

Abstract
A core challenge in biomaterials, with both fundamental significance and technological relevance, concerns the rational design of bioactive microenvironments. Designed properly, peptides can undergo supramolecular assembly into dynamic, physical hydrogels that mimic the mechanical, topological, and biochemical features of native tissue microenvironments. The relatively facile, inexpensive, and automatable preparation of peptides coupled with low batch-to-batch variability motivates the expanded use of assembling peptide hydrogels for biomedical applications. Integral to realizing dynamic peptide assemblies as functional biomaterials for tissue engineering is an understanding of the molecular and macroscopic features that govern assembly, morphology, and biological interactions. In this review, we first discuss the design of assembling peptides, including primary structure (sequence), secondary structure (e.g., α-helix and β-sheets), and molecular interactions that facilitate assembly into multiscale materials with desired properties. Next, we describe characterization tools for elucidating molecular structure and interactions, morphology, bulk properties, and biological functionality. Understanding of these characterization methods enables researchers to access a variety of approaches in this ever-expanding field. Finally, we discuss the biological properties and applications of peptide-based biomaterials for engineering several important tissues. By connecting molecular features and mechanisms of assembling peptides to the material and biological properties, we aim to guide the design and characterization of peptide-based biomaterials for tissue engineering and regenerative medicine.
Keywords: Biomaterials, hydrogels, tissue engineering, peptide, assembly, supramolecular interactions
Graphical abstract
1. Introduction
Whether from biological or synthetic origins, peptides are short segments of linked amino acids whose primary sequence dictates their structure and function. Noncovalent electrostatic, hydrophobic, hydrogen bonding, and pi-pi stacking interactions among the amino acid building blocks, or residues, govern association of individual peptides into dynamic supramolecular structures, such as nanoscale tubes, spheres, ribbons, and fibers [1]. Peptide-based tissue-mimetic hydrogels typically form via intermolecular forces between, and/or entanglement of, nanoscale fibers, which are key topographical features in native extracellular matrix (ECM) microenvironments. Furthermore, assembling peptides direct integration of biocompatible polymers into composite hydrogels, in which both the polymer and peptide components are tunable [2–4]. Unentangled, non-gelling fibers are also useful in the context of regenerative medicine, for example in displaying targeting groups that engage the immune system [5,6]. When designing for biomedical applications, peptides must typically assemble under cytocompatible conditions, such as neutral pH, physiological salt concentrations, and body temperature. The assembly mechanism and resulting properties guide, and sometimes even motivate, the potential uses in vitro and in vivo.
Synthetic peptides are attractive components of biomaterials as they recapitulate topological, chemical, and biological properties of natural proteins, while offering scalable, robust, and economical synthesis. Despite lacking the complex tertiary and quaternary structure of natural proteins, peptides capably direct interactions with cells and facilitate cell-mediated protease remodeling and degradation. Solid-phase peptide synthesis (SPPS) enables the preparation of tailor-made peptide sequences exceeding 50 amino acids in length without the use of complex purification steps associated with protein production [7,8]. Peptides produced through SPPS are chemically defined, and thus amenable to modification and rigid quality control standards. Decoration of peptides during or after SPPS with other functional groups enables assembly, visualization, and biological interactions. Tissue engineers are now able to easily synthesize a whole host of de novo peptides with unique structural and biofunctional properties, including but not limited to proteolytic susceptibility [9–11], cell surface and/or matrix binding [12,13], growth factor binding [14,15], and supramolecular assembly [16–20].
For tissue engineering, peptide-based biomaterials must not only promote cell survival and growth, but also help guide cell behavior. With high water content (typically > 95%), peptide-based hydrogels support diffusion of nutrients, growth factors, and biochemical signals, as well as migration and growth of cells analogous to that in biological tissues. Hydrogel-based tissue engineering resolves some of the numerous difficulties in studying cells and tissues in vivo, such as the inability to determine causal factors due to the high complexity of the environment. Additionally, high interconnectivity between cells and the surrounding extracellular matrix (ECM) in vivo can convolute the roles of specific molecules or proteins. Since biomaterial features can impact epigenetic regulation, metabolic activity [21], protein synthesis, cellular proliferation [22–25], and stem cell differentiation [26–30], among other cellular functions, their characterization is critically important when creating new treatments and therapies.
Peptide-based biomaterials can be designed to create physical hydrogels via non-covalently interacting motifs, allowing the material to flow, and then recover after shear. This shear recovery enables minimally invasive procedures and 3D printing-based tissue manufacturing. While stem cell transplantation is a promising clinical strategy in tissue regenerative therapies, transplantation strategies often suffer from poor cell survival. Hydrogels improve the survival of these cells after needle injection by reducing cell membrane disruption from mechanical forces [31,32].
Peptide materials are well poised to promote regeneration as they meet numerous design criteria: they are easily deliverable via needle, match the compliancy of native tissue, allow infusion of growth factors and other nutrients, and include biological signals to promote cell adhesion, migration, and growth. With tunable supramolecular and biomolecular interactions, the potential of engineered peptide materials as bioactive matrices to promote transplanted or endogenous stem cell activity is immense.
The development of new assembling peptide biomaterials will help meet the demand for tissue-like hydrogels that do not require chemical crosslinking mechanisms and have predictable and reproducible biological effects. This review addresses a core scientific consideration—namely, the intra- and intermolecular interactions dictating peptide structure, matrix formation, and ultimately, biomedical impact. For each of the major classes of assembling peptides, namely β-sheet-forming, helical, and amphiphilic peptides, block copolypeptides, and elastin-like peptide structures, we describe how the primary structure and interactions between peptides and their environments mediates their assembly into useful biomaterials. We present methods to characterize peptide hydrogels and their assembly, including tools for studying the molecular-scale structure and interactions (spectroscopy and calorimetry), nano- and micro-scale morphology (microscopy), and bulk macroscopic properties (rheology). In depth understanding of characterization methods provides newcomers to the peptide material field with a wide array of useful strategies that can answer a variety of specific and useful questions. We then describe several common peptide materials in tissue engineering and regenerative medicine for example applications in brain, muscle, heart, and immunoengineering. Finally, we explore synergistic combinations of assembling peptides and polymers in biomedical applications. Together, these engineered peptide-based materials can help bridge the translational “valley of death” between bench research and bedside medical treatment through their designer, yet bio-inspired, function.
2. Classes of peptide assemblies
In the sections that follow we describe several classes of assembling peptide systems, including amphipathic beta sheets, short aromatic beta sheets, metal ion-induced beta sheets, helices, peptide amphiphiles, block copolypeptides, and elastin-like peptides (Table 1).
Table 1.
Summary of the classes of assembling peptides reviewed here, including the peptide family, the class of assembling peptide to which the family belongs, and selected key references.
| Peptide family | Type | Examples sequences | Ref. |
|---|---|---|---|
| EAK16 | Amphipathic β-sheet | Ac-(AEAEAKAK)2-NH2 | 16, 38 |
| RADA16 | Amphipathic β-sheet | Ac-(RARADADA)2-NH2 | 38, 44, 45 |
| KFE | Amphipathic β-sheet | Ac-(FKFE)3-NH2 | 33, 88, 89 |
| MAX | Amphipathic β-sheet | Ac-VKVKVKVKVDPPTKVKVKVKV-NH2 | 17, 18, 48, 49 |
| E1Y9 | Amphipathic β-sheet | Ac-EYEYKYEYKY-NH2 | 50, 51 |
| Q11 | Amphipathic β-sheet | Ac-QQKFQFQFEQQ-NH2 | 52–55 |
| Diphenylalanine | Short aromatic β-sheet | Ac-FF-COOH | 69, 70, 71,73, 79 |
| Tripeptides | Short aromatic β-sheet | YFD, FYD, DFY, DYF, KYF, KYY, KFF, KYW | 80–82 |
| RAPID | Short aromatic β-sheet | KYFIL, AYFIL, KYFAL | 32, 83 |
| Metal-ion binding | Metal-ion induced β-sheet | Pyrene-CGPC, ILVAGH, GQ(PHGGGWQG)4CG | 35, 97–99 |
| Coiled coils | Helical | Ac-GYK(IAALKEK)2-IAALKEG-NH2, Ac-GYE(IAALEKE)2-IAALEKG-NH2 | 27, 105, 108–111, 114, 115 |
| Collagen-like | Helical | Ac-(GPO)8-GG-NH2, Ac-(GPO)3-GAO-(GPO)4-GG-NH2 | 139–142 |
| Peptide amphiphiles | Lipid-tail β-sheet | Lipid-CCCCGGGJRGD, Lipid-GGGGGGGERGDS | 166–168, 171,172 |
| Block copolypeptides | Multiple | K180L20, K180L40K180 | 181–184 |
| Elastin-like peptides | Intrinsically disordered | VPGPG | 188–190, 192, 193, 201,202 |
2.1. Beta sheet assemblies
β-sheets are naturally occurring structural motifs in which interchain hydrogen bonding facilitates formation of flat peptide sheets or ribbons in solution. Higher order assembly of β-sheets yields fibers and fibrous hydrogels, as well as other hierarchical structures, like the intricate β-barrels found in proteins. Within β-sheets, the side chains of the constituent amino acids extend above and below the plane of the peptide backbone in an alternating fashion. While β-strands, the individual peptide segments comprising β-sheets, are often distant from each other in protein sequences, they interact upon protein folding. Peptides that form β-sheets commonly aggregate in aqueous solution due to their amphipathic nature and form central biomimetic structural components of biomaterials. Studies of β-sheet peptide assemblies contribute substantially to our understanding of both molecular assembly and disease states involving protein aggregation [33–37]. Here, we review the common classes of assembling β-sheet peptides used in biomaterials, noting the origin, sequence features, and environmental effects on these assemblies.
2.1.1. Amphipathic self-assembling peptides (SAPs)
Pioneered by Zhang and coworkers in the early 1990s [16,38–40], amphipathic peptides alternate hydrophobic and hydrophilic amino acids and are among the most widely utilized motifs for synthetic self-assembling β-sheet peptides. In aqueous solution, hydrophobic and electrostatic interactions commonly yield staggered arrangements of peptides (Figure 1A) conducive to dynamic hierarchical assembly. Individual peptides assemble into fibrils (Figure 1B, C), which in turn assemble into macroscopic hydrogels (Figure 1D) through non-covalent fibril interaction and entanglement. Experimentally, diluting concentrated peptide solutions into buffer at a pH and salt concentration that neutralizes charged amino acids typically promotes assembly. The extensive development of amphipathic SAPs has yielded a wide range of sequences, described next, and resulted in the commercialization of these biomimetic materials for cell culture and tissue engineering [41–43].
Figure 1.
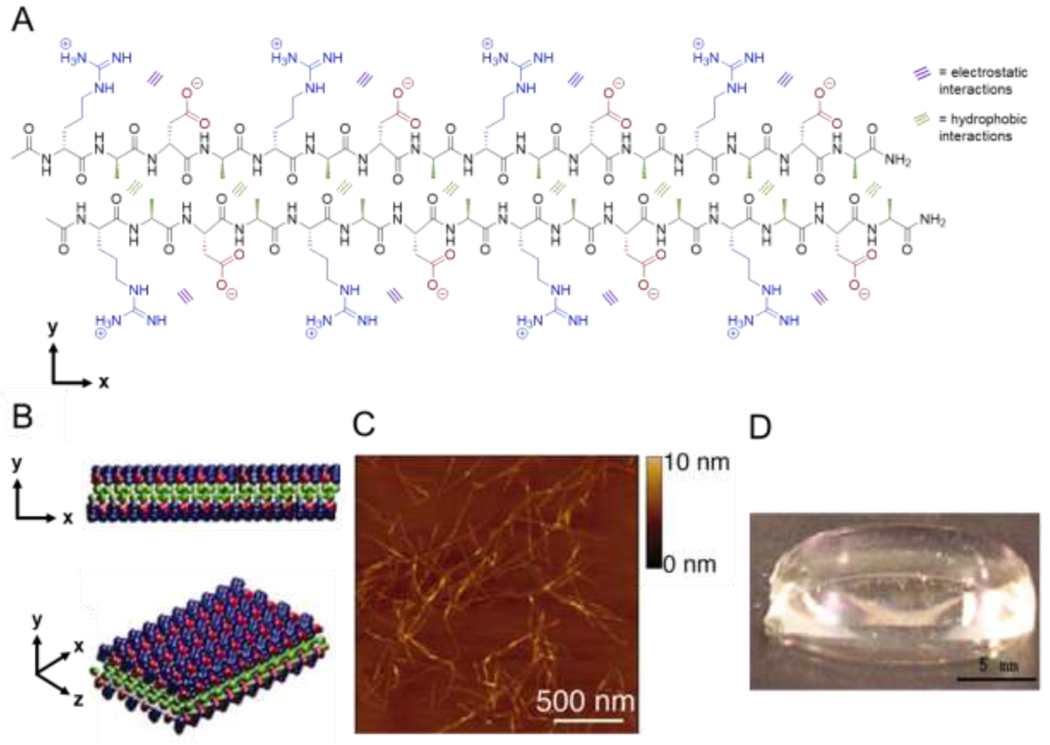
Assembly of amphipathic β-sheet peptides. (A) Intermolecular hydrogen bonding on the peptide backbone, hydrophobic interactions between alanine residues (green), and electrostatic interactions between aspartic acid (red) and arginine (blue) residues direct assembly of the amphipathic peptide RADA16 in aqueous solution. (B) RADA16 nanofibers assemble through hydrophobic interactions of alanine residues between peptides in the core of the nanofiber and electrostatic interactions between the positively and negatively charged side chains of arginine and aspartic acid residues in a checkerboard pattern on the nanofiber surface. (C) Atomic force microscopy image of assembled nanofibers. (D) Macroscopic image of a RADA16 hydrogel formed at 0.1 wt% in PBS at pH 7.5. Adapted with permission from Yokoi et al. [44], Copyright (2005) National Academy of Sciences, U.S.A.
2.1.1.1. EAK16
Discovered in the yeast protein zuotin, the first reported β-sheet amphipathic peptide, EAK16 (AEAEAKAKAEAEAKAK), is ionically self-complementary, with an equal number of cationic lysine (K) and anionic glutamic acid (E) residues alternating with hydrophobic alanine (A) residues [16]. As is common in synthetic peptides derived from natural proteins, EAK16 features an acetylated (Ac) N terminus and an amidated C terminus, indicated as −NH2, to more closely reproduce the sequence as it appears in the protein, i.e., without charges at the termini. Following the addition of salt to an aqueous solution of the peptide, Ac-EAK16-NH2 spontaneously assembles into what was originally termed a stable macroscopic membrane, and later referred to as a hydrogel, composed of interwoven fibrils 10-20 nm in diameter. These hydrogels remain stable at low peptide concentrations (0.6 μM) in a variety of conditions - in the presence of enzymes, heat, and in solutions with pH ranging from 1.5 to 11, and in protein-denaturing solutions of guanidine hydrochloride, sodium dodecyl sulfate (SDS), and urea [16,38]. The remarkable stability of these assemblies is attributed to the combination of electrostatic and hydrophobic interactions [16]. Thereafter, mimicking the molecular design features of EAK led to the development of the RADA16, KFE, MAX, and Q11 self-assembling β-sheet peptides, among others, for biomedical applications.
2.1.1.2. RADA16
Substitution of the lysine and glutamic acid residues in EAK16 with arginine (R) and aspartic acid (D), respectively, yields the class of SAPs known as RAD16 or RADA16 [45]. Similarly to EAK16, RADA16 spontaneously forms stable hydrogels in physiological buffer [38]. Developed to mimic the cell-adhesive peptide motif RGD (G = glycine), RADA16-based biomaterials successfully support attachment of various mammalian cell type [45]. Further, the non-covalent interactions comprising these assemblies impart dynamic shear-thinning and self-healing behavior. RADA16 nanofibers sonicated into smaller fragments reassemble into structures identical to the starting material within 2 hours [44]. Together, the favorable cell attachment properties, high stability, and biocompatibility enabled successful commercialization of RAD16 as PuraMatrix™ peptide hydrogels [43].
2.1.1.3. KFE
Lauffenburger and coworkers first designed KFE12, or (FKFE)3, where F = phenylalanine, as a model amphipathic peptide to probe the fundamental driving forces controlling assembly [33]. Early experiments reduced electrostatic repulsion by increasing ionic strength, allowing hydrophobic interactions to dominate and peptide assembly to occur above a threshold salt concentration. Derjaguin-Landau-Verwey-Overbeek (DLVO) theory, which models the interplay between hydrophobic (van der Waals) forces and electrostatic repulsion, accurately predicts both the threshold salt concentration and assembly of KFE12 in the absence of salt at pH 7.0, where it is charge-neutral [33]. KFE also forms rippled β-sheets (Section 2.1.3) and serves as a model SAP in vaccine adjuvants [46,47].
2.1.1.4. MAX
Schneider, Pochan, and coworkers developed amphipathic peptides composed of β-hairpins, or two interacting β-strands connected by a short turn sequence, that form β-sheet fibrous hydrogels [17]. MAX1 contains alternating hydrophobic valine and hydrophilic, cationic lysine residues in the β-strands, which are connected by the turn sequence VDPPT, where V = valine, a hydrophobic residue, DP = d-proline, P = proline, and T = threonine. At pH 7.4 in aqueous solution, MAX1 adopts a random coil conformation. Raising the pH to 9.0 partially neutralizes the lysine side chains, reducing intrastrand charge repulsion and inducing β-hairpin formation, as observed by circular dichroism and infrared spectroscopy. Reducing pH reverts the peptide to a random coil, affording pH-dependent reversible assembly. MAX1 assembly may also be salt-induced [18], similar to other amphipathic SAPs, with gelation occurring at pH 7.4 and salt concentrations as low as 20 mM. Folded MAX1 hairpins have a valine-rich hydrophobic face and a lysine-rich hydrophilic face; association of the hydrophobic faces leads to bilayer formation, while lateral assembly into fibrils occurs through hydrogen bonding and van der Waals interactions. The resulting fibrils are ca. 3 nm thick, consistent with the length of a hairpin and the predicted strand axis [18]. Via interactions between fibrils (hydrogen bonding and hydrophobic interactions) and fibril entanglement, these fibrils form viscoelastic hydrogels with temperature-and salt-dependent moduli and shear-thinning and self-healing properties. In a sequence termed ‘MAX8’, an anionic glutamic acid replaces one of the MAX1 cationic lysines, and the reduced net charge accelerates gelation [48,49]. Oscillatory rheology and circular dichroism spectroscopy confirm that solutions of MAX8 form β-sheet hydrogels with shear storage moduli G’ of ca. 100 Pa in under a minute upon addition of cell culture media at physiological salt concentration and pH 7.4. In comparison, the onset of β-sheet formation for MAX1 requires ca. 30 min and the resulting hydrogels exhibit a G’ of only 40 Pa after 60 minutes. Salt-induced assembly is a potentially useful alternative to pH-mediated assembly given the known cytotoxic impacts of pH values significantly lower or higher than 7.4. The fast MAX8 gelation kinetics coupled with shear-thinning and self-healing properties allow for homogenous mesenchymal stem cell and hepatocyte encapsulation and good survival after injection (Figure 2 [48]).
Figure 2.
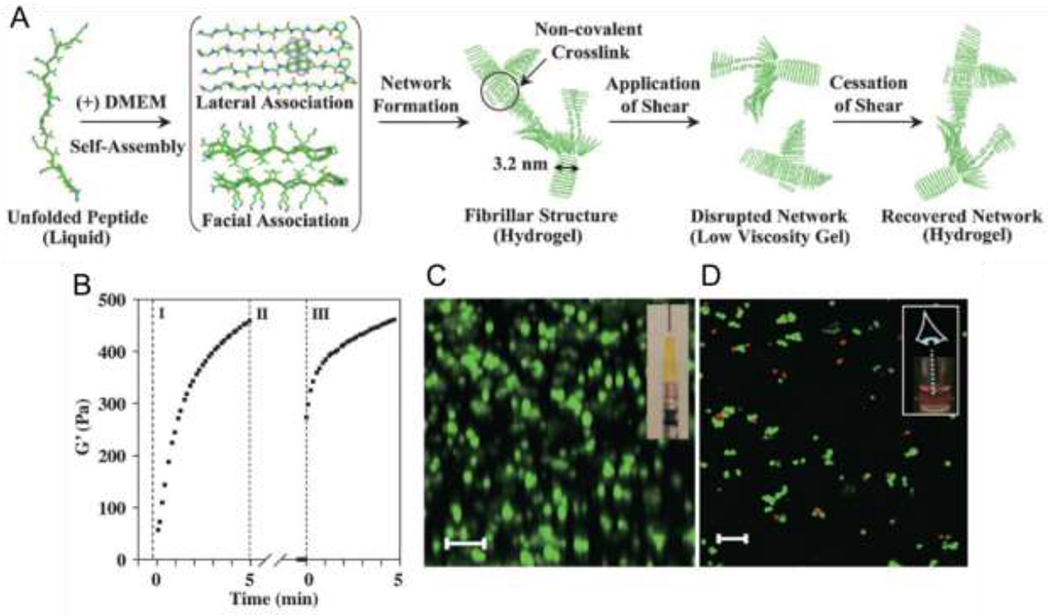
Assembly, hydrogel formation, shear-thinning, and self-healing of MAX8. A) Unfolded MAX8 assembles into β-hairpins that form fibrillar hydrogels upon the addition of salt to aqueous peptide solution. Applied shear disrupts hydrogel networks, which reassemble upon cessation of shear. (B) Recovery of MAX8 hydrogels (0.5 wt%, pH 7.4) from shear stress as analyzed by monitoring G’ as a function of time: region I shows gelation onset under 0.2% strain; region II shows shear-thinning of the hydrogel under 1000% strain; and region III shows recovery following reduction of strain to 0.2%. Frequency = 6 rad*sec−1 for all measurements. (C) Laser scanning confocal microscopy z-stack image shows cells distributed homogenously within the MAX8 hydrogel (viewed along the y axis, from the side of the hydrogel) following syringe injection (inset shows loaded syringe prior to shear-thinning). Scale bar = 100 μm. (D) Laser scanning confocal microscopy z-stack imaging (viewed along the z axis, from the top of the hydrogel) showing cell viability within the MAX8 hydrogel following syringe injection using the Live/Dead assay (Red, dead cells; green, live cells) t = 3 h after delivery. Scale bar = 100 μm. Adapted with permission from Haines-Butterick et al. [59], Copyright (2007) National Academy of Sciences, U.S.A.
2.1.1.5. E1Y9
Mihara and coworkers reported the peptide E1Y9 (sequence: EYEYKYEYKY) in 2012 for cell culture and drug delivery [50,51]. Hydrogelation of this peptide is calcium ion (Ca2+)-dependent and driven mostly by intermolecular interactions between fibers, with only minimal fiber entanglement observed. Injecting solution-state E1Y9 into a Ca2+ solution allows production of gel droplets or strings with control over hydrogel shape. Both 3T3-L1 and PC12 cells adhere to and grow on E1Y9 hydrogels when cultured on string-shaped gels.
2.1.1.6. Q11
In 2003, Collier and Messersmith engineered another major amphipathic SAP, Q11 (QQKFQFQFEQQ; Q = glutamine), as a modular biomaterial platform [52]. Q11-based assemblies are particularly amenable to functionalization via tissue transglutaminase-mediated reactions of glutamine residues with primary amines [53]. This net-neutral peptide sequence features hydrophobic phenylalanine residues and the hydrophilic residues glutamine (neutral), lysine, and glutamic acid. Upon the addition of salt to an aqueous solution, Q11 assembles into a highly entangled gel-like β-sheet network, composed of 4-8 nm wide fibrils. Tissue transglutaminase (tTGase)-mediated conjugation of monodansylcadaverine (MDC), an amine-functionalized fluorescent marker, to both soluble and assembled Q11 proceeded successfully, allowing attachment of up to five MDC dyes per peptide without disrupting nanofiber assembly [52]. The lysine residues within Q11 did not participate in the reaction, presumably due to inaccessibility to tTGase in the assembled fibril. Additionally, native chemical ligation (NCL) [54] of a Q11 variant modified with an N-terminal cysteine and a C-terminal thioester lengthens the peptides and yields hydrogels five times stiffer than unmodified Q11, without significantly changing fibril morphology [55].
The low pH required to process Q11 and other SAPs in the solution state, prior to inducing gelation at physiological pH, previously hindered the application of SAPs for cell encapsulation. By inserting an additional QE motif into the Q11 sequence, the Q11 variant bQ13 remains in solution upon mixing with LNCaP human prostate cancer cells up to pH 9.8 [56]. In comparison, RADA16 and Q11 mixed with LNCaP cells only remain in solution up to pH 3.5, above which premature gelation may occur. The low pH required by RADA16 and Q11 is harmful to cells, as evidenced by the low percentage of LNCaP cells that survive the encapsulation process (50% for RADA16 and 66% for Q11). In contrast, 96% of LNCaP cells survive encapsulation at pH 9.8 in bQ13 hydrogels.
Co-assembly of Q11 with modified Q11 sequences modulates the mechanical and biological properties of the resulting assemblies [57]. Inducing assembly before or after blending Q11 with modified Q11 sequences provides control over fibril composition, i.e., whether they are homogenous (uniformly intermixed peptide fibrils) or heterogeneous (individual fibrils composed of one peptide. Incorporation of cell-adhesive ligands through homogenous co-assembly of Q11 and Q11 modified at the N-terminus with RGD or IKVAV enhances cell attachment, proliferation, and growth without impacting peptide secondary structure or material stiffness and morphology [58].
The peptides KLD-12 [60], FAQ [61,62], CK [62,63], SPG-178 [22], (LE)8 [64], PFD-5 [65,66], MDG1 [67], and VEVSVKVS [64] are further examples of such SAPs in the family of amphipathic peptides with similar design considerations and mechanisms of assembly. Overall, despite the strong dependence of amphipathic SAP assembly on specific conditions that could preclude cell encapsulation or other biomaterials applications, the pros of this class of SAPs often outweigh the cons. Due to the high tunability and variety of material properties that are possible, amphipathic peptide assembly continues to be widely employed in biomaterials systems.
2.1.2. Aromatic short SAPs
Short peptide sequences, with as few as two to five amino acids, also assemble in solution into β-sheet structures. Such short sequences have the advantage of simple, straightforward, and low-cost synthesis. Many of these structures include the aromatic residues tyrosine, tryptophan, and phenylalanine, or synthetic aromatic groups, such as the Fmoc protecting group used in peptide synthesis. Here, we describe the assembly of diphenylalanine, tripeptides, and a class of pentapeptides. For a more in-depth look at short SAPs, we point the reader to a recent review on the assembly, methods of characterization, and current applications of short SAPs [68].
Aspirations to understand amyloid plaque formation characteristic of Alzheimer’s disease motivated early work on assembly of diphenylalanine (FF), taken from the recognition motif of Alzheimer’s disease-relevant amyloid β (Aβ). Reches and Gazit dissolved FF at high concentration (≥100 mg/mL) in 1,1,1,3,3,3 hexafluoro-2-propanol (HFIP), then added the mixture to aqueous solution, resulting in peptide concentrations in the μM range, to induce assembly within seconds into semi-crystalline, hollow nanotubes ca. 100-150 nm in diameter [69]. Studies on synthetic FF analogues, including an uncharged dipeptide with an acetylated N-terminal amine and an amidated C-terminus, as well as several derivatives with N-terminal modifications (Boc-FF, Cbz-FF, and Fmoc-FF), helped elucidate a mechanism of assembly [70]. While all of these FF peptides assemble into nanotubes in aqueous solution, the derivatives with N-terminal modifications form significantly smaller diameter structures. These results indicated that assembly is not driven by electrostatic interactions of the charged termini, but rather by hydrogen bonding and aromatic interactions (i.e., π-π stacking).
Numerous Fmoc-functionalized dipeptides form hydrogels [72], although Fmoc-FF is still the most widely used of these. Together, spectroscopy data and molecular modeling suggest that at neutral pH, β-sheets composed of π-stacked aromatic groups, twist upon association and form ribbons (Figure 3) [71]. Tuning solution pH results in Fmoc-FF assemblies with a range of morphologies [73]. At high pH (>10.2), nearly all of the molecules are ionized and soluble. Between pH 9.5-10.2, some assembly occurs into antiparallel β-sheet fibers with a negative surface charge, which entangle to form a weak hydrogel. Further lowering the pH from 9.5 to 6.2 protonates some of the carboxylic acids and induces the formation of flat ribbons >100 nm in diameter as the fiber surface charge decreases. Ribbon aggregation occurs at pH 5.2, below which macroscopic rodlike structures precipitate out of solution. Due to its tunability and simple synthesis, Fmoc-FF finds applications in drug delivery systems [74,75], ECM mimetic materials [72,76,77], biocatalysis [78], and more, as detailed in a recent review [79].
Figure 3.
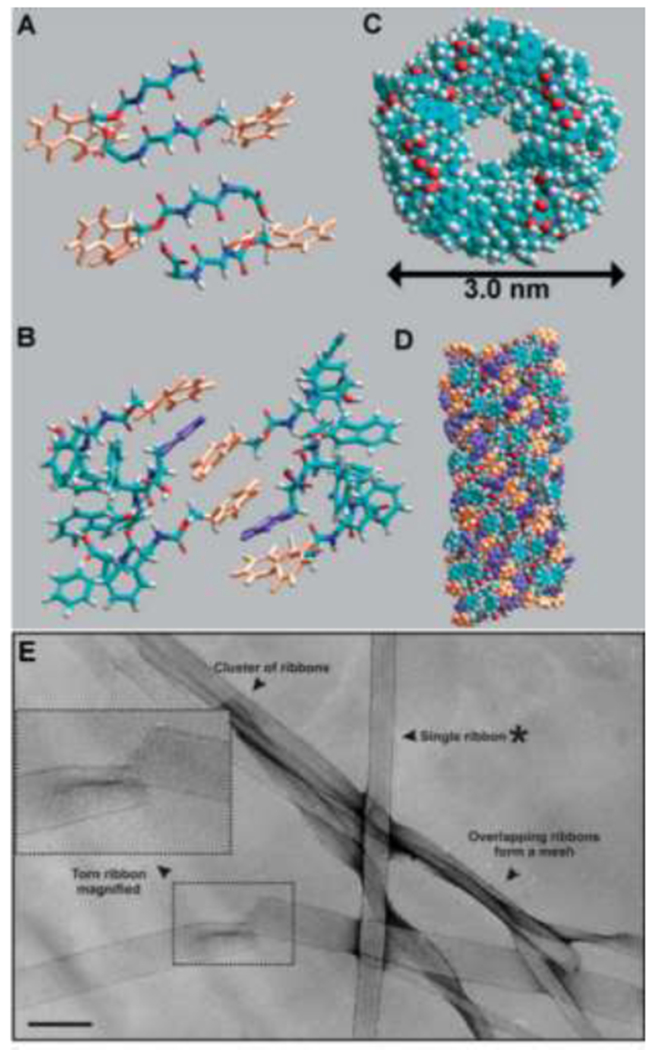
Supramolecular assembly of Fmoc-FF. Combining spectroscopy and molecular models shows that (A) β-sheets initially associate in an anti-parallel manner; and (B) Fmoc groups from adjacent β-sheets interlock by π-π interactions. (C) Top view; and (D) side view of ribbons that form as β-sheets twist to meet the geometrical constraints required to preserve π-π interactions. In A, B, and D, orange rings are fluorenyl groups and purple rings are phenyl groups. (E) TEM image of an Fmoc-FF gel composed of entangled ribbons, proposed to form from multiple Fmoc-FF fibrils. Shear force-induced tears, such as the one enlarged in the inset, show that the ribbons are flat without a hollow interior. Scale bar = 500 Å. Adapted with permission from Smith et al. [71], Copyright (2008) Wiley-VCH GmbH & Weinheim.
Many short peptides containing combinations of aromatic residues, particularly tyrosine (Y) and phenylalanine (F), have at least some propensity to assemble into fibrillar hydrogels. For example, Ulijn and coworkers investigated tripeptides containing Y, F, and aspartic acid (D) residues, and showed the peptides YFD, FYD, DFY, and DYF form distinct structures, while FDY and YDF did not assemble, suggesting that the separation of the aromatic Y and F groups by a hydrophilic charged D residue reduces assembly-promoting π-π interactions [80]. Tuttle, Ulijn, and coworkers developed a computational model to screen the 8000 possible combinations of tripeptides composed of the 20 natural amino acids and identified KYF, KYY, KFF, and KYW (W = tryptophan) to form hydrogels in aqueous solution at neutral pH [81]. Building from this work, Webber and coworkers investigated assembly of tripeptides, varying the arrangement of aromatic residues and the electronegativity of substituents on the phenylalanine ring. They confirmed that separation of hydrophobic residues by even one hydrophilic residue precludes assembly [82].
Using design rules governing solution assembly of short aromatic and amphipathic peptides, Lampe and coworkers designed hydrogel-forming pentapeptides [32,83]. This material platform, termed ‘rapidly assembling pentapeptides for injectable delivery (RAPID)’ hydrogels, features shear-thinning and self-healing properties. The RAPID family of peptides are amphiphilic and feature a central phenylalanine residue, e.g., KYFIL, which forms a mechanically robust hydrogel composed of ca. 40 nm diameter twisted ribbons. Tuning the weight percent and composition of RAPID peptides modulates hydrogel mechanical properties over a broad range (~50-17,000 Pa). For example, changing the concentration of KYFIL from 1.5 to 3 wt % increases the storage modulus from 8 to 17 kPa. Self-healing is rapid, as hydrogels recover 70-80% of initial stiffness within two seconds following application and removal of 100% strain. Similar to the preclusion of assembly for FDY and YDF tripeptides due to separation of the aromatic residues, the RAPID sequence KAFIL, which does not contain two adjacent aromatic residues, does not form fibrils at physiological pH, while KYFIL, AYFIL, and KYFAL form ribbons or fibrils (Figure 4).
Figure 4.
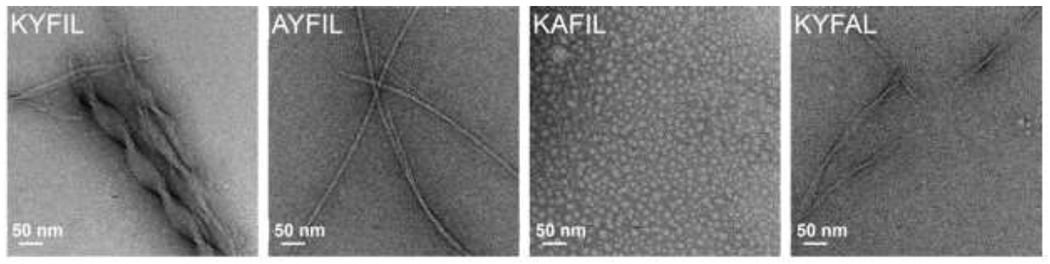
Sequence-dependent assembly of RAPID peptides (1.5 wt% in pH 7.4 PBS). TEM images of four different RAPID peptides highlight the sequence-dependent morphology. KYFIL and KYFAL assemble into hydrogels composed of twisted ribbons, while AYFIL assembles into hydrogels composed of twisted fibrils and KAFIL forms spherical nanostructures in solution without any hydrogel formation, likely because it is the only of these sequences that does not contain two adjacent aromatic residues. Further, the size of the ribbons formed differs by sequence, as KYFIL forms ribbons ca. 40 nm while KYFAL forms ribbons ca. 10 nm. Reproduced with permission from Tang et al. [84], Copyright (2019) American Chemical Society.
2.1.3. Rippled β-sheets
In 1953, one year after first describing the configuration of antiparallel and parallel pleated β-sheets, Pauling and Corey predicted the formation of a structure they termed “rippled” β-sheets from a mixture of d-peptides and l-peptides [85]. In a rippled sheet, adjacent d-peptides and l-peptides typically orient such that the side chains point toward each other, resulting in nested interactions of hydrophobic groups [86]. In a nested configuration, one hydrophobic side chain fits tightly packed in between the hydrophobic side chains of its neighbors. These interactions, coupled with the documented resistance of D-amino acids to enzymatic degradation, render rippled β-sheet structures useful for enhancing both the mechanics and stability of biomaterials. Specific stereochemical interactions, like those driving rippled sheet formation, continue to emerge as a powerful means to control material properties without changing the chemical features of materials, as reported in both the peptide [37,86–89] and synthetic polymer literature [90–92].
Schneider, Pochan, and coworkers demonstrated structural and mechanical differences between hydrogels formed from homochiral and heterochiral mixtures of β-hairpin MAX1 peptides [87]. Introducing the d-enantiomer of MAX1, DMAX1, in increments of 25 mol% lends enzymatic stability to the resulting hydrogels. Surprisingly, oscillatory rheology showed the stiffness of hydrogels formed from 50 mol% DMAX1 (i.e., a 1:1 mixture of the enantiomers) to be four times that of hydrogels composed entirely of MAX1 or DMAX1. Intermediate stoichiometric ratios, namely hydrogels with 25 and 75 mol% DMAX1, exhibit moduli intermediate between that of homochiral and equimolar d-peptide and l-peptide mixtures. A control peptide, identical to MAX1 except for the replacement of three valine residues with isoleucines, exhibits similar assembly kinetics and gel stiffness to that of MAX1 and DMAX1. However, mixtures of DMAX1 with the isoleucine-containing control peptide provide no significant increase in stiffness, indicating that the enhancement in mechanics depends both on sequence and complementary stereochemistry. A later study concluded that the fibrils formed by MAX1 and DMAX1 match Pauling and Corey’s predictions of alternating co-assembly of d-peptides and l-peptides into rippled sheets and that side chain packing plays an important role in the assembly [86]. The complementary chirality of the enantiomers allows the side chains of the valine residues to interact in a nested manner, permitting hydrophobic interactions not possible in a fibril composed of only one enantiomer, thus stiffening the core of the fibril (Figure 5).
Figure 5.
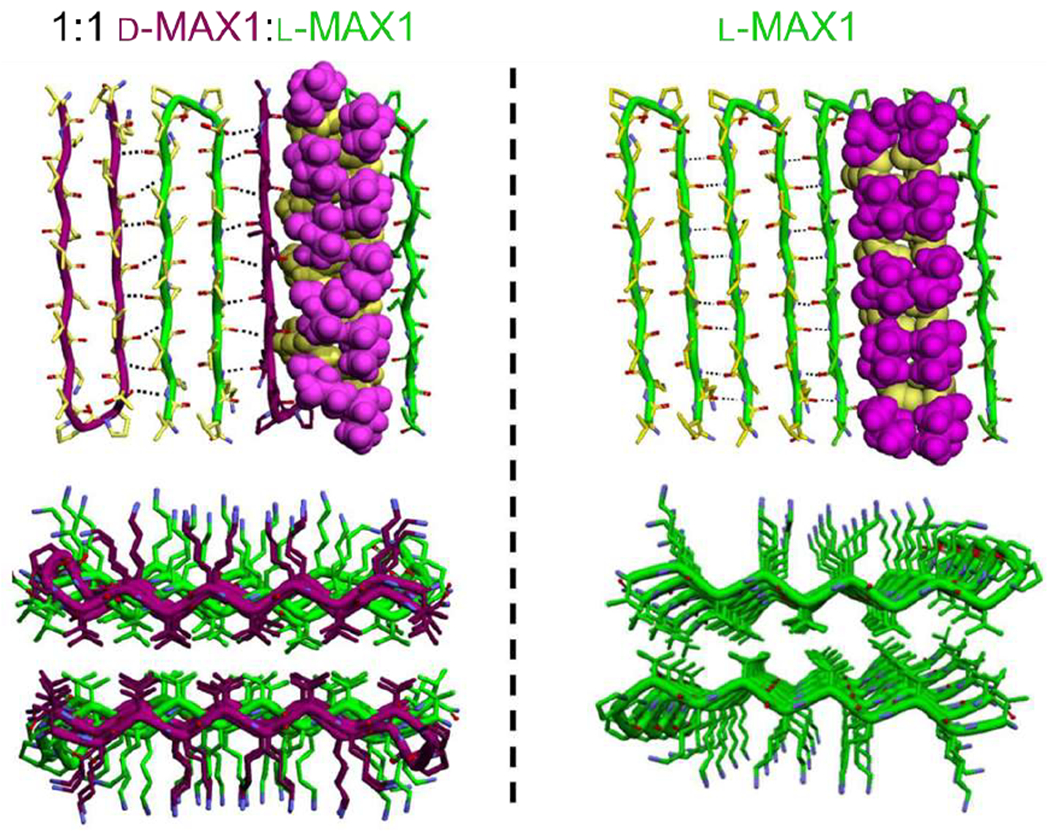
Models of enantiomeric (left) and homochiral (right) fibrils of the rippled sheet forming peptide MAX1. Top images show the orientation of MAX1 β-hairpins in a fibril monolayer. Corey-Pauling-Kulton (CPK) renderings show packing of valine side chains on two adjacent peptides. Bottom images show the packing of side chains as viewed along the long axis of the fibril. Nested interactions in the enantiomeric mixture, particularly between the hydrophobic valine side chains, yield considerably tighter packing than in the side chains in the homochiral fibrils. Adapted with permission from Nagy-Smith et al. [93], Copyright (2017) American Chemical Society.
Furthermore, controlling peptide sequence allows modulation of the bulk mechanical properties. For example, hydrogels formed from enantiomeric mixtures of MAX peptides containing both valine and isoleucine on the hydrophobic face of the hairpin provide only a two-fold increase in stiffness compared to the homochiral gel, while enantiomeric mixtures of MAX peptides incorporating arginine residues on the hydrophilic face yield an eleven-fold increase relative to the analogous homochiral gel. Pairwise interactions of isoleucine are generally less energetically favorable than valine, leading to a smaller stiffness enhancement, while the formation of inter-guanadinyl hydrogen bonds in arginine side chains provides a larger stiffness enhancement.
Nilsson and coworkers reported similar trends using (FKFE)2 peptides [88]. They expected that mixtures of the d- and l-(FKFE)2 peptides would self-sort to produce left-handed and right-handed helical ribbons composed entirely of l-peptides and d-peptides, respectively. However, TEM images revealed narrower, 3.0 nm wide flat nanoribbon fibrils to form from this mixture, in contrast with the wider, 6 nm wide helical nanoribbons formed from the constituent peptides. Circular dichroism and isotope-edited infrared spectroscopy experiments further supported the presence of co-assemblies, and calorimetry showed that binding energy for the enantiomeric pair was more favorable than the analogous homochiral pair. Consistent with the results of Schneider and Pochan with the MAX peptides, 1:1 l/d-(FKFE)2 hydrogels exhibit enhanced stiffness relative to homochiral hydrogels. The rippled sheet fibrils resist proteolytic degradation by chymotrypsin, trypsin, and proteinase K and display a similar degradation profile to d-(FKFE)2 [89]. After a 5-day incubation with these enzymes, minimal d-(FKFE)2 and 1:1 l/d-(FKFE)2 sample degradation occurred, whereas the concentration of l-(FKFE)2 was less than 5% of its initial value.
Most rippled sheet formers reported to date are amphipathic peptides featuring alternating hydrophilic and hydrophobic residues. However, there is also evidence that non-amphipathic peptides can form rippled sheets. For example, d- and l-polyglutamine (polyQ) monomers derived from an amyloid fibril-forming peptide implicated in Huntington’s disease, each seed the aggregation of monomers of the opposite stereochemistry [94]. In another example, d- and l-enantiomers of the 42-residue amyloid β (Aβ) peptide, which is neurotoxic and implicated in Alzheimer’s disease, co-localize in racemic fibrils upon mixing [36]. Enantiomeric mixtures of the 7-residue Aβ fragment Aβ (16-22) also form rippled sheets [37]. While studies on these SAPs did not involve testing hydrogel formation, these examples show that the rippled sheet formation predicted by Pauling and Corey extends beyond highly aggregation-prone amphipathic peptides.
Despite the number of peptide families that form rippled sheets, the prediction may not hold for every peptide. One of the first studies that specifically investigated enantiomeric peptide interactions involved a β-sheet forming fragment of the β2-microglobulin protein K3 (SNFLSCYVSGFHPSDIEVDLLK)[95]. Individually, both the naturally derived l-K3 and the synthetic enantiomer d-K3 form fibrils, which are nearly identical apart from slightly slower d-K3 fibril formation kinetics. Seeding experiments demonstrated that d- and l-K3 do not cross-react, rather, addition of l-peptides results in extension only of preformed l-seed fibrils and addition of d-peptides results in extension only of preformed d-seed fibrils. As another example, the d- and l-enantiomers of the peptide L4K8L4 form nearly identical fibrils with opposite twists, while mixtures of the two enantiomers produce globular aggregates [96]. In both cases, the lack of controlled, β-sheet forming cross-reactivity between d- and l-fibrils seemingly opposes the prediction of Pauling and Corey. However, these results leave open the question of whether the peptides represent an exception to the rippled sheet prediction or if they could form rippled sheets under different conditions. Further exploration of this sequence space may unlock the additional potential of stereochemistry-dependent interactions in SAP-based biomaterials.
2.1.4. Metal ion-induced SAPs
Inspired by natural proteins that bind metals, metal-binding SAPs assemble by coordination and/or electrostatic interactions and allow for subsequent use of the integrated metal ion for mediating reactions, biological processes, and controlled release. In the context of tissue engineering, metal-binding SAPs can capture toxic metals, release therapeutic metals, and probe the role of metals and metal binding events in disease so as to engineer therapeutics. Many of the SAPs in this class are sequences found in or modified from naturally occurring proteins, and contain cysteine or histidine residues, both of which coordinate heavy metal ions. We highlight a few recent examples below, and point readers to an excellent review on metal-triggered SAPs for further information [97].
An important application of metal ion-induced assembly of peptides is the selective capture and detection of metal ions from solution, including cell environments. For example, Lee and coworkers developed ratiometric peptide-based fluorescent probes for Cd(II) [98]. In aqueous conditions, addition of Cd(II) to the pyrene-functionalized peptide, pyrene-CGPC, induces ordered assembly into nanoparticles. Decreases in pyrene monomer emission and increases in multimeric pyrene signal (i.e., excimer emission) indicate Cd(II) capture. Among 17 metal ions tested, Cd(II) alone elicited this ratiometric response from the probe. Together, the disappearance of the S-H stretching band in FT-IR spectroscopy upon the addition of Cd(II) and the loss of Cd(II) binding affinity upon protonation of the C-terminal carboxylate suggest that the C-terminal carboxylate and the thiols on the cysteine residues coordinate with Cd(II). This coordination generates a complex conducive to pyrene π-π stacking interactions and assembly into structured nanoparticles [98]. This pyrene-peptide reporter system detects Cd(II) in live cells and at concentrations as low as 53 nM in urine samples, showcasing the potential for metal ion-induced SAP systems as probes.
A related application of this class of SAPs is the capture and subsequent controlled release of metal ions, for example to release silver in antimicrobial formulations [99]. At pH 8.5, the peptide IH6 (sequence: ILVAGH; where H = histidine and I = isoleucine) adopts a random coil structure in aqueous solution at low concentration (300 μM) and an antiparallel β-sheet conformation at high concentration (≥ 3.0 mM), forming a weak fibrous hydrogel. Addition of silver ions (Ag+, an active component of many sterilizing agents) in a 1:1 molar ratio with IH6 results in a parallel β-sheet structure, with the conformational shift attributed to the coordination of Ag+ with histidine. Silver-containing β-sheet (Ag-IH6) hydrogels display enhanced mechanical strength (G’ = 40 kPa for Ag-IH6 hydrogels vs. 1 kPa in the absence of silver) and feature a more compact fibrous network compared to those prepared at the same concentration from the IH6 peptide alone. The hydrogels enable tunable Ag+ release profiles and exhibit bactericidal activity without harming cocultured human keratinocytes.
Metal ion-induced SAPs also provide insight into the role of metal ions in disease. The aggregation of β-sheet amyloids is a significant driver in a number of diseases, which makes the study of metal ions capable of modulating this aggregation or amyloid formation critical. One example is the interaction of Zn(II) with human prion protein (PrP). A peptide from the octarepeat domain of PrP, PrP58-93 (GQ(PHGGGWGQ)4GG, where W = tryptophan), displays a mixture of random coil and helical character in aqueous solution [35]. ZnCl2 addition leads to the formation of elongated amyloid fibrils that bind Thioflavin T and Congo red dyes, hallmarks of amyloid formation. Correlations between Zn(II) concentration, loss of monomeric peptide, and increase in aggregate fraction provide further of a relationship between Zn(II) and amyloids. Continued investigation of the role of metal ions in assembly of β-sheet amyloids is likely to elucidate pathways of protein misfolding and inform development of future treatments for amyloid diseases.
2.1.5. β-sheet forming peptides: summary and perspective
Among the various β-sheet forming peptide families employed as biomaterials, ample studies demonstrate the utility of the RADA series in multiple contexts and tissue-specific applications (see section 4). The RADA family was among the first reported assembling peptides and contains RAD motifs that resemble the integrin tripeptide RGD; however, more recent systems offer simpler manufacturing and/or added functionality, thus warranting further investigation. Shorter assembling peptides, such as diphenylalanine, numerous tripeptides, and the RAPID series, show promise for reducing material complexity, while still affording well-defined, dynamic fibrous hydrogels amenable to biomolecule conjugation. Moreover, the specific advantage of an integrin-binding region within RADA may be less critical as recent advances in these shorter assembling peptides permit addition of integrin-binding sequences to the assembling sequence. The modularity of peptides that permit conjugation of bioactive motifs adds design flexibility and enables better parsing of the effects of the material building blocks (i.e., the amino acids driving assembly) and the “biological” aspects (i.e., the amino acids driving specific cell-matrix interactions).
2.2. Helical peptide assemblies
Peptides with helical secondary structure often assemble into coiled coils or collagen-mimetic triple helices. Coiled coils contain α-helices, which are 1.6 Å in length per residue and 3.7 residues per turn. Amino acid side chains point outward due to crowding of the interior of the helix with peptide backbone atoms. Yet, collagen and collagen-like peptides feature polyproline II helices (3.1 Å per residue, 3.3 residues per turn). With peptide bonds in the typical trans configuration and a left handed twist, polyproline II helices are far more common than polyproline I helices with more constrained cis peptide bonds and a right-handed twist [100,101]. While intramolecular interactions stabilize individual helices, a combination of intermolecular hydrophobic, electrostatic, and hydrogen bonding interactions stabilize bundles of helical peptides, which can further assemble into fibers and hydrogels. In addition to coiled coil and collagen-like peptides, charged helical peptides assemble in a pH- and salt-dependent way into a variety of micro- and nanoscale-structures [7,102,103]. Compared to β-sheet peptides, where peptides with as few as two amino acids can form fibers, assemblies from helical peptides require substantially longer primary sequences due to the minimal or absent helix stability in short sequences. Yet, the increased length of these sequences provides ample opportunities to install reactive groups and fluorescent labels and to finely modulate the binding strength and specificity, stoichiometry, stimuli-responsive behavior, and higher order assembly. Furthermore, solid-phase peptide synthesis, including microwave-assisted and automated synthesis, enables routine preparation of sequences >50 amino acids in length [7,8]. In this section, we describe the salient features of and design rules governing assembly of helical peptides into coiled coils and collagen-mimetic triple helices, and the higher order assembly of these helical bundles.
2.2.1. Coiled coils
The helical coiled coil is one of the most abundant protein structural motifs found in nature, including in fiber-forming proteins of the extracellular matrix [104,105]. Just two years after the initial description of the protein α-helix [106,107], right handed α-helices (for l-amino acids) were predicted to wrap around each other at an angle of ca. 20° to form a left-handed coiled coil super helix [108]. Most coiled coil-forming peptides contain a repeating sequence of 7 amino acids (i.e., a heptad), denoted (abcdefg)n (Figure 6) [109]. Hydrophobic amino acids in positions a and d together with charged amino acids in positions e and g stabilize coiled coils, while the b, c, and f positions are solvent-exposed and therefore modulate solubility and assembly [27]. Coiled coils can be composed of not just two, but three, four, five [110], and even seven helices [111] per superhelix. Moreover, these superhelical coiled coils may contain just one type of peptide (e.g., homotrimers, or coiled coils containing three copies of the same peptide) or combine different peptides (e.g., heterodimers, or coiled coils formed from two different peptides). Heterochiral coiled coils can also form from mixtures of d- and l-peptides [112], yet require different spacing of hydrophobic residues for optimal packing [113]. We review some of the design rules here for both the primary structure and inducing higher order assembly of coiled coils into fibers and hydrogels, and direct readers to greater detail on coiled coils in reviews by Klok [114], Kros [115], and Collier [105].
Figure 6.
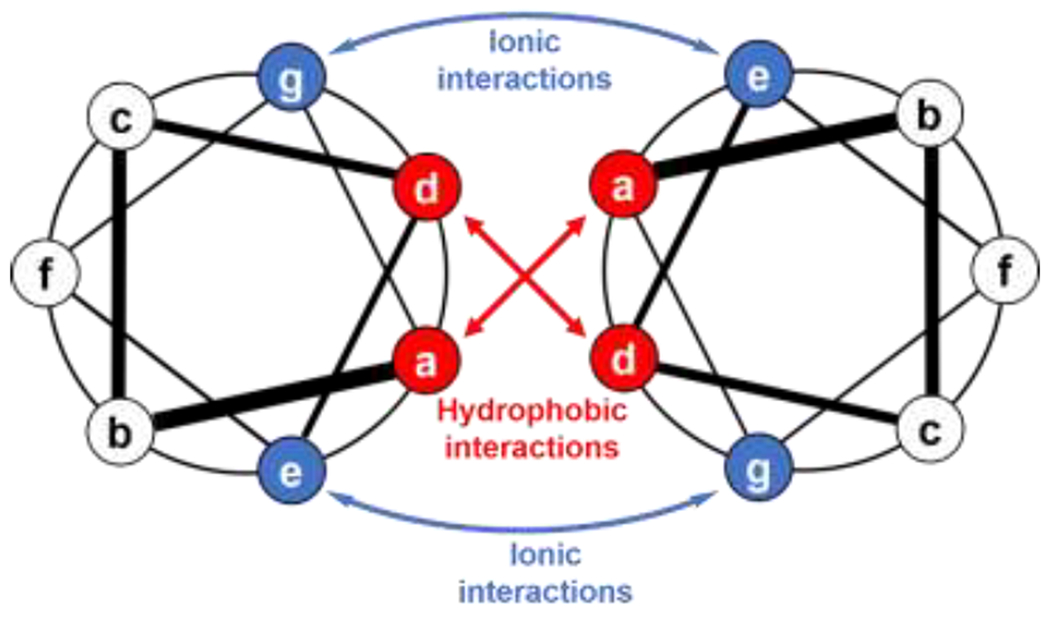
Arrangement of amino acids in a dimeric coiled-coil peptide held together by hydrophobic interactions between residues in the a and d positions of the heptad repeats and electrostatic interactions between e and g residues. Adapted with permission from Mason and Arndt [109], Copyright (2004) Wiley-VCH GmbH & Weinheim.
2.2.1.1. Primary structure considerations
The amino acids in the a and d positions of the heptad form the interior of the coiled coil superhelix, and control stoichiometry (i.e., the number of peptides per coiled coil) and orientation (e.g., parallel vs. antiparallel configurations of chains) within the coiled coil bundle [110]. Typically, hydrophobic interactions hold together these helical peptide bundles [110,116]. Additionally, hydrogen bonding between asparagine (N) side chains in the a and/or d positions promotes specific binding between coiled coil peptides [117–119]. Introducing aromatic and hydrogen bonding tryptophan residues in the a and d positions reduces helicity and hinders higher order assembly into fibrils, while aromatic phenylalanine residues without hydrogen bonding side chains only slightly reduce helicity [120]. The distribution of β-branched amino acids, e.g., isoleucine (I) and valine (V), in which the β-carbon (the side chain carbon adjacent to the carbon on the peptide backbone) is branched, directs packing of coiled coils [110]. Dimers typically form with leucine in the d position and either a β-branched amino acid or asparagine in the a position [110,119,121,122]. Having a β-branched amino acid in the d position hinders dimer formation, while a β-branched amino acid in the a position hinders tetramer formation; β-branched amino acids in both the a and d positions favor trimer formation. For example, placing a leucine (L) in the a position and a β-branched isoleucine (I) in the d position yields tetramers. In the a position, isoleucine (I) residues provide more stability than less hydrophobic valine (V) residues [119,122,123].
Charged residues most frequently occupy positions e and g within coiled coil-forming heptads and stabilize the helical configuration of the individual peptides, promote interactions between peptides within a coiled coil, and facilitate the higher order assembly of coiled coil bundles, for example into fibers. A common scenario is for coiled coil-forming peptides with either cationic lysine (K) residues in both the e and g positions or anionic glutamic acid (E) residues in both the e and g positions to form helical homodimers, in pH ranges that neutralize the charge [124,125]. For example, lysine- and glutamic acid-rich sequences with the heptad repeats (VSSLKSK)n and (VSSLESE)n, respectively, adopt a random coil configuration at neutral pH, yet 1:1 mixtures of these two peptides form heterodimeric coiled coils driven by interchain electrostatic interactions [125]. In another example, heterodimeric coiled coils form at neutral pH from the lysine-rich sequence Ac-GYK(IAALKEK)2-IAALKEG-NH2 (K3) and the glutamic acid-rich sequence Ac-GYE(IAALEKE)2-IAALEKG-NH2 (E3) sequences partially disassemble at pH 5 into homotrimers of the anionic peptide and random coil (unassembled) cationic peptide (Figure 7) [124]. Oppositely charged e and g amino acids within the same peptide, for example those with the heptad repeats (VSSLESK)n [125], (IEALKAE)n [121], and (LAEIEAK)n [126], assemble to form coiled coils at neutral pH, yielding one-component systems for cell encapsulation and other applications.
Figure 7.
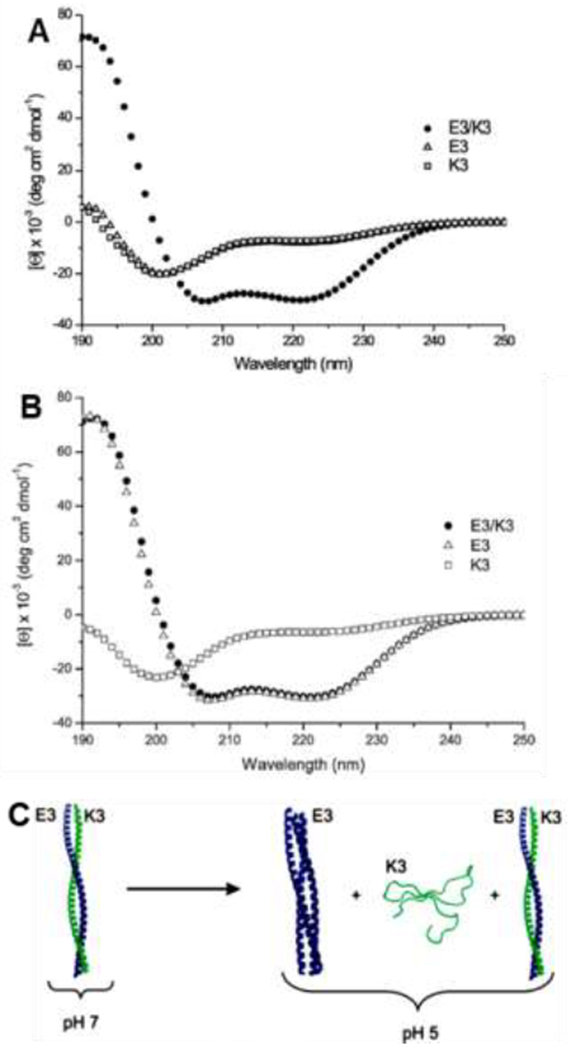
pH-dependent assembly of glutamic acid-containing E3 and lysine-containing K3 peptides. CD spectra of the E3, K3 and their equimolar mixture in 10 mM PBS at (A) pH 7 and (B) pH 5. Both E3 and K3 are random coils while their heterodimeric assembly is helical at pH 7. At pH 5, the heterodimer partially unfolds into E3 trimers and random coil K3 unimers. (C) Schematic of E3 (blue) and K3 (green) peptides self- assembly at pH 7 and pH 5. Adapted with permission from Apostolovic and Klok [124], Copyright (2008) American Chemical Society.
The b, c, and f, positions comprise the solvent-exposed portion of the coiled coil and therefore modulate solubility and higher order assembly of coiled coils into fibers and fibrous hydrogels. Fibers assembled by hydrogen-bonding glutamine (Q) residues in the b, c, and f positions disassemble upon heating, which ruptures hydrogen bonds [27]. However, heating reinforces fibers held together with hydrophobic interactions (b, c, f = alanine, A), consistent with higher temperatures reinforcing hydrophobic interactions since it becomes more favorable to remove structured water. In the b position, replacing hydroxyl-containing serine (S) with hydrophobic, helix-promoting alanine (A) increases helical stability [119,122]. The b, c, and f positions are also ideal places for addition of reactive groups or dyes [117,119].
Length, often described by the number of heptad repeats, determines both helix stability and binding strength between peptides in a coiled coil [121,127]. Typically, coiled coil formation requires 3-4 heptad repeats [122,124]. Binding strength varies appreciably with length: increasing the number of heptad repeats from 3 to 5 decreases thermodynamic dissociation constants from the micromolar to picomolar range [117,128].
Stereochemistry also plays a role in coiled coil formation, though these design rules remain largely unknown. Gellman and coworkers reported the crystal structure of a heterochiral coiled coil, [112] and suggested that, instead of heptad repeats, heterochiral coiled coils may best accommodate sequences with a repeating 11-amino acid pattern [113].
2.2.1.1. Orientation
Peptide sequence modulates the orientation in which peptides pack within coiled coils. Inclusion of an asparagine (N) residue in the a position of one of the central heptads yields parallel orientation of peptides, i.e., with the N and C termini pointing in the same direction [117–119]. The tropomyosin-derived sequence Ac-E-WEEALEKK-LAALESK-LQALEKK-LEALEHG-NH2 forms triple helical bundles, arranged such that two adjacent peptides are parallel and the third antiparallel to the others [129]. In a particularly elegant example of controlling orientation to design precision materials, Gradišar and coworkers engineered a tetrahedron from a single chain of parallel- and antiparallel-forming coiled coil segments [130]. Appending thiol-containing cysteine (C) residues to the N- or C-termini allows determination of peptide orientation within coiled coils by analysis of the disulfide-linked dimers that form under oxidative conditions [110,117,131]. For mixtures of peptides functionalized with N-terminal cysteines, homodimer formation indicates parallel orientation.
2.2.1.2. Higher order assembly
Engineering interactions between coiled coil peptide bundles allows controlled assembly into fibers and fibrous hydrogels that mimic the topological and mechanical properties of the natural extracellular matrix. While the outward-facing b, c, and f residues modulate lateral assembly of coiled coil peptide bundles, chain ends can be designed to promote end-to-end assembly. Woolfson and coworkers staggered coiled coils by patterning the chain ends of each sequence with different charges than in the middle of the sequence [132]. These staggered coiled coils assemble in both longitudinal and lateral directions to produce fibers hundreds of microns in length and ca. 50 nm wide. By adjusting the b, c, and f residues of these sequences, Woolfson and coworkers generated hydrogels [27] and then later expanded this platform to more advanced structures with branches and kinks, as well as systems presenting peptide tags for attaching functional moieties [133–135].
By installing complementary reactive groups on peptide bundles, Pochan and coworkers realized end-to-end assembly of coiled coil peptide bundles [136–138]. Functionalizing the ends of antiparallel homotetrameric bundles with thiol and maleimide groups furnished ‘A-B’-type monomers that polymerize into rod-like nanostructures (Figure 8). Akin to step growth polymerization of A-B monomers, the stoichiometric ratio of the thiol:maleimide groups controls the length of the rods. Blending coiled coil bundles at thiol:maleimide molar ratios of 0.5, 0.88, and 0.94 produced fibrils with average lengths of 145 ± 35 Å, 530 ± 150 Å and 1100 ± 700 Å, respectively [136]. Bundle assembly is temperature sensitive and reversible even after the reaction: increasing the temperature to 90 °C causes the bundles to denature, while reducing the temperature to 25 °C results in reassembly of the bundles and rods [138].
Figure 8.
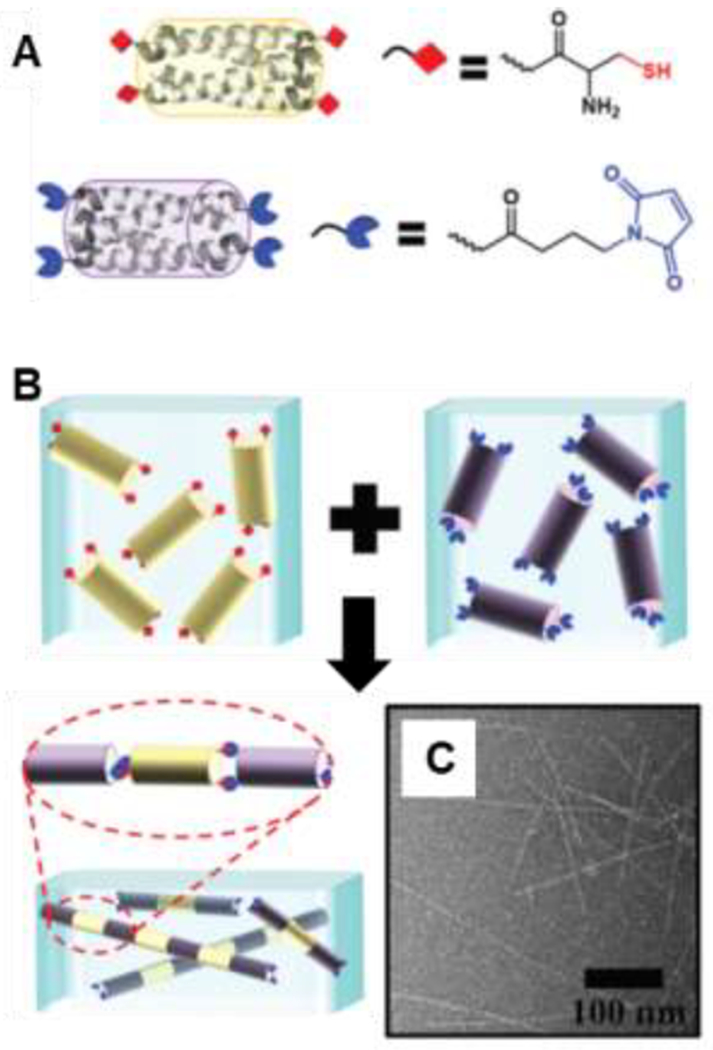
Higher order assembly of helical bundles: (A) Schematic of tetrameric coiled coil bundlemers of thiol-functionalized peptide 1 (yellow) and maleimide-functionalized peptide 2 (blue). (B) Assembly of peptide bundlemers into rigid rod-like chains by thiol-maleimide click chemistry. (C) TEM image of rigid bundlemer rods. Adapted with permission from Sinha et al. [136], Copyright (2019) The Royal Society of Chemistry.
2.2.2. Collagen-like peptides
Collagen is the most abundant protein in the human body and a central element of the extracellular matrix. Three parallel protein strands adopting left-handed polyproline II-type helical configurations (i.e., 3 residues per turn) assemble into right-handed triple helices, where the individual helices expand slightly to 3.3 residues per turn. Pre-organization of the constituent polyproline II helices offsets the entropic cost of assembly, and hydrogen bonding interactions hold together the resulting triple helices [139,140]. After reviewing primary structure contributions to triple helix stability, we discuss molecular features and environmental conditions conducive to aggregation of the triple helices into fibers and fibrous hydrogels. For more information on collagen-like peptides and their assemblies, we direct readers to reviews by Kim [141] and Kiick [142].
2.2.2.1. Factors governing triple helix stability
Collagen-like peptides consist of repeating Gly-X-Y tripeptides, with every third residue occupied by glycine (G) and the X and Y positions typically occupied by proline (P) and hydroxyproline (O). Studies on a number of G-X-Y sequences varying in lengths provide insight into the effects of sequence on triple helix stability. Replacing the glycine residue destabilizes the close-packing in polyproline II helices [139,143]. In the sequence Ac-(GPO)3-G-X-O-(GPO)4-GG-NH2, varying the amino acid in the X position showed proline to confer the most stability, as noted by the high helix melting temperature (Tm = 47 °C), yet alanine and charged residues in the X position also yield stable helices with Tm exceeding 40 °C [139]. Aromatic hydrophobic residues and glycine in the X position are more destabilizing, with the resulting helices melting at 30-35 °C. Similarly, replacing hydroxyproline in the Y position in the sequence Ac-(GPO)3-G-P-Y-(GPO)4-GG-NH2 reduces stability. Raines and coworkers showed that replacing hydroxyproline with fluoroproline in (GPO)10 yields exceptionally stable triple helices, with melting points as high as 90 °C [144].
Stereochemistry of the hydroxyproline or fluoroproline residue contributes appreciably to stability: replacing naturally occurring (4R)-hydroxyproline with (4S)-hydroxyproline or (4S)-fluoroproline disrupts triple helix formation [145,146]. Additionally, enantiomeric mixtures of the collagen-like peptide (PPG)10 enhance thermal stability relative to homochiral analogues [147]. Yet, blends of (dPdPG)10 with (lPlOG)10 do not interact, pointing to the role of other molecular interactions in facilitating stereochemistry-dependent assembly.
Introduction of electrostatic interactions to collagen-like peptides modulates assembly [148–153] and enables formation of heterotrimeric triple helices of a cationic, anionic, and neutral collagen-like peptides [154,155]. Notably, by leveraging electrostatic interactions, it is possible to prepare collagen-like peptide triple helices without hydroxyproline residues, enabling recombinant synthesis of these biomaterials [156]. Combining molecular simulations and experiments recently augmented understanding of the role of electrostatic interactions and non-natural amino acids in assembly of collagen-like peptides [157]. For example, introducing charged or non-natural alkene-modified amino acids reduces the triple helix melting temperature relative to that of (POG)10.
Additional ways to tailor the stability of collagen-like peptides include variation of sequence length and chain end modification. Similar to coiled coil peptides, the stability of collagen-like peptide triple helices increases with peptide length [143,158]. Placing two cysteine residues near the N-terminus of collagen-like peptides affords triple helices bound by a collection of three disulfide bonds termed a ‘cystine knot’ [159]. These triple helices are markedly more stable than analogous triple helices without cysteines, and the cystine knots contribute to the stabilization of hydroxyproline-free triple helices [156]. Placing acetyl groups at the chain ends provides additional stability, attributed to the removal of repulsive electrostatic interactions [143].
2.2.2.2. Higher order assembly of collagen triple helices
In nature, collagen triple helices called tropocollagen assemble in a staggered fashion into fibers [160,161]. Hydrogen bonding, metal-ligand, electrostatic, hydrophobic, and pi-pi stacking interactions between peptide side chains and at the chain ends facilitate lateral and end-to-end assembly of collagen triple helices. The resulting fibers are more stable than the triple helices, as gauged by higher helical melting temperatures, which further increase with peptide length. For example, in aqueous solution at neutral pH and peptide concentrations near 1 mM, (Pro-Hyp-Gly)10 forms fibrillar structures that melt 20 °C higher than the unaggregated triple helices [161]. Increasing temperature accelerates assembly, which occurs most rapidly just below the triple helix melting temperature [161,162]. Triple helices form at neutral pH but not typically at low (<3) or high pH (>10), attributed to fewer hydrogen bonds under the latter conditions. Addition of sugar and replacing Hyp with Gly in the Y position precludes assembly, highlighting the role of hydroxyproline-mediated hydrogen bonding. Replacing a glycine residue with alanine, as occurs in diseases like osteogenesis imperfecta and Alport syndrome, interferes with assembly of (Pro-Hyp-Gly)10 triple helices [162]. However, replacing glycine with hydroxyl-containing serine, promotes fibril formation. Embedding pendent bipyridyl groups in the middle of collagen-like peptides facilitates iron-mediated lateral assembly of triple helices [163,164], while placing metal-binding ligands at the chain ends as well as in the middle of the sequence organizes collagen-like peptides into three-dimensional frameworks [164]. Collagen-like peptides with charged end segments, for example (PRG)4-(POG)4-(EOG)4,[148] assemble into periodic fibers guided by electrostatic interactions between the cationic arginine or lysine residues and anionic glutamic acid or aspartic acid residues [150,151,153]. Hydrophobic and pi-pi stacking interactions also drive end-to-end assembly. Placing electron-rich phenylalanines and electron-deficient pentafluorophenyl-containing residues at the N- and C-termini of (POG)10 enables end-to-end assembly of the collagen triple helices into micron-length fibrils [4,165]. Kloxin and coworkers varied length and implemented electrostatic interactions and aromatic phenylalanine-pentafluorophenyl pairs at the ends of collagen-like peptides to engineer two assembling CMPs with distinct thermal stability (Figure 9). While both peptides form fibers in aqueous solution, peptide 2a is more thermally stable (Tm ~ 62 °C compared to 37 °C for 1a).
Figure 9.
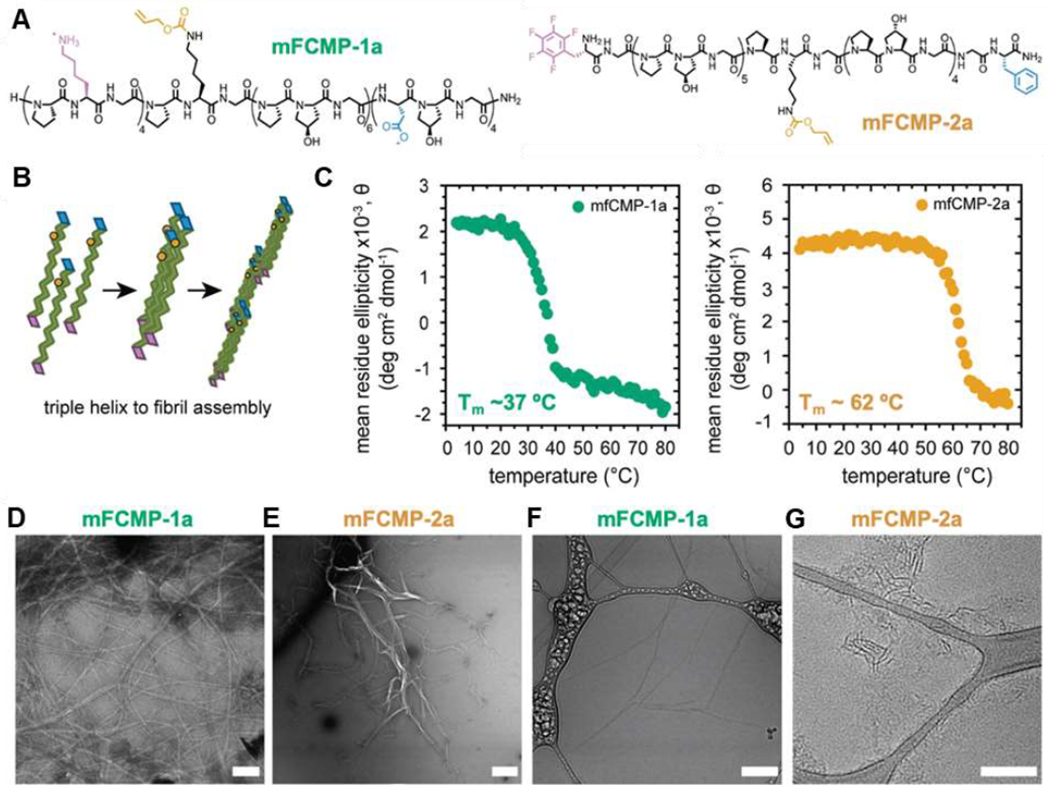
Assembly of collagen-like peptides into triple helices and fibrils: (A) primary structure of multifunctional collagen-mimetic peptides (mFCMPs) 1a and 2a; (B) schematic showing triple helical and higher order assembly of the mFCMPs; (C) CD spectroscopy-based melting temperature measurements, monitoring the ellipticity at 225 nm as a function of temperature, showing mfCMP-2a to melt at higher temperatures than mfCMP-1a; (D, E) TEM and (F, G) cryo-TEM of the mFCMPs. Scale bars = 200 nm. Adapted with permission from Hildebrand et al. [149], Copyright (2020) The Royal Society of Chemistry.
2.3. Peptide amphiphiles
Peptide amphiphiles (PAs) form supramolecular structures that combine the functionality of peptides with the hierarchical organization of lipids. Typically consisting of a long, hydrophobic alkyl chain bound to a peptide, these molecules assemble in aqueous solution into fibers, ribbons, and sphere-like micelles for use in biomedical applications [166–168]. While reports describing attempts to prepare PAs by attachment of lipids to peptides in solution appeared in the early 1980s [169,170], these materials were not widely investigated until the next decade, when Tirrell, Fields, and coworkers utilized a solid-phase peptide synthesis approach to attach a hydrophobic dialkyl glutamate tail to the N-terminus of collagen-derived peptides [171,172]. These collagen-like PAs assemble into triple helical structures with greater thermal stability than collagen-like peptides without hydrophobic tails, as evidenced by 15-20 °C increases in the melting temperature. Their studies laid the groundwork for current PA materials platforms.
In 2001, Stupp and coworkers reported a PA system that forms a bone-like composite of peptide fibers and hydroxyapatite deposits [173]. The design of this material continues to template PAs today, which contain three or, more commonly, four domains (Figure 10) [166,167,174–176]. The first domain is the hydrophobic tail, typically an unbranched alkyl group. In aqueous solution, this tail forms the hydrophobic core of the assembly, enabling surface presentation of the hydrophilic peptide. The second domain often consists of a peptide portion with defined secondary structure, such as a β-sheet forming sequence. Charged amino acid residues, which impart aqueous solubility and modulate gelation behavior, make up the third domain. Often, PAs feature a fourth bioactive signaling epitope domain to bolster the biological function of the material.
Figure 10.
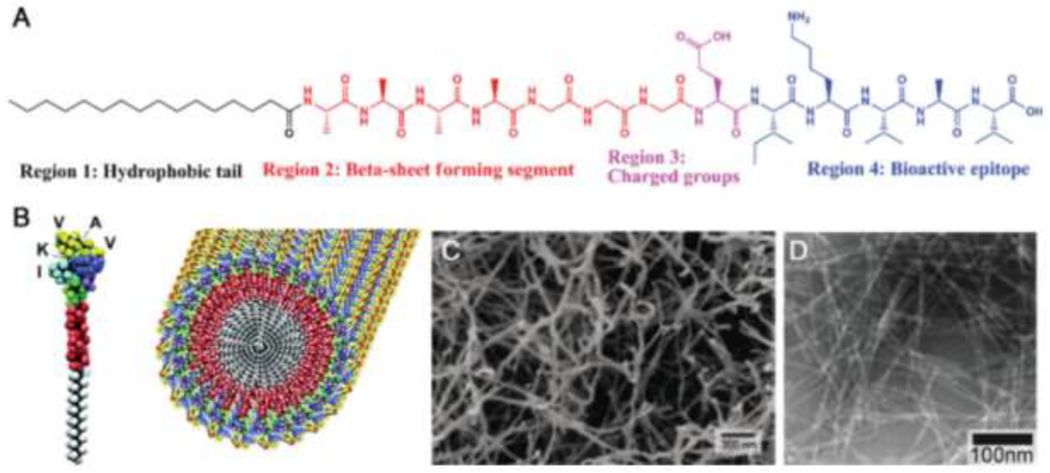
Structure and assembly of PAs: (A) typical structure of a PA, including a hydrophobic tail, β-sheet forming region, charged region, and a bioactive epitope; (B) rendering of a PA with an IKVAV bioactive epitope and the assembly of the IKVAV-presenting PA into nanofibers; (C) scanning electron micrograph of the nanofiber network formed by addition of cell media to an aqueous solution of IKVAV-PA; (D) transmission electron micrograph of the IKVAV-PA nanofiber network. Adapted with permission from Cui et al. [167], Copyright (2010) Wiley-VCH GmbH & Weinheim.
Examples of PA design showcase the versatility of this class of assembling peptides. In the case of the bone-like composite material reported in 2001, the 16-carbon alkyl chain tail yields conical PAs that form fibers in aqueous solution [173]. The peptide portion of the PA (CCCCGGGJRGD, where J is phosphoserine) contains a mineralization-promoting phosphoserine residue and a cell adhesion-promoting RGD motif. Additionally, the CCCC region modulates network strength and the GGG region is a flexible linker. Acidification of the aqueous peptide solution below pH 4 induces assembly into fibrous hydrogels. Raising the pH back to neutral disassembles the hydrogel. We note that, rather than gelation resulting solely from entanglement or interactions between fibers, structuring of surface water on the fibers may also play a role in gelation [177]. Additionally, oxidation of cysteine thiols to form disulfide bridges between adjacent molecules locks the supramolecular structure in place [173]. After oxidation, the PA fibers remain stable at pH 8 for months, whereas unoxidized PAs disassemble within minutes. Further investigation of PAs, varying alkyl tails, peptide sequence, and C-terminal ligands, revealed that each of 12 PAs self-assemble into fibrous networks with tunable surface compositions and assembly conditions, furnishing a functional materials platform for biomedical applications [178].
Assembly of the hydrophobic alkyl tails in aqueous solution, together with interactions between secondary structure-directing peptide segments and electrostatic repulsion of the charged residues in the third domain determine the interfacial curvature, and by extension, the size and shape of PA assemblies. Both the tail length and composition of the hydrophobic structures making up the tail tune assembly [178]. While PAs with tails of ten or more carbons assemble in aqueous solution, PAs with shorter tails do not. Typically, the second domain peptide segment contains hydrophobic amino acids likely to hydrogen bond, often β-sheet forming sequences. Interchain hydrogen bonding is important for the formation of one-dimensional nanostructures (fibers) as opposed to the spherical structures predicted to form from conical PA molecules in water. Accordingly, experiments and molecular simulations show that spherical micelles form upon disruption of hydrogen bonding of PAs by N-methylating amino acids [179]. The electrostatic repulsion between the charged residues provides solubility while simultaneously allowing for on-demand assembly (e.g., upon injection into physiological environment or by adjusting pH). For example, the addition of a glutamic acid residue in the IKVAV-functionalized PA (A4G3EIKVAV) imparts salt-triggered assembly at neutral pH, which is beneficial for encapsulating cells [180].
Mixtures of complementary PAs yield reversible networks that mimic the dynamic mechanical properties of the natural extracellular environment [175,176]. Functionalizing PAs with oligonucleotides, then blending the resulting fibers with PA fibers decorated with complementary oligonucleotides induces rearrangement into fiber bundles driven by oligonucleotide hybridization. Yet, no rearrangement occurs for PAs containing β-sheet motifs that strengthen, but reduce the dynamics of, the assemblies. The hierarchical bundled fibrous morphology of these PA-based biomaterials invokes distinct cell phenotypes compared to analogous dispersed fibers, showcasing the potential of these materials to direct cell behavior.
2.4. Block copolypeptides
Block copolypeptides are large amphiphiles featuring a block of hydrophobic residues connected to a block of hydrophilic residues and can contain 200 or more amino acids per chain. In aqueous solution, block copolypeptides assemble into stable, shear-thinning, and self-healing hydrogels driven by the association of the hydrophobic domains. Deming and coworkers introduced this class of entirely peptidyl amphiphilic SAPs in 2000 [181], demonstrating that diblock copolypeptides of a hydrophobic poly-l-cysteine block and a hydrophilic poly-l-lysine block form aggregates 600-1300 nm in diameter. Thereafter, they showed that block copolypeptides with either poly-l-lysine (K) or poly-l-glutamate (E) as the hydrophilic block and α-helical poly-l-leucine (L) as the hydrophobic block form hydrogels in aqueous solution [182]. For example, the amphiphile K180L20 forms 3 wt% peptide hydrogels with a storage modulus of 12 Pa that are stable up to 90°C and have a minimum gelation concentration of 2.0 wt%. Increasing the hydrophobic content to 20% (K160L40) decreases the minimum gelation concentration to 0.25 wt% and increases the modulus of the 3 wt% gels to 4300 Pa [183]. Further, hydrogel formation depends on secondary structure; the block copolypeptide K190L10, in which the leucine block is too short to form a helix, does not form a hydrogel, whereas those with longer, helix-forming leucine blocks (K180L20, K170L30, and K160L40) form hydrogels [184]. The block copolypeptides discussed thus far all assemble in aqueous solution in the absence of salt, but salt can have a large effect on hydrogel properties due to the role of electrostatics in these assemblies [185].
In contrast to examples of charged synthetic block copolymers [186], which form spherical micelles or vesicles with a hydrophobic core in aqueous solution, Deming’s block copolypeptides assemble into fibers and fibrillar networks. The packing of rigid helices in the hydrophobic domain decreases interfacial curvature, hindering spherical assembly and instead promotes the formation of fibrous hydrogels, with many block copolypeptides gelling below 1.0 wt% [182,183,187]. The hydrophilic polyelectrolyte domain plays an important role in assembly. While the K180L20 copolypeptide forms gels, K80L20 does not, even at concentrations up to 6 wt%. The authors suggest that gels form from peptides with longer charged segments due to electrostatic repulsion that distorts (twists) the packing of helices to generate fibers [183]. Increasing the total length of the block copolypeptides lowers the minimum gelation concentration and increases the modulus of gels, albeit to a smaller extent than changes in the hydrophobic domain. Similarly, triblock copolypeptides having two hydrophilic blocks form stronger hydrogels than their diblock copolypeptides counterparts. These observations taken together suggest a folding mechanism by which interchain electrostatic interactions distort the packing of helical chains, which prefer flat 2D sheets, into fibrillar tapes that entangle to form a gel network [184].
Block copolypeptide hydrogels allow facile tuning of gelation behavior through hydrophilic and hydrophobic block length and composition. This tunability, taken together with the shear thinning, rapid self-healing, and high stability exhibited by these gels, highlights the potential for block copolypeptide use in tissue engineering.
2.5. Elastin-like polypeptides
Elastin-like polypeptides (ELPs) are amphiphilic SAPs that mimic the elasticity, self-assembly, and stability of natural elastins [188–190]. The primary structure of ELPs is derived from the elastin precursor tropoelastin, of which the hydrophobic domain contains 1 to >100 repeating units of the pentapeptide VPGVG. Urry was among the first to study engineered ELP sequences of (VPGVG)n, which upon covalent carbodiimide-mediated crosslinking form fibers and gels with elastic moduli of ca. 213 kPa, similar to native elastin [191].
ELPs feature temperature-responsive behavior [192,193]. Below the transition temperature (Tt), ELPs adopt a random coil structure and remain in solution. Upon increasing the temperature above the Tt, the polypeptide collapses into a β-spiral structure with type II β-turns, a structural motif that reverses the direction of the peptide chain, facilitated by the PG segments. This leads to aggregation and phase separation, producing a polypeptide-rich coacervate phase, similar to synthetic polymers displaying lower critical solution temperature behavior. The release of structured water entropically drives ELP folding above the Tt, enhancing hydrophobic interactions of the valine (V) residues [193]. ELP aggregation is reversible, as lowering the temperature below the Tt will resolubilize the ELP. Urry and coworkers showed that all ELPs of the form (VPGXG)n, where the ‘guest residue’ X may be any amino acid except proline, exhibit this thermo-responsive behavior [194,195]. Substituting proline for X disrupts the type II β-turn [195]. The identity of X modulates the Tt; generally, hydrophobic residues lower Tt [195,196]. ELPs display a remarkably wide range of Tt, with X = tryptophan affording the lowest Tt at −90 °C and X = anionic glutamic acid providing the highest Tt at 250 °C in phosphate-buffered saline [195]. Notably, even one repeating unit of VPGVG is sufficient to induce the thermo-responsive transition, as Ac-GVG-(VPGVG)-NH2 has a Tt at 21 °C [193], and increasing length, or the number of repeats, decreases Tt [196]. Several ELPs, including those with alanine, glycine, and isoleucine as the guest residue, have a Tt near physiological conditions to enable use as ECM scaffolds or drug delivery vehicles [189,197–200].
ELPs can be chemically cross-linked into fibers, hydrogels, and other ordered structures [190,201,202]. However, a recent report indicates that short ELPs with three N-terminal isoleucines and two C-terminal lysines form fibrils without crosslinking, while analogues lacking the additional residues at the termini form only amorphous aggregates [203]. Increasing the number of pentapeptide repeats in the ELP increases fibril length. ELPs may also be combined in hybrids with silk proteins [204], fibronectin [205,206], collagen-like peptides [207], coiled coils [208,209], and other components [210] to impart phase separation and elastic behavior. The tunable Tt of ELPs makes them attractive biomaterials for a variety of applications requiring thermo-responsive behavior, such as gelation at body temperature after injection.
2.6. Classes of assembling peptides: summary and perspectives
Assembly of peptides into fibers or fibrous hydrogels for use in biomaterials relies on a combination of amphiphilic character and adoption of helical or β-sheet secondary structures, among other factors. Since peptide sequence determines secondary structure, it is difficult to directly compare the different classes of peptide biomaterials. Nevertheless, we provide a few considerations for designing biomaterials from helical vs. β-sheet forming peptides. The generation of stable helices requires longer sequences, which add manufacturing and purification challenges owing to the stepwise nature of solid phase peptide synthesis. Yet, longer helical peptides offer an expanded sequence design space and therefore more opportunities to tune molecular interactions, morphology, mechanical properties, and biological function. While the studies we highlighted, among others, established design rules for both helical and β-sheet forming peptides, continued study will provide a deeper understanding of native and engineered sequences that may clarify some guiding principles and enable more tunability in future materials [211–213]. To this end, advanced rational design approaches combining computational and experimental approaches for rational design are particularly impactful [81,138,146,157,214–217]. It is worth noting that while fibers and fibrous hydrogels can form from simple bilayers of β-sheet peptides as the building blocks, fibril formation and gelation from helical peptides, e.g., collagen-like peptides or coiled coils, often requires higher order assembly of helical bundles. Yet, this hierarchical assembly of helical bundles is the basis of structural fibers in mammalian biology, and therefore merits investigation as components of biomaterials. Especially given the increased manufacturing difficulties of longer helical peptides, preparing composites of helical peptide fibers with synthetic polymers, as reported recently by the Kloxin group[149], warrants further investigation.
3. Peptide Characterization Methods
Understanding the physical and chemical characteristics of peptide assemblies is integral to establishing design rules and matching these materials to biomaterials and tissue engineering applications. Peptide hydrogels that replicate the stiffness or morphological structure of a tissue often are the most successful for in vitro and in vivo applications. At the molecular level, nuclear magnetic resonance (NMR) spectroscopy, infrared spectroscopy (IR), and circular dichroism spectroscopy (CD) determine the chemical composition (primary structure) and peptide conformation (secondary structure). Optical, scattering, calorimetric, and imaging techniques provide insight into supramolecular structure and mechanisms of assembly. NMR spectroscopy, fluorescence-based assays, and isothermal titration calorimetry measure intermolecular interactions. Dyes, such as Congo Red and Thioflavin T, bind β-sheet structures to aid in visualization. Scattering techniques (e.g., dynamic light scattering, x-ray scattering, and neutron scattering) together with atomic force microscopy (AFM) and transmission electron microscopy (TEM) provide nanoscale morphological information. Rheology and gravimetric stability measurements yield bulk materials properties. It is important to use multiple of these methods in tandem to generate comprehensive understanding of peptide biomaterials from the molecule up.
3.1. NMR spectroscopy
In addition to confirming molecular structure, NMR spectroscopy provides a valuable label-free means to characterize supramolecular interactions [218–220], such as those driving peptide assembly [221–228]. The direction and magnitude of a chemical shift change, e.g., before and after mixing two components or increasing concentration, provides information about the assembly kinetics and thermodynamics, including rate constants, stoichiometry, and the residues most involved in the interaction. For example, spectra acquired before and after sonication of fluorinated peptides to induce mixing showed a downfield change in chemical shift indicative of conformational changes [227].
In metal-induced assembly of amyloid β (Aβ) fragments, NMR spectroscopy showed the N-terminal Aβ residues 1-28 to be most involved in binding zinc [221] and cadmium [223], as the largest chemical shift changes occurred for protons in this region. By titrating assembling or co-assembling peptides and acquiring spectra after each addition (Figure 11) for the addition of Zn2+ and Cd2+ to Aβ residues 1-16 [223], NMR spectroscopy can help determine the thermodynamic dissociation constant, KD, of an assembling peptide from the changes in chemical shift (in this case of valine methyl proton resonances) and the concentrations of the component(s) [222,229]. The peak shape reveals kinetic information about assemblies relative to the experimental time scale, approximately 22 ms for a 500 MHz NMR spectrometer [230]. In strong complexes, exchange of molecules between the bound and unbound states that is slower than the experimental time scale yields two distinct NMR signals, whereas rapid exchange in weak complexes affords a single peak [228]. Peak broadening indicates that exchange between the free and bound states occurs on the timescale of an experiment. Together with the changes in chemical shift, the degree of NMR signal broadening determined metal binding sites on Aβ [221,223–225].
Figure 11.
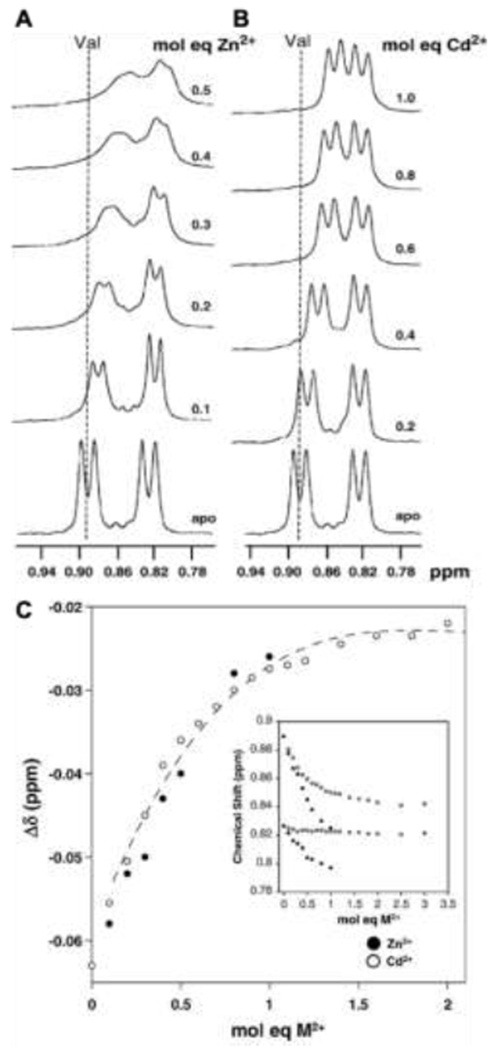
1H NMR spectra of Aβ(1–16) showing valine methyl group resonance shifts during (A) Zn2+ and (B) Cd2+ titration. Increasing molar equivalents of metal ion titrated resulted in larger chemical shift changes, indicating stronger binding. In (C), the plot of valine γCH3 chemical shifts (ppm) against the molar equivalents of Zn2+ and Cd2+ relative to Aβ shows increasing interactions of the peptide with metal up to equimolar ratios, indicating a 1:1 stoichiometry. Insert indicates chemical shifts for the γ and γ’ methyl protons of valine. Adapted with permission from Syme and Viles [223], Copyright (2006) Elsevier.
Providing label-free thermodynamic and kinetic information about peptide assembly, as well as chemical information about the amino acids engaged in supramolecular interactions, NMR spectroscopy is an extremely powerful technique for understanding peptide assembly. The studies highlighted here and in the synthetic materials literature [218–220] showcase the tremendous potential of this technique for informing peptide design for biomaterials.
3.2. Circular dichroism spectroscopy
Circular dichroism (CD) spectroscopy provides information about the secondary structure, stability, and the conformational transitions of peptides upon assembly in solution. The difference in absorbance of left- and right-handed circularly polarized light in the far UV region (190-250 nm) by a peptide solution gives the ellipticity (ɛ) in millidegrees (mdeg). Normalizing ellipticity by residue or peptide concentration affords the mean residue or molar ellipticity (θ), respectively, in mdeg cm−2 dmol [231–234]. Characteristic spectral features for helical, β sheet- and random coil-forming peptides enable determination of secondary structure. The CD spectra of α-helical peptides with natural l-type stereochemistry contains negative peaks at 208 and 222 nm and a positive peak at 193 nm, while the spectra of β sheets contain a negative peak at 218 nm and a positive peak at 195 nm [233]. Random coil spectra have a minimum around 195 nm. Several methods are available for calculating the percentage of a certain secondary structural motif in a given sample. The ellipticity at 222 nm (θ222) is a common measure of peptide helicity; plotting θ222 as a function of temperature allows determination of helix stability by the melting temperature [117,119,234]. Time-dependent measurements of ellipticity provide kinetic information [96]. Greenfield and coworkers published CD spectra methods for determining thermodynamic parameters associated with protein unfolding from [231,232,234,235] that have been adapted to study peptide assembly.
Changes in CD spectral features as a function of time, temperature, pH, concentration, and ratio of co-assembling peptides allow monitoring of self-assembly [236], co-assembly [237,238], and higher-order assembly, including calculation of thermodynamic and kinetic binding constants. Concentration-dependent changes in the melting temperature of solutions with just one type of peptide indicate self-assembly. Klok and coworkers applied CD spectroscopy to study the pH-dependent heterodimeric assembly of coiled coil E3/K3 peptides; the CD-derived association constant was 6.85×107 M−1 which compared reasonably with those obtained from fluorescence resonance energy transfer (4.57×107 M−1) and isothermal titration calorimetry (2.76×107 M−1) [124]. In preparing hydrogel-forming polymer peptide conjugates, CD spectroscopy determines whether peptides retain secondary structure after conjugation [3,239–241].
3.3. Infrared spectroscopy
Like CD spectroscopy, IR spectroscopy probes the secondary structure of peptides. Whereas CD spectroscopy measurements require transparent solutions, IR spectroscopy probes secondary structure in both solution and solid state, allowing studies on peptide-based hydrogels [83,87]. Distinct bonding configurations among peptides with random coil, helical, or β-sheet secondary structures allow determination of secondary structure from characteristic infrared amide I C=O absorptions in the 1550-1700 cm−1 range [242][243]. Absorbance in the 1650-1660 cm−1 range indicates α-helical secondary structure [243][244][83], whereas β-sheet peptides have a characteristic absorbance 1610-1640 cm−1. Identification of parallel vs. antiparallel β-sheets is also possible, since the latter yield an absorbance around 1675-1690 cm−1 [99]. Random coil secondary structures yields amide I absorptions from 1640-1650 and 1660-1685 cm−1 [243].
For characterization of the secondary structure of enantiomeric peptide mixtures [86], IR spectroscopy is particularly useful, as the signals from d- and l-peptides would cancel in CD spectra. Additionally, isotope-edited infrared spectroscopy can gauge whether complementary peptides co-assemble or self-sort [86][37]. Labeling 1-2 amino acids within a peptide with 13C groups changes the vibrational behavior of the peptides in the assembled state due to coupling of the vibrations of the heavier atoms. For example, labeling an l-peptide, and then blending it with an unlabeled d-peptide yielded different absorbances relative to assemblies of each of the individual components, indicating co-assembly of the enantiomers [37]. If no change occurred upon mixing, this result would indicate self-sorting, rather than co-assembly.
3.4. Fluorescence-based methods
3.4.1. Förster resonance energy transfer
Förster resonance energy transfer (FRET) involves energy transfer from an excited molecule (donor) to a nearby acceptor molecule [245–247]. Typically, for FRET to occur, the distance between donor and acceptor molecules must be less than 10 nm, and the emission spectrum of the donor must overlap with the absorption spectra of the acceptor. Exciting the donor and observing acceptor fluorescence indicates FRET is occurring and therefore the donor and acceptor molecules are in close proximity. Alternatively, FRET between a fluorophore and a quencher markedly decreases fluorescence. In the context of peptide assembly, FRET elucidates assembly mechanisms and kinetics. For example, quenching of donor emission accompanied by an increase in acceptor fluorescence characteristic of FRET indicated co-assembly of nitrobenzofuran-labeled l-L4K8L4 and rhodamine B-labeled d-L4K8L4 upon mixing of the two enantiomers at pH 9, but not at pH 7 [96]. Nilsson and coworkers used FRET with the donor fluorophore (5-((2-aminoethyl)amino)naphthalene-1-sulfonic acid) (EDANS) and acceptor quencher 4-(dimethylaminoazo)benzene-4-carboxylic acid (DABCYL) to demonstrate that d- and l-(FKFE)2 co-assemble rather than self-sort into separate fibrils [88]. Further examples of FRET used in SAP systems include monitoring enzymatic cleavage in PAs [248], determining the arrangement (antiparallel vs. parallel) of strands in a β-sheet [249], and comparing the dynamics of polymer-peptide nanotubes in solution and in cells [250]. However, overlap between the broad emission spectra of donor-acceptor pairs, environment-dependent fluorescence, and low signal-to-noise ratios can complicate FRET measurements and analysis [246,251].
3.4.2. Fluorescence Quenching
Fluorescence quenching can refer to any phenomena, including FRET, which diminishes fluorescence intensity. Often, introduction of a quencher reverts the fluorophore to the ground state without emission of fluorescence [252]. For example, in the complexation of the SAP EAK16 with fluorescein-labeled oligodeoxynucleotides (ODNs), the fluorescence quencher potassium iodide was added to an aqueous mixture of the two components to determine whether the ODNs localized inside or remained on the outward surface of the aggregate [253]. In other cases, changes in conformation or aggregation induce quenching. The amino acid tryptophan (W) is intrinsically fluorescent, allowing concentration determination and assembly studies, since aromatic stacking during assembly quenches tryptophan fluorescence [254]. Tryptophan quenching elucidated the assembly mechanism of the peptide Boc-WLWL-OMe (W = tryptophan; L = leucine; OMe = methoxy C-terminus) [255]. While tryptophan fluorescence increased with increasing peptide concentration up to 0.156 mM, further increases in peptide concentration decreased the tryptophan fluorescence, indicating that aromatic stacking begins at that concentration. However, similarly as FRET, sensitivity to the fluorophore environment may limit quenching studies.
3.4.3. Congo red- and Thioflavin-based staining
Introduced in 1885 as a textile dye [256], Congo red provides a valuable tool to study amyloids, protein deposits that aggregate into β-sheet structures and causes adverse long-term outcomes in diseases such as Alzheimer’s [257,258]. Upon binding to amyloids, Congo red emission shifts from 490 nm to 540 nm [259]. Above ca. 5 μM, the dye forms micelle-like structures, which can alter amyloid aggregation and sometimes reduce cytotoxicity [260]. Apart from visualization of fibers and amyloids [16], Congo red also stains collagen, elastin, and other amyloid structures for histological studies [258]. While Congo red assays are relatively simple to implement, the pH-dependent fluorescence and lack of specificity require attention during experiment design [258].
Thioflavin T (ThT), introduced in 1959, is a cationic dye that undergoes both an emission shift, from 445 to 482 nm, and fluorescence intensity enhancement upon binding amyloids [259,261]. This hallmark intensity enhancement eliminates the need for washing and enables in situ observation of fibrilization [261–263]. ThT does not seem to affect amyloid fibrillation kinetics and therefore preserves the structure of peptide hydrogels when imaging [259]. However, ThT binding affinity is sequence dependent, binding preferentially to aromatic residues via pi-pi stacking and exhibiting limited binding to charged residues, particularly cationic residues at low pH due to electrostatic repulsion [259]. Thioflavin S (ThS), has a structure similar to that of ThT but with an additional aromatic group; ThS also labels amyloid structures, though most commonly for diagnosis of amyloid fibril deposits in tissue sections [259], rather than for imaging peptide assemblies.
3.5. Isothermal titration calorimetry (ITC)
ITC provides thermodynamics parameters associated with supramolecular interactions, including those of self-assembling and co-assembling peptides. These experiments involve measuring the heat associated with titration of a ligand (or assembling peptide) into a solution of receptor, or more of the same compound in the case of self-assembling molecules. Plotting the heat input required to hold a solvent-containing reference cell at the same temperature as the sample cell upon injection of a known concentration of ligand produces a thermogram [264,265]. Thermogram analysis yields the binding enthalpy, equilibrium association constant, and the stoichiometry of the complex. Control experiments, namely 1) injection of the peptide ligand (i.e., the peptide in the syringe) into solvent and 2) injection of solvent into the peptide receptor (i.e., the peptide in the sample cell), allow subtraction of heats of dilution [266]. During the binding and control experiments, it is particularly important to ensure the solutions in the syringe and cell are at precisely the same pH, as protonation/deprotonation events can produce substantial heat. Nilsson and coworkers showed enantiomeric mixtures of cationic and anionic peptides have a more favorable binding interaction than the analogous homochiral mixtures [88]. Similarly, Highley and coworkers determined binding energies of coiled coil-forming peptides as a function of length and sequence using ITC [127]. For self-assembling compounds, ITC provides a critical aggregation concentration (CAC), which reflects the propensity of a peptide to assemble, and mechanistic information about the driving forces of assembly. Injecting aqueous solutions of the SAP RADA4 above the CAC into dilute aqueous solution disassembles the peptide during the first several injections and endothermic peaks appear on the thermogram [267]. With further injections of RADA4, the sample cell concentration increases, and the endothermic heats decrease and then transition to exothermic heats of dilution of the peptide assemblies. The CAC is the minimum on a plot of the change in enthalpy with respect to concentration. Performing temperature-dependent experiments and determining the derivative of enthalpy associated with disassembly with respect to temperature gives the heat capacity. In general, exposure of hydrophobic groups to polar media leads to positive heat capacities, whereas hydration of polar groups result in negative heat capacities [267,268]. For example, RADA4 exhibits a positive heat capacity upon titration into 50 mM phosphate buffered saline (PBS, pH 7.4), whereas a more hydrophilic serine-containing RADA4 derivative had a negative heat capacity under the same conditions.
3.6. Scattering
3.6.1. Dynamic light scattering
Dynamic light scattering (DLS) measures the diffusion of particles in solution, allowing calculation of hydrodynamic radius, or size in solution [269–272]. Brownian motion of particles (e.g., peptides or peptide assemblies) in solution produces intensity fluctuations in the particle-scattered light, allowing determination of diffusion coefficients. Provided the solution refractive index, viscosity, and temperature, the Stokes-Einstein equation allows calculation of the hydrodynamic radius. DLS is a bulk measurement, furnishing an average size distribution of particles in solution (or even in hydrogels [273]) to complement nanoscale measurements of size and shape via electron and atomic force microscopy. DLS, in conjunction with Thioflavin T measurements, of the SAP leucine-dehydrophenylalanine showed the size to increase with concentration, halting the measurements above 0.4 wt% as the structures exceed the nanometer scale [263]. The nondestructive nature of DLS enables repeated measurements on the same sample over hours, days, or weeks, including as a function of temperature, for monitoring assembly of peptides and the stability of the resulting nanostructures. DLS can also determine the CAC of peptide assemblies [273]. We note that since the Stokes-Einstein equation assumes spherical particles, the DLS-derived size measurements of fibrillar structures formed frequently from SAPs cannot provide the actual diameter or radius of the fibers, but rather give the average and distribution of nanostructure dimensions [274,275]. Furthermore, nanostructures must be suspended in solution as sedimentation effectively removes material from the path of the light and any quantification.
3.6.2. Small-angle x-ray and neutron scattering
Small-angle x-ray scattering (SAXS) and small-angle neutron scattering (SANS) allow bulk measurements of nanostructure size and shape in solution. Electrons scatter x-rays, while atoms scatter neutrons. In both measurements, the scattering angle corresponds to a length scale, and thereby provides information about the dimensions of structures in the sample. While SAXS can cause more damage to samples than SANS, the shorter acquisition times for SAXS enable time-dependent measurements. In SANS measurements, contrast matching by varying the ratio of H2O:D2O solvent can isolate different components in a sample and provide chemical information. We provide several examples here that highlight the power of these techniques in understanding structural features of peptide assemblies, and direct readers to two excellent reviews that detail the theory and application of small angle scattering techniques to investigate peptide-based biomaterials [276,277]. All of these scattering methods require the use of advanced equipment, typically only accessible in government run national lab facilities.
Using SANS, Schneider and coworkers interrogated peptide hydrogels formed from enantiomeric peptide mixtures [86] after rheology showed these hydrogels to be stiffer than hydrogels formed from the individual peptide enantiomers [87]. Reasoning that hydrogels with a higher cross-link density and a smaller mesh size would yield a higher scattering intensity at a given scattering angle (0.01 Å−1 corresponding to a characteristic length scale of 10 nm), they performed SANS on hydrogels formed from d-peptides, l-peptides, and their enantiomeric mixtures. Similar scattering intensities from all three hydrogels indicated similar mesh sizes and cross-link densities. This finding, together with diffusive wave spectroscopy experiments showing stiffer peptide fibers to form from the enantiomeric mixtures, led to the conclusion that the increased stiffness did not arise from crosslink density, but rather from the stiffness of individual fibers. In another study, SAXS experiments corroborated the formation of sheet-like structures from helical peptides [278]. Additionally, for hairpin β-sheet hydrogels designed to encapsulate chemotherapeutics, SANS experiments verified that encapsulation did not significantly alter the peptide hydrogel nanostructure [279].
3.7. Transmission electron microscopy
Exploiting the ability to focus negatively charged electrons to image at sub-nanometer resolution [280], TEM provides important complementary information to scattering techniques that give average information about nanostructure size and shape. Resolution increases with electron accelerating voltage, but contrast decreases due to fewer interactions with the sample. For the same reason, lower accelerating voltages leads to more sample interactions and energy absorption, leading to sample damage. These factors require balance when imaging soft materials, and typical accelerating voltages range from 80-300 keV [280]. Contrast increases with atomic number due to the higher number of scatterers; staining with heavy metals, for example, increases contrast of organic samples. Negative stains (e.g., uranyl acetate) increase scattering from the background, while positive stains are selective for the sample. Introduction of artifacts upon staining and/or drying samples for imaging under vacuum requires consideration during sample preparation. For peptide assemblies, TEM reveals detailed morphological features and their dimensions, from the pitch of twisted ribbon-like fibers [83] to the periodic structure collagen-like peptide triple helical fibers [4,148]. Cryogenic TEM (cryoTEM) largely alleviates artifacts due to drying, enabling imaging of thin films of sample in vitrified water [281]. Indeed, cryoTEM showed collagen-like peptide fibers to be half the width relative to those in the dry state (Figure 9D–G), suggesting that width increases upon drying [4].
3.8. Atomic force microscopy
Atomic force microscopy (AFM) probes morphology and materials properties at the nanoscale, providing complementary information to bulk measurements of the same properties (e.g., by scattering and rheology). Interaction of the probe (i.e., a reflective cantilever equipped with a tip) with a surface enables topological, chemical, and mechanical mapping with nanoscale resolution in several experimental modes [282]. Contact mode involves measuring the height at which the deflection of a cantilever reaches a setpoint to construct height maps (i.e., height images). However, the constant sample contact can damage soft samples. In similar nanoindentation experiments, measuring the force as a function of distance, given the cantilever spring constant, provides mechanical information [283]. For imaging soft materials, tapping mode is less destructive. In this mode, an oscillating cantilever approaches the surface, where dampened oscillations indicate proximity to the surface and thereby allow acquisition of height images that provide dimensional and morphological information. Differences in oscillations can further provide chemical information about a sample, since interactions between the tip and the surface can introduce a phase shift to the cantilever oscillation; a phage image maps the phase shift as a function of position.
In studying peptide assemblies, AFM confirms fibril morphologies [95,120,284–286], however the small width of fibrils complicates mechanical analysis [86,287]. Schneider and coworkers used AFM height images to quantify fibrils from hairpin β-sheet peptides as approximately 2 nm, consistent with a hairpin bilayer held together with hydrophobic interactions [87]. In characterizing helical peptide assemblies, AFM height images revealed single-layer 2D sheets [278] and the assembly of ca. 10 nm helical bundles into rods [138]. In most cases, sample preparation involves depositing peptide assemblies in solution or as dilute hydrogels, onto silicon wafers or freshly cleaved mica substrates, followed by rinsing with water to remove salt and drying. Similarly as in TEM, imaging in the dry state may distort morphological information, and tip-sample or substrate-sample interactions may introduce additional artifacts. Solution-state AFM imaging allows nanoscale imaging of peptide assemblies in the hydrated state, yet requires covalent or strong non-covalent (e.g., electrostatic) interactions to anchor assemblies to the surface. Therefore, as with any materials characterization, employing a suite of complementary techniques gives the most complete picture of the structure of assembled peptide materials.
3.9. Rheology
Rheology measures the bulk dynamic mechanical properties of peptide biomaterials, including stiffness and elasticity, for comparison to natural tissue mechanics, as well as shear-thinning properties important for designing injectable biomaterials [288,289]. Given the importance of mechanical and flow properties in the design, manufacture, and application of peptide-based biomaterials, rheological characterization is ubiquitous. Applying oscillatory shear to biomaterials and measuring the response yields the shear storage modulus G’, reflective of the solid-like character and stiffness of the sample, and the loss modulus G”, that characterizes the liquid-like behavior and viscous nature of the material. Measuring these moduli as a function of strain, frequency, and temperature in experiments called strain sweeps, frequency sweeps, and temperature sweeps, respectively provides integral information for the storage and use of peptide-based biomaterials in a variety of conditions [18,20,27,288,290]. It is common throughout the peptide [289,291], protein [292], and synthetic hydrogel [293] fields to use the criteria of G’ > G” to indicate gelation [294]. However, this is not an absolute definition for defining a gel; for example, some define gels as materials having G’ at least an order of magnitude greater than G” [295]. A transition to G” > G’ typically indicates gel disassembly or disintegration into a liquid-like material. Therefore, rheology allows monitoring of gel formation, deformation, and disassembly as a function of applied shear, time, and environmental changes [44,67,86]. The ‘cross-over point’ is the condition (shear rate, frequency, temperature) at which G’ = G”. In frequency-dependent experiments, the inverse of the frequency at the cross-over point yields a relaxation time scale for characterizing biomaterial dynamics [20,290]. The moduli of hydrogels depend on peptide charge, concentration, and solvent conditions, among other factors. For example, the storage moduli of cationic amphiphilic self-assembled peptides hydrogels decrease with the number of charged residues in the peptide and increase with increasing salt concentration and ionic strength [296]. Frequency sweeps allow calculation of the hydrogel mesh size using the Boltzmann constant, temperature, and the squared persistence length of fibril divided by the observed G’ at the frequency corresponding to the lowest G” [297]. Measuring viscosity as a function of strain rate describes the time dependency of viscoelastic flow and how materials can flow under strain: for instance, the viscosity at rest (low strain rates) or under significant applied strain, like during bioprinting or injection. Recovery experiments, involving cyclic application and removal of strain that determine the magnitude and response rate of changes in physical properties, are also relevant to injectable, printable, shear thinning and self-healing biomaterials [83,279,284,298–300]. For example, the viscosity of pentapeptide hydrogels decreases linearly with shear rate (shear thinning), presumably due to the disruption of the fibrillar structure (Figure 12C [83]). These hydrogels recover nearly instantaneously upon removal of applied shear over at least 4 cycles (Figure 12D [83]). In the investigation of peptide hydrogels designed to guide mineralization for bone tissue engineering, rheology show that the G’ of peptide hydrogels with a mineralization sequence increases after the addition of mineralization-inducing alkaline phosphatase; control hydrogels with the mineralization sequence reversed exhibit no such increase [67]. Additionally, strain sweeps show that encapsulation of cells in a peptide hydrogel minimally impacts mechanical properties [298]. While shear rheology is more common in characterizing the mechanics and gelation behavior of soft, viscoelastic peptide hydrogels, measuring the response to application of tensile or compressive strain gives the Young’s modulus for materials with more elastic character. Young’s modulus describes deformation in response to a normal force and is well suited to describing elastic materials with extensive linear portions in their stress-strain curves. Assuming the materials are incompressible (i.e., with a Poisson’s ratio of 0.5), Young’s moduli are approximately 3X the shear moduli [301].
Figure 12.
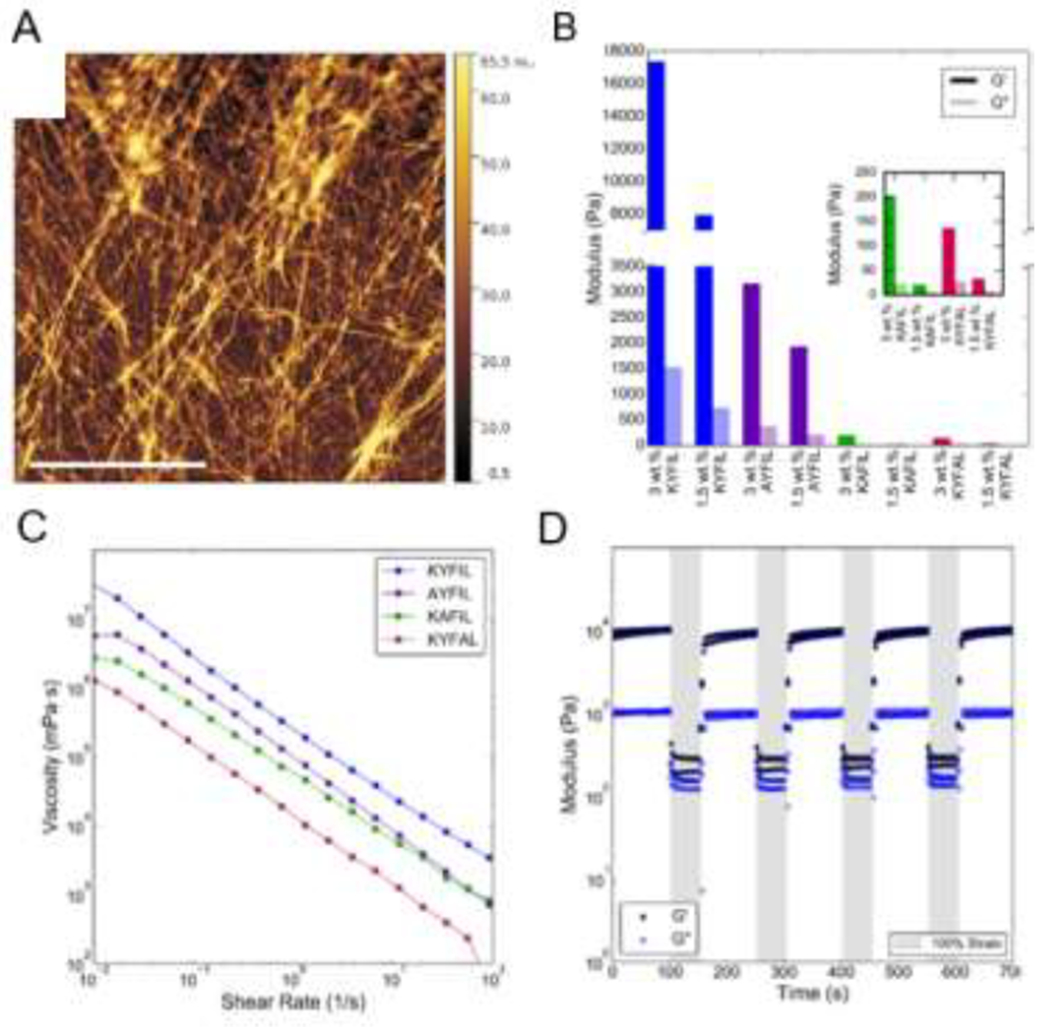
AFM and rheology of peptide hydrogels: (A) AFM height image of an H2 peptide hydrogel, adapted from Scelsi et al. [286], Copyright (2018) Wiley-VCH GmbH & Weinheim; and rheology of RAPID hydrogels, showing a strain sweep (B) to determine the storage (G’) and loss (G”) moduli. Studying hydrogel behavior as a function of shear rate (C) provides the shear-thinning behavior, while cyclically applying and removing strain (D) determines the stress recovery properties, which gauge the injectability of a hydrogel. Adapted with permission from Tang et al. [84], Copyright (2019) American Chemical Society.
3.10. Degradation
The stability of SAP-based biomaterials sets the conditions and time scale for use in tissue engineering and the release of encapsulated therapeutics. Owing to their nature as physical hydrogels, peptide-based biomaterials are inherently susceptible to remodeling via shear forces and dilution, in addition to denaturation (e.g., with hydrogen bond disrupting agents such as urea) and hydrolytic and enzymatic degradation. Peptide fibers can be made to dissolve, break, or slip past each other in response to cellular traction forces or protease secretion. Yet, assembly, gelation, and inclusion of protease-resistant D-peptides impart stability to peptide-based biomaterials. The β-sheet SAP EAK16 forms macroscopic membranes that remain intact against the degrading enzymes α-chymotrypsin, papain, proteinase K, and pronase, in surfactant solution at 90 °C, at high and low pH, and in the presence of hydrogen bond-disrupting guanidine hydrochloride and urea [16]. However, degradation can be beneficial in permitting cell remodeling of the hydrogel microenvironment which may be necessary for cytoskeletal rearrangement, changes in cell morphology, stem and progenitor cell proliferation or differentiation, and cell migration [28,302]. AFM images of d-EAK β-sheet fibrils before and after incubation with trypsin, pepsin, protease K, and pronase showed no evidence of degradation [303]. Peptides and proteins with d-amino acids resist protease degradation [304,305]; indeed Nilsson and coworkers found proteolytic stability of peptide fibrils to increase with d-peptide content, with the stability of fibrils from 1:1 mixtures of d- and l-peptides rivaling that of d-peptide fibrils [89]. These experiments involved incubation of peptide fibrils below the gelation concentration with α-chymotrypsin, trypsin, or proteinase K, followed by disassembly of the remaining fibrils by addition of dimethylsulfoxide. High performance liquid chromatography (HPLC) of the disassembled fibril fraction allows quantification of the concentration of intact peptide. Performing stability measurements below the gelation concentration removes mass transfer limitations imposed by hydrogels and isolates the inherent proteinase stability of the peptides. Integration of d-amino acids into hydrogel-forming diphenylalanine peptides improves stability to proteinase K, but decreases the propensity for assembly [305]. Notably, placement of a single d-amino acid at the C-terminal position imparts more stability than in the N-terminal position [305]. Similarly, Schneider and coworkers designed five hydrogel-forming β-hairpin SAPs with tunable susceptibility to matrix metalloproteinases [306]. Rheology frequency sweeps showed the storage modulus to decrease with time, and isolation of degraded fractions by HPLC for mass spectrometry analysis enabled identification of the fragments.
4. Peptides assemblies in tissue engineering and regenerative medicine
Peptide hydrogels provide niche environments that are tunable to specific tissue needs regarding cell-binding, mechanics, chemical and topological cues, matrix remodeling, and porosity., there are ample ways to engineer injectable and self-healing peptide hydrogels for minimally invasive administration or for 3D printing-based manufacturing [64,83,263,279,284,299]. Self-assembling peptide hydrogels in particular offer benefits in tissue engineering due to their ease of use and customizability, as the sequences can consist of both natural and synthetic amino acids [45,83,215,307] or synthetic polypeptides prepared by ring-opening polymerization of amino acid N-carboxyanhydrides [244,308–311]. Peptide hydrogels serve as biomimetic 3D environments to study cells in vitro and facilitate tissue regeneration in vivo [312,313]. One of the most important factors to consider when designing a peptide hydrogel is biocompatibility, meaning low toxicity to cells and compatibility with the immune system to avoid inflammation and rapid clearance [83,314]. In creating microenvironments for studying cells, peptide hydrogels can impact cell adhesion, morphology, and peptide/protein expression [315–317]. Together, the features of peptide hydrogels render them highly amenable to use for regenerative medicine in a multitude of tissues, including the central nervous system (CNS), muscular tissues, and bone.
Injectable peptide hydrogels are particularly desirable for minimally invasive regenerative medicine. These hydrogels serve multiple purposes: protecting encapsulated cells from shear force during syringe needle injection; preventing cell clumping during injection; maintaining cells at the site of injection and preventing reflux of cells through the needle track; decreasing anoikis (i.e., programmed cell death resulting from matrix detachment); protecting cells from the sometimes hostile environments encountered post-delivery; and providing cues and mechanical support to promote engraftment, migration, growth, and/or stem cell differentiation [32,46,64,315,318]. Shear-thinning and self-healing properties allow the gel to provide a stable microenvironment to cells pre- and post-delivery, while flowing and absorbing forces during injection and protecting cells from shear forces that could otherwise damage cells during delivery [31,32,284]. Furthermore, rapidly assembling (and recovering) hydrogels are particularly beneficial for retaining and protecting encapsulated cells post-injection [4,319]. Engineering favorable interactions between SAP hydrogels and the host cellular environment promotes integration of transplanted and endogenous cells in the tissue site [4,311]. The same viscoelastic shear-thinning and self-healing properties that render SAP hydrogels amenable to injection also enable bioprinting, or the engineering of intricate, precise, hierarchical structures as artificial cell matrices [320]. This biofabrication “bottom up” approach may better replicate in vivo matrices [320]. For instance, PeptiInks, a series of commercial peptide hydrogels designed for 3D printing by Manchester BIOGEL, are cytocompatible and amenable to modification with a variety of motifs from integrin, collagen, and other ECM native protein [320]. Additionally, printable ILVAGK peptide hydrogels support proliferation of a variety of stem cells for regenerative medicine [321].
While some peptide hydrogels are designed for specific tissues as described in tissue-themed sections that follow, others find applications in a wide range of tissues. Biomaterials from the SAPs EFK8, Fmoc-FF, MAX8, RADA, and RAPID are compatible with a wide range of cell types [32,45,48,67,79,322–324]. Apart from supporting cell proliferation and differentiation, SAPs such as RADA16 , I3SLKG, and Fmoc-FF [105,325–329] serve as vessels for drug delivery as reviewed elsewhere [330].
4.1. Neural tissue
The human brain is highly interconnected and complex, containing 100 billion cells with each neuron forming up to 10,000 connections with other neurons [331,332]. The heterogeneity in structure and cell type presents a significant challenge to design biomaterials to mimic and interact beneficially with neural tissue. Structural heterogeneity means that brain tissue varies in stiffness, with Young’s Modulus usually between 0.5 - 2.5 kPa depending on the region [333,334]. Neurons use membrane-based ionic action potentials to signal other neurons chemically at the synapse and are the drivers of brain functionality, necessitating potential biomaterials to support complex electrophysiology [335]. Glial cells provide structure at synapses and remove excess neurotransmitter from the synaptic cleft [336]. Examples of glial cells include oligodendrocytes, which insulate neuronal axons to improve transmission efficiency [337], and astrocytes [317], which provide support and regulate reduction/oxidation in the brain [21]. One of the most studied cell types in neural engineering is the oligodendrocyte progenitor cell (OPC), a migratory proliferative cell with many functions in CNS development and disease [338].
The similar rheological properties of brain tissue and many peptide hydrogels allow cell culture and delivery in environments similar to in vivo conditions, providing simplified systems to study neural tissue development, disease progression, and injury response [31,83,339]. Peptide hydrogels can incorporate high concentrations of neural-relevant bioactive peptide motifs to drive cell growth and differentiation [340]. Incorporating ECM-derived motifs, such as RGD and IKVAV integrin binding peptides, promotes cell adhesion, increases neurite sprouting, and directs neuron growth and migration [180,341–343], which improves neural integration and function. Sequences of note in neural peptide design include RADA16, RAPID peptides, EFK8 variants, and varying peptide amphiphiles.
4.1.1. RADA16
Attributed at least in part to similarity with the integrin binding cell adhesion motif, RGD, RADA hydrogels support differentiation and neurite outgrowth of primary neurons, and, notably, active synaptic connections between neurons [45]. The lack of inflammation and necrosis following implantation into rat leg muscles [45] prompted subsequent investigations of these hydrogels to support neuronal cell growth and promote regeneration of native tissue [328,344]. RADA16 hydrogels increase axonal regeneration of differentiated neural progenitor cells (NPCs) compared to unencapsulated cells when transplanted into an injured spinal cord by protecting the cells post-transplantation from the harsh microenvironment of the injury site [345]. The transplanted gels allow migration of both transplanted and native cells throughout the implant, facilitating integration with surrounding tissue. Additionally, RADA hydrogels decreased traumatic brain injury (TBI)-induced lesion cavity size in rat cortex and spared tissue function in the surrounding region [346]. Hydrogel-assisted transplantation also reduces the host astrocytic inflammatory response relative to unencapsulated cells [346]. RADA-mediated cell delivery also assisted in regenerating a portion of the vagus nerve by promoting nerve myelination important for improving action potential propagation [347].
4.1.2. RAPID
The RAPID family of peptides form hydrogels up to 17 kPa in storage modulus, depending on peptide concentration and sequence. RAPID hydrogels protect cells during syringe-based delivery, a desirable property with the growing use of stem cell-based therapies. Among the RAPID hydrogels, AYFIL exhibits a G’ = 1.5 kPa similar to that of neural tissue (G’ = 0.5 – 2.5 kPa) [83,333,334]. Injecting cell suspended in AYFIL hydrogels dramatically improves the short-term viability of oligodendrocyte progenitor cells (OPCs), mouse myoblasts (C2C12s), human mesenchymal stem cells (hMSCs), and primary mouse lung fibroblasts (MLFs) compared to injection in PBS (Figure 13A,B) [32]. The RAPID hydrogels are cytocompatible, affording high OPC viability over 4 days of 3D cell culture. OPCs extend processes within 2 days of culture in AYFIL hydrogel without bioactive cellular adhesion peptide sequences. Moreover, RAPID, like many peptide sequences, is amenable to modification with cell adhesion motifs such as RGD, IKVAV, and YIGSR, to control cell adhesion and differentiation.
Figure 13.
Tissue engineering applications of RAPID and IKVAV peptide amphiphile (PA) hydrogels. (A,B) RAPID hydrogel encapsulation nearly doubles OPC viability post injection relative to injection in PBS, showing the protective capabilities of shear-thinning RAPID hydrogels and their potential uses in regenerative medicine. Adapted with permission from Tang et al. [32], Copyright (2019) American Chemical Society. (C,D) Culture of neuronal cells in IKVAV PA hydrogels led to larger axon diameters and a greater degree of axonal interconnectivity than cells cultured in PA hydrogels lacking the IKVAV motif. Scale bars are 2 μm. Adapted with permission from Tysseling et al. [341], Copyright (2010) Wiley-VCH GmbH & Weinheim.
4.1.3. FEFEFKFK
Shear thinning, self-healing, nanofibrous hydrogels from the β-sheet forming peptide FEFEFKFK can carry NPCs and serve as scaffolds for spinal cord tissue engineering [298]. While encapsulation of viable bovine neural precursor cell (bNPC) populations in these FEFEFKFK hydrogels over 7 days of encapsulated culture did not substantially impact cell behavior, significant increases in gene expression over 14 days (relative to unencapsulated cells) suggest preservation of the neural progenitor phenotype during culture. ECM protein deposition also increased relative to unencapsulated controls.
4.1.4. Peptide Amphiphiles
IKVAV-functionalized peptide amphiphiles (PA) self-assemble into nanofibers and hydrogels displaying high densities of the laminin epitope IKVAV. The porosity of IKVAV PA hydrogels promotes viability of encapsulated NPCs by allowing cell migration and diffusion of nutrients and oxygen through the networks, with cells surviving as long as 22 days [180]. IKVAV-PA hydrogels with a G’ of ~ 2000 Pa facilitate superior differentiation of NPCs into neurons relative to laminin or solution controls, underscoring the importance of the nanoscopic structure of the hydrogel that imparts mechanical integrity [180,341]. IKVAV PA fibers feature a higher density of IKVAV than naturally found in laminin [348], and the majority of NPC neurite growth occurs along the nanofibers [342]. The effect of the IKVAV motif cannot be overstated, with neurite growth up to three times higher in number and much longer in length in NPCs cultured in IKVAV-PA compared to those in PAs without IKVAV (Figure 13C–D) [342]. The directionality of fibers provides topographical cues to orient cell growth, thereby promoting synapse formation between neurons in culture. Another study showed differentiation of human bone marrow mesenchymal stem cells into neural cells in IKVAV-PA hydrogels, as evidenced by laminin I ECM expression and long processes [349].
Along with uses in cell culture, IKVAV-PA has been studied extensively in vivo. The peptide hydrogel promotes axonal regeneration following spinal cord injury in vivo, increasing functional recovery of motor skills in mice after lesion [350]. IKVAV-PA reduced astrogliosis, neuronal apoptosis, and glial scar formation relative to a PA lacking IKVAV and promoted regeneration of both sensory axons and motor axons [342,350]. Reduction of scar formation and neuronal apoptosis were specifically attributed to the IKVAV-β1-integrin interactions [342,350]. Treatment of contusion injury mice with acellular IKVAV-PA promoted regeneration of axons and serotonergic fibers [341]. While the hydrogel supports impressive neural regeneration, IKVAV-PA did not promote remyelination or increase the local number of neurons in injured sites long term [341]. NPCs transplanted in aligned fiber scaffolds also thrive post transplantation, resulting in cell migration from the peptide scaffold, a highly desired outcome [342]. Aligned scaffolds presumably lead to better neural recovery by mimicking the structure found in mature neural tissue. Together, these results suggest that IKVAV-PA is suited best for axonal regeneration and alignment and requires additional growth extracellular cues to promote full regeneration and recovery in damaged tissue.
Apart from IKVAV-PA, other PAs also form highly functional substrates for neural tissue engineering. Polylysine-based peptide amphiphiles varying in length from one to one hundred lysine residues assemble into nanofibers that resemble fibrous ECM [351]. Encapsulated human iPSC-derived neurons grew neurites 100 μm longer in gels with four lysine repeats (K4, G’ = 80 Pa) compared to those grown in gels from PAs containing ten lysine repeats (K10, G’ = 110 Pa). These results indicate that softer hydrogels with fewer lysine repeats are better suited for promoting the survival of neurons with desired phenotypes. Additionally, recent work from the Stupp lab showed the importance of hierarchical structure on engineering neural cell phenotypes [176]. PA fibers functionalized with complementary DNA strands form supramolecular bundles that promote a reactive phenotype from astrocytes cultured on these materials. Culturing astrocytes on substrates with similar bulk stiffnesses, but comprised only of individual (non-bundled) fibers, does not yield the reactive phenotype.
4.2. Muscular engineering
4.2.1. Skeletal muscle
Skeletal muscle is highly organized, hierarchical, and anisotropic – features that fibrous peptide hydrogels can recapitulate to support growth and differentiation of myocytes for engineering muscle tissue. Myoblasts fuse to form multinucleated myocytes, and aligned myocytes band together to comprise myofibrils and multinucleated skeletal muscle fibers [352–354]. As muscle orients in the direction of contraction, the tissue is highly anisotropic [355–357]. While mature muscle tissue is quite stiff at 23 kPa, the moduli of individual cells and immature muscle can be as low as 10% that of mature muscle. Peptide hydrogels closely mimic the mechanics of softer muscle tissues, enabling use for muscle generation and development. Additional design consideration should be given to: 1) innervation of skeletal muscles by motor neurons, requiring peptide hydrogels to support complex electrophysiology, and 2) blood vessel growth into the tissue, requiring peptide hydrogels to retain structure when subject to shear from flowing liquid [358,359].
Myocytes are most viable in highly organized scaffolds with electrophysiology (i.e., frequent electrical stimulation) analogous to in vivo muscular stimulation [361]. With a sparse ECM, myocytes respond minimally to cell adhesion motifs. Instead, myocytes rely on proximal cells to provide cues and structural support [362]. Biomaterials designed for myocyte culture feature moduli similar to natural soft muscle tissue (G’ = 20 kPa) while allowing intercellular interactions conducive to generating the characteristic hierarchical organization of muscle tissue. On the other hand, skeletal muscle progenitor cells (SMPCs) do not require the aligned cell orientation needed for myocytes, and respond to cell adhesion sequences [363]. Peptide amphiphiles are the major building blocks in peptide hydrogels used in skeletal tissue engineering.
4.2.1.1. PAs
Mouse skeletal muscle progenitor cells cultured in 3-15 kPa PA gels, stiffnesses ideal for developing muscle, remain highly viable (>85%) [25]. Moreover, these gels yield higher levels of muscle progenitor cell alignment compared to cells on unaligned fibers. Encapsulation of growth factors, such as vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF), in PA hydrogels induces myoblast proliferation in vitro. Cell alignment was inversely proportional to cell density, supporting the hypothesis that cell activity and growth leads to increases in PA remodeling, thus decreasing cell access to the scaffold’s oriented nanofibers. PA scaffolds injected in vivo maintain alignment and increase engraftment of myoblasts in injured muscles compared to unencapsulated cells (Figure 14A).
Figure 14.
Tissue engineering applications of PA, RADA, and Q11 SAPs. (A) Encapsulation of myogenic precursor cells in aligned PA scaffolds yielded higher cell viability at all timepoints compared to unencapsulated cells, especially in scaffolds with mid-range (G’ ~9 kPa) and high (G’ ~15 kPa) moduli. C2C12 cells (green) in the PA scaffold (red) grew along the axis of alignment, with the white arrow heads indicating dead cells. Adapted with permission from Sleep et al. [25], Copyright (2017) National Academy of Sciences U.S.A. (B) Application of commercially available PuraStat RADA hydrogels to the surface of cardiac tissue decreased probability of heart failure in mice. Adapted with permission from Giritharan et al. [360], Copyright (2018) BioMed Central Ltd. (C) Mice injected subcutaneously near the shoulder blades with coassemblies of OVA-Q11 and NANP3-Q11 elicited heightened immune responses compared to those receiving separate injections of the two peptides. The Y axis represents the degree of antibody production responses. Adapted with permission from Rudra et al. [315], Copyright (2012) Elsevier.
4.2.1.2. RADA derivatives
Inspired by RADA16, fibrous gels from the β-sheet SAP RLDLRLALRLDLR dubbed SPG-178 have a G’ ~100 Pa, which is notably more compliant than muscle tissue, but still appears to be well suited for myoblast culture [22]. Mouse myoblasts (C2C12 cells) cultured in the hydrogels proliferated ~12.4 fold more than C2C12 cells cultured without peptide over 8 days in a plate. Encapsulated C2C12 cells also elongated by 20% when stretching the gel to 120% of its original length (20% strain), indicating that the hydrogel transmits mechanical forces to encapsulated cells. Relative to unstrained cells, stretched hydrogels yield higher expression of phosphorylated kinase and total extracellular signal-regulated kinase (ERK) than unstretched cells, factors usually expressed in growth signaling pathway in differentiating myoblasts. Together, these results highlight the beneficial impacts of encapsulation in stretched scaffolds for muscle cell growth and differentiation.
4.2.2. Cardiac muscle
While skeletal and cardiac muscle have numerous similarities, such as striation and contractile mechanisms, significant differences in these muscle cell types and tissue structure require distinct biomaterial design criteria for engineering myocardial tissue. The cardiac syncytium is an isotropic connection of mononucleated cells connected by intercalated disks [364]. Numerous gap junctions and adhesion receptors transmit force and electrical signals between myofibrils in the syncytium. This highly interconnected structure ensures rapid transmission of signal responsible for the simultaneous motion of cells in heartbeats. Challenges arise when engineering cardiac tissue as the constant movement of the heart necessitates robust and flexible scaffolds, while the highly interconnected nature of the myocytes requires scaffolds to ensure intercellular connection.
RADA16 hydrogels support proliferation and differentiation of cardiac progenitor cells [365]. Appending the laminin and collagen motifs, YIRGR and TAGSCLRKFSTM, respectively, to RADA16 provides basement membrane extracellular matrix cues naturally presented in the aortic endothelium [366]. Growth of human aortic endothelial cells (HAECs) cultured within functionalized RADA16 scaffolds suggests that the HAECs recognize the cell-binding RADA sequences. The functionalized peptide hydrogels also enhance nitric oxide, laminin 1, and collagen IV release, all of which contribute to healthy myocyte growth and differentiation. In another study, RADA16 modified with QHREDGS, an αvβ3 integrin binding motif, form gels that increase viability of MSCs delivered following myocardial infarction in comparison to unmodified RADA16 [324].
RADA16 is useful for multiple surgical procedures and treatments, such as wound repair, in the heart [367]. Blending PuraMatrix, a commercial variant of RADA16, with mesenchymal stem cells (MSCs) offers an injectable treatment for heart failure [368]. Depositing hydrogel droplets directly onto the epicardial surface of a beating heart forms a coating on the heart that supports growth of transplanted MSCs on the heart. MSC incorporation into the hydrogel droplet coatings enhanced cardiac function at day 28 after administration relative to a hydrogel lacking MSCs, as the gel facilitated MSC regeneration of cardiac tissue. PuraStat®, another commercially available RADA16 formulation, prevented excessive bleeding during heart surgery when applied directly to the heart (Figure 14B) [360].
A peptide amphiphile containing Dopa, a biological adhesive for immobilization of molecules on metal surfaces, enables nanofiber coating on steel surfaces such as cardiac stents [369]. In conjunction with stent implantation, Dopa PAs functionalized with the fibronectin-derived integrin binding motif REDV leads to endothelial cell growth on stainless steel surfaces, an otherwise difficult task due to the stiffness of steel. Cell adhesion, spreading, viability, and proliferation of endothelial cells are greatly increased on steel surfaces coated with PA in comparison to those without.
4.3. Bone engineering
Bone tissue engineering using soft hydrogels is notoriously difficult due to the stiff structures and heterogeneous compositions of bone. Skeletal bones include 1) compact osteons, consisting largely of the mineral hydroxyapatite, the main inorganic component of bone; 2) spongy bone, a less dense osseous structure; and 3) bone marrow, concealed within the osteons [370]. The Young’s Modulus of each tissue at the bone-soft tissue interfaces varies between ~17,900 MPa for osseous tissue [371,372] to ~1 kPa for bone marrow [373]. The cells residing within hard bone structures include osteoblasts that generate hydroxyapatite, osteoclasts that remodel hydroxyapatite, osteocytes that maintain the bone, and undifferentiated bone stem cells known as osteoprogenitor cells [374,375]. Peptide hydrogels are useful for in vitro cell culture, but the low porosity of osteons prevents peptides from interacting with osseous tissue [376]. However, peptides can guide tissue engineering and regeneration of the bone marrow and soft tissues of the bone, as both matrix stiffness and adhesion cues are critical for successful cell differentiation and growth [377]. Additionally, peptides can assist in recalcification of damaged hard bone by guiding regeneration. Peptide hydrogels tuned for bone engineering target both soft and hard tissue to promote growth and differentiation and, as with hydrogels for soft tissues, can vary appreciably in sequence and physical characteristics.
4.3.1. PFD-5
Fibrous hydrogels of the β-sheet peptide PFD-5 (PDFDFDFDFDFDP) form in cell culture medium, and divalent calcium ions provide further stability through electrostatic interactions with the carboxylates on the aspartic acid (D) residues [66]. When encapsulating human fetal osteoblastic (hFOB) cells, PFD-5 initiates bone generation and mineralization in vitro. The peptide also elevates hFOB cell number and alkaline phosphatase (ALP) expression, a marker for hard tissue formation, to levels similar or greater than those in the presence of beta tricalcium phosphate (β-TCP). PFD-5 treatment in vivo increases osteon bone regeneration compared to the no treatment control, with β-TCP-loaded PFD-5 further increasing regeneration of bone as seen by TEM images. Functionalization of PFD-5 with RGD increases osteoblast number compared to the unfunctionalized hydrogel [378].
4.3.2. Functional PAs
Co-assembly of two peptide amphiphiles, one presenting a phosphoserine residue for hydroxyapatite nucleation and the other an RGDS cell adhesion sequence on the nanofiber surface, yields nanofibrous, injectable hydrogels promote mineralization [379]. These hydrogel formulations are capable of syringe injection, remain localized to the injection site 48 hours after implantation in mouse fractures, and generate an artificial bone matrix, mimicking the natural biomineralization process. Compared to PA hydrogels lacking phosphoserine and RGDS, the co-assembled hydrogels afford a nearly two-fold increase in ossified bone in a mouse model.
4.3.3. Amphipathic β-sheet peptides
Glutamic acid-containing (LE)8 and (VEVSVKVS)2 peptides form fibrils that cross-link in the presence of divalent calcium ions into injectable hydrogels [64]. Addition of phosphate initiates mineralization to produce amorphous calcium phosphate within the hydrogels as a precursor to promote bone regeneration. Following injection, mineralization occurs within the composite hydrogels from both peptides in vitro.
The β-hairpin peptide VKVKVKVK-V(D)PPT-KVEVKVKV-MLPHHGA, an amalgam of the MAX8 hairpin peptide and the heptapeptide MLPHHGA that directs mineralization, forms mechanically rigid hydrogels with G’ ~2500 Pa [67]. In solutions with low ionic strength, the peptide remains unfolded due to repulsive electrostatic interactions; addition of CaCl2 and beta glycerophosphate (β-GP) screens these interactions and triggers peptide folding. Upon cleavage of β-GP with naturally occurring alkaline phosphatase to liberate the phosphate for calcium phosphate mineralization within osteons, the hydrogels direct formation of hydroxyapatite induced both biochemically and by cementoblasts, highly differentiated mesenchymal cells that support mineralization [380]. Cementoblasts encapsulated within the hydrogels remained viable for at least 72 hours and deposited calcium phosphate over 9 days of culture.
4.3.4. P11-4
P11-4 is a self-assembling peptide with sequence QQRFEWEFEQQ that forms hydrogels that leverage biomimetic mineralization for treatment of tooth lesions [381]. This 11-amino acid peptide hydrogel assembles into fibrillar scaffolds in solutions with high ionic strength and low pH – conditions that mimic those of oral wounds [382]. Interestingly, P11-4 utilizes natural remineralization induced by saliva mineral content, enabling P11-4 to guide enamel regeneration. Additionally, while many peptide hydrogels have clinical potential, P11-4 is one of the few peptide hydrogels that have progressed through pre-clinical animal trials and into clinical trials (Clinical trials NCT03927794, NCT04776785), illustrating the material’s potential use for future clinical applications.
4.4. Immune engineering
The immune system is an intricate network of cells and tissues throughout the body that prevent, isolate, and eliminate invading pathogens [383]. Peptides designed to interact with the immune system typically target circulatory immune cells responsible for recognizing pathogens and promoting immunity, such as lymphocytes and monocytes, and act as vaccine adjuvants, additives that elevate immune response to vaccinations. Neutrophils act as early responders responsible for phagocytosis, i.e., engulfing material and initiating an immune response, while lymphocytes provide adaptive immunity and monocytes present antigens and can differentiate into macrophages, the activated phenotype of neutrophils [384,385]. Immune cells can easily phagocytose fibrillar, antigen-containing peptide assemblies due to their large size, elevating levels of pathogen recognition and neutralization in the host.
4.4.1. KFKE
Modification of the β-sheet peptide KFE8 (FKFEFKFE) with T-cell binding epitopes can induce an immune response to antigens, effectively vaccinating the host [315,386]. KFE8 supramolecular nanofibers cause antigen-presenting cells to phagocyte the epitope-peptide supramolecular structure at a rate higher than the epitope alone. Fibrillar structure is essential to present epitopes for recognition by T cells. For example, an ovalbumin (OVA) epitope conjugated to FKE8 elicits a heightened immune response compared to that the OVA epitope conjugated to non-assembling peptide sequences [47]. OVA-KFE8/CaCO3 composite microparticles increase epitope uptake and immune response, circumventing the need for additional adjuvant components [387]. Immunization also occurs via oral delivery of OVA-modified FKE8 [387]. Decorating KFE8 fibrils with epitopes of West Nile Virus led to higher survival of mice post infection compared to mice vaccinated with the soluble epitope [46].
4.4.2. Q11
Q11 (QQKFQFQFEQQ) is another β-sheet fibril-forming peptide that acts as an immune adjuvant when fused to a peptide antigen [47]. Appending epitopes to the N-terminus of Q11 displays the epitopes on the nanofiber surface and does not disrupt peptide assembly [315]. Moreover, the epitopes retain functionality in vivo and unmodified Q11 does not elicit an immune response. The antibody production response to epitopes conjugated to peptides depends on the fibrillization state of the peptide, providing another degree of tunability for epitope delivery [47]. OVA-Q11 elicited detectable immune responses over 52 weeks, demonstrating the potential for long-term protection by of Q11-mediated vaccinations (Figure 14C). Similarly, (NANP)3 malaria epitopes conjugated to Q11 elicited T cell-dependent immune responses over 40 weeks [315]. Co-assembly of OVA-Q11 and (NANP)3-Q11 in a 1:1 ratio induced similar responses as separate injections of OVA- and (NANP)3-presenting Q11 fibers at least 16 weeks after injection, highlighting the suitability of these materials for co-vaccinations [315]. Q11 fibers presenting p-nitrophenyl phosphonate (pNP) ligands enable conjugation and presentation of protein epitopes [386]. These fibers elicit robust antibody responses following delivery in mice. Modification of the assembling Q11 sequence by varying charge or hydrophobicity modulates uptake in T-cells [388].
4.4.3. Coil 29
While many examples of peptide-based biomaterials involve β-sheet forming peptides, Coil 29 (QARILEADAEILRAYARILEAHAEILRAQ) forms α-helical nanofibers that can present epitopes and induce robust immune responses [5]. Assembly occurs via arrangement of helical peptides perpendicular to the fiber axis. The C-termini of the peptides cluster near the axis of the nanofiber, while exposed N-termini present conjugated epitopes at the fiber surface.
5. Synergistic combinations of polymers and assembling peptides in functional biomaterials
Peptide-polymer hybrid materials combine the inherent benefits of each component, marrying the sequence-specific functions and biocompatibility of peptides with the tailorability and robust mechanics of polymeric materials [1,389–391]. Appending assembling peptides to polymers, to produce amphiphilic conjugates, directs assembly into an array of micro- and nanoscale structures well suited for tissue engineering and regenerative medicine, among other therapeutic applications [244,392,393]. Reliable bioconjugation and polymer chemistry enable routine preparation of hybrid materials with polymeric and self-assembling or co-assembling peptide components [390,394–399]. In 1999, Kopeček and coworkers reported coiled coil peptides that crosslink poly(hydroxypropylacrylamide) hydrogels [2]. Thereafter, expanding this approach by Kopeček [240,241,400], Klok [401], Collier [3], and others [402] generated a diverse library of biomaterials, incorporating other peptides, e.g., collagen-like peptides and β-sheet peptides [403], and polymers, e.g., poly(ethylene glycol). Recently, Kloxin and coworkers embedded collagen-like peptide fibers within a poly(ethylene glycol) hydrogel to recapitulate the fibrous nanostructure of the extracellular matrix within non-fibrous synthetic polymer hydrogels for 3D cell culture [4]. Placing peptides at polymer chain end(s) yields polymer-peptide block copolymers that associate in solution into defined nanoscale constructs well suited for therapeutic delivery [126,404–410]. Attaching one coiled coil peptide to poly(N-(2-hydroxypropyl)methacrylamide) copolymers and the complementary peptide to methotrexate (MTX) drug conjugates facilitates efficient drug loading into polymer constructs for delivery [411–413]. Appending the peptide LPFFD, similar to a sequence found in the amyloid β peptide implicated in Alzheimer’s disease, to poly(hydroxypropylmethacrylamide) creates a polymer-peptide conjugate that disperses amyloid β aggregates [414]. The conjugate improves on previous peptidyl and peptidomimetic inhibitors of aggregation that suffer from difficulties specifically targeting amyloid β.
6. Concluding remarks: challenges and opportunities in assembling peptide biomaterials
Advances in assembling peptide-based biomaterials enable current capabilities in tissue engineering and regenerative medicine, and will undoubtedly fuel future innovation and impact in this space. Precision design of peptides supports biological and medical use due to reliable formation of fibers and fibrous hydrogels with controlled dimensions and morphology under cytocompatible and physiological conditions [41,415]. This opportunity to review the state-of-the-art in the design, characterization, tunability, and tissue-specific application of peptide-based biomaterial platforms provides a snapshot for the reader in 2021. Inspiration for these materials comes from Nature, rational design, and computationally driven screening and exploration [81,138,146,157,214–217]. Looking forward, engineering peptide-based biomaterials to interact in an increasingly productive and specific manner with living systems, for example, instructing cell growth and differentiation, will further transform tissue engineering and regenerative medicine. We anticipate a future where precision medicine approaches guide the design, synthesis, and testing of peptide-based biomaterials. Simultaneously, tailoring these biomaterials for 3D printing-based biofabrication will propel clinical implementation of these emerging technologies [320,416].
Multi-faceted, multi-scale characterization will be at the forefront of developing and translating the next generation of peptide-based biomaterials, connecting peptide sequence and secondary structure to the morphology, mechanics, and tissue-specific biological function. At the molecular level, chromatography and mass spectrometry confirm the sequence/primary structure and purity of peptides, providing confidence in the structure-property relationships that govern our understanding of peptide biomaterials. Circular dichroism and infrared spectroscopy under different conditions give insight into the molecular conformation or secondary structure, while calorimetry and spectroscopy yield information about molecular interactions. Of these techniques, NMR spectroscopy is notable, as it is a relatively under-utilized label-free method for characterizing molecular interactions of peptides. After assembly into nanoscale structures, scattering provides average dimensional information, whereas microscopy offers complementary information in the form of images of assembled structures. Molecules with environment-dependent optical properties serve as stains for imaging peptide assembly in complex, biologically relevant environments. Then, at the bulk material level, rheology yields details about the mechanics and dynamics. Combining these experimental methods with multiscale molecular simulations brings opportunities to enrich understanding at all levels, from sequence to bulk materials [212,417].
We see challenges and opportunities in tailoring the proteolytic stability, specific interactions, and dynamics of peptide-based biomaterials. While peptides impart structure and biological function to biomaterials, proteolytic stability remains an unaddressed challenge. Devising ways to increase and control stability, such as building on the field’s current understanding of incorporation of d-amino acids and specific interactions between d- and l-peptides [37,86–89], will enable longer term application of these biomaterials. Heteroassembling β-sheet forming peptide pairs also increase the design space, as no such pairs exist in nature [212]. Further, leveraging specific interactions between peptides (e.g., heterodimeric coiled coils,) in combination with photo-patterning techniques holds promise for spatiotemporally controlled presentation of biomolecules [127].
The physical, and thus reversible, nature of the molecular interactions between assembling peptides is ideal for injectable biomaterials and 3DP-based biofabrication; harnessing and tuning these interactions holds promise for engineering peptide-based 3DP inks. Application of shear during printing or injection disrupts these interactions, which reform upon removal of shear. Other stimuli, such as changes in pH and salt concentration, also modulate peptide interactions and allow for triggered assembly, cell encapsulation, and/or release of cells and therapeutics. Tuning the strength of peptide interactions provides compelling opportunities to program the dynamics of biomaterials [175,176]. Given the dynamic nature of the natural extracellular matrix to support tissue growth, engineering cell-driven material rearrangement (i.e., stress relaxation) on biological time scales is an important design criterion [373,418]. While in its infancy, leveraging stereochemistry-driven interactions between peptides provides a compelling way to tailor the stability and thermomechanical properties of peptide assemblies.
In these pursuits, balancing function - biological outcomes, printability, and stability, among others - with material complexity will be important. For example, short β-sheet forming SAPs provide a strikingly simple materials system for forming well-defined, reproducible fibers. Expanding efforts to develop these short assembling peptide platforms will provide opportunities to simplify, scale-up, and reduce costs of manufacturing peptide biomaterials. To these ends, combining relatively complex, functional peptides with simpler, yet biocompatible, mechanically robust, and less expensive polymers merits further development [389]. Feedback between structure-property relationships that establish design rules and rationale to identify new sequences and materials that are well-suited to various applications will be critical going forward.
Statement of significance.
Well-defined topological and mechanical properties of assembled peptides can be used to direct biological responses. Engineering peptide-based extracellular matrices offers immense opportunity for regenerative medicine and tissue engineering. Here we review the molecular-scale features of assembling peptides that result in useful extracellular matrix properties and desired cell interactions. Aiming to inspire researchers approaching this challenge from both the peptide biomaterial design and tissue engineering perspectives, we present characterization tools for understanding the connection between peptide structure and properties and highlight the use of peptide-based biomaterials in neural, orthopedic, cardiac, muscular, and immune engineering applications.
Acknowledgements
This work was supported by the Center for Advanced Biomanufacturing at the University of Virginia, NSF grants 1904198 and 2104723, NIH grant 1T32GM136615, and the Translational Health Research Institute of Virginia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Edwards-Gayle CJC, Hamley IW, Self-assembly of bioactive peptides, peptide conjugates, and peptide mimetic materials, Org. Biomol. Chem 15 (2017) 5867–5876. 10.1039/c7ob01092c. [DOI] [PubMed] [Google Scholar]
- [2].Wang C, Stewart RJ, Kopeček J, Hybrid hydrogels assembled from synthetic polymers and coiled-coil protein domains, Nature. 397 (1999) 417–420. 10.1038/17092. [DOI] [PubMed] [Google Scholar]
- [3].Jing P, Rudra JS, Herr AB, Collier JH, Self-assembling peptide-polymer hydrogels designed from the coiled coil region of fibrin, Biomacromolecules. 9 (2008) 2438–2446. 10.1021/bm800459v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hilderbrand AM, Ford EM, Guo C, Sloppy JD, Kloxin AM, Hierarchically structured hydrogels utilizing multifunctional assembling peptides for 3D cell culture, Biomater. Sci 8 (2020) 1256–1269. 10.1039/c9bm01894h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wu Y, Norberg PK, Reap EA, Congdon KL, Fries CN, Kelly SH, Sampson JH, Conticello VP, Collier JH, A Supramolecular Vaccine Platform Based on α-Helical Peptide Nanofibers, ACS Biomater. Sci. Eng 3 (2017) 3128–3132. 10.1021/acsbiomaterials.7b00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Eskandari S, Guerin T, Toth I, Stephenson RJ, Recent advances in self-assembled peptides: Implications for targeted drug delivery and vaccine engineering, Adv. Drug Deliv. Rev 110–111 (2017) 169–187. 10.1016/j.addr.2016.06.013. [DOI] [PubMed] [Google Scholar]
- [7].Chang LW, Lytle TK, Radhakrishna M, Madinya JJ, Veléz J, Sing CE, Perry SL, Sequence and entropy-based control of complex coacervates, Nat. Commun 8 (2017) 1–7. 10.1038/s41467-017-01249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pedersen SL, Tofteng AP, Malik L, Jensen KJ, Microwave heating in solid-phase peptide synthesis, Chem. Soc. Rev 41 (2012) 1826–1844. 10.1039/c1cs15214a. [DOI] [PubMed] [Google Scholar]
- [9].Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA, Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics, Proc. Natl. Acad. Sci. U. S. A 100 (2003) 5413–5418. 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Patterson J, Hubbell JA, Enhanced proteolytic degradation of molecularly engineered PEG hydrogels in response to MMP-1 and MMP-2, Biomaterials. 31 (2010) 7836–7845. 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- [11].Tokatlian T, Shrum CT, Kadoya WM, Segura T, Protease degradable tethers for controlled and cell-mediated release of nanoparticles in 2- and 3-dimensions, Biomaterials. 31 (2010) 8072–8080. 10.1016/j.biomaterials.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bellis SL, Advantages of RGD peptides for directing cell association with biomaterials, Biomaterials. 32 (2011) 4205–4210. 10.1016/j.biomaterials.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang AY, Leong S, Liang YC, Huang RCC, Chen CS, Yu SM, Immobilization of growth factors on collagen scaffolds mediated by polyanionic collagen mimetic peptides and its effect on endothelial cell morphogenesis, Biomacromolecules. 9 (2008) 2929–2936. 10.1021/bm800727z. [DOI] [PubMed] [Google Scholar]
- [14].Chen TT, Luque A, Lee S, Anderson SM, Segura T, Iruela-Arispe ML, Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells, J. Cell Biol 188 (2010) 595–609. 10.1083/jcb.200906044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].LeSauteur L, Wei L, Gibbs BF, Saragovi HU, Small peptide mimics of nerve growth factor bind TrkA receptors and affect biological responses, J. Biol. Chem 270 (1995) 6564–6569. 10.1074/jbc.270.12.6564. [DOI] [PubMed] [Google Scholar]
- [16].Zhang S, Holmes T, Lockshin C, Rich A, Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane, Proc. Natl. Acad. Sci. U. S. A 90 (1993) 3334–3338. 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schneider JP, Pochan DJ, Ozbas B, Rajagopal K, Pakstis L, Kretsinger J, Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide, J. Am. Chem. Soc 124 (2002) 15030–15037. 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- [18].Ozbas B, Kretsinger J, Rajagopal K, Schneider JP, Pochan DJ, Salt-triggered peptide folding and consequent self-assembly into hydrogels with tunable modulus, Macromolecules. 37 (2004) 7331–7337. 10.1021/ma0491762. [DOI] [Google Scholar]
- [19].Dong H, Paramonov SE, Aulisa L, Bakota EL, Hartgerink JD, Self-assembly of multidomain peptides: Balancing molecular frustration controls conformation and nanostructure, J. Am. Chem. Soc 129 (2007) 12468–12472. 10.1021/ja072536r. [DOI] [PubMed] [Google Scholar]
- [20].Zou L, Addonizio CJ, Su B, Sis MJ, Braegelman AS, Liu D, Webber MJ, Supramolecular Hydrogels via Light-Responsive Homoternary Cross-Links, Biomacromolecules. (2020) acs.biomac.0c00950. 10.1021/acs.biomac.0c00950. [DOI] [PubMed] [Google Scholar]
- [21].Peuchen S, Bolaños JP, Heales SJR, Almeida A, Duchen MR, Clark JB, Interrelationships between astrocyte function, oxidative stress and antioxidant status within the central nervous system, Prog. Neurobiol 52 (1997) 261–281. 10.1016/S0301-0082(97)00010-5. [DOI] [PubMed] [Google Scholar]
- [22].Nagai Y, Yokoi H, Kaihara K, Naruse K, The mechanical stimulation of cells in 3D culture within a self-assembling peptide hydrogel, Biomaterials. 33 (2012) 1044–1051. 10.1016/j.biomaterials.2011.10.049. [DOI] [PubMed] [Google Scholar]
- [23].Genové E, Shen C, Zhang S, Semino CE, The effect of functionalized self-assembling peptide scaffolds on human aortic endothelial cell function, Biomaterials. 26 (2005) 3341–3351. 10.1016/j.biomaterials.2004.08.012. [DOI] [PubMed] [Google Scholar]
- [24].Yamada Y, Patel NL, Kalen JD, Schneider JP, Design of a Peptide-Based Electronegative Hydrogel for the Direct Encapsulation, 3D Culturing, in Vivo Syringe-Based Delivery, and Long-Term Tissue Engraftment of Cells, ACS Appl. Mater. Interfaces 11 (2019) 34688–34697. 10.1021/acsami.9b12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sleep E, Cosgrove BD, McClendon MT, Preslar AT, Chen CH, Sangji MH, Pérez CMR, Haynes RD, Meade TJ, Blau HM, Stupp SI, Injectable biomimetic liquid crystalline scaffolds enhance muscle stem cell transplantation, Proc. Natl. Acad. Sci. U. S. A 114 (2017) E7919–E7928. 10.1073/pnas.1708142114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fernández-Muiños T, Recha-Sancho L, LĈpez-ChicĈn P, Castells-Sala C, Mata A, Semino CE, Bimolecular based heparin and self-assembling hydrogel for tissue engineering applications, Acta Biomater. 16 (2015) 35–48. 10.1016/j.actbio.2015.01.008. [DOI] [PubMed] [Google Scholar]
- [27].Banwell EF, Abelardo ES, Adams DJ, Birchall MA, Corrigan A, Donald AM, Kirkland M, Serpell LC, Butler MF, Woolfson DN, Rational design and application of responsive α-helical peptide hydrogels, Nat. Mater 8 (2009) 596–600. 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA, Degradation-mediated cellular traction directs stem cell fate in covalently crosslinked three-dimensional hydrogels, Nat. Mater 12 (2013) 458–465. 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li L, Eyckmans J, Chen CS, Designer biomaterials for mechanobiology, Nat. Mater 16 (2017) 1164–1168. 10.1038/nmat5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Thomas G, Burnham NA, nn. Camesano TA, Wen Q, Measuring the mechanical properties of living cells using atomic force microscopy, J. Vis. Exp (2013) 50497. 10.3791/50497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Aguado BA, Mulyasasmita W, Su J, Lampe KJ, Heilshorn SC, Improving viability of stem cells during syringe needle flow through the design of hydrogel cell carriers, Tissue Eng. - Part A 18 (2012) 806–815. 10.1089/ten.tea.2011.0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tang JD, Roloson EB, Amelung CD, Lampe KJ, Rapidly Assembling Pentapeptides for Injectable Delivery (RAPID) Hydrogels as Cytoprotective Cell Carriers, ACS Biomater. Sci. Eng 5 (2019) 2117–2121. 10.1021/acsbiomaterials.9b00389. [DOI] [PubMed] [Google Scholar]
- [33].Caplan MR, Moore PN, Zhang S, Kamm RD, Lauffenburger DA, Self-assembly of a β-sheet protein governed by relief of electrostatic repulsion relative to van der Waals attraction, Biomacromolecules. 1 (2000) 627–631. 10.1021/bm005586w. [DOI] [PubMed] [Google Scholar]
- [34].Marini DM, Hwang W, Lauffenburger DA, Zhang S, Kamm RD, Left-Handed Helical Ribbon Intermediates in the Self-Assembly of a ß-Sheet Peptide, Nano Lett. 2 (2002) 295–299. 10.1021/nl015697g. [DOI] [Google Scholar]
- [35].Gielnik M, Pietralik Z, Zhukov I, Szymańska A, Kwiatek WM, Kozak M, PrP (58-93) peptide from unstructured N-terminal domain of human prion protein forms amyloid-like fibrillar structures in the presence of Zn2+ ions, RSC Adv. 9 (2019) 22211–22219. 10.1039/c9ra01510h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dutta S, Foley AR, Warner CJA, Zhang X, Rolandi M, Abrams B, Raskatov JA, Suppression of Oligomer Formation and Formation of Non-Toxic Fibrils upon Addition of Mirror-Image Aβ42 to the Natural l-Enantiomer, Angew. Chemie - Int. Ed 56 (2017) 11506–11510. 10.1002/anie.201706279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Urban JM, Ho J, Piester G, Fu R, Nilsson BL, Rippled β-sheet formation by an amyloid-β fragment indicates expanded scope of sequence space for enantiomeric β-sheet peptide coassembly, Molecules. 24 (2019) 1983. 10.3390/molecules24101983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang S, Lockshin C, Cook R, Rich A, Unusually stable β-sheet formation in an ionic self-complementary oligopeptide, Biopolymers. 34 (1994) 663–672. 10.1002/bip.360340508. [DOI] [PubMed] [Google Scholar]
- [39].Zhang S, Holmes TC, DiPersio CM, Hynes RO, Su X, Rich A, Self-complementary oligopeptide matrices support mammalian cell attachment, Biomaterials. 16 (1995) 1385–1393. 10.1016/0142-9612(95)96874-Y. [DOI] [PubMed] [Google Scholar]
- [40].Zhang S, Self-assembling peptides: From a discovery in a yeast protein to diverse uses and beyond, Protein Sci 29 (2020) 2281–2303. 10.1002/pro.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hainline KM, Fries CN, Collier JH, Progress Toward the Clinical Translation of Bioinspired Peptide and Protein Assemblies, Adv. Healthc. Mater 7 (2018) 1700930. 10.1002/adhm.201700930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Maki J, Gil ES, Spirio L, Combination compositions, US 2018/0369452, 2018.
- [43].Gil ES, Gilbert KP, Synthetic Peptide Hydrogel Formulations for Use As Extracellular Matrix, US 2018/0023049 A1, 2018.
- [44].Yokoi H, Kinoshita T, Zhang S, Dynamic reassembly of peptide RADA16 nanofiber scaffold, Proc. Natl. Acad. Sci. U. S. A 102 (2005) 8414–8419. 10.1073/pnas.0407843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhang S, Holmes TC, DiPersio CM, Hynes RO, Su X, Rich A, Self-complementary oligopeptide matrices support mammalian cell attachment, Biomaterials. 16 (1995) 1385–1393. 10.1016/0142-9612(95)96874-Y. [DOI] [PubMed] [Google Scholar]
- [46].Friedrich BM, Beasley DWC, Rudra JS, Supramolecular peptide hydrogel adjuvanted subunit vaccine elicits protective antibody responses against West Nile virus, Vaccine. 34 (2016) 5479–5482. 10.1016/j.vaccine.2016.09.044. [DOI] [PubMed] [Google Scholar]
- [47].Rudra JS, Sun T, Bird KC, Daniels MD, Gasiorowski JZ, Chong AS, Collier JH, Modulating adaptive immune responses to peptide self-assemblies, ACS Nano. 6 (2012) 1557–1564. 10.1021/nn204530r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ, Schneider JP, Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells, Proc. Natl. Acad. Sci 104 (2007) 7791–7796. 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Branco MC, Nettesheim F, Pochan DJ, Schneider JP, Wagner NJ, Fast dynamics of semiflexible chain networks of self-assembled peptides, Biomacromolecules. 10 (2009) 1374–1380. 10.1021/bm801396e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sawada T, Tsuchiya M, Takahashi T, Tsutsumi H, Mihara H, Cell-adhesive hydrogels composed of peptide nanofibers responsive to biological ions, Polym. J 44 (2012) 651–657. 10.1038/pj.2012.48. [DOI] [Google Scholar]
- [51].Fukunaga K, Tsutsumi H, Mihara H, Self-Assembling Peptides as Building Blocks of Functional Materials for Biomedical Applications, Bull. Chem. Soc. Jpn 92 (2019) 391–399. 10.1246/BCSJ.20180293. [DOI] [Google Scholar]
- [52].Collier JH, Messersmith PB, Enzymatic modification of self-assembled peptide structures with tissue transglutaminase, Bioconjug. Chem 14 (2003) 748–755. 10.1021/bc034017t. [DOI] [PubMed] [Google Scholar]
- [53].Savoca MP, Tonoli E, Atobatele AG, Verderio EAM, Biocatalysis by transglutaminases: A review of biotechnological applications, Micromachines. 9 (2018) 562. 10.3390/mi9110562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Dawson PE, Muir TW, Clark-Lewis I, Kent SBH, Synthesis of Proteins by Native Chemical Ligation, Science (80-. ) 266 (1994) 776–779. [DOI] [PubMed] [Google Scholar]
- [55].Jung JP, Jones JL, Cronier SA, Collier JH, Modulating the mechanical properties of self-assembled peptide hydrogels via native chemical ligation, Biomaterials. 29 (2008) 2143–2151. 10.1016/j.biomaterials.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hainline KM, Gu F, Handley JF, Tian YF, Wu Y, de Wet L, Vander Griend DJ, Collier JH, Self-Assembling Peptide Gels for 3D Prostate Cancer Spheroid Culture, Macromol. Biosci 19 (2019) 1800249. 10.1002/mabi.201800249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gasiorowski JZ, Collier JH, Directed intermixing in multicomponent self-assembling biomaterials, Biomacromolecules. 12 (2011) 3549–3558. 10.1021/bm200763y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jung JP, Nagaraj AK, Fox EK, Rudra JS, Devgun JM, Collier JH, Co-assembling peptides as defined matrices for endothelial cells, Biomaterials. 30 (2009) 2400–2410. 10.1016/j.biomaterials.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ, Schneider JP, Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells, Proc. Natl. Acad. Sci 104 (2007) 7791–7796. 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kisiday J, Jin M, Kurz B, Hung H, Semino C, Zhang S, Grodzinsky AJ, Self-assembling peptide hydrogel fosters chondrocyte extracellular matrix production and cell division: Implications for cartilage tissue repair, Proc. Natl. Acad. Sci. U. S. A 99 (2002) 9996–10001. 10.1073/pnas.142309999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pugliese R, Maleki M, Zuckermann RN, Gelain F, Self-assembling peptides cross-linked with genipin: Resilient hydrogels and self-standing electrospun scaffolds for tissue engineering applications, Biomater. Sci 7 (2019) 76–91. 10.1039/c8bm00825f. [DOI] [PubMed] [Google Scholar]
- [62].Raspa A, Carminati L, Pugliese R, Fontana F, Gelain F, Self-assembling peptide hydrogels for the stabilization and sustained release of active Chondroitinase ABC in vitro and in spinal cord injuries, J. Control. Release 330 (2021) 1208–1219. 10.1016/j.jconrel.2020.11.027. [DOI] [PubMed] [Google Scholar]
- [63].Pugliese R, Marchini A, Saracino GAA, Zuckermann RN, Gelain F, Cross-linked self-assembling peptide scaffolds, Nano Res. 11 (2018) 586–602. 10.1007/s12274-017-1834-6. [DOI] [Google Scholar]
- [64].Nonoyama T, Ogasawara H, Tanaka M, Higuchi M, Kinoshita T, Calcium phosphate biomineralization in peptide hydrogels for injectable bone-filling materials, Soft Matter. 8 (2012) 11531. 10.1039/c2sm26538a. [DOI] [Google Scholar]
- [65].Segman-Magidovich S, Grisaru H, Gitli T, Levi-Kalisman Y, Rapaport H, Matrices of acidic β-sheet peptides as templates for calcium phosphate mineralization, Adv. Mater 20 (2008) 2156–2161. 10.1002/adma.200702125. [DOI] [Google Scholar]
- [66].Amosi N, Zarzhitsky S, Gilsohn E, Salnikov O, Monsonego-Ornan E, Shahar R, Rapaport H, Acidic peptide hydrogel scaffolds enhance calcium phosphate mineral turnover into bone tissue, Acta Biomater. 8 (2012) 2466–2475. 10.1016/j.actbio.2012.04.003. [DOI] [PubMed] [Google Scholar]
- [67].Gungormus M, Branco M, Fong H, Schneider JP, Tamerler C, Sarikaya M, Self assembled bi-functional peptide hydrogels with biomineralization-directing peptides, Biomaterials. 31 (2010) 7266–7274. 10.1016/j.biomaterials.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Hu X, Liao M, Gong H, Zhang L, Cox H, Waigh TA, Lu JR, Recent advances in short peptide self-assembly: from rational design to novel applications, Curr. Opin. Colloid Interface Sci 45 (2020) 1–13. 10.1016/j.cocis.2019.08.003. [DOI] [Google Scholar]
- [69].Reches M, Gazit E, Casting metal nanowires within discrete self-assembled peptide nanotubes, Science (80-. ) 300 (2003) 625–627. 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- [70].Reches M, Gazit E, Self-assembly of peptide nanotubes and amyloid-like structures by charged-termini-capped diphenylalanine peptide analogues, Isr. J. Chem 45 (2005) 363–371. 10.1560/5mc0-v3dx-ke0b-yf3j. [DOI] [Google Scholar]
- [71].Smith AM, Williams RJ, Tang C, Coppo P, Collins RF, Turner ML, Saiani A, Ulijn RV, Fmoc-diphenylalanine self assembles to a hydrogel via a novel architecture based on n-n interlocked β-sheets, Adv. Mater 20 (2008) 37–41. 10.1002/adma.200701221. [DOI] [Google Scholar]
- [72].Jayawarna V, Ali M, Jowitt TA, Miller AF, Saiani A, Gough JE, Ulijn RV, Nanostructured hydrogels for three-dimensional cell culture through self-assembly of fluorenylmethoxycarbonyl-dipeptides, Adv. Mater 18 (2006) 611–614. 10.1002/adma.200501522. [DOI] [Google Scholar]
- [73].Tang C, Smith AM, Collins RF, Ulijn RV, Saiani A, Fmoc-Diphenylalanine Self-Assembly Mechanism Induces Apparent pK a Shifts, Langmuir. 25 (2009) 9447–9453. 10.1021/la900653q. [DOI] [PubMed] [Google Scholar]
- [74].Erdogan H, Yilmaz M, Babur E, Duman M, Aydin HM, Demirel G, Fabrication of Plasmonic Nanorod-Embedded Dipeptide Microspheres via the Freeze-Quenching Method for Near-Infrared Laser-Triggered Drug-Delivery Applications, Biomacromolecules. 17 (2016) 1788–1794. 10.1021/acs.biomac.6b00214. [DOI] [PubMed] [Google Scholar]
- [75].Ischakov R, Adler-Abramovich L, Buzhansky L, Shekhter T, Gazit E, Peptide-based hydrogel nanoparticles as effective drug delivery agents, Bioorganic Med. Chem 21 (2013) 3517–3522. 10.1016/j.bmc.2013.03.012. [DOI] [PubMed] [Google Scholar]
- [76].Jayawarna V, Richardson SM, Hirst AR, Hodson NW, Saiani A, Gough JE, Ulijn RV, Introducing chemical functionality in Fmoc-peptide gels for cell culture, Acta Biomater. 5 (2009) 934–943. 10.1016/j.actbio.2009.01.006. [DOI] [PubMed] [Google Scholar]
- [77].Zhou M, Smith AM, Das AK, Hodson NW, Collins RF, Ulijn RV, Gough JE, Self-assembled peptide-based hydrogels as scaffolds for anchorage-dependent cells, Biomaterials. 30 (2009) 2523–2530. 10.1016/j.biomaterials.2009.01.010. [DOI] [PubMed] [Google Scholar]
- [78].Scott G, Roy S, Abul-Haija YM, Fleming S, Bai S, Ulijn RV, Pickering stabilized peptide gel particles as tunable microenvironments for biocatalysis, Langmuir. 29 (2013) 14321–14327. 10.1021/la403448s. [DOI] [PubMed] [Google Scholar]
- [79].Diaferia C, Morelli G, Accardo A, Fmoc-diphenylalanine as a suitable building block for the preparation of hybrid materials and their potential applications, J. Mater. Chem. B. 7 (2019) 5142–5155. 10.1039/c9tb01043b. [DOI] [PubMed] [Google Scholar]
- [80].Lampel A, McPhee SA, Park HA, Scott GG, Humagain S, Hekstra DR, Yoo B, Frederix PWJM, De Li T, Abzalimov RR, Greenbaum SG, Tuttle T, Hu C, Bettinger CJ, Ulijn RV, Polymeric peptide pigments with sequence-encoded properties, Science (80-. ) 356 (2017) 1064–1068. 10.1126/science.aal5005. [DOI] [PubMed] [Google Scholar]
- [81].Frederix PWJM, Scott GG, Abul-Haija YM, Kalafatovic D, Pappas CG, Javid N, Hunt NT, Ulijn RV, Tuttle T, Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels, Nat. Chem 7 (2015) 30–37. 10.1038/nchem.2122. [DOI] [PubMed] [Google Scholar]
- [82].Sahoo JK, Nazareth C, VandenBerg MA, Webber MJ, Aromatic identity, electronic substitution, and sequence in amphiphilic tripeptide self-assembly, Soft Matter. 14 (2018) 9168–9174. 10.1039/C8SM01994K. [DOI] [PubMed] [Google Scholar]
- [83].Tang JD, Mura C, Lampe KJ, Stimuli-Responsive, Pentapeptide, Nanofiber Hydrogel for Tissue Engineering, J. Am. Chem. Soc 141 (2019) 4886–4899. 10.1021/jacs.8b13363. [DOI] [PubMed] [Google Scholar]
- [84].Tang JD, Mura C, Lampe KJ, Stimuli-Responsive, Pentapeptide, Nanofiber Hydrogel for Tissue Engineering, J. Am. Chem. Soc 141 (2019) 4886–4899. 10.1021/jacs.8b13363. [DOI] [PubMed] [Google Scholar]
- [85].Pauling L, Corey RB, Two Rippled-Sheet Configurations of Polypeptide Chains, and a Note about the Pleated Sheets, Proc. Natl. Acad. Sci 39 (1953) 253–256. 10.1073/pnas.39.4.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Nagy-Smith K, Beltramo PJ, Moore E, Tycko R, Furst EM, Schneider JP, Molecular, Local, and Network-Level Basis for the Enhanced Stiffness of Hydrogel Networks Formed from Coassembled Racemic Peptides: Predictions from Pauling and Corey, ACS Cent. Sci 3 (2017) 586–597. 10.1021/acscentsci.7b00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Nagy KJ, Giano MC, Jin A, Pochan DJ, Schneider JP, Enhanced Mechanical Rigidity of Hydrogels Formed From Enantiomeric Peptide Assemblies, J Am Chem Soc. 133 (2011) 14975–14977. 10.1158/0008-5472.CAN-10-4002.B0NE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Swanekamp RJ, Dimaio JTM, Bowerman CJ, Nilsson BL, Coassembly of enantiomeric amphipathic peptides into amyloid-inspired rippled β-sheet fibrils, J. Am. Chem. Soc 134 (2012) 5556–5559. 10.1021/ja301642c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Swanekamp RJ, Welch JJ, Nilsson BL, Proteolytic stability of amphipathic peptide hydrogels composed of self-assembled pleated B-sheet or coassembled rippled B-sheet fibrils, Chem. Commun 50 (2014) 10133–10136. 10.1039/c4cc04644g. [DOI] [PubMed] [Google Scholar]
- [90].Abdilla A, Dolinski ND, De Roos P, Ren JM, Van Der Woude E, Seo SE, Zayas MS, Lawrence J, Read De Alaniz J, Hawker CJ, Polymer Stereocomplexation as a Scalable Platform for Nanoparticle Assembly, J. Am. Chem. Soc 142 (2020) 1667–1672. 10.1021/jacs.9b10156. [DOI] [PubMed] [Google Scholar]
- [91].Ren JM, Knight AS, Van Ravensteijn BGP, Kohl P, Zerdan RB, Li Y, Lunn DJ, Abdilla A, Qiao GG, Hawker CJ, DNA-Inspired Strand-Exchange for Switchable PMMA-Based Supramolecular Morphologies, J. Am. Chem. Soc 141 (2019) 2630–2635. 10.1021/jacs.8b12964. [DOI] [PubMed] [Google Scholar]
- [92].Tsuji H, Poly(lactic acid) stereocomplexes: A decade of progress, Adv. Drug Deliv. Rev 107 (2016) 97–135. [DOI] [PubMed] [Google Scholar]
- [93].Nagy-Smith K, Beltramo PJ, Moore E, Tycko R, Furst EM, Schneider JP, Molecular, Local, and Network-Level Basis for the Enhanced Stiffness of Hydrogel Networks Formed from Coassembled Racemic Peptides: Predictions from Pauling and Corey, ACS Cent. Sci 3 (2017) 586–597. 10.1021/acscentsci.7b00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kar K, Arduini I, Drombosky KW, Van Der Wel PCA, Wetzel R, D-polyglutamine amyloid recruits l-polyglutamine monomers and kills cells, J. Mol. Biol 426 (2014) 816–829. 10.1016/j.jmb.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wadai H, Yamaguchi KI, Takahashi S, Kanno T, Kawai T, Naiki H, Goto Y, Stereospecific amyloid-like fibril formation by a peptide fragment of β2-microglobulin, Biochemistry. 44 (2005) 157–164. 10.1021/bi0485880. [DOI] [PubMed] [Google Scholar]
- [96].Koga T, Matsuoka M, Higashi N, Structural control of self-assembled nanofibers by artificial p-sheet peptides composed of D- or L-isomer, J. Am. Chem. Soc 127 (2005) 17596–17597. 10.1021/ja0558387. [DOI] [PubMed] [Google Scholar]
- [97].Zou R, Wang Q, Wu J, Wu J, Schmuck C, Tian H, Peptide self-assembly triggered by metal ions, Chem. Soc. Rev 44 (2015) 5200–5219. 10.1039/c5cs00234f. [DOI] [PubMed] [Google Scholar]
- [98].Jung KH, Oh S, Park J, Park YJ, Park SH, Lee KH, A novel fluorescent peptidyl probe for highly sensitive and selective ratiometric detection of Cd(ii) in aqueous and bio-samples: Via metal ion-mediated self-assembly, New J. Chem 42 (2018) 18143–18151. 10.1039/c8nj02298d. [DOI] [Google Scholar]
- [99].Guo Y, Wang S, Du H, Chen X, Fei H, Silver Ion-Histidine Interplay Switches Peptide Hydrogel from Antiparallel to Parallel p-Assembly and Enables Controlled Antibacterial Activity, Biomacromolecules. 20 (2019) 558–565. 10.1021/acs.biomac.8b01480. [DOI] [PubMed] [Google Scholar]
- [100].Kakinoki S, Hirano Y, Oka M, On the stability of polyproline-I and II structures of proline oligopeptides, Polym. Bull 53 (2005) 109–115. 10.1007/s00289-004-0317-6. [DOI] [Google Scholar]
- [101].Adzhubei AA, Sternberg MJE, Makarov AA, Polyproline-II helix in proteins: Structure and function, J. Mol. Biol 425 (2013) 2100–2132. 10.1016/j.jmb.2013.03.018. [DOI] [PubMed] [Google Scholar]
- [102].Priftis D, Leon L, Song Z, Perry SL, Margossian KO, Tropnikova A, Cheng J, Tirrell M, Self-Assembly of α-Helical Polypeptides Driven by Complex Coacervation, Angew. Chemie - Int. Ed 54 (2015) 11128–11132. 10.1002/anie.201504861. [DOI] [PubMed] [Google Scholar]
- [103].Perry SL, Leon L, Hoffmann KQ, Kade MJ, Priftis D, Black KA, Wong D, Klein RA, Pierce CF, Margossian KO, Whitmer JK, Qin J, De Pablo JJ, Tirrell M, Chirality-selected phase behaviour in ionic polypeptide complexes, Nat. Commun 6 (2015). 10.1038/ncomms7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].LUPAS A, Coiled coils: new structures and new functions, Trends Biochem. Sci 21 (1996) 375–382. 10.1016/S0968-0004(96)10052-9. [DOI] [PubMed] [Google Scholar]
- [105].Wu Y, Collier JH, α-Helical coiled-coil peptide materials for biomedical applications, Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 9 (2017) 1–17. 10.1002/wnan.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Pauling L, Corey RB, Two hydrogen-bonded spiral configurations of the polypeptide chain, J. Am. Chem. Soc 72 (1950) 5349. 10.1021/ja01167a545. [DOI] [Google Scholar]
- [107].Pauling L, Corey RB, Branson HR, The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain., Proc. Natl. Acad. Sci. U. S. A 37 (1951) 205–211. 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Crick FHC, The packing of α-helices: simple coiled-coils, Acta Crystallogr. 6 (1953) 689–697. 10.1107/s0365110x53001964. [DOI] [Google Scholar]
- [109].Mason JM, Arndt KM, Coiled coil domains: Stability, specificity, and biological implications, ChemBioChem. 5 (2004) 170–176. 10.1002/cbic.200300781. [DOI] [PubMed] [Google Scholar]
- [110].Harbury PB, Zhang T, Kim PS, Alber T, A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants, Science (80-. ) 262 (1993) 1401–1407. 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- [111].Liu J, Zheng Q, Deng Y, Cheng C, Kallenbach NR, Lu M, A seven-helix coiled coil, Proc. Natl. Acad. Sci 103 (2006) 15457–15462. https://doi.org/10.1073_pnas.0604871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Mortenson DE, Steinkruger JD, Kreitler DF, Perroni DV, Sorenson GP, Huang L, Mittal R, Yun HG, Travis BR, Mahanthappa MK, Forest KT, Gellman SH, High-resolution structures of a heterochiral coiled coil, Proc. Natl. Acad. Sci. U. S. A 112 (2015) 13144–13149. 10.1073/pnas.1507918112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Kreitler DF, Yao Z, Steinkruger JD, Mortenson DE, Huang L, Mittal R, Travis BR, Forest KT, Gellman SH, A Hendecad Motif Is Preferred for Heterochiral Coiled-Coil Formation, J. Am. Chem. Soc 141 (2019) 1583–1592. 10.1021/jacs.8b11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Apostolovic B, Danial M, Klok HA, Coiled coils: Attractive protein folding motifs for the fabrication of self-assembled, responsive and bioactive materials, Chem. Soc. Rev 39 (2010) 3541–3575. 10.1039/b914339b. [DOI] [PubMed] [Google Scholar]
- [115].Marsden HR, Kros A, Self-assembly of coiled coils in synthetic biology: inspiration and progress, Angew. Chemie - Int. Ed 49 (2010) 2988–3005. 10.1002/anie.200904943. [DOI] [PubMed] [Google Scholar]
- [116].Hu JC, O’Shea EK, Kim PS, Sauer RT, Sequence requirements for coiled-coils: Analysis with λ repressor-GCN4 leucine zipper fusions, Science (80-. ) 250 (1990) 1400–1403. 10.1126/science.2147779. [DOI] [PubMed] [Google Scholar]
- [117].Thomas F, Boyle AL, Burton AJ, Woolfson DN, A set of de novo designed parallel heterodimeric coiled coils with quantified dissociation constants in the micromolar to sub-nanomolar regime, J. Am. Chem. Soc 135 (2013) 5161–5166. 10.1021/ja312310g. [DOI] [PubMed] [Google Scholar]
- [118].Oshaben KM, Horne WS, Tuning assembly size in peptide-based supramolecular polymers by modulation of subunit association affinity, Biomacromolecules. 15 (2014) 1436–1442. 10.1021/bm5000423. [DOI] [PubMed] [Google Scholar]
- [119].Aronsson C, Danmark S, Zhou F, Öberg P, Enander K, Su H, Aili D, Self-sorting heterodimeric coiled coil peptides with defined and tuneable self-assembly properties, Sci. Rep 5 (2015) 1–10. 10.1038/srep14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Solemanifar A, Nguyen TAH, Laycock B, Shewan HM, Donose BC, Creasey RCG, Assessing the effect of aromatic residue placement on the α-helical peptide structure and nanofibril formation of 21-mer peptides, Mol. Syst. Des. Eng 5 (2020) 521–531. 10.1039/c9me00082h. [DOI] [Google Scholar]
- [121].Jeffrey YS, Hodges RS, Kay CM, Effect of Chain Length on the Formation and Stability of Synthetic α-Helical Coiled Coils, Biochemistry. 33 (1994) 15501–15510. 10.1021/bi00255a032. [DOI] [PubMed] [Google Scholar]
- [122].Litowski JR, Hodges RS, Designing heterodimeric two-stranded α-helical coiled-coils. Effects of hydrophobicity and α-helical propensity on protein folding, stability, and specificity, J. Biol. Chem 277 (2002) 37272–37279. 10.1074/jbc.M204257200. [DOI] [PubMed] [Google Scholar]
- [123].Zhu B-Y, Zhou ME, Kay CM, Hodges RS, Packing and hydrophobicity effects on protein folding and stability: Effects of β-branched amino acids, valine and isoleucine, on the formation and stability of two-stranded a-helical coiled coils/leucine zippers, Protein Sci. 2 (1993) 383–394. 10.1002/pro.5560020310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Apostolovic B, Klok HA, pH-sensitivity of the E3/K3 heterodimeric coiled coil, Biomacromolecules. 9 (2008) 3173–3180. 10.1021/bm800746e. [DOI] [PubMed] [Google Scholar]
- [125].Graddis TJ, Myszka DG, Chaiken IM, Controlled Formation of Model Homo- and Heterodimer Coiled Coil Polypeptides, Biochemistry. 32 (1993) 12664–12671. 10.1021/bi00210a015. [DOI] [PubMed] [Google Scholar]
- [126].Vandermeulen GWM, Tziatzios C, Klok HA, Reversible self-organization of poly(ethylene glycol)-based hybrid block copolymers mediated by a de novo four-stranded α-helical coiled coil motif, Macromolecules. 36 (2003) 4107–4114. 10.1021/ma034124i. [DOI] [Google Scholar]
- [127].Grewal MG, Gray VP, Letteri RA, Highley CB, User-defined, temporal presentation of bioactive molecules on hydrogel substrates using supramolecular coiled coil complexes, Biomater. Sci 22 (2021) 154. 10.1039/D1BM00016K. [DOI] [PubMed] [Google Scholar]
- [128].De Crescenzo G, Litowski JR, Hodges RS, O’Connor-McCourt MD, Real-time monitoring of the interactions of two-stranded de novo designed coiled-coils: Effect of chain length on the kinetic and thermodynamic constants of binding, Biochemistry. 42 (2003) 1754–1763. 10.1021/bi0268450. [DOI] [PubMed] [Google Scholar]
- [129].Lovejoy B, Choe S, Cascio D, McRorie D, DeGrado W, Eisenberg D, Crystal structure of a synthetic triple-stranded alpha-helical bundle, Science (80-. ) 259 (1993) 1288–1293. 10.1126/science.8446897. [DOI] [PubMed] [Google Scholar]
- [130].Gradišar H, Božič S, Doles T, Vengust D, Hafner-Bratkovič I, Mertelj A, Webb B, Šali A, Klavžar S, Jerala R, Design of a single-chain polypeptide tetrahedron assembled from coiled-coil segments, Nat. Chem. Biol 9 (2013) 362–366. 10.1038/nchembio.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].O’Shea EK, Rutkowski R, Kim PS, Evidence that the leucine zipper is a coiled coil, Science (80-. ) 243 (1989) 538–542. 10.1126/science.2911757. [DOI] [PubMed] [Google Scholar]
- [132].Pandya MJ, Spooner GM, Sunde M, Thorpe JR, Rodger A, Woolfson DN, Sticky-end assembly of a designed peptide fiber provides insight into protein fibrillogenesis, Biochemistry. 39 (2000) 8728–8734. 10.1021/bi000246g. [DOI] [PubMed] [Google Scholar]
- [133].Ryadnov MG, Woolfson DN, Engineering the morphology of a self-assembling protein fibre, Nat. Mater 2 (2003) 329–332. 10.1038/nmat885. [DOI] [PubMed] [Google Scholar]
- [134].Woolfson DN, Mahmoud ZN, More than just bare scaffolds: Towards multi-component and decorated fibrous biomaterials, Chem. Soc. Rev 39 (2010) 3464–3479. 10.1039/c0cs00032a. [DOI] [PubMed] [Google Scholar]
- [135].Mahmoud ZN, Grundy DJ, Channon KJ, Woolfson DN, The non-covalent decoration of self-assembling protein fibers, Biomaterials. 31 (2010) 7468–7474. 10.1016/j.biomaterials.2010.06.041. [DOI] [PubMed] [Google Scholar]
- [136].Sinha NJ, Wu D, Kloxin CJ, Saven JG, Jensen GV, Pochan DJ, Polyelectrolyte character of rigid rod peptide bundlemer chains constructed: Via hierarchical self-Assembly, Soft Matter. 15 (2019) 9858–9870. 10.1039/c9sm01894h. [DOI] [PubMed] [Google Scholar]
- [137].Zhang HV, Polzer F, Haider MJ, Tian Y, Villegas JA, Kiick KL, Pochan DJ, Saven JG, Computationally designed peptides for self-assembly of nanostructured lattices, Sci. Adv 2 (2016). 10.1126/sciadv.1600307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Wu D, Sinha N, Lee J, Sutherland BP, Halaszynski NI, Tian Y, Caplan J, Zhang HV, Saven JG, Kloxin CJ, Pochan DJ, Polymers with controlled assembly and rigidity made with click-functional peptide bundles, Nature. 574 (2019) 658–662. 10.1038/s41586-019-1683-4. [DOI] [PubMed] [Google Scholar]
- [139].Persikov AV, Ramshaw JAM, Kirkpatrick A, Brodsky B, Amino acid propensities for the collagen triple-helix, Biochemistry. 39 (2000) 14960–14967. 10.1021/bi001560d. [DOI] [PubMed] [Google Scholar]
- [140].Shoulders MD, Raines RT, Collagen structure and stability, Annu. Rev. Biochem 78 (2009) 929–958. 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Yu SM, Li Y, Kim D, Collagen mimetic peptides: Progress towards functional applications, Soft Matter. 7 (2011) 7927–7938. 10.1039/c1sm05329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Luo T, Kiick KL, Collagen-like peptides and peptide-polymer conjugates in the design of assembled materials, Eur. Polym. J 49 (2013) 2998–3009. 10.1016/j.eurpolymj.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Persikov AV, Ramshaw JAM, Brodsky B, Prediction of collagen stability from amino acid sequence, J. Biol. Chem 280 (2005) 19343–19349. 10.1074/jbc.M501657200. [DOI] [PubMed] [Google Scholar]
- [144].Holmgren SK, Bretscher LE, Taylor KM, Raines RT, A hyperstable collagen mimic, Chem. Biol 6 (1999) 63–70. 10.1016/S1074-5521(99)80003-9. [DOI] [PubMed] [Google Scholar]
- [145].Bretscher LE, Jenkins CL, Taylor KM, DeRider ML, Raines RT, Conformational stability of collagen relies on a stereoelectronic effect, J. Am. Chem. Soc 123 (2001) 777–778. 10.1021/ja005542v. [DOI] [PubMed] [Google Scholar]
- [146].DeRider ML, Wilkens SJ, Waddell MJ, Bretscher LE, Weinhold F, Raines RT, Markley JL, Collagen stability: Insights from NMR spectroscopic and hybrid density functional computational investigations of the effect of electronegative substituents on prolyl ring conformations, J. Am. Chem. Soc 124 (2002) 2497–2505. 10.1021/ja0166904. [DOI] [PubMed] [Google Scholar]
- [147].Xu F, Khan IJ, McGuinness K, Parmar AS, Silva T, Murthy NS, Nanda V, Self-assembly of left- and right-handed molecular screws, J. Am. Chem. Soc 135 (2013) 18762–18765. 10.1021/ja4106545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Rele S, Song Y, Apkarian RP, Qu Z, Conticello VP, Chaikof EL, D-periodic collagen-mimetic microfibers J. Am. Chem. Soc 129 (2007) 14780–14787. 10.1021/ja0758990. [DOI] [PubMed] [Google Scholar]
- [149].Hilderbrand AM, Ford EM, Guo C, Sloppy JD, Kloxin AM, Hierarchically structured hydrogels utilizing multifunctional assembling peptides for 3D cell culture, Biomater. Sci 8 (2020) 1256–1269. 10.1039/c9bm01894h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].O’Leary LER, Fallas JA, Bakota EL, Kang MK, Hartgerink JD, Multi-hierarchical self-assembly of a collagen mimetic peptide from triple helix to nanofibre and hydrogel, Nat. Chem 3 (2011) 821–828. 10.1038/nchem.1123. [DOI] [PubMed] [Google Scholar]
- [151].Sarkar B, Oleary LER, Hartgerink JD, Self-assembly of fiber-forming collagen mimetic peptides controlled by triple-helical nucleation, J. Am. Chem. Soc 136 (2014) 14417–14424. 10.1021/ja504377s. [DOI] [PubMed] [Google Scholar]
- [152].Jiang T, Xu C, Zuo X, Conticello VP, Structurally homogeneous nanosheets from self-assembly of a collagen-mimetic peptide, Angew. Chemie - Int. Ed 53 (2014) 8367–8371. 10.1002/anie.201403780. [DOI] [PubMed] [Google Scholar]
- [153].Tanrikulu IC, Forticaux A, Jin S, Raines RT, Peptide tessellation yields micrometre-scale collagen triple helices, Nat. Chem 8 (2016) 1008–1014. 10.1038/nchem.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Gauba V, Hartgerink JD, Surprisingly high stability of collagen ABC heterotrimer: Evaluation of side chain charge pairs, J. Am. Chem. Soc 129 (2007) 15034–15041. 10.1021/ja075854z. [DOI] [PubMed] [Google Scholar]
- [155].Russell LE, Fallas JA, Hartgerink JD, Selective assembly of a high stability AAB collagen heterotrimer, J. Am. Chem. Soc 132 (2010) 3242–3243. 10.1021/ja909720g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Krishna OD, Kiick KL, Supramolecular assembly of electrostatically stabilized, hydroxyproline-lacking collagen-mimetic peptides, Biomacromolecules. 10 (2009) 2626–2631. 10.1021/bm900551c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Hilderbrand A, Taylor P, Stanzione F, LaRue M, Chen G, Jayaraman A, Kloxin A, Combining simulations and experiments for the molecular engineering of multifunctional collagen mimetic peptide-based materials, Soft Matter. (2021). 10.1039/D0SM01562H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Raman SS, Parthasarathi R, Subramanian Y, Ramasami T, Role of length-dependent stability of collagen-like peptides, J. Phys. Chem. B. 112 (2008) 1533–1539. 10.1021/jp0728297. [DOI] [PubMed] [Google Scholar]
- [159].Barth D, Musiol HJ, Schütt M, Fiori S, Milbradt AG, Renner C, Moroder L, The role of cystine knots in collagen folding and stability, part I. Conformational properties of (Pro-Hyp-Gly)5 and (Pro-(4S)-FPro-Gly)5 model trimers with an artificial cystine knot, Chem. - A Eur. J 9 (2003) 3692–3702. 10.1002/chem.200304917. [DOI] [PubMed] [Google Scholar]
- [160].Buehler MJ, Nature designs tough collagen: Explaining the nanostructure of collagen fibrils, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 12285–12290. 10.1073/pnas.0603216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Kar K, Amin P, Bryan MA, Persikov AV, Wang AMYH, Brodsky B, Self-association of collagen triple helix peptides into higher order structures, J. Biol. Chem 281 (2006) 33283–33290. 10.1074/jbc.M605747200. [DOI] [PubMed] [Google Scholar]
- [162].Kar K, Wang Y-H, Brodsky B, Sequence dependence of kinetics and morphology of collagen model peptide self-assembly into higher order structures, Protein Sci. 17 (2008) 1086–1095. 10.1110/ps.083441308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Przybyla DE, Chmielewski J, Metal-triggered radial self-assembly of collagen peptide fibers, J. Am. Chem. Soc 130 (2008) 12610–12611. 10.1021/ja804942w. [DOI] [PubMed] [Google Scholar]
- [164].Pires MM, Przybyla DE, Chmielewski J, A metal-collagen peptide framework for three-dimensional cell culture, Angew. Chemie - Int. Ed 48 (2009) 7813–7817. 10.1002/anie.200902375. [DOI] [PubMed] [Google Scholar]
- [165].Cejas MA, Kinney WA, Chen C, Leo GC, Tounge BA, Vinter JG, Joshi PP, Maryanoff BE, Collagen-related peptides: Self-assembly of short, single strands into a functional biomaterial of micrometer scale, J. Am. Chem. Soc 129 (2007) 2202–2203. 10.1021/ja066986f. [DOI] [PubMed] [Google Scholar]
- [166].Matson JB, Zha RH, Stupp SI, Peptide self-assembly for crafting functional biological materials, Curr. Opin. Solid State Mater. Sci 15 (2011) 225–235. 10.1016/j.cossms.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Cui H, Webber MJ, Stupp SI, Self-Assembly of Peptide Amphiphiles: From Molecules to Nanostructures to Biomaterials, Pept. Sci 94 (2010) 1–18. 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Webber MJ, Berns EJ, Stupp SI, Supramolecular nanofibers of peptide amphiphiles for medicine, Isr. J. Chem 53 (2013) 530–554. 10.1002/ijch.201300046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Jain RK, Gupta CM, Anand N, Synthesis of Peptidylglycophospholipids, novel derivatives of muramyl-dipeptide, Tetrahedron Lett. 22 (1981) 2317–2320. [Google Scholar]
- [170].Thompson NL, Brian AA, McConnell HM, Covalent linkage of a synthetic peptide to a fluorescent phospholipid and its incorporation into supported phospholipid monolayers, Biochim. Biophys. Acta 772 (1984) 10–19. [DOI] [PubMed] [Google Scholar]
- [171].Berndt P, Fields GB, Tirrell M, Synthetic Lipidation of Peptides and Amino Acids: Monolayer Structure and Properties, J. Am. Chem. Soc 117 (1995) 9515–9522. 10.1021/ja00142a019. [DOI] [Google Scholar]
- [172].Yu YC, Berndt P, Tirrell M, Fields GB, Self-assembling amphiphiles for construction of protein molecular architecture, J. Am. Chem. Soc 118 (1996) 12515–12520. 10.1021/ja9627656. [DOI] [Google Scholar]
- [173].Hartgerink JD, Beniash E, Stupp SI, Self-Assembly and Mineralization of Peptide-Amphiphile Nanofibers, Science (80-. ) 294 (2001) 1684–1688. [DOI] [PubMed] [Google Scholar]
- [174].Hendricks MP, Sato K, Palmer LC, Stupp SI, Supramolecular Assembly of Peptide Amphiphiles, Acc. Chem. Res 50 (2017) 2440–2448. 10.1021/acs.accounts.7b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Wester JR, Lewis JA, Freeman R, Sai H, Palmer LC, Henrich SE, Stupp SI, Supramolecular Exchange among Assemblies of Opposite Charge Leads to Hierarchical Structures, J. Am. Chem. Soc 142 (2020) 12216–12225. 10.1021/jacs.0c03529. [DOI] [PubMed] [Google Scholar]
- [176].Freeman R, Han M, Álvarez Z, Lewis JA, Wester JR, Stephanopoulos N, McClendon MT, Lynsky C, Godbe JM, Sangji H, Luijten E, Stupp SI, Reversible self-assembly of superstructured networks, Science (80-. ) 362 (2018) 808. 10.1126/science.aat6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Ortony JH, Qiao B, Newcomb CJ, Keller TJ, Palmer LC, Deiss-Yehiely E, Olvera De La Cruz M, Han S, Stupp SI, Water Dynamics from the Surface to the Interior of a Supramolecular Nanostructure, J. Am. Chem. Soc 139 (2017) 8915–8921. 10.1021/jacs.7b02969. [DOI] [PubMed] [Google Scholar]
- [178].Hartgerink JD, Beniash E, Stupp SI, Peptide-amphiphile nanofibers: A versatile scaffold for the preparation of self-assembling materials, Proc. Natl. Acad. Sci 99 (2002) 5133–5138. 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Paramonov SE, Jun H-W, Hartgerink JD, Self-assembly of peptide-amphiphile nanofibers: The roles of hydrogen bonding and amphiphilic packing, J. Am. Chem. Soc 128 (2006) 7291–7298. 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- [180].Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, Stupp SI, Selective Differentiation of Neural Progenitor Cells by High-Epitope Density Nanofibers, Science (80-. ) 303 (2004) 1352–1355. 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- [181].Cha JN, Stucky GD, Morse DE, Deming TJ, Biomimetic synthesis of ordered silica structures mediated by block copolypeptides, Nature. 403 (2000) 289–292. 10.1038/35002038. [DOI] [PubMed] [Google Scholar]
- [182].Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Deming TJ, Rapidly recovering hydrogel scaffolds from self-assembling diblock copolypeptide amphiphiles, Nature. 417 (2002) 424–428. 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- [183].Breedveld V, Nowak AP, Sato J, Deming TJ, Pine DJ, Rheology of block copolypeptide solutions: Hydrogels with tunable properties, Macromolecules. 37 (2004) 3943–3953. 10.1021/ma049885f. [DOI] [Google Scholar]
- [184].Deming TJ, Polypeptide hydrogels via a unique assembly mechanism, Soft Matter. 1 (2005) 28–35. 10.1039/b500307e. [DOI] [PubMed] [Google Scholar]
- [185].Nowak AP, Breedveld V, Pine DJ, Deming TJ, Unusual Salt Stability in Highly Charged Diblock Co-polypeptide Hydrogels, J. Am. Chem. Soc 125 (2003) 15666–15670. 10.1021/ja0381050. [DOI] [PubMed] [Google Scholar]
- [186].Jacquin M, Muller P, Cottet H, Theodoly O, Self-assembly of charged amphiphilic diblock copolymers with insoluble blocks of decreasing hydrophobicity: From kinetically frozen colloids to macrosurfactants, Langmuir. 26 (2010) 18681–18693. 10.1021/la103391p. [DOI] [PubMed] [Google Scholar]
- [187].Bellomo EG, Wyrsta MD, Pakstis L, Pochan DJ, Deming TJ, Stimuli-responsive polypeptide vesicles by conformation-specific assembly, Nat. Mater 3 (2004) 244–248. 10.1038/nmat1093. [DOI] [PubMed] [Google Scholar]
- [188].Daamen WF, Veerkamp JH, van Hest JCM, van Kuppevelt TH, Elastin as a biomaterial for tissue engineering, Biomaterials. 28 (2007) 4378–4398. 10.1016/j.biomaterials.2007.06.025. [DOI] [PubMed] [Google Scholar]
- [189].Chilkoti A, Dreher MR, Meyer DE, Raucher D, Targeted drug delivery by thermally responsive polymers, Adv. Drug Deliv. Rev 54 (2002) 613–630. 10.1016/S0169-409X(02)00041-8. [DOI] [PubMed] [Google Scholar]
- [190].Li L, Charati MB, Kiick KL, Elastomeric polypeptide-based biomaterials, Polym. Chem 1 (2010) 1160–1170. 10.1039/b9py00346k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Urry DW, Characterization of Soluble Peptides of Elastin by Physical Techniques, 1982. 10.1016/0076-6879(82)82096-X. [DOI] [PubMed] [Google Scholar]
- [192].Urry DW, Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers, J. Phys. Chem. B 101 (1997) 11007–11028. 10.1021/jp972167t. [DOI] [Google Scholar]
- [193].Reiersen H, Clarke AR, Rees AR, Short elastin-like peptides exhibit the same temperature-induced structural transitions as elastin polymers: Implications for protein engineering, J. Mol. Biol 283 (1998) 255–264. 10.1006/jmbi.1998.2067. [DOI] [PubMed] [Google Scholar]
- [194].Urry DW, Luan CH, Parker TM, Gowda DC, Prasad KU, Reid MC, Safavy A, Temperature of Polypeptide Inverse Temperature Transition Depends on Mean Residue Hydrophobicity, J. Am. Chem. Soc 113 (1991) 4346–4348. 10.1021/ja00011a057. [DOI] [Google Scholar]
- [195].Urry DW, Gowda DC, Parker TM, Luan C-H, Reid MC, Harris CM, Pattanaik A, Harris RD, Hydrophobicity scale for proteins based on inverse temperature transitions, Biopolymers. 32 (1992) 1243–1250. 10.1002/bip.360320913. [DOI] [PubMed] [Google Scholar]
- [196].Nuhn H, Klok HA, Secondary structure formation and LCST behavior of short elastin-like peptides, Biomacromolecules. 9 (2008) 2755–2763. 10.1021/bm800784y. [DOI] [PubMed] [Google Scholar]
- [197].Di Zio K, Tirrell DA, Mechanical properties of artificial protein matrices engineered for control of cell and tissue behavior, Macromolecules. 36 (2003) 1553–1558. 10.1021/ma0256587. [DOI] [Google Scholar]
- [198].Shamji MF, Betre H, Kraus VB, Chen J, Chilkoti A, Pichika R, Masuda K, Setton LA, Development and characterization of a fusion protein between thermally responsive elastin-like polypeptide and interleukin-1 receptor antagonist: Sustained release of a local antiinflammatory therapeutic, Arthritis Rheum. 56 (2007) 3650–3661. 10.1002/art.22952. [DOI] [PubMed] [Google Scholar]
- [199].Tang JD, Caliari SR, Lampe KJ, Temperature-Dependent Complex Coacervation of Engineered Elastin-like Polypeptide and Hyaluronic Acid Polyelectrolytes, Biomacromolecules. 19 (2018) 3925–3935. 10.1021/acs.biomac.8b00837. [DOI] [PubMed] [Google Scholar]
- [200].Meco E, Zheng WS, Sharma A, Lampe KJ, Guiding oligodendrocyte precursor cell maturation with urokinase plasminogen activator-degradable elastin-like protein hydrogels, Biomacromolecules. (2020). 10.1021/acs.biomac.0c00828. [DOI] [PubMed] [Google Scholar]
- [201].Urry DW, Okamoto K, Harris RD, Hendrix CF, Long MM, Synthetic, Crosslinked Polypentapeptide of Tropoelastin: An Anisotropic, Fibrillar Elastomer, Biochemistry. 15 (1976) 4083–4089. 10.1021/bi00663a026. [DOI] [PubMed] [Google Scholar]
- [202].Gourgas O, Muiznieks LD, Bello DG, Nanci A, Sharpe S, Cerruti M, Cross-Linked Elastin-like Polypeptide Membranes as a Model for Medial Arterial Calcification, Biomacromolecules. 20 (2019) 2625–2636. 10.1021/acs.biomac.9b00417. [DOI] [PubMed] [Google Scholar]
- [203].Cao M, Shen Y, Wang Y, Wang X, Li D, Self-Assembly of Short Elastin-like Amphiphilic Peptides: Effects of temperature, molecular hydrophobicity and charge distribution, Molecules. 24 (2019). 10.3390/molecules24010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Huang W, Rollett A, Kaplan DL, Silk-elastin-like protein biomaterials for the controlled delivery of therapeutics, Expert Opin. Drug Deliv 12 (2015) 779–791. 10.1517/17425247.2015.989830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Panitch A, Yamaoka T, Fournier MJ, Mason TL, Tirrell DA, Design and Biosynthesis of Elastin-like Artificial Extracellular Matrix Proteins Containing Periodically Spaced Fibronectin CS5 Domains, Macromolecules. 32 (1999) 1701–1703. 10.1021/ma980875m. [DOI] [Google Scholar]
- [206].Heilshorn SC, DiZio KA, Welsh ER, Tirrell DA, Endothelial cell adhesion to the fibronectin CS5 domain in artificial extracellular matrix proteins, Biomaterials. 24 (2003) 4245–4252. 10.1016/S0142-9612(03)00294-1. [DOI] [PubMed] [Google Scholar]
- [207].Luo T, Kiick KL, Noncovalent Modulation of the Inverse Temperature Transition and Self-Assembly of Elastin-b-Collagen-like Peptide Bioconjugates, J. Am. Chem. Soc 137 (2015) 15362–15365. 10.1021/jacs.5b09941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Haghpanah JS, Yuvienco C, Civay DE, Barra H, Baker PJ, Khapli S, Voloshchuk N, Gunasekar SK, Muthukumar M, Montclare JK, Artificial protein block copolymers blocks comprising two distinct self-assembling domains, ChemBioChem. 10 (2009) 2733–2735. 10.1002/cbic.200900539. [DOI] [PubMed] [Google Scholar]
- [209].Olsen AJ, Katyal P, Haghpanah JS, Kubilius MB, Li R, Schnabel NL, O’Neill SC, Wang Y, Dai M, Singh N, Tu RS, Montclare JK, Protein Engineered Triblock Polymers Composed of Two SADs: Enhanced Mechanical Properties and Binding Abilities, Biomacromolecules. 19 (2018) 1552–1561. 10.1021/acs.biomac.7b01259. [DOI] [PubMed] [Google Scholar]
- [210].Le DHT, Sugawara-Narutaki A, Elastin-like polypeptides as building motifs toward designing functional nanobiomaterials, Mol. Syst. Des. Eng 4 (2019) 545–565. 10.1039/c9me00002j. [DOI] [Google Scholar]
- [211].Seroski DT, Dong X, Wong KM, Liu R, Shao Q, Paravastu AK, Hall CK, Hudalla GA, Charge guides pathway selection in β-sheet fibrillizing peptide co-assembly, Commun. Chem 3 (2020). 10.1038/s42004-020-00414-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Shao Q, Wong KM, Seroski DT, Wang Y, Liu R, Paravastu AK, Hudalla GA, Hall CK, Anatomy of a selectively coassembled β-sheet peptide nanofiber, Proc. Natl. Acad. Sci 117 (2020) 4710–4717. 10.1073/pnas.1912810117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Wong KM, Wang Y, Seroski DT, Larkin GE, Mehta AK, Hudalla GA, Hall CK, Paravastu AK, Molecular complementarity and structural heterogeneity within co-assembled peptide β-sheet nanofibers, Nanoscale. 12 (2020) 4506–4518. 10.1039/c9nr08725g. [DOI] [PubMed] [Google Scholar]
- [214].Condon JE, Jayaraman A, Development of a Coarse-Grained Model of Collagen-Like Peptide (CLP) for Studies of CLP Triple Helix Melting, J. Phys. Chem. B 122 (2018) 1929–1939. 10.1021/acs.jpcb.7b10916. [DOI] [PubMed] [Google Scholar]
- [215].Wang Y, An Y, Shmidov Y, Bitton R, Deshmukh SA, Matson JB, A combined experimental and computational approach reveals how aromatic peptide amphiphiles self-assemble to form ion-conducting nanohelices, Mater. Chem. Front (2020). 10.1039/D0QM00369G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Zheng T, Bulacu M, Daudey G, Versluis F, Voskuhl J, Martelli G, Raap J, Sevink GJA, Kros A, Boyle AL, A non-zipper-like tetrameric coiled coil promotes membrane fusion, RSC Adv. 6 (2016) 7990–7998. 10.1039/c5ra26175a. [DOI] [Google Scholar]
- [217].Haider MJ, Zhang HV, Sinha N, Fagan JA, Kiick KL, Saven JG, Pochan DJ, Self-assembly and soluble aggregate behavior of computationally designed coiled-coil peptide bundles, Soft Matter. 14 (2018) 5488–5496. 10.1039/C8SM00435H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Ema T, Tanida D, Sakai T, Versatile and practical macrocyclic reagent with multiple hydrogen-bonding sites for chiral discrimination in NMR, J. Am. Chem. Soc 129 (2007) 10591–10596. 10.1021/ja073476s. [DOI] [PubMed] [Google Scholar]
- [219].Zou L, Braegelman AS, Webber MJ, Dynamic Supramolecular Hydrogels Spanning an Unprecedented Range of Host-Guest Affinity, ACS Appl. Mater. Interfaces (2019) 8–13. 10.1021/acsami.8b22151. [DOI] [PubMed] [Google Scholar]
- [220].Zheng K, He C, Nour HF, Zhang Z, Yuan T, Traboulsi H, Mazher J, Trabolsi A, Fang L, Olson MA, Augmented polyhydrazone formation in water by template-assisted polymerization using dual-purpose supramolecular templates, Polym. Chem (2020). 10.1039/c9py01476d. [DOI] [Google Scholar]
- [221].Gaggelli E, Janicka-Klos A, Jankowska E, Kozlowski H, Migliorini C, Molteni E, Valensin D, Valensin G, Wieczerzak E, NMR studies of the Zn2+ interactions with rat and human β-amyloid (1-28) peptides in water-micelle environment, J. Phys. Chem. B (2008). 10.1021/jp075168m. [DOI] [PubMed] [Google Scholar]
- [222].Danielsson J, Pierattelli R, Banci L, Gräslund A, High-resolution NMR studies of the zinc-binding site of the Alzheimer’s amyloid β-peptide, FEBS J. (2007). 10.1111/j.1742-4658.2006.05563.x. [DOI] [PubMed] [Google Scholar]
- [223].Syme CD, Viles JH, Solution 1H NMR investigation of Zn2+ and Cd 2+ binding to amyloid-beta peptide (Aβ) of Alzheimer’s disease, Biochim. Biophys. Acta - Proteins Proteomics (2006). 10.1016/j.bbapap.2005.09.012. [DOI] [PubMed] [Google Scholar]
- [224].Abdrakhmanov R, Blokhin D, Usachev K, Karataeva F, Klochkov V, NMR Studies of the Mn2+ Interactions with Amyloid Peptide Aβ13-23 in Water Environment, Bionanoscience. (2017). 10.1007/s12668-016-0317-7. [DOI] [Google Scholar]
- [225].Hou L, Zagorski MG, NMR reveals anomalous copper(II) binding to the amyloid Aβ peptide of Alzheimer’s disease, J. Am. Chem. Soc (2006). 10.1021/ja046032u. [DOI] [PubMed] [Google Scholar]
- [226].Maity S, Kumar P, Haldar D, Sonication-induced instant amyloid-like fibril formation and organogelation by a tripeptide, Soft Matter. (2011). 10.1039/c1sm05277b. [DOI] [Google Scholar]
- [227].Maity S, Das P, Reches M, Inversion of Supramolecular Chirality by Sonication-Induced Organogelation, Sci. Rep (2015). 10.1038/srep16365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Becker W, Bhattiprolu KC, Gubensäk N, Zangger K, Investigating Protein–Ligand Interactions by Solution Nuclear Magnetic Resonance Spectroscopy, ChemPhysChem. (2018). 10.1002/cphc.201701253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Williamson MP, Using chemical shift perturbation to characterise ligand binding, Prog. Nucl. Magn. Reson. Spectrosc (2013). 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- [230].Bryant RG, The NMR time scale, J. Chem. Educ 60 (1983) 933. 10.1021/ed060p933. [DOI] [Google Scholar]
- [231].Greenfield NJ, Using circular dichroism spectra to estimate protein secondary structure, Nat. Protoc 1 (2007) 2876–2890. 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Greenfield NJ, Analysis of the kinetics of folding of proteins and peptides using circular dichroism, Nat. Protoc 1 (2007) 2891–2899. 10.1038/nprot.2006.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Greenfield N, Fasman GD, Computed Circular Dichroism Spectra for the Evaluation of Protein Conformation, Biochemistry. 8 (1969) 4108–4116. 10.1021/bi00838a031. [DOI] [PubMed] [Google Scholar]
- [234].Greenfield NJ, Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions, Nat. Protoc 1 (2007) 2527–2535. 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Greenfield NJ, Applications of circular dichroism in protein and peptide analysis, TrAC - Trends Anal. Chem 18 (1999) 236–244. 10.1016/S0165-9936(98)00112-5. [DOI] [Google Scholar]
- [236].Zimenkov Y, Dublin SN, Ni R, Tu RS, Breedveld V, Apkarian RP, Conticello VP, Rational design of a reversible pH-responsive switch for peptide self-assembly, J. Am. Chem. Soc 128 (2006) 6770–6771. 10.1021/ja0605974. [DOI] [PubMed] [Google Scholar]
- [237].Behanna HA, Donners JJJM, Gordon AC, Stupp SI, Coassembly of amphiphiles with opposite peptide polarities into nanofibers, J. Am. Chem. Soc 127 (2005) 1193–1200. 10.1021/ja044863u. [DOI] [PubMed] [Google Scholar]
- [238].Okesola BO, Lau HK, Derkus B, Boccorh DK, Wu Y, Wark AW, Kiick KL, Mata A, Covalent co-assembly between resilin-like polypeptide and peptide amphiphile into hydrogels with controlled nanostructure and improved mechanical properties, Biomater. Sci 8 (2020) 846–857. 10.1039/c9bm01796h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Ding L, Jiang Y, Zhang J, Klok HA, Zhong Z, PH-Sensitive Coiled-Coil Peptide-Cross-Linked Hyaluronic Acid Nanogels: Synthesis and Targeted Intracellular Protein Delivery to CD44 Positive Cancer Cells, Biomacromolecules. 19 (2018) 555–562. 10.1021/acs.biomac.7b01664. [DOI] [PubMed] [Google Scholar]
- [240].Wu K, Yang J, Koňák Č, Kopečková P, Kopeček J, Novel synthesis of HPMA copolymers containing peptide grafts and their self-assembly into hybrid hydrogels, Macromol. Chem. Phys 209 (2008) 467–475. 10.1002/macp.200700486. [DOI] [Google Scholar]
- [241].Radu-Wu LC, Yang J, Wu K, Kopecek J, Self-Assembled Hydrogels from Poly[N-(2-hydroxypropyl)methacrylamide] Grafted with β-Sheet Peptides, Biomacromolecules. (2009) 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Yang H, Yang S, Kong J, Dong A, Yu S, Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy, Nat. Protoc 10 (2015) 382–396. 10.1038/nprot.2015.024. [DOI] [PubMed] [Google Scholar]
- [243].Seo J, Hoffmann W, Warnke S, Huang X, Gewinner S, Schöllkopf W, Bowers MT, Von Helden G, Pagel K, An infrared spectroscopy approach to follow β-sheet formation in peptide amyloid assemblies, Nat. Chem 9 (2017) 39–44. 10.1038/nchem.2615. [DOI] [PubMed] [Google Scholar]
- [244].Fan J, Li R, Wang H, He X, Nguyen TP, Letteri RA, Zou J, Wooley KL, Multi-responsive polypeptide hydrogels derived from: N -carboxyanhydride terpolymerizations for delivery of nonsteroidal anti-inflammatory drugs, Org. Biomol. Chem 15 (2017) 5145–5154. 10.1039/c7ob00931c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Förster T, Zwischenmolekulare Energiewanderung und Fluoreszenz, Ann. Phys 437 (1948) 55–75. [Google Scholar]
- [246].Sun Y, Rombola C, Jyothikumar V, Periasamy A, Förster resonance energy transfer microscopy and spectroscopy for localizing protein-protein interactions in living cells, Cytom. Part A 83 (2013) 780–793. 10.1002/cyto.a.22321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Sekar RB, Periasamy A, Fluorescence resonance energy transfer ( FRET ) microscopy imaging of live cell protein localizations, J. Cell Biol 160 (2003) 629–633. 10.1083/jcb.200210140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Acar H, Samaeekia R, Schnorenberg MR, Sasmal DK, Huang J, Tirrell MV, LaBelle JL, Cathepsin-Mediated Cleavage of Peptides from Peptide Amphiphiles Leads to Enhanced Intracellular Peptide Accumulation, Bioconjug. Chem 28 (2017) 2316–2326. 10.1021/acs.bioconjchem.7b00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Bansode ND, Sonar MV, Ganesh KN, A nanofiber assembly directed by the non-classical antiparallel p-structure from 4S-(OH) proline polypeptide, Chem. Commun 52 (2016) 4884–4887. 10.1039/c6cc00838k. [DOI] [PubMed] [Google Scholar]
- [250].Rho JY, Brendel JC, MacFarlane LR, Mansfield EDH, Peltier R, Rogers S, Hartlieb M, Perrier S, Probing the Dynamic Nature of Self-Assembling Cyclic Peptide–Polymer Nanotubes in Solution and in Mammalian Cells, Adv. Funct. Mater 28 (2018) 1704569. 10.1002/adfm.201704569. [DOI] [Google Scholar]
- [251].Leavesley SJ, Rich TC, Overcoming Limitations of FRET Measurements, Cytom. A 89 (2016) 325–327. 10.1002/cyto.a.22851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Silvius JR, Nabi IR, Fluorescence-quenching and resonance energy transfer studies of lipid microdomains in model and biological membranes (Review), Mol. Membr. Biol 23 (2006) 5–16. 10.1080/09687860500473002. [DOI] [PubMed] [Google Scholar]
- [253].Wang M, Law M, Duhamel J, Chen P, Interaction of a self-assembling peptide with oligonucleotides: Complexation and aggregation, Biophys. J 93 (2007) 2477–2490. 10.1529/biophysj.106.102624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Ghisaidoobe ABT, Chung SJ, Intrinsic tryptophan fluorescence in the detection and analysis of proteins: A focus on förster resonance energy transfer techniques, Int. J. Mol. Sci 15 (2014) 22518–22538. 10.3390/ijms151222518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Pandit G, Roy K, Agarwal U, Chatterjee S, Self-Assembly Mechanism of a Peptide-Based Drug Delivery Vehicle, ACS Omega. 3 (2018) 3143–3155. 10.1021/acsomega.7b01871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Steensma DP, Historical Perspective “‘Congo’“ Red Out of Africa?, 2001. http://www.basf-ag.basf.de/en/daten/geschichte/ (accessed June 23, 2020).
- [257].Ashburn TT, Han H, McGuinness BF, Lansbury PT, Amyloid probes based on Congo Red distinguish between fibrils comprising different peptides, Chem. Biol 3 (1996) 351–358. 10.1016/S1074-5521(96)90118-0. [DOI] [PubMed] [Google Scholar]
- [258].Yakupova EI, Bobyleva LG, Vikhlyantsev IM, Bobylev AG, Congo Red and amyloids: History and relationship, Biosci. Rep 39 (2019). 10.1042/BSR20181415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Groenning M, Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status, J. Chem. Biol 3 (2010) 1–18. 10.1007/s12154-009-0027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Lendel C, Bolognesi B, Wahlström A, Dobson CM, Gräslund A, Detergent-like interaction of congo red with the amyloid β peptide, Biochemistry. 49 (2010) 1358–1360. 10.1021/bi902005t. [DOI] [PubMed] [Google Scholar]
- [261].Biancalana M, Koide S, Molecular mechanism of Thioflavin-T binding to amyloid fibrils, Biochim. Biophys. Acta - Proteins Proteomics 1804 (2010) 1405–1412. 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Evgrafova Z, Rothemund S, Voigt B, Hause G, Balbach J, Binder WH, Synthesis and Aggregation of Polymer-Amyloid β Conjugates, Macromol. Rapid Commun 41 (2020). 10.1002/marc.201900378. [DOI] [PubMed] [Google Scholar]
- [263].Thota CK, Yadav N, Chauhan VS, A novel highly stable and injectable hydrogel based on a conformationally restricted ultrashort peptide, Sci. Rep 6 (2016) 1–12. 10.1038/srep31167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Freyer MW, Lewis EA, Isothermal Titration Calorimetry: Experimental Design, Data Analysis, and Probing Macromolecule/Ligand Binding and Kinetic Interactions, Methods Cell Biol. 84 (2008) 79–113. 10.1016/S0091-679X(07)84004-0. [DOI] [PubMed] [Google Scholar]
- [265].Kabiri M, Unsworth LD, Application of isothermal titration calorimetry for characterizing thermodynamic parameters of biomolecular interactions: Peptide self-assembly and protein adsorption case studies, Biomacromolecules. 15 (2014) 3463–3473. 10.1021/bm5004515. [DOI] [PubMed] [Google Scholar]
- [266].Archer WR, Schulz MD, Isothermal titration calorimetry: practical approaches and current applications in soft matter, Soft Matter. 16 (2020) 8760–8774. 10.1039/d0sm01345e. [DOI] [PubMed] [Google Scholar]
- [267].Kabiri M, Bushnak I, McDermot MT, Unsworth LD, Toward a mechanistic understanding of ionic self-complementary peptide self-assembly: Role of water molecules and ions, Biomacromolecules. 14 (2013) 3943–3950. 10.1021/bm401077b. [DOI] [PubMed] [Google Scholar]
- [268].Hildebrand A, Garidel P, Neubert R, Blume A, Thermodynamics of Demicellization of Mixed Micelles Composed of Sodium Oleate and Bile Salts, Langmuir. 20 (2004) 320–328. 10.1021/la035526m. [DOI] [PubMed] [Google Scholar]
- [269].Stetefeld J, McKenna SA, Patel TR, Dynamic light scattering: a practical guide and applications in biomedical sciences, Biophys. Rev 8 (2016) 409–427. 10.1007/s12551-016-0218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Tarasov SG, Gaponenko V, Howard OMZ, Chen Y, Oppenheim JJ, Dyba MA, Subramaniam S, Lee Y, Michejda C, Tarasova NI, Structural plasticity of a transmembrane peptide allows self-assembly into biologically active nanoparticles, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 9798–9803. 10.1073/pnas.1014598108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Carvalho PM, Felício MR, Santos NC, Gonçalves S, Domingues MM, Application of light scattering techniques to nanoparticle characterization and development, Front. Chem 6 (2018) 237. 10.3389/fchem.2018.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Chu B, Laser Light Scattering, Annu. Rev. Phys. Chem 21 (1970) 145–174. 10.1146/annurev.pc.21.100170.001045. [DOI] [Google Scholar]
- [273].Rochas C, Geissler E, Measurement of dynamic light scattering intensity in gels, Macromolecules. 47 (2014) 8012–8017. 10.1021/ma501882d. [DOI] [Google Scholar]
- [274].Von Maltzahn G, Vauthey S, Santoso S, Zhang S, Positively charged surfactant-like peptides self-assemble into nanostructures, Langmuir. 19 (2003) 4332–4337. 10.1021/la026526+. [DOI] [Google Scholar]
- [275].Liu Y, Zhang L, Wei W, Effect of noncovalent interaction on the self-assembly of a designed peptide and its potential use as a carrier for controlled bFGF release, Int. J. Nanomedicine 12 (2017) 659–670. 10.2147/IJN.S124523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Hyland LL, Taraban MB, Yu YB, Using small-angle scattering techniques to understand mechanical properties of biopolymer-based biomaterials, Soft Matter. 9 (2013) 10218–10228. 10.1039/c3sm51209f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Guilbaud JB, Saiani A, Using small angle scattering (SAS) to structurally characterise peptide and protein self-assembled materials, Chem. Soc. Rev 40 (2011) 1200–1210. 10.1039/c0cs00105h. [DOI] [PubMed] [Google Scholar]
- [278].Magnotti EL, Hughes SA, Dillard RS, Wang S, Hough L, Karumbamkandathil A, Lian T, Wall JS, Zuo X, Wright ER, Conticello VP, Self-Assembly of an α-Helical Peptide into a Crystalline Two-Dimensional Nanoporous Framework, J. Am. Chem. Soc 138 (2016) 16274–16282. 10.1021/jacs.6b06592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Sun JEP, Stewart B, Litan A, Lee SJ, Schneider JP, Langhans SA, Pochan DJ, Sustained release of active chemotherapeutics from injectable-solid β-hairpin peptide hydrogel, Biomater. Sci 4 (2016) 839–848. 10.1039/c5bm00538h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Franken LE, Grünewald K, Boekema EJ, Stuart MCA, A Technical Introduction to Transmission Electron Microscopy for Soft-Matter: Imaging, Possibilities, Choices, and Technical Developments, Small. 16 (2020). 10.1002/smll.201906198. [DOI] [PubMed] [Google Scholar]
- [281].Cui H, Hodgdon TK, Kaler EW, Abezgauz L, Danino D, Lubovsky M, Talmon Y, Pochan DJ, Elucidating the assembled structure of amphiphiles in solution via cryogenic transmission electron microscopy, Soft Matter. 3 (2007) 945–955. 10.1039/b704194b. [DOI] [PubMed] [Google Scholar]
- [282].Marrese M, Guarino V, Ambrosio L, Atomic Force Microscopy: A Powerful Tool to Address Scaffold Design in Tissue Engineering, J. Funct. Biomater 8 (2017) 7. 10.3390/jfb8010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Drira Z, Yadavalli VK, Nanomechanical measurements of polyethylene glycol hydrogels using atomic force microscopy, J. Mech. Behav. Biomed. Mater 18 (2013) 20–28. 10.1016/jjmbbm.2012.09.015. [DOI] [PubMed] [Google Scholar]
- [284].Ren P, Li J, Zhao L, Wang A, Wang M, Li J, Jian H, Li X, Yan X, Bai S, Dipeptide Self-assembled Hydrogels with Shear-Thinning and Instantaneous Self-healing Properties Determined by Peptide Sequences, ACS Appl. Mater. Interfaces 12 (2020) 21433–21440. 10.1021/acsami.0c03038. [DOI] [PubMed] [Google Scholar]
- [285].Kang MK, Colombo JS, D’Souza RN, Hartgerink JD, Sequence effects of self-assembling multidomain peptide hydrogels on encapsulated SHED cells, Biomacromolecules. 15 (2014) 2004–2011. 10.1021/bm500075r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Scelsi A, Bochicchio B, Smith A, Workman VL, Castillo Diaz LA, Saiani A, Pepe A, Tuning of hydrogel stiffness using a two-component peptide system for mammalian cell culture, J. Biomed. Mater. Res. - Part A 107 (2019) 535–544. 10.1002/jbm.a.36568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Sweers KKM, Bennink ML, Subramaniam V, Nanomechanical properties of single amyloid fibrils, J. Phys. Condens. Matter 24 (2012). 10.1088/0953-8984/24/24/243101. [DOI] [PubMed] [Google Scholar]
- [288].Yan C, Pochan DJ, Rheological properties of peptide-based hydrogels for biomedical and other applications, Chem. Soc. Rev 39 (2010) 3528–3540. 10.1039/b919449p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Sathaye S, Mbi A, Sonmez C, Chen Y, Blair DL, Schneider JP, Pochan DJ, Rheology of peptide- and protein-based physical hydrogels: Are everyday measurements just scratching the surface?, Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 7 (2015) 34–68. 10.1002/wnan.1299. [DOI] [PubMed] [Google Scholar]
- [290].Appel EA, Biedermann F, Rauwald U, Jones ST, Zayed JM, Scherman OA, Supramolecular cross-linked networks via host-guest complexation with cucurbit[8]uril, J. Am. Chem. Soc 132 (2010) 14251–14260. 10.1021/ja106362w. [DOI] [PubMed] [Google Scholar]
- [291].Lin F, Yu J, Tang W, Zheng J, Defante A, Guo K, Wesdemiotis C, Becker ML, Peptide-Functionalized Oxime Hydrogels with Tunable Mechanical Properties and Gelation Behavior, Biomacromolecules. 14 (2013) 3749–3758. 10.1021/BM401133R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].Ikeda S, Nishinari K, On solid-like rheological behaviors of globular protein solutions, Food Hydrocoll. 15 (2001) 401–406. 10.1016/S0268-005X(01)00052-2. [DOI] [Google Scholar]
- [293].Young RJ, Lovell PA, Introduction to Polymers, 3rd ed., CRC Press, 2011. https://www.routledge.com/Introduction-to-Polymers/Young-Lovell/p/book/9780849339295. [Google Scholar]
- [294].Yoshioka H, Mikami M, Mori Y, Tsuchida E, A Synthetic Hydrogel with Thermoreversible Gelation. I. Preparation and Rheological Properties, 10.1080/10601329409349722. 31 (2008) 113–120. 10.1080/10601329409349722. [DOI] [Google Scholar]
- [295].Guenet JM, Physical aspects of organogelation: A point of view, Gels. 7 (2021). 10.3390/gels7020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Sarkar B, Siddiqui Z, Nguyen PK, Dube N, Fu W, Park S, Jaisinghani S, Paul R, Kozuch SD, Deng D, Iglesias-Montoro P, Li M, Sabatino D, Perlin DS, Zhang W, Mondal J, Kumar VA, Membrane-Disrupting Nanofibrous Peptide Hydrogels, ACS Biomater. Sci. Eng 5 (2019) 4657–4670. 10.1021/acsbiomaterials.9b00967. [DOI] [PubMed] [Google Scholar]
- [297].Branco MC, Pochan DJ, Wagner NJ, Schneider JP, Macromolecular diffusion and release from self-assembled β-hairpin peptide hydrogels, Biomaterials 30 (2009) 1339–1347. 10.1016/j.biomaterials.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Wan S, Borland S, Richardson SM, Merry CLR, Saiani A, Gough JE, Self-assembling peptide hydrogel for intervertebral disc tissue engineering, Acta Biomater 46 (2016) 29–40. 10.1016/j.actbio.2016.09.033. [DOI] [PubMed] [Google Scholar]
- [299].Lindsey S, Piatt JH, Worthington P, Sönmez C, Satheye S, Schneider JP, Pochan DJ, Langhans SA, Beta hairpin peptide hydrogels as an injectable solid vehicle for neurotrophic growth factor delivery, Biomacromolecules 16 (2015) 2672–2683. 10.1021/acs.biomac.5b00541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Sinthuvanich C, Haines-Butterick LA, Nagy KJ, Schneider JP, Iterative design of peptide-based hydrogels and the effect of network electrostatics on primary chondrocyte behavior, Biomaterials 33 (2012) 7478–7488. 10.1016/j.biomaterials.2012.06.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Hogrebe NJ, Gooch KJ, Direct influence of culture dimensionality on human mesenchymal stem cell differentiation at various matrix stiffnesses using a fibrous self-assembling peptide hydrogel, J. Biomed. Mater. Res. Part A 104 (2016) 2356–2368. 10.1002/jbm.a.35755. [DOI] [PubMed] [Google Scholar]
- [302].Kim IL, Mauck RL, Burdick JA, Hydrogel design for cartilage tissue engineering: A case study with hyaluronic acid, Biomaterials. 32 (2011) 8771–8782. 10.1016/j.biomaterials.2011.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Luo Z, Zhao X, Zhang S, Self-organization of a chiral D-EAK16 designer peptide into a 3D nanofiber scaffold, Macromol. Biosci 8 (2008) 785–791. 10.1002/mabi.200800003. [DOI] [PubMed] [Google Scholar]
- [304].Chen X, Fan Z, Chen Y, Fang X, Sha X, Retro-inverso carbohydrate mimetic peptides with annexin1-binding selectivity, are stable in vivo, and target tumor vasculature, PLoS One. 8 (2013) 8–15. 10.1371/journal.pone.0080390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Li X, Du X, Li J, Gao Y, Pan Y, Shi J, Zhou N, Xu B, Introducing d -amino acid or simple glycoside into small peptides to enable supramolecular hydrogelators to resist proteolysis, Langmuir. 28 (2012) 13512–13517. 10.1021/la302583a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Giano MC, Pochan DJ, Schneider JP, Controlled biodegradation of Self-assembling p-hairpin Peptide hydrogels by proteolysis with matrix metalloproteinase-13, Biomaterials. 32 (2011) 6471–6477. 10.1016/j.biomaterials.2011.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Restuccia A, Seroski DT, Kelley KL, O’Bryan CS, Kurian JJ, Knox KR, Farhadi SA, Angelini TE, Hudalla GA, Hierarchical self-assembly and emergent function of densely glycosylated peptide nanofibers, Commun. Chem 2 (2019) 1–12. 10.1038/s42004-019-0154-z. [DOI] [Google Scholar]
- [308].Deming TJ, Preparation and development of block copolypeptide vesicles and hydrogels for biological and medical applications, Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 6 (2014) 283–297. 10.1002/wnan.1262. [DOI] [PubMed] [Google Scholar]
- [309].Zhang S, Alvarez DJ, Sofroniew MV, Deming TJ, Design and Synthesis of Nonionic Copolypeptide Hydrogels with Reversible Thermoresponsive and Tunable Physical Properties, Biomacromolecules. 16 (2015) 1331–1340. 10.1021/acs.biomac.5b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Wollenberg AL, O’Shea TM, Kim JH, Czechanski A, Reinholdt LG, Sofroniew MV, Deming TJ, Injectable polypeptide hydrogels via methionine modification for neural stem cell delivery, Biomaterials. 178 (2018) 527–545. 10.1016/j.biomaterials.2018.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Cai L, Liu S, Guo J, Jia YG, Polypeptide-based self-healing hydrogels: Design and biomedical applications, Acta Biomater. (2020). 10.1016/j.actbio.2020.07.001. [DOI] [PubMed] [Google Scholar]
- [312].Choe S, Bond CW, Harrington DA, Stupp SI, McVary KT, Podlasek CA, Peptide amphiphile nanofiber hydrogel delivery of sonic hedgehog protein to the cavernous nerve to promote regeneration and prevent erectile dysfunction, Nanomedicine Nanotechnology, Biol. Med 13 (2017) 95–101. 10.1016/j.nano.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Hosoyama K, Lazurko C, Muñoz M, McTiernan CD, Alarcon EI, Peptide-based functional biomaterials for soft-tissue repair, Front. Bioeng. Biotechnol 7 (2019) 205. 10.3389/fbioe.2019.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Kretsinger JK, Haines LA, Ozbas B, Pochan DJ, Schneider JP, Cytocompatibility of self-assembled β-hairpin peptide hydrogel surfaces, Biomaterials. 26 (2005) 5177–5186. 10.1016/j.biomaterials.2005.01.029. [DOI] [PubMed] [Google Scholar]
- [315].Rudra JS, Mishra S, Chong AS, Mitchell RA, Nardin EH, Nussenzweig V, Collier JH, Self-assembled peptide nanofibers raising durable antibody responses against a malaria epitope, Biomaterials. 33 (2012) 6476–6484. 10.1016/j.biomaterials.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Liu R, Hudalla GA, Using self-assembling peptides to integrate biomolecules into functional supramolecular biomaterials, Molecules. 24 (2019) 1450. 10.3390/molecules24081450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Lopez-Silva TL, Leach DG, Azares A, Li IC, Woodside DG, Hartgerink JD, Chemical functionality of multidomain peptide hydrogels governs early host immune response, Biomaterials. 231 (2020) 119667. 10.1016/j.biomaterials.2019.119667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Li Y, Rodrigues J, Tomás H, Injectable and biodegradable hydrogels: Gelation, biodegradation and biomedical applications, Chem. Soc. Rev 41 (2012) 2193–2221. 10.1039/c1cs15203c. [DOI] [PubMed] [Google Scholar]
- [319].Liu C, Zhang Q, Zhu S, Liu H, Chen J, Preparation and applications of peptide-based injectable hydrogels, RSC Adv. 9 (2019) 28299–28311. 10.1039/c9ra05934b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Raphael B, Khalil T, Workman VL, Smith A, Brown CP, Streuli C, Saiani A, Domingos M, 3D cell bioprinting of self-assembling peptide-based hydrogels, Mater. Lett 190 (2017) 103–106. 10.1016/j.matlet.2016.12.127. [DOI] [Google Scholar]
- [321].Loo Y, Lakshmanan A, Ni M, Toh LL, Wang S, Hauser CAE, Peptide Bioink: Self-Assembling Nanofibrous Scaffolds for Three-Dimensional Organotypic Cultures, Nano Lett. 15 (2015) 6919–6925. 10.1021/acs.nanolett.5b02859. [DOI] [PubMed] [Google Scholar]
- [322].Sheikholeslam M, Wheeler SD, Duke KG, Marsden M, Pritzker M, Chen P, Peptide and peptide-carbon nanotube hydrogels as scaffolds for tissue & 3D tumor engineering, Acta Biomater. 69 (2018) 107–119. 10.1016/j.actbio.2017.12.012. [DOI] [PubMed] [Google Scholar]
- [323].Haines-Butterick L, Salick DA, Pochan DJ, Schneider JP, In vitro assessment of the pro-inflammatory potential of β-hairpin peptide hydrogels, Biomaterials. 29 (2008) 4164–4169. 10.1016/j.biomaterials.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [324].Cai H, Wu F-Y, Wang Q-L, Xu P, Mou F-F, Shao S-J, Luo Z-R, Zhu J, Xuan S-S, Lu R, Guo H-D, Self-assembling peptide modified with QHREDGS as a novel delivery system for mesenchymal stem cell transplantation after myocardial infarction, FASEB J. 33 (2019) 8306–8320. 10.1096/fj.201801768RR. [DOI] [PubMed] [Google Scholar]
- [325].Liu J, Zhang L, Yang Z, Zhao X, Controlled release of paclitaxel from a self-assembling peptide hydrogel formed in situ and antitumor study in vitro., Int. J. Nanomedicine 6 (2011) 2143–2153. 10.2147/ijn.s24038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Paladini F, Meikle ST, Cooper IR, Lacey J, Perugini V, Santin M, Silver-doped self-assembling di-phenylalanine hydrogels as wound dressing biomaterials, J. Mater. Sci. Mater. Med 24 (2013) 2461–2472. 10.1007/s10856-013-4986-2. [DOI] [PubMed] [Google Scholar]
- [327].Chen C, Zhang Y, Hou Z, Cui X, Zhao Y, Xu H, Rational Design of Short Peptide-Based Hydrogels with MMP-2 Responsiveness for Controlled Anticancer Peptide Delivery, Biomacromolecules. 18 (2017) 3563–3571. 10.1021/acs.biomac.7b00911. [DOI] [PubMed] [Google Scholar]
- [328].Gelain F, Unsworth LD, Zhang S, Slow and sustained release of active cytokines from self-assembling peptide scaffolds, J. Control. Release 145 (2010) 231–239. 10.1016/j.jconrel.2010.04.026. [DOI] [PubMed] [Google Scholar]
- [329].Koutsopoulos S, Zhang S, Two-layered injectable self-assembling peptide scaffold hydrogels for long-term sustained release of human antibodies, J. Control. Release 160 (2012) 451–458. 10.1016/j.jconrel.2012.03.014. [DOI] [PubMed] [Google Scholar]
- [330].Sis MJ, Webber MJ, Drug Delivery with Designed Peptide Assemblies, Trends Pharmacol. Sci 40 (2019) 747–762. 10.1016/j.tips.2019.08.003. [DOI] [PubMed] [Google Scholar]
- [331].Ghysen A, Dambly-Chaudiere C, A genetic programme for neuronal connectivity, Trends Genet. 16 (2000) 221–226. 10.1016/S0168-9525(99)01969-1. [DOI] [PubMed] [Google Scholar]
- [332].Azevedo FAC, Carvalho LRB, Grinberg LT, Farfel JM, Ferretti REL, Leite REP, Filho WJ, Lent R, Herculano-Houzel S, Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain, J. Comp. Neurol 513 (2009) 532–541. 10.1002/cne.21974. [DOI] [PubMed] [Google Scholar]
- [333].Cheng S, Clarke EC, Bilston LE, Rheological properties of the tissues of the central nervous system: A review, Med. Eng. Phys 30 (2008) 1318–1337. 10.1016/j.medengphy.2008.06.003. [DOI] [PubMed] [Google Scholar]
- [334].Medberry CJ, Crapo PM, Siu BF, Carruthers CA, Wolf MT, Nagarkar SP, Agrawal V, Jones KE, Kelly J, Johnson SA, Velankar SS, Watkins SC, Modo M, Badylak SF, Hydrogels derived from central nervous system extracellular matrix, Biomaterials. 34 (2013) 1033–1040. 10.1016/j.biomaterials.2012.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [335].Verschure PFMJ, Pennartz CMA, Pezzulo G, The why, what, where, when and how of goal-directed choice: Neuronal and computational principles, Philos. Trans. R. Soc. B Biol. Sci 369 (2014). 10.1098/rstb.2013.0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [336].Henn FA, Hamberger A, Glial cell function: uptake of transmitter substances., Proc. Natl. Acad. Sci. U. S. A 68 (1971) 2686–2690. 10.1073/pnas.68.11.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Simons M, Trajkovic K, Neuron-glia communication in the control of oligodencrocyte function and myelin biogenesis, J. Cell Sci 119 (2006) 4381–4389. 10.1242/jcs.03242. [DOI] [PubMed] [Google Scholar]
- [338].Fontenas L, Welsh TG, Piller M, Coughenour P, Gandhi AV, Prober DA, Kucenas S, The Neuromodulator Adenosine Regulates Oligodendrocyte Migration at Motor Exit Point Transition Zones, Cell Rep. 27 (2019) 115–128.e5. 10.1016/j.celrep.2019.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [339].Koss KM, Unsworth LD, Neural tissue engineering: Bioresponsive nanoscaffolds using engineered self-assembling peptides, Acta Biomater. 44 (2016) 2–15. 10.1016/j.actbio.2016.08.026. [DOI] [PubMed] [Google Scholar]
- [340].Hong A, Aguilar MI, Del Borgo MP, Sobey CG, Broughton BRS, Forsythe JS, Self-assembling injectable peptide hydrogels for emerging treatment of ischemic stroke, J. Mater. Chem. B 7 (2019) 3927–3943. 10.1039/c9tb00257j. [DOI] [Google Scholar]
- [341].Tysseling VM, Sahni V, Pashuck ET, Birch D, Hebert A, Czeisler C, Stupp SI, Kessler JA, Self-assembling peptide amphiphile promotes plasticity of serotonergic fibers following spinal cord injury, J. Neurosci. Res 88 (2010) 3161–3170. 10.1002/jnr.22472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [342].Pan L, North HA, Sahni V, Jeong SJ, Mcguire TL, Berns EJ, Stupp SI, Kessler JA, β1-Integrin and Integrin Linked Kinase Regulate Astrocytic Differentiation of Neural Stem Cells, PLoS One. 9 (2014) e104335. 10.1371/journal.pone.0104335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [343].Sun Y, Li W, Wu X, Zhang N, Zhang Y, Ouyang S, Song X, Fang X, Seeram R, Xue W, He L, Wu W, Functional Self-Assembling Peptide Nanofiber Hydrogels Designed for Nerve Degeneration, ACS Appl. Mater. Interfaces 8 (2016) 2348–2359. 10.1021/acsami.5b11473. [DOI] [PubMed] [Google Scholar]
- [344].Francis NL, Zhao N, Calvelli HR, Saini A, Gifford JJ, Wagner GC, Cohen RI, Pang ZP, Moghe PV, Peptide-Based Scaffolds for the Culture and Transplantation of Human Dopaminergic Neurons, Tissue Eng. - Part A 26 (2020) 193–205. 10.1089/ten.tea.2019.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [345].Guo J, Su H, Zeng Y, Liang YX, Wong WM, Ellis-Behnke RG, So KF, Wu W, Reknitting the injured spinal cord by self-assembling peptide nanofiber scaffold, Nanomedicine Nanotechnology, Biol. Med 3 (2007) 311–321. 10.1016/j.nano.2007.09.003. [DOI] [PubMed] [Google Scholar]
- [346].Guo J, Leung KKG, Su H, Yuan Q, Wang L, Chu TH, Zhang W, Pu JKS, Ng GKP, Wong WM, Dai X, Wu W, Self-assembling peptide nanofiber scaffold promotes the reconstruction of acutely injured brain, Nanomedicine Nanotechnology, Biol. Med 5 (2009) 345–351. 10.1016/j.nano.2008.12.001. [DOI] [PubMed] [Google Scholar]
- [347].Yoshimatsu M, Nakamura R, Kishimoto Y, Yurie H, Hayashi Y, Kaba S, Ohnishi H, Yamashita M, Tateya I, Omori K, Recurrent laryngeal nerve regeneration using a self-assembling peptide hydrogel, Laryngoscope. (2019) lary.28434. 10.1002/lary.28434. [DOI] [PubMed] [Google Scholar]
- [348].Farrukh A, Ortega F, Fan W, Marichal N, Paez JI, Berninger B, del Campo A, Salierno MJ, Bifunctional Hydrogels Containing the Laminin Motif IKVAV Promote Neurogenesis, Stem Cell Reports. 9 (2017) 1432–1440. 10.1016/j.stemcr.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [349].Ji W, Álvarez Z, Edelbrock AN, Sato K, Stupp SI, Bioactive Nanofibers Induce Neural Transdifferentiation of Human Bone Marrow Mesenchymal Stem Cells, ACS Appl. Mater. Interfaces 10 (2018) 41046–41055. 10.1021/acsami.8b13653. [DOI] [PubMed] [Google Scholar]
- [350].Tysseling-Mattiace VM, Sahni V, Niece KL, Birch D, Czeisler C, Fehlings MG, Stupp SI, Kessler JA, Self-assembling nanofibers inhibit glial scar formation and promote axon elongation after spinal cord injury, J. Neurosci 28 (2008) 3814–3823. 10.1523/JNEUROSCI.0143-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [351].Godbe JM, Freeman R, Burbulla LF, Lewis J, Krainc D, Stupp SI, Gelator Length Precisely Tunes Supramolecular Hydrogel Stiffness and Neuronal Phenotype in 3D Culture, ACS Biomater. Sci. Eng 6 (2020) 1196–1207. 10.1021/acsbiomaterials.9b01585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [352].Clarke M, Muscle sliding filaments, Nat. Rev. Mol. Cell Biol 9 (2010) s7–s7. 10.1038/nrm2581. [DOI] [Google Scholar]
- [353].Gillies AR, Lieber RL, Structure and function of the skeletal muscle extracellular matrix, Muscle and Nerve. 44 (2011) 318–331. 10.1002/mus.22094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [354].Mukund K, Subramaniam S, Skeletal muscle: A review of molecular structure and function, in health and disease, Wiley Interdiscip. Rev. Syst. Biol. Med 12 (2020). 10.1002/wsbm.1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [355].Mathur AB, Collinsworth AM, Reichert WM, Kraus WE, Truskey GA, Endothelial, cardiac muscle and skeletal muscle exhibit different viscous and elastic properties as determined by atomic force microscopy, J. Biomech 34 (2001) 1545–1553. 10.1016/S0021-9290(01)00149-X. [DOI] [PubMed] [Google Scholar]
- [356].Morrow DA, Haut Donahue TL, Odegard GM, Kaufman KR, Transversely isotropic tensile material properties of skeletal muscle tissue, J. Mech. Behav. Biomed. Mater 3 (2010) 124–129. 10.1016/j.jmbbm.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [357].Breuner CW, Berk SA, Physiological Ecology of Rocky Intertidal Organisms: A Synergy of Concepts, Integr. Comp. Biol 59 (2019) 243–250. 10.1093/ICB. [DOI] [PubMed] [Google Scholar]
- [358].Van der Kloot W, Cohen IS, End-plate potentials in a model muscle fiber. Corrections for the effects of membrane potential on currents and on channel lifetimes, Biophys. J 45 (1984) 905–911. 10.1016/S0006-3495(84)84237-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [359].Purves D, Augustine GJ, Fitzpatrick D, Katz LC, LaMantia A-S, McNamara JO, Williams SM, The Motor Unit, (2001). [Google Scholar]
- [360].Giritharan S, Salhiyyah K, Tsang GM, Ohri SK, Feasibility of a novel, synthetic, self-assembling peptide for suture-line haemostasis in cardiac surgery, J. Cardiothorac. Surg 13 (2018) 68. 10.1186/s13019-018-0745-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [361].Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, Genesis, Modulation, and Regeneration of Skeletal Muscle, (2002). https://www.ncbi.nlm.nih.gov/books/NBK26853/ (accessed August 2, 2020).
- [362].Louch WE, Sheehan KA, Wolska BM, Methods in cardiomyocyte isolation, culture, and gene transfer, J. Mol. Cell. Cardiol 51 (2011) 288–298. 10.1016/j.yjmcc.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [363].Hosoyama T, Van Dyke J, Suzuki M, Applications of skeletal muscle progenitor cells for neuromuscular diseases, Am. J. Stem Cells 1 (2012) 253–263. [PMC free article] [PubMed] [Google Scholar]
- [364].Carmeliet E, Conduction in cardiac tissue. Historical reflections, Physiol. Rep 7 (2019). 10.14814/phy2.13860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [365].Tokunaga M, Liu ML, Nagai T, Iwanaga K, Matsuura K, Takahashi T, Kanda M, Kondo N, Wang P, Naito AT, Komuro I, Implantation of cardiac progenitor cells using self-assembling peptide improves cardiac function after myocardial infarction, J. Mol. Cell. Cardiol 49 (2010) 972–983. 10.1016/j.yjmcc.2010.09.015. [DOI] [PubMed] [Google Scholar]
- [366].Narmoneva DA, Oni O, Sieminski AL, Zhang S, Gertler JP, Kamm RD, Lee RT, Self-assembling short oligopeptides and the promotion of angiogenesis, Biomaterials. 26 (2005) 4837–4846. 10.1016/j.biomaterials.2005.01.005. [DOI] [PubMed] [Google Scholar]
- [367].Wang W, Liu G, Liu M, Li X, Mechanisms underlying the action of self-assembling short-peptide nano-fiber gel scaffold materials in the aesthetic repair of burn wounds, Mater. Express 10 (2020) 454–459. 10.1166/mex.2020.1640. [DOI] [Google Scholar]
- [368].Ichihara Y, Kaneko M, Yamahara K, Koulouroudias M, Sato N, Uppal R, Yamazaki K, Saito S, Suzuki K, Self-assembling peptide hydrogel enables instant epicardial coating of the heart with mesenchymal stromal cells for the treatment of heart failure, Biomaterials. 154 (2018) 12–23. 10.1016/j.biomaterials.2017.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [369].Ceylan H, Tekinay AB, Guler MO, Selective adhesion and growth of vascular endothelial cells on bioactive peptide nanofiber functionalized stainless steel surface, Biomaterials. 32 (2011) 8797–8805. 10.1016/J.BIOMATERIALS.2011.08.018. [DOI] [PubMed] [Google Scholar]
- [370].Clarke B, Normal bone anatomy and physiology.,. Clin. J. Am. Soc. Nephrol 3 Suppl 3 (2008) S131. 10.2215/CJN.04151206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [371].Hart NH, Nimphius S, Rantalainen T, Ireland A, Siafarikas A, Newton RU, Mechanical basis of bone strength: Influence of bone material, bone structure and muscle action, J. Musculoskelet. Neuronal Interact 17 (2017) 114–139. [PMC free article] [PubMed] [Google Scholar]
- [372].Morgan EF, Unnikrisnan GU, Hussein AI, Bone Mechanical Properties in Healthy and Diseased States, Annu. Rev. Biomed. Eng 20 (2018) 119–143. 10.1146/annurev-bioeng-062117-121139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [373].Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee HP, Lippens E, Duda GN, Mooney DJ, Hydrogels with tunable stress relaxation regulate stem cell fate and activity, Nat. Mater 15 (2016) 326–334. 10.1038/nmat4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [374].Mohamed AMFS, An overview of bone cells and their regulating factors of differentiation, Malaysian J. Med. Sci 15 (2008) 4–12. [PMC free article] [PubMed] [Google Scholar]
- [375].Bonewald LF, The Role of the Osteocyte in Bone and Nonbone Disease, Endocrinol. Metab. Clin. North Am 46 (2017) 1–18. 10.1016/j.ecl.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [376].Guo Y, qiu Zhang C, cheng Zeng Q, xin Li R, Liu L, xin Hao Q, hong Shi C, zheng Zhang X, xian Yan Y, Mechanical strain promotes osteoblast ECM formation and improves its osteoinductive potential, Biomed. Eng. Online 11 (2012) 80. 10.1186/1475-925X-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [377].Leiva O, Leon C, Kah Ng S, Mangin P, Gachet C, Ravid K, The role of extracellular matrix stiffness in megakaryocyte and platelet development and function, Am. J. Hematol 93 (2018) 430–441. 10.1002/ajh.25008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [378].Green H, Ochbaum G, Gitelman-Povimonsky A, Bitton R, Rapaport H, RGD-presenting peptides in amphiphilic and anionic β-sheet hydrogels for improved interactions with cells, RSC Adv. 8 (2018) 10072–10080. 10.1039/c7ra12503h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [379].Mata A, Geng Y, Henrikson KJ, Aparicio C, Stock SR, Satcher RL, Stupp SI, Bone regeneration mediated by biomimetic mineralization of a nanofiber matrix, Biomaterials. 31 (2010) 6004–6012. 10.1016/j.biomaterials.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [380].Bao X, Liu Y, Han G, Zuo Z, Hu M, The effect on proliferation and differentiation of cementoblast by using sclerostin as inhibitor, Int. J. Mol. Sci 14 (2013) 21140–21152. 10.3390/ijms141021140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [381].Welk A, Ratzmann A, Reich M, Krey KF, Schwahn C, Effect of self-assembling peptide P11-4 on orthodontic treatment-induced carious lesions, Sci. Reports 2020 101 10 (2020) 1–10. 10.1038/s41598-020-63633-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [382].Philip N, State of the Art Enamel Remineralization Systems: The Next Frontier in Caries Management, Caries Res. 53 (2019) 284–295. 10.1159/000493031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [383].Chaplin DD, Overview of the immune response, J. Allergy Clin. Immunol 125 (2010) S3. 10.1016/j.jaci.2009.12.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [384].Nicholson LB, The immune system, Essays Biochem. 60 (2016) 275–301. 10.1042/EBC20160017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [385].Marshall JS, Warrington R, Watson W, Kim HL, An introduction to immunology and immunopathology, Allergy, Asthma Clin. Immunol 14 (2018) 49. 10.1186/s13223-018-0278-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [386].Hudalla GA, Modica JA, Tian YF, Rudra JS, Chong AS, Sun T, Mrksich M, Collier JH, A Self-Adjuvanting Supramolecular Vaccine Carrying a Folded Protein Antigen, Adv. Healthc. Mater 2 (2013) 1114–1119. 10.1002/adhm.201200435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [387].Snook JD, Chesson CB, Peniche AG, Dann SM, Paulucci A, Pinchuk IV, Rudra JS, Peptide nanofiber-CaCO3 composite microparticles as adjuvant-free oral vaccine delivery vehicles, J. Mater. Chem. B 4 (2016) 1640–1649. 10.1039/c5tb01623a. [DOI] [PubMed] [Google Scholar]
- [388].Wen Y, Waltman A, Han H, Collier JH, Switching the Immunogenicity of Peptide Assemblies Using Surface Properties, ACS Nano. 10 (2016) 9274–9286. 10.1021/acsnano.6b03409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [389].Radvar E, Azevedo HS, Supramolecular Peptide / Polymer Hybrid Hydrogels for Biomedical Applications, Macromol. Biosci 1800221 (2019) 1–16. 10.1002/mabi.201800221. [DOI] [PubMed] [Google Scholar]
- [390].Shu JY, Panganiban B, Xu T, Peptide-Polymer Conjugates: From Fundamental Science to Application, Annu. Rev. Phys. Chem 64 (2013) 631–657. 10.1146/annurev-physchem-040412-110108. [DOI] [PubMed] [Google Scholar]
- [391].Hamley IW, Castelletto V, Self-Assembly of Peptide Bioconjugates: Selected Recent Research Highlights, Bioconjug. Chem 28 (2017) 731–739. 10.1021/acs.bioconjchem.6b00284. [DOI] [PubMed] [Google Scholar]
- [392].Naik SS, Ray JG, Savin DA, Temperature- and pH-responsive self-assembly of poly(propylene oxide)-b-poly(lysine) block copolymers in aqueous solution, Langmuir. 27 (2011) 7231–7240. 10.1021/la200882f. [DOI] [PubMed] [Google Scholar]
- [393].Gebhardt KE, Ahn S, Venkatachalam G, Savin DA, Role of secondary structure changes on the morphology of polypeptide-based block copolymer vesicles, J. Colloid Interface Sci 317 (2008) 70–76. 10.1016/j.jcis.2007.09.048. [DOI] [PubMed] [Google Scholar]
- [394].Gauthier MA, Klok HA, Peptide/protein-polymer conjugates: Synthetic strategies and design concepts, Chem. Commun (2008) 2591–2611. 10.1039/b719689j. [DOI] [PubMed] [Google Scholar]
- [395].Hamley IW, PEG-peptide conjugates, Biomacromolecules. 15 (2014) 1543–1559. 10.1021/bm500246w. [DOI] [PubMed] [Google Scholar]
- [396].Deacon SPE, Apostolovic B, Carbajo RJ, Schott AK, Beck K, Vicent MJ, Pineda-Lucena A, Klok HA, Duncan R, Polymer coiled-coil conjugates: Potential for development as a new class of therapeutic “molecular Switch,” Biomacromolecules. 12 (2011) 19–27. 10.1021/bm100843e. [DOI] [PubMed] [Google Scholar]
- [397].Sohdi AA, Campbell D, Topham PD, Polymer-peptide conjugate hydrogels; towards controlled drug delivery, Chiang Mai J. Sci 39 (2012) 351–372. [Google Scholar]
- [398].Mei Y, Beers KL, Byrd HCM, VanderHart DL, Washburn NR, Solid-Phase ATRP Synthesis of Peptide-Polymer Hybrids, J. Am. Chem. Soc 126 (2004) 3472–3476. 10.1021/ja039583d. [DOI] [PubMed] [Google Scholar]
- [399].Fisher SA, Baker AEG, Shoichet MS, Designing Peptide and Protein Modified Hydrogels: Selecting the Optimal Conjugation Strategy, J. Am. Chem. Soc 139 (2017) 7416–7427. 10.1021/jacs.7b00513. [DOI] [PubMed] [Google Scholar]
- [400].Yang J, Xu C, Kopečková P, Kopeček J, Hybrid hydrogels self-assembled from HPMA copolymers containing peptide grafts, Macromol. Biosci 6 (2006) 201–209. 10.1002/mabi.200500208. [DOI] [PubMed] [Google Scholar]
- [401].Ding L, Jiang Y, Zhang J, Klok HA, Zhong Z, PH-Sensitive Coiled-Coil Peptide-Cross-Linked Hyaluronic Acid Nanogels: Synthesis and Targeted Intracellular Protein Delivery to CD44 Positive Cancer Cells, Biomacromolecules. 19 (2018) 555–562. 10.1021/acs.biomac.7b01664. [DOI] [PubMed] [Google Scholar]
- [402].Stahl PJ, Romano NH, Wirtz D, Yu SM, PEG-based hydrogels with collagen mimetic peptide-mediated and tunable physical cross-links, Biomacromolecules. 11 (2010) 2336–2344. 10.1021/bm100465q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [403].Elder AN, Dangelo NM, Kim SC, Washburn NR, Conjugation of β-sheet peptides to modify the rheological properties of hyaluronic acid, Biomacromolecules. 12 (2011) 2610–2616. 10.1021/bm200393k. [DOI] [PubMed] [Google Scholar]
- [404].Vandermeulen GWM, Tziatzios C, Duncan R, Klok HA, PEG-based hybrid block copolymers containing α-helical coiled coil peptide sequences: Control of self-assembly and preliminary biological evaluation, Macromolecules. 38 (2005) 761–769. 10.1021/ma0485538. [DOI] [Google Scholar]
- [405].Ang J, Jung BT, Keten S, Xu T, Sub-20 nm Stable Micelles Based on a Mixture of Coiled-Coils: A Platform for Controlled Ligand Presentation, Biomacromolecules. 18 (2017) 3572–3580. 10.1021/acs.biomac.7b00917. [DOI] [PubMed] [Google Scholar]
- [406].Collier JH, Messersmith PB, Self-assembling polymer-peptide conjugates: Nanostructural tailoring, Adv. Mater 16 (2004) 907–910. 10.1002/adma.200306379. [DOI] [Google Scholar]
- [407].Luo T, He L, Theato P, Kiick KL, Thermoresponsive self-assembly of nanostructures from a collagen-like peptide-containing diblock copolymera, Macromol. Biosci 15 (2015) 111–123. 10.1002/mabi.201400358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [408].Dong H, Shu JY, Dube N, Ma Y, Tirrell MV, Downing KH, Xu T, 3-Helix micelles stabilized by polymer springs, J. Am. Chem. Soc 134 (2012) 11807–11814. 10.1021/ja3048128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [409].Shu JY, Tan C, DeGrado WF, Xu T, New design of helix bundle peptide-polymer conjugates, Biomacromolecules. 9 (2008) 2111–2117. 10.1021/bm800113g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [410].Sahin E, Kiick KL, Macromolecule-induced assembly of coiled-coils in alternating multiblock polymers, Biomacromolecules. 10 (2009) 2740–2749. 10.1021/bm900474k. [DOI] [PubMed] [Google Scholar]
- [411].Apostolovic B, Deacon SPE, Duncan R, Klok HA, Hybrid polymer therapeutics incorporating bioresponsive, coiled coil peptide linkers, Biomacromolecules. 11 (2010) 1187–1195. 10.1021/bm901313c. [DOI] [PubMed] [Google Scholar]
- [412].Apostolovic B, Klok HA, Copolymerization behavior of N -(2-Hydroxypropyl)methacrylamide and a methacrylated coiled-coil peptide derivative, Biomacromolecules. 11 (2010) 1891–1895. 10.1021/bm100533g. [DOI] [PubMed] [Google Scholar]
- [413].Apostolovic B, Deacon SPE, Duncan R, Klok HA, Cell uptake and trafficking behavior of non-covalent, coiled-coil based polymer-drug conjugates, Macromol. Rapid Commun 32 (2011) 11–18. 10.1002/marc.201000434. [DOI] [PubMed] [Google Scholar]
- [414].Jiang X, Halmes AJ, Licari G, Smith JW, Song Y, Moore EG, Chen Q, Tajkhorshid E, Rienstra CM, Moore JS, Multivalent Polymer-Peptide Conjugates: A General Platform for Inhibiting Amyloid Beta Peptide Aggregation, ACS Macro Lett. 8 (2019) 1365–1371. 10.1021/acsmacrolett.9b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [415].Li J, Xing R, Bai S, Yan X, Recent advances of self-assembling peptide-based hydrogels for biomedical applications, Soft Matter. 15 (2019) 1704–1715. 10.1039/C8SM02573H. [DOI] [PubMed] [Google Scholar]
- [416].Chen J, Zou X, Self-assemble peptide biomaterials and their biomedical applications, Bioact. Mater 4 (2019) 120–131. 10.1016/j.bioactmat.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [417].Seroski DT, Dong X, Wong KM, Liu R, Shao Q, Paravastu AK, Hall CK, Hudalla GA, Charge guides pathway selection in β-sheet fibrillizing peptide co-assembly, Commun. Chem 3 (2020). 10.1038/s42004-020-00414-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [418].Gough JE, Saiani A, Miller AF, Peptide hydrogels: mimicking the extracellular matrix, Bioinspired, Biomim. Nanobiomaterials 1 (2012) 4–12. 10.1680/bbn.11.00007. [DOI] [Google Scholar]