

This cohort study investigated the genetic background and the natural history of a large international cohort of dilated cardiomyopathy presenting in individuals older than 60 years to assess the importance of genetic testing in those patients.
Key Points
Question
Is dilated cardiomyopathy diagnosed in individuals older than 60 years characterized by a specific genetic background and have similar clinical characteristics to the known epidemiology?
Findings
In this cohort study of 184 patients with late-onset dilated cardiomyopathy, compared with the current epidemiology, a high genetic variation burden was identified, largely due to titin-truncating variants. Patients with a positive genetic test had higher mortality than genotype-negative patients.
Meanings
These data suggest that late-onset dilated cardiomyopathy may be a specific subgroup of patients and support the use of extensive genetic testing in older patients because it may provide important etiologic and prognostic information.
Abstract
Importance
Dilated cardiomyopathy (DCM) is frequently caused by genetic factors. Studies identifying deleterious rare variants have predominantly focused on early-onset cases, and little is known about the genetic underpinnings of the growing numbers of patients with DCM who are diagnosed when they are older than 60 years (ie, late-onset DCM).
Objective
To investigate the prevalence, type, and prognostic impact of disease-associated rare variants in patients with late-onset DCM.
Design, Setting, and Participants
A population of patients with late-onset DCM who had undergone genetic testing in 7 international tertiary referral centers worldwide were enrolled from March 1990 to August 2020. A positive genotype was defined as the presence of pathogenic or likely pathogenic (P/LP) variants.
Main Outcomes and Measures
The study outcome was all-cause mortality.
Results
A total of 184 patients older than 60 years (103 female [56%]; mean [SD] age, 67 [6] years; mean [SD] left ventricular ejection fraction, 32% [10%]) were studied. Sixty-six patients (36%) were carriers of a P/LP variant. Titin-truncating variants were the most prevalent (present in 46 [25%] of the total population and accounting for 46 [69%] of all genotype-positive patients). During a median (interquartile range) follow-up of 42 (10-115) months, 23 patients (13%) died; 17 (25%) of these were carriers of P/LP variants, while 6 patients (5.1%) were genotype-negative.
Conclusions and Relevance
Late-onset DCM might represent a distinct subgroup characterized by and a high genetic variation burden, largely due to titin-truncating variants. Patients with a positive genetic test had higher mortality than genotype-negative patients. These findings support the extended use of genetic testing also in older patients.
Introduction
Dilated cardiomyopathy (DCM) is a nonischemic, nontoxic, noninfectious, or nonvalvular heart muscle disease characterized by left ventricular or biventricular systolic dysfunction.1,2 Typical onset of the disease is in the fourth or fifth decade of life and it affects predominantly male individuals with a male-to-female ratio of 3:1.2 Although a specific cause might be unknown in a non-negligible number of patients, to date, more than one-third of familial forms and approximately 25% of sporadic cases have positive genetic testing for pathogenic (P) or likely pathogenic (LP) variants.3,4,5,6 Recent evidence highlights the importance of specific genotypes in determining the prognosis of patients with DCM, either alone or in combination with environmental factors.7,8,9,10,11
Contemporary analyses highlight that DCM is increasingly diagnosed in older patients12 and a late-onset (ie, age >60 years) form of DCM could differ from more typical-onset DCM presentations.13 However, the genetic background and the prognostic impact of genetic characterization among patients with DCM presenting who are older than 60 years are largely unknown.
The aim of the present study was to investigate the genetic background and the natural history of a large international cohort of individuals with DCM presenting in individuals older than 60 years to assess the importance of genetic testing in those patients.
Methods
Study Population
For the present study, we included all consecutive patients with DCM older than 60 years at the time of diagnosis (ie, late-onset DCM), with available genetic testing analysis, from 7 international tertiary centers worldwide: Cardiovascular Department, Azienda Sanitaria Universitaria Integrata Giuliano Isontina, University of Trieste, Trieste, Italy (coordinating center); University of Colorado Denver, Anschutz Medical Campus, Aurora (coordinating center); Victor Chang Cardiac Research Institute, Sydney, Australia; Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts; Pauley Heart Center, Virginia Commonwealth University Health System, Richmond; Stanford University Medical Center, Stanford, California; and Guys and St. Thomas Hospital, London, UK. Ethnicy was self-reported, where available. The 60-years age cutoff was selected according to the literature and the United Nations and World Health Organization.13,14 Patients were enrolled from March 1990 to August 2020. This study was approved by independent institutional ethics boards. Written consent was waived because this study used retrospective audit data.
DCM was defined as the presence of impaired left ventricular ejection fraction (ie, <50%) after exclusion of significant extrinsic factors such as history of significant hypertension, ischemic heart disease, valvular disease, extrinsic factors (including excessive alcohol intake or chemotherapy), advanced systemic disease affecting short-term prognosis, congenital heart diseases, persistent tachyarrhythmias, and active myocarditis.15,16,17 Late-onset DCM was defined as the diagnosis of DCM, with left ventricular systolic dysfunction requiring medical attention, after the 60th year of age. Family history was extensively investigated and a pedigree of 3 or more generations collected when possible. All familial cases were defined by the presence of at least 1 other affected family member or an unexplained sudden death in a first-degree relative of a patient with DCM.18 All patients underwent extensive clinical and echocardiographic evaluation, with measurement of left ventricular ejection fraction according to current international guidelines and received timely guideline-directed medical and device treatment.19,20 The study outcome measure was all-cause mortality.
Genetic Analysis and Cluster Classification
Genetic testing was done using next-generation DNA sequencing of multigene DCM panels for established causative DCM genes, as previously reported.21,22 Gene variants were classified as P/LP using accepted algorithms including the American College of Medical Genetics and Genomics standards.23 To maintain a conservative approach, all variants of uncertain significance were excluded from the present analysis. In keeping with the literature, titin (TTN) missense variations were also excluded from the analysis.4,17,24,25
To increase the statistical power of genotype-phenotype association analysis, patients with rare variants in genes belonging to the same subcellular compartment or with similar functions were clustered together in groups, as previously described.4,22 Selected genes sharing a common gene ontology function/biological process, a common subcellular compartment and with consolidated scientific evidence were included in the clusters. Owing to their distinct characteristics, TTN, lamin A/C (LMNA), filamin C (FLNC), and RNA-binding motif 20 (RBM20) were considered as separate groups.17,26,27,28,29 Finally, the following 9 variant clusters were defined as (1) no pathogenic variants, (2) TTN-truncating variants (TTNtv; only affecting N2B/N2BA isoforms), (3) LMNA, (4) structural cytoskeleton Z-disk, (5) desmosomal genes, (6) motor sarcomeric genes, (7) FLNC, (8) RBM20, and (9) other DCM genes. As previously described, desmin (DES), dystrophin (DMD), nexilin (NEXN), nebulin (NEBL), and LIM domain binding 3 (LDB3) were included in the structural cytoskeleton Z -disk group; plakophilin-2 (PKP2), desmocollin-2 (DSC2), desmoglein-2 (DSG2), catenin alpha-3 (CTNNA3), desmoplakin (DSP), and junction plakoglobin (JUP) were grouped in the desmosomal genes; myosin heavy chain 7 (MYH7), actin alpha cardiac muscle 1 (ACTC1), and troponin T2 (TNNT2) were classified into the motor sarcomeric genes group.4
Statistical Analysis
Variables were expressed, as appropriate, as mean (SD), median (interquartile range), or counts and percentage. Comparisons between groups were made by the analysis of variance or the t test on continuous variables, using the Brown-Forsythe statistic when the assumption of equal variances did not hold or by the nonparametric Mann-Whitney test when necessary. The χ2 test or the Fisher exact test was applied for discrete variables. Survival curves for all-cause mortality were estimated and compared between groups using the log-rank test. Bonferroni correction was applied to account for multiple comparisons, and, as some patients were grouped as families, family clustering was considered. No adjustments for missing data were needed as less than 10% missing in key variables. Prespecified subgroup analyses according to continent of enrollment, proband vs familial cases, and sex differences were also conducted. SPSS statistical software version 26 (IBM) and the R version 1.2.5033 (R Foundation) were used for statistical analyses. Two-sided P values were statistically significant at .05 or lower according to Bonferroni correction.
Results
Study Population
A total of 184 patients with late-onset DCM from Australia, Italy, the UK, and the US were included in the study. Baseline characteristics of the patients are summarized in Table 1 and in eTable 1 in the Supplement. The mean (SD) age at diagnosis was 67 (6) years (eFigure 1 in the Supplement), patients had a mean (SD) left ventricular ejection fraction of 32% (10%), and patients were mostly diagnosed as part of family screening (120 [65%]). A total of 48 patients (30%) were in New York Heart Association classes III or IV. Most patients were of European ancestry (168 [98%]). Data on other race and ethnic groups were not available.
Table 1. Baseline Characteristics of the Study Population.
| Characteristic | No. (%) | P value | Available data, % | ||
|---|---|---|---|---|---|
| Total cohort | Genetic negative | Genetic positive | |||
| No. of patients | 184 | 118 | 66 | NA | NA |
| Age, mean (SD), y | 67 (6) | 67 (7) | 67 (6) | .89 | 100 |
| Sex | |||||
| Male | 81 (44) | 56 (47) | 25 (38) | .22 | 100 |
| Female | 103 (56) | 62 (53) | 41 (62) | ||
| European ancestrya | 168 (98) | 108 (97) | 60 (98) | .18 | 94 |
| Family history | 120 (65) | 69 (59) | 51 (76) | .01 | 100 |
| NYHA III/IV | 48 (30) | 28 (27) | 20 (33) | .48 | 85 |
| LVEF %, mean (SD) | 32 (10) | 32 (9) | 31 (11) | .84 | 91 |
| LVEF <35% | 107 (62) | 70 (64) | 37 (58) | .53 | 91 |
| LVEDD, mean (SD) | 61 (9) | 62 (9) | 60 (8) | .12 | 83 |
| RAAS-I | 129 (87) | 83 (86) | 46 (89) | .62 | 82 |
| β-Blockers | 127 (84) | 83 (84) | 44 (83) | .78 | 82 |
Abbreviations: LVEDD, left ventricular end diastolic diameter; LVEF, left ventricular ejection fraction; NA, not applicable; NYHA, New York Heart Association; RAAS-I, renin angiotensin aldosterone system inhibitors.
Data on other race and ethnic groups are not available.
Genetic Background
The genetic diagnostic yield was 36% (ie, 66 patients were carriers of P/LP variants). The variants most frequently observed were TTNtv (n = 46), collectively affecting 25% of the patients (n = 46) and accounting for 69% of patients with positive genetic testing (n = 46). The remaining cases (n = 20) had other P/LP variants (motor sarcomeric genes, 5 [8%]; LMNA, 4 [6%]; FLNC, 4 [6%]; desmosomal genes, 4 [6%]; and other variants, 2 [3%]; Figure 1). Details of variants are reported in eTable 2 in the Supplement.
Figure 1. Diagnostic Yield and Genetic Landscape in the Late-Onset Dilated Cardiomyopathy Population.

FLNC indicates filamin C; LMNA, lamin A/C; TTNtv, titin-truncating variants.
Although clinical characteristics were mostly comparable, patients with positive genetic testing more frequently had a family history of DCM compared with those with negative genetic testing (51 [76%] vs 69 [59%], respectively; Table 1). Interestingly, no differences were found between baseline characteristics of patients with TTNtv compared with carriers of other P/LP variants (Table 2).
Table 2. Baseline Characteristics of Variation Positive Cohort.
| Characteristic | No. (%) | P value | ||
|---|---|---|---|---|
| Total cohort | TTNtv | Other | ||
| No. of patients | 66 | 46 | 20 | NA |
| Age, mean (SD), y | 67 (6) | 67 (7) | 68 (5) | .68 |
| Sex | ||||
| Male | 25 (38) | 20 (44) | 5 (25) | .15 |
| Female | 41 (62) | 26 (56) | 15 (75) | |
| European ancestrya | 60 (98) | 44 (100) | 16 (94) | .11 |
| Family history | 51 (76) | 35 (76) | 16 (80) | .73 |
| NYHA III/IV | 20 (33) | 15 (37) | 5 (29) | .56 |
| LVEF %, mean (SD) | 31 (11) | 31 (11) | 31 (12) | .96 |
| LVEF <35% | 37 (58) | 24 (55) | 13 (68) | .31 |
| LVEDD, mean (SD) | 60 (8) | 59 (9) | 61 (7) | .46 |
| RAAS-I | 46 (89) | 34 (92) | 12 (80) | .22 |
| β-Blockers | 44 (83) | 32 (84) | 12 (80) | .71 |
Abbreviations: LVEDD, left ventricular end diastolic diameter; LVEF, left ventricular ejection fraction; NA, not applicable; NYHA, New York Heart Association; RAAS-I, renin angiotensin aldosterone system inhibitors; TTNtv, titin-truncating variants.
Data on other race and ethnic groups are not available.
Sex Differences
Female sex accounted for 56% (n = 103) of patients (Figure 2). Interestingly, the percentage of female individuals was consistent in both patients with positive and negative genetic testing (41 [62%] vs 62 [57%], respectively) and among carriers of TTNtv and other P/LP variants (26 [56%] and 15 [75%], respectively; Figure 2). Both male and female individuals presented with similar baseline characteristics and phenotypic expression of the disease (eTable 3 in the Supplement).
Figure 2. Sex Prevalence in the Total, Titin-Truncating Variants (TTNtv), and Other Genetic Variant Cohorts.

Outcomes
Over a median (interquartile range) follow-up of 42 (10-115) months, the primary outcome measure of all-cause mortality was met in 23 patients (13%; incidence of 2.4 events per 100 patients per year). Of those, 6 patients (5%) had negative genetic testing whereas 17 (25%) were carriers of P/LP variants (Figure 3A). Furthermore, patients with negative genetic testing had significantly better prognosis compared with those with either TTNtv or carriers of other P/LP variants. Remarkably, no different survival trends were observed between TTNtv carriers and carriers of other P/LP variants (Figure 3A). Adverse events were equally distributed among male and female individuals with TTNtv. Conversely, although not significant, most deaths occurred in female carriers of other P/LP variants and male individuals with negative genetic testing (Figure 3B).
Figure 3. Long-term Prognosis of Late-Onset Dilated Cardiomyopathy (DCM) According to Genetic Testing Results and Sex Distribution of Adverse Events .
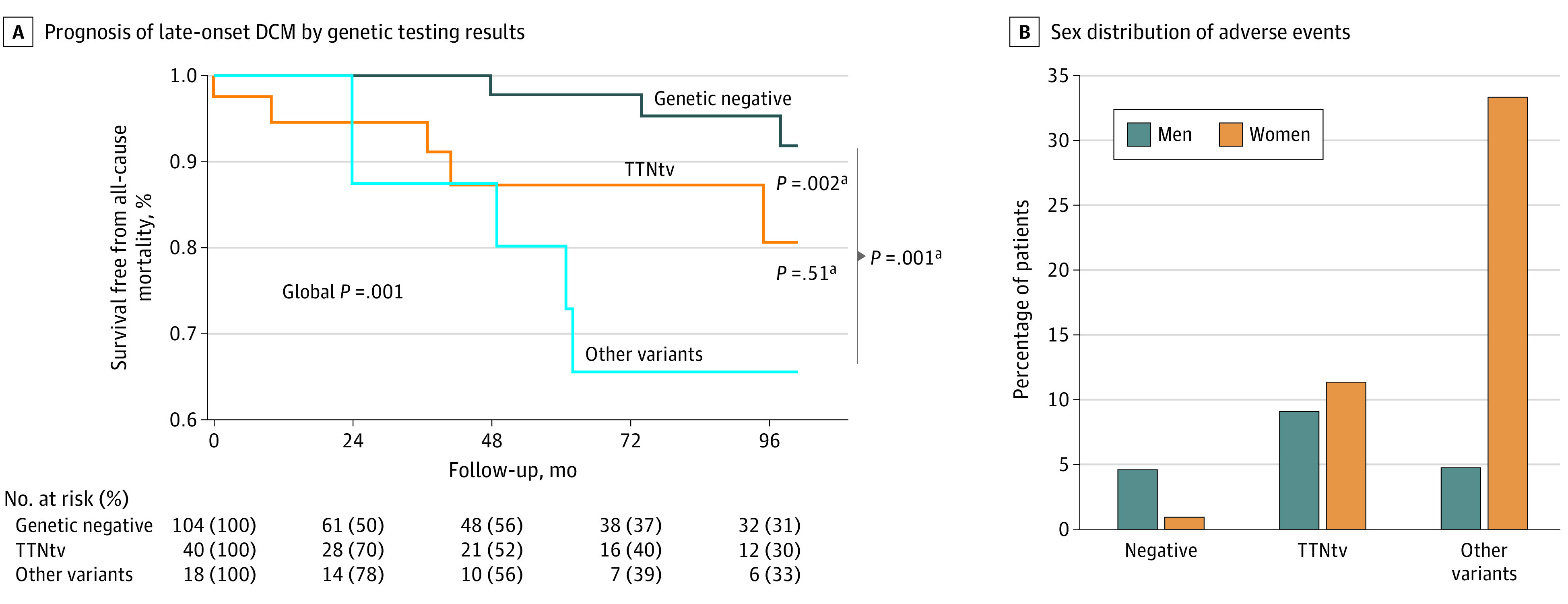
TTNtv indicates titin-truncating variants.
aSignificant after Bonferroni correction.
Subgroup Analyses
Continents
When the population was analyzed according to the continent of origin, patients from the Australian cohort presented more frequently with familial forms compared with those in the US or Europe (63 [97%] vs 32 [63%] vs 25 [37%], respectively). However, clinical characteristics were largely comparable (eTable 4 in the Supplement) and the genetic landscape was consistent across continents (eFigure 2 in the Supplement).
Familial vs Sporadic Cases
All patients presented with diagnosis of DCM and left ventricular dysfunction. Sporadic patients had a lower positive genetic yield compared with familial patients with DCM (15 [23%] vs 51 [43%], respectively), although the distribution of genetic variants was comparable between sporadic and familial cases (eTable 5 in the Supplement). Sporadic DCM presented with more advanced phenotypes compared with familial patients with DCM, but prognosis of familial cases was significantly worse compared with sporadic ones (eFigure 3 in the Supplement).
Probands and Genes
Besides a younger age, clinical characteristics were comparable between probands and relatives (eTable 6 in the Supplement). Clinical characteristics were comparable when considered single genes rather than clusters of genes (eTable 7 in the Supplement).
Sex
The higher prevalence of female sex was confirmed worldwide both in the total population and in the carriers of P/LP variants (eFigure 4 in the Supplement). It is noteworthy that the positive genetic yield and distribution of genetic variants were comparable between male and female individuals (positive genetic yield, 41 female individuals [40%] vs 25 male individuals [31%]; TTNTv, 26 female individuals [25%] vs 20 male individuals [25%]) (eTable 3 in the Supplement). Similarly, long-term prognosis was comparable between sexes (eFigure 2 in the Supplement).
Discussion
Main Findings
To our knowledge, this analysis is the first report to study patients with DCM with onset of the disease 60 years or older and characterizes the genetic landscape and natural history of this population. In the present study, a total of 184 patients from 7 centers were analyzed, making this the largest available analysis of patients with late-onset DCM worldwide. Our results highlight that patients with late-onset DCM should be considered as a distinct subgroup of patients with DCM. This is characterized by a slightly higher prevalence of female sex and a different genetic landscape compared with the current epidemiology of DCM in younger patients, showing a significant larger proportion of TTNtv carriers.3,30 Our findings are consistent across the 7 study cohorts worldwide. Furthermore, we found that patients with positive genetic testing have significantly worse outcomes compared with those with negative genetic testing, regardless of the implicated genes involved (Figure 3A).
This analysis is timely and important owing to the aging population. Thus, it is imperative to characterize patients with later onset of cardiovascular conditions for tailored management.31 Contemporary studies on DCM report that the mean age of presentation is significantly higher compared with the past, with important prognostic considerations.12,13 A personalized medicine approach based on the patient’s genotype has been proposed by international consensus statements designed to tailor medical and device treatment for specific conditions, such as LMNA, FLNC, PLN, and desmosome variations.32,33 Furthermore, specific TTNtv can predict more pronounced left ventricular reverse remodeling during follow-up.5,17,34 This is particularly important also for device indications. However, while the long-term benefit of genetic testing in relatively young people is more established, the diagnostic yield of genetic testing was considered low in older patients35 and, therefore, its cost-effectiveness might be diluted if applied to older people.
From our analysis, 36% of patients with late-onset of DCM had positive genetic testing, which was comparable with the established positive genetic yield in younger patients.3,30 This has important clinical and prognostic implications. Indeed, patients with positive genetic testing in our population had a significantly worse prognosis, and they may have benefited from more tailored management, including device implantation. Although 60% of patients were familial cases, clinical characteristics were comparable between sporadic and familial cases (eTable 5 in the Supplement) and between probands and relatives (eTable 6 in the Supplement). Indication for familial screening might help earlier diagnosis of relatives, improving their outcomes. Therefore, after careful exclusion of environmental factors causing systolic dysfunction, genetic testing might be indicated also in this subgroup of patients with DCM, who hitherto have not been routinely tested.
Genetic Landscape of the Late-Onset DCM
In our population, 25% of patients with late-onset DCM are carriers of TTNtv, which represents more than two-thirds of those with positive genetic testing (46 [69%]). Notably, the prevalence of TTNtv in the late-onset DCM group is almost double that reported in the literature among patients with DCM diagnosed in the third to fifth decade of life.3,30 Interestingly, phenotypic characteristics of patients with late-onset DCM were comparable between TTNtv carriers and carriers of other P/LP variants, suggesting a more homogeneous population than usually found in younger patients with DCM.
The enrichment of TTNtvs in our population supports the known interaction between TTNtv and environmental factors that promotes the onset of a dilated phenotype.7,8,9,29 The later onset of the disease may be secondary to either the delayed penetrance of TTNtv compared with other more aggressive variants or to a need for a multiple-hit effect by accumulation of environmental factors over time, thus preventing this genetic form to present earlier in life. Indeed, while it is known that penetrance of TTNtv increases with age,7 the penetrance is lower earlier in life. Similarly, it has been described that extrinsic factors, such as alcohol or pregnancy, synergistically interact to affect the onset of DCM and are important prognostic factors in those patients.7,8,9 In older patients, multiple environmental factors might have synergistically played alongside the genetic background, thus contributing to the phenotype. However, our population was thoroughly characterized and potential secondary extrinsic causes of the disease were excluded. In the future, detailed analyses are warranted to investigate the role and interplay of multiple environmental factors in promoting the DCM phenotypes in patients with pathogenic variants.
Outcomes
Patients with genetically determined late-onset DCM showed worse long-term outcomes than those with negative genetic testing, while no difference was observed between TTNtv and other classes of variant genes (Figure 3A). The event rate for late-onset patients is higher than those observed in younger cohorts, perhaps driven by competing noncardiovascular causes of death.12,17,22 It is well known that patients with TTNtv have milder phenotype and better prognosis compared with those with more aggressive variants, such as LMNA or FLNC. However, in patients with later onset of the disease, this prognostic difference softens.17,26,27,28,29 It cannot be excluded that patients with more aggressive variations might have experienced an adverse outcome earlier in life, whereas a further insult was necessary to develop the phenotype in patients with TTNtv. This underlines the need to pursue a proper etiological characterization of DCM, including genetic testing, regardless of the age at onset as the pathogenic variants might affect prognosis.
Late-Onset DCM: A Different Disease?
The results of this study suggest that late-onset DCM may represent a specific population with different pathophysiology, characterized by increased prevalence of TTNtv. The role of TTNtv as modifiers of environmental factors rather than a mendelian gene has been previously hypothesized.8,9,17 In our study, aging and its associated multiple comorbidities could be the trigger for the overt DCM phenotype in older patients with TTNtv. Furthermore, the finding of a predominance of the female sex is remarkable because it is the opposite of the sex prevalence in the younger DCM population.2,3,12,21,30 Of note, outcomes of female individuals appear more pronounced in patients with variants other than TTNtv where women had a nonsignificantly higher percentage of deaths (Figure 3B). This finding may indicate the presence of specific sex-related molecular/metabolic mechanisms, including sex hormones, yet to be fully elucidated. Similarly, it is possible that men are usually diagnosed earlier in life owing to a more aggressive disease or specific high-penetrant variants, which develop a phenotype at earlier stage. Furthermore, the similar distribution of adverse events between sexes in TTNtv carriers supports the concept of TTN as a modifier gene. In these patients, the occurrence of an environmental factor or aging itself, acting as a second hit, might have contributed to the development of a clinically overt phenotype. Future analyses are warranted to elucidate the possible sex effect on the late-onset DCM population.
Limitations
Owing to the retrospective nature of the study, the highly select nature of the cohort, and the different indications for genetic testing, some selection biases of the population cannot be entirely excluded. Although the exact date of the onset of the disease is difficult to ascertain because DCM is often characterized by asymptomatic left ventricular dysfunction, we relied on the date of diagnosis when patients were older than 60 years. More granular data on comorbidities and additional clinical and echocardiographic data, as well as data on cardiac magnetic resonance and biomarkers, were not available for each patient and limiting the analysis to patients with the aforementioned data might have introduced further selection bias. Although less than a half of patients were diagnosed because of family screening programs, enrichment for genetic DCM cannot be entirely excluded. Similarly, most patients were of European ancestry. Further studies are required to confirm these data in patients with different race and ethnic groups. Furthermore, the prevalence of female sex in this population might be secondary to less aggressive variations in this cohort and the known worse outcomes of male individuals with DCM. Further studies comparing the characteristics and genetic background of younger patients with those with late-onset DCM might be required to elucidate this issue. Owing to the relatively low event rate in patients with DCM, robust multivariable analyses were not possible. Lastly, follow-up data and specific causes of death were not available for this analysis. Prospective and unbiased cohorts are warranted in the future to confirm these findings and drive tailored and personalized treatment.
Conclusions
Late-onset DCM is a distinct subgroup of patients with nonischemic DCM characterized by genetic testing yield comparable with younger patients, a high prevalence of TTNtv, and a worse prognosis among gene variation carriers. The high prevalence of TTNtv variants may suggest a specific gene-environment interaction, favoring a later onset of the disease. Therefore, genetic testing might be considered in patients with DCM diagnosed when they are older than 60 years.
eTable 1. Contribution of each Center and baseline characteristics
eTable 2. List and classification of rare variants
eTable 3. Baseline characteristics according to sex
eTable 4. Baseline characteristics according to Continent of enrolment
eTable 5. Baseline characteristics according to family history
eTable 6. Baseline characteristics of Probands and Relatives
eTable 7. Baseline characteristics according to gene involved
eFigure 1. Distribution of age at diagnosis
eFigure 2. Genetic Background of the Late-Onset DCM (> 60 years old) across continents
eFigure 3. Long-term prognosis of late-onset DCM in sporadic and familial cases (upper panel) and according to sex (lower panel)
eFigure 4. Sex prevalence across continents in the whole population (upper panels) and in the genetic positive cohort (lower panels)
References
- 1.Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270-276. doi: 10.1093/eurheartj/ehm342 [DOI] [PubMed] [Google Scholar]
- 2.Merlo M, Cannatà A, Gobbo M, Stolfo D, Elliott PM, Sinagra G. Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail. 2018;20(2):228-239. doi: 10.1002/ejhf.1103 [DOI] [PubMed] [Google Scholar]
- 3.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531-547. doi: 10.1038/nrcardio.2013.105 [DOI] [PubMed] [Google Scholar]
- 4.Dal Ferro M, Stolfo D, Altinier A, et al. Association between mutation status and left ventricular reverse remodelling in dilated cardiomyopathy. Heart. 2017;103(21):1704-1710. doi: 10.1136/heartjnl-2016-311017 [DOI] [PubMed] [Google Scholar]
- 5.Moretti M, Merlo M, Barbati G, et al. Prognostic impact of familial screening in dilated cardiomyopathy. Eur J Heart Fail. 2010;12(9):922-927. doi: 10.1093/eurjhf/hfq093 [DOI] [PubMed] [Google Scholar]
- 6.Akinrinade O, Ollila L, Vattulainen S, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J. 2015;36(34):2327-2337. doi: 10.1093/eurheartj/ehv253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ware JS, Cook SA. Role of titin in cardiomyopathy: from DNA variants to patient stratification. Nat Rev Cardiol. 2018;15(4):241-252. doi: 10.1038/nrcardio.2017.190 [DOI] [PubMed] [Google Scholar]
- 8.Ware JS, Amor-Salamanca A, Tayal U, et al. Genetic etiology for alcohol-induced cardiac toxicity. J Am Coll Cardiol. 2018;71(20):2293-2302. doi: 10.1016/j.jacc.2018.03.462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ware JS, Li J, Mazaika E, et al. ; IMAC-2 and IPAC Investigators . Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med. 2016;374(3):233-241. doi: 10.1056/NEJMoa1505517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tayal U, Ware JS, Lakdawala NK, Heymans S, Prasad SK. Understanding the genetics of adult-onset dilated cardiomyopathy: what a clinician needs to know. Eur Heart J. 2021;42(24):2384-2396. doi: 10.1093/eurheartj/ehab286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hershberger RE, Cowan J, Jordan E, Kinnamon DD. The complex and diverse genetic architecture of dilated cardiomyopathy. Circ Res. 2021;128(10):1514-1532. doi: 10.1161/CIRCRESAHA.121.318157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merlo M, Cannatà A, Pio Loco C, et al. Contemporary survival trends and aetiological characterization in non-ischaemic dilated cardiomyopathy. Eur J Heart Fail. 2020;22(7):1111-1121. doi: 10.1002/ejhf.1914 [DOI] [PubMed] [Google Scholar]
- 13.Halliday BP, Gulati A, Ali A, et al. Sex- and age-based differences in the natural history and outcome of dilated cardiomyopathy. Eur J Heart Fail. 2018;20(10):1392-1400. doi: 10.1002/ejhf.1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Integrated care for older people: guidelines on community-level interventions to manage declines in intrinsic capacity. World Health Organization; 2017. [PubMed] [Google Scholar]
- 15.Pinto YM, Elliott PM, Arbustini E, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37(23):1850-1858. doi: 10.1093/eurheartj/ehv727 [DOI] [PubMed] [Google Scholar]
- 16.Cannatà A, Fabris E, Merlo M, et al. Sex differences in the long-term prognosis of dilated cardiomyopathy. Can J Cardiol. 2020;36(1):37-44. doi: 10.1016/j.cjca.2019.05.031 [DOI] [PubMed] [Google Scholar]
- 17.Akhtar MM, Lorenzini M, Cicerchia M, et al. Clinical phenotypes and prognosis of dilated cardiomyopathy caused by truncating variants in the TTN gene. Circ Heart Fail. 2020;13(10):e006832. doi: 10.1161/CIRCHEARTFAILURE.119.006832 [DOI] [PubMed] [Google Scholar]
- 18.Mestroni L, Maisch B, McKenna WJ, et al. ; Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy . Guidelines for the study of familial dilated cardiomyopathies. Eur Heart J. 1999;20(2):93-102. doi: 10.1053/euhj.1998.1145 [DOI] [PubMed] [Google Scholar]
- 19.Ponikowski P, Voors AA, Anker SD, et al. ; ESC Scientific Document Group . 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37(27):2129-2200. doi: 10.1093/eurheartj/ehw128 [DOI] [PubMed] [Google Scholar]
- 20.Lang RM, Badano LP, Mor-Avi V, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2015;28(1):1-39.e14. doi: 10.1016/j.echo.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 21.Mazzarotto F, Tayal U, Buchan RJ, et al. Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation. 2020;141(5):387-398. doi: 10.1161/CIRCULATIONAHA.119.037661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gigli M, Merlo M, Graw SL, et al. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2019;74(11):1480-1490. doi: 10.1016/j.jacc.2019.06.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akinrinade O, Heliö T, Lekanne Deprez RH, et al. Relevance of titin missense and non-frameshifting insertions/deletions variants in dilated cardiomyopathy. Sci Rep. 2019;9(1):4093. doi: 10.1038/s41598-019-39911-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Begay RL, Graw S, Sinagra G, et al. ; Familial Cardiomyopathy Registry . Role of titin missense variants in dilated cardiomyopathy. J Am Heart Assoc. 2015;4(11):4. doi: 10.1161/JAHA.115.002645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasselberg NE, Haland TF, Saberniak J, et al. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur Heart J. 2018;39(10):853-860. doi: 10.1093/eurheartj/ehx596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parikh VN, Caleshu C, Reuter C, et al. Regional variation in RBM20 causes a highly penetrant arrhythmogenic cardiomyopathy. Circ Heart Fail. 2019;12(3):e005371. doi: 10.1161/CIRCHEARTFAILURE.118.005371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Begay RL, Graw SL, Sinagra G, et al. Filamin C truncation mutations are associated with arrhythmogenic dilated cardiomyopathy and changes in the cell-cell adhesion structures. JACC Clin Electrophysiol. 2018;4(4):504-514. doi: 10.1016/j.jacep.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619-628. doi: 10.1056/NEJMoa1110186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haas J, Frese KS, Peil B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36(18):1123-35a. doi: 10.1093/eurheartj/ehu301 [DOI] [PubMed] [Google Scholar]
- 31.Cannatà A, Camparini L, Sinagra G, Giacca M, Loffredo FS. Pathways for salvage and protection of the heart under stress: novel routes for cardiac rejuvenation. Cardiovasc Res. 2016;111(2):142-153. doi: 10.1093/cvr/cvw106 [DOI] [PubMed] [Google Scholar]
- 32.Felker GM, Thompson RE, Hare JM, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000;342(15):1077-1084. doi: 10.1056/NEJM200004133421502 [DOI] [PubMed] [Google Scholar]
- 33.Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16(11):e301-e372. doi: 10.1016/j.hrthm.2019.05.007 [DOI] [PubMed] [Google Scholar]
- 34.Asselbergs FW, Sammani A, Elliott P, et al. ; Cardiomyopathy & Myocarditis Registry Investigators Group . Differences between familial and sporadic dilated cardiomyopathy: ESC EORP cardiomyopathy & myocarditis registry. ESC Heart Fail. 2021;8(1):95-105. doi: 10.1002/ehf2.13100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lakdawala NK, Thune JJ, Colan SD, et al. Subtle abnormalities in contractile function are an early manifestation of sarcomere mutations in dilated cardiomyopathy. Circ Cardiovasc Genet. 2012;5(5):503-510. doi: 10.1161/CIRCGENETICS.112.962761 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. Contribution of each Center and baseline characteristics
eTable 2. List and classification of rare variants
eTable 3. Baseline characteristics according to sex
eTable 4. Baseline characteristics according to Continent of enrolment
eTable 5. Baseline characteristics according to family history
eTable 6. Baseline characteristics of Probands and Relatives
eTable 7. Baseline characteristics according to gene involved
eFigure 1. Distribution of age at diagnosis
eFigure 2. Genetic Background of the Late-Onset DCM (> 60 years old) across continents
eFigure 3. Long-term prognosis of late-onset DCM in sporadic and familial cases (upper panel) and according to sex (lower panel)
eFigure 4. Sex prevalence across continents in the whole population (upper panels) and in the genetic positive cohort (lower panels)