlate with age of onset, which can occur from the prenatal period to well into adulthood1–3. Biallelic mutation of NPC1, which encodes the lysosomal transmembrane protein NPC1, is causative for this disease4. NPC1 performs an essential role in lysosomal cholesterol trafficking, and loss of function of the NPC1 gene causes accumulation of unesterified cholesterol in late endosomes/lysosomes as well as abnormally high glycosphingolipid levels, especially in the central nervous system (CNS)5–9. Loss of NPC1 function also results in accumulation of lipids, which gives rise to enlarged, lipid-laden macrophages that are present in peripheral tissues, notably lung, liver and spleen10–15.
Prenatal NPC1 disease is rare and has a very poor prognosis, with splenomegaly, hepatomegaly, and fetal ascites seen as the most common phenotypes16. In neonatal NPC1 patients, cholestatic jaundice and liver dysfunction occur frequently; while some neonatal patients exhibit transient hepatosplenomegaly that resolves by 4 months of age, up to 10% of others exhibit rapidly progressing liver dysfunction that is fatal1,17. While these phenotypes can lead to neonatal diagnosis, diagnosis is frequently missed until later ages, when the neurological abnormalities that are the classical primary presentation in NPC1 disease arise. Neurological phenotypes can present during infantile ages as symptoms of hypotonia and motor delay, which are frequently identified due to increased clumsiness and learning difficulties in school1,18. In the juvenile stage, other neurological signs often arise, such as gait disturbance and ataxia accompanied by fine-motor skill impairment; these are related to cerebellar neurodegeneration, in particular Purkinje neuron loss19. Adult onset NPC1 disease can present with more common psychiatric manifestations such as cognitive decline, bipolar disorder or schizophrenia, often resulting in missed or delayed diagnosis20–23. Previous reports have shown that the age of onset, in particular neurological symptom onset, often correlates with lifespan/disease progression in NPC1 patients1,24. Therefore, characterizing a patient’s age of neurological onset is essential for predicting disease course as well as identifying candidates who would benefit from earlier treatment interventions. In addition, a method that quantitatively scores neurological severity in NPC1 disease across 17 clinical phenotype domains and can be adjusted to age of onset has recently been developed, and this system can be used to monitor neurological disease status and subsequent progression25,26.
Currently, there are very limited treatment options available for NPC disease, and most potential therapies are undergoing clinical trials27. Of note, these treatments primarily target the CNS. The oligosaccharide derivative 2-hydroxypropyl-β-cyclodextrin (HPβCD) has been shown to delay disease progression and increase lifespan in the Npc1m1N mouse model of NPC1 disease28–33. A recently completed phase 1-2a clinical trial (NCT01747135, ClinicalTrials.gov) extended these results to individuals with NPC1 disease and suggested that intrathecal treatment with HPβCD slowed disease progression26. However, a subsequent 12-month, mock-controlled clinical trial has not demonstrated efficacy (NCT02534844, ClinicalTrials.gov). The glucosyl ceramide synthase inhibitor N-butyldeoxynojirimycin (miglustat, Zavesca) is correlated with reduced glycosphingolipid levels, stabilized neurological phenotypes, and significantly reduced mortality risk in NPC134–37, and is the only treatment that is approved for NPC disease worldwide38. However, the FDA has not approved its use for NPC disease, thus miglustat is limited to off-label use for NPC in the United States. Recent studies have proposed that the heat-shock protein co-inducer arimoclomol may have potential therapeutic benefit for neurological disorders, including NPC139 (NCT02612129, ClinicalTrials.gov). Several studies using these potential therapies suggest that treatment at early stages of the disease may be more beneficial than at later stages29,37,40,41. Therefore, the ability to predict NPC1 disease severity, progression, and individual treatment response could greatly improve disease management.
Previously, we found that LysoTracker Red (LysoTracker) staining in fibroblasts showed promise as a marker that correlated with the age of onset of any NPC1-associated symptom, and showed the highest correlation with age of onset of neurological symptoms42. LysoTracker staining can be used as a cellular biomarker in circulating B cells of NPC1 patients to quantitate the enlargement of acidic, endolysosomal storage compartments that occurs because of accumulated unesterified cholesterol43. LysoTracker has also been used in the lysosomal storage disorder Gaucher disease to identify abnormally large endolysosomal compartments44. In addition, further understanding of the gene expression changes that arise in NPC1 disease could improve prediction of disease course, as this knowledge could lead to discovery of NPC1 genetic modifiers, biomarkers, and novel pathways involved in disease progression. However, as is the case for other orphan disorders, the small NPC1 patient population has previously been a limiting factor in gathering comprehensive genomic and transcriptomic datasets. In this study, we analyzed clinical data for 41 patients enrolled in an NPC1 natural history study along with quantitative LysoTracker staining and RNA gene expression of primary fibroblasts from these patients. Our results validate and extend previous data showing that LysoTracker levels correlate with age of onset, with the highest correlation to neurological onset, and also correlate with disease severity. Additionally, we used genome-wide expression analyses to identify 127 genes that significantly correlated with LysoTracker staining, disease severity, or age of onset. Hierarchical clustering analysis of these significantly correlated genes indicated distinct gene expression profiles across the cohort, suggesting genes and pathways that may contribute to phenotypic heterogeneity and varied response to HPβCD treatment. This comprehensive dataset provides an important resource to guide future studies on NPC disease biomarker validation and modifier identification, thus furthering our understanding of NPC1 disease biology in the setting of genetic and phenotypic heterogeneity.
Results
Quantitative LysoTracker measurements in NPC1 fibroblasts are reproducible
As part of an NPC1 natural history study at the National Institutes of Health (NCT00344331, Clinical Trials.gov), primary skin fibroblasts were collected by skin biopsy from 41 individuals with NPC1. This expanded a fibroblast set from an NPC1 patient cohort that was previously published by our group42 and included 24 of the 27 patients in the previous study along with 17 additional patients. The demographics of these 41 individuals are described in Table 1.
Table 1.
NPC1 patient demographics.
| Age of onset (years) | Age of first neurological symptom (years) | Age-adjusted neurological severity score | LysoTracker levels (fold change) | Presenting symptom | First neurological symptom | Sex | Pathogenic DNA variants | Predicted protein changes |
|---|---|---|---|---|---|---|---|---|
| N/A | N/A | 0.1 | 9.7 | None | None reported | M* | c.410C>T, c.2000C>T | p.T137M, p.S667L |
| − 0.4** | N/A | 0.0 | 43.1 | Hyperecholic intestines, IUGR | None reported | F | c.3182T>C, c.1628C>T | p.I1061T, p.P543L |
| 0 | N/A | 2.0 | 30.9 | Splenomegaly | None reported | M | c.3182T>C, c.3182T>C | p.I1061T, p.I1061T |
| 0 | N/A | 0.0 | 38.3 | Jaundice, liver failure† | None reported | M | c.3182T>C, c.681T>G | p.I1061T, p.C227W |
| 0.1 | N/A | 2.4 | 36.7 | Jaundice | None reported | M | c.3182T>C, c.3107C>T | p.I1061T, p.T1036M |
| 0.5 | N/A | 0.1 | 9.9 | Splenomegaly | None reported | F | c.2932C>T, c.3246-2A>G | p.R978C, (predicted abnormal splicing) |
| 1 | N/A | 0.1 | 7.1 | Hepatosplenomegaly | None reported | F* | c.665A>G, c.1402T>G | p.N222S, p.C468G |
| 2 | N/A | 0.1 | 7.8 | Splenomegaly | None reported | F* | c.665A>G, c.1402T>G | p.N222S, p.C468G |
| − 0.4** | 1.2 | 0.8 | 24.1 | Fetal ascites | Not walking at 14 months | F | c.3182T>C, c.3182T>C | p.I1061T, p.I1061T |
| 0 | 1 | 8.0 | 66.6 | Jaundice | Developmental plateau | M | c.3565_3566insG, 2008_2011delTGCT | p.E1189Gfs*69, p.C670Pfs*12 |
| 0 | 1.5 | 5.1 | 47.9 | Hepatosplenomegaly | Loss of gross motor skills, dysphagia | M | c.2978delG, c.3591+4delA | p.G993Efs*4, (possible splicing effect) |
| 0 | 3 | 2.2 | 17.9 | Jaundice, splenomegaly | Clumsy, dysarthria | M | c.3182T>C, c.3182T>C | p.I1061T, p.I1061T |
| 0 | 3.5 | 2.0 | 16.0 | Hepatosplenomegaly, jaundice | Vertical gaze palsy | M | c.3182T>C, c.3182T>C | p.I1061T, p.I1061T |
| 0 | 8 | 1.2 | 12.2 | Splenomegaly | Learning disability, fine motor ataxia | F* | c.3107C>T, c.2861C>T | p.T1036M, p.S954L |
| 0.01 | 7 | 2.0 | 32.9 | Jaundice | Vertical gaze palsy | M | c.3182T>C, c.3281T>C | p.I1061T, p.I1094T |
| 0.3 | 2 | 1.3 | 44.1 | Hepatosplenomegaly | Clumsiness, speech delay | M | c.3439G>T, c.3742_3745delCTCA | p.G1146V, p.L1248Vfs*3 |
| 0.5 | 1.5 | 4.5 | 68.0 | Splenomegaly | Gross motor delay | M | c.2516T>G, c.3259T>C | p.I839R, p.F1087L |
| 0.7 | 2 | 2.5 | 29.5 | Splenomegaly | Clumsiness, possibly vertical gaze palsy | F | c.3493G>A, c.3741_3744delACTC | p.V1165M, p.L1248Vfs*3 |
| 1 | 3 | 1.4 | 38.4 | Splenomegaly | Fine motor limitation | M | c.3182T>C, c.3556C>G | p.I1061T, p.R1186G |
| 1.7 | 1.7 | 6.7 | 26.9 | Developmental delay | Gross motor delay, speech delay | F | c.57+1G>T, Unknown | (possible splicing effect), Unknown |
| 2 | 2 | 1.0 | 20.9 | Splenomegaly | Clumsiness | F* | c.1920delG, c.1554—1009G>A | p.H641Tfs*2, (predicted abnormal splicing) |
| 2 | 2 | 1.7 | 19.8 | Splenomegaly | Clumsiness | F* | c.1920delG, c.1554—1009G>A | p.H641Tfs*2, (predicted abnormal splicing) |
| 3 | 3 | 6.7 | 55.7 | Fine motor ataxia | Abnormal gait, fine motor skills | F | c.2979dupA, c.2103C>G | p.D994Rfs*13, p.N701K |
| 3 | 3 | 1.5 | 10.5 | Learning disability | Learning disability | F* | c.410C>T, c.2000C>T | p.T137M, p.S667L |
| 3 | 9 | 1.3 | 10.5 | Splenomegaly | Learning disability | M* | c.3107C>T, c.2861C>T | p.T1036M, p.S954L |
| 4 | 5 | 0.9 | 20.4 | Hepatosplenomegaly | School difficulties | F | c.3182T>C, c.3019C>G or C>T | p.I1061T, p.F1167C |
| 5 | 5 | 1.7 | 21.6 | Vertical gaze palsy | Vertical gaze palsy | F | c.3182T>C, c.3182T>C | p.I1061T, p.I1061T |
| 5 | 5 | 2.0 | 13.5 | Developmental delay | Vertical gaze palsy | F | c.2201G>T, c.2201G>T | p.S734I, p.S734I |
| 5 | 10 | 0.8 | 22.0 | Hepatosplenomegaly | Clumsiness, hearing loss | F | c.2474A>G, c.289_291dupTGT | p.Y825C, p.C97_P98insC |
| 6 | 6 | 1.2 | 13.3 | Learning disability | Learning disability | M | c.2861C>T, Unknown | p.S954L, Unknown |
| 6 | 6 | 2.2 | 8.1 | Seizures | Seizures | M | c.1211G>A, c.3019C>G | p.R404Q, p.P1007A |
| 7 | 7 | 0.8 | 9.8 | Clumsiness | Clumsiness | F* | c.1552C>T, c.2594C>T | p.R518W, p.S865L |
| 7 | 7 | 1.3 | 10.4 | Learning disability | Learning disability | F* | c.1552C>T, c.2594C>T | p.R518W, p.S865L |
| 8 | 8 | 1.2 | 28.3 | Learning delay | Learning delay, hearing loss | M | 6 poss. cDNA changes, c.3662delT | p.F842L, p.F1221Sfs*21 |
| 8 | 8 | 1.4 | 6.5 | Learning disability | Learning disability | F | c.3182T>C, c.3019C>G or C>T | p.I1061T, p.P1007A |
| 10 | 10 | 1.0 | 8.5 | Learning delay | Clumsiness, learning disability | F | c.1211G>A, c.2861C>T, c.1123A>G | p.R404Q, p.S954L, p.T375A |
| 11 | 11 | 0.9 | 11.2 | Learning disability | Learning disability | F | c.3182T>C, c.743G>T | p.I1061T, p.G248V |
| 12 | 12 | 0.6 | 13.9 | Unsteady gait | Abnormal gait | M | c.743 G>T, c.3410_3411insA | p.G248V, p.N1137Kfs*121 |
| 17 | 17 | 0.9 | 4.8 | Clumsiness | Clumsiness | M | c.3019C>G or C>T, Unknown | p.P1007A, Unknown |
| 18 | 18 | 0.7 | 20.8 | Psychosis | Psychosis | F | c.3176G>A, c.3742_3745delCTCA | p.R1059Q, p.L1248Vfs*3 |
| 18 | 18 | 0.6 | 8.0 | Depression | Psychiatric symptoms | F | c.3182T>C, c.2861C>T | p.I1061T, p.S954L |
*Part of a sibling pair.
**Negative values indicates age of onset in utero.
†Subsequently received a liver transplant.
LysoTracker levels were measured in each of the 41 fibroblast cell lines (measured as fold change over background, Table 1) using experimental methods as previously described42. Pairwise analysis of the LysoTracker levels for the 24 patient cell lines included in both studies showed a highly significant correlation with previous measurements (Spearman r = 0.76, p < 0.0001, Fig. 1), demonstrating the reproducibility of LysoTracker staining in cultured primary fibroblasts from individuals with NPC1.
Figure 1.
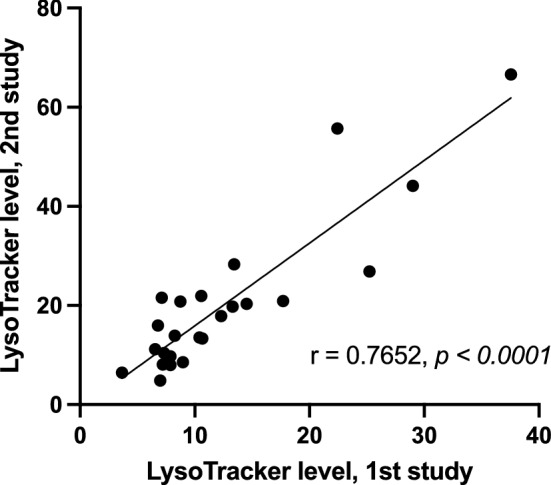
LysoTracker measurements of fibroblasts from individuals with NPC1 are reproducible. Primary fibroblast cell lines established from 24 individuals with NPC1 were previously stained with LysoTracker, and these LysoTracker levels were measured by FACS for each cell line42. Several years later, fibroblasts from these 24 cell lines were again subjected to the same LysoTracker staining protocol. Measurements from both studies were paired for each patient, and correlation analysis showed significant correlation of the paired measurements for these NPC1 cell lines (Spearman r = 0.7652, p < 0.0001). Line was calculated using simple linear regression.
Fibroblast LysoTracker level significantly correlates with age of onset and disease severity
Three general clinical parameters were ascertained for each individual: age of onset (including onset of any NPC1-associated symptoms such as hepatosplenomegaly, jaundice and all neurological phenotypes), age of first neurological symptom, and age-adjusted neurological severity score at the time of diagnosis (see “Materials and methods” section)25,26. Pairwise correlations between the 3 clinical phenotypes and LysoTracker staining levels were examined across the patient cohort, resulting in multiple significant correlations. LysoTracker level was inversely correlated with the age of onset (− 0.57, p = 0.0001, Fig. 2A), and showed an even greater correlation when specifically analyzing the age of first neurological symptom (− 0.66, p < 0.0001, Fig. 2B). In addition, LysoTracker level was directly correlated with neurological severity score (0.45, p = 0.0032, Fig. 2C). The significant correlations of LysoTracker levels with age of onset and neurological severity score at diagnosis suggest that LysoTracker levels could potentially serve as a valid biomarker for predicting disease onset and severity. These results confirm and extend data from the patient cohort previously described by our group42.
Figure 2.

Fibroblasts from individuals with NPC1 disease show LysoTracker staining that significantly correlates with age of onset and disease severity. (a) LysoTracker level, measured as fold-change increase over background, is inversely correlated with the age of symptom onset. These age of onset data points included any NPC1-associated phenotypes, such as hepatosplenomegaly, jaundice and neurological abnormalities. (b) LysoTracker level also shows inverse correlation with the age of first neurological symptom, and this correlation is greater than that seen for any symptom onset. (c) LysoTracker level correlates with the age-adjusted neurological severity score (see “Materials and methods” section for score description). For all 3 graphs, each point represents a single NPC individual, and log10-transformed values are presented and were used for statistical analyses. P values indicate significant correlation using Spearman’s correlation; lines were calculated using simple linear regression.
Correlation of genome-wide expression with disease onset, severity, and cellular phenotypes
RNA was isolated from the 41 NPC1 primary fibroblast cell lines as well as parallel cultures of each cell line treated with HPβCD for 24 h. RNA sequencing (RNA-Seq) data were generated from all samples as previously described45. Expression levels of each gene were independently assessed for correlation with the three previously described clinical parameters (age of onset, age of first neurological symptom, and neurological severity score) and two cellular phenotypes (baseline LysoTracker level and change in LysoTracker level after HPβCD treatment, see “Materials and methods” section). These analyses identified 127 genes with expression profiles that significantly correlated with at least one clinical or cellular phenotype (Supplementary Table 1). Ten genes were correlated with age of onset, 40 genes were correlated with the age of first neurological symptom, 43 genes were correlated with neurological severity score, 21 genes were correlated with LysoTracker levels in untreated cells, and 27 genes were correlated with change in LysoTracker level following HPβCD treatment. Most genes did not overlap among these correlations, as only 14 genes correlated with two phenotypes (Supplementary Table 1). This suggests that the severity of NPC1 disease phenotypes is impacted by numerous genes/pathways rather than a small set of genes. Pathway enrichment analysis of all 127 genes aligned with the known role of NPC1 protein in cholesterol and glycosphingolipid homeostasis: the top 6 canonical pathways involved cholesterol synthesis, and the top 3 molecular and cellular functions were Lipid Metabolism, Small Molecule Biochemistry, and Vitamin and Mineral Metabolism.
The 127 significantly correlated genes could include candidate drug targets for NPC1 treatment or potential biomarkers that could be used for disease progression monitoring in diagnosed NPC1 patients. Examination of this gene list for protein biomarkers showed that GPNMB is the only previously suggested NPC1 disease biomarker present within the significantly correlated gene set45,46 (Supplementary Table 2). Additionally, biomarker analysis (Ingenuity Pathway Analysis) identified 7 of the 127 genes as highly relevant biomarkers, each of which is detectable in human blood: GPNMB, HBEGF, HMGCR, PIK3CA, RSF1, STK11, and TLR4. To identify putative drug targets, the 127 gene list was analyzed at the Drug-Gene Interaction Database (https://www.dgidb.org/)47. Thirty-four genes (27%) are deemed "clinically actionable" or are part of the druggable genome, and an additional 33 (26%) are in gene categories that are potentially druggable (e.g. kinases, enzymes; Supplementary Table 3). Therefore, over half (53%) of the significantly correlated genes represent potential targets for drug treatment. Additionally, genes that modify NPC1-related pathways could also be included in the significantly correlated gene set. While no lysosomal storage disorder genes were present48, 26/127 (20.5%) genes were associated with neurological disease in two or more patients (see functional information, Supplementary Table 1). These genes could represent novel pathways that impact neurological NPC1 disease progression.
Several notable genes that were highly correlated with neurological onset showed significantly different expression between patients grouped by age of onset (Fig. 3). AHI1 (Fig. 3A) encodes a protein that localizes to primary cilia and is mutated in the rare ciliopathy Joubert syndrome 3 (OMIM #608629), which exhibits multiple neurological phenotypes, including cerebellar vermis hypoplasia and ataxia49–52. In addition, Ahi1 was previously reported to show differential expression in lobule X in comparison to lobules III and VI of mouse cerebellum in both WT and Npc1 mutants53, consistent with important cerebellar functions for this gene, and of interest given the differential anterior to posterior Purkinje cell degeneration seen across cerebellar lobules in NPC1 disease54. ROCK2 (Fig. 3B) encodes a serine-threonine kinase that acts downstream of Rho to control neuronal cytoskeletal changes and regulate dendrite structure and function55,56. ROCK2 has been proposed as a possible target for treatment of many neurological diseases, including Alzheimer disease57,58, and also has putative roles in regulating cholesterol transport and cholesterol synthesis via the SREBP2 pathway59,60. PQLC2 (Fig. 3C; a.k.a. SLC66A1, LAAT-1) encodes a lysosomal cationic amino acid transporter which recruits a heterotrimeric signaling complex composed of C9orf72, SMCR8, and WDR41 to the lysosome. Together these proteins perform critical roles in maintaining correct amino acid levels and regulating lysosomal function in response to mTORC1 and TLR signaling61–64. CLCN6 (Fig. 3D) encodes a transmembrane Cl−/H+ exchanger predominantly found on late endosomes and highly expressed in the nervous system. Heterozygous mutation of CLCN6 is associated with a rare neurological disorder that exhibits childhood-onset neurodegeneration with hypotonia, respiratory insufficiency, and brain imaging abnormalities65. A missense mutation of CLCN6 has been seen in a single patient with the early infantile epileptic encephalopathy West syndrome, and this sequence change causes autophagosome accumulation and blockage of autophagosome-lysosome fusion66. Additionally, a Clcn6 mouse mutant exhibits defects in lysosomal storage and mild, slowly progressing neurological abnormalities67,68.
Figure 3.

Gene expression significantly correlates with age of neurological disease onset. Four representative genes with significant correlation with the age of first neurological symptom are shown: (a) AHI1, (b) ROCK2, (c) PQLC2, and (d) CLCN6. The graphs show scatter plots of expression for each gene in each patient (variance stabilized read counts, y-axis) plotted against the age of first neurological symptom for each patient (N = 33). Patients are divided into four groups based on age of onset categories that have been previously described86, as follows: early infantile (2 months–2 years, N = 9), late infantile (3–6 years, N = 10), juvenile (7–15 years, N = 11), and adolescent/adulthood (> 15 years, N = 3). AHI1 and ROCK2 show direct correlation with age of first neurological symptom, while PQLC2 and CLCN6 show inverse correlation. Gene expression values are significantly different across age groups, as calculated by Welch ANOVA (p < 0.05 for all genes) followed by Dunnett’s multiple comparisons test (p values as indicated; *p < 0.05, **p < 0.01, ***p < 0.001).
Hierarchical clustering of genes correlated with age of first neurological symptom reveals shared gene expression patterns among subsets of NPC1 fibroblasts
Hierarchical clustering was performed using variance stabilized, normalized read counts for 121 of the 127 significantly correlated genes across the NPC1 patient cohort (six genes were removed due to low variance stabilized count levels; see Materials and methods). Despite variability across the dataset, clustering grouped distinct subsets of genes together, in particular the genes that correlated with the age of first neurological symptom or with change in LysoTracker level following HPβCD treatment (Supplementary Fig. 1). Given the clinical importance of the age of first neurological symptom, hierarchical clustering was performed using the subset of 37 genes correlated with age of first neurological symptom. This separated patient cell lines into two groups (Fig. 4, Branches A and B) and genes into two groups with relatively high or low expression in Branch B individuals (Fig. 4, rows). Interestingly, Branch B included patients with lower neurological severity scores, later disease onset, and lower LysoTracker levels. Branch A was larger and showed more variability, but it included a patient subset with the highest neurological severity scores and LysoTracker levels, all of whom had early disease onset (Fig. 4, left side of Branch A). Furthermore, this Branch A patient subset showed inverse expression of the two gene groups in comparison to Branch B. Therefore, gene expression profiles for the 37 genes correlated with the age of first neurological symptom not only revealed distinct expression patterns among subsets of patients, but also generally grouped patients by LysoTracker levels, age of first neurological symptom, and neurological severity score.
Figure 4.

Genes correlated with age of first neurological symptom show distinct gene expression patterns among NPC1 patient subgroups. The heatmap shows hierarchical clustering of variance stabilized read counts of the 37 genes significantly correlated with the age of first neurological symptom. Patient cell lines (columns) were grouped into two high-order branches by clustering analysis, labeled Branch A and Branch B. Genes (rows) clustered into two branches, and the patient cell line Branches A and B show inverse expression of these two gene groups. Hierarchical clustering of all significantly correlated genes is shown in Supplementary Fig. 1. Score: neurological severity scores for each patient are shown in the yellow to orange heatmap, with darker colors indicating higher scores/greater severity. Onset: age of first neurological symptom is shown in the blue heatmap, with darker blues indicating older ages (EI = early infantile, LI = late infantile, J = juvenile, A/A = adolescent/adult). The bar graph indicates LysoTracker levels (LTR) in untreated fibroblasts.
A subset of significantly correlated genes change expression following HPβCD treatment
Animal studies and human clinical trial data indicate that HPβCD treatment may alleviate disease phenotypes in NPC126,28–33. Since RNA-Seq data were collected from each patient using paired fibroblast cultures that were either untreated or treated with HPβCD, we could calculate expression changes in each gene that resulted from HPβCD treatment across the cohort (ΔHPβCD values). Hierarchical clustering using these ΔHPβCD values illustrated that most of the significantly correlated genes were not changing (Supplementary Fig. 2), suggesting that HPβCD is not affecting many potentially clinically relevant genes, including those that are correlated with the age of first neurological symptom.
However, a notable cluster of 24 genes did show expression changes, with negative ΔHPβCD values that indicated decreased expression following HPβCD treatment (Supplementary Fig. 2, Fig. 5). This cluster is enriched for genes encoding proteins involved in cholesterol synthesis and lysosomal functions. Only a subset of clustered patient cell lines showed negative ΔHPβCD values for these genes (Fig. 5, Branch D, for decreased expression), and this cluster included cell lines from patients with the highest LysoTracker levels and neurological severity scores. The remaining patient cell lines with unchanged or increased expression of these genes clustered separately (Fig. 5, Branch U, for unchanged/upregulated) and exhibited lower neurological severity scores and LysoTracker levels. Interestingly, 2 cell lines from patients who previously showed improvement in clinical parameters in response to HPβCD treatment in a clinical trial were included in Branch D (Fig. 5 and Supplementary Fig. 2, “R” labels)26. In contrast, 3 cell lines which clustered together in Branch U were derived from patients who did not show improvement in response to HPβCD treatment in the same study (Fig. 5 and Supplementary Fig. 2, “N” labels)26.
Figure 5.

Hierarchical clustering reveals reduced expression of genes following HPβCD treatment in a subset of NPC1 patients. Heatmap values are the difference between variance stabilized read counts for cells after HPβCD treatment and for untreated cells (ΔHPβCD). Heatmap colors indicate reduced expression following HPβCD treatment (blue), unchanged expression (white), and increased expression following HPβCD treatment (red). This figure displays clustering of a subgroup of 24 genes that showed reduced expression; hierarchical clustering of ΔHPβCD values for all significantly correlated genes is shown in Supplementary Fig. 2. Branch D and Branch U are the two main dendrogram branches for patient cell lines identified by clustering analysis. Branch D (downregulated, blue color) includes cell lines with broadly reduced levels of the notable gene cluster; Branch U (upregulated/unchanged) contains the remaining cell lines with unchanged or upregulated levels of this gene cluster. R = responder, N = non-responder; these labels refer to patients who were identified in a previously reported clinical trial to show phenotypic stability/improvement in response to HPβCD treatment or no improvement, respectively26. Score: neurological severity scores for each patient are shown in the yellow to orange heatmap, with darker colors indicating higher scores/greater severity. Onset: age of onset (visceral or neurological) is shown in the blue heatmap, with darker blues indicating older ages (EI = early infantile, LI = late infantile, J = juvenile, A/A = adolescent/adult, PRE = presymptomatic). The bar graph indicates LysoTracker levels (LTR) in untreated fibroblasts.
Discussion
NPC1 is a rare disease with variable phenotypic presentation, thus necessitating unconventional approaches to gather comprehensive data from the limited number of individuals with NPC1. This study used primary fibroblasts from a cohort of NPC1 patients enrolled in an NIH natural history study to demonstrate the reproducibility of LysoTracker measurements and thus validate LysoTracker as a reliable marker of acidic cellular compartments in NPC1 cells, and to show that LysoTracker staining significantly correlates with the clinical parameters of age of onset and disease severity. Importantly, LysoTracker levels in fibroblasts, a non-neural tissue, correlate with neurological symptom onset, showing that this measure of easily accessible peripheral tissue may be a reliable indicator of neurological disease.
This study also correlated expression of specific genes with clinical age of onset, neurological disease severity, and LysoTracker levels. The significantly correlated gene set presented in this study provides potential candidates for NPC1 biomarkers, of relevance given the urgent need for biomarkers to monitor NPC1 disease progression and treatment efficacy. This dataset may also contain genes which modify NPC1 disease phenotypes. Variations in modifier genes and pathways may explain why some patients with the same NPC1 mutation exhibit heterogeneous phenotypes and disease progression17,24,69–71. In support of this hypothesis, varied phenotypic severity in Npc1 mouse models on different inbred strain backgrounds suggests the presence of modifiers, and comparative analyses between the 127 significantly correlated gene set and these studies might shed light on interesting modifier candidates11,30,72–77. For example, previous studies of the Npc1em1Pav mouse model identified two strain-specific modifier regions, and the mouse homologs of 4 of the 127 correlated genes map to these two regions (Slc39a10, Nbeal1, Daw1 and Rsf1), with two of these genes harboring strain-specific missense mutations (Rsf1 and Nbeal1)72. Overall, the transcriptomic data in this study will provide a more comprehensive understanding of the cellular pathways perturbed by loss of NPC1 function, which may suggest new treatment avenues for NPC1 disease.
These gene expression differences reflect changes within the NPC1 patient population, and not in comparison to NPC1+/+ cells, which has been previously studied by other groups78–80. In fact, our significantly correlated gene list showed very little overlap with the genes that were previously published as significantly different from NPC1+/+ cells. This suggests that expression levels in NPC1 patient cells may overlap the range of expression seen in control cells, but modest expression changes could still have a modifier effect among NPC1 patients that is only revealed in the context of reduced NPC1. Indeed, moderate gene expression differences were seen across much of our significantly correlated gene set, therefore the possibility of false positives is a notable limitation of this gene set. In future studies, these genes should be independently validated using cells from a different set of NPC1 patients. This gene list provides an important initial starting point for understanding correlations of pathway and gene expression differences, and it should not be taken as a definitive, final list of candidate modifiers.
Although NPC1 individuals in this study showed heterogeneity of gene expression, hierarchical clustering of the significantly correlated genes revealed gene expression patterns that were shared among subsets of patients. The differential expression of genes associated with neurological phenotypes, including noteworthy genes with previously described roles in brain function and development (Figs. 3 and 4, Supplementary Table 1), was especially intriguing and may warrant further studies. Interestingly, the grouping of patient subsets by expression of these genes aligned with LysoTracker levels, neurological severity score, and age of onset (Fig. 4), suggesting that these gene expression differences have the potential to stratify patients into subsets based on distinct cellular pathology and disease severity. While it is premature to state that these data may represent gene signatures that would correlate with and predict NPC1 disease progression, future examination of these different expression patterns of genes and pathways and their relationship with NPC1 disease phenotypes may lead to better understanding of disease progression and facilitate discovery of new treatments.
Hierarchical clustering of the changes in gene expression after HPβCD treatment showed little to no change in most genes that correlated with clinical or cellular NPC1 phenotypes (Supplementary Fig. 2). This emphasizes the fact that current treatments such as HPβCD do not affect genes that may represent important pathways, in particular genes relevant for neurological phenotypes, and suggests that these genes/pathways may be priority novel targets for future studies on enhanced disease treatment. However, hierarchical clustering did reveal a subset of patient fibroblasts with decreased expression of a cluster of genes encoding cholesterol enzymes and lysosomal proteins. This suggests these NPC1 patient cells are responding to cholesterol redistribution, which fits with the proposed actions of HPβCD to mobilize cholesterol to the ER, restore autophagic flux, and permit secretion of lysosomal material32,81–83. This gene cluster also included CLCN6, HS1BP3, and ULK3, genes that were correlated with the age of neurological onset. Of note, this analysis showed these changes only occurred in some patient cell lines, and sizable differences in gene expression changes in response to HPβCD in individual patients were observed. Similar to what was seen for the genes correlated with age of neurological onset, these differences aligned to some extent with disease severity, age of onset, and cellular pathology. It is interesting to note that 2 patients previously shown as responders to HPβCD treatment26 showed decreased expression of this gene set, while 3 patients that previously did not show an HPβCD response showed separate clustering, with unchanged or slightly increased expression of these genes. Overall, the broad, gene expression-based view of each NPC1 patient cell line in this study provides a first glimpse at the transcriptomic complexity of patient response in the context of varied disease progression and severity, and it suggests that future studies with a larger patient cohort will be able to delve more deeply into the gene expression patterns that may underlie NPC1 disease heterogeneity.
In summary, these results provide strong evidence that fibroblast LysoTracker level may predict NPC1 disease course and severity. This information could be used to greatly facilitate disease diagnosis and management. Collection of fibroblast LysoTracker levels could result in earlier treatment as well as guide patient selection in clinical trials, identifying patients who may show a more aggressive disease progression. Furthermore, the gene set identified here suggests that differential gene expression patterns may reflect disease severity and response to therapeutic intervention, supporting future analyses to better understand these gene expression differences to help direct NPC1 treatments.
Materials and methods
NPC1 patient cell lines, LysoTracker staining, HPβCD treatment, and RNA-Seq analysis
Primary skin fibroblasts were obtained with written informed consent from NPC1 patients or guardians as part of a natural history study of NPC1 disease at the National Institutes of Health, which was approved by the Eunice Kennedy Shriver National Institute of Child Health and Human Development Institutional Review Board (NCT00344331, ClinicalTrials.gov). All methods were performed in accordance with the relevant guidelines and regulations set forth by the National Institutes of Health and the Eunice Kennedy Shriver National Institute of Child Health and Human Development. A variety of NPC1 mutations were present among the cohort of 41 patients used in this study (Table 1).
Fibroblast culture conditions, LysoTracker Red DND-99 staining (Invitrogen, catalog #L7528), RNA isolation, treatment with 300 µM of HPβCD (Sigma-Aldrich, Catalog no. C0926), and RNA-Seq analysis were all described in detail previously45. LysoTracker levels (LysoTracker fold-change, which was calculated as the ratio of the mean of LysoTracker levels in stained divided by unstained samples) were analyzed by Fluorescence-activated cell sorter (FACS) analysis, as previously published42. Previous studies demonstrated LysoTracker levels decrease in NPC1 cells following HPβCD treatment45,84,85, thus the change in LysoTracker after HPβCD treatment was calculated as the following ratio: [stained HPβCD-treated LysoTracker level minus unstained]/[stained untreated LysoTracker level minus unstained]. Duplicate biological replicates were analyzed on three different days, totaling 6 independent samples per cell line. Sequence data used for the RNA-Seq analyses are available through dbGaP, study accession number phs002392.
RNA-seq analysis
RNA-Seq data from all samples was analyzed as previously described45. Briefly, reads were aligned to the GRCh38 human reference using HISAT2, reads were counted in GENCODE release 28 annotated genes using featureCounts v1.6.4, and DESeq2 v1.22.1 was used for variance-stabilized transformation, which converts counts into a log-like scale. The normalized, variance-stabilized counts were then correlated with clinical parameters and cellular phenotypes. Since there is only a single sample for each cell line, no statistical significance can be assigned to the effect on a single gene in a single cell line, therefore all counts (or differences, depending on the analysis) were collectively used to correlate to clinical parameters and cellular phenotypes.
Statistical analyses
Pairwise Spearman’s correlations were determined among the following 5 variables for each NPC1 patient/fibroblast cell line: LysoTracker levels, change in LysoTracker levels following HPβCD treatment, age of onset (visceral or neurological), age of first neurological symptom, and age-adjusted neurological severity score at first evaluation. This score incorporates measurements of 17 clinical neurological phenotypes, and then adjusts this aggregate score to age by dividing by the age at initial evaluation25. A higher age-adjusted neurological severity score indicates more severe disease phenotypes.
For correlation calculations, Spearman’s rho (rs) values were determined for each gene in relation to each clinical parameter or cellular phenotype. Additionally, p-values were determined for the rs values of each gene/covariate pair, with the Benjamini–Hochberg correction applied to control the false discovery rate (FDR). A gene was considered significantly correlated with the parameter or phenotype if the FDR of the correlation was < 0.1. Correlations and p-value corrections were performed in the R statistical programming language (version 3.5.1).
All other statistical analyses were performed using Prism software (GraphPad).
Pathway enrichment and hierarchical clustering analysis
Pathway enrichment analysis was done using Ingenuity Pathway Analysis (Qiagen IPA). Hierarchical clustering was performed using Morpheus (https://software.broadinstitute.org/morpheus) with a distance measure of one minus Pearson’s correlation coefficient and clustering of both rows and columns. Six of the 127 genes had variance stabilized counts at the lower limit of detection in more than 50% of the patient cell lines (ENSG00000237708, ENSG00000242314, ENSG00000255945, FAM153A, H19, and HLA-DQB2). Therefore, these genes were removed from the hierarchical clustering analyses. To calculate the changes in gene expression after HPβCD treatment (ΔHPβCD) for clustering analysis, the variance stabilized count value for each gene in untreated cells was subtracted from that of HPβCD-treated cells.
Supplementary Information
Acknowledgements
The authors wish to thank members of the Pavan lab for helpful discussions and scientific input. This research was supported by the Intramural Research Programs of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (ZIA HD008989) and the National Human Genome Research Institute (NHGRI; 1ZIAHG000068-15) at the National Institutes of Health (NIH). JLR-G was supported by an NHGRI Intramural Research Training Award, the NIH Oxford-Cambridge Scholars Program, and the Medical Scientist Training Program from the University of Wisconsin-Madison School of Medicine and Public Health (3T32GM008692). FMP is a Wellcome Trust Investigator in Science and a Royal Society Wolfson Research Merit Award holder. This study was also supported by the Ara Parseghian Medical Research Fund and Niemann–Pick Canada.
Author contributions
J.L.R.-G., D.E.W.-C., F.M.P., W.J.P., and J.L.R.-G. conceptualized the experiments. J.L.R.-G., D.E.W.-C., N.L.J., F.M.P., R.K.D., W.J.P., and J.L.R.-G. planned the experimental methodology. L.L.B., D.E.W.-C., N.L.J., N.Y.F., R.K.D., and J.L.R.-G. performed formal analysis, and L.L.B., R.K.D., and J.L.R.-G. performed data curation. Original manuscript draft was written by L.L.B., D.E.W.-C., W.J.P., and J.L.R.-G., and figures and were designed and created by L.L.B., D.E.W.-C., and J.R.L.-G. All authors reviewed and edited the manuscript. W.J.P. and J.L.R.-G. supervised the work, and funding was acquired by F.D.P. and W.J.P.
Competing interests
William J Pavan, Jorge L. Rodriguez-Gil, and Forbes D. Porter are patent co-inventors: “Compositions and methods for predicting age of onset of a lysosomal storage disease or a disease associated with a lysosomal defect.” United States Patent number 9983200, dated May 29, 2018. Laura L. Baxter, Dawn E. Watkins-Chow, Nicholas L. Johnson, Nicole Y. Farhat, Frances M. Platt, and Ryan K. Dale declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
William J. Pavan, Email: bpavan@mail.nih.gov
Jorge L. Rodriguez-Gil, Email: jrgil@stanford.edu
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-06112-y.
References
- 1.Vanier MT. Niemann–Pick disease type C. Orphanet J. Rare Dis. 2010;5:16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mengel E, et al. Differences in Niemann–Pick disease Type C symptomatology observed in patients of different ages. Mol. Genet. Metab. 2017;120:180–189. doi: 10.1016/j.ymgme.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Patterson MC, et al. Recommendations for the detection and diagnosis of Niemann–Pick disease type C: An update. Neurol. Clin. Pract. 2017;7:499–511. doi: 10.1212/CPJ.0000000000000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carstea ED, et al. Niemann–Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Saha P, Li J, Blobel G, Pfeffer SR. Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2. Proc. Natl. Acad. Sci. USA. 2016;113:10079–10084. doi: 10.1073/pnas.1611956113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanier MT. Complex lipid trafficking in Niemann–Pick disease type C. J. Inherit. Metab. Dis. 2015;38:187–199. doi: 10.1007/s10545-014-9794-4. [DOI] [PubMed] [Google Scholar]
- 7.Infante RE, et al. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. USA. 2008;105:15287–15292. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cologna SM, Rosenhouse-Dantsker A. Insights into the molecular mechanisms of cholesterol binding to the NPC1 and NPC2 proteins. Adv. Exp. Med. Biol. 2019;1135:139–160. doi: 10.1007/978-3-030-14265-0_8. [DOI] [PubMed] [Google Scholar]
- 9.Pfeffer SR. NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J. Biol. Chem. 2019;294:1706–1709. doi: 10.1074/jbc.TM118.004165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez-Gil JL, et al. Hepatocellular carcinoma as a complication of Niemann–Pick disease type C1. Am. J. Med. Genet. A. 2021 doi: 10.1002/ajmg.a.62382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Praggastis M, et al. A murine Niemann–Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 2015;35:8091–8106. doi: 10.1523/JNEUROSCI.4173-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez-Gil JL, et al. NPC1 deficiency in mice is associated with fetal growth restriction, neonatal lethality and abnormal lung pathology. J. Clin. Med. 2019;9:12. doi: 10.3390/jcm9010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beltroy EP, Richardson JA, Horton JD, Turley SD, Dietschy JM. Cholesterol accumulation and liver cell death in mice with Niemann–Pick type C disease. Hepatology. 2005;42:886–893. doi: 10.1002/hep.20868. [DOI] [PubMed] [Google Scholar]
- 14.Ramirez CM, et al. Ontogenic changes in lung cholesterol metabolism, lipid content, and histology in mice with Niemann–Pick type C disease. Biochim. Biophys. Acta. 2014;1841:54–61. doi: 10.1016/j.bbalip.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roszell BR, et al. Pulmonary abnormalities in animal models due to Niemann–Pick type C1 (NPC1) or C2 (NPC2) disease. PLoS ONE. 2013;8:e67084. doi: 10.1371/journal.pone.0067084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spiegel R, et al. The clinical spectrum of fetal Niemann–Pick type C. Am. J. Med. Genet. A. 2009;149A:446–450. doi: 10.1002/ajmg.a.32642. [DOI] [PubMed] [Google Scholar]
- 17.Vanier MT, Millat G. Niemann–Pick disease type C. Clin. Genet. 2003;64:269–281. doi: 10.1034/j.1399-0004.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- 18.Patterson MC, et al. Recommendations for the diagnosis and management of Niemann–Pick disease type C: An update. Mol. Genet. Metab. 2012;106:330–344. doi: 10.1016/j.ymgme.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 19.Geberhiwot T, et al. Consensus clinical management guidelines for Niemann–Pick disease type C. Orphanet J. Rare Dis. 2018;13:50. doi: 10.1186/s13023-018-0785-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sevin M, et al. The adult form of Niemann–Pick disease type C. Brain. 2007;130:120–133. doi: 10.1093/brain/awl260. [DOI] [PubMed] [Google Scholar]
- 21.Imrie J, et al. Niemann–Pick disease type C in adults. J. Inherit. Metab. Dis. 2002;25:491–500. doi: 10.1023/a:1021259403196. [DOI] [PubMed] [Google Scholar]
- 22.Wraith JE, et al. Niemann–Pick type C Suspicion Index tool: analyses by age and association of manifestations. J. Inherit. Metab. Dis. 2014;37:93–101. doi: 10.1007/s10545-013-9626-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonnot, O., Klunemann, H. H., Velten, C., Torres Martin, J. V. & Walterfang, M. Systematic review of psychiatric signs in Niemann–Pick disease type C. World J. Biol. Psychiatry20, 320–332. 10.1080/15622975.2018.1441548 (2019). [DOI] [PubMed]
- 24.Imrie J, Heptinstall L, Knight S, Strong K. Observational cohort study of the natural history of Niemann–Pick disease type C in the UK: A 5-year update from the UK clinical database. BMC Neurol. 2015;15:257. doi: 10.1186/s12883-015-0511-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yanjanin NM, et al. Linear clinical progression, independent of age of onset, in Niemann–Pick disease, type C. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010;153B:132–140. doi: 10.1002/ajmg.b.30969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ory DS, et al. Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann–Pick disease, type C1: A non-randomised, open-label, phase 1–2 trial. Lancet. 2017;390:1758–1768. doi: 10.1016/S0140-6736(17)31465-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pallottini V, Pfrieger FW. Understanding and treating Niemann–Pick Type C disease: Models matter. Int. J. Mol. Sci. 2020;21:8979. doi: 10.3390/ijms21238979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camargo F, et al. Cyclodextrins in the treatment of a mouse model of Niemann–Pick C disease. Life Sci. 2001;70:131–142. doi: 10.1016/s0024-3205(01)01384-4. [DOI] [PubMed] [Google Scholar]
- 29.Davidson CD, et al. Chronic cyclodextrin treatment of murine Niemann–Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE. 2009;4:e6951. doi: 10.1371/journal.pone.0006951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu B, Li H, Repa JJ, Turley SD, Dietschy JM. Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res. 2008;49:663–669. doi: 10.1194/jlr.M700525-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Liu B, et al. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc. Natl. Acad. Sci. USA. 2009;106:2377–2382. doi: 10.1073/pnas.0810895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peake KB, Vance JE. Normalization of cholesterol homeostasis by 2-hydroxypropyl-beta-cyclodextrin in neurons and glia from Niemann–Pick C1 (NPC1)-deficient mice. J. Biol. Chem. 2012;287:9290–9298. doi: 10.1074/jbc.M111.326405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramirez CM, et al. Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann–Pick type C1 mouse and markedly prolongs life. Pediatr. Res. 2010;68:309–315. doi: 10.1203/PDR.0b013e3181ee4dd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fecarotta S, et al. Long term follow-up to evaluate the efficacy of miglustat treatment in Italian patients with Niemann–Pick disease type C. Orphanet J. Rare Dis. 2015;10:22. doi: 10.1186/s13023-015-0240-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patterson MC, et al. Treatment outcomes following continuous miglustat therapy in patients with Niemann–Pick disease Type C: A final report of the NPC Registry. Orphanet J. Rare Dis. 2020;15:104. doi: 10.1186/s13023-020-01363-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stein VM, et al. Miglustat improves purkinje cell survival and alters microglial phenotype in feline Niemann–Pick disease type C. J. Neuropathol. Exp. Neurol. 2012;71:434–448. doi: 10.1097/NEN.0b013e31825414a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Curelaru S, Zehavi Y, Almagor T, Spiegel R. Favorable outcomes following early onset oral miglustat in early infantile Niemann Pick Type C. Mol. Genet. Metab. Rep. 2021;27:100739. doi: 10.1016/j.ymgmr.2021.100739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wraith JE, et al. Recommendations on the diagnosis and management of Niemann–Pick disease type C. Mol. Genet. Metab. 2009;98:152–165. doi: 10.1016/j.ymgme.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 39.Kirkegaard, T. et al. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med.8, 355ra118. 10.1126/scitranslmed.aad9823 (2016). [DOI] [PMC free article] [PubMed]
- 40.Vite, C. H. et al. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann–Pick type C1 disease. Sci. Transl. Med.7, 276ra226. 10.1126/scitranslmed.3010101 (2015). [DOI] [PMC free article] [PubMed]
- 41.Liu B, et al. Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res. 2010;51:933–944. doi: 10.1194/jlr.M000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodriguez-Gil JL, et al. A somatic cell defect is associated with the onset of neurological symptoms in a lysosomal storage disease. Mol. Genet. Metab. 2013;110:188–190. doi: 10.1016/j.ymgme.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.te Vruchte D, et al. Relative acidic compartment volume as a lysosomal storage disorder-associated biomarker. J. Clin. Invest. 2014;124:1320–1328. doi: 10.1172/JCI72835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Westbroek W, et al. A new glucocerebrosidase-deficient neuronal cell model provides a tool to probe pathophysiology and therapeutics for Gaucher disease. Dis. Model Mech. 2016;9:769–778. doi: 10.1242/dmm.024588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodriguez-Gil JL, et al. Transcriptome of HPbetaCD-treated Niemann–Pick disease type C1 cells highlights GPNMB as a biomarker for therapeutics. Hum. Mol. Genet. 2021;30:2456–2468. doi: 10.1093/hmg/ddab194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marques AR, et al. Gpnmb is a potential marker for the visceral pathology in Niemann–Pick Type C disease. PLoS ONE. 2016;11:e0147208. doi: 10.1371/journal.pone.0147208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freshour, S. L. et al. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 49, D1144–D1151. 10.1093/nar/gkaa1084 (2021). [DOI] [PMC free article] [PubMed]
- 48.Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat. Rev. Dis. Primers. 2018;4:27. doi: 10.1038/s41572-018-0025-4. [DOI] [PubMed] [Google Scholar]
- 49.Dixon-Salazar T, et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am. J. Hum. Genet. 2004;75:979–987. doi: 10.1086/425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferland RJ, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat. Genet. 2004;36:1008–1013. doi: 10.1038/ng1419. [DOI] [PubMed] [Google Scholar]
- 51.Hsiao YC, Munoz-Estrada J, Tuz K, Ferland RJ. The transition zone protein AHI1 regulates neuronal ciliary trafficking of MCHR1 and its downstream signaling pathway. J. Neurosci. 2021;41:3932–3943. doi: 10.1523/JNEUROSCI.2993-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsiao YC, et al. Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum. Mol. Genet. 2009;18:3926–3941. doi: 10.1093/hmg/ddp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin, K. B. et al. Identification of novel pathways associated with patterned cerebellar purkinje neuron degeneration in Niemann–Pick disease, Type C1. Int. J. Mol. Sci.21. 10.3390/ijms21010292 (2019). [DOI] [PMC free article] [PubMed]
- 54.Higashi Y, Murayama S, Pentchev PG, Suzuki K. Cerebellar degeneration in the Niemann–Pick type C mouse. Acta Neuropathol. 1993;85:175–184. doi: 10.1007/BF00227765. [DOI] [PubMed] [Google Scholar]
- 55.Duffy P, et al. Rho-associated kinase II (ROCKII) limits axonal growth after trauma within the adult mouse spinal cord. J. Neurosci. 2009;29:15266–15276. doi: 10.1523/JNEUROSCI.4650-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monnier PP, Sierra A, Schwab JM, Henke-Fahle S, Mueller BK. The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol. Cell Neurosci. 2003;22:319–330. doi: 10.1016/s1044-7431(02)00035-0. [DOI] [PubMed] [Google Scholar]
- 57.Weber AJ, Herskowitz JH. Perspectives on ROCK2 as a therapeutic target for Alzheimer's disease. Front. Cell Neurosci. 2021;15:636017. doi: 10.3389/fncel.2021.636017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koch JC, et al. ROCK2 is a major regulator of axonal degeneration, neuronal death and axonal regeneration in the CNS. Cell Death Dis. 2014;5:e1225. doi: 10.1038/cddis.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ricker E, et al. Serine-threonine kinase ROCK2 regulates germinal center B cell positioning and cholesterol biosynthesis. J. Clin. Invest. 2020;130:3654–3670. doi: 10.1172/JCI132414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Q, et al. Rho-associated coiled-coil-containing kinase 2 deficiency in bone marrow-derived cells leads to increased cholesterol efflux and decreased atherosclerosis. Circulation. 2012;126:2236–2247. doi: 10.1161/CIRCULATIONAHA.111.086041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Talaia, G., Amick, J. & Ferguson, S. M. Receptor-like role for PQLC2 amino acid transporter in the lysosomal sensing of cationic amino acids. Proc. Natl. Acad. Sci. USA118. 10.1073/pnas.2014941118 (2021). [DOI] [PMC free article] [PubMed]
- 62.Amick, J., Tharkeshwar, A. K., Talaia, G. & Ferguson, S. M. PQLC2 recruits the C9orf72 complex to lysosomes in response to cationic amino acid starvation. J. Cell Biol.219. 10.1083/jcb.201906076 (2020). [DOI] [PMC free article] [PubMed]
- 63.Liu B, Du H, Rutkowski R, Gartner A, Wang X. LAAT-1 is the lysosomal lysine/arginine transporter that maintains amino acid homeostasis. Science. 2012;337:351–354. doi: 10.1126/science.1220281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jezegou A, et al. Heptahelical protein PQLC2 is a lysosomal cationic amino acid exporter underlying the action of cysteamine in cystinosis therapy. Proc. Natl. Acad. Sci. USA. 2012;109:E3434–3443. doi: 10.1073/pnas.1211198109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Polovitskaya MM, et al. A recurrent gain-of-function mutation in CLCN6, encoding the ClC-6 Cl(-)/H(+)-exchanger, causes early-onset neurodegeneration. Am. J. Hum. Genet. 2020;107:1062–1077. doi: 10.1016/j.ajhg.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He H, et al. West syndrome caused by a chloride/proton exchange-uncoupling CLCN6 mutation related to autophagic-lysosomal dysfunction. Mol. Neurobiol. 2021;58:2990–2999. doi: 10.1007/s12035-021-02291-3. [DOI] [PubMed] [Google Scholar]
- 67.Pressey SN, et al. Distinct neuropathologic phenotypes after disrupting the chloride transport proteins ClC-6 or ClC-7/Ostm1. J. Neuropathol. Exp. Neurol. 2010;69:1228–1246. doi: 10.1097/NEN.0b013e3181ffe742. [DOI] [PubMed] [Google Scholar]
- 68.Poet M, et al. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc. Natl. Acad. Sci. USA. 2006;103:13854–13859. doi: 10.1073/pnas.0606137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benussi A, et al. Phenotypic heterogeneity of Niemann–Pick disease type C in monozygotic twins. J. Neurol. 2015;262:642–647. doi: 10.1007/s00415-014-7619-x. [DOI] [PubMed] [Google Scholar]
- 70.Walterfang M, et al. Gender dimorphism in siblings with schizophrenia-like psychosis due to Niemann–Pick disease type C. J. Inherit. Metab. Dis. 2009;32(Suppl 1):S221–226. doi: 10.1007/s10545-009-1173-1. [DOI] [PubMed] [Google Scholar]
- 71.Millat G, et al. Niemann–Pick C1 disease: Correlations between NPC1 mutations, levels of NPC1 protein, and phenotypes emphasize the functional significance of the putative sterol-sensing domain and of the cysteine-rich luminal loop. Am. J. Hum. Genet. 2001;68:1373–1385. doi: 10.1086/320606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rodriguez-Gil, J. L. et al. Genetic background modifies phenotypic severity and longevity in a mouse model of Niemann–Pick disease type C1. Dis. Model. Mech.13. 10.1242/dmm.042614 (2020). [DOI] [PMC free article] [PubMed]
- 73.Miyawaki, S., Yoshida, H., Mitsuoka, S., Enomoto, H. & Ikehara, S. A mouse model for Niemann–Pick disease. Influence of genetic background on disease expression in spm/spm mice. J. Hered.77, 379–384. 10.1093/oxfordjournals.jhered.a110265 (1986). [DOI] [PubMed]
- 74.Zhang J, Erickson RP. A modifier of Niemann Pick C 1 maps to mouse chromosome 19. Mamm. Genome. 2000;11:69–71. doi: 10.1007/s003350010013. [DOI] [PubMed] [Google Scholar]
- 75.Calderon JF, Klein AD. Controversies on the potential therapeutic use of rapamycin for treating a lysosomal cholesterol storage disease. Mol. Genet. Metab. Rep. 2018;15:135–136. doi: 10.1016/j.ymgmr.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marshall CA, et al. In Niemann–Pick C1 mouse models, glial-only expression of the normal gene extends survival much further than do changes in genetic background or treatment with hydroxypropyl-beta-cyclodextrin. Gene. 2018;643:117–123. doi: 10.1016/j.gene.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 77.Parra J, et al. Npc1 deficiency in the C57BL/6J genetic background enhances Niemann–Pick disease type C spleen pathology. Biochem. Biophys. Res. Commun. 2011;413:400–406. doi: 10.1016/j.bbrc.2011.08.096. [DOI] [PubMed] [Google Scholar]
- 78.Hetmańczyk-Sawicka K, et al. Changes in global gene expression indicate disordered autophagy, apoptosis and inflammatory processes and downregulation of cytoskeletal signalling and neuronal development in patients with Niemann–Pick C disease. Neurogenetics. 2020;21:105–119. doi: 10.1007/s10048-019-00600-6. [DOI] [PubMed] [Google Scholar]
- 79.De Windt A, et al. Gene set enrichment analyses revealed several affected pathways in Niemann–Pick disease type C fibroblasts. DNA Cell Biol. 2007;26:665–671. doi: 10.1089/dna.2006.0570. [DOI] [PubMed] [Google Scholar]
- 80.Reddy JV, Ganley IG, Pfeffer SR. Clues to neuro-degeneration in Niemann–Pick type C disease from global gene expression profiling. PLoS ONE. 2006;1:e19. doi: 10.1371/journal.pone.0000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen FW, Li C, Ioannou YA. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS ONE. 2010;5:e15054. doi: 10.1371/journal.pone.0015054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rosenbaum AI, Zhang G, Warren JD, Maxfield FR. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann–Pick type C mutant cells. Proc. Natl. Acad. Sci. USA. 2010;107:5477–5482. doi: 10.1073/pnas.0914309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dai S, et al. Methyl-beta-cyclodextrin restores impaired autophagy flux in Niemann–Pick C1-deficient cells through activation of AMPK. Autophagy. 2017;13:1435–1451. doi: 10.1080/15548627.2017.1329081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kondo Y, et al. In vitro evaluation of 2-hydroxyalkylated beta-cyclodextrins as potential therapeutic agents for Niemann–Pick Type C disease. Mol. Genet. Metab. 2016;118:214–219. doi: 10.1016/j.ymgme.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 85.Soga M, et al. HPGCD outperforms HPBCD as a potential treatment for Niemann–Pick disease type C during disease modeling with iPS cells. Stem Cells. 2015;33:1075–1088. doi: 10.1002/stem.1917. [DOI] [PubMed] [Google Scholar]
- 86.Vanier MT. Niemann–Pick diseases. Handb. Clin. Neurol. 2013;113:1717–1721. doi: 10.1016/B978-0-444-59565-2.00041-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.