

Abstract
Immune‐mediated inflammatory diseases (IMIDs) are a diverse group of complex inflammatory diseases that result from dysregulated immune pathways and can involve any system of the human body. Inflammatory bowel disease (IBD) is one such disease involving the gastrointestinal (GI) system. With high prevalence in the West and increasing incidence in newly industrialized countries, IBD poses a significant burden on health care. IMIDs of the GI system other than IBD can have similar clinical features, causing diagnostic and therapeutic challenges. Although these disorders share a common pathophysiology, the defects can occur anywhere in the complex network of cytokines, inflammatory mediators, and innate and adaptive systems, leading to unregulated inflammation. Precise knowledge about them will help determine the possible targeted therapy. Thus, it is essential to distinguish these disorders from IBD. This review describes various IMIDs of the GI tract that mimic IBD.
Keywords: Crohn's disease, monogenic disorders, ulcerative colitis
Immune‐mediated inflammatory diseases (IMIDs) of the gastrointestinal tract including inflammatory bowel disease (IBD) are a diverse group of complex inflammatory diseases that share similar pathophysiology. IMIDs other than IBD can have similar clinical features causing diagnostic and therapeutic challenges. Thus, it is essential to distinguish these disorders from IBD. This review describes various IMIDs of the gastrointestinal tract that mimic IBD.
Introduction
Immune‐mediated inflammatory diseases (IMIDs) encompass a group of complex immune‐mediated diseases characterized by dysregulation of immune pathways and can involve any system of the human body. IMIDs involving the gastrointestinal (GI) tract are a group of disorders that share similar pathophysiology and can have similar clinical manifestations, sometimes causing significant clinical confusion. These disorders include inflammatory bowel disease (IBD), coeliac disease, GI vasculitis, eosinophilic gastroenteritis, certain monogenic disorders, sarcoidosis, immune checkpoint inhibitor‐induced colitis (ICI colitis), and microscopic colitis. IBD is commonest among them, causing a significant burden on the healthcare system. IBD comprises two major phenotypes: Crohn's disease (CD) and ulcerative colitis (UC). Even though our understanding of etiopathogenesis has improved, it is not very clear yet, and it was hypothesized that it occurs as a result of intricate interactions among environmental factors, such as the gut microbiome, in a genetically susceptible individual. 1 Intestinal homeostasis is maintained by the gut microbiome, and intestinal epithelial integrity as well as immune tolerance by gut immunity, which prevents abnormal activation of immune cells despite exposure to millions of foreign antigens daily. 2 In addition, various immune checkpoints and barriers are in place throughout the GI tract, which prevent abnormal immune activation. A breach in any of these barriers triggers the innate and adaptive immune system, resulting 3 in abnormal sustained immune response, thus causing intestinal damage. IMIDs of the GI tract result from defects in specific immune pathways and mimic IBD. With increasing knowledge and availability of sophisticated technologies, our understanding of the pathogenesis of intestinal IMIDs has improved remarkably. In this review, we describe various IMIDs of the GI tract which are similar to IBD and their pathogenesis (Table 1).
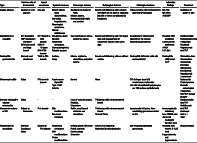
Table 1.
Disease characteristics and differences among different immune‐mediated intestinal diseases
| Type | Common site of involvement | Age of occurrence | Systemic features | Endoscopic features | Radiological features | Histological features | Laboratory findings | Treatment |
|---|---|---|---|---|---|---|---|---|
| Coeliac disease | Duodenum and jejunum | Any age |
Growth failure, bone abnormalities, Dermatitis herpetiformis, Neurological symptoms, Vitamin deficiencies |
Scalloping of duodenal folds, Mosaic pattern, Decreased fold height and number |
Bowel wall thickening with stricture formation can be seen in ulcerative jejunitis or when complicated by lymphoma | Villous atrophy with increased intra epithelial lymphocytes |
IgA tTG IgA EMA Anti‐DGP |
Avoidance of gluten in diet |
| Gastrointestinal vasculitis |
BD: Ileocolonic HSP: Duodenum and ileum SVV: both small and large bowel |
BD: Second to third decade HSP: 3–15 years SVV: Fifth decade |
Oro‐genital ulcers Sinusitis Respiratory symptoms, Glomerulonephritis, Palpable purpura |
Oval punched‐out ulcers, erythema, edema | Bowel wall thickening with the target sign and engorgement of mesenteric vessels with comb sign |
Acute/chronic inflammation Necrosis of the vessels Necrotizing granulomas |
Elevated CRP, creatinine; Urinary active sediments ANCA HLA B51 |
Corticosteroids Cyclophosphamide Thiopurines Rituximab Mepolizumab Anti‐TNF |
| Eosinophilic gastroenteritis | Stomach and duodenum | Third to fifth decade |
Asthma, Atopy |
Edema, erythema, ulcerations, polypoidal lesions | Bowel wall thickening with or without ascites | Eosinophilic infiltration with ≥30 eosinophils/hpf | Eosinophilia >500 cells/mm3 |
Elimination diets Prednisolone Budesonide Cromolyn Montelukast Omalizumab Mepolizumab Lirentelimab |
| Microscopic colitis | Colon | Fifth to sixth decade |
Autoimmune thyroiditis Type 1 DM Arthritis |
Normal | None |
CC: Collagen band ≥10 micrometers in diameter LC: ≥20 intraepithelial lymphocytes per 100 surface epithelial cells |
Budesonide Cholestyramine Bismuth subsalicylate Thiopurines Anti TNF therapy Surgery |
|
| Immune checkpoint‐induced enterocolitis | Colon | ‐ | ‐ |
Loss of vascular pattern, Friability, Ulcerations |
Thickening with enhancement of bowel |
Corticosteroids Anti‐TNF Vedolizumab |
||
| Monogenic variants of IBD | Colonic or ileocolonic | First decade |
Skin manifestations Recurrent infections |
Colitis, small bowel strictures and ulcerations |
Thickening with enhancement of bowel, Perianal fistula |
Lymphocytic infiltrates, Non‐necrotizing granulomas similar to CD |
Immunoglobulin deficiency Low FOXP3 cells Abnormal neutrophil function tests |
Anti TNF Abatacept Anti‐IL‐1 HSCT |
| Gastrointestinal sarcoidosis |
Stomach Esophagus Colon |
Second to fifth decade |
Respiratory symptoms Skin manifestations Ocular manifestations |
Diffuse gastric infiltration Pyloric stenosis Gastric polyps Ulceration |
Bowel wall thickening Abdominal lymphadenopathy |
Non‐caseating epithelioid granulomas |
Elevated ACE, vitamin D 1,25 level Hypercalcemia, and hypercalciuria |
Glucocorticoids |
ACE, angiotensin converting enzyme; ANCA, antineutrophil cytoplasmic antibody; Anti‐DGP, anti deamidated gliadin peptide antibody; Anti IL1, anti interleukin‐1 antibody; Anti‐TNF, anti‐tumor necrosis factor antibody; BD, Behcet's disease; CC, collagenous colitis; CD, Crohn's disease; CRP, C‐reactive protein; FOXP3, Forkhead box P3; HLA, human leukocyte antigen; HSCT, hematopoietic stem cell transplantation; HSP, Henoch‐Schönlein purpura; IBD, inflammatory bowel disease; IgA EMA, anti endomysial antibody; IgA tTG, tissue transgutaminase antibody; LC, lymphocytic colitis; SVV, small vessel vasculitis.
Monogenic disorders presenting as IBD
Increasing availability and declining cost of next‐generation sequencing methods have made it possible to recognize Mendelian disorders that mimic IBD. 4 Conventional adult‐onset IBD is considered a polygenic disorder with a possible link to 240 genetic loci belonging to approximately 300 genes. However, their penetrance is low. 5 In comparison, more than 50 different sets of genes have been reported that present with features similar to adult‐onset IBD with Mendelian inheritance. 6 Even though some of the genes implicated in monogenic IBD variants have incomplete penetrance, it is still higher than the penetrance of genes implicated in conventional adult‐onset IBD, such as NOD2, ATG16L1, IRGM, IL23R, CARD9, RNF186, and so on. 7 These Mendelian disorders constitute a small fraction of IBD cases and present during early childhood, 8 and as the age of presentation increases, the incidence of these monogenic variants decreases. These Mendelian disorders can be classified into different functional groups based on the pathways involved, including disorders of epithelial barrier integrity, disorders of immune dysregulation, autoinflammatory disorders, disorders of immunodeficiency, and innate immune defects including phagocyte killing. As mutations involved in these disorders can affect more than one pathway, specific differentiation of a disorder into one of these groups is not accurate, and many of these disorders can have features of immunodeficiency.
Disorders of epithelial integrity
Intestinal epithelium not only acts as a physical barrier but also helps in maintaining immune tolerance. IBD‐like phenotype can be observed in disorders of epithelial barrier defects as seen in dystrophic epidermolysis bullosa, Kindler syndrome, familial diarrhea due to activating dominant guanylate cyclase C mutation (GUCY2C), X‐linked ectodermal dysplasia and immunodeficiency, and ADAM‐17 deficiency. 9 Most of these syndromes primarily present with skin manifestations during early childhood. 10 For example, hypomorphic mutations in the IKBKG gene encoding nuclear factor κB (NFκB) essential modulator protein (NEMO) lead to intestinal inflammation in X‐linked ectodermal dysplasia. 11 Disorders of collagen VII defects can also present with CD‐like phenotype, indicating the role of collagen in maintaining epithelial barrier integrity. Other disorders involving the regulation of epithelial polarity such as Tetratricopeptide Repeat Domain 7A (TTC7A) are also associated with severe combined immunodeficiency (SCID)‐like presentation and intestinal inflammation presenting in early life. 12 Other examples include congenital sodium diarrhea (CSD) and congenital tufting enteropathy caused by the epithelial adhesion genes EPCAM and SPINT2, respectively. Mutations in SLC9A3 and GUCY2C also lead to congenital sodium diarrhea. 13 These patients are predisposed to the development of IBD in later life. 14
Another group of disorders that are predominantly reported from Asian countries such as China, Japan, and Korea includes chronic nonspecific multiple small intestinal ulcers (CNSU) or cryptogenic multifocal ulcerous stenosing enteritis (CMUSE). 15 , 16 These disorders involve defects in the prostaglandin pathway. Variants of the SLCO2A1 gene have been shown to be associated with circumferential ulcers in the ileum and protein‐losing enteropathy, resulting in a condition known as chronic enteropathy associated with SLCO2A1 (CEAS). 17 , 18 The SLCO2A1 gene is involved in the transport of PGE2 into the cytoplasm and its subsequent destruction. Mutations in this gene lead to excess PGE2 levels and inflammation. The exact pathophysiology of this condition is not yet established, but elevated PGE2 may disrupt the integrity of the epithelial barrier and then predispose to inflammation. Another variant of SLCO2A1 is also associated with similar presentation and with primary hypertrophic osteoarthropathy (PHO). 19 CEAS is more frequently observed in females than males, whereas PHO‐associated enteropathy is more common in males. A similar condition associated with mutations in PLA2G4A involved in prostaglandin metabolism also gives rise to similar enteropathy presenting with small intestinal ulcers. 20
Disorders of immune dysregulation
Interleukin 10, an anti‐inflammatory cytokine, and its receptor defects are associated with very early onset IBD‐like phenotype with a predisposition to perianal and rectovaginal fistulae and presentation during the first year of life. Defects in the autoimmune regulator gene (AIRE), which is responsible for central tolerance, result in autoimmune polyendocrinopathy candidiasis with ectodermal dystrophy (APECED). It is a multisystem autoimmune disease that manifests by severe, intractable diarrhea and failure to thrive in the first decade of life. Histopathological examination shows small intestinal villous atrophy. 21 Diseases associated with JAK–STAT dysfunction and STAT3 and STAT1 gain of function are also associated with lymphoproliferation and IBD‐like presentation. 22
Autoinflammatory syndromes
Disorders of excessive inflammatory macrophage activation, such as phospholipase Cγ2 defects or familial Mediterranean fever, X‐linked lymphoproliferative syndrome types 1 and 2 or familial hemophagocytic lymphohistiocytosis (HLH) type 5, Hermansky–Pudlak syndrome (types 1, 4 and 6), and mevalonate kinase deficiency, can present with IBD‐like features. 23 Mutations in X‐linked inactivator of apoptosis (XIAP) result in a rare, severe form of primary immunodeficiency disorder (X‐linked lymphoproliferative syndrome 2) characterized by HLH, which is associated with severe fistulizing perianal colonic phenotype at an early age.
Disorders of immunodeficiency
There are approximately 350 primary immunodeficiency disorders (PIDDs) with distinct molecular etiologies; among them, approximately one‐third have GI manifestations, 22 which could be GI infections, IBD‐like inflammation, or the development of GI malignancies. Even though these PIDDs are predisposed to infections due to defective microbial killing, they may also be associated with hyper‐inflammatory responses manifesting as IBD. In addition, most of these disorders are associated with the involvement of other systems, and GI manifestations occur during the disease course. 24
B‐cell disorders: Immunodeficiency disorders involving B‐cell defects such as common variable immunodeficiency (CVID), hyper‐IgM syndrome, and agammaglobulinemia can have bowel involvement similar to IBD. However, pure immunoglobulin deficiency or B‐cell defects will not predispose to intestinal inflammation. For example, despite BTK‐associated agammaglobulinemia being associated with a severe lack of immunoglobulins, it is less frequently associated with colitis than CVID subsets with less severe immunoglobulin deficiency. 24
T‐cell disorders: Regulatory Treg cells (T‐cells) are most important for maintaining immunotolerance in the intestines. Various genes affect Treg cell function, defects of which predispose to uncontrolled inflammation presenting as IBD‐like phenotype. The most important genes are FOXP3, CTLA4, LRBA, IL2Rα, BACH2, and TGFB1. 25 All these genes affect Treg cell function either directly or indirectly. Immune dysregulation, polyendocrinopathy, enteropathy, X‐linked (IPEX) syndrome is prototype disorder in this category, which results from mutations in the FOXP3 gene. 26 IPEX is characterized by multisystem autoimmune manifestations with reduced Treg cells. The typical presentation is severe, intractable diarrhea in infancy. The haploinsufficiency of the CTLA4 gene causes a syndrome named CHAI (CTLA‐4 haploinsufficiency with autoimmune infiltration) characterized by lymphadenopathy, splenomegaly, hypogammaglobulinemia, lymphocytic infiltration of multiple nonlymphoid organs (especially the intestines), and autoimmunity. 27 CTLA4 is an inhibitory receptor expressed on effector T‐cells and Treg cells and mediates peripheral immunotolerance. Mutations in another protein closely associated with CTLA‐4 known as LRBA (lipopolysaccharide‐responsive vesicle trafficking, beige‐like anchor protein) cause LATAIE (LRBA deficiency with autoantibodies, Treg defects, autoimmune infiltration, and enteropathy). 28 LRBA binds with the CTLA‐4 and protects it from destruction. BACH2 is a transcriptional regulatory protein required for Treg cell development. Mutations of BACH2 gene cause a syndrome known as BACH2‐related immunodeficiency and autoimmunity (BRIDA), which presents with IBD‐like phenotype and predisposes to infections. 2 Mutation in TGF β1 also resembles FOXP3 deficiency in the first few months of life. Wiskott–Aldrich syndrome (WAS) is a rare X‐linked primary immunodeficiency characterized by the triad of eczema, thrombocytopenia, and susceptibility to infections due to combined immunodeficiency. 29 Patients with WAS exhibit a spectrum of autoimmune manifestations such as hemolytic anemia, vasculitis, inflammatory arthritis, neutropenia, and IBD.
Neutrophil disorders: Neutrophils play an essential role in protecting intestinal epithelial integrity from invading pathogens. 30 Chronic granulomatous disease (CGD) is a prototype of these disorders, and others include glycogen storage disease type 1b, congenital neutropenia, and leucocyte adhesion deficiency‐1. Defects in genes such as WAS, LRBA, BTK, CD40LG, or FOXP3 can also have underlying neutrophilic functional defects, but primarily they are regulatory T‐cell disorders. CGD is characterized by impaired NADPH oxidase (PHOX) complex generation leading to defective microbicidal respiratory burst. 31 Clinical presentation includes skin lesions, susceptibility to bacterial and fungal infections, and inflammatory GI manifestations usually presenting after the first decade of life. 32 The most common CGD mutation occurs in the CYBB gene with X‐linked inheritance; other mutations involving CYBA, NCF1, NCF2, and NCF4 are associated with less severe phenotypes. X‐linked CGD is more likely to present as an IBD phenotype. 33 , 34
Monogenic IBD variants presenting in adults
The majority of the monogenic IBD variants manifest in the first decade of life, but some mutations can present after the second decade but rarely in adulthood (Table 2). Usually, patients with monogenic disorders have multisystem involvement along with intestinal symptoms. Other features apart from young age of onset, refractoriness to conventional therapy, growth failure, multisystemic symptoms such as skin, hair, endocrine involvement, recurrent childhood infections, hemophagocytic lymphohistiocytosis, and similar manifestations in first‐degree relatives also favor monogenic disorders. A subset of patients with congenital sodium diarrhea can present with IBD‐like phenotype in late adolescence or adulthood. 14 Hermansky–Pudlak syndrome type 1, 4, or 6 can develop Crohn's phenotype at any age, including early childhood, but most often in adolescence or young adulthood. However, they present with characteristic oculocutaneous albinism and bleeding diathesis. 35 , 36 Rare variants of the XIAP gene can present with intestinal inflammation without XLP2‐like manifestations. 37 Mutations in SLC37A4 cause glycogen storage disorder 1b and present with hepatosplenomegaly, failure to thrive, and IBD‐like phenotype in the first two decades of life. Congenital neutropenia syndrome caused by mutations in G6PC3 can also predispose to developing IBD‐like phenotype manifested in adult life. 38 In a nationwide French registry of fourteen patients with congenital neutropenia syndrome, five had features of IBD, and most of them were diagnosed in the second decade of life. 39 Similarly, mutations in the ICOS and LRBA genes can present with CVID‐like phenotype in young adults, and other disorders such as CGD and WAS can also present after adolescence. 40 Mutations in SLCO2A1 and PLA2G4A involve defects in prostaglandin metabolism and present with small intestinal ulcerations. Deficiency of CD55 (decay‐accelerating factor) leads to CHAPLE (CD55 deficiency with hyperactivation of complement, angiopathic thrombosis, and protein‐losing enteropathy) syndrome, which is characterized by hyperactivation of complement, vascular thrombosis, protein‐losing enteropathy, and intestinal ulcers. 41
Table 2.
Monogenic inflammatory bowel disease (IBD) variants that can be present in adults
| Type | Phenotype | Associated features | Lab abnormalities |
|---|---|---|---|
| LRBA and ICOS | UC/CD | Recurrent infections, hepatosplenomegaly, lymphadenopathy | Hypogammaglobulinemia |
| XIAP (XLP2) | Crohn's‐like granulomatous colitis in males |
Perianal fistula; HLH; Splenomegaly; Cholangitis; Skin abscesses; Arthritis, EBV and CMV infections; |
Hypogammaglobinemia |
|
CGD CYBB CYBA NCF1 NCF2 NCF4 |
Crohn's‐like colitis |
Recurrent infections for catalase‐positive organisms; Perianal fistula; Gastric outlet obstruction; Eczema |
Decreased neutrophil oxidative burst study; Elevated IgG |
| LAD1 (ITGB2) | Crohn's‐like with stenosis/stricturing phenotype |
Neutrophilia Recurrent infections Delayed separation of umbilical cord; Poor wound healing; Folliculitis; Ulcers of skin; Peridontitis; Gingivitis |
Absent CD11/CD18 complex expression |
| SLCO2A1 | Small‐bowel stricturing disease | Primary hypertrophic osteoarthropathy | — |
| PLA2G4A | Small‐bowel stricturing disease | — | — |
| WAS | UC‐like colitis in males |
Thrombocytopenia; Atopic dermatitis; Autoimmune hemolytic anemia; Arthritis; Bacterial/viral infections; Lymphoreticular malignancy |
Microthrombocytopenia; Variable lymphopenia; Absent class‐switched memory B‐cells; High IgE; Low IgG, IgA, and IgM |
| CD 55 (CHAPEL syndrome) | Enterocolitis with ulcers in terminal ileum |
Early‐onset, protein‐losing enteropathy; Primary intestinal obstruction; Edema; Malabsorption; Recurrent infections; Angiopathic thromboemboli |
Absent CD55 expression on lymphocytes |
| G6P3C | Crohn's‐like with strictures; Oral and genital aphthous ulcerations |
Cutaneous vascular malformation; Cardiac defects; Urogenital developmental defects; Bleeding tendency |
Severe neutropenia; T‐cell lymphopenia; Elevated IgG |
| SLC37A4 |
Crohn's‐like colitis with ulcerations; Strictures; Perianal fistula |
Hepatosplenomegaly; Nephrocalcinosis; Folliculitis |
Neutropenia and/or neutrophil dysfunction with predisposition to infections; Hypoglycemia; Hyperuricemia; Hyperlipidemia |
| SLC9A3 and GUCY2C (congenital sodium diarrhea) | IBD‐like small intestinal involvement in adults | Elevated liver enzymes and cholestasis |
High stool sodium levels Low serum sodium levels, Metabolic acidosis, Alkaline fecal pH, Low urinary sodium excretion |
CD, Crohn's disease; HLH, hemophagocytic lymphohistiocytosis; UC, ulcerative colitis.
Approach to patients with monogenic disorders and management
Monogenic IBD phenotypes are rarely encountered, but they can be challenging when one comes across such cases. Treating physicians should be vigilant and identify these cases when risk factors are present. Peripheral blood counts, immunoglobulin profile, and lymphocyte assays help in identifying common immunodeficiency disorders such as CVID and lymphocyte disorders. Neutrophil oxidase burst assays can be carried out if CGD is suspected (Table 3). Targeted gene analysis can be done to confirm the diagnosis if any one of them is positive. If first‐line investigations are normal and monogenic IBD is strongly suspected, whole exome or genome sequencing can identify the underlying genetic defects (Fig. 1). Monogenic disorders are present early in life and are difficult to treat, being sometimes refractory to immunosuppressive drugs including biologics. Targeting the underlying defective immune pathway might be beneficial. Some of the monogenic disorders, such as mutations of IL‐10, can be effectively cured by hematopoietic stem cell transplantation (HSCT). However, HSCT may not be curative in every case. For example, mutations resulting in epithelial defects such as TTC7A have a high risk of recurrence even after HSCT. Patients with mevalonate kinase deficiency, IL‐10 mutations or CGD are associated with excess IL‐1b. Hence treatment with IL‐1b receptor antagonists might be beneficial. Similarly, abatacept, a CTLA‐4 fusion protein, has been used in disorders with LRBA mutation. Replacement of immunoglobulin in immunodeficiency disorders such as CVID and the use of eculizumab in CHAPEL syndrome are helpful in treating these disorders.
Table 3.
Laboratory abnormalities in monogenic disorders
| Investigation | Genetic disorder |
|---|---|
| Elevated IgE/eosinophilia | IPEX, WAS, IKBKG, DOCK8 |
| Immunoglobulin profile | LBRA, CVID, agammaglobulinemia |
| Lymphocyte subset analysis | CVID, agammaglobulinemia |
| DHR test | CGD |
| Flow cytometric analysis of XIAP expression by NK cells and lymphocytes | XIAP |
| Flow cytometric analysis of FOXP3 expression by CD4 T‐cells | IPEX |
CGD, chronic granulomatous disease; CVID, common variable immunodeficiency; IPEX, immune dysregulation, polyendocrinopathy, enteropathy, X‐linked; WAS, Wiskott–Aldrich syndrome; XIAP, X‐linked inactivator of apoptosis.
Figure 1.
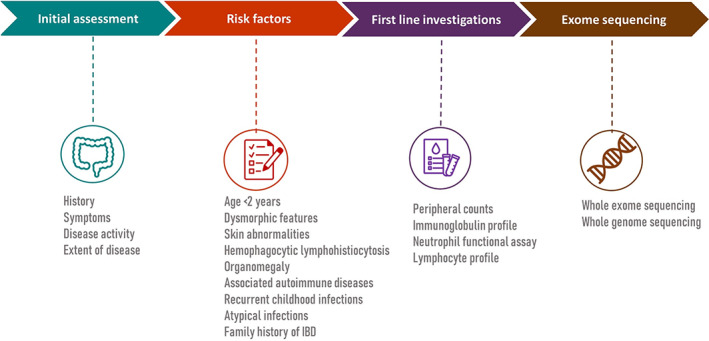
Steps in the assessment and investigations of patients with suspected monogenic inflammatory bowel disease (IBD)‐like phenotype
Coeliac disease
Coeliac disease (CeD), also known as gluten‐sensitive enteropathy, is an immune‐mediated disease with similar pathogenesis as that of IBD, but unlike IBD, the environmental and genetic factors responsible for disease pathogenesis are known. The global prevalence of CeD based on serology in the general population is 1.4%, with the highest prevalence in Western countries. 42 The prevalence of biopsy‐proven CeD is estimated to be lower (0.7%) than serology‐based estimates. 42 CeD results from immune‐mediated intestinal injury triggered by dietary ingestion of gluten protein in genetically predisposed individuals. There is a clear genetic association between individuals possessing HLA‐DQ2 and DQ8 and CeD. 43 Proline‐rich gluten proteins in wheat, rye, and barley are the key drivers of intestinal inflammation. Gluten proteins are poorly digested by enterocytes, and transported across the intestinal epithelium by paracellular mechanism to be presented to gluten‐specific CD4 cells by antigen‐presenting cells (APC) expressing HLA‐DQ2 and DQ8. These HLA molecules bind more avidly to negatively charged gluten proteins after modification by the tissue transglutaminase (tTG) enzyme. 44 Although CeD was previously thought to be a childhood disease, its onset can occur at any age. CeD is characterized by diarrhea, symptoms of malabsorption (anemia, vitamin deficiencies, bloating, and steatorrhea), or sometimes predominant extraintestinal manifestations such as growth failure, bone abnormalities, osteoporosis, neurological symptoms, infertility, and association with other autoimmune diseases (autoimmune hepatitis, type 1 diabetes, autoimmune thyroiditis). 45 CeD is diagnosed by the presence of serological markers (IgA tTG, anti‐endomysial antibody, anti‐deamidated gliadin peptide antibodies) along with evidence of villous atrophy of duodenal epithelium with the exception in children, where more than 10 times elevation of IgA tTG is sufficient to make a diagnosis. 46 Endoscopy may show scalloped duodenal folds, mosaic pattern, as well as decreased duodenal fold height and number. However, normal endoscopy does not rule out CeD. With overlapping features, CeD can mimic IBD, especially in children. However, observational studies have shown that these disorders could coexist in the same patient.
In a meta‐analysis of observational studies by Sanchez et al., there was an increased risk of IBD in CeD and an increased risk of CeD in IBD. 47 Patients with CeD are also at increased risk of developing microscopic colitis. Management includes avoidance of gluten‐containing food products and nutritional supplementation of deficient nutrients and vitamins. Less than 5% of patients do not respond despite adherence to a gluten‐free diet, referred to as refractory CeD, which is categorized into type 1 and type 2. 48
Vasculitis‐related enteropathy
Vasculitis is characterized by inflammation of blood vessels and can involve any organ system with protean clinical manifestations depending on the size of the vessels involved. Vasculitis can be divided into large‐, medium‐, and small‐vessel vasculitis and other minor groups according to the revised Chapel Hill Consensus, 2012. 49 The extent and severity of GI involvement vary depending on the type of vasculitis, and GI involvement usually occurs along with the involvement of other systems. Clinical manifestations vary from mild abdominal pain and diarrhea to acute intestinal perforation. 50 , 51 , 52 The underlying pathophysiology is diffuse ischemia of intestines due to inflammation and thickening of mesenteric vessels or necrotizing inflammation involving small intramural and submucosal vessels causing ulceration and perforation. It has been shown that involvement of the GI system is associated with poor prognosis in patients with systemic vasculitis. 53 Large (Takayasu arteritis and giant‐cell arteritis) and medium (polyarteritis nodosa and Kawasaki disease) vessel vasculitis predominantly present with intestinal ischemia due to transmural necrotizing inflammation of mesenteric vessels leading to abdominal pain. Granulomatosis with polyangiitis (GPA), microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis (EGPA) manifest with GI symptoms in 5–30% of cases. 54 Clinical symptoms include abdominal pain, diarrhea, and hematochezia. Endoscopic features include deep punched‐out ulcers as well as edematous and erythematous mucosa. Radiological imaging findings are usually nonspecific, ranging from multifocal or diffuse bowel‐wall thickening to mesenteric vascular engorgement. 55 Histopathology of GPA and EGPA typically shows necrotizing granulomas with neutrophilic infiltrates and eosinophilic infiltrates in EGPA. 56 Without treatment, mortality is more than 80%, especially with multiorgan involvement. 57 Treatment includes immunosuppression in the form of glucocorticoids, and in severe cases with multiorgan involvement, higher immunosuppression with cyclophosphamide is recommended. 54
Behcet's disease (BD) is another multisystem vasculitis involving all vessels and clinically presents with recurrent oral and genital ulcers. According to Chapel Hill Classification, 2012, 51 it is classified under variable vessel vasculitis. GI involvement can be seen in 10–15% of cases. The incidence of BD is highest in Turkey, with a prevalence of 80–370 cases per 100 000 population, followed by Asia and the Middle Eastern countries, including Israel, Saudi Arabia, and Iran. 58 BD commonly presents in the second and third decade of life with a variable male‐to‐female ratio depending on the geographical location. HLA‐B5101 is strongly associated with BD, and high concordance in twins has been observed. 59 Recent studies have shown an association of mutations in IL10, IL19, IL23R, and IL12B2 with BD. 58 Abdominal pain, diarrhea with or without bleeding, fatigue, and weight loss are common manifestations along with recurrent oral and genital ulcers and ocular involvement, and patients with BD are at high risk of developing thrombosis. Perianal involvement is extremely rare. The mucosal ulceration is typically localized to the ileocecal area with large (>1 cm), deep, punched‐out volcano ulcers with a high risk of perforation. 60 Common patterns of involvement are localized single ulcer (67%) or localized multiple ulcers (27%), but multi‐segmental or diffuse colonic involvement is rare (6%). 61 Histopathology shows nonspecific inflammation with no granuloma. Radiological imaging studies show features similar to CD. Diagnosis is made by either the ICBD (International Criteria for Behcet's Disease) criteria or the International Study Group (ISG) diagnostic criteria. 62 , 63 However, ISG criteria do not include intestinal symptoms. Medical management constitutes corticosteroids, immunomodulators, and anti‐TNF agents. In severe cases with perforation, surgery is recommended. The young age of onset (<25 years), history of laparotomy, and volcano‐shaped ulcers on colonoscopy predict surgery. 64 Other systemic vasculitides such as IgA vasculitis can also involve the GI system with predominant duodenal and small intestinal involvement, presenting during childhood with abdominal pain, intussusception, hemorrhage, and perforation. 65 Kawasaki disease is another medium‐vessel vasculitis commonly seen in children, but with uncommon GI involvement, presenting with mesenteric ischemia‐like features.
Eosinophilic gastroenteritis
Eosinophilic gastroenteritis is a rare eosinophil‐mediated inflammatory disorder of unknown etiology, infrequently suspected and challenging to diagnose, and mimics other GI disorders such as IBD. Eosinophils are an essential part of the innate immune system of the GI tract, and eosinophilic infiltration of the GI mucosa is commonly seen in many disorders, including parasitic infections, IBD, hypereosinophilic syndrome, and drug hypersensitivity. Eosinophilic GI disorders (EGIDs) are a group of disorders that encompass eosinophilic esophagitis (EoE), eosinophilic gastroenteritis (EGE), and eosinophilic colitis (EC). 66 Even though predominant symptoms are localized to one segment of the bowel, eosinophilic infiltration can be simultaneously seen in other segments of the intestine, as shown in a case series by de Chambrun et al., where 80% of patients with EoE had eosinophilic infiltration of the colon on histopathologic examination. 67 The prevalence of EGE is estimated to be 1 in 100 000 and commonly seen in the third to fifth decade of life, although it can be present as early as in infancy and as late as in the eighth decade. 68 There is a slight male preponderance, with a male/female ratio of 3:2. The exact pathophysiology is not clear, but the role of T‐helper (Th)‐2 cytokines and eosinophilic mediators such as interleukin (IL)‐5 and eotaxins has been demonstrated. 69 Approximately 25–70% of patients have either a personal or family history of atopy. Peripheral eosinophilia (eosinophils >500/microL) is seen in around 70–80% of cases, and the presence of very high peripheral eosinophilic counts indicate underlying primary eosinophilic disorder. 66 EGE is characterized by eosinophilic infiltration of all layers of the GI tract, and according to classification by Klein et al., EGE is divided into three types: mucosal, muscular, and serosal. Clinical presentation depends on the predominant layer involved. 70 Mucosal disease is the most common form occurring in approximately 50% of cases and presents with abdominal pain, malabsorption, and protein‐losing enteropathy, whereas muscular disease presents as intestinal obstruction and serosal disease presents with eosinophilic ascites and pleural effusion. 71 , 72 The stomach and proximal intestines are the most common sites of involvement (40–80%). Endoscopy findings vary from normal mucosa to erythema, friability, fine granularity, ulcerations, nodules, and strictures. Histopathology shows infiltration by eosinophils (>30/hpf), but the mere presence of eosinophils does not confirm the diagnosis of EGE. Three different clinical courses have been reported from a northern French cohort, which includes an initial flare followed by remission (44%), intermittent exacerbations (36%), and a chronic continuous course (21%). 67 The latter is predominantly seen in mucosal disease (80%). Treatment includes corticosteroids (prednisolone 0.5–1 mg/kg/day) with a good clinical response of about 50–90% with a slow taper over a period of 6–8 weeks. Budesonide can also be used as an alternate to prednisolone. Azathioprine can be used as a steroid‐sparing agent in patients with the steroid‐dependent disease. In refractory cases, biologics like omalizumab (anti‐IgE) or mepolizumab (anti‐IL‐5), or anti‐TNF agents (infliximab and adalimumab) can be tried. 73 A phase II RCT of antolimab (an anti‐siglec‐8 antibody) by Dellon et al. showed promising results, with 95% decrease in eosinophils on histopathology and significant improvement in symptoms in patients with EGE and EoE. 74 A study by Gonsalves et al. showed 100% histological remission in 15 patients with EGE using an elemental diet formula, indicating the role of food allergens. 75
Microscopic colitis
Microscopic colitis (MC) is an inflammatory disease of the large intestine characterized by chronic watery diarrhea, with normal or near‐normal endoscopic pictures and typical histopathological findings. MC is frequently missed or confused with diarrhea‐predominant irritable bowel syndrome (IBS), and the symptoms can result in severe impairment in the quality of life. A nationwide cohort study in Sweden found that patients with MC were at increased risk of death with a 10‐year absolute risk difference of 3.4% compared with the population without MC, which may be to be due to a higher burden of comorbidities. 76 The incidence increases with age, with a peak incidence in sixth and seventh decades of life. 77 The etiopathogenesis is unclear but similar to IBD, and the role of gut microbiota and defect in epithelial integrity in genetically predisposed individuals have been proposed. 78 A recent study demonstrated dysbiosis with an abundance of Haemophilus parainfluenzae, Veillonella parvula, and other Veillonella unclassified species and a low abundance of Alistipes putredinis compared with healthy controls. 78 However, the causal relationship is not known. The major subtypes include collagenous colitis (CC) and lymphocytic colitis (LC). Histopathological changes can be patchy or diffuse, with major changes seen in the right colon rather than the left colon. CC is characterized by a collagenous band of size >10 μm in the lamina propria associated with increased intraepithelial lymphocytes (IELs) (<20/100 colonocytes), whereas LC is characterized by a collagenous band of size <10 μm and IELs >20/100 colonocytes. With these histopathological findings, MC can mimic early changes of CD or UC. However, endoscopic findings can differentiate both conditions. A retrospective study showed that combined biopsies from ascending and descending colon are sufficient to diagnose MC in 100% cases. 79 It has been shown that symptoms correlate with cellularity rather than collagen band thickness. 80 Smoking, the use of drugs such as proton pump inhibitors (PPIs), nonsteroidal anti‐inflammatory agents (NSAIDs), and selective serotonin reuptake inhibitors (SSRIs), and infection with Clostridium concious have been shown to predispose MC. Other disorders such as bile salt diarrhea, celiac disease, and IBS can coexist with MC. 81 , 82 Kane et al. developed and validated a scoring system based on parameters such as age ≥50 years, use of proton pump inhibitors or nonsteroidal anti‐inflammatory drugs, female sex, absence of abdominal pain, and weight loss. The score ranged from −8 to +38, and a score ≥8 identified patients with MC with >90% sensitivity. However, the specificity was less, and they showed that this score could identify potential patients, thereby reducing the need for unnecessary colonoscopies and biopsies. 83 Various randomized trials and meta‐analyses showed approximately 80% clinical response with budesonide after induction, but prednisolone has not been shown to be effective. 84 Budesonide is also effective for maintaining remission. However, relapse rates are high on withdrawing. 85 Thiopurines, anti‐TNFs, and vedolizumab are recommended only if there is no response to budesonide. 86
Immune checkpoint inhibitor‐induced colitis
Immune checkpoint inhibitors (ICIs) target immune checkpoints such as cytotoxic T‐lymphocyte associated protein‐4 (CTLA‐4) and programmed death receptor/ligand (PD/L). CTLA‐4 is an inhibitory receptor that competes with CD28 in attachment to CD80/86 on antigen‐presenting cells, thereby negatively regulating effector T‐cells, and playing an essential role in peripheral tolerance. Antibodies to CTLA‐4 block its interaction with CD80/86, leading to the activation of the CD28‐mediated co‐stimulatory pathway, boosting anti‐tumor T‐cell response. 87 PD receptors are located on various cells, including effector T‐cells, and they inhibit signals downstream to T‐cell receptor (TCR). CTLA‐4 regulates T‐cells in lymph nodes, whereas PD‐L1 regulates T‐cells located in peripheral tissues. 88 Anti‐CTLA‐4 (ipilimumab), anti‐PD‐1 (nivolumab, pembrolizumab), and anti‐PD‐L1 (atezolizumab, avelumab, and durvalumab) antibodies have been approved for use in melanoma, non‐small‐cell lung carcinoma (NSCLC), hepatocellular carcinoma, classic Hodgkin's disease, gastric or gastro‐esophageal junction malignancies, head and neck squamous cell carcinoma, and urothelial carcinoma. Many adverse events due to nonspecific immune activation have also been reported with their increasing use, collectively known as immune‐related adverse events (irAEs). Immune activation leads to T‐cell‐induced inflammation in various organ systems, including the skin, GI, liver, endocrine glands, and lungs. Immune‐mediated colitis (IMC) is one of the major irAEs and requires urgent therapy depending on the grade of severity. Clinical, radiological, and histopathological manifestations resemble those of IBD. 89 Major risk factors for IEC include the dose, type of agent, use of NSAIDs, pre‐existing IBD, and the type of tumor, and recent association with a certain type of microbiota has also been reported with increased risk of IMC. Colitis has been more frequently observed in patients receiving a higher dose of ipilimumab (10 mg/kg compared to 3 mg/kg dose; 5 vs 2%). 90 Diarrhea and colitis are more frequently associated with CTLA‐4 inhibitors compared to PD‐L1 inhibitors. IMC usually develops after a median duration of 4–8 weeks, but can develop even after 2 years following infusion. The clinical presentation is similar to IBD, with diarrhea, abdominal pain, hematochezia and, in severe cases, bowel perforation. Endoscopy shows nonspecific features such as erythema, erosion, ulceration, and luminal bleeding, and these changes can be diffuse or patchy. Histopathology shows cryptitis, distortion, ulceration, and pericryptal granulomas, similar to the changes seen in IBD. 91 The most common site of involvement is the left colon, but in 12% cases isolated ileal involvement can be seen. 92 Early endoscopic finding or fecal calprotectin before initiation of ICI therapy does not predict the risk of developing IMC. 92 Some studies have shown a more potent anti‐tumor effect with ICIs in patients who develop irAEs, suggesting an adequate immune response. 93 There is an increased risk of developing recurrence (23%) of IMC following the reintroduction of ICI after recovering from the first episode. 90 Colonization by Firmicutes, such as Faecalibacterium prausnitzii L2‐6 and the butyrate‐producing bacterium Gemmiger formicilis, has been associated with good anti‐tumor response. 94 Management is more or less similar to that of IBD, and it depends on the severity of IMC. 95 Low‐grade colitis (grade 1 and 2) is managed with oral steroids, and if there is no improvement, intravenous methylprednisolone (1 mg/kg/day) is advised. High‐grade colitis (grade 3 and 4) is managed with intravenous methylprednisolone, and if there is no response within 3–5 days, infliximab should be considered. 92 Recent studies have demonstrated the role of the integrin inhibitor vedolizumab in colitis refractory to infliximab. 96 A recent meta‐analysis has also shown a pooled response rate of more than 80% with infliximab and vedolizumab and a response rate of 60% with steroids. 97 There are case reports of successful treatment of IMC with fecal microbial transplantation. 98
Gastrointestinal sarcoidosis
Sarcoidosis is a systemic granulomatous disease of unknown etiology characterized by the formation of non‐necrotizing granulomas. 99 Thoracic involvement is most commonly seen, and GI involvement can be incidental, with only <1% presenting with symptomatic GI sarcoidosis. 100 There is an oro‐anal gradient in the incidence of GI sarcoidosis, with common sites of involvement being upper GI tract (stomach and esophagus). Involvement of the small intestine and colon can be seen in up to 15–20% of cases of GI sarcoidosis. 101 The pathogenesis of sarcoidosis is not clear, but it is hypothesized that abnormal activation of T‐cells secondary to unknown antigen leads to granulomatous inflammation. The granulomas have a central follicle composed of epithelioid and CD4 helper T‐cells with TH1 differentiation surrounded by a ring of fibroblasts, B‐cells, and CD8 T‐lymphocytes. 99 Clinical presentation includes diarrhea, weight loss, and abdominal pain. Endoscopy shows edematous, ulcerated mucosa, or polypoidal lesions in stomach and intestines. Radiological investigation reveals segmental thickening with enhancement. Diagnosis is made by demonstrating non‐necrotizing granulomas on histopathology. Pulmonary and thoracic involvement favors sarcoidosis. Corticosteroids constitute the treatment of choice.
Approach
The GI system is the largest lymphoid organ in our body. Despite exposure to thousands of foreign antigens and millions of bacteria in the intestinal lumen, immunotolerance is maintained to prevent abnormal activation of inflammation. Disruption of homeostasis by any one of the mechanisms described previously in this review results in inflammation. All those disorders discussed in this review result from the activation of different immune system pathways, and their clinical, endoscopic, histological features overlap, creating confusion. One should think with an open mind and look for other possibilities, especially in patients not responding to conventional therapies. A detailed clinical history and examination would provide valuable information in identifying the underlying etiology. In most cases, additional radiological, endoscopic, and histopathological investigations will help in arriving at a correct diagnosis. It is sometimes difficult to pin‐point the diagnosis, which requires further sophisticated investigations like T‐ and B‐cell immunophenotyping, candidate gene analysis, and exome sequencing. Although most of these disorders are managed on similar lines with immunosuppressants, there are specific differences in management depending upon the underlying immune pathway involved. Investigations such as immunoglobulin profile, T‐ and B‐cell immunophenotyping, and functional neutrophil assays can help identify the underlying immunodeficiency. The recent addition of candidate gene sequencing and whole‐exome sequencing to the diagnostic armamentarium has helped in identifying the underlying defective immune pathways that can be targeted.
Conclusion
IBD is a prototype of immune‐mediated inflammatory diseases of the GI tract that share common pathophysiology and clinical manifestations with other disorders. These disorders closely mimic each other, causing significant dilemmas at times. Newer next‐generation sequencing methods have unravelled the key genetic mutations and immune pathways, which have helped to develop newer targeted therapies. The presence of atypical features such as very young age of onset, refractoryness to biologics, and involvement of other organ systems should raise the suspicion, and the diagnosis should be revisited. Immunoglobulin assays, lymphocyte profiling, and neutrophilic functional assays should be carried out in patients with suspected underlying immunodeficiency disorder. Whole‐exome/genome sequencing and targeted gene analysis should be reserved for cases with a strong suspicion of monogenic disorders with IBD‐like phenotype. Understanding the underlying immune mechanisms can help in identifying the etiology and individualize therapy.
Declaration of conflict of interest: None.
References
- 1. de Souza HSP, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016; 13: 13–27. [DOI] [PubMed] [Google Scholar]
- 2. Abdel‐Motal UM, Al‐Shaibi A, Elawad M, Lo B. Zero tolerance! A perspective on monogenic disorders with defective regulatory T cells and IBD‐like disease. Immunol. Rev. 2019; 287: 236–40. [DOI] [PubMed] [Google Scholar]
- 3. Garrett WS, Gordon JI, Glimcher LH. Homeostasis and Inflammation in the Intestine. Cell. 2010; 140: 859–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Graham DB, Xavier RJ. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature. 2020; 578: 527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park SC, Jeen YT. Genetic studies of inflammatory bowel disease‐focusing on asian patients. Cells. 2019; 8(5): 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nameirakpam J, Rikhi R, Rawat SS, Sharma J, Suri D. Genetics on early onset inflammatory bowel disease: an update. Genes Dis. 2020; 7: 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uhlig HH, Schwerd T. From genes to mechanisms: the expanding spectrum of monogenic disorders associated with inflammatory bowel disease. Inflamm. Bowel Dis. 2016; 22: 202–12. [DOI] [PubMed] [Google Scholar]
- 8. Ouahed J, Spencer E, Kotlarz D et al. Very early onset inflammatory bowel disease: a clinical approach with a focus on the role of genetics and underlying immune deficiencies. Inflamm. Bowel Dis. 2020; 26: 820–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kern JS, Herz C, Haan E et al. Chronic colitis due to an epithelial barrier defect: the role of kindlin‐1 isoforms. J. Pathol. 2007; 213: 462–70. [DOI] [PubMed] [Google Scholar]
- 10. Blaydon DC, Biancheri P, Di W‐L et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl. J. Med. 2011; 365: 1502–8. [DOI] [PubMed] [Google Scholar]
- 11. Miot C, Imai K, Imai C et al. Hematopoietic stem cell transplantation in 29 patients hemizygous for hypomorphic IKBKG/NEMO mutations. Blood. 2017; 130: 1456–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kammermeier J, Lucchini G, Pai S‐Y et al. Stem cell transplantation for tetratricopeptide repeat domain 7A deficiency: long‐term follow‐up. Blood. 2016; 128: 1306–8. [DOI] [PubMed] [Google Scholar]
- 13. Janecke AR, Heinz‐Erian P, Yin J et al. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum. Mol. Genet. 2015; 24: 6614–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Janecke AR, Heinz‐Erian P, Müller T. Congenital sodium diarrhea: a form of intractable diarrhea, with a link to inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 2016; 63: 170–6. [DOI] [PubMed] [Google Scholar]
- 15. Singh A. Cryptogenic multifocal ulcerating stenosing enteropathy (CMUSE) and/or chronic non‐specific multiple ulcers of the small intestine (CNSU) and non‐granulomatous ulcerating jejunoileitis (NGUJI). Curr. Gastroenterol. Rep. 2019; 21: 53. [DOI] [PubMed] [Google Scholar]
- 16. Umeno J, Esaki M, Hirano A et al. Clinical features of chronic enteropathy associated with SLCO2A1 gene: a new entity clinically distinct from Crohn's disease. J. Gastroenterol. 2018; 53: 907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang H, Wang X, Ou D et al. Four variants of SLCO2A1 identified in three chinese patients with chronic enteropathy associated with the SLCO2A1 gene. Dig. Dis. Sci. 2021; 66: 2992–3001. [DOI] [PubMed] [Google Scholar]
- 18. Hosoe N, Ohmiya N, Hirai F et al. Chronic enteropathy associated with SLCO2A1 gene [CEAS]‐characterisation of an enteric disorder to be considered in the differential diagnosis of Crohn's disease. J. Crohns Colitis. 2017; 11: 1277–81. [DOI] [PubMed] [Google Scholar]
- 19. Wang Q, Li Y‐H, Lin G et al. Primary hypertrophic osteoarthropathy related gastrointestinal complication has distinctive clinical and pathological characteristics: two cases report and review of the literature. Orphanet J. Rare Dis. 2019; 14: 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang J, Kim JS, Kim AY et al. Cryptogenic multifocal ulcerous stenosing enteritis: radiologic features and clinical behavior. World J. Gastroenterol. 2017; 23: 4615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahonen P, Myllärniemi S, Sipilä I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy‐candidiasis‐ectodermal dystrophy (APECED) in a series of 68 patients. N. Engl. J. Med. 1990; 322: 1829–36. [DOI] [PubMed] [Google Scholar]
- 22. Glocker E, Grimbacher B. Inflammatory bowel disease: is it a primary immunodeficiency? Cell. Mol. Life Sci. 2012; 69: 41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glocker E‐O, Speckmann C. Chapter 26 ‐ autoinflammatory diseases predominantly affecting the gastrointestinal tract. In: Sullivan KE, Stiehm ER (eds). Stiehm's Immune Deficiencies. Amsterdam: Academic Press, 2014; 573–84. [Google Scholar]
- 24. Hartono S, Ippoliti MR, Mastroianni M, Torres R, Rider NL. Gastrointestinal disorders associated with primary immunodeficiency diseases. Clin Rev Allergy Immunol. 2019; 57: 145–65. [DOI] [PubMed] [Google Scholar]
- 25. Pazmandi J, Kalinichenko A, Ardy RC, Boztug K. Early‐onset inflammatory bowel disease as a model disease to identify key regulators of immune homeostasis mechanisms. Immunol. Rev. 2019; 287: 162–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park JH, Lee KH, Jeon B et al. Immune dysregulation, polyendocrinopathy, enteropathy, X‐linked (IPEX) syndrome: a systematic review. Autoimmun. Rev. 2020; 19: 102526. [DOI] [PubMed] [Google Scholar]
- 27. Chen CB, Tahboub F, Plesec T, Kay M, Radhakrishnan K. A review of autoimmune enteropathy and its associated syndromes. Dig. Dis. Sci. 2020; 65: 3079–90. [DOI] [PubMed] [Google Scholar]
- 28. Lo B, Fritz JM, Su HC, Uzel G, Jordan MB, Lenardo MJ. CHAI and LATAIE: new genetic diseases of CTLA‐4 checkpoint insufficiency. Blood. 2016; 128: 1037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ohya T, Yanagimachi M, Iwasawa K et al. Childhood‐onset inflammatory bowel diseases associated with mutation of Wiskott‐Aldrich syndrome protein gene. World J. Gastroenterol. 2017; 23: 8544–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012; 5: 354–66. [DOI] [PubMed] [Google Scholar]
- 31. Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore). 2000; 79: 170–200. [DOI] [PubMed] [Google Scholar]
- 32. Marciano BE, Rosenzweig SD, Kleiner DE et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004; 114: 462–8. [DOI] [PubMed] [Google Scholar]
- 33. Winkelstein JA, Marino MC, Johnston RB et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore). 2000; 79: 155–69. [DOI] [PubMed] [Google Scholar]
- 34. Roos D, Kuhns DB, Maddalena A et al. Hematologically important mutations: X‐linked chronic granulomatous disease (third update). Blood Cells Mol. Dis. 2010; 45: 246–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Leusse A, Dupuy E, Huizing M et al. Ileal Crohn's disease in a woman with Hermansky‐Pudlak syndrome. Gastroenterol. Clin. Biol. 2006; 30: 621–4. [DOI] [PubMed] [Google Scholar]
- 36. Girot P, Berre CL, Maissin AD, Freyssinet M, Trang‐Poisson C, Bourreille A. Crohn's‐like acute severe colitis associated with Hermansky‐Pudlak syndrome: a case report. World J. Gastroenterol. 2019; 25: 1031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zeissig Y, Petersen B‐S, Milutinovic S et al. XIAP variants in male Crohn's disease. Gut. 2015; 64: 66–76. [DOI] [PubMed] [Google Scholar]
- 38. Bolton C, Burch N, Morgan J et al. Remission of inflammatory bowel disease in glucose‐6‐phosphatase 3 deficiency by allogeneic haematopoietic stem cell transplantation. J. Crohns Colitis. 2020; 14: 142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Desplantes C, Fremond ML, Beaupain B et al. Clinical spectrum and long‐term follow‐up of 14 cases with G6PC3 mutations from the French Severe Congenital Neutropenia Registry. Orphanet J. Rare Dis. 2014; 10: 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dupuis‐Girod S, Medioni J, Haddad E et al. Autoimmunity in Wiskott‐Aldrich syndrome: risk factors, clinical features, and outcome in a single‐center cohort of 55 patients. Pediatrics. 2003; 111(5 Pt 1): e622–7. [DOI] [PubMed] [Google Scholar]
- 41. Ozen A, Comrie WA, Ardy RC et al. CD55 deficiency, early‐onset protein‐losing enteropathy, and thrombosis. N. Engl. J. Med. 2017; 377: 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Singh P, Arora A, Strand TA et al. Global prevalence of celiac disease: systematic review and meta‐analysis. Clin. Gastroenterol. Hepatol. 2018; 16: 823–836.e2. [DOI] [PubMed] [Google Scholar]
- 43. Lundin KE, Scott H, Hansen T et al. Gliadin‐specific, HLA‐DQ(alpha 1*0501,beta 1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J. Exp. Med. 1993; 178: 187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schuppan D. Current concepts of celiac disease pathogenesis. Gastroenterology. 2000; 119: 234–42. [DOI] [PubMed] [Google Scholar]
- 45. Volta U, Caio G, Stanghellini V, De Giorgio R. The changing clinical profile of celiac disease: a 15‐year experience (1998‐2012) in an Italian referral center. BMC Gastroenterol. 2014; 18: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lebwohl B, Rubio‐Tapia A. Epidemiology, presentation, and diagnosis of celiac disease. Gastroenterology. 2021; 160: 63–75. [DOI] [PubMed] [Google Scholar]
- 47. Pinto‐Sanchez MI, Seiler CL, Santesso N et al. Association between inflammatory bowel diseases and celiac disease: a systematic review and meta‐analysis. Gastroenterology. 2020; 159: 884–903.e31. [DOI] [PubMed] [Google Scholar]
- 48. van Gils T, Nijeboer P, van Wanrooij RL, Bouma G, Mulder CJJ. Mechanisms and management of refractory coeliac disease. Nat. Rev. Gastroenterol. Hepatol. 2015; 12: 572–9. [DOI] [PubMed] [Google Scholar]
- 49. Jennette JC, Falk RJ, Bacon PA et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013; 65: 1–11. [DOI] [PubMed] [Google Scholar]
- 50. Shaikh FM, Sabu CB, Peirce TH, Naqvi SA. Extensive intestinal ischaemic necrosis in Wegener's granulomatosis. Gut. 2006; 55: 1368–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Geraghty J, Mackay IR, Smith DC. Intestinal perforation in Wegener's granulomatosis. Gut. 1986; 27: 450–1, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haworth SJ, Pusey CD. Severe intestinal involvement in Wegener's granulomatosis. Gut. 1984; 25: 1296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guillevin L, Lhote F, Gayraud M et al. Prognostic factors in polyarteritis nodosa and Churg‐Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore). 1996. Jan; 75(1): 17–28. [DOI] [PubMed] [Google Scholar]
- 54. Soowamber M, Weizman AV, Pagnoux C. Gastrointestinal aspects of vasculitides. Nat. Rev. Gastroenterol. Hepatol. 2017; 14: 185–94. [DOI] [PubMed] [Google Scholar]
- 55. Ha HK, Lee SH, Rha SE et al. Radiologic features of vasculitis involving the gastrointestinal tract. Radiographics. 2000; 20: 779–94. [DOI] [PubMed] [Google Scholar]
- 56. Chetty R, Serra S. A pragmatic approach to vasculitis in the gastrointestinal tract. J. Clin. Pathol. 2017; 70: 470–5. [DOI] [PubMed] [Google Scholar]
- 57. Noth I, Strek ME, Leff AR. Churg‐Strauss syndrome. Lancet. 2003; 361: 587–94. [DOI] [PubMed] [Google Scholar]
- 58. Yazısız V. Similarities and differences between Behçet's disease and Crohn's disease. World J. Gastrointest. Pathophysiol. 2014; 5: 228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mizuki N, Ohno S, Ando H et al. A strong association between HLA‐B*5101 and Behçet's disease in Greek patients. Tissue Antigens. 1997; 50: 57–60. [DOI] [PubMed] [Google Scholar]
- 60. Ebert EC. Gastrointestinal manifestations of Behçet's disease. Dig. Dis. Sci. 2009; 54: 201–7. [DOI] [PubMed] [Google Scholar]
- 61. Skef W, Hamilton MJ, Arayssi T. Gastrointestinal Behçet's disease: a review. World J. Gastroenterol. 2015; 21: 3801–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. International Team for the Revision of the International Criteria for Behçet's Disease (ITR‐ICBD) . The International Criteria for Behçet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J. Eur. Acad. Dermatol. Venereol. 2014; 28: 338–47. [DOI] [PubMed] [Google Scholar]
- 63. Criteria for diagnosis of Behçet's disease . International study group for Behçet's disease. Lancet. 1990; 335: 1078–80. [PubMed] [Google Scholar]
- 64. Hisamatsu T, Hayashida M. Treatment and outcomes: medical and surgical treatment for intestinal Behçet's disease. Intest Res. 2017; 15: 318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Esaki M, Matsumoto T, Nakamura S et al. GI involvement in Henoch‐Schönlein purpura. Gastrointest. Endosc. 2002; 56: 920–3. [DOI] [PubMed] [Google Scholar]
- 66. Pineton de Chambrun G, Dufour G, Tassy B et al. Diagnosis, natural history and treatment of eosinophilic enteritis: a review. Curr. Gastroenterol. Rep. 2018; 20: 37. [DOI] [PubMed] [Google Scholar]
- 67. Pineton de Chambrun G, Gonzalez F, Canva J‐Y et al. Natural history of eosinophilic gastroenteritis. Clin. Gastroenterol. Hepatol. 2011; 9: 950–956.e1. [DOI] [PubMed] [Google Scholar]
- 68. Talley NJ, Shorter RG, Phillips SF, Zinsmeister AR. Eosinophilic gastroenteritis: a clinicopathological study of patients with disease of the mucosa, muscle layer, and subserosal tissues. Gut. 1990; 31: 54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J. Allergy Clin. Immunol. 2004; 113: 11–28. [DOI] [PubMed] [Google Scholar]
- 70. Klein NC, Hargrove RL, Sleisenger MH, Jeffries GH. Eosinophilic gastroenteritis. Medicine (Baltimore). 1970; 49: 299–319. [DOI] [PubMed] [Google Scholar]
- 71. Rienzo‐Madero B d, Kajomovitz‐Bialostozky D, Pelaez‐Luna M, Quijano‐Orvananos F. Mo1361 – eosinophilic enteritis presenting as intestinal obstruction: a case report. Gastroenterology. 2019; 156: S–1464. [DOI] [PubMed] [Google Scholar]
- 72. Biswas S, Hoo W, Katsoulas N, Munro J, Oke O. Eosinophilic enteritis: a rare cause of abdominal pain. Int. J. Colorectal Dis. 2007; 22: 87–8. [DOI] [PubMed] [Google Scholar]
- 73. Abou Rached A, El Hajj W. Eosinophilic gastroenteritis: approach to diagnosis and management. World J. Gastrointest. Pharmacol. Ther. 2016; 7: 513–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dellon ES, Peterson KA, Murray JA et al. 932 histologic and symptomatic improvement across multiple forms of eosinophilic gastrointestinal diseases in enigma, a randomized, double‐blind, placebo‐controlled trial of antolimab (AK002). Gastroenterology. 2020; 158: S‐186. [Google Scholar]
- 75. Gonsalves N, Doerfler B, Zalewski A et al. 229 results from the element study: prospective study of elemental diet in eosinophilic gastroenteritis nutrition trial. Gastroenterology. 2020; 158: S–43. [Google Scholar]
- 76. Khalili H, Bergman D, Roelstraete B et al. Mortality of Patients With Microscopic Colitis in Sweden. Clin. Gastroenterol. Hepatol. 2020; 18: 2491–2499.e3. [DOI] [PubMed] [Google Scholar]
- 77. Burke KE, D'Amato M, Ng SC, Pardi DS, Ludvigsson JF, Khalili H. Microscopic colitis. Nat. Rev. Dis. Primers. 2021; 7: 1–17. [DOI] [PubMed] [Google Scholar]
- 78. Morgan DM, Cao Y, Miller K et al. Microscopic Colitis Is Characterized by Intestinal Dysbiosis. Clin. Gastroenterol. Hepatol. 2020; 18: 984–6. [DOI] [PubMed] [Google Scholar]
- 79. Virine B, Chande N, Driman DK. Biopsies from ascending and descending colon are sufficient for diagnosis of microscopic colitis. Clin. Gastroenterol. Hepatol. 2020; 18: 2003–9. [DOI] [PubMed] [Google Scholar]
- 80. Lee E, Schiller LR, Vendrell D, Ana CAS, Fordtran JS. Subepithelial collagen table thickness in colon specimens from patients with microscopic colitis and collagenous colitis. Gastroenterology. 1992; 103: 1790–6. [DOI] [PubMed] [Google Scholar]
- 81. Green PHR, Yang J, Cheng J, Lee AR, Harper JW, Bhagat G. An Association Between Microscopic Colitis and Celiac Disease. Clin. Gastroenterol. Hepatol. 2009; 7: 1210–6. [DOI] [PubMed] [Google Scholar]
- 82. Kamp EJCA, Kane JS, Ford AC. Irritable bowel syndrome and microscopic colitis: a systematic review and meta‐analysis. Clin. Gastroenterol. Hepatol. 2016; 14: 659–668.e1. [DOI] [PubMed] [Google Scholar]
- 83. Kane JS, Rotimi O, Everett SM, Samji S, Michelotti F, Ford AC. Development and validation of a scoring system to identify patients with microscopic colitis. Clin. Gastroenterol. Hepatol. 2015; 13: 1125–31. [DOI] [PubMed] [Google Scholar]
- 84. Miehlke S, Heymer P, Bethke B et al. Budesonide treatment for collagenous colitis: a randomized, double‐blind, placebo‐controlled, multicenter trial. Gastroenterology. 2002; 123: 978–84. [DOI] [PubMed] [Google Scholar]
- 85. Stewart MJ, Seow CH, Storr MA. Prednisolone and budesonide for short‐ and long‐term treatment of microscopic colitis: systematic review and meta‐analysis. Clin. Gastroenterol. Hepatol. 2011; 9: 881–90. [DOI] [PubMed] [Google Scholar]
- 86. Pardi DS, Loftus EV, Tremaine WJ, Sandborn WJ. Treatment of refractory microscopic colitis with azathioprine and 6‐mercaptopurine. Gastroenterology. 2001; 120: 1483–4. [DOI] [PubMed] [Google Scholar]
- 87. Sansom DM. CD28, CTLA‐4 and their ligands: who does what and to whom? Immunology. 2000; 101: 169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Quezada SA, Peggs KS. Exploiting CTLA‐4, PD‐1 and PD‐L1 to reactivate the host immune response against cancer. Br. J. Cancer. 2013; 108: 1560–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Cañete F, Mañosa M, Lobatón T et al. Nivolumab‐induced immune‐mediated colitis: an ulcerative colitis look‐alike‐report of new cases and review of the literature. Int. J. Colorectal Dis. 2019; 34: 861–5. [DOI] [PubMed] [Google Scholar]
- 90. Soularue E, Lepage P, Colombel JF et al. Enterocolitis due to immune checkpoint inhibitors: a systematic review. Gut. 2018; 67: 2056–67. [DOI] [PubMed] [Google Scholar]
- 91. Abu‐Sbeih H, Wang Y. Management Considerations for Immune Checkpoint Inhibitor–Induced Enterocolitis Based on Management of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2020; 26: 662–8. [DOI] [PubMed] [Google Scholar]
- 92. Bellaguarda E, Hanauer S. Checkpoint inhibitor–induced colitis. Am. J. Gastroenterol. 2020; 115: 202–10. [DOI] [PubMed] [Google Scholar]
- 93. Zhou X, Yao Z, Yang H, Liang N, Zhang X, Zhang F. Are immune‐related adverse events associated with the efficacy of immune checkpoint inhibitors in patients with cancer? A systematic review and meta‐analysis. BMC Med. 2020; 18: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Frankel AE, Deshmukh S, Reddy A et al. Cancer immune checkpoint inhibitor therapy and the gut microbiota. Integr. Cancer Ther. 2019; 18: 1534735419846379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Abu‐Sbeih H, Wang Y. Gut microbiome and immune checkpoint inhibitor‐induced enterocolitis. Dig. Dis. Sci. 2020; 65: 797–9. [DOI] [PubMed] [Google Scholar]
- 96. Bergqvist V, Hertervig E, Gedeon P et al. Vedolizumab treatment for immune checkpoint inhibitor‐induced enterocolitis. Cancer Immunol. Immunother. 2017; 66: 581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ibraheim H, Baillie S, Samaan MA et al. Systematic review with meta‐analysis: effectiveness of anti‐inflammatory therapy in immune checkpoint inhibitor‐induced enterocolitis. Aliment. Pharmacol. Ther. 2020; 52: 1432–52. [DOI] [PubMed] [Google Scholar]
- 98. Wang Y, Ma W, Abu‐Sbeih H, Jiang Z‐D, DuPont HL. Fecal microbiota transplantation (FMT) for immune checkpoint inhibitor induced–colitis (IMC) refractory to immunosuppressive therapy. J. Clin. Oncol. 2020; 38(15_suppl): 3067–7. [Google Scholar]
- 99. Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011; 305: 391–9. [DOI] [PubMed] [Google Scholar]
- 100. Mayock RL, Bertrand P, Morrison CE, Scott JH. Manifestations of sarcoidosis. Analysis of 145 patients, with a review of nine series selected from the literature. Am. J. Med. 1963; 35: 67–89. [DOI] [PubMed] [Google Scholar]
- 101. Brito‐Zerón P, Bari K, Baughman RP, Ramos‐Casals M. Sarcoidosis involving the gastrointestinal tract: diagnostic and therapeutic management. Am. J. Gastroenterol. 2019; 114: 1238–47. [DOI] [PubMed] [Google Scholar]