

SUMMARY
Disruption of retromer-dependent endosomal trafficking is considered pathogenic in late-onset Alzheimer’s disease (AD). Here, to investigate this disruption in the intact brain, we turn to a genetic mouse model where the retromer core protein VPS35 is depleted in hippocampal neurons, and then we replete VPS35 using an optimized viral vector protocol. The VPS35 depletion-repletion studies strengthen the causal link between the neuronal retromer and AD-associated neuronal phenotypes, including the acceleration of amyloid precursor protein cleavage and the loss of synaptic glutamate receptors. Moreover, the studies show that the neuronal retromer can regulate a distinct, dystrophic, microglia morphology, phenotypic of hippocampal microglia in AD. Finally, the neuronal and, in part, the microglia responses to VPS35 depletion were found to occur independent of tau. Showing that the neuronal retromer can regulate AD-associated pathologies in two of AD’s principal cell types strengthens the link, and clarifies the mechanism, between endosomal trafficking and late-onset sporadic AD.
In brief
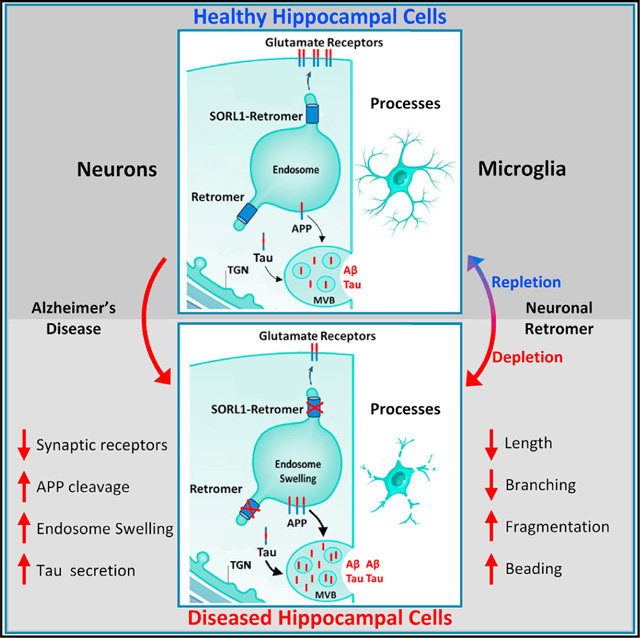
Alzheimer’s cellular pathologies in the hippocampal formation are typified by a neuronal phenotype and a distinct, dystrophic, microglial phenotype. Qureshi et al. show that retromer-dependent endosomal recycling in hippocampal neurons, a pathogenic biological pathway, can regulate both cellular phenotypes and find that this regulation is largely independent of tau.
Graphical Abstract
INTRODUCTION
The neuron’s early endosome, a central trafficking station of the endolysosomal network, has proven to be an organelle fundamental to Alzheimer’s disease (AD) (Nixon, 2005). A combination of genetic (Karch and Goate, 2015) and functional (Small et al., 2017) studies have implicated endosomal trafficking as a pathogenic biological pathway in the disease. More specifically, recent studies have suggested that the endosome’s recycling routes might mediate AD’s pathophysiology (Small and Petsko, 2020). Endosomal recycling critically depends on retromer, a dedicated trafficking complex that is considered a “master conductor” of endosomal trafficking (Seaman, 2005; Burd and Cullen, 2014; Kovtun et al., 2018). Retromer core proteins are deficient in the hippocampal formation of patients with AD, and depleting VPS35, the core’s central protein, has emerged as the most reliable approach for disrupting retromer-dependent endosomal recycling (Small and Petsko, 2015). Nevertheless, attempts to model VPS35 depletion in the mammalian brain have been hindered because VPS35 knockout mice are embryonic lethal.
To overcome this challenge, we have recently developed a mouse model in which VPS35 is genetically depleted selectively in hippocampal and forebrain neurons, allowing survival into adulthood (Simoes et al., 2020). In parallel, we have optimized a VPS35 repletion strategy using AAV9-VPS35, overcoming the autoregulation observed for VPS35 expression, and showing that exogenous VPS35 can bind other retromer core proteins critical for retromer function.
We then combined the VPS35 neuronal-selective knockout (VPS35 nsKO) mice with the AAV9-VPS35 protocol and completed an extensive series of VPS35 depletion-repletion studies to strengthen the mechanistic link between retromer-dependent endosomal recycling and AD’s known cellular phenotypes. For neurons, we relied on prior studies that have associated retromer dysfunction with the pathogenic cleavage of the amyloid precursor protein (APP) and the loss of hippocampal-enriched glutamate receptors (Temkin et al., 2017), which typify AD-associated amyloid and synaptic pathology.
Besides neurons, glia and in particular microglia are now considered a second brain cell type phenotypic of AD’s cellular pathologies (Salter and Stevens, 2017; Hansen et al., 2018; Takatori et al., 2019; Streit et al., 2020), which play a dominant role in modulating how the brain responds to AD’s triggering neuronal-based pathologies. We therefore took advantage of our VPS35 depletion-repletion paradigm to ask whether the neuronal retromer will not only regulate AD-associated neuronal phenotypes but might also regulate glial phenotypes.
RESULTS
The effects of neuronal VPS35 depletion on neurons and glia
By crossing mice expressing loxP-flanked VPS35 (VPS35fl/fl) with mice expressing Cre recombinase under the Camk2α promoter, we generated VPS35 nsKO mice (Figure 1A), in whom VPS35 is selectively depleted in forebrain and hippocampal neurons (Simoes et al., 2020).
Figure 1. The effects of neuronal VPS35 depletion on hippocampal neurons and glia.
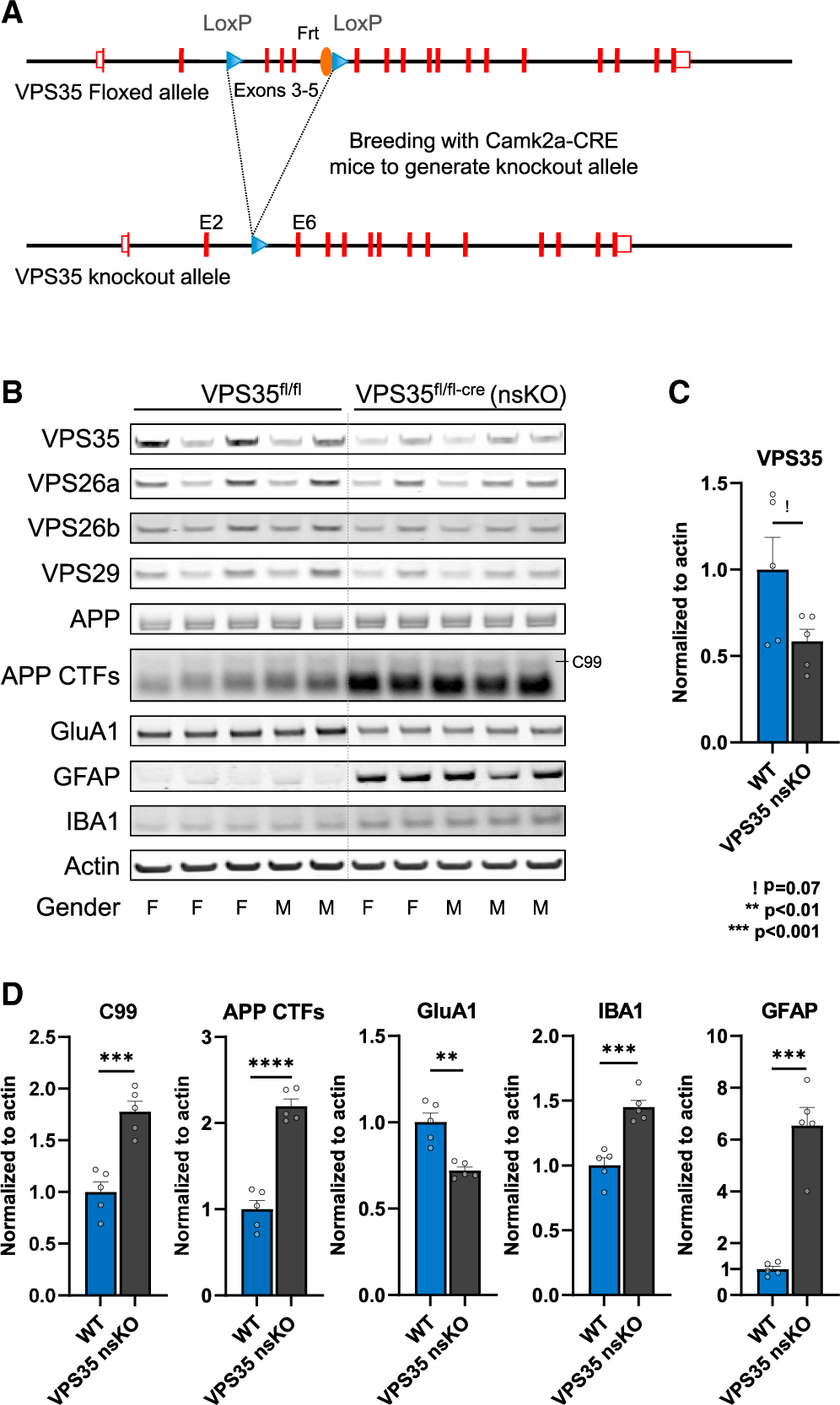
(A) Neuronal-selective VPS35 knockout mice were generated by crossing mice expressing loxP-flanked VPS35 (exons 3–5) (VPS35fl/fl) with mice expressing Cre recombinase under the Camk2α promoter. Camk2a-CRE mice were obtained from Jackson Laboratory (Stock No: 005,359).
(B) Immunoblots from of the CA1 region of WT (VPS35fl/fl, n = 5) and VPS35 nsKO (n = 5) mice (3 months of age) showing VPS35, VPS26a, VPS26b, VPS29, full-length APP, total C-terminal fragments of APP (APP CTFs), GluA1, GFAP, IBA1, and actin protein levels.
(C) Bar graph represents actin-normalized levels of VPS35 and (D) β-secretase-derived C-terminal fragment of APP (C99), total C-terminal fragments of APP (APP CTFs), and glutamate receptor ionotropic AMPA1 (GluA1), and the microglial markers ionized calcium-binding adapter molecule 1 (IBA1) and glial fibrillary acidic protein (GFAP).
See also Figure S1. WT = VPS35fl/fl, n = biological replicates. Data are represented as mean ± SEM. ! p = 0.07, **p < 0.01, ***p < 0.001.
The dorsal CA1 hippocampal region was micro-dissected in 3-month-old VPS35 nsKO mice and wild-type (WT = VPS35fl/fl) littermates, an age when previous studies have established that there is no cell or synaptic loss in these mice (Simoes et al., 2020). By immunoblotting, VPS35 depletion was found to result in AD-associated indicators of neuronal dysfunction, including an accumulation of the C-terminal fragments (CTFs) of APP (t = 9.08, p = 0.00002) (when normalized to either actin or APP) and a reduction in the glutamate receptor GluA1 (t = 4.93, p = 0.0011) (Figures 1B–1D and S1).
Additionally, neuronal VPS35 depletion was observed to cause an increase in the astrocytic marker glial fibrillary acidic protein (GFAP) (t = 7.8, p = 0.00005) and an increase in the microglial marker IBA1 (t = 5.69, p = 0.0005) (Figure 1D).
Optimizing an in vivo VPS35 repletion protocol
Next, we turned to viral vector technology to replete VPS35. We C-terminally tagged human VPS35 with green fluorescent protein (GFP) and delivered AAV9-VPS35-GFP (Figure S2) to mouse neuronal cultures. Immunoblot levels of endogenous and exogenous VPS35 were measured across three doses. Interestingly, a pattern consistent with autoregulation was observed (Figure 2A), whereby the higher the levels of exogenous VPS35 expressed, the lower the levels of endogenous VPS35 were detected. A significant increase in total VPS35 was observed at both the middle dose (t = −10.74, p < 0.001) and the highest dose (t = −26.02, p < 0.001), but not at the lowest dose (t = −1.53, p = 0.199).
Figure 2. The effects of AAV9-VPS35 on the endogenous neuronal retromer.
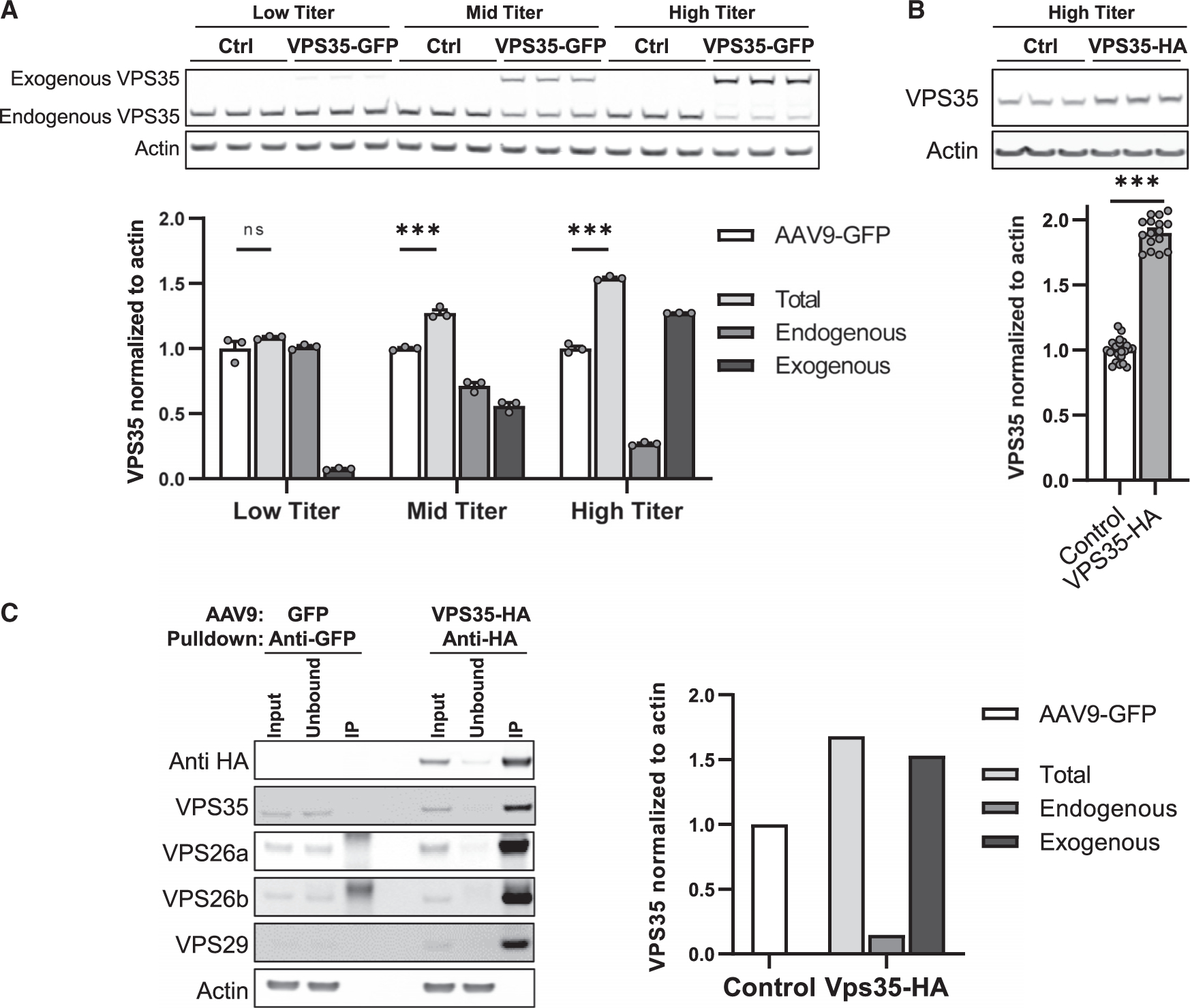
(A) Immunoblots showing expression levels of exogenous and endogenous VPS35 after delivery of three doses of AAV9-VPS35-GFP in primary neuronal culture (5E+9 vg/well, 2E+10 vg/well, 1E+11 vg/well), and after delivery of AAV9-GFP as a control. Bar graphs show the mean levels of total VPS35 (light gray bars), endogenous VPS35 (medium gray bars), exogenous VPS35 (black bars). Mean level of VPS35 with AAV9-GFP expression (white bars) is shown as control (n = 3).
(B) Representative immunoblots showing expression levels following AAV9-VPS35-HA at 1E+11 vg/well (n = 16). Bar graphs show mean levels, normalized by actin; AAV9-GFP and AAV9-EV (empty vector) were used as controls (n = 23).
(C) Immunoblots showing that anti-HA antibody co-immunoprecipitated endogenous retromer core proteins VPS26a, VPS26b, and VPS29. Bar graphs represent levels of VPS35, total (light gray; input), endogenous VPS35 (medium gray; unbound), and exogenous VPS35 (black; unbound subtracted from input) extracted from the co-immunoprecipitation data (right three bands on blots). Control bar (white; input from the control reaction (left three bands on the blots)). All primary neurons were cultured from C57BL/6J mice at postnatal day 0.
See also Figure S2. n = biological replicates. Data are represented as mean ± SEM. ***p < 0.001, ns = not significant.
Because VPS35 acts as a scaffold to which other retromer proteins bind, we next tagged the human VPS35 with HA, which is smaller than GFP and less likely to interfere with VPS35’s scaffold function. We find that AAV9-VPS35-HA, delivered at the high dose, caused higher total VPS35 expression than AAV9-VPS35-GFP (Figure 2B). A co-immunoprecipitation study was performed using anti-HA antibody, showing that exogenously expressed VPS35 pulls down endogenous VPS29 and VPS26 (Figure 2C), the other two retromer proteins that comprise the retromer’s heterotrimeric core complex (Seaman, 2005). These studies establish that exogenous VPS35 binds other endogenous retromer core proteins, forming a complex that is critical for retromer’s function.
To determine the optimum in vivo AAV9-VPS35 delivery, we completed a series of experiments (two to six animals per experiment), delivering AAV9-VPS35-HA intracranially to the dorsal CA1 region of the hippocampus of wild-type (C57BL/6J) mice in varying doses and volumes (Figures S3A and S3B). Brain slices were generated for fluorescent immunohistochemistry and probed with anti-HA antibody to map the degree and distribution of expression, and neuronal and nuclear markers were used to assay potential toxicity. In parallel, tissue samples from the CA1 were micro-dissected and processed for immunoblotting to quantify expression levels (Figure 3A, right panel).
Figure 3. The effects of AAV9-VPS35 on the endogenous hippocampal retromer.

(A) Schematic representation (left panel) of the protocol of AAV9 delivery into the hippocampal CA1 region. All animals were injected at 3 months and aged to 6 months of age. At 6 months of age, brains were processed for fluorescent IHC and biochemistry (as illustrated in the right panel).
(B) Coronal brain sections immunofluorescently stained 3 months after delivery of AAV9-VPS35-HA into right CA1 and AAV9-GFP into the left CA1, showing expression distribution of exogenous VPS35 (anti-HA). Insets represent higher magnification images of indicated subregions.
(C) Brain sections stained with the neuronal marker, NeuN, and the nuclear marker, Hoechst, showed no changes in the neuronal layers compared to the contralateral CA1.
(D) Immunoblots showing VPS35 overexpression and levels of beta III tubulin (N-tubulin), NeuN, glutamate receptor ionotropic AMPA1 alpha 1 (GluA1), full length APP (APP), total C-terminal fragments of APP (APP CTFs), and actin in the CA1 regions injected with AAV9 empty vector (EV) or VPS35-HA (n = 4).
(E) Immunoblots showing that anti-HA antibody co-immunoprecipitated endogenous retromer core proteins VPS26a, VPS26b, and VPS29. Bar graphs represent levels of VPS35, total (light gray = input), endogenous VPS35 (medium gray = unbound), and exogenous VPS35 (black = unbound subtracted from input) extracted from the co-immunoprecipitation data (right three bands on blots). Control bar (white = input from the control reaction [left three bands on the blots]) (n = 4). All data in this figure acquired from C57BL/6J mice. See also Figure S3. n = biological replicates. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01.
Using the optimum delivery protocol, with the highest subtoxic dose of the vector (Figure 3A, left panel), we injected AAV9-VPS35-HA to 3-month-old mice that were then aged to 6 months. Results showed a broad expression distribution across the dorsal hippocampus, with a significant increase in VPS35 (t = 8.74, p = 0.003) and with no evidence of toxicity (Figures 3B–3D). As in neuronal culture, a co-immunoprecipitation study with anti-HA antibody showed that exogenous VPS35 downregulates the corresponding endogenous protein, and that exogenous VPS35 binds the two other retromer core proteins (Figure 3E).
The effects of VPS35 depletion-repletion on hippocampal neurons
Using the optimized protocol, AAV9-VPS35-HA (hereinafter referred to as AAV9-VPS35) was delivered to the right CA1 region of the VPS35 nsKO mice, and AAV9-GFP was delivered to the contralateral CA1 as a control (Figure 4C). In addition, VPS35fl/fl mice were evaluated as non-injected controls. All VPS35 nsKO mice were injected at 3 months of age and aged to 6 months. In the no-surgery control conditions, AD’s neuronal phenotypes identified in 3-month-old VPS35 nsKO mice, were still detectable at 6 months of age as measured by APP CTFs (t = −15.89, p = 0.00009) and by GluA1 levels (t = 9.70, p = 0.0006) (Figure 4B). Three months after 3-month-old VPS35 nsKO mice received AAV9-VPS35 or AAV9-GFP injections, we observed a normalization of APP CTFs (t = 3.88, p = 0.018) and a normalization of GluA1 levels with AAV9-VPS35, as shown by immunoblotting (t = −6.45, p = 0.003; Figures 4A and 4B) and confirmed by fluorescent immunohistochemistry (IHC) (Figure 4C).
Figure 4. AAV9-VPS35 normalizes APP and glutamate receptor defects in hippocampal neurons.
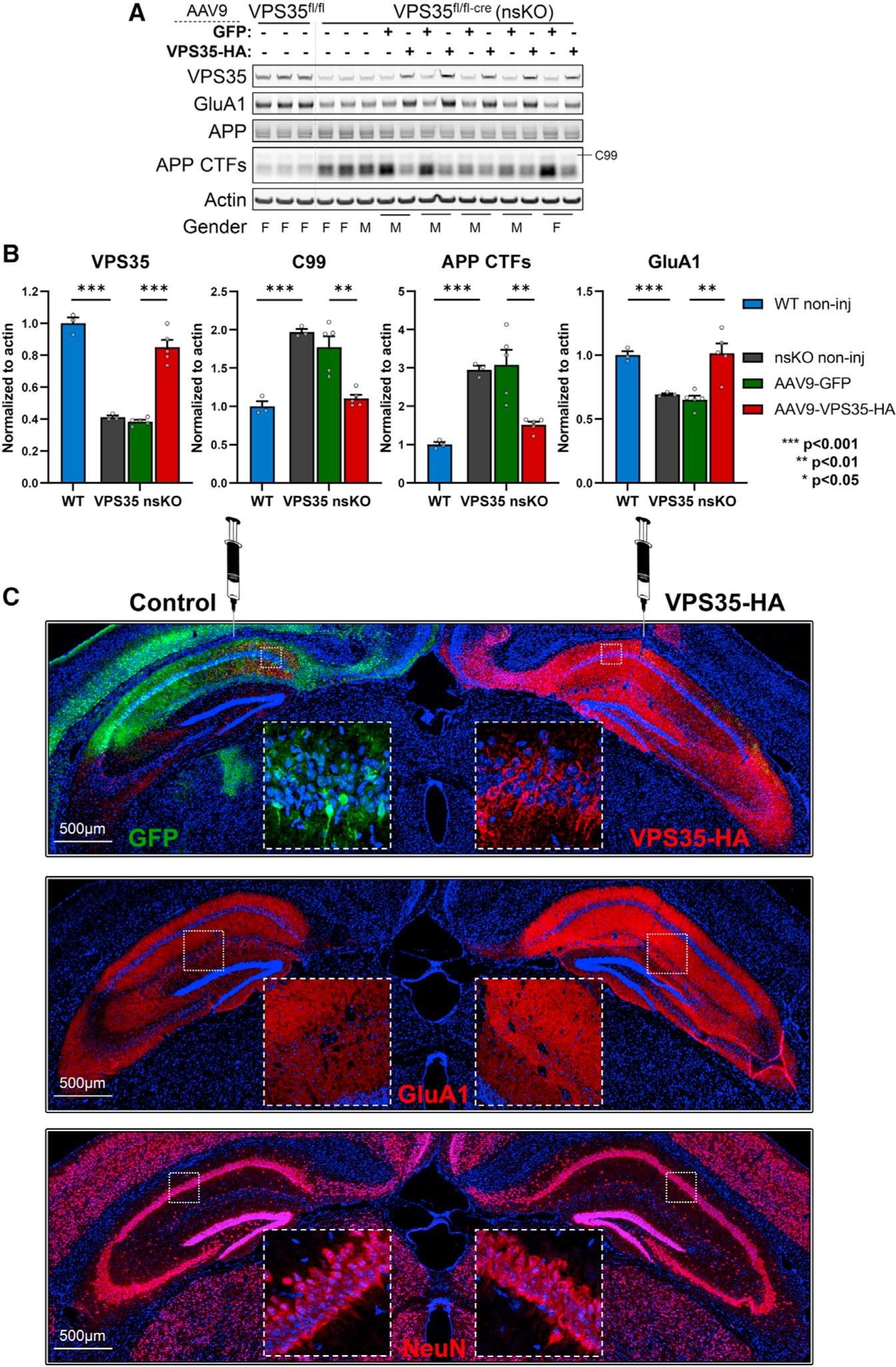
(A) Immunoblots from the CA1 region of WT (VPS35fl/fl, n = 3) and VPS35 nsKO (n = 8) mice (6 months of age) showing VPS35, GluA1, full-length APP, total C-terminal fragments of APP (APP CTFs) and actin protein levels. AAV9-GFP and AAV9-VPS35-HA were injected in left and right CA1 respectively in five of the VPS35 nsKO mice at 3 months.
(B) Bar graphs represent actin-normalized levels of VPS35 (left graph), β-secretase derived C-terminal fragment of APP (C99), total C-terminal fragments of APP (APP CTFs; middle graph), and glutamate receptor ionotropic AMPA1 (GluA1; right graph). WT = VPS35fl/fl.
(C) Coronal brain sections immunofluorescently stained with anti-HA and anti-GFP, 3 months after AAV9-VPS35-HA into right CA1 and AAV9-GFP into the left CA1 of VPS35 nsKO mouse (upper panel), showing increase in GluA1 expression in the right hippocampus (middle panel), with no detectable changes in neuronal staining (lower panel).
See also Figure S1. n = biological replicates. Data are represented as mean ± SEM. **p < 0.01, ***p < 0.001.
Retromer is considered a “master conductor” of endosomal trafficking (Burd and Cullen, 2014) because it regulates the two pathways whereby cargo is recycling out of endosomes: from the endosome back to the cell surface, and from the endosome back to the TGN (trans-Golgi network). APP and GluA1 are neuronal endosomal cargo that typify the first pathway, and based on older (Small and Petsko, 2015) and newer studies (Mishra et al., 2021), SORL1 (sortilin-related receptor1) can act a retromer-receptor that allows the retromer coat to interact with this cargo. We, therefore, took advantage of our depletion-repletion paradigm to investigate SORL1. First, we needed to establish the expression profile of SORL1 at the protein level, since mRNA studies have suggested that SORL1 mRNA might be differentially expressed in glia (Li et al., 2019b). Consistent with the fact that there is often a mRNA-protein expression mismatch (Buccitelli and Selbach, 2020), an immunofluorescence map of SORL1 protein expression in WT mice confirms previous cell culture observations (Thonberg et al., 2017), showing that SORL1 protein is differentially expressed in neurons (Figures 5A and 5B). Next, we show that glial SORL1 is not reliably upregulated in response to neuronal VPS35 depletion (Figure 5E). A reliable effect was, however, observed within CA1 neurons, with larger SORL1-positive puncta in VPS35-depleted neurons, an effect that was partially rescued upon VPS35 repletion (Figures 5F and 5G). This likely reflects the fact that VPS35 depletion is known to cause enlarged endosomes (Bhalla et al., 2012), a cytopathogenic hallmark of AD (Cataldo et al., 2000), and that retromer plays a role in transporting SORL1 from the endosome to the cell surface (Christensen et al., 2020).
Figure 5. AAV9-VPS35 normalizes SORL1 and cathepsin D defects in hippocampal neurons.
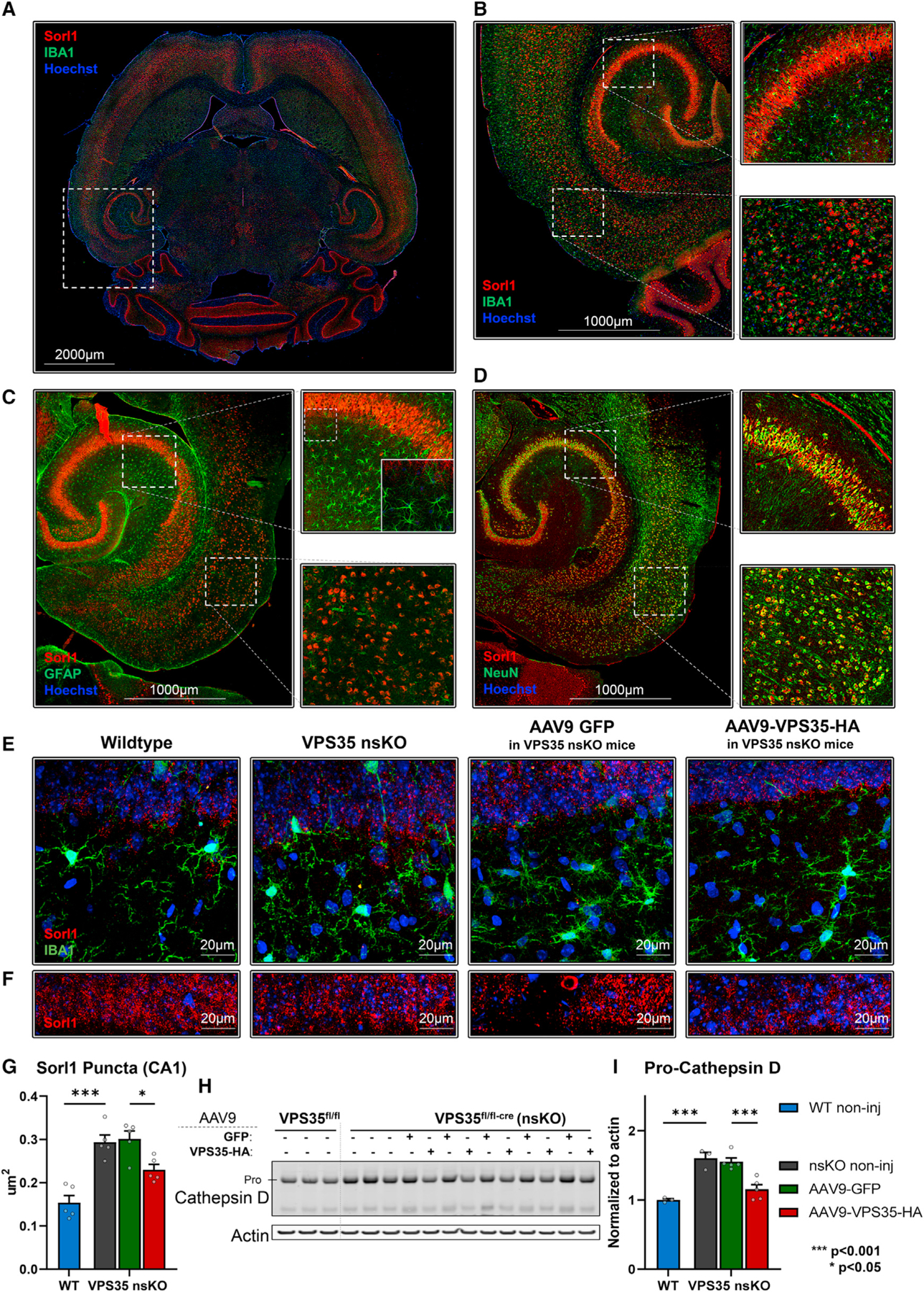
(A) Axial brain section immunofluorescently stained with Sorl1 and co-stained with microglial marker IBA1.
(B and C) (B) Hippocampal and EC areas of the mouse brain showing a lack of colocalization between Sorl1 and IBA1, and (C) Sorl1 and GFAP.
(D) Strong colocalization was observed with neuronal marker NeuN in the cortex and hippocampus.
(E) 63x z-projection images showing Sorl1 and IBA1 co-staining in wild-type (VPS35fl/fl, n = 5), VPS35 nsKO (n = 5), VPS35 nsKO mice with AAV9 GFP injection (n = 5), VPS35 nsKO mice with AAV9 VPS35-HA injection (n = 5).
(F) 63x z-projection images showing Sorl1 staining in wild-type (VPS35fl/fl, n = 5), VPS35 nsKO (n = 5), VPS35 nsKO mice with AAV9 GFP injection (n = 5), VPS35 nsKO mice with AAV9 VPS35-HA injection (n = 5).
(G) Quantification of Sorl1 puncta size for (F).
(H) Immunoblots from the CA1 region of WT (VPS35fl/fl, n = 3) and VPS35 nsKO (n = 8) mice (6 months of age) showing cathepsin D and actin protein levels. AAV9-GFP and AAV9-VPS35-HA were injected in left and right CA1 respectively in five of the VPS35 nsKO mice at 3 months.
(I) Bar graphs represent actin-normalized levels of Pro-cathepsin D (upper band).
See also Figure S4. n = biological replicates. Data are represented as mean ± SEM. *p < 0.05, ***p < 0.001.
Finally, we used the paradigm to investigate whether neuronal VPS35 can regulate retromer’s second pathway, an important functional question since this pathway is ultimately critical for the maturation and delivery of the lysosomal protease cathepsin D (CTSD) (Benes et al., 2008), required for normal lysosomal function. From other studies, it is known that retromer transports CTSD receptors back to the TGN, those that transport CTSD to the endosome leading to the conversion of pro-CTSD to mature-CTSD. Thus, an increase in pro-CTSD is considered a reliable indicator of impaired retromer-dependent trafficking (Benes et al., 2008; Rojas et al., 2008; Han et al., 2021), and ultimately lysosomal dysfunction. Indeed, immunoblotting tissue micro-dissected from the CA1 showed that compared with WT mice, VPS35 depletion leads to an increase in the pro-CTSD, which was partially rescued by VPS35 repletion (Figures 5H and 5I). In VPS35 nsKO conditions mature-CSTD was also increased, but the ratio of pro-CTSD/mature-CTSD was still significantly increased in the VPS35 depletion and rescued with VPS35 repletion (Figure S4). We note that the ratio was also reduced in the AAV9-GFP condition, but this was driven by a significant increase in mature-CTSD (Figure S4), with no change in pro-CTSD, which might reflect the apparent cross-reactivity of the antibody to GFP because of immunoblot overlap of GFP and mature-CTSD bands. Despite this outstanding issue regarding the GFP overexpression condition, we can conclude that VPS35 in CA1 neurons regulates both of the retromer-dependent endosomal pathways. Moreover, it expands the pathophysiology of retromer dysfunction in hippocampal neurons to include both endosomal and lysosomal dysfunction.
The effects of VPS35 depletion-repletion on hippocampal glia
In the no-surgery control conditions, the gliosis observed at 3 months in VPS35 nsKO mice was sustained at 6 months with an increase of GFAP (t = −2.74, p = 0.052), IBA1 (t = −8.24, p = 0.001), and another microglial marker CD11b (t = −6.65, p = 0.002) (Figures 6A and 6C).
Figure 6. AAV9-VPS35 partially normalizes the responses of hippocampal glia.
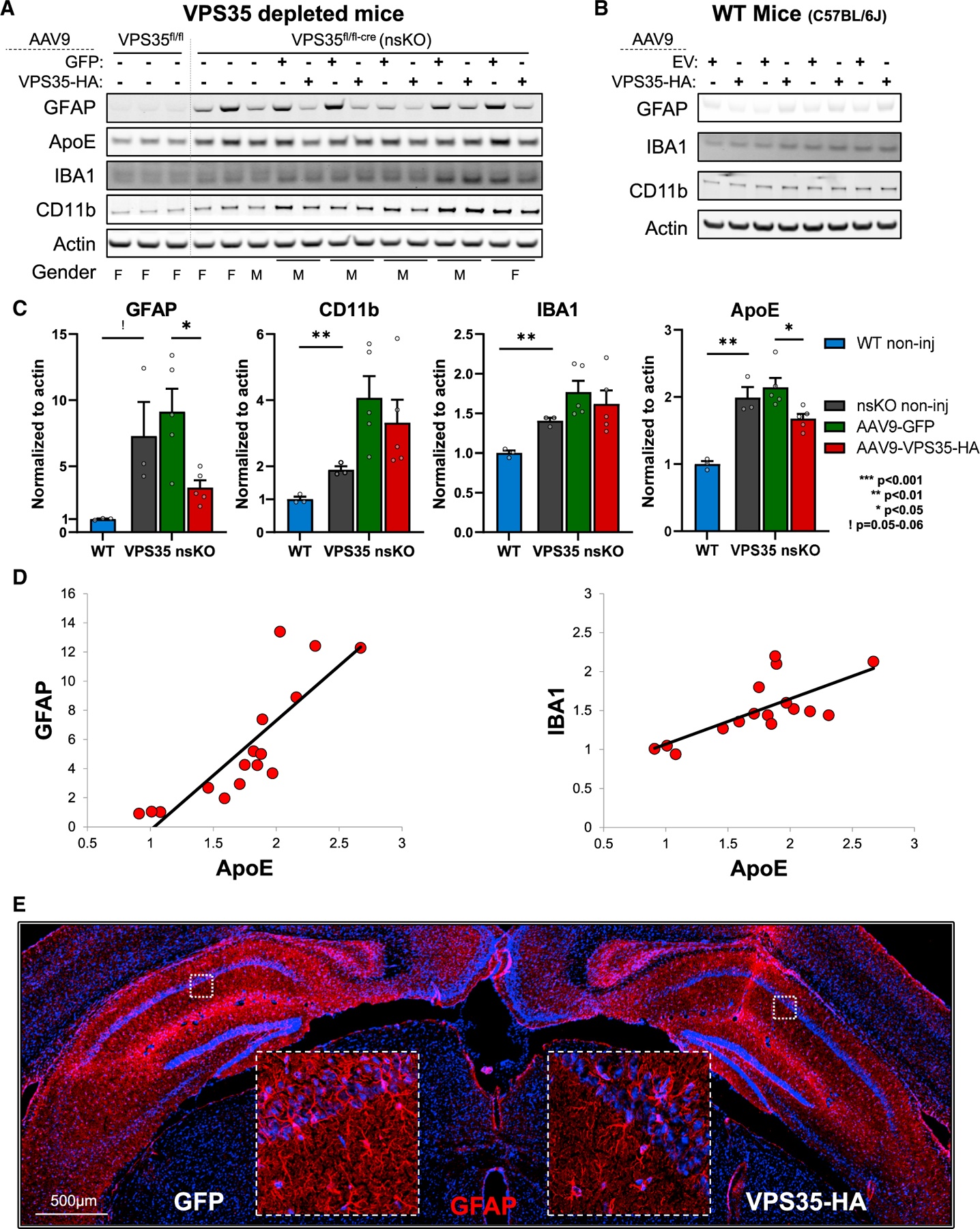
(A) Immunoblots from the CA1 region of WT (VPS35fl/fl, n = 3) and VPS35 nsKO (n = 8) mice (6 months of age) were probed for astrocyte marker glial fibrillary acidic protein (GFAP), apolipoprotein E (APOE), and the microglial markers ionized calcium-binding adapter molecule 1 (IBA1) and CD11 antigen-like family member B (CD11b). AAV9-GFP and AAV9-VPS35-HA were injected in left and right CA1 respectively in five of the VPS35 nsKO mice at 3 months. VPS35 expression in these animals is shown in Figures 4A and 4B.
(B) Immunoblots from the CA1 region (WT = C57BL/6J) injected with AAV9 empty vector (EV) or VPS35-HA probed for astrocyte and microglial markers. VPS35 expression in these animals is shown in Figure 3D.
(C) Bar graphs represent actin-normalized levels of the three glial markers and APOE from panel (A) VPS35 nsKO mice displayed a dramatic increase in gliosis, which was partially normalized by VPS35 repletion (WT = VPS35fl/fl).
(D) Scatterplots showing the correlation between normalized APOE and glial markers.
(E) Coronal brain section immunofluorescently stained, 3 months after AAV9-VPS35-HA injection into right CA1 and AAV9-GFP into the left CA1 of VPS35 nsKO mouse showing a decrease in GFAP expression in the right hippocampus, confirming the lower astrocytic signal detected in immunoblots with VPS35-HA overexpression.
See also Figure S5. n = biological replicates. Data are represented as mean ± SEM. ! p = 0.052, *p < 0.05, **p < 0.01.
Three months after 3-month old VPS35 nsKO mice received AAV9-VPS35, we observed a partial normalization of the astrocytic phenotype, as shown by GFAP immunoblotting (t = 3.39, p = 0.028) and by GFAP fluorescence via IHC (Figures 6A, 6C, and 6E). While a slight change in the microglial markers was observed, this was not statistically significant (IBA1, t = 1.98, p = 0.11; CD11b, t = 1.57, p = 0.19). Importantly, exogenous VPS35 expression in wild-type (C57BL/6J) mice had minimal effects on astrocytes and microglia at 3 months post-surgery (Figure 6B).
Given the glial phenotypes we decided to investigate the AD-related protein apolipoprotein E (APOE), expressed by astrocytes as well as microglia (Zhang et al., 2014, 2016). We find that APOE is also elevated in VPS35 nsKO mice (t = −6.02, p = 0.004) (Figures 6A and 6C) and partially normalized by AAV9-VPS35 repletion (t = 2.98, p = 0.018). APOE is produced by both astrocytes and microglia, and indeed we find that across all conditions APOE levels strongly correlated with GFAP levels (β = 0.84, P < 0.0001), but also with IBA1 levels (β = 0.72, P = 0.002) (Figure 6D). By multiple regression, including both GFAP and IBA1 in the model, we find that the relationship of each to APOE is independent of the other (GFAP, β = 0.46, P < 0.0001; IBA1, β = 0.28, P = 0.0015), suggesting that both glial cell types contribute to the observed effects.
Finally, since various AAV vectors themselves can increase both astrocytic and microglial markers (Ciron et al., 2006; Perez et al., 2020), we performed co-labeling via IHC and confirmed the results of previous studies (Castle et al., 2016; Hudry et al., 2016; Schuster et al., 2014; Aschauer et al., 2013; Hinderer et al., 2014; Samaranch et al., 2012), showing that AAV9, when injected directly into the adult brain parenchyma, is not expressed in microglia and minimally expressed in astrocytes (Figure S5).
The effects of VPS35 depletion-repletion on the morphology of hippocampal microglia
To better characterize microglial changes after VPS35 depletion-repletion, we completed an extensive morphological investigation using Sholl analysis, a standard automated method (Sholl, 1953) that can characterize and quantify the complexity and extent of microglial processes (Ferreira et al., 2014; Morrison and Filosa, 2013; Heindl et al., 2018; Sipe et al., 2016).
The number of microglial cells in the stratum radiatum of the CA1 hippocampal region in the WT and VPS35 nsKO mice did not differ (Figure S6A). By plotting the number of intersecting branches of the processes against the radial distance from the microglia cell body (Figure 7A), we find that VPS35 depletion dramatically alters microglia morphology (Figures 7A and S6B). The alteration is characterized by a decrease in the spatial extension of the microglia processes (leftward shift of the plot Figure 7A; and quantified in Figure 7C [left], t = 7.82, p = 2.52 × 10−12) and a decrease in the number of branching throughout their shortened processes (downward shift of the plot Figure 7A; and quantified in Figure 7C [mid], t = 6.15, p = 1.11 × 10−8, and Figure 7C [right], t = 7.73, p = 3.9 × 10−12).
Figure 7. Neuronal VPS35 regulates an Alzheimer’s-associated microglial morphological phenotype.
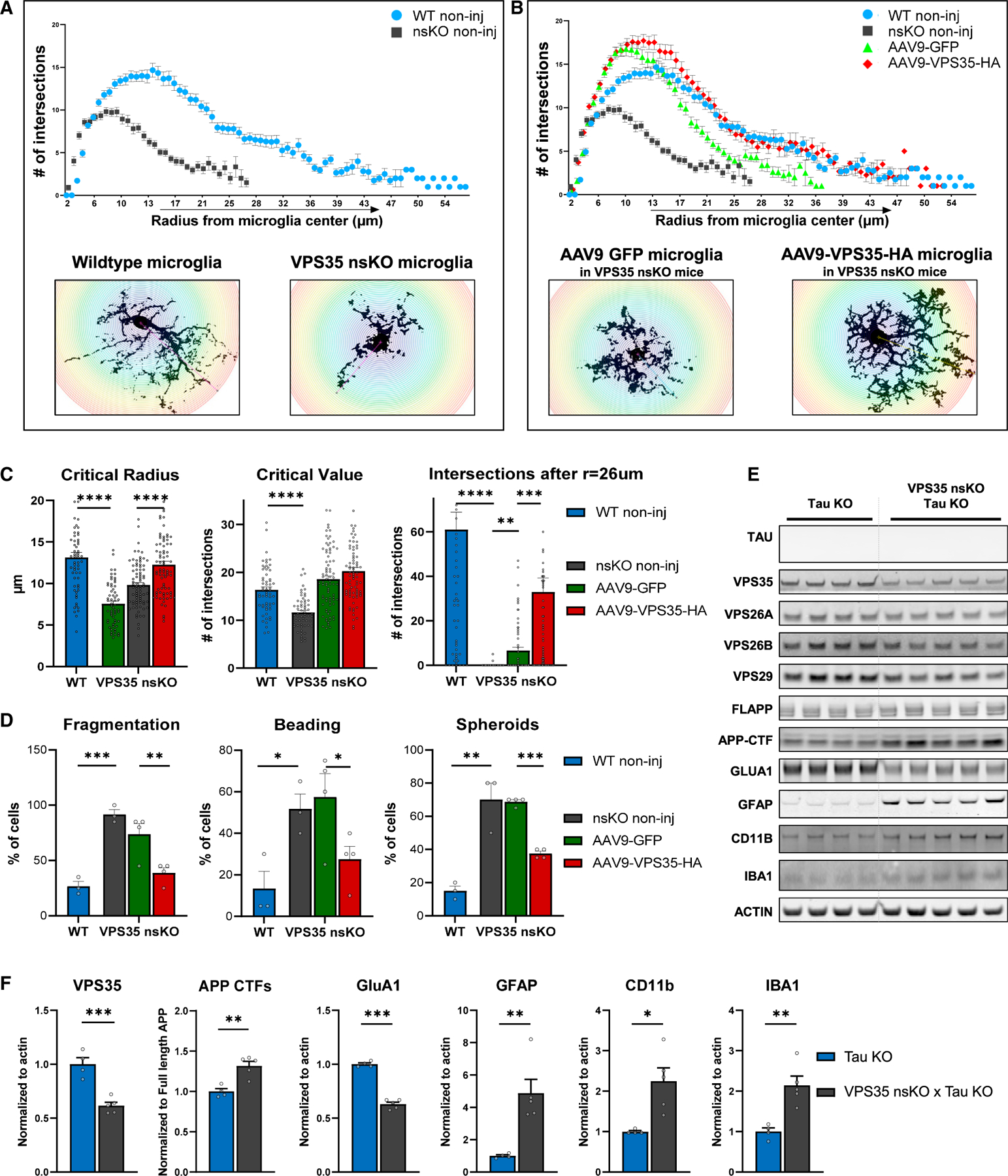
(A) Brains from 6-month-old WT (VPS35fl/fl, n = 3, two females and one male) and VPS35 nsKO mice (n = 3, two females and one male) were harvested and the dorsal hippocampus region was stained with microglial marker IBA1. Microglial morphology was analyzed using ImageJ’s Sholl plugin (n = 60 per condition, 20 cells per animals). Number of branches intersecting the Sholl shells were plotted against the radius of the respective Sholl shell to generate curves. Representative images of microglial cells are included in insets.
(B) Data from (A) are combined with microglial analysis from brains of 6-month-old VPS35 nsKO mice (n = 4, three females and one male) that were injected at 3 months of age with AAV9-GFP and AAV9-VPS35-HA in left and right dorsal CA1, respectively. Brains were harvested and the dorsal hippocampus region was stained with microglial marker IBA1. Microglial morphology was analyzed using ImageJ’s Sholl plugin (n = 80 per condition, 20 from left, and 20 from right dorsal hippocampus, total 40 cells per animal). Number of branches intersecting the Sholl shells were plotted against the radius of the respective Sholl shell to generate curves. Representative images of microglial cells are included in insets.
(C) Critical radius, defined as the radius of a Sholl shell from the cell center (left), at which the critical value occurs, and (middle) critical value, which is the highest number of branches intersecting a Sholl shell. Right: Number of total intersections per cell, after radius of 26 μm from the center of the cell body, was calculated and analyzed.
(D) Left: The percentage of total microglial cells with fragmented processes. Mid; The percentage of total microglial cells with beaded processes. Right: The percentage of total microglial cells with spheroid formation.
The number of animals for these studies (C and D) is the same as described in (A) and (B). 20 microglial cells were analyzed from each of the 14 dorsal hippocampi included in the study: total = 280 microglial cells. See also Figure S6.
(E) Immunoblots from of the CA1 region of Tau KO animals (Tau KO + VPS35fl/fl, n = 4) and Tau KO + VPS35 nsKO (n = 5) mice (6–8 months of age) were probed for VPS35, VPS26A, VPS26B, VPS29, full-length APP, total C-terminal fragments of APP (APP CTFs), GluA1, astrocyte marker glial fibrillary acidic protein (GFAP), and the microglial markers ionized calcium-binding adapter molecule 1 (IBA1) and CD11 antigen-like family member B (CD11b) and actin protein levels. (F) Bar graphs represent actin-normalized levels of VPS35, total C-terminal fragments of APP (APP CTFs), GluA1, GFAP, CD11b, and IBA1 from (E). n = biological replicates. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Two key morphological effects were observed after VPS35 repletion. First, the spatial extent of the microglia processes was normalized, and for most of the distal processes, there was normalization of the branching number, as indicated by the complete overlap of the WT and AAV9-VPS35 plots after a radius of 17 μm from the cell body (Figure 7B). The second effect was an above-normal increase in branching number in processes closer to the cell body (Figures 7B and 7C [mid]). Informatively, this second effect was also observed after AAV9-GFP injections, and thus is likely the effect of the viral vector, as previously described (Mingozzi and High, 2013; Mastakov et al., 2002; Samaranch et al., 2014; Pluvinage and Wyss-Coray, 2017).
The observed alterations in microglia are similar to the dysmorphic microglia observed in the hippocampus of AD brains (Navarro et al., 2018; Streit et al., 2009, 2020; Davies et al., 2017; Shahidehpour et al., 2021), diametrically distinct from the hypertrophic microglia morphology observed in the cortex and typically associated with amyloid plaques (Navarro et al., 2018). Besides the loss of length and branching of microglia processes, as described earlier, AD-associated dysmorphic microglia in the hippocampus are characterized by the loss of fragmentation, beading, and spheroid formation (Navarro et al., 2018). We therefore additionally characterized our microglia dataset to determine whether they phenocopy this dysmorphic phenotype. We found that compared to WT brains, these phenotypic features are recapitulated with VPS35 depletion (fragmentation, t = −10.4, p = 0.0005; beading, t = −3.47, p = 0.026; and spheroid formation, t = −5.28, p = 0.006, Figure 7D) and were all at least partially normalized by AAV9-VPS35 (fragmentation, t = 7.0, p = 0.006; beading, t = 5.55, p = 0.011; and spheroid formation, t = 13.1, p = 0.0009, Figure 7D).
Collectively, results from the depletion-repletion paradigm show that VPS35 in hippocampal neurons can regulate a dysmorphic microglial morphology, phenotypic of the microglia morphology observed in the hippocampus of AD brains.
The effects of VPS35 depletion-repletion occur independent of tau
Neurons are known to signal with microglia, but since this signaling can reflect a complex interplay of proteins and lipids secreted from neurons and also potentially with neuronal and synaptic debris that occurs with synaptic dysfunction (Wilton et al., 2019; Bazan et al., 2005), isolating the precise neuronal signals that message with microglia is extremely challenging.
Nevertheless, in a recent study (Simoes et al., 2020), where we used CSF proteomics to essentially establish the secretome of the VPS35 nsKO mice, an elevation in CSF tau was the most reliable effect. Elevation in CSF tau is an early event in AD’s tau pathology (Barthelemy et al., 2020), and tau pathology occurs first and foremost in the hippocampus when and where there are relatively few amyloid plaques. Moreover, the AD-associated microglia response in the hippocampus has been proposed to be linked to tau pathology (Shahidehpour et al., 2021; Streit et al., 2020; Navarro et al., 2018). It is thus plausible to hypothesize that secreted tau in the hippocampus might be involved in the microglial response we observe in the hippocampus of the VPS35 nsKO mice.
To test this hypothesis, we first generated a mouse model, by crossing the VPS35 nsKO mice with Tau KO mice (VPS35 nsKO X Tau KO mice), established a breeding colony, and aged them to 6 months of age. Remarkably, even in the absence of tau, neuronal VPS35 depletion still resulted in a similar microglial, and astrocytic, response (Figures 7E and 7F). Besides replicating the primary findings, this result refutes the hypothesis that tau secretion, at least by itself, is playing a dominant role in the microglial response in the hippocampus of VPS35 nsKO mice.
Interestingly, in the absence of tau, VPS35 depletion also still resulted in the neuronal phenotypes, an elevation of APP CTFs, and a loss of GluA1. Since previous studies have shown that VPS35 depletion results in GluA1 loss in the absence of APP (Temkin et al., 2017), we can conclude that loss of GluA1, and the synaptic pathology that ensues, occurs in the setting of retromer dysfunction independent of both AD-associated amyloid or tau pathology.
DISCUSSION
The link between retromer-dependent endosomal recycling and AD-associated pathologies has been shown by a range of studies that have manipulated retromer in different model systems (Small and Petsko, 2015). To date, however, most in vivo mouse studies have been performed in transgenic mice engineered to overexpress human mutations in APP and PSEN1/2 (Wen et al., 2011; Li et al., 2019a) or FTD-associated mutations in the tau gene MAPT (Vagnozzi et al., 2019; Carosi et al., 2020). While these studies are useful in showing that retromer can accelerate preexisting amyloid and tau pathologies, they do not model the common sporadic form of AD, in which these mutations do not occur. Moreover, recent studies suggest that retromer defects in sporadic AD can occur independent of, and upstream, to either amyloid or tau pathology (Small and Petsko, 2020).
Taken together with previous studies, the current findings show that dysfunction in retromer-dependent endosomal recycling, in neurons of the hippocampal formation, can recapitulate many of AD’s cardinal pathologies, and that it does so upstream to preexisting amyloid or tau pathology. Evidence for amyloid pathology is based on how retromer is shown to regulate the amyloidogenic processing of APP, whose intraneuronal fragments are now understood to be neurotoxic in the disease (Kwart et al., 2019; Jiang et al., 2010; Lauritzen et al., 2012). Synaptic pathology is another key neuronal phenotype of AD, but this slowly evolving pathology begins as synaptic dysfunction that is thought to percolate years before the overt and wholesale killing of synapses and then neurons (Selkoe, 2002). A loss of glutamate receptors is what typifies this early synaptic stage of neurodegeneration (Yasuda et al., 1995; Wakabayashi et al., 1999; Qu et al., 2021), and we extend previous work in the hippocampal slice (Temkin et al., 2017) to show that this phenotype is regulated by retromer in the intact hippocampus. With respect to tau pathology, retromer has been linked to intraneuronal tauopathy in mouse models (Carosi et al., 2021; Vagnozzi et al., 2019) or in IPS neurons derived from patients with AD (Young et al., 2018), but only on the background of preexisting intraneuronal pathology. It is now understood that accelerated tau secretion represents an early and specific manifestation of tau pathology in AD that is distinct from other tauopathies (Zetterberg, 2017), which occurs via active unconventional secretion (Merezhko et al., 2018). In a previous proteomic screen of the CSF of VPS35 nsKO mice (Simoes et al., 2020), we found that the neuronal retromer can phenocopy the accelerated secretion component of tau pathology.
While AD-associated pathologies begin in neurons, Alois Alzheimer himself recognized the importance of both neuronal and glial pathologies (Alzheimer, 1906, 1907), and recent studies have highlighted the particular relevance of microglia both to the AD’s cellular phenomenology and its pathophysiology (Salter and Stevens, 2017; Hammond et al., 2019; Hong et al., 2016a, 2016b; De Strooper and Karran, 2016; Hansen et al., 2018). Our studies show that the neuronal retromer in the hippocampal formation can regulate a dystrophic morphological phenotype of microglia, a morphological phenotype observed in the hippocampal formation, notable for its dearth of extracellular amyloid plaques (Navarro et al., 2018; Davies et al., 2017; Tischer et al., 2016; Shahidehpour et al., 2021). In fact, dystrophic microglia are diametrically distinct from hypertrophic microglia, characterized by an exuberant overgrowth of microglial processes, typically found in the cortex surrounding amyloid plaques and likely reflective of hyperactive phagocytic clearance of extracellular aggregates (Navarro et al., 2018).
Thus, while not a perfect phenocopy of AD’s cellular pathologies, these findings support the conclusion that disrupting retromer-dependent endosomal recycling can by itself recapitulate the early stages of the disease’s key manifestations. The findings also suggest that retromer knockout mice represent an in vivo model of late-onset sporadic AD. Notably, only a single endogenous protein is depleted in these models, which contrasts to many other AD models that often have multiple exogenous proteins transgenically co-expressed at non-physiological levels.
The neuronal-selective retromer depleted models also instantiate a cellular crosstalk between neurons, the cell type where AD begins, and microglia, a cell type that is implicated in AD genetic risk by modulating disease onset and progression. This cellular dialogue is now considered at the nexus of AD pathophysiology. We recognize that AAV9 vectors were injected into the hippocampal parenchyma to replete VPS35. Nevertheless, a range of previous reports (Castle et al., 2016; Hudry et al., 2016; Schuster et al., 2014; Aschauer et al., 2013; Hinderer et al., 2014; Samaranch et al., 2012), and our own evidence, show that AAV9, when injected in brain parenchyma, does not typically infect microglia or astrocytes to express the vectored plasmids, so we can fairly conclude that the source of the glial-based observations emanates from neurons. By showing a microglial response to neuronal VPS35 depletion even when tau is knocked out, we refuted our hypothesis that neuronal tau secretion might initiate this cellular crosstalk. Furthermore, documenting that the neuronal pathologies were resistant to knocking out tau supports the hypothesis that AD’s neuronal and microglia pathology might occur independent of, and in parallel to, tau pathology (Small and Petsko, 2020).
This sets the stage for future studies to isolate the specific mechanisms by which retromer depletion in neurons signal to microglia, astrocytes, and other non-neuronal cells. We have previously found that at the age that we observe initial microglia changes in the hippocampus of VPS35 nsKO mice, there is no evidence of neuronal cell death (Simoes et al., 2020), suggesting that when retromer function is disrupted in neurons there must be a secreted factor that leads to the observed alterations in glia. Indeed, neurons actively signal with microglia, but this signaling can reflect a complex interplay of proteins and lipids secreted from neurons (Hammond et al., 2019; Ransohoff, 2016; Song and Colonna, 2018). Besides microglia morphology, microglial molecular expression is a second approach for testing for a phenocopy between mouse models and AD, and it might inform the search for inter-cellular signaling. Ongoing single cell transcriptomics studies from human brain samples are starting to establish the precise gene-expression profiles associated with AD’s morphological changes in the hippocampus and other brain regions (Keren-Shaul et al., 2017; Hansen et al., 2018; Hickman et al., 2018; Chen and Colonna, 2021; Deczkowska et al., 2018). Once these profiles are firmly established in AD, future studies will be able to test whether the neuronal retromer can regulate this molecular phenotype, which might offer clues on region-specific intra-cellular signaling at different stages of disease progression.
Mechanistically, showing that disrupting retromer-dependent endosomal recycling can regulate both AD-associated neuronal and microglial phenotypes strengthens and clarifies a causal link between this trafficking route and AD. Therapeutically, demonstrating that AD-associated neuronal and microglial phenotypes can be rescued by retromer viral vectors in adulthood implies that upregulating retromer function in patients might ameliorate AD-associated pathologies across multiple brain cells.
Limitations of the study
The main limitation of this study is that the conclusion that the neuronal retromer can regulate neuronal and microglial phenotypes observed in AD has been shown only in a mouse model of late-onset AD. Mechanistic conclusions about AD are best achieved in AD patients. Notably, studies have already provided the proof-of-concept that retromer can be pharmacologically targeted (Young et al., 2018; Mecozzi et al., 2014), and recent studies have established a CSF biomarker of retromer-dependent neuronal dysfunction (Simoes et al., 2020). Thus, future clinical trials might be able to formally test our mechanistic conclusions and address this limitation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Scott A. Small, sas68@columbia.edu.
Materials availability
Plasmids generated in this study are available on request, for research purpose only, but may require an MTA from Columbia University.
The AAVs generated are not available due to limited quantity and availability, however the full instructions on how to make them are provided in STAR Methods.
Mice generated in this are available on request, for research purpose only, but may require an MTA from Columbia University.
Experimental model and subject details
Adult mice (greater than 3 months of age) both male and female were used in our study. All animals were kept in groups before 3 months of age and kept single housed after 3 months (or post-surgery). Animals were assigned randomly to experimental groups. All experiments involving mice were approved by the Institutional Animal Care and Use Committee of Columbia University.
Wild-type C57BL/6J mice were obtained from Jackson labs (Stock No: 000,664).
VPS35 floxed mice were generated at the Center for Mouse Genome Modification at UConn Health. Homologous recombination was performed in mouse embryonic stem cells (mES), which was derived from an F1(C57BL/6j x 129sv) embryo, targeting the VPS35 gene at exons 2 to 6. The recombined gene had LoxP sites before exons 3 and after exon 5. G418 and Gancyclovir selection and nested long-range PCR were used to identify targeted clones. Targeted ES cells were aggregated into morula to generate chimeric mice. Next the chimeric mice were bred with ROSA26-Flpe (C57BL/6j background) to remove the Frt-flanked PGKneo cassette. Neuronal-selective VPS35 knockout mice were generated by crossing mice expressing loxP-flanked VPS35 (exons 3–5) (VPS35fl/fl – Background 75% C57BL/6J & 25% 129sv) with mice expressing Cre recombinase under the Camk2α promoter. First generation obtained was inbred to get VPS35 nsKO mice. Camk2a-CRE mice were obtained from Jackson labs (Stock No: 005,359). One group of animals was sacrificed at 3 to 3.5 months of age, referenced as 3 months in the text. Another group of animals was sacrificed at 6 to 6.5 months of age and is referenced as 6-month group.
To generate VPS35 nsKO + Tau KO mice, we have crossed our VPS35 fl/fl Camk2a-CRE mice with Htau mice. Htau mice only express human tau since the mouse tau is knocked out by introduction of an eGFP gene (Andorfer et al., 2003). Since Htau is maintained in heterozygous state as a result of this breeding we also get mice that are Tau KO with and without VPS35 neuronal knockout. We used these animals to investigate the gliosis phenotype in the setting of Tau KO.
Primary mouse neuronal culture
Postnatal day 0 primary mouse cortical and hippocampal neuronal cultures from C57BL/6J mice were implemented as described previously (Bhalla et al., 2012). Cultures were kept at 37°C in 5% CO2 incubator. Neurons (1.2E+06 per well) were transduced with various doses of AAV9 (5E+9 vg/well, 2E+10 vg/well & 1E+11 vg/well; Multiplicity of infection of 4k, 17k, & 83k respectively), 7 days after plating in a six-well plate. The culture was maintained for 3 weeks after transduction (4 weeks total). At day 28 the neurons were lysed using RIPA buffer with protease and phosphatase inhibitors.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
METHOD DETAILS
Design and preparation of the retromer constructs
DNA constructs expressing the human retromer proteins tagged with HA or GFP at the C-terminus were designed and synthesized using Thermofisher’s GeneArt portal. mRNA sequence for VPS35 was acquired from the National Center for Biotechnology Information. The constructs were then subcloned into pcDNA 3.1 (+) Hygro vectors and eventually into AAV9 transfer plasmids for viral production. The constructs contained either one of the tags (EGFP or HA) at the C-terminus. The EGFP tagged proteins contained a polyglycine (6G) linker to reduce steric hindrance. EGFP sequence was obtained from pcDNA3-EGFP plasmid (Addgene plasmid 13,031).
AAV9 production
DNA constructs were used to drive expression of transgenes such as VPS35 and GFP (Klein et al., 2002). The expression cassette was flanked with adeno-associated virus serotype 2 (AAV2) terminal repeats. The cytomegalovirus early enhancer element, chicken beta-actin hybrid promoter (“CAG” promoter) was used. The transgene was followed by the woodchuck hepatitis virus post-transcriptional regulatory element and the bovine growth hormone polyadenylation signal (Figure S2). EV control was generated by deleting the GFP sequence. Retromer transgenes were individually subcloned into the construct in lieu of the GFP: VPS35-HA and VPS35-GFP. Each of these constructs was individually packaged into a recombinant adeno-associated virus vector, AAV9, by described methods (Wang et al., 2010) using capsid and helper plasmid DNA from the University of Pennsylvania (Gao et al., 2002). The AAV9 preparations were filter-sterilized using Millex syringe filters (Millipore) at the end of the procedure and then stored frozen in aliquots. The tittering method was for encapsidated vector genomes per ml by a slot-blot method against a standard curve using the Amersham ECL Direct Nucleic Acid Labeling and Detection Systems (GE Healthcare Bio-Sciences).
Co-immunoprecipitation
Co-immunoprecipitation was performed as described previously (Maccarrone et al., 2017; Ghorbani et al., 2014) with some modifications. Briefly, recombinant Protein G Agarose beads (ThermoFisher cat# 15,920,010) were washed three times with PBS, centrifuged at 1,500 g (2 min) and incubated with 3.5–5 ug of primary antibody (anti-HA, ab9110 & anti-GFP, ab290) overnight with constant rocking at 4°C. Next day the antibody coated beads were washed with cold PBS (4°C) and pelleted by spinning at 1,500 g for 2 min.
Cells (or tissue) were washed twice with cold PBS and homogenized in IP buffer (50mM Tris, 100mM EDTA, 150mM NaCl, pH 7.3, protease inhibitors, phosphatase inhibitors and 0.5% Triton X). Protein concentration was determined using Pierce BCA protein assay kit (ThermoFisher cat # 23,225). 300–700 ug of protein was prepared in 1,100 μL of IP buffer. Of this 1,100 μL, 100 μL was saved as total lysate (input) and 1,000 μL was incubated with antibody coated agarose beads overnight at 4°C. Next day the samples (containing beads) were centrifuged at 1,500 g for 2 min at 4°C; the supernatant was collected and labeled as the unbound fraction. Beads were washed five times with cold PBS. After the last wash PBS was completely aspirated and proteins were eluted from the beads and samples were prepared for loading onto electrophoresis gels by adding 150 μL −1X sample buffer (IP buffer 100 μL buffer +35 μL of 4X NuPAGE™ LDS Sample Buffer ThermoFisher cat# NP0007, +15 μL of 10X Reducing Agent ThermoFisher Cat# NP0009). After agitating the samples gently, they were incubated at 50°C on water bath for 30 min, mixing well every 10 min. The samples were incubated at 90°C for 5 min before loading onto NuPAGE® Bis-Tris 4–12% gels.
Western blots
Proteins from neuronal cultures and the mouse brain CA1 region were isolated as described previously (Kirby et al., 2015). Lysates from the samples were run on NuPAGE® Bis-Tris 4–12% gels, transferred onto nitrocellulose membranes using iblot and were probed with antibodies. Primary antibodies targeting the following proteins were used for probing; HA-tag (ab9110, Abcam, 1:2k), VPS35 (ab57632, Abcam, 1:1k), VPS26a (ab23892, Abcam, 1:500), VPS26b (NBP1–92575, Novus, 1:500) VPS29 (NBP1–85288, Novus, 1:500), VPS29 (sab2501105, Sigma-Aldrich, 1:500) and β-actin (ab6276, Abcam), beta III tubulin (neurotubulin) (ab18207, Abcam, 1:2k), NeuN (ab177487, Abcam 1:1k), GluA1 (mab2263, Millipore, 1:1k), APP & APP-CTF (ab32136, Abcam, 1:10K & 1:2k respectively), GFAP (ab68428, Abcam, 1:10k), IBA1 (ab178846, Abcam, 1:500), CD11b (ab133357, Abcam, 1:500), cathepsin D (ab19555, Abcam, 1:2k), SORL1 (anti-LR11 BD-611861, BD bioscience 1:1k). The primary antibodies were selected based on extensive citations by previous studies; most of the antibodies used are monoclonal antibodies with minimal non-specific signal and several of them have been validated by KO testing. To confirm the microglial phenotype, we used antibodies against two proteins that are highly expressed in microglia (IBA1 and CD11b) and are commonly used as markers of microglial to study the microglial morphology in the brain; these proteins, however, are also expressed in peripheral macrophages. IRDye® 800 or 680 antibodies (LI-COR) were used as secondary with dilutions of 1:10k for 800CW, 1:15k for 680RD, and 1:25k for 680LT antibodies. Western blots were scanned using the Odyssey imaging system as described previously (Eaton et al., 2013).
Stereotactic injection of AAV9 in mouse hippocampus
All animal procedures were approved by Columbia University IACUC. AAV9 viral particles were injected into the CA1 region of the mouse brain as described previously (Cetin et al., 2007) with slight modifications. Dorsal CA1 region was chosen because of association of the hippocampus with memory formation (Wittenberg and Tsien, 2002) and also because this region is relatively close to the skull in mice and can be very precisely targeted. Briefly, the surgical procedure was carried out in an aseptic environment. Animals were anesthetized using ketamine / xylazine. Fur was clipped and the site cleaned with betadine scrub followed by 70% ethanol. Eyes were lubricated with an application of sterile, bland ophthalmic ointment. Preemptive analgesia was induced by carprofen and marcaine injections. Mouse was placed on infra-red warming pad in the stereotaxic frame connected to an automatic injection pump. Surgeon wore proper protective equipment and sterile gloves. Using sterile scissors, skin was cut, and bone exposed. A burr hole was placed in the skull at the experimental coordinates calculated using landmarks (bregma and lambda). The dura mater was pierced using a sterile needle and the syringe needle lowered to the desired z coordinate.
AAV9 VPS35-HA virus was injected through a 28g needle attached to a Hamilton syringe at a rate of 0.2 μL/min over a period of 5 −10 min (1–3 μL volume). For unilateral injections control AAV9 was injected on the contralateral side, for bilateral injections control AAV9 was injected in littermate animals. Optimal dose was identified using a dose response ranging from 4E+9 to 1E+11 vector genomes. The needle was retracted 5 min after the end of injection. Surgical wound was closed/sutured by a combination of skin glue and sterile stitches. Post-surgery, 1 mL of normal saline was injected via intraperitoneal route, to minimize dehydration. Infra-red warming pad was used during post op recovery. Carprofen injection was given once daily for 3 days post op. Animals were monitored continuously until ambulating and once daily for the first 3 days and then twice a week until day 14 post op. Sutures were removed on day 10–14 post op. Animals were injected at 3 to 3.5 months of age, and the brains were harvested 3 months post-surgery, for analysis. Brains from animals used for Sorl1 expression and localization study, were harvested 7 months post-surgery.
Perfusion and tissue processing
After the survival period, the animals were perfused with saline, the brains extracted, and the site of injection (dorsal CA1) was micro-dissected out of the brain for biochemical analysis. The dorsal part of CA1 was micro-dissected specifically because the spread of injected virus, and exogenous VPS35 expression was maximal in this region. Animals selected for fluorescent IHC analysis were perfused with normal saline followed by 4% PFA and 30% sucrose. The brains were embedded in optimal cutting temperature (OCT Tissue-tek Cat# 62,550–12) and frozen at −80°C. Frozen blocks of brains in OCT were sectioned at a Leica CM3050 S cryostat set at 12 microns thickness. Sections were transferred onto Superfrost Plus Microscope Slides (Fisher Cat# 12–550-15) and stored at −80°C. For microglial morphological analysis, frozen blocks of brains in OCT were sectioned at a Leica CM3050 S cryostat set at 40 microns thickness. Sections were transferred onto Surgipath X-tra Adhesive Microscope slides (Leica Ref# 3,800,200) and stored at −80°C.
Fluorescent Immunohistochemistry (IHC)
Standard staining and microscopy techniques were used, as described previously (Qureshi et al., 2018; Niklas et al., 2011; Klein et al., 2002; Fu et al., 2019) with some modifications. All washes were done with PBS (1X-PBS containing 0.01% Sodium Azide). Briefly, antigen retrieval was performed using citric acid buffer (10mM sodium citrate, 1.9mM citric acid, pH6.0) as described previously (Shi et al., 1993; Pellicer and Sundblad, 1994; Kanai et al., 1998; Brown and Chirala, 1995). Buffer in a coplin jar was preheated in a microwave (900W, MCD790SW) before immersing the slides. The slides were then heated for 10 min in intervals of 3 + 2 + 3 + 2 min with monitoring/replenishing of the buffer level in between the intervals. Buffer and slides were allowed to cool down at room temperature for 30min. Slides were washed once and incubated in 0.03% H2O2 in water for 30min. After one wash sections were permeabilized using 0.01% digitonin (wt/vol) for 10 min and then blocked in 5% donkey serum in PBS (vol/vol) for 1.5–3 h, followed by incubation in primary antibodies overnight at 4°C, and for an additional 1 h at room temperature the next morning. Primary antibodies were prepared in 1% donkey serum (in PBS) for the following proteins: GFP (ab13970, Abcam, 1:200), HA-tag (ab9110, Abcam, 1:250 & ab18181, Abcam, 1:150), NeuN (ab177487, Abcam, 1:500), GluA1 (ab183797, Abcam, 1:100), GFAP (ab7260, Abcam, 1:500), IBA1, (ab178846, Abcam, 1:250), Sorl1 (anti-LR11 BD-611861, BD bioscience 1:1k). The next day after five washes, the sections were incubated for 2 h (overnight for Sorl1 and IBA1) with secondary antibody at room temperature. Secondary antibodies (ThermoFisher) conjugated with alexa fluor dyes were used at a dilution of 1:250 in 1% donkey serum (in PBS). After three washes Hoechst 33,258 nuclear stain (ThermoFisher H3569, 1:1,500 for 5 min) was used to counter stain the nuclei. After another five washes coverslips were mounted onto the slides with ProLong Gold Antifade Mountant (ThermoFisher P36934). For microglial morphological analysis 40μm section were stained with IBA1 as described above with the following changes: PBS-T (0.3% Triton X-100) was used instead of PBS for all washes, antibodies were prepared in 1% donkey serum in PBS-T, sections were blocked in 5% donkey serum in PBS-T (vol/vol) for 3 h instead of 1.5 h, primary antibody incubation was overnight at 4°C and one additional hour at room temperature the next morning. Secondary antibody incubation was overnight at 4°C.
For microglial structural analysis, 20X objective was used for broad overview images, and 63x z-stack images were taken throughout entire tissue depth 40μm with 0.75μm steps. All images for this analysis were taken from the same region of the dorsal hippocampus across all animals. 20 microglial cells per hemi-brain per animal were analyzed using ImageJ’s Sholl plugin as described previously (Sholl, 1953; Ferreira et al., 2014; Morrison and Filosa, 2013; Heindl et al., 2018; Sipe et al., 2016). Briefly Z-stack images of microglia were pre-processed for Sholl analysis by first isolating the cell and all its processes, slice by slice and then made into a maximum intensity z-projection. The z-projection was then thresholded into a binary image. To correct for any loss of signal through the thresholding process, the ImageJ’s “close” function from the “binary” submenu was used to connect processes separated by one pixel. Sholl analysis was run on this z-projection with the following settings: start radius was the smallest radius from which processes began, end radius was set to the distal end of the longest process, step-size was 0.744μm, 3 samples were taken per radius. The number of intersections per radius generated by this analysis were plotted against the radius of the Sholl shell to generate the curves in Figure 7. The Sholl plugin also gave us Critical Radius and Critical Value. The radius of the Sholl shell with the highest number of intersecting branches was defined as the critical radius, and the number of branches intersecting that shell as critical value. In addition to this we defined a variable as the total number of intersections in each cell after radius of 26 μm from the center of the cell body. The 63x z-stack images were also used to create 3D projections in ImageJ and these 3D projections along with the z-stack were used to manually analyze the cells for fragmentation, beading and spheroid formation. A fragmented cell was defined as any cell where more than 50% of its processes were fragmented. Similarly, a beaded cell was defined as any cell where more than 50% of its processes had punctate IBA1 staining. A spheroid was defined as a spherical swelling of a process, which has an approximate diameter of more than double the diameter of the original process. Numbers of microglial cells were counted manually using ImageJ Cell Counter plugin in 20x confocal microscope field (0.16 mm2 area). Microglial surface rendering was done using ImageJ 3D viewer plugin. Sorl1 puncta were measured on ImageJ using Analyze Particles. 63x stacks were pre-processed by subtracting background using a rolling ball radius of 15.0 pixels. The images were then thresholded and converted into a binary stack. Background noise was further removed by using Binary Close and Remove Outliers (radius 0.5 pixels, threshold 50). Particles were analyzed for all sizes and all circularities. Puncta sizes were then averaged across all areas measured.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using Microsoft Excel and SPSS. For group comparisons, the Student’s t-test, independent two-sample t-tests, or paired t-tests were used where appropriate. For relationships among variables, the Pearson correlation was used to determine bivariate correlations, and a multiple regression model was used to determine if multiple variables independently correlate with a third variable. All data are presented as means; the error bars indicate standard error of the mean. All bar graphs were created in GraphPad Prism 8.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| HA | Abcam | Cat# ab9110; RRID:AB_307019 |
| GFP | Abcam | Cat# ab290; RRID:AB_303395 |
| VPS35 | Abcam | Cat# ab57632; RRID:AB_946126 |
| VPS26a | Abcam | Cat# ab23892; RRID:AB_2215043 |
| VPS26b | Novus | Cat# NBP1-92575; RRID:AB_11020053 |
| VPS29 | Novus | Cat# NBP1-85288; RRID:AB_11009921 |
| VPS29 | Sigma-Aldrich | Cat# sab2501105; RRID:AB_10602838 |
| β-actin | Abcam | Cat# ab6276; RRID:AB_2223210 |
| beta III tubulin | Abcam | Cat# ab18207; RRID:AB_444319 |
| NeuN | Abcam | Cat# ab177487; RRID:AB_2532109 |
| GluA1 | Millipore | Cat# mab2263; RRID:AB_1711819 |
| APP & APP-CTF | Abcam | Cat# ab32136; RRID:AB_2289606 |
| GFAP | Abcam | Cat# ab68428; RRID:AB_1209224 |
| IBA1 | Abcam | Cat# ab178846; RRID:AB_2636859 |
| CD11b | Abcam | Cat# ab133357; RRID:AB_2650514 |
| Cathepsin D | Abcam | Cat# ab19555; RRID:AB_444974 |
| SORL1 | BD bioscience | Cat# BD-611861; RRID:AB_399341 |
| GFP (IHC) | Abcam | Cat# ab13970; RRID:AB_300798 |
| GluA1 (IHC) | Abcam | Cat# ab183797; RRID:AB_2728702 |
| GFAP (IHC) | Abcam | Cat# ab7260; RRID:AB_305808 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Recombinant Protein G Agarose beads | ThermoFisher | 15920010 |
| LDS Sample Buffer | ThermoFisher | NP0007 |
| 10X Reducing Agent | ThermoFisher | NP0009 |
| Hoechst 33258 nuclear stain | ThermoFisher | H3569 |
| ProLong Gold Antifade Mountant | ThermoFisher | P36934 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Pierce BCA protein assay kit | ThermoFisher | 23225 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| VPS35 nsKO mice | This paper and Simoes et al. 2020 | N/A |
| VPS35 nsKO + Htau mice | This paper | N/A |
| VPS35 nsKO + Tau KO mice | This paper | N/A |
| C57BL/6J mice | Jackson Labs | 000664 |
|
| ||
| Plasmids and Viral Vectors | ||
|
| ||
| pcDNA3.1 VPS35-HA | This paper | N/A |
| pcDNA3.1 VPS35-GFP | This paper | N/A |
| AAV9 VPS35-HA | This paper | N/A |
| AAV9 VPS35-GFP | This paper | N/A |
| AAV9 GFP | This paper | N/A |
| AAV9 Empty Vector | This paper | N/A |
|
| ||
| Other | ||
|
| ||
| optimal cutting temperature (OCT) | Tissue-tek | 62550-12 |
| Superfrost™ Plus Microscope Slides | Fisher | 12-550-15 |
| Surgipath X-tra Adhesive Microscope slides | Leica | 3800200 |
Highlights.
Endosomal trafficking is a pathogenic biological pathway in Alzheimer’s disease (AD)
Retromer conducts specific endosomal recycling routes found defective in AD
Neuronal and microglial phenotypes characterize AD-associated hippocampal pathology
Retromer in hippocampal neurons regulates neuronal and microglial phenotypes
ACKNOWLEDGMENTS
This study was partly supported by an NIH grant AG008702 and an anonymous foundation to S.A.S. This paper is dedicated to the memory of R.L.K.
Footnotes
DECLARATION OF INTERESTS
S.A.S., G.A.P., and B.S. are on the Scientific Advisory Board of Retromer Therapeutics; G.A.P. is also on the Scientific Advisory Boards of MeiraGTx, AnnovisBio, Amicus Therapeutics, and Proclara Biosciences. B.S. is also on the advisory board of Annexon Biosciences and Neumora. Y.H.Q. is consulting for Retromer Therapeutics. Last, S.A.S., G.A.P., and Y.H.Q. are co-inventors on patent “Stabilization of retromer for the treatment of Alzheimer’s disease and other neurodegenerative disorders” owned by Columbia University. S.A.S., S.S., and Y.H.Q. are co-inventors on patent “Precision targeted retromer therapeutics for the treatment of neurodegenerative diseases and disorders” owned by Columbia University.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110262.
REFERENCES
- Alzheimer A (1906). Über eine eigenartige Erkrankung der Hirnrinde. “A Characteristic Disease of the Cerebral Cortex”. Neurologisches Centralblatt 25, 1134. [Google Scholar]
- Alzheimer A (1907). Über eine eigenartige Erkrankung der Hirnrinde., 64, 146–148. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-gerichtliche Medizin 64, 146–148. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, De Silva R, Tucker KL, Barde YA, Duff K, and Davies P (2003). Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 86, 582–590. [DOI] [PubMed] [Google Scholar]
- Aschauer DF, Kreuz S, and Rumpel S (2013). Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS One 8, e76310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelemy NR, LI Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS, Buckles V, Fagan AM, Perrin RJ, Goate AM, et al. (2020). A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat. Med. 26, 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG, Marcheselli VL, and Cole-Edwards K (2005). Brain response to injury and neurodegeneration: endogenous neuroprotective signaling. Ann. N. Y Acad. Sci. 1053, 137–147. [DOI] [PubMed] [Google Scholar]
- Benes P, Vetvicka V, and Fusek M (2008). Cathepsin D–many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 68, 12–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla A, Vetanovetz CP, Morel E, Chamoun Z, Di Paolo G, and Small SA (2012). The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol. Dis. 47, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RW, and Chirala R (1995). Utility of microwave-citrate antigen retrieval in diagnostic immunohistochemistry. Mod. Pathol. 8, 515–520. [PubMed] [Google Scholar]
- Buccitelli C, and Selbach M (2020). mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 21, 630–644. [DOI] [PubMed] [Google Scholar]
- Burd C, and Cullen PJ (2014). Retromer: a master conductor of endosome sorting. Cold Spring Harb Perspect. Biol. 6, a016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carosi JM, Denton D, Kumar S, and Sargeant TJ (2021). Retromer dysfunction at the nexus of tauopathies. Cell Death Differ 28, 884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carosi JM, Hein LK, Van Den Hurk M, Adams R, Milky B, Singh S, Bardy C, Denton D, Kumar S, and Sargeant TJ (2020). Retromer regulates the lysosomal clearance of MAPT/tau. Autophagy 17, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle MJ, Turunen HT, Vandenberghe LH, and Wolfe JH (2016). Controlling AAV tropism in the nervous system with natural and engineered capsids. Methods Mol. Biol. 1382, 133–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, and Nixon RA (2000). Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 157, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin A, Komai S, Eliava M, Seeburg PH, and Osten P (2007). Stereotaxic gene delivery in the rodent brain. Nat. Protoc. 1, 3166–3173. [DOI] [PubMed] [Google Scholar]
- Chen Y, and Colonna M (2021). Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J. Exp. Med. 218, e20202717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SK, Narimatsu Y, Simoes S, Goth C, Vaegter CB, Small SA, Cahalan MD, and Andersen OM (2020). Endosomal trafficking is required for glycosylation and normal maturation of the Alzheimer’s-associated protein sorLA. bioRxiv. 10.1101/2020.07.12.199885. [DOI] [Google Scholar]
- Ciron C, Desmaris N, Colle MA, Raoul S, Joussemet B, Verot L, Ausseil J, Froissart R, Roux F, Cherel Y, et al. (2006). Gene therapy of the brain in the dog model of Hurler’s syndrome. Ann. Neurol. 60, 204–213. [DOI] [PubMed] [Google Scholar]
- Davies DS, Ma J, Jegathees T, and Goldsbury C (2017). Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol. 27, 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, and Karran E (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615. [DOI] [PubMed] [Google Scholar]
- Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, and Amit I (2018). Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173, 1073–1081. [DOI] [PubMed] [Google Scholar]
- Eaton SL, Roche SL, Llavero Hurtado M, Oldknow KJ, Farquharson C, Gillingwater TH, and Wishart TM (2013). Total protein analysis as a reliable loading control for quantitative fluorescent Western blotting. PLoS One 8, e72457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira TA, Blackman AV, Oyrer J, Jayabal S, Chung AJ, Watt AJ, Sjöström PJ, and Van Meyel DJ (2014). Neuronal morphometry directly from bitmap images. Nat. Methods 11, 982–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Possenti A, Freer R, Nakano Y, Villegas NCH, Tang M, Cauhy PVM, Lassus BA, Chen S, Fowler SL, et al. (2019). A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat. Neurosci. 22, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, and Wilson JM (2002). Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. U S A. 99, 11854–11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorbani M, Mohamadpour AH, Mehri S, Abnous K, and Hassanzadeh-Khayyat M (2014). Proteomics screening of molecular targets of granulocyte colony stimulating factor in the mouse brain and PC12 cell line. Life Sci. 108, 44–47. [DOI] [PubMed] [Google Scholar]
- Hammond TR, Marsh SE, and Stevens B (2019). Immune signaling in neurodegeneration. Immunity 50, 955–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Goldstein LA, Hou W, Watkins SC, and Rabinowich H (2021). Involvement of CASP9 (caspase 9) in IGF2R/CI-MPR endosomal transport. Autophagy 17, 1393–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Hanson JE, and Sheng M (2018). Microglia in Alzheimer’s disease. J. Cell Biol 217, 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindl S, Gesierich B, Benakis C, Llovera G, Duering M, and Liesz A (2018). Automated morphological analysis of microglia after stroke. Front Cell Neurosci 12, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman S, Izzy S, Sen P, Morsett L, and El Khoury J (2018). Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderer C, Bell P, Vite CH, Louboutin JP, Grant R, Bote E, Yu H, Pukenas B, Hurst R, and Wilson JM (2014). Widespread gene transfer in the central nervous system of cynomolgus macaques following delivery of AAV9 into the cisterna magna. Mol. Ther. Methods Clin. Dev. 1, 14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, LI S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. (2016a). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Dissing-Olesen L, and Stevens B (2016b). New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 36, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E, Martin C, Gandhi S, Gyorgy B, Scheffer DI, MU D, Merkel SF, Mingozzi F, Fitzpatrick Z, Dimant H, et al. (2016). Exosome-associated AAV vector as a robust and convenient neuroscience tool. Gene Ther. 23, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, and Nixon RA (2010). Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. U S A. 107, 1630–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai K, Nunoya T, Shibuya K, Nakamura T, and Tajima M (1998). Variations in effectiveness of antigen retrieval pretreatments for diagnostic immunohistochemistry. Res. Vet. Sci. 64, 57–61. [DOI] [PubMed] [Google Scholar]
- Karch CM, and Goate AM (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A unique microglia type Associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- Kirby ED, Kuwahara AA, Messer RL, and Wyss-Coray T (2015). Adult hippocampal neural stem and progenitor cells regulate the neurogenic niche by secreting VEGF. P.N.A.S. USA 112, 4128–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Hamby ME, Gong Y, Hirko AC, Wang S, Hughes JA, King MA, and Meyer EM (2002). Dose and promoter effects of adeno-associated viral vector for green fluorescent protein expression in the rat brain. Exp. Neurol. 176, 66–74. [DOI] [PubMed] [Google Scholar]
- Kovtun O, Leneva N, Bykov YS, Ariotti N, Teasdale RD, Schaffer M, Engel BD, Owen DJ, Briggs JAG, and Collins BM (2018). Structure of the membrane-assembled retromer coat determined by cryo-electron tomography. Nature 561, 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwart D, Gregg A, Scheckel C, Murphy E, Paquet D, Duffield M, Fak J, Olsen O, Darnell R, and Tessier-Lavigne M (2019). A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP beta-CTFs, not abeta. Neuron 104, 256–270. [DOI] [PubMed] [Google Scholar]
- Lauritzen I, Pardossi-Piquard R, Bauer C, Brigham E, Abraham JD, Ranaldi S, Fraser P, St-George-Hyslop P, Le Thuc O, Espin V, et al. (2012). The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 32, 16243–16255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JG, Chiu J, and Pratico D (2019a). Full recovery of the Alzheimer’s disease phenotype by gain of function of vacuolar protein sorting 35. Mol. Psychiatry 25, 2630–2640. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE, et al. (2019b). Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 101, 207–223.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone G, Bonfiglio JJ, Silberstein S, Turck CW, and Martins-De-Souza D (2017). Characterization of a protein interactome by Co-immunoprecipitation and shotgun mass spectrometry. Methods Mol. Biol. 1546, 223–234. [DOI] [PubMed] [Google Scholar]
- Mastakov MY, Baer K, Symes CW, Leichtlein CB, Kotin RM, and During MJ (2002). Immunological aspects of recombinant adeno-associated virus delivery to the mammalian brain. J. Virol. 76, 8446–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, Schneider RT, Petsko GA, Ringe D, and SMALL SA (2014). Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 10, 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merezhko M, Brunello CA, Yan X, Vihinen H, Jokitalo E, Uronen RL, and Huttunen HJ (2018). Secretion of tau via an unconventional non-vesicular mechanism. Cell Rep 25, 2027–2035 e4. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, and High KA (2013). Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 122, 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Knupp A, Szabo M, Kinoshita C, Hailey DW, Wang Y, and Young JE (2021). The Alzheimer’s gene SORL1 is a key regulator of endosomal recycling in human neurons. bioRxiv. 10.1101/2021.07.26.453861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison HW, and Filosa JA (2013). A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J. Neuroinflammation 10, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro V, Sanchez-Mejias E, Jimenez S, Munoz-Castro C, Sanchez-Varo R, Davila JC, Vizuete M, Gutierrez A, and Vitorica J (2018). Microglia in Alzheimer’s disease: activated, dysfunctional or degenerative. Front Aging Neurosci. 10, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niklas J, Melnyk A, Yuan Y, and Heinzle E (2011). Selective permeabilization for the high-throughput measurement of compartmented enzyme activities in mammalian cells. Anal. Biochem. 416, 218–227. [DOI] [PubMed] [Google Scholar]
- Nixon RA (2005). Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol. Aging 26, 373–382. [DOI] [PubMed] [Google Scholar]
- Pellicer EM, and Sundblad A (1994). [Antigen retrieval by microwave oven with buffer of citric acid]. Medicina (B Aires) 54, 129–132. [PubMed] [Google Scholar]
- Perez BA, Shutterly A, Chan YK, Byrne BJ, and Corti M (2020). Management of neuroinflammatory responses to AAV-mediated gene therapies for neurodegenerative diseases. Brain Sci. 10, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluvinage JV, and Wyss-Coray T (2017). Microglial barriers to viral gene delivery. Neuron 93, 468–470. [DOI] [PubMed] [Google Scholar]
- Qu W, Yuan B, Liu J, Liu Q, Zhang X, Cui R, Yang W, and Li B (2021). Emerging role of AMPA receptor subunit GluA1 in synaptic plasticity: implications for Alzheimer’s disease. Cell Prolif 54, e12959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi YH, Patel VM, Berman DE, Kothiya MJ, Neufeld JL, Vardarajan B, Tang M, Reyes-Dumeyer D, Lantigua R, Medrano M, et al. (2018). An Alzheimer’s linked loss-of-function CLN5 variant impairs Cathepsin D maturation consistent with a retromer trafficking defect. Mol. Cell Biol 38, e00011–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 777–783. [DOI] [PubMed] [Google Scholar]
- Rojas R, Van Vlijmen T, Mardones GA, Prabhu Y, Rojas AL, Mohammed S, Heck AJ, Raposo G, Van Der Sluijs P, and Bonifacino JS (2008). Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol 183, 513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, and Stevens B (2017). Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027. [DOI] [PubMed] [Google Scholar]
- Samaranch L, Salegio EA, San Sebastian W, Kells AP, Foust KD, Bringas JR, Lamarre C, Forsayeth J, Kaspar BK, and Bankiewicz KS (2012). Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum. Gene Ther. 23, 382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranch L, Sebastian WS, Kells AP, Salegio EA, Heller G, Bringas JR, Pivirotto P, Dearmond S, Forsayeth J, and Bankiewicz KS (2014). AAV9-mediated expression of a non-self protein in nonhuman primate central nervous system triggers widespread neuroinflammation driven by antigen-presenting cell transduction. Mol. Ther. 22, 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster DJ, Dykstra JA, Riedl MS, Kitto KF, Belur l.R., Mcivor RS, Elde RP, Fairbanks CA, and Vulchanova L (2014). Biodistribution of adeno-associated virus serotype 9 (AAV9) vector after intrathecal and intravenous delivery in mouse. Front Neuroanat. 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN (2005). Recycle your receptors with retromer. Trends Cell Biol 15, 68–75. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. [DOI] [PubMed] [Google Scholar]
- Shahidehpour RK, Higdon RE, Crawford NG, Neltner JH, Ighodaro ET, Patel E, Price D, Nelson PT, and Bachstetter AD (2021). Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol. Aging 99, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SR, Chaiwun B, Young L, Cote RJ, and Taylor CR (1993). Antigen retrieval technique utilizing citrate buffer or urea solution for immunohistochemical demonstration of androgen receptor in formalin-fixed paraffin sections. J. Histochem. Cytochem. 41, 1599–1604. [DOI] [PubMed] [Google Scholar]
- Sholl DA (1953). Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat. 87, 387–406. [PMC free article] [PubMed] [Google Scholar]
- Simoes S, Neufeld JL, Triana-Baltzer G, Moughadam S, Chen EI, Kothiya M, Qureshi YH, Patel V, Honig LS, Kolb H, and Small SA (2020). Tau and other proteins found in Alzheimer’s disease spinal fluid are linked to retromer-mediated endosomal traffic in mice and humans. Sci. Transl Med. 12, eaba6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe GO, Lowery RL, Tremblay M, Kelly EA, Lamantia CE, and Majewska AK (2016). Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 7, 10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, and Petsko GA (2015). Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat. Rev. Neurosci. 16, 126–132. [DOI] [PubMed] [Google Scholar]
- Small SA, and Petsko GA (2020). Endosomal recycling reconciles the Alzheimer’s disease paradox. Sci. Transl Med. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Simoes-Spassov S, Mayeux R, and Petsko GA (2017). Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer’s disease. Trends Neurosci. 40, 592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WM, and Colonna M (2018). The identity and function of microglia in neurodegeneration. Nat. Immunol. 19, 1048–1058. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, and Bechmann I (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 118, 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Khoshbouei H, and Bechmann I (2020). Dystrophic microglia in late-onset Alzheimer’s disease. Glia 68, 845–854. [DOI] [PubMed] [Google Scholar]
- Takatori S, Wang W, Iguchi A, and Tomita T (2019). Genetic risk factors for alzheimer disease: emerging roles of microglia in disease pathomechanisms. In Reviews on Biomarker Studies in Psychiatric and Neurodegenerative Disorders, GUEST PC, ed. (Cham: Springer International Publishing; ), pp. 83–116. [DOI] [PubMed] [Google Scholar]
- Temkin P, Morishita W, Goswami D, Arendt K, Chen L, and Malenka R (2017). The retromer supports AMPA receptor trafficking during LTP. Neuron 94, 74–82 e5. [DOI] [PubMed] [Google Scholar]
- Thonberg H, Chiang HH, Lilius L, Forsell C, Lindström AK, Johansson C, Björkström J, Thordardottir S, Sleegers K, Van Broeckhoven C, et al. (2017). Identification and description of three families with familial Alzheimer disease that segregate variants in the SORL1 gene. Acta Neuropathol. Commun. 5, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer J, Krueger M, Mueller W, Staszewski O, Prinz M, Streit WJ, and Bechmann I (2016). Inhomogeneous distribution of Iba-1 characterizes microglial pathology in Alzheimer’s disease. Glia 64, 1562–1572. [DOI] [PubMed] [Google Scholar]
- Vagnozzi AN, Li JG, Chiu J, Razmpour R, Warfield R, Ramirez SH, and Pratico D (2019). VPS35 regulates tau phosphorylation and neuropathology in tauopathy. Mol. Psychiatry 6. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wakabayashi K, Narisawa-Saito M, Iwakura Y, Arai T, Ikeda K, Takahashi H, and Nawa H (1999). Phenotypic down-regulation of glutamate receptor subunit GluR1 in Alzheimer’s disease. Neurobiol. Aging 20, 287–295. [DOI] [PubMed] [Google Scholar]
- Wang DB, Dayton RD, Henning PP, Cain CD, Zhao LR, Schrott LM, Orchard EA, Knight DS, and Klein RL (2010). Expansive gene transfer in the rat CNS rapidly produces amyotrophic lateral sclerosis relevant sequelae when TDP-43 is overexpressed. Mol. Ther. 18, 2064–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Tang FL, Hong Y, Luo SW, Wang CL, He W, Shen C, Jung JU, Xiong F, Lee DH, et al. (2011). VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J. Cell Biol 195, 765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton DK, Dissing-Olesen L, and Stevens B (2019). Neuron-glia signaling in synapse elimination. Annu. Rev. Neurosci. 42, 107–127. [DOI] [PubMed] [Google Scholar]
- Wittenberg GM, and Tsien JZ (2002). An emerging molecular and cellular framework for memory processing by the hippocampus. Trends Neurosci. 25, 501–505. [DOI] [PubMed] [Google Scholar]
- Yasuda RP, Ikonomovic MD, Sheffield R, Rubin RT, Wolfe BB, and Armstrong DM (1995). Reduction of AMPA-selective glutamate receptor subunits in the entorhinal cortex of patients with Alzheimer’s disease pathology: a biochemical study. Brain Res. 678, 161–167. [DOI] [PubMed] [Google Scholar]
- Young JE, Fong LK, Frankowski H, Petsko GA, Small SA, and Goldstein LSB (2018). Stabilizing the retromer complex in a human stem cell model of Alzheimer’s disease reduces TAU phosphorylation independently of amyloid precursor protein. Stem Cell Rep. 10, 1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg H (2017). Review: tau in biofluids - relation to pathology, imaging and clinical features. Neuropathol. Appl. Neurobiol. 43, 194–199. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.