

Abstract
Dopamine D3 receptors (D3R) play a critical role in neuropsychiatric conditions including substance use disorders (SUD). Recently, we reported a series of N-(3-hydroxy-4-(4-phenylpiperazin-1yl)butyl)-1H-indole-2-carboxamide analogues as high affinity and selective D3R lead molecules for the treatment of opioid use disorders (OUD). Further optimization led to a series of analogues that replaced the 3-OH with a 3-F in the linker between the primary pharmacophore (PP) and secondary pharmacophore (SP). Among the 3-F-compounds, 9b demonstrated the highest D3R binding affinity (Ki = 0.756 nM) and was 327-fold selective for D3R over D2R. In addition, modification of the PP or SP with a 3,4-(methylenedioxy)phenyl group was also examined. Further, an enantioselective synthesis as well as chiral HPLC methods were developed to give enantiopure R- and S-enantiomers of the four lead compounds. Off-target binding affinities, functional efficacies, and metabolic profiles revealed critical structural components for D3R selectivity as well as drug-like features required for development as pharmacotherapeutics.
Graphical Abstract
INTRODUCTION
In the past decade, prescription opioid abuse and misuse has been a major contributor to mortality in the United States, resulting in a serious national public health crisis.1,2 Nearly 12 million Americans used prescription opioids nonmedically in 2016, with >42 000 people dying from an opioid overdose.1,3,4 The federal response to the opioid crisis includes research support toward the development of nonaddictive analgesics and additional medications to treat opioid use disorders (OUD).5
Medication for addiction treatment in the form of opioid substitution therapies (e.g., methadone or buprenorphine) has proven effective for OUD with new formulations adding to the arsenal of medication options.6−8 Further, the opioid antagonist, naloxone is used to prevent overdose. However, methadone and buprenorphine continue to be underutilized due to lack of availability in underserved communities or to undesirable and serious side effects such as respiratory depression, constipation, and abuse liability.9,10 Recently, the nonopioid α2A adrenergic receptor agonist, lofexidine, was approved by the U.S. Food and Drug Administration as the first medication for use in reducing symptoms associated with opioid withdrawal in adults.11 However, lofexidine has no effect on the addictive liability of opioid analgesics.12,13
Extensive preclinical and clinical studies suggest that all drugs of abuse ultimately activate the mesolimbic dopaminergic pathway to produce rewarding effects that can lead to addiction.14 Specifically, mediation of dopamine (DA) signaling through dopamine D1-like (D1R and D5R) and D2-like (D2R, D3R, D4R) G protein-coupled receptors plays a critical role in the pleasurable effects of addictive drugs. However, in some people, frequent or chronic stimulation of DA can induce compulsive and repetitive drug seeking, which can lead to substance use disorders (SUD).15,16 D3R are highly localized in mesolimbic regions of the brain and are critically involved in motivation to drug seeking.17,18 Thus, the discovery of selective D3R antagonists and partial agonists has been the focus of many research programs interested in the development of medications to treat neuropsychiatric conditions including SUD.18−24
Over the past 2 decades, many D3R-selective ligands with high affinity have been discovered, affording critical tools for understanding mechanisms underlying addiction.21,25−28 In addition, selective D3R antagonists have been extensively investigated preclinically and have shown promising results in rodent models of drug self-administration, relapse-like behaviors, and conditioned place preference.23,29,30 Nevertheless, to date, advancement to human clinical studies has been limited to only a few selective D3R antagonists, including GSK598,809 (1, Figure 1).31,32 However, the findings that 1 produced hypertension in dogs when coadministered with cocaine diminished prospects for its further development as a cocaine use disorder therapeutic.33
Figure 1.

Chemical structures of lead D3R antagonists/partial agonists.
The advancement of bitopic ligands that include a substituted phenyl piperazine as the primary pharmacophore (PP) and various arylcarboxamides as the secondary pharmacophore (SP) linked with a variety of substituted alkyl chains have provided excellent tools for investigating D3R in animal models of psychostimulant abuse.34−40 For example, the first enantioselective D3R antagonist (R)-PG64841 (2, Figure 1) was a recent lead molecule in this class that showed promise in rodent models, but not in nonhuman primates, possibly due to metabolic instability.26 Modification of the 3-OH in the linking chain between the PP and the SP with a F-group led to BAK2–6637 (3, Figure 1) where the (R)- enantiomer was also the eutomer,38 as was also reported for R-2.41
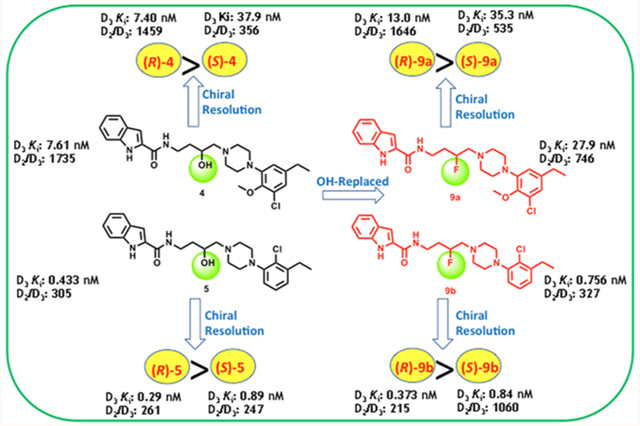
The high-resolution crystal structure of D3R in complex with the D2R/D3R antagonist, eticlopride,42 provided a platform for further optimization leading to two D3R-selective analogues, VK4–116 (4) and VK4–40 (5) in Figure 1.39 These structural analogues showed high affinity and selective binding at D3R over D2R and excellent metabolic stability in mouse, rat, and nonhuman primate liver microsomes.30 In addition, lead compound 4 dose-dependently inhibited the acquisition and maintenance of oxycodone self-administration, lowering the break-point for oxycodone self-administration under a progressive-ratio schedule of reinforcement and inhibited oxycodone extinction response and reinstatement of oxycodone-seeking behaviors.30 Moreover, pretreatment with 4 dose-dependently reduced naloxone-precipitated conditioned place aversion in rats chronically treated with oxycodone but had no effect on the antinociceptive actions of oxycodone, tested in a hot plate assay.30 Herein we report further investigation of the PP and SP in these bitopic molecules and substitution of the linking chain with 3-F. We had previously shown that replacing the 3-OH with a 3-F in the linking chain resulted in some of the most selective D3R antagonists/partial agonists reported,37,38 and we also predicted that this substitution might increase metabolic stability. Chiral resolution of the lead molecules (Figure 2) was pursued in order to further optimize this template and provide new pipeline compounds for development.
Figure 2.

Drug design strategy.
CHEMISTRY
In Scheme 1, the synthesis of the 3-F-analogues 9a,b and 10a,b began with the secondary alcohol intermediates 6a,b synthesized using our previously published strategy.39 Replacement of the 3-OH group with F was carried out in the presence of the fluorinating agent diethylaminosulfur trifluoride (DAST) to generate the 3-F intermediates (7a,b).37,38 Subsequently, removal of the phthalimide protecting group yielded primary amine intermediates (8a,b) in quantitative yields, which underwent amide coupling with indole- or benzofuran-2-carboxylic acids in the presence of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) to generate the 3-F-analogues 9a,b and 10a,b. In Scheme 2, to further extend SAR, we synthesized 13a,b with the 3,4-(methylenedioxy) moiety as the SP, by using the primary amine intermediates 12a,b (generated in a single step starting with 11a, 11b) via a general amidation procedure. The 3,4-(methylenedioxy)phenyl group was selected as a SP to see how another biologically relevant heterocyclic moiety would affect the profile of these compounds. This moiety exists in other CNS active agents (e.g., MDMA), and we wanted to expand the SAR to determine if this bicyclic ring structure would affect D3R selectivity. As we have noted previously, the PP typically affects the ligand affinity for D2R and D3R, but the SP and linker are key to D3R selectivity.25,40
Scheme 1.

Synthesis of 3-Fluoro Analoguesa
aReagents and conditions: (a) DAST, anhydrous DCM, −78 °C to rt, overnight; (b) hydrazine, EtOH, reflux, overnight; (c) indole-2-carboxylic acid for 9a, 9b; benzfuran-2-carboxylic acid for 10a, 10b, EDC/HOBt, DIPEA, CHCl3/DMF, 0 °C to rt 8 h.
Scheme 2.

Synthesis of 3,4-(Methylenedioxy) SP Analoguesa
aReagents and conditions: (a) (1) 19, 2-PrOH reflux, 3 h, (2) hydrazine, reflux, overnight; (b) benzo[d][1,3]dioxole-5-carboxylic acid, EDC/ HOBt, DIPEA, CHCl3/DMF, 0 °C to rt, 8 h.
Likewise, as for the SP, we extended SAR by replacing the substituted phenyl rings in the PP of compounds 2−5 with the 6-ethylbenzo[d][1,3]dioxol moiety. We thought this may be an interesting replacement for the more classic substituted 2,3-diCl-phenyl substitution in previous series of D3R-selective antagonists.25,26 In Scheme 3, the synthesis began with the nitration of benzo[d][1,3]dioxole-5-carbaldehyde 14 in fuming nitric acid to give a nitro intermediate 15. Conversion of 15 to the alkene 16 was accomplished under Wittig reaction conditions, and the nitro group as well as the alkene were reduced via catalytic hydrogenation to yield aniline 17 in one pot. Compound 17 was reacted with bis(2-chloroethyl)amine to generate the novel piperazine PP, 18. The primary amine 20 was prepared via epoxide opening of 2-(2-oxiran-2-yl)ethyl)isoindoline-1,3-dione 19 with piperazine 18, followed by deprotection of the phthalimide using hydrazine in the same step. Coupling of 20 with indole- or benzofuran-2-carboxylic acids gave the 6-ethylbenzo[d][1,3]dioxol analogues 21 and 22, respectively.
Scheme 3.

Synthesis of 3,4-(Methylenedioxy) PP Analoguesa
aReagents and conditions: (a) Fuming HNO3, 0 °C to rt, 2 h; (b) methyltriphenylphosphonium bromide, lithium tert-butoxide, THF −78 to 0 °C to rt, 8 h; (c) 10% Pd/C, H2, 50 psi, EtOAc, 45 min; (d) bis(2-chloroethyl)amine·HCl, diethylene glycol monoethyl ether, 150 °C, 7 h; (e) (1) 2-PrOH, reflux, 3 h, (2) hydrazine, reflux, overnight; (f) indole-2-carboxylic acid for 21; benzfuran-2-carboxylic acid for 22, EDC/HOBt, DIPEA, CHCl3/DMF, 0 °C to rt 8 h.
Synthesis of the (R)- and (S)-enantiomers of lead compounds 4 and 5 are shown in Scheme 4. Enantioselective synthesis was achieved by applying similar procedures described above. Briefly, the chiral primary amine intermediates (R)-12a,b and (S)-12a,b were generated by epoxide opening of chiral 2-(2-oxiran-2-yl)ethyl)isoindoline-1,3-dione (R)-1939 and (S)-19,38 respectively, with N-arylpiperazine intermediates 11a,b followed by deprotection of the phthalimide. Subsequently, (R)-12a,b and (S)-12a,b were coupled with indole 2-carboxylic acid to afford the enantiopure (R)-4,5 and (S)-4,5 (HPLC: >98% ee, chromatograms in Figures S1 and S2). Alternatively, (R)-4,5 and (S)-4,5 were also prepared using chiral preparative HPLC from their corresponding racemates achieving >99% ee. The enantiomers of the 3-F-analogues (R)-9a,b and (S)-9a,b were synthesized starting from 11a,b which were reacted with enantiopure phthalimide intermediates (R)-19 and (S)-19 to yield chiral hydroxy intermediates (R)-6a,b and (S)-6a,b (Scheme 5). Further replacement of the chiral 3-OH group with F was achieved in the presence of the fluorinating agent DAST to generate the 3-F intermediates (R)-7a,b and (S)-7a,b, retaining the absolute configuration at the chiral center, as previously observed.38 Subsequently, deprotection of phthalimide group yielded chiral primary amine intermediates (R)-8a,b and (S)-8a,b in quantitative yields that underwent amide coupling with indole carboxylic acids in the presence of EDC and hydroxybenzotriazole (HOBt) to generate the enantiomers of 3-F-analogues (R)-9a,b and (S)-9a,b (Scheme 5). Alternatively, (R)-9b and (S)-9b were also prepared using chiral preparative HPLC from corresponding racemate 9b resulting in >99% ee for each enantiomer (Figures S3 and S4).
Scheme 4.

Synthesis of (R)- and (S)-4 and 5a
aReagents and conditions: (a) (1) 2-PrOH reflux, 3 h, (2) hydrazine, reflux, overnight; (b) indole-2-carboxylic acid, EDC/HOBt, DIPEA, CHCl3/DMF, 0 °C to rt, 8 h.
Scheme 5.

Synthesis of (R)- and (S)-3-Fluoro Analoguesa
aReagents and conditions: (a) 2-PrOH, reflux, 3 h; (b) DAST, anhydrous DCM, −78 °C to rt, overnight; (c) hydrazine, EtOH, reflux, overnight; (d) Indole-2-carboxylic acid, EDC/HOBt, DIPEA, CHCl3/DMF, 0 °C to rt, 8 h.
PHARMACOLOGICAL RESULTS AND DISCUSSION
Dopamine D2R and D3R Binding Analysis.
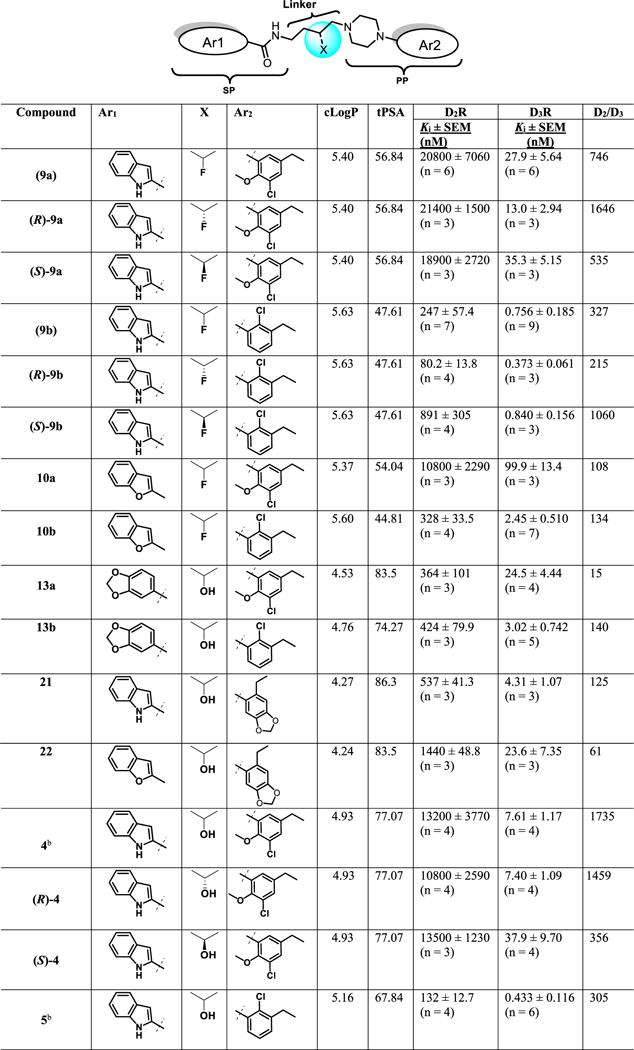
The radioligand [3H]N-methylspiperone was employed in all the competition binding experiments using membrane preparations from stably transfected HEK293 cells expressing human D2L and D3 dopamine receptors to determine binding affinities and D2R/D3R selectivities (Table 1). All new analogues showed moderate to high D3R binding affinities in the range of Ki = 0.290−99.9 nM. D2R binding affinities were generally lower and thus D2R/D3R selectivities ranged from 15 to 1735-fold.
Table 1.
Chemical Structures and Human D2-Like Receptor Binding Affinity Data of Newly Developed Analoguesa
|
|
The values represent the arithmetic mean ± SEM of triplicate determinations from at least three independent experiments. IC50 values for each compound were determined from dose−response curves, and Ki values were calculated by the Cheng−Prusoff equation43 using GraphPad Prism version 6.00 for Macintosh. n = number of independent experiments, each performed in triplicate.
Compound reported in ref 39.
As previously discovered, replacing the 3-OH group in the linker with a 3-F atom, and retaining the PP of 4, resulted in compounds 9a and 10a, where the SPs were either the indole2-carboxamide or the benzofuran-2-carboxamide, respectively, with moderately high affinity and selectivity for D3R. Of note, 9a, the analogue with the indole carboxamide as the SP, was 746-fold D3R selective over D2R. In contrast, the analogues of 5, 9b, and 10b had relatively higher D3R affinities compared to 4, but D2R affinities were also improved leading to reduced selectivity, as previously seen with 5. For example, the indole analogue 9b had the highest affinity in the 3-F series (Ki = 0.756 nM) but was less selective (D2R/D3R = 327) than its analogue, 9a (D2R/D3R = 746).
Replacing the SP with a benzo[d][1,3]dioxole-5-carboxamide scaffold, resulted in lower D3R binding affinities for 13a and 13b compared to their indole analogues, 4 and 5, respectively. Moreover, D2R affinity increased for 13a (D2R Ki = 364 nM) compared to 4 resulting in lower D3R-selectivity, further exemplifying the indole as a privileged SP structure.
Replacing the PP with 6-ethylbenzo[d][1,3]dioxol-piperazine resulted in compounds 21 and 22 that were relatively well tolerated at D3R, but again D2R affinities were improved leading to less D3R-selective compounds. Likewise, the indole analogue 21 showed higher D3R affinity than the benzofuran analogue, 22, with D3R Ki = 4.31 nM and 23.6 nM, respectively. Interestingly, we have previously shown that the PP plays a more prominent role in affinity,40 but in this case the PP is identical and the SPs are affecting affinity. These data demonstrate that the entire bitopic molecule must be taken into account when assessing affinities and selectivities. Further the chemical composition of each PP, SP, and linker plays a critical and sometimes differing role in the pharmacological profiles of these unique molecules. Although these heterocyclic modifications to the SP and PP resulted in D3R-selective analogues, none were superior to the lead molecules 4 and 5.
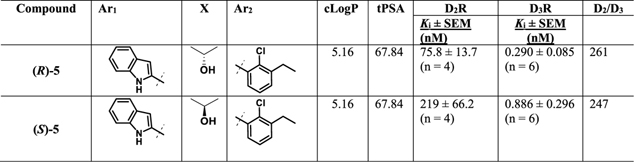
As the lead molecules 4 and 5 are the racemates, it was important to resolve the chiral center in the linking chain in order to identify which enantiomer would be taken on for further development. The binding affinities for the racemates of 4 and 5 were comparable to those previously reported (Table 1). (R)-4 exhibited very similar D3R binding affinity to the racemate with a Ki = 7.40 nM and was similarly selective over D2R (1459-fold). Whereas, (S)-4 was the distomer with a Ki = 37.9 nM, (~5-fold enantioselectivity). As noted with (R)2, D3R enantioselectivity was modest; D2R affinity showed essentially no enantioselectivity. As previously described, (±)-5 showed subnanomolar binding affinity at D3R (Ki = 0.433 nM) and (R)-5 also exhibited similarly high D3R binding affinity (Ki = 0.290 nM). (S)-5 (Ki = 0.886 nM) was also the distomer, although its binding affinity was only reduced by ~3-fold compared to (R)-5. Further, both the R and S enantiomers of 5 were less selective at D3R over D2R when compared to similar enantiomers of 4, although still in the 300-fold range.
In comparison, the 3-F analogues with an indole-3-carboxamide as the SP demonstrated high D3R affinity, and thus it was of interest to resolve the racemates 9a and 9b as potential pipeline lead molecules. As previously shown, both the R-enantiomers (R)-9a,b were the eutomers at D3R. (R)-9a exhibited ~2-fold higher binding affinity at D3R (Ki = 13.0 nM) compared to (±)-9a and ~1650-fold selectivity over D2R (2-fold higher than (±)-9a). Whereas, (S)-9a showed reduced D3R binding affinity and selectivity (Ki = 35.3 nM, 535-fold over D2R). Moreover, (R)-9b exhibited subnanomolar affinity at D3R (Ki = 0.373 nM). Interestingly, (S)-9b also displayed subnanomolar binding affinity at D3R with Ki = 0.840 nM and was >1000-fold selective over D2R. Thus, the selectivity profile of (S)-9b at D3R over D2R was dramatically increased and ~5-fold higher than either (±)-9b (327-fold) or (R)-9b (215-fold).
In summary, (R)-9a and (R)-9b with the 3-F in the linking chain showed similar binding affinities and selectivity profiles to their corresponding 3-OH analogues, (R)-4 and (R)-5. Whereas, (S)-9a and (S)-9b showed similar binding affinities to (S)-4 and (S)-5 respectively; however, (S)-9b exclusively showed higher selectivity over D2R and was approximately 4fold more selective than (S)-5, demonstrating the critical role of substitution and stereochemistry of the linking chain in the binding profile of this series of molecules. Of note, cLogP and PSA values were obtained for all final compounds and are listed in Table 1. In general, these values suggest that most of these compounds are highly lipophilic, which is typical of bitopic compounds selective for D3R.
Functional and Off-Target Data.
The in vitro functional efficacies of a subset of 3-F-analogues 9a,b and 10a,b and R- and S-enantiomers of both 4 and 5 were examined for stimulation or inhibition of quinpirole-stimulated mitogenesis in CHO cells (Table 2). These data revealed that all the 3-F analogues were D3R antagonists, as were (R)- and (S)-4 and (R)-5. However, (S)-5 was a relatively low efficacy partial agonist, like (±)-5.39 This difference in efficacies between enantiomers was also observed with previous lead compound 2, where (R)-2 was an antagonist and (S)-2 was a weak partial agonist at D3R.41
Table 2.
Functional Data for Selected Compounds Using Stimulation or Inhibition of Quinpirole-Stimulated Mitogenesis in CHO Cells with Human Dopamine D3Ra
| cmpd | agonist EC50± SEM, nM | % stimulation | antagonist IC50± SEM, nM |
|---|---|---|---|
| 9a | >5400 | 2.7 | 510 ± 130 |
| 9b | >6800 | 5.4 | 95 ± 31 |
| 10a | >4000 | 5.1 | 490 ± 150 |
| 10b | >7500 | 15.5 | 450 ± 160 |
| (R)-4 | >3200 | 17.7 | 350 ± 110 |
| (S)-4 | >6800 | 17.0 | 1020 ± 200 |
| (R)-5 | >2200 | 20.2 | 18.7 ± 6.6 |
| (S)-5 | 46 ± 17 | 25.9 | 660 ± 260 |
Data were obtained through the NIDA Addiction Treatment Discovery Program Contract (ADA12013) with Oregon Health & Science University.
In addition to binding data for D2R and D3R, the same set of compounds was tested for binding affinities at serotonin 5HT1A, 5-HT2A, and 5-HT2C receptors. As depicted in Table 3, none of these analogues showed high affinity binding to any of the 5-HT receptor subtypes tested compared to their D3R affinities. Compound 10b showed the highest affinities of the 3-F analogues, across the 5-HT receptor subtypes, and was a low potency partial agonist at 5HT1A (EC50 = 351 nM). (R)- and (S)-5 showed higher affinities across the 5-HT receptor subtypes than (R)- and (S)-4; however, selectivities for D3R remained high ((R)-4, 5-HT1A/D3R = 469, 5-HT2A/D3R = 324, 5-HT2c/D3R = 211, and (S)-4, 5-HT1A/D3R = 149, 5-HT2A/D3R = 228, 5-HT2c/D3R = 170).
Table 3.
In Vitro Binding and Functional Data for Selected Compounds at 5-HT1A, 5-HT2A, and 5-HT2C Receptorsa
| cmpd | 5-HT1A[3H]-8-OH-DPAT Ki ± SEM, nM | 5-HT2A[125I]DOI Ki ± SEM, nM |
5-HT2C[125I]DOI Ki± SEM, nM |
5-HT1A[35S]GTPγS binding |
||
|---|---|---|---|---|---|---|
| agonist EC50± SEM, nM | % stimulation | |||||
| 9a | 9500 ± 3000 | 6600 ± 1400 | >8900 | NDb | ||
| 9b | 910 ± 120 | 620 ± 120 | 580 ± 160 | NDb | ||
| 10a | 2660 ± 570 | 2990 ± 530 | 1780 ± 390 | NDb | ||
| 10b | 117 ± 14 | 336 ± 19 | 464 ± 36 | 351 ± 84 | 43 | |
| (R)-4 | 2800 ± 340 | 1933 ± 20 | 1260 ± 320 | NDb | ||
| (S)-4 | 4980 ± 270 | 7600 ± 1800 | 5670 ± 930 | NDb | ||
| (R)-5 | 248 ± 18 | 21.3 ± 6.6 | 50 ± 13 | 1050 ± 260 | 74 | |
| (S)-5 | 131 ± 12 | 940 ± 130 | 1110 ± 360 | 2060 ± 580 | 69 | |
Data were obtained through the NIDA Addiction Treatment Discovery Program Contract (ADA12013) with Oregon Health & Science University.
ND = Not determined. Functional assays for each receptor were not conducted if the Ki value for the binding assay was >250 nM for 5-HT receptors.
Rat Liver Microsome Metabolic Stability.
Test compounds including 3-F analogues (9a, 10a, 9b, 10b) and 4, 5 as well as the R-, S- enantiomers of 9a, 9b, 4, and 5 were screened for phase I metabolic stability in rat liver microsomes following our previously described procedure.39 As seen in Figure 3A, of all the four 3-F analogues, 9a was the most stable with ~75% intact remaining, whereas 9b, 10b, and 10a showed lower stability with 38%, 34%, and 12% parent remaining, respectively. Individual R- and S- enantiomers of 9a and 9b (Figure 3B) showed higher stability with 57−70% intact remaining. Lastly, compounds 4 and 5 as well as their R- and S-enantiomers were found to be metabolically stable with 57− 86% remaining at 60 min.
Figure 3.

Phase I metabolic stability of 3-F analogues (9a, 9b, 10a, 10b), 4, 5, and R-, S-enantiomers of 9a, 9b, 4, and 5 in rat liver microsomes fortified with NADPH. The R- and S-enantiomers of 9a, 9b, 4, and 5 were found to be stable in rat liver microsomes with >50% intact remaining at 60 min.
Pharmacokinetics of R- and S-Enantiomers in Rats.
Owing to the promising in vitro metabolic stability and potency of the (R)- and (S)-enantiomers of 4 and 5, pharmacokinetic evaluation was conducted in rats following oral (PO) administration at a 10 mg/kg dose. As shown in Figure 4, all compounds were orally available, with (R)- and (S)-4 achieving a maximum plasma concentration (Cmax) of 454.4 ± 58.8 pmol/mL and 226.4 ± 28.9 pmol/mL, respectively, at 4 h (Tmax) post administration. In contrast, brain concentrations were higher with Cmax values of 1796.6 ± 228 pmol/g and 959.9 ± 53.3 pmol/g for the (R)-4 and (S)-4 enantiomers, respectively. Similarly, the overall exposure calculated from area under the curve (AUC0‑t) was higher for both the enantiomers in brain versus plasma, with a corresponding brain to plasma (b/p) ratio of 3.8 and 4.1 for (R)-4 and (S)-4, respectively.
Figure 4.

Pharmacokinetic evaluation of (R)- and (S)-enantiomers of 4 and 5 in rats following oral administration (10 mg/kg). Plasma and brain concentration vs time profile of (R)- and (S)-enantiomers of 4 and 5; concentration data is expressed as mean ± SD, n = 3, per time-point. Pharmacokinetic parameters are expressed as mean ± SD. Brain to plasma ratios are calculated from mean AUCbrain to AUCplasma. Data for (R)-5 is adapted from previously reported data.44
Likewise, (R)-5 and (S)-5 achieved a plasma Cmax of 305.5 ± 112.8 pmol/mL and 600.6 ± 108.1 pmol/mL and a brain Cmax of 3850 ± 513.7 pmol/g and 3666.7 ± 312.7 pmol/g, respectively. The corresponding plasma exposures (AUC0-t) for (R)-5 and (S)-5 were 1985 ± 278.6 h pmol/mL and 2678 ± 207.5 h pmol/mL, and brain exposures were 21 608 ± 1411 h pmol/g and 16 665 ± 1205 h pmol/g, respectively. Significant brain/plasma ratios for (R)-5 (~11) and (S)-5 (~6) suggests excellent brain penetrability.
Notably, both (R)- and (S)-4 and showed 12- and 88-fold higher brain levels, respectively, versus their D3R Ki values (Table 1) at 8 h post drug administration. Moreover (R)- and (S)-5 showed nearly 1000- and 7000-fold higher levels at 8 h versus their respective D3R Ki values (Table 1). Given this, the 10 mg/kg oral dose may serve as an effective starting dose in rodent self-administration studies, although this will need further confirmation.
CONCLUSION
Novel 3-F and 3,4-(methylenedioxy) analogues of lead molecules 4 and 5 were synthesized and evaluated for binding affinities at D2R and D3R; all modifications reported herein resulted in compounds with high D3R affinities and selectivities. Although modification of the PP or SP with a 3,4-(methylenedioxy) group was generally well tolerated, none of these analogues proved superior to 4 and 5 in terms of D3R binding affinities or selectivities. The R- and S-enantiomers of both 4 and 5 were resolved by two methods: (1) enantioselective synthesis using resolved chiral intermediates and (2) chiral preparative HPLC. The R-enantiomers of both the compounds were the eutomers and displayed D3R and D2R affinities comparable to the racemates, with the S-enantiomers having lower affinities at D3R and thus lower selectivities. R-4, (S)-4, and (R)-5 were D3R antagonists, whereas (S)-5 was a low efficacy partial agonist, a change in efficacy previously observed with (R)- and (S)-2. All four enantiomers displayed significant metabolic stability in rat liver microsomes and very high brain to plasma ratios when administered orally in rats. Finally, inconsequential off target binding affinities further support the development of lead candidates (R)-4 and (R)-5 as potential pharmacotherapeutics to treat OUD.
Replacement of the 3-OH in the linking chain between the PP and SP resulted in a series of interesting 3-F-analogues. Compounds 9a,b, direct analogues of 4 and 5, proved to have the highest D3R binding and selectivity profiles among the 3-F-analogues, and separation of enantiomers was achieved through enantioselective synthesis or chiral preparative chromatography in >99% ee. The D3R over D2R > 1000 selectivity profile of (S)-9b was notable and further emphasized the importance of the linker in the binding profiles of these compounds. Interestingly, D2R affinity was decreased by >10-fold between the (R)- and (S)-enantiomers, which has not been observed previously. The introduction of 3-F into the linker also resulted in metabolic stability in rat liver microsomes, suggesting these analogues will serve as future in vivo tools for further investigation into the role of D3R in SUD and as potential medication candidates to treat opioid use disorders.
EXPERIMENTAL METHODS
The reaction conditions and yields were not optimized. Anhydrous solvents and many reagents were purchased from Sigma-Aldrich Co. and were used without further purification. All other chemicals and reagents were purchased from LLC, AK Scientific, TCI America, Acros Organics, Maybridge, and Alfa Aesar. All amine final products with a 3-F linker were converted into the oxalate salts, and the other compounds were converted into HCl salts. Spectroscopic data and yields refer to the free base form of all compounds. Teledyne ISCO CombiFlash Rf or flash column chromatography was performed using silica gel (EMD Chemicals, Inc.; 230−400 mesh, 60 Å). 1H, 13C and 19F NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer. 1H chemical shifts are reported as parts per million (δ ppm) relative to tetramethylsilane (0.00 ppm) as an internal standard. Coupling constants are measured in Hz. Chemical shifts for 13C NMR spectra are reported as parts per million (δ ppm) relative to deuterated solvents. 19F spectra chemical shifts are reported relative to CDCl3/CFCl3 as internal standard. Chemical shifts, multiplicities, and coupling constants (J) have been reported and calculated using Vnmrj Agilent-NMR 400MR or MNova 9.0. Gas chromatography/mass spectrometry (GC/MS) data were acquired (where obtainable) using an Agilent Technologies (Santa Clara, CA) 6890N GC equipped with an HP-5MS column (cross-linked 5% PH ME siloxane, 30 m × 0.25 mm i.d. × 0.25 μm film thickness) and a 5973 mass-selective ion detector in electron-impact mode. Ultrapure grade helium was used as the carrier gas at a flow rate of 1.2 mL/min. The injection port and transfer line temperatures were 250 and 280 °C, respectively, and the oven temperature gradient used was as follows: the initial temperature (100 °C) was held for 3 min and then increased to 295 °C at 15 °C/ min over 13 min and finally maintained at 295 °C for 10 min. Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA), and the results agree within ±0.4% of calculated values. HRMS (mass error within 5 ppm) and MS/MS fragmentation analysis were performed on a LTQ-Orbitrap Velos (Thermo-Scientific, San Jose, CA) coupled with an electrospray ionization (ESI) source in positive ion mode. High-pressure liquid chromatography (HPLC) analysis was performed using an Agilent system coupled with a diode array detector (DAD). Separation of the analyte, purity and enantiomeric excess determinations was achieved employing one of the following different methods. HPLC Method A: CHIRALCEL OZ-H (Daicel Corporation CPI Company) column (4.6 mm × 250 mm, 5 μm). The mobile phase used (1 mL/min flow rate) was composed of 0.2% diethylamine in 2-propanol and hexanes with gradient elution, starting with 10% of 2-propanol linearly increasing to 30%, total run time was 30 min. The DAD detector wavelength was set at 254 nm, and the column temperature was maintained at 40 °C. A Sample of 20 μL with concentration of 0.5 mg/mL was injected for the analytical method. HPLC Method B: CHIRALPAK AD-H (Daicel Corporation CPI Company) column (4.6 mm × 250 mm, 5 μm). The mobile phase used (1 mL/min flow rate) was composed of 10% 2-propanol in hexanes with isocratic elution. The total run time was 80 min, the DAD detector wavelength was set at 254 nm, and the column temperature was maintained at 40 °C. A sample of 20 μL with a concentration of 0.5 mg/mL was injected for analytical method. Preparative HPLC Method C: CHIRALCEL OZ-H (Daicel Corporation CPI Company) semipreparative column (20 mm × 250 mm, 5 μm) performed on an Agilent Technologies HP series 1200 HPLC. The mobile phase used (10 mL/min flow rate) was composed of 10% 2-propanol in hexanes with isocratic resolution. The total run time was 50 min and DAD detector wavelength was set at 254 nm. A sample of 0.6 mL with a concentration of 10 mg/mL was injected for preparative separation. Preparative HPLC Method D: CHIRALPAK AD-H (Daicel Corporation CPI Company) semipreparative column (21 mm × 250 mm, 5 μm). The mobile phase used (18 mL/min flow rate) was composed of 10% 2-propanol in hexanes with isocratic resolution. The total run time was 60 min and DAD detector wavelengths were set at 254 and 280 nm. A sample of 5.0 mL with a concentration of 10 mg/mL was injected for preparative separation. cLogP and polar surface area (PSA) values were calculated using ChemDraw Professional Ultra 15.0. Melting point determination was conducted using an OptiMelt automated melting point system and are uncorrected. Optical rotations were determined using a Jasco DIP-370 polarimeter. Based on NMR, HPLC, and combustion analysis data, all final compounds are ≥95% pure, unless otherwise stated.
General Amidation Procedure.
The appropriate aryl carboxylic acid (1.2 mmol) was dissolved in mixture of CHCl3 and dimethylformamide (8:2) and cooled to 0 °C. To this solution was added EDCI (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) HCl (1.3 mmol) and HOBt (hydroxybenzotriazole) (1.2 mmol mmol).45 After 30 min, the aryl amine (1.0 mmol) was added at 0 °C. To this reaction mixture was added DIPEA (N,N-diisopropylethylamine) (1.5 mmol), and the mixture was continuously stirred at room temperature for 8 h. The reaction progress was monitored by thin layer chromatography (TLC). After completion of the reaction, the mixture was basified to pH 9 with saturated NaHCO3 water solution. The product was extracted with CHCl3 (50 mL × 4). The combined organic fractions were evaporated to afford the crude amide product. All final products were purified using flash column chromatography eluting with CHCl3 and acetone solvent systems, as described.
(R)-N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)3-hydroxybutyl)-1H-indole-2-carboxamide (R-4).
The compound was prepared from indole-2-carboxylic acid (0.22 g, 1.33 mmol) and (R)-12a (0.38 g, 1.11 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product as a colorless solid (0.274 g, 51% yield). 1H NMR (400 MHz, CDCl3) δ 9.33 (s, 1H), 7.64 (d, J = 7.9 Hz, 1H), 7.43 (t, J = 8.1 Hz, 1H), 7.40 (s, 1H), 7.32−7.22 (m, 1H), 7.13 (t, J = 7.6 Hz, 1H), 6.85 (d, J = 8.4 Hz, 2H), 6.62 (s, 1H), 3.91 (m, 2H), 3.84 (s, 3H), 3.58−3.41 (m, 1H), 3.16 (br s, 4H), 2.85 (s, 2H), 2.56 (dd, J = 15.1, 7.6 Hz, 4H), 2.45 (d, J = 6.4 Hz, 2H), 1.82 (m, 1H), 1.64 (m, 1H), 1.21 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 161.52, 146.26, 145.95, 140.77, 136.06, 131.15, 128.30, 127.72, 124.25, 122.33, 121.73, 120.52, 116.67, 111.87, 101.88, 66.43, 63.75, 59.09, 53.69, 50.39, 38.05, 33.26, 28.47, 15.40. Mp 194−195 °C. Anal. (C26H33ClN4O3) C, H, N. [α]23D −27.89 (CHCl3, c 0.23). (R-4) was analyzed by HPLC Method A, retention time = 11.004 min and enantiomeric excess was >99% (Figure S1, panel B).
(S)-N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)3-hydroxybutyl)-1H-indole-2-carboxamide (S-4).
The compound was prepared from indole-2-carboxylic acid (0.127 g, 0.78 mmol) and (S)-12a (0.225 g, 0.658 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product as a colorless solid (0.176 g, 55% yield). 1H NMR (400 MHz, CDCl3) δ 9.42 (s, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.43−7.40 (m, 2H), 7.31−7.22 (m, 1H), 7.13 (t, J = 7.5 Hz, 1H), 6.89−6.81 (d, J = 2.1 Hz, 2H), 6.62 (d, J = 1.5 Hz, 1H), 4.02−3.86 (m, 2H), 3.85 (s, 3H), 3.50 (m, 1H), 3.15 (br s, 4H), 2.84 (m, 2H), 2.55 (m, 4H), 2.48−2.34 (d, J = 2.1 Hz, 2H), 1.91−1.77 (m, 1H), 1.64 (m, 1H), 1.21 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 161.47, 146.41, 145.76, 140.76, 136.16, 131.14, 128.16, 127.61, 124.18, 122.44, 121.84, 120.38, 116.67, 111.77, 101.69, 66.48, 63.74, 58.98, 53.68, 50.39, 37.96, 33.10, 28.46, 28.21, 15.42. Mp 191−192 °C. Anal. (C26H33ClN4O3) C, H, N. [α]23D +34.54 (CHCl3, c 0.16). (S-4) was analyzed by HPLC Method A, retention time = 18.356 min and enantiomeric excess was >99% (Figure S1, panel C).
(R)-N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-hydroxybutyl)-1H-indole-2-carboxamide ((R)-5).
The compound was prepared from indole-2-carboxylic acid (0.64 g, 3.95 mmol) and (R)-12b (1.12 g, 3.59 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product as a pale-pink solid (0.98 g, 60% yield). 1H NMR (400 MHz, CDCl3) δ 9.44 (s, 1H), 7.64 (d, J = 7.7 Hz, 1H), 7.44 (d, J = 7.9 Hz, 2H), 7.33− 7.21 (m, 1H), 7.15 (m, 2H), 6.95 (dd, J = 17.8, 7.6 Hz, 2H), 6.85 (s, 1H), 4.01−3.82 (m, 2H), 3.51 (m, 1H), 3.07 (br s, 4H), 2.89 (m, 2H), 2.78 (dd, J = 14.8, 7.3 Hz, 2H), 2.63 (m, 2H), 2.52−2.40 (m, 2H), 1.83 (m, 1H), 1.64 (m, 1H), 1.23 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 161.54, 149.26, 143.23, 136.18, 131.14, 128.72, 127.64, 126.78, 124.22, 124.16, 121.70, 120.49, 117.97, 111.85, 101.83, 66.45, 63.70, 51.63, 38.09, 33.26, 27.46, 14.02. The HCl salt was precipitated from acetone. Mp 233−234 °C. Anal. (C25H31ClN4O2·HCl) C, H, N. [α]23D −45.29 (CHCl3, c 0.23). (R5) was analyzed by HPLC Method A, retention time = 14.698 min and enantiomeric excess was >98% (Figure S2, panel B).
(S)-N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-hydroxybutyl)-1H-indole-2-carboxamide (S-5).
The compound was prepared from indole-2-carboxylic acid (0.50 g, 3.14 mmol) and (S)-12b (0.89 g, 2.86 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product as a pale-yellow solid (0.78 g, 59% yield). 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 1H), 7.64 (d, J = 7.8 Hz, 1H), 7.45 (dd, J = 8.3, 0.7 Hz, 2H), 7.33−7.21 (m, 1H), 7.14 (dt, J = 7.9, 4.3 Hz, 2H), 6.95 (ddd, J = 18.5, 7.8, 1.4 Hz, 2H), 6.86 (s, 1H), 4.07−3.82 (m, 2H), 3.51 (m, 2H),3.07 (br s, 3H), 2.89 (m, 2H), 2.78 (q, J = 7.5 Hz, 2H), 2.62 (m, 2H), 2.51−2.38 (m, 2H), 1.84 (ddd, J = 13.5, 5.6, 3.3 Hz, 1H), 1.73−1.55 (m, 1H), 1.24 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 161.56, 149.36, 143.16, 136.20, 131.02, 128.66, 127.76, 126.90, 124.20, 124.15, 121.92, 120.48, 117.97, 111.92, 101.82, 66.40, 63.70, 51.64, 38.11, 33.17, 27.47, 14.08. The HCl salt was precipitated from acetone. Mp 237−238 °C. Anal. (C25H31ClN4O2·HCl) C, H, N. [α]23D +48.13 (CHCl3, c 0.25). (S-5) was analyzed by HPLC Method A, retention time = 31.560 min) and enantiomeric excess was >99% (Figure S2, panel C).
(R)-2-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-hydroxybutyl)isoindoline-1,3-dione ((R)-6a).
(R)-2-(2-(oxiran-2-yl)ethyl)isoindoline-1,3-dione, (R)-19 (0.66 g, 3.04 mmol) was added to a solution of 11a (0.61 g, 2.43 mmol) in 2-propanol (15 mL) and stirred at reflux for 3 h. The crude product was purified by flash chromatography using 35% EtOAc in hexanes as eluent to give the desired product as a brown oil in 70% yield.38 1H NMR (400 MHz, CDCl3) δ 7.85 (br s, 2H), 7.72 (br s, 2H), 6.84 (s, 1H), 6.60 (s, 1H), 3.97−3.68 (m and s, 3H and 3H), 3.11 (br s, 4H), 2.78 (m, 2H), 2.67−2.50 (m, 4H), 2.49−2.27 (m, 2H), 1.79 (d, J = 6.5 Hz, 2H), 1.60 (br s, 1H), 1.20 (t, J = 6.8 Hz, 3H).
(S)-2-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-hydroxybutyl)isoindoline-1,3-dione ((S)-6a).
(S)-2-(2-(oxiran-2-yl)ethyl)isoindoline-1,3-dione, (S)-19 (0.79 g, 3.13 mmol) was added to a mixture of 11a (0.84 g, 3.91 mmol) in 2-propanol (15 mL) and stirred at reflux for 3 h. The crude product was purified by flash chromatography using 40% EtOAc in hexanes as eluent to give the desired product as brown oil in 65% yield.37 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 3.2 Hz, 2H), 7.71 (d, J = 2.5 Hz, 2H), 6.84 (s, 1H), 6.60 (s, 1H), 3.90 (m, 2H), 3.82 (s, 3H), 3.77 (m, 1H), 3.11 (br s, 4H), 2.78 (m, 2H), 2.54 (m, 4H), 2.47−2.30 (m, 2H), 1.79 (m, 2H), 1.60 (br s, 1H), 1.20 (t, J = 7.6 Hz, 3H).
(R)-2-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-hydroxybutyl)isoindoline-1,3-dione ((R)-6b).
The compound was prepared starting from 11b (0.70 g, 3.22 mmol) following the same procedure described for ((R)-6a). The crude product was purified by flash chromatography using 40% EtOAc in hexanes as eluent to give the desired product as brown oil in 75% yield. 1H NMR (400 MHz, CDCl3) δ 7.85 (br s, 2H), 7.72 (br s, 2H), 7.15 (t, J = 7.3 Hz, 1H), 6.93 (dd, J = 17.7, 7.6 Hz, 2H), 4.00−3.70 (m, 3H), 3.03 (br s, 4H), 2.91−2.69 (m, 4H), 2.59 (br s, 2H), 2.51−2.30 (m, 2H), 1.79 (m, 2H), 1.60 (br s, 1H) 1.22 (t, J = 7.1 Hz, 3H).
(S)-2-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-hydroxybutyl)isoindoline-1,3-dione ((S)-6b).
The compound was prepared starting from (S)-19 (0.90 g, 4.14 mmol) and 11b (0.74 g, 3.31 mmol) following the same procedure described for ((S)6a). The product was purified by flash column chromatography using 40% EtOAc in hexanes, as the eluent, to give the desired product in 85% yield, as pale-yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.92− 7.79 (m, 2H), 7.77−7.65 (m, 2H), 7.15 (t, J = 7.5 Hz, 1H), 6.92 (dd, J = 17.9, 7.6 Hz, 2H), 4.03−3.82 (m, 2H), 3.78 (m, 1H), 3.03 (br s, 4H), 2.89−2.67 (m, 4H), 2.59 (m, 2H), 2.50−2.30 (m, 2H), 1.79 (q, J = 6.5 Hz, 2H), 1.60 (br s, 1H), 1.23 (t, J = 7.8 Hz, 3H).
2-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-fluorobutyl)isoindoline-1,3-dione (7a).
To compound 6a (1.68 g, 3.56 mmol) was added anhydrous dichloromethane (DCM) and the reaction mixture was cooled to −78 °C. Maintaining the same temperature, DAST (0.86 g, 0.84 mL, 5.34 mmol) dissolved in anhydrous DCM was added dropwise, and the reaction was stirred at 20 °C for 8 h.36,37 Thin layer chromatography was used to monitor the progress of the reaction. Once the reaction was completed, ice cold water was added to the reaction mixture and the product was extracted with ethyl acetate (EtOAc) and purified by flash column chromatography using 20% EtOAc in hexanes, as the eluent, to give the desired product in 42% yield, as brown oil. 1H NMR (400 MHz, CDCl3) δ 7.95−7.80 (m, 2H), 7.79−7.62 (m, 2H), 6.82 (s, 1H), 6.57 (s, 1H), 4.92−4.66 (m, 1H), 3.97−3.74 (m and s, 2H and 3H), 3.64− 3.41 (m, 2H), 3.11 (d, J = 3.0 Hz, 4H), 2.80−2.59 (m, 4H), 2.54 (td, J = 7.5, 4.4 Hz, 2H), 2.18−1.78 (m, 2H), 1.20 (dt, J = 12.1, 5.0 Hz, 3H).
(R)-2-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)3-fluorobutyl)isoindoline-1,3-dione ((R)-7a).
The compound was prepared starting from (R)-6a (5.83 g, 1.238 mmol) following the same procedure described for 7a. The product was purified by flash column chromatography using 30% EtOAc in hexanes, as the eluent, to give the desired product in 85% yield, as a pale-yellow solid. 1H NMR (400 MHz, CDCl3) was identical to the one described for 7a.
(S)-2-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)3-fluorobutyl)isoindoline-1,3-dione ((S)-7a).
The compound was prepared starting from (S)-6a (0.44 g, 0.93 mmol) following the same procedure described for 7a. The product was purified by flash column chromatography using 40% EtOAc in hexanes, as the eluent, to give the desired product in 70% yield, as a pale-yellow solid. 1H NMR (400 MHz, CDCl3) was identical to the one described for 7a.
2-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-fluorobutyl)isoindoline-1,3-dione (7b).
The compound was prepared starting from 6b (2.45 g, 5.56 mmol) following the same procedure described for 7a. The product was purified by flash column chromatography using 20% EtOAc in hexanes, as the eluent, to give the desired product in 73% yield, as a pale-yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.90−7.80 (m, 2H), 7.77−7.66 (m, 2H), 7.14 (t, J = 7.8 Hz, 2H), 6.92 (ddd, J = 14.9, 7.8, 1.5 Hz, 1H), 4.94−4.65 (m, 1H), 4.12 (q, J = 7.2 Hz, 2H), 3.95−3.76 (m, 2H), 3.04 (s, 2H), 2.74 (ddd, J = 13.8, 12.9, 7.4 Hz, 2H), 2.64−2.44 (m, 2H), 2.20−1.92 (m, 4H), 1.58 (s, 2H), 1.24 (t, J = 16.2, 7.5 Hz, 3H).
(R)-2-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-fluorobutyl)isoindoline-1,3-dione ((R)-7b).
The compound was prepared starting from (R)-6b (0.65 g, 1.47 mmol) following the same procedure described for 7a. The product was purified by flash column chromatography using 30% EtOAc in hexanes, as the eluent, to give the desired product as a pale-yellow solid in 62% yield. 1H NMR (400 MHz, CDCl3) was identical to the one described for 7b.
(S)-2-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-fluorobutyl)isoindoline-1,3-dione ((S)-7b).
The compound was prepared starting from (S)-6b (1.42 g, 3.20 mmol) following the same procedure described for 7a. The product was purified by flash column chromatography using 40% EtOAc in hexanes, as the eluent, to give the desired product as a pale-yellow solid in 80% yield. 1H NMR (400 MHz, CDCl3) was identical to the one described for 7b.
4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-fluorobutan-1-amine (8a).
Hydrazine (0.19 g, 0.128 mL, 1.47 mmol) was added to a solution of compound 7a (0.70 g, 1.47 mmol) in ethanol (EtOH) (15 mL) and stirred at reflux overnight. The solvent was evaporated, and the reaction mixture was basified with 20% aqueous K2CO3 solution (15 mL) and extracted in CHCl3 (2 × 15 mL). The organic layers were combined, dried on anhydrous Na2SO4, filtered, and concentrated to afford a yellow oil in 98% yield, which was sufficiently pure to be used in the next step without further purification.
(R)-4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-fluorobutan-1-amine ((R)-8a).
The compound was prepared starting from (R)-7a (0.60 g, 1.26 mmol) following the same procedure described for 8a. The crude product was used in the next step without further purification.
(S)-4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-fluorobutan-1-amine ((S)-8a).
The compound was prepared starting from (S)-7a (0.345 g, 0.72 mmol) following the same procedure described for 8a. The crude product was used in the next step without further purification.
4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-fluorobutan-1amine (8b).
The compound was prepared starting from 7b (1.80 g, 4.05 mmol) following the same procedure described for 8a. The compound was obtained as a brown oil in 94% yield, which was sufficiently pure to be used in the next step without further purification.
(R)-4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-fluorobutan1-amine ((R)-8b).
The compound was prepared starting from (R)-7b (0.67 g, 1.51 mmol) following the same procedure described for 8a. The crude product was used in the next step without further purification.
(S)-4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-fluorobutan1-amine ((S)-8b).
The compound was prepared starting from (S)-7b (1.27 g, 2.86 mmol) following the same procedure described for 8a. The crude product was used in the next step without further purification.
N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-fluorobutyl)-1H-indole-2-carboxamide (9a).
The compound was prepared from indole-2-carboxylic acid (0.13 g, 0.80 mmol) and 8a (0.25 g, 0.72 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product as a yellow oil (0.23 g, 65% yield). 1H NMR (400 MHz, CDCl3) δ 9.16 (s, 1H), 8 (s, 1H), 7.66 (t, J = 8.7 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.34−7.22 (m, 1H), 7.15 (dd, J = 13.2, 6.1 Hz, 1H), 6.84 (dd, J = 7.6, 1.7 Hz, 1H), 6.60 (d, J = 2.0 Hz, 1H), 6.55 (s, 1H), 4.90 (m, 1H), 3.83 (s, 3H), 3.79−3.58 (m, 2H), 3.15 (br s, 2H), 2.95 (s, 1H), 2.92−2.84 (m, 2H), 2.75−2.64 (m, 3H), 2.56 (dt, J = 15.2, 5.6 Hz, 2H), 2.02 (dd, J = 17.8, 14.3 Hz, 2H), 1.59 (s, 2H), 1.20 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 161.70, 146.41, 145.68, 140.77, 136.29, 130.37, 128.18, 127.68, 124.53, 122.49, 121.76, 120.53, 116.84,111.63, 101.80, 92.43, 90.23, 61.82, 58.64, 54.22, 50.26, 36.44, 33.26, 28.34, 15.03. 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.88 (m, 1F). The oxalate salt was precipitated from acetone. Mp 198−199 °C. Anal. (C26H32ClFN4O2·1.2C2H2O4·H2O) C, H, N. 9a was analyzed by HPLC Method B, retention time = 60.434 and 62.026 min and purity was determined to be >98% (Figure S3, panel A).
(R)-N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)3-fluorobutyl)-1H-indole-2-carboxamide((R)-9a).
The compound was prepared from indole-2-carboxylic acid (0.14 g, 0.90 mmol) and (R)-8a (0.26 g, 0.75 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 35% acetone/CHCl3 as eluent to give the desired product as pale-yellow solid (0.235 g, 64.0% yield). 1H NMR (400 MHz, CDCl3), 13C NMR (101 MHz, CDCl3), and 19F NMR (376 MHz, CDCl3/CFCl3) were identical to the ones described for 9a. The oxalate salt was precipitated from acetone. Mp 202−203 °C. Anal.(C26H32ClFN4O2·C2H2O4·0.5H2O) C, H, N. [α]23D +11.69 (CHCl3, c 1.0). (R)-9a was analyzed by HPLC Method B, retention time = 62.022 min and enantiomeric excess was >99% (Figure S3).
(S)-N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)3-fluorobutyl)-1H-indole-2-carboxamide ((S)-9a).
The compound was prepared from indole-2-carboxylic acid (0.29 g, 1.82 mmol) and (S)-8a (0.57 g, 1.65 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product (0.54 g, 66% yield). 1H NMR (400 MHz, CDCl3), 13C NMR (101 MHz, CDCl3), and 19F NMR (376 MHz, CDCl3/CFCl3) were identical to the ones described for 9a. The oxalate salt was precipitated from acetone. Mp 202−203 °C. Anal. (C26H32ClFN4O2·C2H2O4·H2O) C, H, N. [α]23D −9.69 (MeOH, c 1.0), (S)-9a was analyzed by HPLC Method B, retention time = 60.353 min and enantiomeric excess was >99% (Figure S3, panel B).
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-fluorobutyl)1H-indole-2-carboxamide (9b).
The compound was prepared from indole-2-carboxylic acid (0.34 g, 2.10 mmol) and 8b (0.60 g, 1.91 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/ CHCl3 as eluent to give the desired product as a yellow solid (0.80 g, 91% yield). 1H NMR (400 MHz, CDCl3) δ 9.11 (s, 1H), 7.86 (s, 1H), 7.49 (s, 1H), 7.28 (s, 1H), 7.19−7.06 (m, 1H), 7.06−6.93 (m, 1H), 6.85−6.73 (m, 2H), 6.68 (s, 1H), 6.48 (s, 1H), 4.76 (m, 1H), 3.53 (d, J = 25.1 Hz, 2H), 2.92 (br s, 2H), 2.80 (ddd, J = 6.7, 3.6, 1.8 Hz, 3H), 2.73 (ddd, J = 5.1, 3.6, 1.8 Hz, 3H), 2.67−2.54 (m, 1H), 1.85 (s, 2H), 1.51 (s, 3H), 1.16−0.98 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 161.68, 149.42, 143.22, 136.16, 130.50, 128.69, 127.62, 126.88, 124.31, 124.10, 121.84, 120.67, 117.97, 111.80, 101.97, 92.45, 90.77, 62.22, 62.01, 54.00, 51.27, 33.20, 32.99, 27.45, 14.08. 19F NMR (376 MHz, CDCl3/CFCl3) δ −182.303 to −182.513 (m, 1F). The oxalate salt was precipitated from acetone. Mp 188−189 °C. Anal. (C25H29ClFN3O2·C2H2O4·0.5H2O) C, H, N. 9b was analyzed by HPLC Method B, retention time = 45.655 and 49.301 min and purity was determined to be >98% (Figure S4, panel A).
(R)-N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-fluorobutyl)-1H-indole-2-carboxamide ((R)-9b).
The compound was prepared from indole-2-carboxylic acid (0.20 g, 1.28 mmol) and (R)-8b (0.32 g, 1.02 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product colorless solid (0.32 g, 68.0% yield). 1H NMR (400 MHz, CDCl3), 13C NMR (101 MHz, CDCl3), and 19F NMR (376 MHz, CDCl3/CFCl3) were identical to the ones described for 9b. The oxalate salt was precipitated from acetone. Mp 193−194 °C. Anal. (C25H30ClFN4O· C2H2O4) C, H, N. [α]23D +9.18 (CHCl3, c 1.1). (R)-9b was analyzed by HPLC Method B, retention time = 47.907 min and enantiomeric excess was >99% (Figure S4, panel C).
(S)-N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-fluorobutyl)-1H-indole-2-carboxamide ((S)-9b).
The compound was prepared from indole-2-carboxylic acid (0.38 g, 2.37 mmol) and (S)-8b (0.74 g, 1.97 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product in (0.815 g, 90% yield). 1H NMR (400 MHz, CDCl3), 13C NMR (101 MHz, CDCl3), and 19F NMR (376 MHz, CDCl3/CFCl3) were identical to the ones described for 9b. The oxalate salt was precipitated from acetone. Mp 195−196 °C. Anal. (C25H30ClFN4O·C2H2O4) C, H, N. [α]23D −9.87 (MeOH, c 0.8). (S)-9b was analyzed by HPLC Method B, retention time = 46.085 min and enantiomeric excess was >99% (Figure S4, panel B).
N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-fluorobutyl)benzofuran-2-carboxamide (10a).
The compound was prepared from benzofuran-2-carboxylic acid (0.13 g, 0.80 mmol) and 8a (0.25 g 0.727 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product as a pale pink oil (0.25 g, 70% yield). 1H NMR (400 MHz, CDCl3) δ 7.72−7.64 (m, 1H), 7.51−7.44 (m, 2H), 7.44−7.36 (m, 1H), 7.33− 7.26 (m, 1H), 7.01 (m, 1H), 6.85 (d, J = 2.0 Hz, 1H), 6.61 (d, J = 2.0 Hz, 1H), 4.89 (m, 1H), 3.84 (s, 3H), 3.74−3.61 (m, 2H), 3.16 (br s, 3H), 2.79−2.64 (m, 4H), 2.59−2.48 (m, 2H), 2.24−1.97 (m, 2H), 1.59 (br s, 3H), 1.19 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.00, 154.65, 148.55, 146.26, 146.01, 140.71, 128.23, 127.61, 126.79, 123.60, 122.72, 122.33, 116.60, 111.69, 110.41, 106.50, 92.03, 90.30, 61.99, 59.24, 54.36, 50.18, 35.88, 33.14, 28.53, 15.39. 19F NMR (376 MHz, CDCl3/CFCl3) δ−182.153 to −182.403 (m, 1F). The oxalate salt was precipitated from acetone. Mp 179−180 °C. Anal. (C26H31ClFN3O3·1.1C2H2O4·H2O) C, H, N.
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-fluorobutyl)benzofuran-2-carboxamide (10b).
The compound was prepared from benzofuron-2-carboxylic acid (0.33 g, 2.10 mmol) and 8b (0.60 g, 1.91 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desire product as a pale-yellow solid (0.78 g 90% yield). 1H NMR (400 MHz, CDCl3) δ 7.67 (ddd, J = 7.8, 1.2, 0.7 Hz, 1H), 7.48 (ddd, J = 5.4, 4.5, 3.0 Hz, 2H), 7.41 (ddd, J = 8.3, 7.2, 1.3 Hz, 1H), 7.32−7.26 (m, 1H), 7.15 (t, J = 7.8 Hz, 1H), 7.01 (s, 1H), 6.93 (ddd, J = 15.4, 7.8, 1.5 Hz, 2H). 5.03− 4.75 (m, 1H), 3.75−3.61 (m, 2H), 3.08 (br s, 4H), 2.83−2.71 (m, 6H), 2.18−1.88 (m, 2H), 2.61−2.69 (m, 1H), 1.61 (br s, 2H), 1.27− 1.13 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 158.92, 154.70, 149.48, 148.62, 143.22, 128.59, 127.50, 126.81, 124.02, 123.68, 122.71, 117.81, 111.69, 110.39, 92.14, 90.36, 62.21, 61.96, 54.09, 51.40, 35.94, 35.88, 33.17, 32.92, 27.34, 14.08. 19F NMR (376 MHz, CDCl3/CFCl3) δ −182.530 to −182.660 (m, 1F). The oxalate salt was precipitated from acetone. Mp 175−176 °C. Anal. (C25H29ClFN3O2·1.3C2H2O4·H2O) C, H, N.
4-Amino-1-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)butan-2-ol (12a).
2-(2-(Oxiran-2-yl)ethyl)isoindoline-1,3-dione 19 (0.76 g, 3.54 mmol)46 was added to a solution of 11a (0.90 g, 3.54 mmol) in 2-propanol (15 mL) and stirred at reflux for 3 h. The formation of 2-(4-(4-(3-chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-hydroxybutyl)isoindoline-1,3-dione intermediate was confirmed by thin layer chromatography. The reaction mixture was cooled to 20 °C, hydrazine was added, and the reaction mixture was heated to reflux, overnight. The solvent was evaporated, and the residue basified to pH 9, with 20% aq K2CO3 solution (20 mL), followed by extraction with CHCl3. The combined organic layers were dried on anhydrous Na2SO4, filtered, and the solvent was evaporated. The crude product was used in the following step without further purification.
(R)-4-Amino-1-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin1-yl)butan-2-ol ((R)-12a).
The compound was prepared starting from (R)-19 (0.528 g, 2.40 mmol)38 and 11a (0.53 g, 2.4 mmol) following the same procedure described for 12a. The crude product was used in the following step without further purification.
(S)-4-Amino-1-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin1-yl)butan-2-ol ((S)-12a).
The compound was prepared starting from 11a (0.30 g, 1.20 mmol) and (S)-19 (0.28 g, 1.30 mmol)38 following the same procedure described for 12a. The crude product was used in the following step without further purification.
4-Amino-1-(4-(2-chloro-3-ethylphenyl)piperazin-1-yl)butan-2-ol (12b).
The compound was prepared starting from 11b (0.43 g, 1.92 mmol) following the same procedure described for 12a. The crude product was used in the following step without further purification.
(R)-4-Amino-1-(4-(2-chloro-3-ethylphenyl)piperazin-1-yl)butan2-ol ((R)-12b).
The compound was prepared starting from (R)-19 (1.24 g, 5.72 mmol) and 11b (1.28 g, 5.70 mmol) following the same procedure described for 12a. The crude product was used in the following step without further purification.
(S)-4-Amino-1-(4-(2-chloro-3-ethylphenyl)piperazin-1-yl)butan2-ol ((S)-12b).
The compound was prepared starting from 11b (0.85 g, 3.82 mmol) and (S)-19 (0.83 g, 3.82 mmol)38 following the same procedure described for 12a. The crude product was used in the following step without further purification.
N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-hydroxybutyl)benzo[d] [1,3]dioxole-5-carboxamide (13a).
The compound was prepared from piperonylic acid (0.25 g, 1.51 mmol) and 12a (0.43 g, 1.25 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 40% acetone/CHCl3 as eluent to give the desired product as a colorless solid (0.34 g, 55% yield). 1H NMR (400 MHz, CDCl3) δ 7.38−7.27 (m, 2H), 7.21 (s, 1H), 6.90−6.76 (m, 2H), 6.61 (d, J = 1.9 Hz, 1H), 6.02 (s, 2H), 3.95−3.79 (m and s, 2H and 3H), 3.51−3.33 (m, 1H), 3.15 (br s, 4H), 2.86 (m, 2H), 2.55 (m, 4H), 2.44 (dd, J =10.3, 5.3 Hz, 2H), 1.85 (m, 1H), 1.62– 1.58 (m, 2H), 1.20 (dd, J = 9.2, 6.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.50, 149.93, 147.84, 146.36, 145.72, 140.80, 140.07, 128.83, 128.21, 122.32, 121.47, 116.66, 107.93, 107.52, 101.56, 66.55, 63.74, 59.10, 50.29, 38.39, 36.47, 36.38, 33.26, 28.46, 15.37. The HCl salt was precipitated from acetone. Mp 183−184 °C. Anal. (C25H32ClN3O5·HCl·1.5H2O) C, H, N. HRMS = 490.21023 [M + H+].
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-hydroxybutyl)benzo[d][1,3]dioxole-5-carboxamide (13b).
The compound was prepared from piperonylic acid (0.23 g, 1.41 mmol) and 12b (0.40 g, 1.28 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/ CHCl3 as eluent to give the desired product as colorless solid (0.445 g, 75% yield). 1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 8.3 Hz, 2H), 7.30 (s, 1H), 7.16 (t, J = 7.7 Hz, 1H), 6.96 (d, J = 7.3 Hz, 1H), 6.92 (d, J = 8.0 Hz, 1H), 6.81 (d, J = 8.0 Hz, 1H), 6.01 (s, 2H), 3.99− 3.78 (m, 2H), 3.50−3.34 (m, 1H), 3.07 (br s, 4H), 2.88 (m, 2H),2.77 (q, J = 7.5 Hz, 2H), 2.62 (m, 2H), 2.52−2.35 (m, 2H), 1.88− 1.71 (m, 1H), 1.68−1.50 (m, 1H), 1.31−1.13 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 166.45, 150.06, 149.34, 147.84, 143.31, 129.04, 128.71, 126.88, 124.14, 121.47, 117.96, 107.91, 107.57, 101.54, 66.63, 63.69, 51.59, 38.54, 33.24, 27.45, 14.06. The HCl salt was precipitated from acetone. Mp 198−199 °C. Anal. (C24H30ClN3O2·HCl·0.75H2O) C, H, N.
6-Nitrobenzo[d][1,3]dioxole-5-carbaldehyde (15).
3,4-(Methylenedioxy)-benzaldehyde 14 (5.0 g, 33.3 mmol) was added portion-wise to a mixture of dichloroethane and fuming HNO3 (90%, 2 equiv) at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for an additional 2 h. Thin layer chromatography was used to monitor the progress of reaction. Ice cold water (50 mL) was added, the mixture was extracted with CHCl3, and washed with brine. The organic phase was dried on anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by flash chromatography using 20% EtOAc in hexanes as eluent, to give the desired product as a yellow solid in 85% yield. 1H NMR (400 MHz, CDCl3) δ 10.30 (s, 1H), 7.53 (s, 1H), 7.34 (s, 1H), 6.22 (s, 2H). GC/MS (electron ionization, EI) m/z 195 (M+), (retention time = 8.258 min).
5-Nitro-6-vinylbenzo[d][1,3]dioxole (16).
Lithium tert-butoxide (LiOtBu) (4.46 g, 55.8 mmol) was added portionwise to a suspension of methyltriphenylphosphonium bromide (7.90 g, 22.3 mmol) in anhydrous THF (100 mL) at −78 °C, under an argon atmosphere. The reaction mixture was allowed to warm to room temperature and stirred for 3 h. The formation of yellow precipitate was observed, then a solution of 15 (5.0 g, 25.6 mmol) in anhydrous THF (50 mL) was added dropwise over 30 min at 0 °C. The reaction mixture was maintained at room temperature and stirred for an additional 8 h. The reaction mixture was quenched with addition of saturated aq NH4Cl solution (50 mL), and the THF was removed under vacuum. The crude product was extracted with EtOAc. The organic layers were combined, dried on anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by flash chromatography using 5% Et2O in hexanes as eluent to give the desired product as a yellow solid in 85% yield. 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 8.6 Hz, 1H),7.48 (s, 1H), 6.98 (s, 1H), 6.12 (s, 2H), 5.61 (d, J = 17.2 Hz, 1H), 5.41 (d, J = 10.6 Hz, 1H). GC/MS (EI) m/z 193 (M+), (retention time = 8.039 min).
6-Ethylbenzo[d][1,3]dioxol-5-amine (17).
A mixture of 16 (3.17 g, 14.8 mmol) and 10% Pd/C (0.350 g) in ethyl acetate (30 mL) was shaken on a Parr hydrogenator apparatus, under an atmosphere of hydrogen gas (H2, 45 psi) at room temperature, for 45 min. The reaction mixture was filtered through a Celite pad and evaporated under vacuum. The crude product was purified by flash chromatography using 2% MeOH/CHCl3 as eluent to give the desired product as a yellow oil in 80% yield. 1H NMR (400 MHz, CDCl3) δ 6.71−6.49 (m, 1H), 6.28 (d, J = 2.0 Hz, 1H), 5.84 (d, 2H), 3.44 (br s, 2H), 2.43 (q, J = 7.5 Hz, 2H), 1.203 (t, J = 7.5 Hz, 3H). GC/MS (EI) m/z 165 (M+), (retention time = 7.138 min).
1-(6-Ethylbenzo[d][1,3]dioxol-5-yl)piperazine (18).
A mixture of 17 (2.8 g, 15.13 mmol) and bis(2-chloroethyl)amine HCl (2.69 g, 15.1 mmol) in diethylene glycol monomethyl ether (4.0 mL) was heated at 150 °C for 7 h under argon atmosphere. The reaction mixture was cooled to room temperature, ice cold water was added, and the mixture was basified with 2 M aq NaOH to pH 8−9, followed by extraction with EtOAc. The combined organic layers were washed twice with ice cold water, dried on anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by column chromatography using 5% MeOH/CHCl3 as eluent to give the desired product as a brown solid in 70% yield. 1H NMR (400 MHz, CDCl3) δ 6.71 (d, J = 9.8 Hz, 1H), 5.92 (s, 1H), 5.30 (s, 2H), 3.49 (s, 1H), 3.36 (m, 4H), 3.11 (m, 4H), 2.59 (dd, J = 14.9, 7.3 Hz, 2H), 1.16 (t, J = 7.4 Hz, 3H) ppm. GC/MS (EI) m/z 234 (M+), (retention time = 9.833 min).
4-Amino-1-(4-(6-ethylbenzo[d][1,3]dioxol-5-yl)piperazin-1-yl)butan-2-ol (20).
The compound was prepared starting from 18 (0.32 g, 1.38 mmol) and 19 (0.36 g, 1.66 mmol).46 The crude product was used in the following steps without further purification.
N-(4-(4-(6-Ethylbenzo[d][1,3]dioxol-5-yl)piperazin-1-yl)-3-hydroxybutyl)-1H-indole-2-carboxamide (21).
The compound was prepared from indole-2-carboxylic acid (0.12 g 0.78 mmol) and 20 (0.21 g, 0.654 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product as a yellow solid (0.15 g, 50% yield). 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.43 (m, 2H), 7.33−7.26 (m, 1H), 7.13 (t, J = 7.5 Hz, 1H), 6.84 (s, 1H), 6.71 (s, 2H), 5.90 (s, 2H), 3.92 (m, 2H), 3.48 (m, 1H), 2.84 (m, 4H), 2.65−2.60 (dd, J = 20.5, 13.1 Hz, 2H and br s, 1H), 2.44 (d, J = 9.6 Hz, 2H), 1.85 (m, 1H), 1.64−1.61 (m, 4H), 1.17 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 161.38, 145.67, 144.75, 144.11, 136.03, 133.13, 131.12, 127.81, 124.26, 121.87, 120.54, 111.78, 108.62, 102.02, 101.74, 100.85, 66.51, 63.74, 53.22, 38.09, 33.24, 23.52, 15.30. The HCl salt was precipitated from acetone. Mp 133−135 °C. Anal. (C26H32N4O4·HCl·H2O) C, H, N.
N-(4-(4-(6-Ethylbenzo[d][1,3]dioxol-5-yl)piperazin-1-yl)-3-hydroxybutyl)benzofuran-2-carboxamide (22).
The compound was prepared from benzfuran-2-carboxylic acid (0.128 g, 0.79 mmol) and 20 (0.215 g, 0.66 mmol) according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product as a brown oil (0.18 g, 58% yield). 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 8.3 Hz, 2H), 7.45−7.40 (m, 2H),7.28 (m, 1H), 6.71 (s, 2H), 5.90 (s, 2H), 4.00−3.77 (m, 3H), 3.60− 3.43 (m, 1H), 2.86−2.80 (m, 6H), 2.65−2.60 (dt, J = 24.3, 12.1 Hz, 2H and br s, 1H), 2.46−2.43 (m, 1H), 1.83 (m, 1H), 1.72−1.47 (m, 2H), 1.17 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 158.95, 154.77, 149.03, 145.66, 144.79, 144.09, 133.13, 127.66, 126.63, 123.54, 122.59, 111.79, 110.01, 108.61, 102.02, 100.84, 65.88, 63.82, 53.21, 37.37, 33.64, 23.52, 15.29. The HCl salt was precipitated from acetone. Mp 127−128 °C. Anal. (C26H31N3O5·HCl·1.5H2O) C, H, N.
Radioligand Binding Assays.
Binding at dopamine D2-like receptors was determined similarly to previously described methods.39 Membranes were prepared from HEK293 cells expressing human D2LR or D3R grown in a 50:50 mix of DMEM and Ham’s F12 culture media, supplemented with 20 mM HEPES, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 1× antibiotic/antimycotic, 10% heatinactivated fetal bovine serum, and 200 μg/mL hygromycin (Life Technologies, Grand Island, NY) and kept in an incubator at 37 °C and 5% CO2. Upon reaching 80−90% confluence, cells were harvested using premixed Earle’s Balanced Salt Solution (EBSS) with 5 mM EDTA (Life Technologies) and centrifuged at 3 000 rpm for 10 min at 21 °C. The supernatant was removed, and the pellet was resuspended in 10 mL of hypotonic lysis buffer (5 mM MgCl2·6 H2O, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 14 500 rpm (~25 000g) for 30 min at 4 °C. The pellet was then resuspended in fresh EBSS binding buffer made from 8.7 g/L Earle’s Balanced Salts without phenol red (US Biological, Salem, MA), 2.2 g/L sodium bicarbonate, pH to 7.4. A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration and membranes were diluted to 500 μg/mL and stored in a −80 °C freezer for later use.
Radioligand competition binding experiments were conducted using thawed membranes on the test day, and each test compound was diluted into 10 half-log serial dilutions using 30% DMSO vehicle, starting from a 1 mM or 100 μM concentration. When it was necessary to assist solubilization of the drugs at the highest tested concentration, 0.1% acetic acid (final concetration v/v) was added alongside the vehicle. Previously frozen membranes were diluted in fresh EBSS to a 200 μg/mL (for hD2LR or hD3R) stock for binding. Radioligand competition experiments were conducted in 96-well plates containing 300 μL of fresh binding buffer, 50 μL of diluted test compound, 100 μL of membranes (20 μg/well total protein for hD2LR and hD3R, and 50 μL of [3H]N-methylspiperone radioligand diluted in binding buffer (0.4 nM final concentration; PerkinElmer). Nonspecific binding was determined using 10 μM (+)-butaclamol (Sigma-Aldrich, St. Louis, MO) and total binding was determined with 30% DMSO vehicle. All compound dilutions were tested in triplicate and the reaction incubated for 1 h at rt. The reaction was terminated by filtration through PerkinElmer Uni-Filter-96 GF/B, presoaked for 1 h in 0.5% polyethylenimine, using a Brandel 96-Well Plates Harvester Manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed 3 times with 3 mL (3 × 1 mL/well) of icecold binding buffer. A volume of 65 μL of PerkinElmer MicroScint 20 Scintillation Cocktail was added to each well, and filters were counted using a PerkinElmer MicroBeta Microplate Counter. IC50 values for each compound were determined from dose−response curves, and Ki values were calculated using the Cheng−Prusoff equation.43 When a complete inhibition could not be achieved at the highest tested concentrations, Ki values have been extrapolated by constraining the bottom of the dose−response curves (= 0% residual specific binding) in the nonlinear regression analysis. These analyses were performed using GraphPad Prism version 6.00 for Macintosh (GraphPad Software, San Diego, CA). All the results were rounded to three significant figures. Ki values were determined from at least 3 independent experiments and are reported as mean ± SEM.
Rat Microsomal Stability Assay.
The phase I metabolic stability assay was conducted in rat liver microsomes as previously described with minor modifications.39 In brief, the reaction was carried out with 100 mM potassium phosphate buffer, pH 7.4, in the presence of a NADPH regenerating system, (compound final concentration was 10 μM and 0.5 mg/mL microsomes). Positive controls for phase I metabolism (buprenorphine) were also evaluated. Compound disappearance was monitored over time using a liquid chromatography and tandem mass spectrometry (LC/MS/MS) method. All reactions were performed in triplicate.
Chromatographic analysis was performed using an Accela ultrahigh-performance system consisting of an analytical pump and an autosampler coupled with a TSQ Vantage mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). Chromatographic separation was achieved at ambient temperature using Agilent Eclipse Plus column (100 mm × 2.1 mm i.d.) packed with a 1.8 μm C18 stationary phase. The mobile phase consisted of 0.1% formic acid in acetonitrile and 0.1% formic acid in H2O with gradient elution, starting with 10% (organic) linearly increasing to 99% (0−2 min), maintaining at 99% (2−2.5 min), and re-equilibrating to 10% by 2.7 min. The total run time for each analyte was 4.5 min. The mass transitions used for compounds for LC/MS/MS analysis are given in Table S2.
Pharmacokinetics in Rats.
Male Long−Evans rats (Charles River Laboratories, Raleigh, NC, 3 animals per time point × 7 time points × 4 compounds = 84 in total) were used to study pharmacokinetic profiles of R- and S-enantiomers of 4 and 5 in rats following oral administration (10 mg/kg). i.v. catheterization of the right external jugular vein for blood sampling was performed under sodium pentobarbital (60 mg/kg, i.p.) anesthesia, utilizing standard aseptic surgical techniques as we reported previously.30 After recovery from surgery, each animal received a single p.o. dose of 10 mg/kg of R- or S-enantiomers of 4 or 5. The dose administered of each test compound was based on the findings in our behavioral assays reported previously.30 Blood samples were collected via a jugular vein catheter prior to dosing and after dosing at different time points (0,0.5, 1, 2, 4, 8, and 24 h after p.o. administration). After blood sampling at each time point, animals were euthanized with an overdose (100 mg/kg) of pentobarbital and then brains were taken out. Blood samples were centrifuged to obtain plasma, and all the plasma and brain samples were stored at −20 °C until analysis.
Bioanalysis in Plasma and Brain Samples.
R/S-4 or R/S-5 concentrations in plasma and brain samples were analyzed by LC/ MS/MS. Briefly, analytes were extracted from plasma samples by protein precipitation using acetonitrile and from brain samples by placing weighed tissue into 2 times w/v of acetonitrile and homogenizing. The homogenates were centrifuged at 10 000 rpm for 10 min, and the resultant supernatant was used as the sample solution. Standard curves (0.01−50 μM) were prepared using naivë plasma and brain tissue homogenates. Standards, QCs, and samples (25 μL) were mixed with 100 μL of acetonitrile containing 0.5 μM Losartan (internal standard; IS) in low retention microcentrifuge tubes. The mixture was vortexed for 1 min and centrifuged at 10 000 rpm for 10 min at 5 °C. A volume of 50 μL of supernatant was transferred to 250 μL polypropylene autosampler vials, mixed with 50 μL of water, and sealed with a Teflon cap. A volume of 3 μL was injected onto the ultraperformance liquid chromatography (UPLC) instrument coupled with a mass spectrometer for quantitative analysis.
Chromatographic analysis was performed using an Accela ultrahigh-performance system consisting of an analytical pump and an autosampler coupled with a TSQ Vantage mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). Separation of the analyte was achieved at ambient temperature using the Agilent Eclipse Plus column (100 mm × 2.1 mm, 3 μm, i.d.) packed with a 1.8 μm C18 stationary phase. The mobile phase was composed of 0.1% formic acid in acetonitrile and 0.1% formic acid in H2O with gradient elution. The total run time for each analyte was 4.5 min. The [M + H]+ ion transitions of R/S 4 m/z 143.930, 213.003; R/S 5 m/z 143.991, 213.100; and the internal standard m/z 423.03 > 180.086, 207.086 were used for analysis
Calibration curves were computed using the ratio of the peak area of analyte to the internal standard by using a quadratic regression with 1/x weighting. The parameters of each calibration curve were used to back-calculate concentrations and to obtain values for unknown samples by interpolation.
Supplementary Material
ACKNOWLEDGMENTS
Support for this research was provided by the National Institute on Drug Abuse-Intramural Research Program ZIA DA 000424 and Johns Hopkins Drug Discovery (Grant P30MH075673 to B.S.S.). All procedures involving rats were approved by the Animal Care and Use Committee (IACUC) of the U.S. National Institute on Drug Abuse (NIDA) and were consistent with The Guide for the Care and Use of Laboratory Animals, 8th edition (National Research Council, 2011). We thank Dr. Chloe Jordan and Dr. Guo-hua Bi (Addiction Biology Unit, NIDA-IRP) for the PK studies and Dr. Ludovic Muller (Structural Biology Core, NIDA-IRP) for the high-resolution mass spectrometry analyses. We thank Dr. Daryl A. Guthrie for editing an earlier version of this manuscript.
ABBREVIATIONS USED
- DA
dopamine
- SAR
structure activity relationship
- TM
transmembrane
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- OBS
orthosteric binding site
- SBP
secondary binding pocket
- 5-HT
5-hydroxytryptamine (serotonin)
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b00607.
Elemental analysis results, additional metabolism data and HPLC spectra of 4, 5, 9a, and 9b with their corresponding enantiomers (PDF)
SMILES files of all the compounds (CSV)
The authors declare no competing financial interest.
REFERENCES
- (1).Gomes T; Tadrous M; Mamdani MM; Paterson JM; Juurlink DN The Burden of Opioid-Related Mortality in the United States. JAMA. Netw. Open 2018, 1, No. e180217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lyden J; Binswanger IA The United States Opioid Epidemic. Semin. Perinatol 2019, 43, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Center for Behavioral Health Statistics and Quality. Behavioral Health Trends in the United States: Results from the 2014 National Survey on Drug Use and Health, HHS Publication No. SMA 15–4927, NSDUH Series H-50, 2015; https://www.samhsa.gov/data/sites/default/files/NSDUH-FRR1-2014/NSDUH-FRR1-2014.pdf (accessed July 26, 2019).
- (4).Florence CS; Zhou C; Luo F; Xu L. The Economic Burden of Prescription Opioid Overdose, Abuse, and Dependence in the United States, 2013. Med. Care 2016, 54, 901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Volkow ND; Koroshetz WJ The Role of Neurologists in Tackling the Opioid Epidemic. Nat. Rev. Neurol 2019, 15, 301. [DOI] [PubMed] [Google Scholar]
- (6).Haight BR; Learned SM; Laffont CM; Fudala PJ; Zhao Y; Garofalo AS; Greenwald MK; Nadipelli VR; Ling W; Heidbreder C; et al. Efficacy and Safety of A Monthly Buprenorphine Depot Injection For Opioid Use Disorder: A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2019, 393, 778–790. [DOI] [PubMed] [Google Scholar]
- (7).Bonhomme J; Shim RS; Gooden R; Tysu D; Rust G. Opioid Addiction and Abuse in Primary Care Practice: A Comparison of Methadone and Buprenorphine as Treatment Options. J. Natl. Med. Assoc 2012, 104, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ma J; Bao YP; Wang RJ; Su MF; Liu MX; Li JQ; Degenhardt L; Farrell M; Blow FC; Ilgen M; Shi J; Lu L. Effects of Medication-Assisted Treatment on Mortality Among Opioids Users: A Systematic Review and Meta-Analysis. Mol. Psychiatry 2018, DOI: 10.1038/s41380-018-0094-5. [DOI] [PubMed] [Google Scholar]
- (9).Dyer KR; White JM Patterns of Symptom Complaints in Methadone Maintenance Patients. Addiction. 1997, 92, 1445–1455. [PubMed] [Google Scholar]
- (10).Vignali C; Stramesi C; Morini L; Pozzi F; Groppi A. Methadone-Related Deaths. A Ten Year Overview. Forensic Sci. Int 2015, 257, 172–176. [DOI] [PubMed] [Google Scholar]
- (11).Gorodetzky CW; Walsh SL; Martin PR; Saxon AJ; Gullo KL; Biswas K. A Phase III, Randomized, Multi-Center, Double Blind, Placebo Controlled Study of Safety And Efficacy of Lofexidine For Relief of Symptoms in Individuals Undergoing Inpatient Opioid Withdrawal. Drug Alcohol Depend. 2017, 176, 79− 88. [DOI] [PubMed] [Google Scholar]
- (12).Gowing L; Farrell M; Ali R; White JM Alpha(2)Adrenergic Agonists for the Management of Opioid Withdrawal. Cochrane Database. Syst. Rev 2016, 3, CD002024. [Google Scholar]
- (13).Fishman M; Tirado C; Alam D; Gullo K; Clinch T; Gorodetzky CW Safety and Efficacy of Lofexidine for Medically Managed Opioid Withdrawal: A Randomized Controlled Clinical Trial. J. Addict. Med 2019, 13, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Volkow ND; Wang GJ; Fowler JS; Tomasi D. Addiction Circuitry in the Human Brain. Annu. Rev. Pharmacol. Toxicol 2012, 52, 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Koob GF; Volkow ND Neurocircuitry of Addiction. Neuropsychopharmacology 2010, 35, 217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Volkow ND; Wang GJ; Fowler JS; Tomasi D; Telang F. Addiction: Beyond Dopamine Reward Circuitry. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 15037–15042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sokoloff P; Le Foll B. The Dopamine D3 Receptor, A Quarter Century Later. Eur. J. Neurosci 2017, 45, 2–19. [DOI] [PubMed] [Google Scholar]
- (18).Heidbreder CA; Newman AH Current Perspectives on Selective Dopamine D(3) Receptor Antagonists as Pharmacotherapeutics for Addictions and Related Disorders. Ann. N. Y. Acad. Sci 2010, 1187, 4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Newman AH; Grundt P; Nader MA Dopamine D3 Receptor Partial Agonists and Antagonists as Potential Drug Abuse Therapeutic Agents. J. Med. Chem 2005, 48, 3663–3679. [DOI] [PubMed] [Google Scholar]
- (20).Leggio GM; Bucolo C; Platania CB; Salomone S; Drago F. Current Drug Treatments Targeting Dopamine D3 Receptor. Pharmacol. Ther 2016, 165, 164–177. [DOI] [PubMed] [Google Scholar]
- (21).Cortes A; Moreno E; Rodriguez-Ruiz M; Canela EI; Casado V. Targeting The Dopamine D3 Receptor: an Overview of Drug Design Strategies. Expert Opin. Drug Discovery 2016, 11, 641− 664. [DOI] [PubMed] [Google Scholar]
- (22).Pich EM; Collo G. Pharmacological Targeting of Dopamine D3 Receptors: Possible Clinical Applications of Selective Drugs. Eur. Neuropsychopharmacol 2015, 25, 1437–1447. [DOI] [PubMed] [Google Scholar]
- (23).Micheli F; Bacchi A; Braggio S; Castelletti L; Cavallini P; Cavanni P; Cremonesi S; Dal Cin M; Feriani A; Gehanne S; Kajbaf M; Marchio L; Nola S; Oliosi B; Pellacani A; Perdona E; Sava A; Semeraro T; Tarsi L; Tomelleri S; Wong A; Visentini F; Zonzini L; Heidbreder C. 1,2,4-Triazolyl 5-Azaspiro[2.4]Heptanes: Lead Identification and Early Lead Optimization of A New Series of Potent and Selective Dopamine D3 Receptor Antagonists. J. Med. Chem 2016, 59, 8549–8576. [DOI] [PubMed] [Google Scholar]
- (24).Hackling AE; Stark H. Dopamine D3 Receptor Ligands With Antagonist Properties. ChemBioChem 2002, 3, 946–961. [DOI] [PubMed] [Google Scholar]
- (25).Keck TM; Burzynski C; Shi L; Newman AH Beyond Small-Molecule SAR: Using the Dopamine D3 Receptor Crystal Structure to Guide Drug Design. Adv. Pharmacol 2014, 69, 267–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Keck TM; John WS; Czoty PW; Nader MA; Newman AH Identifying Medication Targets for Psychostimulant Addiction: Unraveling the Dopamine D3 Receptor Hypothesis. J. Med. Chem 2015, 58, 5361–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Micheli F; Heidbreder C. Dopamine D3 Receptor Antagonists: A Patent Review (2007 – 2012). Expert Opin. Ther. Pat 2013, 23, 363–381. [DOI] [PubMed] [Google Scholar]
- (28).Lober S; Hubner H; Tschammer N; Gmeiner P. Recent Advances In The Search for D3- and D4-Selective Drugs: Probes, Models and Candidates. Trends Pharmacol. Sci 2011, 32, 148–157. [DOI] [PubMed] [Google Scholar]
- (29).Wager TT; Chappie T; Horton D; Chandrasekaran RY; Samas B; Dunn-Sims ER; Hsu C; Nawreen N; Vanase-Frawley MA; O’Connor RE; Schmidt CJ; Dlugolenski K; Stratman NC; Majchrzak MJ; Kormos BL; Nguyen DP; Sawant-Basak A; Mead AN Dopamine D3/D2 Receptor Antagonist PF-4363467 Attenuates Opioid Drug-Seeking Behavior without Concomitant D2 Side Effects. ACS Chem. Neurosci 2017, 8, 165–177. [DOI] [PubMed] [Google Scholar]
- (30).You ZB; Bi GH; Galaj E; Kumar V; Cao J; Gadiano A; Rais R; Slusher BS; Gardner EL; Xi ZX; Newman AH Dopamine D3R Antagonist VK4−116 Attenuates Oxycodone Self-Administration and Reinstatement Without Compromising its Antinociceptive Effects. Neuropsychopharmacology 2019, 44, 1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Murphy A; Nestor LJ; McGonigle J; Paterson L; Boyapati V; Ersche KD; Flechais R; Kuchibatla S; Metastasio A; Orban C; Passetti F; Reed L; Smith D; Suckling J; Taylor E; Robbins TW; Lingford-Hughes A; Nutt DJ; Deakin JF; Elliott R. Acute D3 Antagonist GSK598809 Selectively Enhances Neural Response During Monetary Reward Anticipation in Drug and Alcohol Dependence. Neuropsychopharmacology 2017, 42, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Di Ciano P; Mansouri E; Tong J; Wilson AA; Houle S; Boileau I; Duvauchelle T; Robert P; Schwartz JC; Le Foll B. Occupancy of Dopamine D2 And D3 Receptors By a Novel D3 Partial Agonist BP1.4979: A [(11)C]-(+)-PHNO PET Study in Humans. Neuropsychopharmacology 2019, 44, 1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Appel NM; Li SH; Holmes TH; Acri JB Dopamine D3 Receptor Antagonist (GSK598809) Potentiates the Hypertensive Effects of Cocaine in Conscious, Freely-Moving Dogs. J. Pharmacol. Exp. Ther 2015, 354, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Powell GL; Bonadonna JP; Vannan A; Xu K; Mach RH; Luedtke RR; Neisewander JL Dopamine D3 Receptor Partial Agonist LS-3−134 Attenuates Cocaine-Motivated Behaviors. Pharmacol., Biochem. Behav 2018, 171, 46–53. [DOI] [PubMed] [Google Scholar]
- (35).Hayatshahi HS; Xu K; Griffin SA; Taylor M; Mach RH; Liu J; Luedtke RR Analogues of Arylamide Phenylpiperazine Ligands To Investigate the Factors Influencing D3 Dopamine Receptor Bitropic Binding and Receptor Subtype Selectivity. ACS Chem. Neurosci 2018, 9, 2972. [DOI] [PubMed] [Google Scholar]
- (36).Boateng CA; Bakare OM; Zhan J; Banala AK; Burzynski C; Pommier E; Keck TM; Donthamsetti P; Javitch JA; Rais R; Slusher BS; Xi ZX; Newman AH High Affinity Dopamine D3 Receptor (D3R)-Selective Antagonists Attenuate Heroin Self-Administration in Wild-Type but not D3R Knockout Mice. J. Med. Chem 2015, 58, 6195–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Banala AK; Levy BA; Khatri SS; Furman CA; Roof RA; Mishra Y; Griffin SA; Sibley DR; Luedtke RR; Newman AH N-(3-Fluoro-4-(4-(2-Methoxy Or 2,3-Dichlorophenyl)Piperazine-1-Yl)Butyl)Arylcarboxamides as Selective Dopamine D3 Receptor Ligands: Critical Role of the Carboxamide Linker for D3 Receptor Selectivity. J. Med. Chem 2011, 54, 3581–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kumar V; Banala AK; Garcia EG; Cao J; Keck TM; Bonifazi A; Deschamps JR; Newman AH Chiral Resolution and Serendipitous Fluorination Reaction for the Selective Dopamine D3 Receptor Antagonist BAK2−66. ACS Med. Chem. Lett 2014, 5, 647− 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kumar V; Bonifazi A; Ellenberger MP; Keck TM; Pommier E; Rais R; Slusher BS; Gardner E; You ZB; Xi ZX; Newman AH Highly Selective Dopamine D3 Receptor (D3R) Antagonists and Partial Agonists Based on Eticlopride and the D3R Crystal Structure: New Leads for Opioid Dependence Treatment. J. Med. Chem 2016, 59, 7634–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Newman AH; Beuming T; Banala AK; Donthamsetti P; Pongetti K; LaBounty A; Levy B; Cao J; Michino M; Luedtke RR; Javitch JA; Shi L. Molecular Determinants of Selectivity and Efficacy at The Dopamine D3 Receptor. J. Med. Chem 2012, 55, 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Newman AH; Grundt P; Cyriac G; Deschamps JR; Taylor M; Kumar R; Ho D; Luedtke RR N-(4-(4-(2,3-Dichloroor 2-Methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with Functionalized Linking Chains as High Affinity and Enantioselective D3 Receptor Antagonists. J. Med. Chem 2009, 52, 2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chien EYT; Liu W; Zhao Q; Katritch V; Won Han G; Hanson MA; Shi L; Newman AH; Javitch JA; Cherezov V; Stevens RC Structure of the Human Dopamine D3 Receptor in Complex with A D2/D3 Selective Antagonist. Science 2010, 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cheng Y; Prusoff WH Relationship Between The Inhibition Constant (K1) and The Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of An Enzymatic Reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (44).Jordan CJ; Humburg B; Rice M; Bi G-H; You Z-B; Shaik AB; Cao J; Bonifazi A; Gadiano A; Rais R; Slusher B; Newman AH; Xi Z-X The Highly Selective Dopamine D3R Antagonist, R-VK4−40, Attenuates Oxycodone Reward and Augments Analgesia in Rodents. Neuropharmacology 2019, 158, 107597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kamal A; Shaik AB; Rao BB; Khan I; Bharath Kumar G; Jain N. Design and Synthesis of Pyrazole/Isoxazole linked Arylcinnamides as Tubulin Polymerization Inhibitors and Potential Antiproliferative Agents. Org. Biomol. Chem 2015, 13, 10162–10178. [DOI] [PubMed] [Google Scholar]
- (46).Grundt P; Prevatt KM; Cao J; Taylor M; Floresca CZ; Choi JK; Jenkins BG; Luedtke RR; Newman AH Heterocyclic Analogues of N-(4-(4-(2,3-Dichlorophenyl)Piperazin-1-Yl)Butyl)Arylcarboxamides with Functionalized Linking Chains as Novel Dopamine D3 Receptor Ligands: Potential Substance Abuse Therapeutic Agents. J. Med. Chem 2007, 50, 4135–4146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.