

Abstract
In aqueous solution, many biochemical reaction pathways involve reaction of an aldehyde with an amine, which progresses through generally unstable hydrated and dehydrated Schiff base intermediates that often are unobservable by conventional NMR. There are 4 states in the relevant equilibrium: (1) gem-diol ↔ (2) aldehyde ↔ (3) hemiaminal ↔ (4) Schiff base. We show that for the reaction between protein amino groups and DOPAL, a highly toxic metabolite of dopamine, the 1H resonances of both the hemiaminal and the dehydrated Schiff base can be observed by CEST NMR, even when their populations fall below 0.1 %. CEST NMR reveals the quantitative exchange kinetics between reactants and Schiff base intermediates, explaining why the Schiff base NMR signals are rarely observed. In full agreement with results previously reported for the Parkinson’s disease implicated α-synuclein, we observe that reactivity of DOPAL with Nα-amino groups is far greater than with lysine Nε-amines. In the presence of O2, the Schiff base DOPAL-peptide intermediates rapidly react with remaining free DOPAL to irreversibly form dicatechol pyrrole adducts.
Keywords: Amides, Amines, CEST, DOPAL, Reaction intermediates, Schiff base, Synuclein
Graphical Abstract
Modified CEST NMR methodology allowed detection of highly transient, very low populated Schiff base intermediates, not observable by conventional NMR, but key to product formation.
As reported by Hugo Schiff in 1864,[1] nucleophilic attack on a carbonyl carbon atom by an amine can lead to a meta-stable hemiaminal (also known as carbinolamine) intermediate, whose subsequent dehydration generates a Schiff base (Figure 1A). Schiff base formation is reversible, and the excess water present in physiological conditions can skew the equilibrium towards the starting materials due to the dehydration nature of the reaction. Therefore, their presence and biological importance is commonly established by cyanoborohydride reduction to stable amines. The transient nature of a Schiff base intermediate makes this structure commonly not directly observable by 1H NMR spectroscopy, unless stabilized by intramolecular hydrogen bonding or locked in by covalent ring formation.[2–3]
Figure 1.
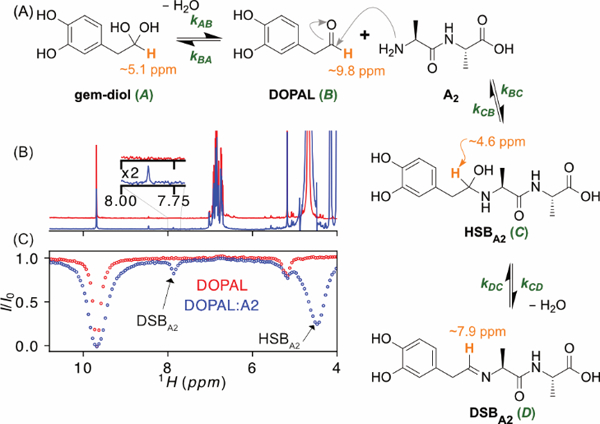
Formation of DOPAL-dialanine Schiff base adduct. (A) Reaction scheme. The 1H used as a reporter for the four states is shown in orange. (B) Superimposed 1H NMR spectra of 2 mM DOPAL in the absence (red) and presence (blue) of 10 mM A2 measured at 600 MHz, in 10 mM sodium phosphate D2O buffer, pD 7.9, and T = 35 °C. (C) CEST attenuation profiles of the DOPAL aldehyde signal, obtained using B1 = 30 Hz, τcest = 5 s, on the samples with (blue) and without (red) A2, stepping the CEST frequency in 30 Hz increments from 4 to 11 ppm. Intensity dips assigned to the hydrated (carbinolamine) and dehydrated Schiff base positions are marked.
We previously showed that the primary, highly toxic dopamine metabolite, 3,4-dihydroxyphenylacetaldehyde or DOPAL, reacts with lysine sidechain amino groups to form dicatechol pyrrole lysine (DCPL) adducts,[4] that can further react to form covalent intermolecular isoindole linkages.[5] This pathway is at the core of the “catechol-aldehyde hypothesis”, which provides a plausible link between DOPAL and the presence of α-synuclein-containing aggregates in dopaminergic neurons of Parkinson’s disease patients.[6–9] The first step in the reaction pathway of the DCPL adduct formation involves a Schiff base linkage between the DOPAL aldehyde and the Lys ε-amino group.[9–12] Mass spectrometry confirmed the presence of DOPAL Schiff base adducts, but typically involved trapping by cyanoborohydride, and made it difficult to assess their populations and lifetimes under physiological conditions.[4, 11–12] No direct NMR evidence has been reported for Schiff base DOPAL-Lysine reaction intermediates. The absence of a 1H NMR signal for the Schiff base intermediate could result from its low population, or from line broadening caused by a chemical exchange process, e.g. between the hydrated and dehydrated Schiff base, on a micro- to millisecond time scale. Here, we set out to resolve the causes for the absence of the Schiff base 1H signals in conventional NMR spectra but show that they can be characterized quantitatively by chemical-exchange-saturation-transfer (CEST) NMR.
CEST NMR has been used extensively to characterize low populations of sparsely populated “excited states” in proteins in dynamic equilibrium with their major “ground state”,[13–15] and recently was shown to also be particularly powerful for identifying elusive reaction intermediates in organocatalysis, as well as for identifying predicted but previously unobserved species in silicon hydride anion clusters.[16–17] CEST is based on the original saturation transfer experiments by Forsén and Hoffman[18] and the method is widely used to encode chemical information in magnetic resonance imaging (MRI).[19–20] CEST NMR is carried out by systematically stepping the frequency of a probing radiofrequency (RF) field. We show that, once such measurements have identified the location of a hidden resonance, additionally stepping the amplitude and duration of the RF field at such a position makes quantitative analysis of the kinetic process highly robust.
In this study, we report the NMR observation of the carbinolamine and Schiff base 1H resonances in the reaction between DOPAL and both the N-acetyl-Lys sidechain ε-amine as well as the N-terminal α-amine of dialanine (A2). Anaerobic conditions were used to prevent progression of the reaction to form dicatechol pyrrole products. Populations of, in particular, the Lys Cε Schiff base adduct relative to the summed populations of the dynamic equilibrium between DOPAL in its aldehyde and hydrated, gem-diol (short for geminal diol) forms remains very low (<0.1%) under physiological pH and temperature conditions. Higher population of the Cα amine Schiff base adduct (ca 1%) is consistent with its prior observation by mass spectrometry.[11, 21] Varying the RF field strength and duration of the CEST irradiation provides direct access to the forward and reverse reaction rates, thereby yielding Arrhenius activation energies from measurement of their temperature dependencies.
CEST-based NMR analysis of Schiff base intermediates is investigated for the reaction of DOPAL with two model systems: the dipeptide L-Alanyl-L-alanine (A2), and Nα-acetylated lysine (AcL) as a mimic for the 15 nucleophilic lysine residues in α-synuclein. As the α-methylene protons of DOPAL slowly exchange with deuterons of the D2O solvent, and to suppress differential J-coupling and relaxation behavior, they were fully exchanged (overnight incubation) with deuterium prior to all measurements. We first focus on the DOPAL-A2 reaction. The DOPAL aldehyde is in equilibrium with its hydrated, gem-diol form, and the equilibrium then involves four states (Figure 1A): the gem-diol, DOPAL, the hydrated Schiff base (HSBA2) formed upon reaction of DOPAL with A2, and the dehydrated Schiff base (DSBA2).
With kAB and kBA not impacted by the presence of A2, we first characterize the equilibrium between the aldehyde and gem-diol forms of DOPAL (Figure 1A) by 1H-CEST. Typically, CEST is carried out by recording a set of NMR spectra while stepwise changing the frequency of a weak, saturating RF-field of strength B1 (applied for a duration τcest) across a region where the low-intensity peak of the minor state is expected to resonate. Chemical exchange between major and minor states during the irradiation period then will attenuate intensity of the major site if the frequency of the CEST RF-field falls sufficiently close to that of the minor species to saturate its z magnetization. The method is equally applicable when directly observing the minor state, as applies for observation of the DOPAL aldehyde proton, which exists in a 32:68 equilibrium with its gem-diol form (5.17 ppm; Figure 1B). The width and amplitude of the aldehyde intensity dip, observed when the RF field is applied in the vicinity of the gem-diol resonance (red trace in Figure 1C), allows extraction of the forward and reverse rates of exchange, and of the chemical shifts.[13] As we show here, if the chemical shifts are known a priori, a robust and fast method to obtain populations and rates fixes the CEST RF frequency at that of the minor species, while stepwise varying B1 or τcest. For CEST studies of small molecules, which can have very long T1 relaxation times under O2-free conditions, this can become particularly important.
With the saturating field fixed at the gem-diol resonance, attenuation of the aldehyde as a function of τcest (Figure S1) is analyzed by fitting observed intensities to the Bloch-McConnell equations for a system of a single spin exchanging between two states (A: gem-diol; B: aldehyde). With the populations, pA and pB accurately known at each temperature from integration of their resonances in the 1D NMR spectrum, the exchange rate is then the only adjustable parameter (see SI). The individual rate constants for the gem-diol equilibrium are derived from using kAB = pBkex, kBA = pAkex (Figure S1). Measurements at temperatures, T, ranging from 20 to 45 °C then yield the activation energies from the Arrhenius equation:
| [1] |
where is the corresponding rate A is the fitted pre-exponential factor, and R is the universal gas constant. Activation energies are = 18.7 ± 0.2 kcal/mol and = 11.9± 0.1 kcal.mol−1 for the forward and reverse directions, respectively.
Next, we investigate Schiff base formation between DOPAL and A2 (Scheme 1; Figure 1A) for a sample containing 2 mM DOPAL and 10 mM A2. A very weak resonance assigned to the Schiff base (DSBA2) is present in the 1H NMR spectrum (blue inset, Figure 1B, and Figure S2), but the HSBA2 resonance is not observed (Figure 1B and Figure S2). This latter resonance is revealed by the 1H-CEST profile, which shows a deep, broad intensity dip slightly upfield of the residual HDO frequency (Figure 1C and Figure S2B). To investigate the kinetics, 1H-CEST data were recorded with the saturation B1 field applied at the now known HSBA2 and DSBA2 frequencies, while observing the intensity of the aldehyde resonance (Figure 2). Saturation profiles are shown as a function of saturation time (0.1 s – 15 s; Figure 2 A–B), B1 RF field strength (3 Hz – 100 Hz; Figure 2 C–D) and offset around the HSBA2 resonance frequency (Figure 2 E), followed by analysis using a four-state-exchange Bloch-McConnell equation (SI eq S5) to describe Scheme 1. Details of the fitting procedure are presented in the Supporting Information.
Figure 2. Schiff base formation between DOPAL and dialanine.
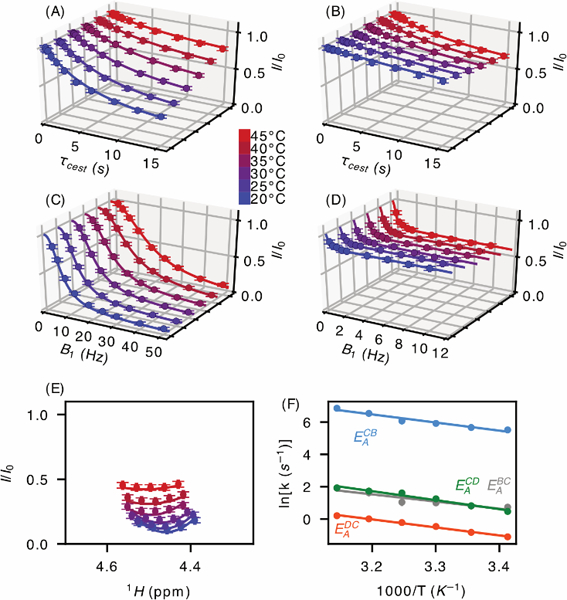
Saturation profiles of the aldehyde resonance in the presence of 10 mM A2 upon irradiation of the hydrated (A, C) and dehydrated (B, D) Schiff base resonance for temperatures ranging from 20 to 45 ºC. (A) & (B) were recorded with B1 = 10 Hz and τcest ranging from 0.1 to 15 s. (C) & (D) were recorded with τcest = 30 s and B1 from 3 to 100 Hz. (E) was recorded with B1 = 30 Hz and τcest = 5 s, for 5 offset frequencies around the HSBA2 resonance. (F) Activation energies calculated for the DOPAL/Schiff base reaction (EABC = 8.8 ± 1.6 kcal/mol and EACB = 10.0 ± 1.1 kcal/mol) and the hemiaminal/Schiff base reaction (EACD = 11.0 ± 0.9 kcal/mol and EADC = 9.8 ± 0.4 kcal/mol).
With the gem-diol/aldehyde interconversion rates known from the above measurements on free DOPAL, the direct observation of the DSBA2 resonance (blue spectrum in Figure 1B) allows us to fix the ratio pD/pB ≈ 4% at all temperatures. For the fitting procedure, the resonance frequencies of the gem-diol, aldehyde, and DSBA2 proton were obtained from the regular 1D NMR spectrum, while the chemical shift of HSBA2 at each temperature was rapidly and precisely determined using the 1H-CEST approach, applied over a very limited range of frequencies around the approximately known position (Figure S2B). Subsequently, only three variables (kBC, kCB, kCD) require fitting at each temperature (Figure S3) and their temperature dependence yields the activation energies (Figure 2 F).
The above reaction scheme applies to the solution of DOPAL and dialanine under anaerobic conditions. If no special precautions are taken to remove O2, the Schiff base adduct will react with free DOPAL to form a relatively stable dicatechol pyrrole dialanine (DCPdA) product (SI Figure S4). Formation of its pyrrole ring presumably involves oxidization steps and intermediates similar to those in the Paal-Knorr mechanism,[22–23] fully analogous to formation of dicatechol pyrrole lysine, reported previously.[4] Mass spectrometry and HMBC NMR data collected in methanol, where DCPdA is stable for days, were used to validate its structure (SI Figure S5, S6).
Next, we use CEST NMR to investigate the reaction with lysine ε-amino groups, using Nα-acetylated lysine (AcL) as a mimic for the 15 Lys residues in α-synuclein, whose reaction with DOPAL has been implicated in Parkinson’s disease.[6–9] (Figure 3A). In contrast to the DOPAL reaction with A2, neither the Schiff base nor its hydrated hemiaminal form (DSBAcL and HSBAcL, respectively) are directly observable in the DOPAL:AcL NMR spectrum (Figure 3B). However, the HSBAcL yields a strong 1H-CEST signal at 20ºC, slightly upfield but partly overlapping with the gem-diol 1H resonance (Figure 3C). The effect of saturating the DSBAcL resonance at 7.82 ppm also was observed by 1H-CEST but was much weaker and only visible above room temperature. These 1H-CEST data were recorded with a weaker saturating field of 6 Hz to minimize direct saturation of the aldehyde resonance, and narrower frequency windows centered around the resonance frequency of each state were used after a preliminary coarser CEST scan of the full spectrum. Such profiles were recorded with 30 Hz RF field and τCEST = 5 s (HSBAcL) and 6 Hz RF field and τCEST = 15 s (DSBAcL) to identify the approximate positions. The HSBAcL CEST signal progressively broadens with temperature and was no longer observable above 35 ºC (Figure S7C). With δHSBAcL and δDSBAcL known, measurement of the 1H CEST effect as a function of B1 and τcest was used to obtain quantitative rates (Figure S7). Analysis again involved a four-state exchange system, with state A the gem-diol, B the directly observed aldehyde, C the HSBAcL and D the DSBAcL. In contrast to the DOPAL-A2 reaction, the ratio could not be determined by inspection of the conventional 1H NMR spectrum, therefore requiring fitting of four exchange rates: kBC, kCB, kCD, and kDC (Figure 3), with results summarized in Table S2.
Figure 3. Schiff base formation between DOPAL and Nα-Acetyl Lysine.
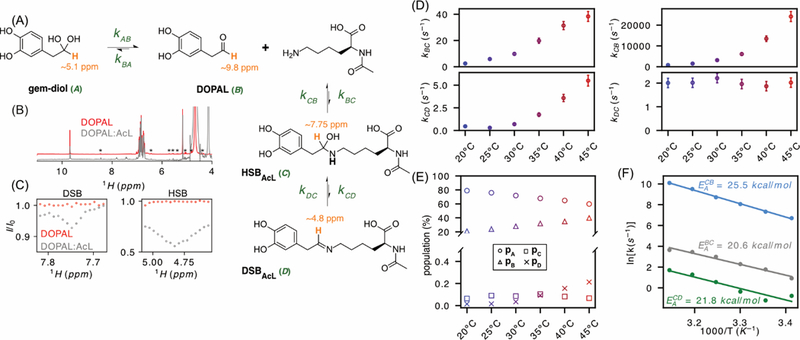
(A) Reaction scheme. (B) 1H spectra of 2 mM DOPAL in the absence (red) and presence (gray) of 30 mM AcL. Asterisks correspond to impurities in our starting material. (C) Zoomed region of 1H-CEST on DSB (Left) measured with B1 = 6 Hz, τCEST = 15 s at 40ºC and HSB (Right) measured with B1 = 30 Hz, τCEST = 5 s at 20 °C. Both measurements were carried out on a 2mM DOPAL sample in absence (red) and presence (gray) of 30 mM AcL. (D) Rate constants fitted as a function of temperature: kBC (top left), kCB (top right), kCD (top left), kDC (top left). (E) Populations derived from the rate constants as a function of temperature: pA (○), pB (△), pC (□), pD (×). Uncertainties in these populations are estimated at ±0.3% (pA and pB) and ±0.02% (pC and pD). (F) Activation energies calculated for the DOPAL/Schiff base reaction (EABC = 20.6 ± 1.6 kcal/mol and EACB = 25.5 ± 0.5 kcal/mol) and the hemiaminal/Schiff base reaction (EACD = 21.8 ± 3 kcal/mol). kDC has no significant temperature dependence, and EADC was smaller than 3 kcal/mol.
Equilibrium populations confirm quantitatively that α-amines are much less favored to forming Schiff base adducts than Lys ε-amino groups. At near-physiological pH and temperature, the apparent dissociation constant for Schiff base formation with A2 (Kd ≈ 0.5 M) is high, but much lower than for the Lys ε-amine (Kd ≈ 10 M). We attribute this difference to the higher electronegativity of Cα compared to Lys Cε, which also is responsible for the much lower pKa value of α- compared to ε-amines.
Our work shows that 1H-CEST NMR is remarkably sensitive to detection of even very small (≤0.1%) fractions of transient reaction intermediates, and that adaptation of this CEST experiment can provide quantitative rate information over a very wide kinetic range. The more than 10-fold lower population of Schiff base adduct with Lys ε-amino groups compared to α-amines explains why the former is difficult to observe experimentally. Nevertheless, even the very small, transient population of Schiff base adduct with Lys ε-amines is sufficiently large to be trapped by reaction with free DOPAL, then forming the previously reported DCPL, capable of protein cross linking.
Experimental Section
Sample Preparation.
3,4-Dihydroxyphenylacetaldehyde (DOPAL) (VDM Biochemicals) was dissolved in deuterated methanol (Sigma-Aldrich) at a concentration of 300 mM. The prepared DOPAL stocks were aliquoted and stored at −80 °C. For NMR measurements, the DOPAL stock was then diluted to a concentration of 2 mM in 10 mM sodium phosphate dissolved in D2O, pD 7.9. The sample was deoxygenated by sparging nitrogen gas through the sample for 3 h. The sample was subsequently equilibrated for 8 h at 37 °C to allow for complete exchange of the DOPAL methylene protons with deuterons, prior to recording of NMR spectra. For the reactions of DOPAL with A2 and AcL, either 10 mM A2 (Sigma-Aldrich) or 30 mM Nα-acetyl lysine (Sigma-Aldrich) was added to the deuterium exchanged DOPAL solution, which was again deoxygenated by sparging with nitrogen before NMR data acquisition.
NMR Spectroscopy.
All NMR measurements were carried out at 600 MHz on a Bruker Avance spectrometer equipped with a cryogenic probe and pulsed field gradients. Full details regarding data analysis are presented in the Supplementary Information.
Supplementary Material
Acknowledgements
We thank J. L. Baber and J. Ying for technical support, and J. M. Courtney, T. Kakeshpour, A. J. Robertson, D. A. Torchia and V. Tugarinov for valuable discussions. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Schiff H, Annalen der Chemie und Pharmacie 1864, 131, 118–119. [Google Scholar]
- [2].Chan-Huot M, Sharif S, Tolstoy PM, Toney MD, Limbach HH, Biochemistry 2010, 49, 10818–10830. [DOI] [PubMed] [Google Scholar]
- [3].Sharif S, Denisov GS, Toney MD, Limbach HH, J. Am. Chem. Soc. 2006, 128, 3375–3387. [DOI] [PubMed] [Google Scholar]
- [4].Werner-Allen JW, DuMond JF, Levine RL, Bax A, Angew. Chem. Int. Ed. 2016, 55, 7374–7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Werner-Allen JW, Monti S, DuMond JF, Levine RL, Bax A, Biochemistry 2018, 57, 1462–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Blaschko H, Pharmacological Reviews 1952, 4, 415–458. [PubMed] [Google Scholar]
- [7].Panneton WM, Kumar VB, Gan Q, Burke WJ, Galvin JE, PLoS One 2010, 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Goldstein DS, Kopin IJ, Sharabi Y, Pharmacol. Ther. 2014, 144, 268–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Monzani E, Nicolis S, Dell’Acqua S, Capucciati A, Bacchella C, Zucca FA, Mosharov EV, Sulzer D, Zecca L, Casella L, Angew. Chem. Int. Ed. 2019, 58, 6512–6527. [DOI] [PubMed] [Google Scholar]
- [10].Plotegher N, Berti G, Ferrari E, Tessari I, Zanetti M, Lunelli L, Greggio E, Bisaglia M, Veronesi M, Girotto S, Dalla Serra M, Perego C, Casella L, Bubacco L, Scientific Reports 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rees JN, Florang VR, Eckert LL, Doorn JA, Chem. Res. Toxicol. 2009, 22, 1256–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Follmer C, Coelho-Cerqueira E, Yatabe-Franco DY, Araujo GDT, Pinheiro AS, Domont GB, Eliezer D, J. Biol. Chem. 2015, 290, 27660–27679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vallurupalli P, Bouvignies G, Kay LE, J. Am. Chem. Soc. 2012, 134, 8148–8161. [DOI] [PubMed] [Google Scholar]
- [14].Gladkova C, Schubert AF, Wagstaff JL, Pruneda JN, Freund SMV, Komander D, EMBO J. 2017, 36, 3555–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hansen AL, Kay LE, Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E1705–E1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lorenz C, Hastreiter F, Hioe J, Lokesh N, Gartner S, Korber N, Gschwind RM, Angew. Chem. Int. Ed. 2018, 57, 12956–12960. [DOI] [PubMed] [Google Scholar]
- [17].Lokesh N, Seegerer A, Hioe J, Gschwind RM, J. Am. Chem. Soc. 2018, 140, 1855–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Forsen S, Hoffman RA, J. Chem. Phys. 1963, 39, 2892–2901. [Google Scholar]
- [19].Ward KM, Aletras AH, Balaban RS, J. Magn. Reson. 2000, 143, 79–87. [DOI] [PubMed] [Google Scholar]
- [20].van Zijl PCM, Yadav NN, Magn. Reson. Med. 2011, 65, 927–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jinsmaa Y, Florang VR, Rees JN, Mexas LM, Eckert LL, Allen EMG, Anderson DG, Doorn JA, Chem.-Biol. Interact. 2011, 192, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Amarnath V, Anthony DC, Amarnath K, Valentine WM, Wetterau LA, Graham DG, J. Org. Chem. 1991, 56, 6924–6931. [Google Scholar]
- [23].Kornienko A, La Clair JJ, Nat. Prod. Rep. 2017, 34, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.