

Abstract
Recombinant adeno-associated virus (rAAV) vectors are increasingly being used for clinical gene transfer and have shown great potential for the treatment of several monogenic disorders. However, contaminant DNA from producer plasmids can be packaged into rAAV alongside the intended expression cassette-containing vector genome. The consequences of this are unknown. Our analysis of rAAV preps revealed abundant contaminant sequences upstream of the AAV replication (Rep) protein driving promoter, P5, on the Rep-Cap producer plasmid. Characterization of P5-associated contaminants after infection showed transfer, persistence, and transcriptional activity in AAV-transduced murine hepatocytes, in addition to in vitro evidence suggestive of integration. These contaminants can also be efficiently translated and immunogenic, revealing previously unrecognized side effects of rAAV-mediated gene transfer. P5-associated contaminant packaging and activity were independent of an inverted terminal repeat (ITR)-flanked vector genome. To prevent incorporation of these potentially harmful sequences, we constructed a modified P5-promoter (P5-HS), inserting a DNA spacer between an Rep binding site and an Rep nicking site in P5. This prevented upstream DNA contamination regardless of transgene or AAV serotype, while maintaining vector yield. Thus, we have constructed an rAAV production plasmid that improves vector purity and can be implemented across clinical rAAV applications. These findings represent new vector safety and production considerations for rAAV gene therapy.
Keywords: Adeno-associated virus, gene therapy, rAAV, P5, vectorology, contamination, viral manufacturing
Graphical abstract
Brimble and colleagues describe a previously unknown safety consideration for clinical rAAV-mediated gene therapy that exists regardless of AAV serotype, vector cassette, or target disease. They also present an easily implementable vector design strategy to mitigate this concern.
Introduction
The relative ease and versatility of recombinant adeno-associated virus (rAAV) production, coupled with its robust transduction of non-dividing cells, has made AAV a popular gene delivery tool. Clinical successes in the treatment of hemophilia and other monogenic disorders have helped to increase interest in AAV-mediated gene transfer as a treatment strategy.1, 2, 3, 4, 5, 6 However, concerns about short- and long-term toxicities exist even as AAV-based therapies advance clinically. Dose-dependent liver toxicity has been observed sporadically after systemic rAAV administration.2,4,7,8 While in nearly all cases these symptoms resolve, albeit with therapeutic efficacy loss if untreated, three X-linked myotubular myopathy patients with pre-existing liver pathology experienced severe liver failure and died after receiving 3 × 1014 vg/kg rAAV,9 with an additional patient death in this trial after a 1.3 × 1014 vg/kg infusion.10 Thus, although rAAV therapies are providing significant clinical benefit, improving their safety remains a top priority.
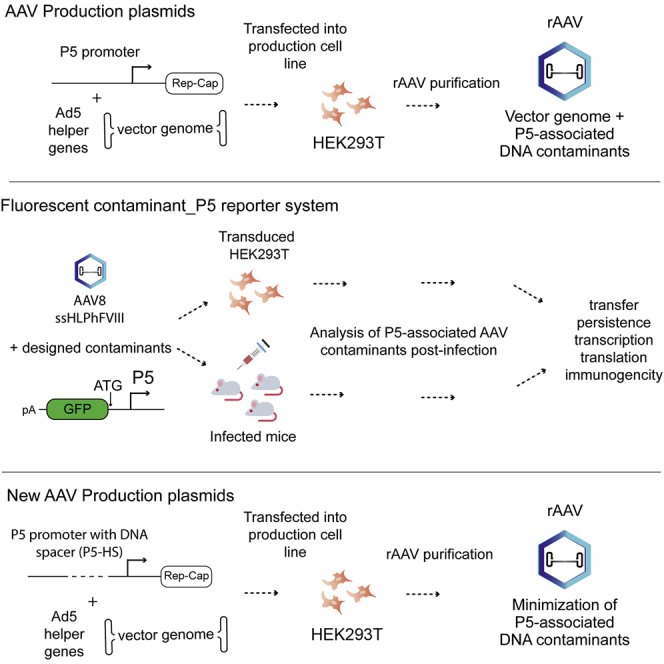
In addition to vector genomes, rAAV particles contain non-vector contaminating DNA,11, 12, 13, 14, 15, 16, 17 and the impact of this on vector safety is unknown. Prior research into rAAV contaminants has focused on design interventions, such as increasing production plasmid DNA size or the use of DNA minicircles in the production process to reduce the level of contaminants.12,14,16,17 However, there has been comparatively little investigation into the effects of DNA impurities on transduced cells and tissues. This work investigated the origin and consequences of contaminating DNA derived from rAAV production plasmids and vector design strategies for their reduction, focusing specifically on contaminants associated with the commonly used AAV P5 promoter.
Results
DNA contaminants associated with the P5 promoter are abundantly packaged into rAAV
The wild-type AAV genome comprises four overlapping same-frame replication (Rep) genes and three overlapping same-frame capsid (Cap) genes (Figure 1A). Additional alternate reading frame proteins membrane-associated accessory protein (MAAP) and AAV assembly protein (AAP) exist embedded within the capsid gene region.18,19 Expression of these proteins is driven by three promoters: P5, P19, and P40. P5 drives the large Rep proteins Rep78 and Rep68, P19 drives the short Rep proteins Rep52 and Rep40, and P40 drives expression of the capsid genes VP1, VP2 and VP3. This 4.7kb genome is flanked by inverted terminal repeats (ITRs). In rAAV production, the ITRs flank an expression cassette of interest that is packaged into the rAAV virion. The replication and capsid genes are, in most cases, delivered on a separate plasmid construct. The AAV2 P5 promoter is used in most clinical human embryonic kidney 293T (HEK293T)-based rAAV production systems. As in wild-type AAV, P5 transcribes Rep78 and Rep68 in rAAV production. P5 can achieve this positioned either in its wild-type location relative to Rep or directly downstream of the capsid gene, with promoter activity reading through the plasmid backbone (Figure 1A). qPCR analysis of rAAV preparations demonstrated the presence of contaminant sequences from upstream of P5 on the Rep-Cap production plasmid packaged within rAAV virions (Figure 1B). Sequences closer to P5 were more abundant than more remote sequences. This was also true for contaminant sequences originating from the vector-cassette plasmid, outside of the ITRs (Figure 1C), consistent with prior reports.14,20 Next-generation sequencing (NGS) of rAAV preps revealed enrichment of sequences upstream of P5 regardless of the relative positioning of P5 in the production plasmid (Figure 1D). Quantitative assessment of NGS data from a good manufacturing practice (GMP) produced and purified AAV8 factor VIII (FVIII) vector prep revealed 87% of reads mapped to the therapeutic rAAV expression cassette, while 13% of reads mapped to other producer-plasmid sequences, of which 4% overall mapped to the P5-containing Rep-Cap plasmid (Figure 1E).
Figure 1.

Detection of transcriptionally active contaminant sequences associated with the P5-promoter packaged into AAV
(A) Schematic depicting WT (top) and recombinant (bottom) AAV genome organizations. ITR, inverted terminal repeat; RBS, Rep-binding-site; Rep, replication genes; Cap, capsid genes. Arrows denote transcriptional start sites. (B) qPCR detection of contaminant producer plasmid amplicons at increasing distance upstream of the P5 promoter Rep binding site in AAV8ssHLPhFVIII (red) and AAV8scLP1hFIXcoPadua (blue) preparations. Error bars indicate SEM. (C) qPCR detection of contaminant producer plasmid amplicons at increasing distance outside of a WT ITR in an AAV8scLP1hFIX co-preparation. Error bars indicate SEM. (D) Circos plot mapping the location of Rep-Cap plasmid reads from NGS of AAV8ssHLPhFVIII in which the P5 promoter of the Rep-Cap plasmid was positioned either directly upstream of the Rep gene (left) or moved downstream of the capsid gene (right). (E) Pie chart of NGS read contributions of producer plasmid sequences to a clinical (GMP) grade AAV8ssHLPhFVIII preparation. Left chart: light blue, reads corresponding to the ssHLPhFVIII cassette; orange, vector genome plasmid contaminants; dark blue + expanded chart, Rep-Cap plasmid contaminants (gray, AAV Rep gene contaminants; yellow, rest of Rep-Cap plasmid contaminants).
P5-associated AAV contaminants persist in infected cells and are transcriptionally active
To determine the presence, persistence, and activity of P5-associated contaminants after rAAV-mediated transduction, male C57BL/6J mice were injected with a high dose (8.4 × 1011 vg) of AAV8_ssHLP-hFVIII via tail vein. Livers were analyzed by fluorescence in situ hybridization (FISH) at 1 week and 4 months after vector administration. RNA FISH determined that contaminant transcript from the P5-containing plasmid was detectable within hepatocyte nuclei at both time points (Figure 2A). Slides were then treated with RNase, which abolished the signal. Subsequent DNA FISH probing resulted in detection of contaminant sequences within hepatocyte nuclei. Uninfected C57BL/6J mouse hepatocytes did not exhibit detectable signal at any stage of the process, confirming probe specificity. NGS from mouse livers 4 months after infection showed that of DNA sequence reads mapping to the production plasmids, 90.1% ± 0.6 corresponded to the vector cassette and 9.94% corresponded to contaminant DNA sequences; RNA from these sources were 96.6% ± 0.5 and 3.4%, respectively (Figure 2B).
Figure 2.

P5-associated AAV contaminants are transcriptionally active after infection
(A) Sequential RNA-DNA nuclear FISH assay of mouse hepatocytes 1 week after 8.4 × 1011vg AAV8ssHLPhFVIII injection (top) or uninfected C57/BL6J (bottom). Red indicates probe detecting contaminant Rep-Cap plasmid sequences. Blue indicates DAPI staining. White arrows identify contaminant sequence hybridization. RNA FISH (left), RNA FISH after RNase treatment (middle), DNA FISH (right). (B) Pie chart of NGS read contributions mapping to produce plasmid contaminant 4 months after 8.4 × 1011vg injection with AAV8ssHLPhFVIII from mouse liver DNA (top) and mouse liver RNA (bottom). Reads mapped as in (Figure 1D).
P5-associated contaminants in rAAV can be translated and immunogenic
Previous studies of wild-type AAV have revealed bidirectional P5 promoter activity, driving expression of small upstream RNAs.21,22 Transfection of a plasmid with GFP-encoding DNA positioned directly upstream of P5 resulted in GFP expression, notably greater than expression generated when the GFP-encoding DNA was placed upstream of ITR sequences (Figure 3A). To determine whether transcriptional activity from P5 was driving RNA and protein expression of its associated rAAV contaminants, we constructed a fluorescence-based system where reporter sequences were positioned upstream of P5 and thus packaged as contaminants in rAAV (Figure S1). AAV8 preparations packaging an FVIII expression cassette using REP-CAP plasmids containing an upstream GFP cassette (AAV8GFP_P5F8), an upstream GFP cassette in reverse orientation (AAV8RevGFP_P5F8), or no upstream cassette (AAV8Empty_P5F8) were used to produce virus. NGS of purified preps confirmed packaging of the upstream sequences as rAAV contaminants (Figure S2). HEK293T cells were then transduced with a dose range of rAAV. After 72 h, GFP-positive cells were observed in HEK293T cells transduced with AAV8GFP_P5F8, but not AAV8RevGFP_P5F8 or AAV8Empty_P5F8 (Figure 3B). A time course of infection showed a rise in detectable GFP expression from 24 h to 3 days (Figure 3C), suggesting expression of the protein from DNA and not artifactual direct protein transfer by pseudotransduction.23 Expression was mostly gone by 7 days in the rapidly dividing HEK293T cells. Interestingly, however, passaged cells retained small but stable GFP-positive populations, implying contaminant integration (Figure S3).
Figure 3.

P5-associated rAAV contaminants can be translated and immunogenic after infection
(A) Bar graph depicting transcriptional activity of forward and reverse direction of AAV2 P5 promoter and outside of AAV2ITRs after plasmid transfection of 293T cells. GFP cassette positioned relative to P5: upstream (blue), downstream (orange), upstream but reverse orientation (green), no GFP cassette (red). GFP upstream of AAV2 ITR: wild-type (gray), self-complementary (purple). Error bars indicate SEM. (B) Bar chart showing quantification of GFP-expressing 293T cells by FACS 72 h after transduction with AAV8GFP_P5F8 (red), AAV8RevGFP_P5F8 (orange), or AAV8Empty_P5F8 (blue) (n = 3). Error bars indicate SEM. (C) Time course graph depicting detectable GFP-positive cells in post-infection samples (5 × 106 MOI) with AAV8GFP_P5F8 (red), AAV8RevGFP_P5F8 (orange), or AAV8Empty_P5F8 (blue). Error bars indicate SEM. (D) Immunohistochemistry (IHC) images of C57BL/6J mouse liver sections stained for GFP protein 1 week after 9.4 × 1011vg infection with AAV8GFP_P5F8 (top left), AAV8RevGFP_P5F8 (top right), AAV8Empty_P5F8 (bottom left), or 2 × 1010vg AAV8-GFP (bottom right) (n = 5). (E) Representative FACS plots and bar chart depicting GFP reactive tetramer positive splenic CD8+ T cells from BALB/cJ mice 9 days after 1 × 1012 vg infection with AAV8GFP_P5F8 or AAV8Empty_P5F8 (nonparametric t test n = 5, p = 0.0079). Error bars indicate SEM.
Next, we evaluated protein translation from P5-associated contaminants in vivo. C57BL/6J mice were injected with 9.4 × 1011 vg of AAV8GFP_P5F8, AAV8RevGFP_P5F8, or AAV8Empty_P5F8 or 2 × 1010 vg of an AAV8 carrying a standard GFP expression cassette (AAV8-GFP). Livers were harvested 1 week after infection. Immunohistochemical staining for GFP revealed substantial numbers of GFP-expressing hepatocytes in AAV8GFP_P5F8 and AAV8-GFP-treated mice, but none in AAV8RevGFP_P5F8- or AAV8Empty_P5F8-infected mice (Figures 3D and S4). Furthermore, tetramer staining of splenocytes from mice infected with 1 × 1012 vg of either AAV8GFP_P5F8 or AAV8Empty_P5F8 revealed GFP-reactive CD8+ T cells in AAV8GFP_P5F8-infected mice, but not mice that received AAV8Empty_P5F8 (Figures 3E and S5). Combined, these results suggest that P5-associated contaminants have the potential to be transcribed, translated, and immunogenic after vector administration.
P5-associated contaminants are incorporated into rAAV independently of an ITR-flanked expression cassette
To test whether P5-associated contaminants were being incorporated into rAAV independently of an expression cassette, we used adenovirus serotype 5 (Ad5) helper genes and an Rep-Cap plasmid with a GFP cassette directly upstream of P5 to produce AAV8 capsids in HEK293T cells in the absence of an ITR-flanked expression cassette (AAV8GFP_P5NV) (Figure 4A.) When purified, alkaline gel analysis of AAV8GFP_P5NV showed DNA fragments ranging in sizes up to, but not exceeding, the packaging capacity of AAV, with AAV8GFP_P5F8 packaging an oversized FVIII cassette known to produce a smeared DNA size pattern used as a positive control (Figure 4B). To assess if P5-associated contaminant activity was also independent of the presence of an expression cassette, HEK293T cells were infected with 5 × 106 MOI AAV8GFP_P5F8, AAV8RevGFP_P5F8, AAV8Empty_P5F8, or an equivalent GFP copy delivery of AAV8GFP_P5NV. Cells were examined for GFP expression at 72 h after infection and showed that cells infected with AAV8GFP_P5NV displayed an equivalent percentage of GFP-expressing cells compared with AAV8GFP_P5F8 (p = 0.72) and significantly greater GFP-expressing cells than AAV8RevGFP_P5F8, AAV8Empty_P5F8, or uninfected HEK293T cells (p < 0.001) (Figure 4C).
Figure 4.

P5-associated rAAV contaminants are incorporated and translated independent of an ITR-flanked expression cassette
(A) Schematic of AAV8GFP_P5NV production methodology. (B) Alkaline gel of viral DNA from AAV8GFP_P5NV and AAV8GFP_P5F8 virions (1 kb DNA ladder). (C) Graph of GFP expressing HEK293T cells 72 h after transduction with AAV8GFP_P5NV (purple), AAV8GFP_P5F8 (red), AAV8RevGFP_P5F8 (orange), or AAV8Empty_P5F8 (blue) (one-way ANOVA with Tukey's multiple comparisons test). Error bars indicate SEM. (D) IHC images of C57BL/6J mouse liver sections stained for GFP protein 1 week after 2.7 × 1011vg infection with AAV8GFP_P5NV (left), AAV8Empty_P5F8 (middle), or AAV8-GFP (right) (n = 5).
To validate this independent activity of P5-associated rAAV contaminants in vivo, C57BL/6J mice were injected with 2.7e11vg AAV8GFP_P5NV, AAV8Empty_P5F8, or AAV8-GFP. At 1 week after infection, livers were harvested and stained for GFP protein. As expected, mice infected with AAV8GFP_P5NV resulted in readily detectable GFP-positive cells, whereas the AAV8Empty_P5F8-infected mice did not show any positive cells (Figures 4D and S6).
A spacer sequence inserted into the P5 promoter P5-HS can minimize incorporation of associated contaminants into AAV
The P5 promoter contains an Rep binding site (RBS) near an REP nicking site within a YY1 binding motif, which, because of proximity to one another, may be the cause of Rep-mediated packaging of P5-adjacent contaminant sequences (Figure S7A). We hypothesized that disrupting this proximity might maintain the critical function of P5 in rAAV production, while eliminating upstream contamination. Therefore, we constructed P5 promoters with the nick site removed (P5Δnick) or physically separated from the P5 RBS by spacer sequences of either 5 bp or 100 bp (P5-HS) (Figures 5A and S7B–S7D). All designs significantly reduced contamination (Figure 5B). However, while nick site removal adversely affected yield, P5-HS promoters produced rAAV titers equivalent to the standard P5 configuration (p > 0.9999) (Figure 5C).
Figure 5.

Development and characterization of the P5-HS promoter, a system to minimize Rep-Cap plasmid contamination of AAV
(A) Schematic of P5-HS design detailing position of DNA spacer sequence relative to P5 REP binding site and YY1+1 binding site (red). Forward-facing arrow depicts transcriptional start site. (B and C) Graphs showing relative upstream contamination from Rep78/68 promoter sequence (B) and relative titer (C), detected by qPCR, in AAV8ssHLPhFVIII preps produced with P5Δnick (purple square), P5-HS5 (red triangle), and P5-HS100 (orange triangle) promoters compared with unmodified P5 (blue circle) (one-way ANOVA with Dunnett's multiple comparisons test. n = 5; ∗∗p = 0.015; ∗∗∗∗p < 0.0001). Error bars indicate SEM. (D and E) Pie charts (D) and REP-CAP plasmid Circos plots (E) of NGS reads from cell factory scale AAV8ssHLPhFVIII (top) and AAV8scLP1hFIXco (bottom) produced with an Rep-Cap plasmid containing the P5 promoter (left) or the P5-HS5 promoter (right). (F and G) Graph of ELISA data of circulating factor VIII (F) and factor IX (G) protein from blood of C57/BL6J mice infected with 2 × 1010vg of AAV8 produced with Rep-Cap plasmids containing P5 (blue) or P5-HS (red) promoters (t test with Welch's correction. p > 0.5). Error bars indicate SEM. (H and I) Comparison (one-way ANOVA with Sidak's multiple comparisons test) of AAV1 (blue) and AAV2 (red) produced with P5 (dark) or P5-HS5 (light) by qPCR examining (H) titer (n = 3; p > 0.7) and (I) upstream contamination (n = 3; ∗∗p < 0.025). Error bars indicate SEM.
To test whether the P5-HS system could be applied to large-scale AAV production, the 5-bp P5-spacer (P5-HS5) was tested in cell factories. AAV8-FVIII and AAV8-FIX were produced using a plasmid in which Rep78/68 was driven by either P5 or P5-HS5. Purified rAAV was analyzed for contaminant abundance. Contaminant amplicons upstream of P5-HS5 were lower than P5 in both contexts, whereas titer was equivalent (Figure S8). NGS reads mapped to the producer plasmids showed a reduction of contamination upstream of P5 and an overall purity increase (Figures 5D and 5E). To ensure that P5-HS did not impact derived rAAV potency, mice were infected with 2 × 1010vg of AAV8-FVIII or AAV8-FIX preps produced from P5 or P5-HS5 plasmids. Circulating human clotting factors were measured and shown to be equivalent (Figures 5F and 5G). To ascertain if P5-HS could produce vector of serotypes besides AAV8, P5-HS5 was cloned into additional Rep-Cap plasmids (AAV1 and AAV2), used to produce FVIII, and measured for titer and contamination. As expected, P5-HS5 helped produce AAV1 and AAV2 to equivalent titers as P5 and upstream contamination was significantly reduced in P5-HS-produced vectors of both serotypes (Figures 5H and 5I).
P5-HS retains promoter regulation and reduces detectable contaminant translation
To assess whether the modifications made to the P5 promoter influenced transcriptional activity, fluorescent reporter cassettes were constructed (Figure 6A). These cassettes drove GFP in the forward orientation and mCherry in the reverse. HEK293T cells were transfected with mCherry/GFP constructs for P5, P5-HS5, and P5-HS100 alone, with an Rep78-expressing plasmid, or with an Ad5 helper plasmid and examined for expression 48 h after transfection. In the reverse direction, equivalent mCherry expression was observed across P5, P5-HS5, and P5-HS100 promoters (Figure 6B). Interestingly, in the forward direction, P5-HS5 and P5-HS100 appeared to have significantly lower activity than P5 (Figure 6C). However, the relative repression and induction by Rep78 and the Ad5 helper, as compared with their respective promoters alone, was equivalent across the three constructs (Figure 6D). Finally, given that the P5-HS promoters did not reduce the potential for reverse activity in their sequence, we examined the functional reduction in contaminant activity in P5-HS-produced AAV. We constructed a version of the contaminant reporter Rep-Cap plasmid that contained the P5-HS5 promoter and produced FVIII (AAV8GFP_P5−HS5F8). HEK293T cells were infected with 1 × 106 MOI of AAV8GFP_P5−HS5F8, AAV8GFP_P5F8, or AAV8Empty_P5F8. fluorescence-activated cell sorting (FACS) analysis 72 h after infection showed that AAV8GFP_P5−HS5F8 resulted in significantly reduced GFP-positive cells and was not significantly different from AAV8Empty_P5F8 and uninfected HEK293T cells (Figure 6E).
Figure 6.

P5-HS retains P5 promoter characteristics and minimizes detectable contaminant translation
(A) Schematic of promoter activity reporter construct design. (B-E) Comparison (one-way ANOVA with Tukey's multiple comparisons test) of (B) reverse promoter activity from P5 (blue), P5-HS5 (red), and P5-HS100 (orange) (n = 3; p > 0.98); (C) forward promoter activity from P5 (blue), P5-HS5 (red), and P5-HS100 (orange) (n = 3; ∗∗∗∗p < 0.0001); (D) forward promoter activity from P5 (blue), P5-HS5 (red), and P5-HS100 (orange) in presence of REP78 (n = 3; p > 0.99) or adenoviral helper genes (n = 3; p > 0.29); and (E) GFP-expressing HEK293T cells 72 h after transduction with 1 × 106 MOI AAV8ssHLPhFVIII produced with AAV8GFP_P5F8 (red), AAV8GFP_P5−HS5F8 (purple), and AAV8Empty_P5F8 (blue) (n = 5; ∗∗∗∗p < 0.0001). Error bars indicate SEM.
Discussion
The majority of rAAV production methodologies in HEK293/293T cells use the AAV2 P5 promoter. The identification of contaminating P5-associated transcripts at clinically relevant doses of rAAV highlights a novel issue with AAV-mediated gene transfer that has safety implications regardless of capsid serotype or therapeutic DNA transgene. Precise sequences of AAV production plasmids and designs vary among groups, a feature likely to extend to the plasmid backbone sequences upstream of P5, which often contain bacterially derived sequences and antibiotic resistance genes. Given this likely divergence in sequence identity across purified rAAV, it would be difficult to make robust conclusions about the overall effect of this contaminant activity on an infected cell for a specific preparation until it was independently assessed. As such, a reporter system was used to show the potential consequences of sequence transfer. We have demonstrated that the sequence transfer from a standard AAV production schema is sufficient to elicit protein translation to an extent that can elicit contaminant-specific immune recognition in vivo under these conditions. Considering these results, further research should be undertaken to evaluate the post-infection presence, activity, and translation potential of P5-associated contaminants in current and future clinical AAV products that use the intact P5 promoter in the production methodology.
This study focused on AAV contaminants associated with the P5 promoter. However, there are other sources of DNA contaminants. In addition to contaminants incorporated from AAV production plasmids, there are lower-abundance contaminating sequences, such as from 293T host cell DNA, incomplete vector genomes, or hybrid ITR flanked contaminants originating from low-level recombination.24,25 Further work using highly sensitive assays will be needed to ascertain what, if any, post-infection consequence these sequences could potentially have.
Our study of P5-associated AAV contaminants has focused primarily on the liver. However, it will be important to assess this phenotype in other cell contexts to understand the full implications of contaminant activity. Engineering efforts have yielded AAV capsid variants that can target a variety of human cell types.26, 27, 28, 29 Furthermore, recent developments have allowed for multiple administrations of AAV in preclinical models.30, 31, 32, 33 If translated clinically, repeated rAAV treatments would increase the total received dose of both therapeutic transgene and any DNA contaminants, potentially amplifying any negative effects therein. While vector design can limit expression of the intended transgene to the target tissue, the tissue specificity of the promoter activity derived from the reverse P5 sequence is at present uncharacterized. Systemic injection with a P5-produced rAAV vector would result in the transfer of these contaminant sequences to many tissues, posing a risk for aberrant transcripts at any site that, like the liver, exhibited promoter activity from the reverse P5 sequence.
The transcriptional activity of the P5 promoter within these contaminants poses an additional risk. Despite existing largely episomally, it is known that a small proportion of ITR containing rAAV genomes integrate chromosomally after administration.34 Indeed, integrated sequences corresponding to vector backbone contaminants have been previously detected.35 Owing to the robust bidirectional transcriptional activity of P5 noted in this study, if P5-associated contaminants were to become integrated proximal to a proto-oncogene, there would be the potential for transcriptional dysregulation regardless of the orientation of the integration event. We observed in our experiments a small population of cells that retained expression of GFP in continually passaged cells infected with AAV8GFP_P5F8. This is suggestive of integration; however, we did not characterize integration sites in this study, and so this question remains open. Further investigation on the potential for P5-associated contaminants in rAAV to integrate and any profile of such integration will help determine the level of risk generated by the transfer of these sequences.
It should be noted that a prior study into AAV contaminants did not detect transcription of contaminant sequences in mouse livers when assayed.12 The results of our study are not incongruous with these prior results. The amplicons used for post-infection qRT-PCR in the 2008 study did not specifically look for contamination directly upstream of the P5 promoter and focused primarily on assessing if transcription of the capsid gene could be detected. In the present study, it has been observed that sequences directly upstream of the P5 promoter are subject to transcription, which would be many kilobases from the capsid sequence. However, if the P5 promoter is positioned directly downstream of the capsid gene, as is the case with many publicly available plasmids (Table S1), capsid gene sequence would be subject to incorporation, and antisense transcript of capsid gene sequence would likely be detected.
The P5 promoter, while used almost ubiquitously in HEK293-based AAV production, has been successfully replaced in the baculoviral AAV production system. In baculoviral AAV production, a promoter of baculoviral origin (ΔIE1 or the P10 promoter) drives Rep78 expression.36,37 However, in the 293-based system, previous attempts to switch out the P5 promoter for heterologous constitutive promoter sequences have been found to disrupt the balance between the shorter forms of Rep (Rep52/48) driven by the P19 promoter and the larger forms of Rep(Rep78/68) driven from the P5 promoter, yielding low levels of AAV production.38,39 With the P5-HS promoter, we have demonstrated a methodology to minimize contaminants from the Rep-Cap plasmid that will be easy to implement into plasmid transfection-based rAAV production and will not suffer from low yield issues.
In summary, these data provide direct evidence that DNA contaminants in rAAV vector preps can be expressed in vector-treated animals and that subtle details of production system design can influence their abundance. Although it has been previously argued that rAAV DNA contaminants are transcriptionally inert,12 we demonstrate that contaminants associated with the P5 promoter can be transcribed, express protein, and establish antigen-specific CD8+ T cell immunity. Additionally, we show that the P5 promoter can be modified to minimize the abundance of such contaminants. These findings should be carefully considered when designing clinical rAAV manufacturing systems and interpreting immune-mediated adverse safety events observed after treatment with rAAV-based products.
Materials and methods
Vectors and sequences
P5ΔAT promoter sequence was synthesized by Genewiz and cloned into AAV2/8 Rep-Cap backbone through NotI + SalI sticky end cloning. GFP cassettes were cloned into an AAV2/8 Rep-Cap backbone by SpeI + XbaI sticky end cloning to yield forward and reverse direction constructs. P5-HS5 spacer was cloned into AAV2/1 2/2 and 2/8 capsids by NEB site directed mutagenesis kit (Catalog #E0554S). SDM primer sequences were gcaacCTCCATTTTGAAGCGGGAGG and ACCCTGCGTGCTCACTCG. Spacer sequences were generated by random sequence generator (FaBox).40 All cloned and modified constructs were full plasmid sequence verified by Massachusetts General Hospital CCIB DNA core.
AAV production, purification, and titer
Large-scale AAV8 production and purification
Scale-up AAV8 factor IX (FIX) and FVIII vectors were produced by two plasmid transfections in CellSTACK culture chambers (Corning Catalog #CLS3271). Before transfection, adherent human embryonic kidney 293T cells (ATCC CRL-3216) were cultured in DMEM (Lonza Bio Whittaker Catalog #12-733Q) supplemented with 10% fetal bovine serum (Fisher Scientific Catalog #SH3007103) and 2 mM L-glutamine (Corning Catalog #MT25005CI) at 37°C 10% CO2.
Transfection plasmids were as follows: an Rep-Cap plasmid with AAV2_8 was used to provide the replication and capsid genes. scLP1hFIXco+helpV3 provided a self-complementary FIX vector genome and adenoviral helper genes. ssHLPhFVIIIv3 provided a single-stranded FVIII vector genome with adenoviral helper genes.
Transfection plasmids were resuspended in DMEM and passed through a 0.2 μm filter into DMEM containing PEIpro (Polyplus Catalog #1,150,015). DNA-PEI mixture was incubated for 15 min and mixed with 1,000 mL of media from the CellSTACK culture chamber. Transfection mixture in media was then poured back into the CellSTACK culture chamber and incubated at 37°C 10% CO2.
Cells were harvested from the CellSTACK culture chamber 2 days after transfection and resuspended in 35 mL phosphate-buffered saline (PBS) in a 50-mL conical flask. Cells were lysed by five freeze-thaw cycles in a dry ice/ethanol bath and water bath at 37°C in turn. Cell lysate solution was spun down at 2500 rpm for 10 min in a tabletop centrifuge. Supernatant was transferred to a fresh 50-mL conical tube and treated with 25 U/mL Benzonase (Millipore-Sigma Catalog #E1014) in presence of 1 mM MgCl2 at 37°C for 1 h. Treated supernatants were passed through a 0.2-μM filter, diluted in PBS, and run on through the POROS CaptureSelect AAVX resin (Thermo Fisher Scientific Catalog #A36740). Flow rate on column was set to 2 mL/min. Column was equilibrated with 5 column volumes (CV) PBS pH 7.4; subsequently, AAV diluted supernatant was applied to column. The column was then washed with 15 CV PBS pH 7.4 and eluted with 5 CV of 0.1 M glycine HCl, pH 2.7 followed by 5 CV of PBS pH 7.4. Elution fractions of 4 mL were collected and neutralized with 1 M pH 8.8 Tris-HCl. Fractions were titered by qPCR, and peak fractions were combined. Eluted AAV was concentrated by 100 kDa Amicon filter (Millipore-Sigma Catalog #C7715) and supplemented with 0.25% human albumin (Sigma-Aldrich Catalog #A6608), followed by filtration using a 0.2-μm filter. Virus was stored at 4°C for short-term use and at −80°C for long-term use.
Small-scale AAV production and sample preparation for contaminant analysis
Small-scale AAV production transfections were performed in 6-well plates (Corning Catalog #3516). HEK293T cells were seeded at 800,000 cells per well. Cells were transfected 24 h after seeding. At 7 days after transfection, 2 μL of supernatant from the transfection well was collected and pretreated with 500 U DNase I in NEBuffer3 (1×) in a reaction volume of 100 μL. rAAV vectors were incubated at 37°C for 1 h. Reactions were neutralized with 2 μL 0.5 M EDTA pH 8 and incubated at 98°C for 10 min. Solution was cooled, and capsid proteins were denatured via addition of 2 μL 10% SDS, 2 μL of 20 mg/mL proteinase K (Ambion Catalog #AM2546) and incubation at 55°C for 1 h. Proteinase K was inactivated at 98°C for 1 h, and 94 μL of 0.01% Pluronic F-68 (Gibco Catalog #24,040,032) was added to make a 100-fold sample dilution. Samples were then analyzed for production efficiency by qPCR.
qPCR titration of AAV virus and contaminant amplicons
Vector genome titer and contaminant amplicon titers were assessed by qPCR using the final sample dilution of 1 × 10−4 against linearized serially diluted plasmid standard with SYBR Green (Applied Biosystems Catalog #4,309,155). All dilutions were made in the presence of 0.01% (v/v) Pluronic F-68 in nuclease free water. Viral titer was quantitated with a 7500 Real-Time PCR System (Applied Biosystems) using the default settings. Plate to plate variation in contaminant titer experiments was accounted for via measurement and normalization of the vector genome titer calculated from each plate to a predetermined value.
NGS analysis of AAV virus
There were 1 × 1011vg AAV genomes diluted in PBS to a volume of 200 μL. AAV capsids were denatured at 95°C for 15 min. Samples were cooled on ice, and 117 μL of 100% EtOH was added to precipitate DNA. Virus was then run through QIAamp MinElute Virus spin kit (QIAGEN Catalog #57,704) and eluted in 25 μL ddH2O. Purified DNA fragments were then subjected to NGS in the Hartwell Center at St Jude Children's Research Hospital using Nextera XT: 100 bp, 10 million reads (100 million for Figure 1C) sequencing (Illumina Miseq). The paired-end reads were trimmed, filtered against quality (Phred-like Q20 or greater) and length (50 bp or longer), and aligned to the AAV production plasmid sequences to identify contaminating and expression cassette DNA, using the Map Reads to Reference tool in CLC Genomics Workbench v12.0.1 (Qiagen). After removing duplicate mapped reads, pie charts were created on percentage of read count by analyzing the proportion of producer plasmid contaminant DNA compared with the FVIII or FIX expression cassette DNA. Reads that did not map to the AAV production plasmids were not included in the analysis. For analysis of post-infection hepatocytes, Kapa hyper and total stranded sequencing was performed on purified DNA and RNA, respectively. Then, 100 bp, 100 million reads were collected (Illumina Nextseq 550). Sequence reads that corresponded to the C57/BL6 mouse genome, aligned to a mouse reference sequence GRCm38 (UCSC mm10), were discarded from the analysis, and the remaining reads were mapped back to the AAV producer plasmids. Mapping result was analyzed with IGV genome browser (Broad Institute) and visualized with Circos plots (http://www.circos.ca/).41
Alkaline gel
An 0.8% agarose gel was prepared in ddH20 with 50 mM NaOH and 1 mM EDTA. Gel was submerged in alkaline running buffer (50 mM NaOH and 1 mM EDTA). Virus to be assayed was diluted to required concentration with PBS to a volume of 25 μL. Sample was then mixed with 8.5 μL loading buffer (20% glycerol, 1.2% SDS, 2.5 M NaOH, 50 mM EDTA) and loaded onto gel and run at 15 V for 18 h at 4°C. The gel was then incubated in neutralization buffer (0.1 M Tris-HCL, pH 8.0), rinsed in ddH2O, and imaged (Alpha Imager HP – Protein Simple).
In vitro studies
Cell culture
HEK293T cells were cultured in DMEM media (Corning Catalog #15-013-CV) supplemented with 10% FBS (GE Hyclone Catalog #SH30071.03) and 2 mM L-glutamine (Corning Catalog #25-005-CI), referred to herein as D10 media. Cultured cells were incubated at 37°C and 10% CO2. Cells were passaged by the aspiration of media, followed by a wash with PBS without calcium or magnesium (Lonza BioWhittaker Catalog #17-516F) and addition of 0.5% trypsin (Corning Catalog #25-052-CI) for 5 min. Trypsinized cells were neutralized by D10 media and split to the required concentration into a D10-containing flask or dish. Cells tested negative for mycoplasma, and cell line identity was confirmed by short tandem repeat (STR) analysis.
Transductions
Transduction of HEK293T cells with AAV was performed in 24-well plates. Cells were seeded at 50,000 cells per well. Cells were transduced 24 h after seeding. Required quantities of purified AAV were diluted to the volume of the lowest concentration virus of the experimental group in PBS. Virus was added directly into wells containing cultured cells in 1 mL D10 media.
Transfections
Transfection of HEK293T cells was performed in 6-well plates. Cells were seeded at 250,000 cells per well. Cells were transfected 24 h after seeding. Then, 500 ng of each plasmid was mixed 1:10 with PEIpro in DMEM, incubated at room temperature for 15 min, and transfected into wells containing 2 mL D10 media.
FACS
Media were aspirated from cells in a 24-well tissue culture plate and rinsed with 1XPBS (Lonza Biowhittaker Catalog #17-516F). Then, 150 μL of trypsin was added to each required well. Plates were incubated at 37°C 10% CO2 for 5 min. Trypsin was neutralized with 1 mL D10 media. Neutralized cells were transferred to FACS tubes (Corning Catalog #352,054). Then, 2 mL PBS was added to each tube, and tubes were centrifuged at 400g for 5 min. Supernatant was aspirated, and 3 mL of PBS was added to each tube. Tubes were again centrifuged at 400g for 5 min. Supernatant was removed, and cells were resuspended in PBS. Cells were placed on ice until being run on a BD LSR Fortessa FACS machine. There were 10,000 events collected per sample. FCS files were analyzed by FlowJo v.10.
In vivo studies
All animal procedures were carried out in compliance with the insitutional review board at St. Jude Children's Research Hospital. For in vivo contamination studies, male C57BL/6J mice were injected via tail vein with 8 × 1011vg AAV8. Upon termination of the experiment, livers were harvested and snap frozen in liquid nitrogen and stored at −80°C. Hepatic genomic DNA and RNA were isolated using commercial purification kits (QIAGEN Catalog #158,667 #74,104). Genomic DNA was treated with RNase A. RNA was treated with DNase.
For in vivo GFP_P5 flanker studies in Figures 3E and 4D, C57BL/6J mice were injected via tail vein with 9.4 × 1011vg and 2.7 × 1011vg of AAV8, respectively. One week after injection livers were harvested and fixed in 10% formalin, 4 μm sections were cut for slides. Slides were stained for GFP protein by immunohistochemistry. Slides were imaged on EVOS FL Auto (Life Technologies) microscope, on brightfield at 20× magnification.
For therapeutic protein expression studies, C57BL/6J mice were injected via tail vein with 2 × 1010vg of AAV. Circulating clotting factor levels of FVIII or FIX were assessed by ELISA on plasma. (Diagnostica Stago Catalog #00280 #00943). ELISA analysis was performed on BioTek plate reader.
For contaminant immunogenicity studies, BALB/cJ mice were injected via tail vein with 1 × 1012vg of AAV8. At 9 days post-infection, spleens were harvested from mice and placed in HBSS (Thermo Fisher Catalog #14,175,095). Tissue was homogenized, filtered through a 100-μm filter (Corning Catalog #352,360), and spun down at 500g for 5 min. Supernatant was discarded, and cells were resuspended in 5 mL of ACK (ammonium-chloride-potassium) lysis buffer and incubated for 5 min at 37°C to remove red blood cells. Following incubation, the cell solution was neutralized with 40 mL HBSS, spun down at 500g, and resuspended in PBS for counting. Isolated splenocytes were plated in 96-well plates at 5 million cells per well in a volume of 150 μL PBS. Plates were spun down at 500g, supernatant was discarded, and cells resuspended in 150 μL of pre-titrated GFP (H-2Kd EGFP200–208 HYLSTQSAL) tetramer stain. Splenocytes were incubated in the dark and at room temperature for 90 min, spun down at 500g, and resuspended in 200 μL PBS twice, spun down again at 500g, then resuspended in 50 μL Fc block (BD Biosciences Catalog #553,142), and incubated at room temperature in darkness for 10 min. Without washing or removing the Fc block, 50 μL of Live/Dead stain (Tonbo Catalog #13-0870-T100) and pre-titrated surface antibody cocktail were added to each well (CD8-FITC, Tonbo Catalog #35-0081-U100; B220-BV421, BioLegend Catalog #103,240; F4/80-BV421, BioLegend Catalog #123,132). Cells were incubated at 4°C in darkness for 30 min, washed in PBS, and spun down at 500g twice, and resuspended in 150 μL of fluorescence-activated cell sorting buffer. Cells were placed on ice until being run on a BD LSR Fortessa FACS machine. There were 4,000,000 events collected per well. FCS files were analyzed by FlowJo v.10.
FISH experiments
Fresh liver samples were cut with a razor blade to expose a fresh edge and touched onto slides (Thermo-Scientific Catalog #5,991,051). Slides were incubated in 1% PFA in PBS plus 0.05% NP-40 for 5 min, then in 1% PFA in PBS for 5 min, and then in 70% EtOH for 5 min before transfer to storage in 70% EtOH at −20°C.
Fixed slides were RNA hybridized by dehydration in EtOH. Slides were sequentially treated for 2 min each in 70%, 80%, and 100% EtOH solutions. Slides were dried and probed for either vector genome RNA or contaminant RNA. Denatured probe for Rep-Cap plasmid contaminating DNA was prepared by nick translation and resuspended in buffer comprising 50% formamide, 2× saline sodium citrate, and 10% dextran sulfate.42 Rep-Cap contaminant probe sequence is described in Figure S8. Probe was applied to slides and hybridized at 37°C overnight. Slides were washed in 50% formamide and 2× saline sodium citrate for 5 min at 37°C. Slides were mounted in mounting medium (Vector Labs Catalog #H-1000-10). RNA FISH images were collected and analyzed as previously described.43
After RNA FISH analysis, slides were treated with RNase A (Sigma Catalog #R4642). Probe was applied to slides and hybridized at 37°C overnight. Slides were washed in 50% formamide and 2× saline sodium citrate for 5 min at 37°C. Slides were mounted in mounting medium.
For sequential DNA-FISH, slides were treated in 4% PFA, 0.5% Tween 20, and 0.5% NP-40 for 10 min at RT, and then treated in 0.2N HCl, 0.5% Triton X 100 for 10 min on ice. Following fixation, slides were denatured in 70% formamide 2× saline sodium citrate at 80°C for 10 min. Slides were then dehydrated in a graded alcohol series for 2 min each as for RNA FISH. Denatured probe (same labeled DNA as was used for RNA detection) was then applied to the slides and hybridized overnight at 37°C.42 Washing and mounting of slides is the same as for RNA FISH. DNA FISH images were collected and analyzed as previously described.43
Graphs and statistical analysis
Figures were prepared in Adobe Illustrator. Graphs and statistics were generated in GraphPad Prism 8.0. and R studio. Error bars represent SEM. Pie charts were generated in Microsoft Excel.
Data sharing statement
Original data, resources, and protocols available on request by email: Andrew.Davidoff@stjude.org.
DNA and RNA sequencing fastQ files uploaded to NCBI sequence read archive: PRJNA741271.
Acknowledgments
We thank Bryan Piras and Michael Meagher from the St. Jude Children's Research Hospital Good Manufacturing Practices (GMP) facility for providing GMP-grade rAAV8-hFVIII vector and Dr. Jun Yang from the Department of Surgery of St. Jude Children's Research Hospital for critical reading of the manuscript. This study was supported in part by the National Cancer Institute of the National Institutes of Health under Award Number P30 CA021765.
Author contributions
M.A.B., J.T.G., A.C.N., and A.M.D. designed the research. M.A.B., P.-H.C., S.M.W., I.L.R., A.S., Y.S., J.Z., and C.L.M. performed the research. M.A.B., P.-H.C., S.M.W., I.L.R., A.S., Y.S., J.Z., and C.L.M. collected the data. Y.-D.W., M.V., and P.G.T. contributed vital new reagents or analytical tools. M.A.B., P.-H.C., C.L.M., Y.-D.W., M.V., and A.S. analyzed and interpreted the data. M.A.B. and A.S. performed the statistical analysis. M.A.B. and A.M.D. wrote the manuscript.
Declaration of interests
M.A.B., P.-H.C., C.L.M., and A.M.D. are listed inventors on a provisional patent that includes the P5-HS design described in this article. A.M.D. and A.C.N. are listed inventors on patents relating to FVIII and FIX construct designs for AAV gene therapy and are entitled to royalty income from their licensing. A.C.N. acts as an advisor for Freeline Therapeutics, BioMarin Pharmaceutical, and Generation Bio; is a founder of, has a sponsored research agreement with, and owns equity in Freeline Therapeutics; and is a consultant to several biopharmaceutical companies. J.T.G. possesses equity in Vertex Pharmaceuticals and is entitled to royalty income from patents related to gene therapy technology.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.01.008.
Supplemental information
References
- 1.Nathwani A.C., Tuddenham E.G.D., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Jaquilmac Pie A., Harrington C., et al. Adeno-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nathwani A.C., Reiss U.M., Tuddenham E.G.D., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennicelli J., Wright J.F., Komaromy A., Jacobs J.B., Hauck B., Zelenaia O., Mingozzi F., Hui D., Chung D., Rex T.S., et al. Reversal of blindness in animal models of leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol. Ther. 2008;16:458–465. doi: 10.1038/sj.mt.6300389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K., et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 5.Mendell J.R., Al-Zaidy S.A., Rodino-Klapac L.R., Goodspeed K., Gray S.J., Kay C.N., Boye S.L., Boye S.E., George L.A., Salabarria S., et al. Current clinical applications of in vivo gene therapy with AAVs. Mol. Ther. 2021;29:464–488. doi: 10.1016/j.ymthe.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasi K.J., Rangarajan S., Mitchell N., Lester W., Symington E., Madan B., Laffan M., Russell C.B., Li M., Pierce G.F., et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia. A. N. Engl. J. Med. 2020;382:29–40. doi: 10.1056/NEJMoa1908490. [DOI] [PubMed] [Google Scholar]
- 7.Konkle B.A., Walsh C.E., Escobar M.A., Josephson N.C., Young G., von Drygalski A., McPhee S.W.J., Samulski R.J., Bilic I., de la Rosa M., et al. BAX 335 hemophilia B gene therapy clinical trial results: potential impact of CpG sequences on gene expression. Blood. 2021;137:763–774. doi: 10.1182/blood.2019004625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J., et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.High-dose AAV gene therapy deaths. Nat. Biotechnol. 2020;38:910. doi: 10.1038/s41587-020-0642-9. [DOI] [PubMed] [Google Scholar]
- 10.Johnson V. Astellas pauses gene therapy trial for X-linked myotubular myopathy. 2021. https://www.genetherapylive.com/view/astellas-pauses-gene-therapy-trial-x-linked-myotubular-myopathy genetherapylive.
- 11.Penaud-budloo M., Lecomte E., Le A., Cogne B., Roulet A., Lindenbaum P., Moullier P., Ayuso E. Accurate identification and quantification of DNA species by next-generation sequencing in adeno-associated viral vectors produced in insect cells. Hum. Gene. Ther. Methods. 2017;28:148–162. doi: 10.1089/hgtb.2016.185. [DOI] [PubMed] [Google Scholar]
- 12.Hauck B., Murphy S.L., Smith P.H., Qu G., Liu X., Zelenaia O., Mingozzi F., Sommer J.M., High K.A., Wright J.F. Undetectable transcription of cap in a clinical AAV vector: implications for preformed capsid in immune responses. Mol. Ther. 2009;17:144–152. doi: 10.1038/mt.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chadeuf G., Ciron C., Moullier P., Salvetti A. Evidence for encapsidation of prokaryotic sequences during recombinant adeno-associated virus production and their in vivo persistence after vector delivery. Mol. Ther. 2005;12:744–753. doi: 10.1016/j.ymthe.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Schnödt M., Schmeer M., Kracher B., Krüsemann C., Espinosa L.E., Grünert A. DNA minicircle technology improves purity of adeno-associated viral vector preparations. Mol. Ther. Nucleic Acids. 2016;5:1–11. doi: 10.1038/mtna.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lecomte E., Saleun S., Bolteau M., Guy-Duché A., Adjali O., Blouin V., Penaud-Budloo M., Ayuso E. The SSV-seq 2.0 PCR-free method improves the sequencing of adeno-associated viral vector genomes containing GC-rich regions and homopolymers. Biotechnol. J. 2021;16:e2000016. doi: 10.1002/biot.202000016. [DOI] [PubMed] [Google Scholar]
- 16.Halbert C.L., Metzger M.J., Lam S.L., Miller A.D. Capsid-expressing DNA in AAV vectors and its elimination by use of an oversize capsid gene for vector production. Gene Ther. 2011;18:411–417. doi: 10.1038/gt.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z., Halbert C.L., Lee D., Butts T., Tapscott S.J., Storb R., Miller A.D. Elimination of contaminating cap genes in AAV vector virions reduces immune responses and improves transgene expression in a canine gene therapy model. Gene Ther. 2014;21:363–370. doi: 10.1038/gt.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonntag F., Schmidt K., Kleinschmidt J.A. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc. Natl. Acad. Sci. U S A. 2010;107:10220–10225. doi: 10.1073/pnas.1001673107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogden P.J., Kelsic E.D., Sinai S., Church G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science. 2019;366:1139–1143. doi: 10.1126/science.aaw2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Penaud-Budloo M., Lecomte E., Guy-Duché A., Saleun S., Roulet A., Lopez-Roques C., Tournaire B., Cogné B., Léger A., Blouin V., et al. Accurate identification and quantification of DNA species by next-generation sequencing in adeno-associated viral vectors produced in insect cells. Hum. Gene Ther. Methods. 2017;28:148–161. doi: 10.1089/hgtb.2016.185. [DOI] [PubMed] [Google Scholar]
- 21.Stutika C., Gogol-Döring A., Botschen L., Mietzsch M., Weger S., Feldkamp M., Chen W., Heilbronn R. A comprehensive RNA sequencing analysis of the adeno-associated virus (AAV) type 2 transcriptome reveals novel AAV transcripts, splice variants, and derived proteins. J. Virol. 2016;90:1278–1289. doi: 10.1128/JVI.02750-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stutika C., Mietzsch M., Gogol-Döring A., Weger S., Sohn M., Chen W., Heilbronn R., Kotin R., Siniscalco M., Samulski R., et al. Comprehensive small RNA-seq of adeno-associated virus (AAV)-Infected human cells detects patterns of novel, non-coding AAV RNAs in the absence of cellular miRNA regulation. PLoS One. 2016;11:e0161454. doi: 10.1371/journal.pone.0161454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexander I.E., Russell D.W., Miller A.D. Transfer of contaminants in adeno-associated virus vector stocks can mimic transduction and lead to artifactual results. Hum. Gene Ther. 1997;8:1911–1920. doi: 10.1089/hum.1997.8.16-1911. [DOI] [PubMed] [Google Scholar]
- 24.Tran N.T., Heiner C., Weber K., Weiand M., Wilmot D., Xie J., Wang D., Brown A., Manokaran S., Su Q., et al. AAV-genome population sequencing of vectors packaging CRISPR components reveals design-influenced heterogeneity. Mol. Ther. - Methods Clin. Dev. 2020;18:639–651. doi: 10.1016/j.omtm.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y., Cooper R., Kiladjian A., Bergelson S., Feschenko M. A digestion-free method for quantification of residual host cell DNA in rAAV gene therapy products. Mol. Ther. Methods Clin. Dev. 2019;13:526–531. doi: 10.1016/j.omtm.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maguire C.A., Gianni D., Meijer D.H., Shaket L.A., Wakimoto H., Rabkin S.D., Gao G., Sena-Esteves M. Directed evolution of adeno-associated virus for glioma cell transduction. J. Neurooncol. 2010;96:337–347. doi: 10.1007/s11060-009-9972-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li W., Zhang L., Johnson J.S., Zhijian W., Grieger J.C., Ping-Jie X., Drouin L.M., Agbandje-McKenna M., Pickles R.J., Samulski R.J. Generation of novel AAV variants by directed evolution for improved CFTR delivery to human ciliated airway epithelium. Mol. Ther. 2009;17:2067–2077. doi: 10.1038/mt.2009.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang L., Jiang J., Drouin L.M., Agbandje-McKenna M., Chen C., Qiao C., Pu D., Hu X., Wang D.-Z., Li J., et al. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc. Natl. Acad. Sci. U S A. 2009;106:3946–3951. doi: 10.1073/pnas.0813207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Landegger L.D., Pan B., Askew C., Wassmer S., Galvin A., Taylor R., Forge A., Stankovic K.M., Holt J.R., Vandenberghe L.H., et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotechnol. 2017;35:280–284. doi: 10.1038/nbt.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salas D., Kwikkers K.L., Zabaleta N., Bazo A., Petry H., Van Deventer S.J., Aseguinolaza G.G., Ferreira V. Immunoadsorption enables successful rAAV5-mediated repeated hepatic gene delivery in nonhuman primates. Blood Adv. 2019;3:2632–2641. doi: 10.1182/bloodadvances.2019000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mingozzi F., Chen Y., Murphy S.L., Edmonson S.C., Tai A., Price S.D., Metzger M.E., Zhou S., Wright J.F., Donahue R.E., et al. Pharmacological modulation of humoral immunity in a nonhuman primate model of AAV gene transfer for hemophilia B. Mol. Ther. 2012;20:1410–1416. doi: 10.1038/mt.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meliani A., Boisgerault F., Hardet R., Marmier S., Collaud F., Ronzitti G., Leborgne C., Costa Verdera H., Simon Sola M., Charles S., et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 2018;9:4098. doi: 10.1038/s41467-018-06621-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majowicz A., Salas D., Zabaleta N., Rodríguez-Garcia E., González-Aseguinolaza G., Petry H., Ferreira V. Successful repeated hepatic gene delivery in mice and non-human primates achieved by sequential administration of AAV5ch and AAV1. Mol. Ther. 2017;25:1831–1842. doi: 10.1016/j.ymthe.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deyle D.R., Russell D.W. Adeno-associated virus vector integration. Curr. Opin. Mol. Ther. 2009;11:442–447. [PMC free article] [PubMed] [Google Scholar]
- 35.Dalwadi D.A., Calabria A., Tiyaboonchai A., Posey J., Naugler W.E., Montini E., Grompe M. AAV integration in human hepatocytes. Mol. Ther. 2021;29:2898–2909. doi: 10.1016/j.ymthe.2021.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urabe M., Ding C., Kotin R.M. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther. 2002;13:1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- 37.Chen H. Intron splicing-mediated expression of AAV rep and cap genes and production of AAV vectors in insect cells. Mol. Ther. 2008;16:924–930. doi: 10.1038/mt.2008.35. [DOI] [PubMed] [Google Scholar]
- 38.Ogasawara Y., Urabe M., Ozawa K. The use of heterologous promoters for adeno-associated virus (AAV) protein expression in AAV vector Production.pdf. Microbiol. Immunol. 1998;42:177–185. doi: 10.1111/j.1348-0421.1998.tb02269.x. [DOI] [PubMed] [Google Scholar]
- 39.Li J., Samulski R.J., Xiao X. Role for highly regulated rep gene expression in adeno- associated virus vector production. J. Virol. 1997;71:5236–5243. doi: 10.1128/jvi.71.7.5236-5243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villesen P. FaBox: an online toolbox for FASTA sequences. Mol. Ecol. Notes. 2007;7:965–968. [Google Scholar]
- 41.Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Namekawa S.H., Payer B., Huynh K.D., Jaenisch R., Lee J.T. Two-step imprinted X inactivation: repeat versus genic silencing in the mouse. Mol. Cell. Biol. 2010;30:3187–3205. doi: 10.1128/MCB.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paulk N.K., Pekrun K., Zhu E., Nygaard S., Li B., Xu J., Chu K., Leborgne C., Dane A.P., Haft A., et al. Bioengineered AAV capsids with combined high human liver transduction in vivo and unique humoral seroreactivity. Mol. Ther. 2018;26:289–303. doi: 10.1016/j.ymthe.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.