

Abstract
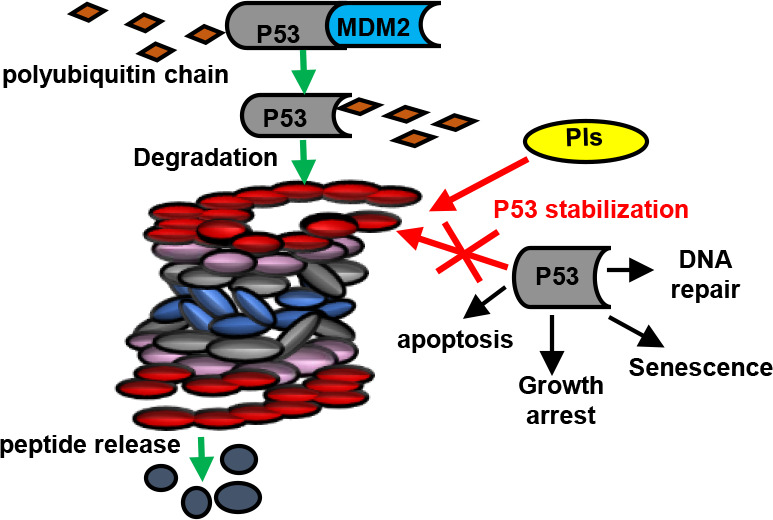
Protein p53 is degraded by the 26S proteasome, a protein complex that breaks down cellular proteins. Degradation begins with activation of the protein ubiquitin (Ub) by the ubiquitin-activating E1 enzymes, ubiquitin-conjugating E2 enzymes, and ubiquitin E3 ligases, linking Ub or the polyubiquitin chain to p53 and marking it for degradation by the 26S proteasome. E3 ubiquitin ligases participate in this process and regulate p53 stability. There are compounds that inhibit the 26S proteasome and interfere at the p53 level, and some of these inhibitors are used to treat cancer and other diseases and can stabilize tumor suppressor proteins through the p53 pathway. This review discusses how the ubiquitin–proteasome system, p53, and these compounds are related.
1. Ubiquitin–Proteasome System
The 26S proteasome is a macromolecular complex comprising two particles: the regulatory particle (19S) and the catalytic particle (20S), formed by multiple subunits. This complex degrades 80% of the cellular proteins that participate in several signaling pathways, cell cycle regulation, transcription, apoptosis, signal transduction, stress response, and antigen presentation.1
The 19S particle recognizes ubiquitinated substrates through some subunits (Rpn1, Rpn10, and Rpn13), processes them, and sends them to the catalytic site for degradation.2 This particle consists of six ATPase subunits (Rpt1–Rpt6) and 13 non-ATPase subunits (Rpn1–3, Rpn5–Rpn13, and Rpn15), which form two subcomplexes: the “lid” and the “base” proteasomes (Figure 1).2
Figure 1.

UPS and deubiquitinating enzymes. (1) Ubiquitin (Ub)-activating E1 enzyme transfers ubiquitin to the ubiquitin-conjugating E2 enzyme, which, together with ubiquitin E3 ligase, transfers Ub to the target protein, forming the polyubiquitinated substrate. (2) 19S Ub receptors (Rpn1, Rpn10, and Rpn13) recognize PS deubiquitinating enzymes (Rpn11, UCHL5, and USP14 deubiquitinate PS) and degrade it inside 20S, and the β1, β2, and β5 subunits perform the catalysis (blue). (3) Polyubiquitin chain is cleaved by the deubiquinating enzymes (DUBs), which keep the Ub pool inside the cell. Six ATPase subunits (Rpt1–Rpt6) and non-ATPase subunits are in purple and red, respectively.
The 20S particle is formed by α and β rings, aligned in a sequence of α1−α7 and β1−β7 rings. The β subunits are in the center of the subcomplex, and the β1, β2, and β5 subunits have catalytic activities, namely, caspase-like (CL), trypsin-like (TL), and chymotrypsin-like (CT-L) activities. The α subunits are the entry “gate” for proteins that will be degraded, and these subunits regulate substrate entry into the catalytic chamber1 (Figure 1).
Ubiquitin (Ub) is a protein formed by 76 amino acids. The 26S proteasome requires Ub-dependent post-translational modification to recognize proteins marked for proteolysis; this is called the ubiquitin–proteasome system (UPS). The consecutive action of three types of enzymes—ubiquitin-activating E1 enzymes, ubiquitin-conjugating E2 enzymes, and ubiquitin E3 ligases—promotes Ub conjugation1 (Figure 1). Ubiquitin E3 ligases are classified into three families, according to their catalytic domains: HECT, which has a cysteine active site and forms an intermediate thioester bond with Ub, transferring it to the substrate; RING, which has two conserved residues, cysteine and histidine, that bind to Zn2+ and which positions Ub and transfers it directly to the substrate; and RING-between-RING (RBR), which uses the two mechanisms mentioned above.3
Ub binding to the substrate can be mono- or polyubiquitinated; in the latter case, lysine residues on different Ub are linked to form the polyubiquitin chain. The Ct Gly 76 group of the first Ub of the chain binds to the lysine residue of the substrate to form an isopeptide bond.2
Ubiquitination is a reversible post-translational modification. Some enzymes cleave Ub that are linked together or that are linked to protein substrates. These enzymes are called deubiquitinating enzymes (DUBs), and they maintain the intracellular Ub “pool” by recycling Ub4 (Figure 1).
2. p53 and Its Degradation by the 26S Proteasome
Stress-related alterations in cellular homeostasis activate protein p53, which suppresses tumors that regulate apoptosis, senescence, and cell cycle progression.5 In 50% of cancers, cells have mutations in the p53 gene due to metabolic stress, so p53 can no longer inhibit tumor cell growth;6 in other words, p53 is not active anymore, so it cannot repress the transcription of target tumoral genes.5 Mutations in the p53 gene take place at chromosome 17 and are frequent in cancer cells. Mutations in the locus include somatic missense mutations in exons 5–8, somatic nonsense mutations, and absence of p53 expression due to alterations in epigenetic molecular mechanisms. In this context, mutations in the p53 gene in cancer cells cause mutations and aberrations in the p53 downstream effectors, alter the p53 activation pathways, and interfere with mouse double-minute 2 (MDM2) expression.5
A total of 600 E3 ligases are known to date.7 Some of these E3 ligases interact with p53 and render it stable, while others do not. E6AP was the first E3 ligase to be discovered to interact with p53. Together with protein HPV16E and under certain physiological conditions in patients infected with high-risk HPV, E6AP degrades p53 with the aid of the 26S proteasome.8
MDM2 or human double-minute 2 (HDM2), another negative p53 regulator, was discovered8 in normal cells and induces cellular homeostasis. However, under cellular stress caused by different conditions (alterations in metabolism and cell cycle caused by diseases), there is an imbalance in cells.6 Depending on the MDM2 levels in the cell, p53 can have different post-translational modifications (PTM). At low MDM2 levels, p53 is monoubiquitinated and translocated from the nucleus to the cytoplasm to induce the apoptotic pathway.6
High MDM2 levels are observed in various types of cancer, including osteosarcoma and esophageal carcinoma. Such high MDM2 levels induce p53 polyubiquitination and degradation, inhibiting the p53 tumor suppressor activity.9
As mentioned above, p53 stability is achieved through interaction between p53 and MDM2. p53 turnover is regulated mainly by MDM2. High p53 protein levels transcriptionally activate the MDM2 gene through p53 interaction with the p53-responsive element located in the MDM2 intron gene.9 This raises MDM2 levels, consequently polyubiquitinating p53. Next, p53 is degraded by the 26S proteasome, and p53 levels decrease. In this way, p53 levels remain in equilibrium in normal cells. This negative feedback cycle is regulated mainly by proteins MDM4, HAUSP/USP7, ARF, Pirh2, MSL, and COP1; the latter three are E3 ligases.9
However, MDM2 destabilization can also occur and consequently promote p53 stabilization.8 An E3 RNF12 ligase complexes with MDM2, which is degraded by 26S proteasome; in this way, RNF12 stabilizes p53 indirectly. On the contrary, E3 ligase RNF31, discovered in experiments carried out with breast cancer tissues, stabilizes MDM2, which was confirmed by RNF31 depletion in mammalian cells (Figure 2).8
Figure 2.

p53 stability. Schematic representation of how MDM2 levels in normal cells affect p53 stability and how interaction between MDM2 and the E3 ligase RNF12 also stabilizes p53.
MDM2 forms complexes with other E4 ligases, interfering in p53 stability. MDM4 is an E4 ligase6 that kinetically enhances p53 polyubiquitination by MDM2. MDM4 recognizes monoubiquitinated p53 and interacts with an E2 ligase.9 As in the case of MDM2-knockout mice, MDM4-knockout mice show embryogenic lethality, and MDM4 and MDM2 form a complex during embryogenesis. The complex guides conjugation of multiple ubiquitin units in the lysine residue of the substrate. Therefore, MDM4 assists the polyubiquitination function exerted by MDM2 in p53, inhibiting the p53-mediated transcriptional transactivation.9 Another function of MDM4 is to prevent MDM2 autoubiquitination. Together, these processes negatively regulate p53 activity.6 MDM4 is overexpressed in tumors of the stomach, lung, breast, retina, and colon.9
UBE4B is an E4 ligase that interacts with p53, leading to p53 polyubiquitination. In vivo studies have demonstrated that UBE4B gene deletion causes embryo death.10 In addition to E3 ligases, subunit Rpn10, linked to the proteasome, increases the ratio between poly- and monoubiquitinated p53, increasing the interaction between p53 and MDM2.10
In contrast to MDM2, MSL2 ubiquitinates p53 lysine residues and does not degrade p53; in fact, the MSL2 interaction with p53 increases p53 levels in the cytoplasm. This interaction exposes the p53 amino acid residues, translocating p53 from the nucleus to the cytoplasm, followed by MSL2 release by p53 (Figure 2).9
Proteins COP1 and Pirh2 inhibit p53 independently of MDM2. Both proteins may lead to p53 breakdown by UPS.9 COP1 has significant functions as an oncogene under certain conditions or as a tumor suppressor under others.6 COP1 forms a complex with other proteins, degrades such proteins,7 and is essential for regulating DNA repair, cell proliferation, and apoptosis. COP1 is regulated by its gene, with a promoter controlled by p53 levels in cells. COP1 is highly expressed in adenocarcinomas and breast cancers9 and is present in the nucleus and cytoplasm.6
In 2004, transfection and binding assays in U20S cells (human female bone; source disease, bone osteosarcoma) indicated that p53 is a COP1 ligand. Subsequently, through miR-944 assays in HCT116 cells (human colon carcinoma), research groups showed that COP1 downregulation induces p53 activation and hence cell apoptosis. However, immunohistochemical analysis did not detect increased p53 in COP1 −/– and COP1 hypo/+ embryos or COP1 hypo/hypo mice because there is a counterbalance by increased c-JUN.7
COP1 is involved in diverse cancers: lymphoma, melanoma, glioma, breast cancer, lung cancer, colorectal cancer, hepatocellular carcinoma, leukemia, renal cell carcinoma, and ovarian cancer. Because it is connected to post-translational modifications by Ub in many proteins, namely, p53, c-Jun (proto-oncogene or transcription factor), E26 transformation-specific (ETS), β-catenin, signal transducers and activators of transcription 3 (STAT3), metastasis-associated protein (MTA1), p27, and CCAAT/enhancer binding protein β (14-3-3σ C/EBPα), these are linked in many biochemistry pathways.7
Studies have demonstrated that COP1 is highly expressed in hepatocellular carcinoma (HCC). COP1 action in this cancer was confirmed by silencing of HepG2 and Huh7 cancer cells’ COP1 siRNA, causing apoptosis of these cells.7
Cells maintain p53 levels irrespective of MDM2 in different ways. Other proteins, including Pirh2, also perform this role.9 This occurs through two p53 binding sites, the N-terminal, which interacts with the DNA-binding domain, and the C-terminal, which binds to a tetramerization domain, specific to the p53 tetrameric form.9 Another function of Pirh2 is to interact with p53 tetramers, to inhibit p53 activity through transcriptional control; nevertheless, Pirh2 has little or no in vitro activity toward monomeric or dimeric p53.8
When calmodulin-dependent kinase II (CaMK II) phosphorylates Pirh2, the latter loses its stability. Consequently, p53 ubiquitination is reduced. Pirh2 inactivation takes place during the cell cycle in the G2/M phase transition, but Pirh2 activity increases in the G1/S phase transition,8 raising p53 levels.
Five Pirh2 isoforms exist, but only Pirh2A acts as a RING E3 ligase to ubiquitinate p53 in a negative feedback loop and to activate PirhA expression. Pirh2A functions as an oncogene, but in some cancers, it acts as a tumor suppressor, as shown by studies in which Pirh2A downregulation worsens cancer patients’ clinical conditions.6
P53 acts in cell senescence, that is, when the cell has no more proliferative capacity. p38 mitogen-activated protein kinases (p38MAPK) are critical for senescence-associated secretory phenotypes (SASP), involved with secretion of several growth factors that lead to tissue repair. p53 suppresses p38MAPK, so it has antagonistic effects on senescence.11
However, p53 is downregulated at a later senescence stage. The E3 ligase SCF (SKP1-CUL1-F-box) (SCFFBXO22) forms a complex with the lysine demethylase enzyme KDM4A, which is highly expressed in senescence. This complex induces protein p16, also known as cyclin-dependent kinase inhibitor 2A, maintaining the cell cycle arrest irreversibility. Moreover, this complex promotes methylated p53 degradation by the 26S proteasome through p53 polyubiquitination at Lys 48 in the C-terminal domain (CTD).11
Experiments using cells exposed to shRNA Fbxo22 have shown significantly increased levels of p53 and p21, also known as cyclin-dependent kinase inhibitor 1, but further Fbxo22 addition decreases p21 and p53 levels. Recent studies have shown that protein PHD finger protein 20 (PHF20) binds to dimethylated p53, stabilizing and activating it. When Fbxo22 is depleted from RPE cells, interaction between Flag-PHF22 and p53 increases. The same occurred in experiments involving a mutation in the amino acids in the KDM4A TUDOR domain. Together with the use of a classic 20S proteasome inhibitor (MG132), studies have shown that p53 binds to and is degraded by this complex.11
Other E3 ligases include carboxy-terminus of Hsc70 interacting protein (CHIP), ARF-binding protein 1 (ARF-BP1), Ring Finger Protein 1 (RING1), and F-box/WD repeat-containing protein 7 (F-box protein). All of them participate in p53 degradation by the 26S proteasome.
CHIP is a cochaperone E3 ligase with three essential domains; two interact with the heat shock enzymes Hsp70/90 and the C-terminal U-box domain (E3 ligase) that links to E2 ligase from the Ubc4/5 family, ubiquitinates p53, and degrades it.12 Studies have shown that CHIP regulates p53 through three processes. The first involves ubiquitin, preferably when p53 is bound to HSP70/Hsc70; heat shock protein HSP 90 (Hsp90-beta) inhibits this pathway. This process is linked to abnormal cell physiological conditions or stress. For the second process, in addition to modifying p53 via ubiquitin, CHIP interferes with p53 phosphorylation through p53 interaction with the Daxx protein.12 This blocks phosphorylation at Ser46, which is necessary to maintain the p53 apoptotic activity. The protein involved in inhibiting this process is not yet known. In the third process, CHIP interacts with Mortalin and affects its interaction with ubiquitinated p53, thereby increasing p53 degradation by the proteasome. Protein UBXN2A inhibits this process linked to cancer cells.12
Depending on the tissue where ARF-BP1 is found, it can either lead to p53 degradation or exert an antagonistic function by inhibiting MDM2, so it is tissue-specific.6,8 ARF-BP1 is involved with other substrates such as Myc proto-oncogene, BCL2 prosurvival, and induced myeloid leukemia cell differentiation protein Mcl-1. As previously mentioned, some substrates of this E3 ligase are tumor suppressors, whereas others are involved in cell survival and are highly expressed in lung, prostate, and breast cancers.6
By using a two-hybrid yeast system, Zhang and collaborators discovered an interaction between ARF-BP1 and MDM2. These researchers cotransformed a vector (human ARF cDNA fragment fused to the yeast Gal4 DNA binding domain) into HF7c yeast cells with a cDNA library of two human HeLa hybrids. Subsequently, they sequenced the positive plasmids and suggested that the two proteins interact, as later confirmed in a cell-free system. This research team showed that ARF, MDM2, and p53 form a ternary complex discovered by immunoprecipitation assay in HeLa cells, transfected with plasmids that express these three proteins. Western blot revealed that p53 and ARF interact in cells that do not express MDM2, while there is no interaction between the two proteins in the presence of MDM2.13
Ring finger protein 1 (RING1) is part of the transcriptional repression complex 1 (PRC1), and FBW7α is part of the SCF complex, as well as other E3 ligases; these proteins destabilize p53. Two processes are known—RING1 is overexpressed in hepatocellular carcinoma tissues, and FBW7α is frequently mutated in cancer and regulates several tumorigenic substrates (notch, c-Jun, c-Myc, and G1/S-specific cyclin-E1 cyclin E).6
Transcriptional repressor E4F1 (which is part of the polycomb repressive complex 1) influences various metabolic pathways and mitochondrial functions. For example, E4F1 activates p53 in a dependent manner during the p53-dependent cell cycle arrest. Also, E4F1 activates p53 during cell cycle arrest in lysine residues other than MDM2, preventing p53 degradation. Research into E4F1 mutant mice has shown embryogenic death due to mitotic and apoptotic cell defects. Mice with skin and epidermis exposed to hydroxy-tamoxifen present E4F1e inactivation and exhibit hyperplasia and hyperkeratosis with skin lesions.8
2.1. Tripartite-Motif-Containing Protein Family
The tripartite-motif-containing proteins (TRIM) family, which contains an N-terminal RING, is the main family regulating p53 and is essential for many functions: metabolism, immunity autophagy, and tumorigenesis.6
TRIM25 is an oncogene that is overexpressed in breast and ovarian cancers that regulates p53, and its mechanism has been elucidated recently. Western blot of cells exposed to β-estradiol shows elevated p53 and MDM2 levels in a codependent manner, which does not occur when cells are transfected with TRIM25 siRNA. Although TRIM25 increases the p53 level, the p53 transcriptional and DNA damage response activities are inhibited.14
p53 acetylation increases in HCT116 cells transfected with small interfering TRIM25 RNAs and in TRIM25 −/– mouse embryos. Consequently, the p53 apoptotic activity increases and so does gene transcription. Unlike MDM2, TRIM25 prevents p53 degradation. This occurs through TRIM25 linking to the p53/MDM2 complex, which interferes in the association of p300 (histone acetyltransferase) and MDM2 and thus prevents polyubiquitination.14
This reduces endogenous p21 which, together with p53, interrupts the cell cycle in response to DNA damage. Decreased TRIM25 expression raises the levels of both p21 and protein BAX. BAX binds to the mitochondrial membrane, releasing cytochrome c and activating caspases. When TRIM25 expression decreases, caspase-3 is activated, PARP is cleaved, and cancer cells die (HCTT116).14
Experiments with tandem affinity purification-tagged p53 have shown that TRIM24, an E3 ligase, interacts with15 and phosphorylates p53, leading to p53 degradation by the proteasome and hence inhibiting p53 activity. The first tests using TRIM24 RNAi and performed on Drosophila showed that TRIM24 inhibits p53,15 which was confirmed in TRIM24 KO (knockout) mice and by the fact that TRIM24 mice tend to develop hepatocellular carcinomas.6 Later trials on Drosophila indicated that TRIM24 has MDM2-independent activity given that Drosophila lacks the gene encoding this protein.10 Research conducted in patient tissues showed that high TRIM24 levels are a poor prognosis in patients with breast and other cancers.6
Some TRIM proteins (TRIM13 and TRIM19) stabilize p53 because they interact with MDM2 and lead to MDM2 degradation by the 26S proteasome. Little is known about TRIM13 and TRIM19, but TRIM19 forms a complex with protein L11 (nucleolar protein) and acts on the nucleus.10,12
TRIM28 silencing in mammalian cells increases p53 activity and the competition between TRIM28 with ARF proteins to bind to MDM2, which reduces formation of the MDM2–TRIM28 dimer. Several types of cancer express high levels of this TRIM protein, and TRIM28 KO mice have embryonic lethality, which is consistent with results indicating that high TRIM28 levels worsen the survival of patients with cancer.6,10,12
TRIM69 ubiquitinates and regulates p53 in human epithelial cells in response to ultraviolet B irradiation, a risk factor for cataract development. In TRIM69 KO mice, TRIM69 has protective effects on the mouse hippocampus after consumption of a high-fat diet, inhibiting apoptosis and inflammation.6
On the other hand, TRIM71 ubiquitinates p53 during stem cell development and neurogenesis. TRIM71 KO mice suffer embryogenic lethality due to neural tube development problems. In ovarian cancer, TRM71 binds to a p53 mutant and inhibits the mutant’s p53 target gene activation, suppressing the growth of ovarian cancer in mouse xenograft models.6
3. Specific 26S Proteasome Inhibitors and Their Involvement with the Tumor Suppressor p53
Studies on the 26S proteasome focus on compounds that act in this complex and can be used to treat diseases, such as cancer.16 These compounds are called proteasomal inhibitors (PIs), act on 20S or 19S, and interfere with protein p53. One of these compounds is bortezomib, which is part of the first generation of PIs approved by the Food and Drug Administration (FDA) in 2003 to treat lymphomas and multiple myelomas (MM). Compounds like ixazomib, approved by the FDA in 2015, and carfilzomib, approved by the FDA in 2012, are part of the second generation of PIs.16
PIs cause cellular effects including accumulation of polyubiquitinated proteins and protein aggregates that promote stress in the endoplasmic reticulum and nondegradation of the transcription factor NF-κβ, responsible for cellular apoptosis control, and of some proteins such as p53 and the BCl-2 family16 (Figure 3).
Figure 3.

Inhibition of 26S proteasome. Inhibition of the proteasome causes various cellular changes, such as stress in the endoplasmic reticulum, mitochondrial injury, activation of c-Jun N-terminal kinases (JNK), accumulation of polyubiquitinated proteins, p53 stabilization, and apoptosis.
Inhibitors used to treat cancer cases do not inhibit the three catalytic subunits of 20S simultaneously. Thus, inhibition of β1 or β2 and β5 is sufficient to activate cell death in hematological malignancies, for example. PIs have been rarely compared, so it has not been established whether differences in clinical results obtained with distinct PIs are due to inhibition of a specific 20S catalytic subunit, dosing schedules, patient-related factors, or other causes.17,18
After the FDA approved the use of bortezomib, it became the gold standard for myeloma treatment in association with lenalidomide and dexamethasone. Later, carfilzomib was also approved, and today it represents the first-line MM treatment in conjunction with the previously mentioned drugs.19,20
Research published in 2013 showed that new PIs, analogues of MG132 and bortezomib, stabilize p53, maintaining its pathways intact; however, cytotoxic activity occurs in cancer cells treated with these inhibitors.21 MG132 analogues were administered to mouse embryonic fibroblasts from transgenic p53 +/+ and p53 −/– littermates, and results showed that proteasomal inhibition caused p53 to accumulate. Experimental tests carried out during this research clarified that p53 acts as a cell death cascade downstream mediator.21
3.1. Bortezomib
Bortezomib (BTZ) is a dipeptide bearing the amino acids phenylalanine and leucine linked to a boronic acid. BTZ inhibits the catalytic activity of the β5 subunit of the 20S proteasome18 to culminate in mitochondrial membrane depolarization and apoptosis. Apoptosis is caused by increased intracellular p27 and p53, with consequent increase in pro-apoptotic factors such as phorbol-12-myristate-13-acetate-induced protein (NOXA) and B-cell lymphoma 2 (BCl-2).22 BTZ also decreases the activity of NF-κB, accumulates proteins with errors in their ternary and quaternary structure because of protein inactivation due to degradation by the 26S proteasome, activates c-Jun/N-terminal kinase, and establishes cell cycle inhibitors.22
In some cases, cancer cells rapidly develop resistance to BTZ by (i) promoting selective regulation of specific 19S subunits and reducing the entry of proteins to be degraded by the proteasome and (ii) causing mutations in the 19S β5 subunit gene in patients with MM and in the 19S α5 subunit gene in patients with prostate cancer.18
Current prostate cancer treatments that inhibit androgen production are effective in 80% of patients. However, some prostate cancers do not respond to conventional treatment, and the use of effective chemotherapy drugs should be chosen. Thus, searching for new effective drugs for these types of cancer is necessary.24
Research into prostate cancer cells (PC-3) exposed to BTZ alone or in combination with other chemotherapeutic agents (irinotecan and etoposide) has determined both the cytotoxic effect of the inhibitor and the inhibitory growth of these cells. On the basis of the results, BTZ is more potent than the two other analyzed compounds. Still, its association with etoposide is more effective in PC-3 cells compared to these compounds alone.24
In addition to the types of cancer mentioned above, BTZ has also been tested in a clinical trial of anti-N-methyl-d-aspartate receptor encephalitis, an autoimmune inflammatory disease of the central nervous system that affects adults and children. Currently, adult patients, who do not respond to the treatment recommended for the disease, can be treated with BTZ. A case study of an 8 year old child treated with BTZ has shown disease regression.23
3.2. Ixazomib
Like BTZ, ixazomib is a borate peptide that binds to the 20S β5 subunit. Ixazomib has greater selectivity, faster dissociation, and higher activity compared to BTZ in cancerous tissues.18
The FDA approved the use of ixazomib to treat MM, so some studies have analyzed this molecule to treat other types of cancer. The common therapy for colon cancer uses chemotherapeutic agents that damage DNA but have marked side effects, making the discovery of new molecules that reduce side effects crucial. In this context, ixazomib is an option because it induces apoptosis in HCT116, DLD1, and HT29 cancer cells in vitro. Treatment of HCT116 cells with ixazomib activates apoptosis by the caspase-dependent pathway, with different p53 statuses and decreased cell proliferation being detected. Caspase activation does not depend on p53, and the extrinsic pathway is activated in cancer cells by caspase-8 via DR5 cell death receptors.16−18 In the case of BTZ, the ixazomib apoptotic activity is due to p53 stability and consequent caspase-3, -8, and -9 activation.18
3.3. Carfilzomib
Carfilzomib (CFZ) inhibits the 20S β5 activity and has fewer side effects, greater chemical stability, and more selective mechanism of action than BTZ.19 The CFZ chemical interaction with the β subunits involves van der Waals bridges with the S1, S3, and S4 β5 pockets and with the S3 and S4 β2 pockets. CFZ is part of the second generation of PIs and bears chemical modifications. Unlike BTZ, CFZ is a tetrapeptide with an epoxyketone terminal group, which confers this molecule with a different pharmacological profile compared to first generation inhibitors.24
CFZ causes cancer cell apoptosis in different ways. It acts in the mitochondrial membrane depolarization to culminate in cytochrome c release, increased pro-apoptotic factor Noxa levels, and activation of the caspase-3 and -7 pathways and of the enzymatic-Jun-N-terminal kinase.23 Currently, CFZ is also being tested to treat other diseases and not only cancers.
Pulmonary arterial hypertension (PAH) is a lethal disease characterized by abnormal cell growth of the pulmonary veins. PAH distorts the right ventricle (RV) of the heart, leading to its failure. Thus, studying molecules that enhance vascular remodeling and increase lung vasodilation is necessary. Previous studies have shown that 26S proteasome inhibitors suppress pulmonary arterial cell growth. One study using PAH model rats treated with CFZ has shown cellular apoptosis involving p53, induced by nuclear protein 1. Increased p53 levels are associated with remodeling of collagen and elastic fibers of the pulmonary veins due to caspase-3 cleavage and protein downregulation to the antiapoptotic Bcl-x.25
3.4. Other 26S Proteasome Inhibitors
Oprozomib (second generation of PIs) is a tripeptide epoxyketone analogous to CFZ and has been modified to improve drug absorption. Just like CFZ, oprozomib irreversibly binds to the 26S proteasome. The mechanisms through which oprozomib induces apoptosis have been identified as caspase-3, -8, and -9 activation and poly(ADP-ribose) polymerase (PARP) cleavage. Also, oprozomib probably blocks angiogenesis during MM progression.16
Researchers have concluded that oprozomib inhibits the growth of various types of cancer in preclinical and clinical trials of head and neck cancers, hematological malignancies, mesothelioma, and Waldenstrom’s macroglobulinemia. Furthermore, this compound has been approved by the FDA for MM and RMM treatment. However, it is not yet administered in patients with lung cancer.18
In vitro tests in lung cancer cells have shown that oprozomib action does not depend on p53, but that p53 levels are maintained, and that p53 and its transcriptional targets p21, PUMA, and Noxa are stabilized. Oprozomib activates caspase-3 and PARP, a receptor that acts on the p53 pathway, cleaves caspase-3, and initiates apoptosis. The functions of p53 in the apoptosis pathway are not known. Still, in the cytoplasm, p53 is known to bind to proteins of the BCL-2 family including pro-apoptotic (Bax, BCl-2, PUMA, Noxa, and Bcl-2-like protein 11) and antiapoptotic proteins.16−18
Delanzomib is another recent inhibitor that has been tested on cervical cancer cells. This compound increases the transcriptional levels and protein expressions of p21, p53, PUMA, and Noxa. Whereas BTZ induces side effects such as sensory peripheral thrombocytopenia, neuropathy, and postural hypotension, this new compound is believed to have minor side effects for patients.16,18
4. Concluding Remarks
Many studies have shown that 26S proteasome inhibitors can be used to treat different types of cancer. These drugs stabilize p53 and alter the mitochondrial pathway, leading to cancer cell apoptosis. In cases in which the chemotherapy chosen to treat a specific type of cancer does not have the desired effect, these inhibitors could be used as an alternative treatment.
Current research has focused on the development of new inhibitors from existing compounds through replacement of chemical radicals in their structure, leading to the development of new analogues. Another perspective is the discovery of new molecules that inhibit the 26S proteasome from already existing compound libraries.
High quantities of the 26S proteasome are present in cancer cells and infectious disease cells because these cells have accelerated metabolism. This increases the affinity of these cells for inhibitors in relation to normal cells, which has been a focus for the development of compounds that inhibit protein degradation by the proteasome. Therefore, proteasome inhibitors hold promise for discovering new drugs that could increase the survival of patients with terminal cancers and which could be an alternative for cases that are resistant to the selected drug treatment.
Acknowledgments
This work was supported by grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) #2016/06769-2 to V.R.
Biographies

Andressa Barban do Patrocinio is an associate researcher at the University of Franca, Brazil. She received a bachelor’s degree in Pharmacy-Biochemistry from Universidade Estadual Paulista Júlio de Mesquita Filho-UNESP (2001), master’s (2005) and doctorate (2018) degrees in Sciences with an emphasis in Biochemistry from Ribeirão Preto Medical School-University of São Paulo. Current research focuses on the following topics: 26S proteasome (protein degradation systems) and its inhibitors. Experience in the following techniques: in vitro culture of parasites for testing new drugs against neglected tropical diseases; real time PCR; Western blotting; enzymatic activity assays; protein isolation by chromatography; protein expression in biotechnological systems; scanning, transmission and confocal electron microscopy; and enzyme-linked immunosorbent assay (ELISA). Participation includes projects for the development of biopharmaceuticals aimed at neglected diseases, especially schistosomiasis.

Vanderlei Rodrigues received a bachelor’s degree at Medicine from Ribeirão Preto Medical School- University of São Paulo (1974) and a bachelor’s at Ciências Biologicas from Universidade de São Paulo (1970), master's (1978) and doctorate at Biochemistry (1983) from Ribeirão Preto Medical School- University of São Paulo. Post-doctorate work was performed at the National Institute for Medical Research, NIMR, Inglaterra (1985–1987), and at The Ohio State University in the United States (1990–1990), with experience in Biochemistry, focusing on Molecular Biology, acting on the following subjects: schistosoma mansoni and 26S proteasome.

Lizandra Guidi Magalhães is an associate professor at the University of Franca, Brazil. She received a bachelor’s degree in biomedicine (2003) and master’s and doctorate degrees in immunology from Ribeirão Preto Medical School-University of São Paulo, respectively, in 2005 and 2009. She is involved in identifying new compounds from natural products against neglected tropical diseases with focus on schistosomiasis, leishmamiasis, and trypanosomiasis.
Author Contributions
All authors have read and agree to the published version of the manuscript. Conceptualization, A.B.P.; data curation, A.B.P.; writing—original draft preparation, A.B.P.; writing—review and editing A.B.P., L.G.M., and V.R.; supervision, L.G.M.
The authors declare no competing financial interest.
References
- Rousseau A.; Bertolotti A. Regulation of Proteasome Assembly and Activity in Health and Disease. Nat. Rev. Mol. Cell Biol. 2018, 19 (11), 697–712. 10.1038/s41580-018-0040-z. [DOI] [PubMed] [Google Scholar]
- Sakata E.; Eisele M. R.; Baumeister W. Molecular and Cellular Dynamics of the 26S Proteasome. Biochim. Biophys. Acta - Proteins Proteomics 2021, 1869 (3), 140583. 10.1016/j.bbapap.2020.140583. [DOI] [PubMed] [Google Scholar]
- Ebner P.; Versteeg G. A.; Ikeda F. Ubiquitin Enzymes in the Regulation of Immune Responses. Crit. Rev. Biochem. Mol. Biol. 2017, 52 (4), 425–460. 10.1080/10409238.2017.1325829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- do Patrocínio A. B.; Cabral F. J.; de Paiva T. H.; Magalhães L. G.; de Lima Paula L. A.; Brigato O. M.; Guerra-Sá R.; Rodrigues V. Deubiquitinating Enzymes as Possible Drug Targets for Schistosomiasis. Acta Trop. 2021, 217, 105856. 10.1016/j.actatropica.2021.105856. [DOI] [PubMed] [Google Scholar]
- Bixby D.; Kujawski L.; Wang S.; Malek S. N. The Pre-Clinical Development of MDM2 Inhibitors in Chronic Lymphocytic Leukemia Uncovers a Central Role for P53 Status in Sensitivity to MDM2 Inhibitor-Mediated Apoptosis. Cell Cycle 2008, 7 (8), 971–979. 10.4161/cc.7.8.5754. [DOI] [PubMed] [Google Scholar]
- Bang S.; Kaur S.; Kurokawa M. Regulation of the P53 Family Proteins by the Ubiquitin Proteasomal Pathway. Int. J. Mol. Sci. 2020, 21 (1), 261. 10.3390/ijms21010261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Liu Y.; Pan S.; Xie S.; Wang Z.-w.; Zhu X. Role of the COP1 Protein in Cancer Development and Therapy. Semin. Cancer Biol. 2020, 67, 43–52. 10.1016/j.semcancer.2020.02.001. [DOI] [PubMed] [Google Scholar]
- Love I. M.; Grossman S. R. It Takes 15 to Tango: Making Sense of the Many Ubiquitin Ligases of P53. Genes and Cancer 2012, 3 (3–4), 249–263. 10.1177/1947601912455198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza M.; Mandani G.; Momand J. The MDM2 Gene Family. Biomol. Concepts 2014, 5 (1), 9–19. 10.1515/bmc-2013-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock A. K.; Vousden K. H. The Role of Ubiquitin Modification in the Regulation of P53. Biochim. Biophys. Acta - Mol. Cell Res. 2014, 1843 (1), 137–149. 10.1016/j.bbamcr.2013.05.022. [DOI] [PubMed] [Google Scholar]
- Johmura Y.; Sun J.; Kitagawa K.; Nakanishi K.; Kuno T.; Naiki-Ito A.; Sawada Y.; Miyamoto T.; Okabe A.; Aburatani H.; Li S.; Miyoshi I.; Takahashi S.; Kitagawa M.; Nakanishi M. SCF Fbxo22-KDM4A Targets Methylated P53 for Degradation and Regulates Senescence. Nat. Commun. 2016, 7, 1–12. 10.1038/ncomms10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sane S.; Rezvani K. Essential Roles of E3 Ubiquitin Ligases in P53 Regulation. Int. J. Mol. Sci. 2017, 18 (2), 442. 10.3390/ijms18020442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Xiong Y.; Yarbrough W. G. ARF Promotes MDM2 Degradation and Stabilizes P53: ARF-INK4a Locus Deletion Impairs Both the Rb and P53 Tumor Suppression Pathways. Cell 1998, 92 (6), 725–734. 10.1016/S0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Elabd S.; Hammer S.; Solozobova V.; Yan H.; Bartel F.; Inoue S.; Henrich T.; Wittbrodt J.; Loosli F.; Davidson G.; Blattner C. TRIM25 Has a Dual Function in the P53/Mdm2 Circuit. Oncogene 2015, 34 (46), 5729–5738. 10.1038/onc.2015.21. [DOI] [PubMed] [Google Scholar]
- Pant V.; Lozano G. Limiting the Power of P53 through the Ubiquitin Proteasome Pathway. Genes Dev. 2014, 28 (16), 1739–1751. 10.1101/gad.247452.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeten M. S. F.; Cloos J.; Jansen G. Positioning of Proteasome Inhibitors in Therapy of Solid Malignancies. Cancer Chemother. Pharmacol. 2018, 81 (2), 227–243. 10.1007/s00280-017-3489-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse A.; Besse L.; Kraus M.; Mendez-Lopez M.; Bader J.; Xin B. T.; de Bruin G.; Maurits E.; Overkleeft H. S.; Driessen C. Proteasome Inhibition in Multiple Myeloma: Head-to-Head Comparison of Currently Available Proteasome Inhibitors. Cell Chem. Biol. 2019, 26 (3), 340–351.e3. 10.1016/j.chembiol.2018.11.007. [DOI] [PubMed] [Google Scholar]
- Tundo G. R.; Sbardella D.; Santoro A. M.; Coletta A.; Oddone F.; Grasso G.; Milardi D.; Lacal P. M. Since January 2020 Elsevier Has Created a COVID-19 Resource Centre with Free Information in English and Mandarin on the Novel Coronavirus COVID- 19. The COVID-19 Resource Centre Is Hosted on Elsevier Connect, the Company ’ s Public News and Information. 2020, No. January.
- Strifler S.; Knop S. The Role of Carfilzomib in Treatment of Newly Diagnosed Multiple Myeloma. Futur. Oncol. 2018, 14 (30), 3123–3134. 10.2217/fon-2018-0040. [DOI] [PubMed] [Google Scholar]
- Saavedra-García P.; Martini F.; Auner H. W. Proteasome Inhibition in Multiple Myeloma: Lessons for Other Cancers. Am. J. Physiol. - Cell Physiol. 2020, 318 (3), C451–C462. 10.1152/ajpcell.00286.2019. [DOI] [PubMed] [Google Scholar]
- Neilsen P. M.; Pehere A. D.; Pishas K. I.; Callen D. F.; Abell A. D. New 26S Proteasome Inhibitors with High Selectivity for Chymotrypsin-like Activity and P53-Dependent Cytotoxicity. ACS Chem. Biol. 2013, 8 (2), 353–359. 10.1021/cb300549d. [DOI] [PubMed] [Google Scholar]
- Robak P.; Robak T. Bortezomib for the Treatment of Hematologic Malignancies: 15 Years Later. Drugs R D 2019, 19 (2), 73–92. 10.1007/s40268-019-0269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordani R.; Micalizzi C.; Giacomini T.; Gastaldi M.; Franciotta D.; Fioredda F.; Buratti S.; Morana G.; Pirlo D.; Renna S.; Castagnola E.; Risso M.; Lanteri P.; Vari M. S.; Mancardi M. M. Bortezomib-Responsive Refractory Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Pediatr. Neurol. 2020, 103, 61–64. 10.1016/j.pediatrneurol.2019.09.004. [DOI] [PubMed] [Google Scholar]
- Aras B.; Yerlikaya A. Bortezomib and Etoposide Combinations Exert Synergistic Effects on the Human Prostate Cancer Cell Line PC-3. Oncol. Lett. 2016, 11 (5), 3179–3184. 10.3892/ol.2016.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Ibrahim Y. F.; Das D.; Zungu-Edmondson M.; Shults N. V.; Suzuki Y. J. Carfilzomib Reverses Pulmonary Arterial Hypertension. Cardiovasc. Res. 2016, 110 (2), 188–199. 10.1093/cvr/cvw047. [DOI] [PMC free article] [PubMed] [Google Scholar]