

SUMMARY
Macrophages display phenotypic plasticity and can be induced by hepatitis B virus (HBV) to undergo either M1-like pro-inflammatory or M2-like anti-inflammatory polarization. Here, we report that M1-like macrophages stimulated by HBV exhibit a strong HBV-suppressive effect, which is diminished in M2-like macrophages. Tran-scriptomic analysis reveals that HBV induces the expression of interleukin-1β (IL-1β) in M1-like macrophages, which display a high oxidative phosphorylation (OXPHOS) activity distinct from that of conventional M1-like macrophages. Further analysis indicates that OXPHOS attenuates the expression of IL-1β, which suppresses the expression of peroxisome proliferator-activated receptor α (PPARα) and forkhead box O3 (FOXO3) in hepatocytes to suppress HBV gene expression and replication. Moreover, multiple HBV proteins can induce the expression of IL-1β in macrophages. Our results thus indicate that macrophages can respond to HBV by producing IL-1β to suppress HBV replication. However, HBV can also metabolically reprogram macrophages to enhance OXPHOS to minimize this host antiviral response.
Graphical Abstract
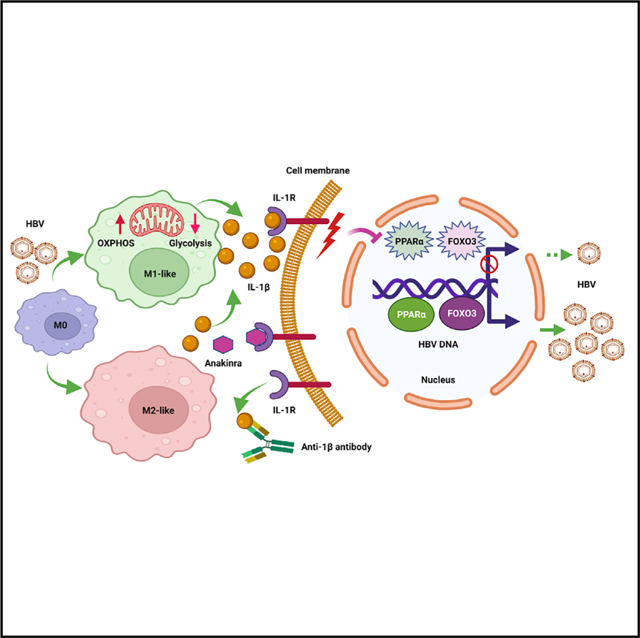
In brief
Li et al. show that HBV can stimulate macrophages to undergo M1- or M2-like polarization and reprogram mitochondrial metabolism. Both M1- and M2-like macrophages can suppress HBV gene expression and viral replication, with the former exhibiting a stronger effect, by releasing IL-1β to downregulate the transcription factors PPARα and FOXO3.
INTRODUCTION
Hepatitis B virus (HBV) can cause acute and chronic hepatitis as well as hepatocellular carcinoma. It is a small DNA virus with a genome size of about 3.2 kb. The HBV genome encodes four genes named S, C, X, and P genes. The S gene codes for viral envelope proteins named surface antigens (HBsAg). The C gene codes for the core protein that forms the viral core particle (i.e., the core antigen or HBcAg) and a related protein named the precore protein. The precore protein is the precursor of the serum e antigen (HBeAg), which has immunomodulatory functions (Tsai et al., 2018; Yang et al., 2019). The X gene codes for a regulatory protein known as the X protein (HBx), and the P gene codes for the viral DNA polymerase.
Macrophages are important immune cells that display phenotypic plasticity, allowing them to exhibit different functions (Krenkel and Tacke, 2017). Macrophages can undergo M1 pro-inflammatory or M2 anti-inflammatory polarization (Cambier et al., 2014; Tsai et al., 2018). M1 macrophages have a high glycolytic activity and a low oxidative phosphorylation (OXPHOS) activity and express pro-inflammatory cytokines, whereas M2 macrophages have the opposite metabolic activities and express an increased level of anti-inflammatory cytokines (Liu et al., 2021; Mills et al., 2016; Tannahill et al., 2013). We previously studied the effect of HBV on hepatic macrophages, also known as Kupffer cells (KCs), by using mice as a model. We produced HBV-negative mice from hemizygous HBV transgenic dams that carried the 1.3-mer overlength HBV genome. We termed these HBV-negative mice “transgenic-derived mice” or TGD mice in brief. We found that KCs isolated from TGD mice would undergo M2-like anti-inflammatory polarization when they were exposed to HBV in the presence of HBeAg. In contrast, KCs isolated from control mice that were born to HBV-negative dams would undergo M1-like pro-inflammatory polarization under the same experimental condition (Tian et al., 2016). These results indicated that HBV could induce KCs to undergo either M1-like or M2-like polarization depending on whether KCs had been exposed to maternal HBV antigens.
Although M2-like KCs induced by HBV could suppress HBV-specific CD8+ T cells to result in HBV persistence in mice (Tian et al., 2016; Xu et al., 2014), whether they can directly affect HBV replication in hepatocytes is unclear. In this article, we studied the effects of M1- and M2-like macrophages on HBV replication in hepatocytes. Our results indicated that M1-like macrophages stimulated by HBV could directly suppress HBV replication, and this HBV-suppressive activity was diminished in M2-like macrophages. Our further analysis indicated that the HBV-suppressive effect of macrophages was mediated by interleukin-1β (IL-1β), which inhibited the expression of the nuclear receptor peroxisome proliferator-activated receptor α (PPARα) and the transcription factor forkhead box O3 (FOXO3) to suppress HBV gene expression. Surprisingly, our results also indicated that HBV could metabolically reprogram M1-like macrophages to stimulate their OXPHOS activities to attenuate the expression of IL-1β.
RESULTS
Suppression of HBV replication by M1- and M2-like macrophages
To determine the possible effect of M1- and M2-like macrophages on HBV replication in hepatocytes, we isolated KCs from TGD and control mice and co-cultured them with hepatocytes isolated from HBV transgenic mice. The purity of KCs was confirmed by flow cytometry (Figure S1A). As shown in Figure 1A, KCs isolated from control and TGD mice expressed similar levels of IL-1β, CD163, and IL-10 before co-culturing. However, 3 days after co-culturing, KCs isolated from control mice expressed an increased level of IL-1β but not CD163 or IL-10, whereas those isolated from TGD mice expressed an increased level of CD163 and IL-10 but only a slightly increased level of IL-1β. IL-1β is a marker of M1-like macrophages, and CD163 and IL-10 are markers of M2-like macrophages. These results were consistent with our previous report, which indicated that KCs isolated from control and TGD mice would undergo M1- and M2-like polarizations, respectively, when stimulated by HBV in the presence of HBeAg (Tian et al., 2016). For comparison, we also treated KCs isolated from control mice with lipopolysaccharides (LPSs), which increased the expression of IL-1β and reduced the expression of CD163 and IL-10. To determine the effect of KCs on HBV replication in hepatocytes, we lysed hepatocytes for western blot, northern blot, and Southern blot analyses 3 days after co-culturing. As shown in Figure 1B, KCs isolated from control mice could significantly reduce the levels of HBsAg, HBV core protein, HBV RNAs, and HBV DNA replicative intermediates (RIs) in hepatocytes. KCs isolated from TGD mice could also reduce the levels of HBV proteins, RNAs, and RI DNA, although to a lesser degree. The analysis of HBsAg, HBeAg, and HBV DNA in the incubation media on days 1 and 3 after co-culturing generated similar results (Figures S1B and S1C). To ensure that the effect of KCs on HBV was not specific to hepatocytes isolated from our HBV transgenic mice, we also isolated hepatocytes from control mice that had been hydrodynamically injected with the 1.3-mer HBV genomic DNA (i.e., pHBV1.3mer) for the co-culturing experiment. As shown in Figure 1C, similar results were obtained, and KCs from both control and TGD mice could reduce HBV protein and RNA levels in mouse hepatocytes, with the former exhibiting a stronger suppressive effect on HBV.
Figure 1. M1- and M2-like macrophages displayed different HBV-suppressive activities.
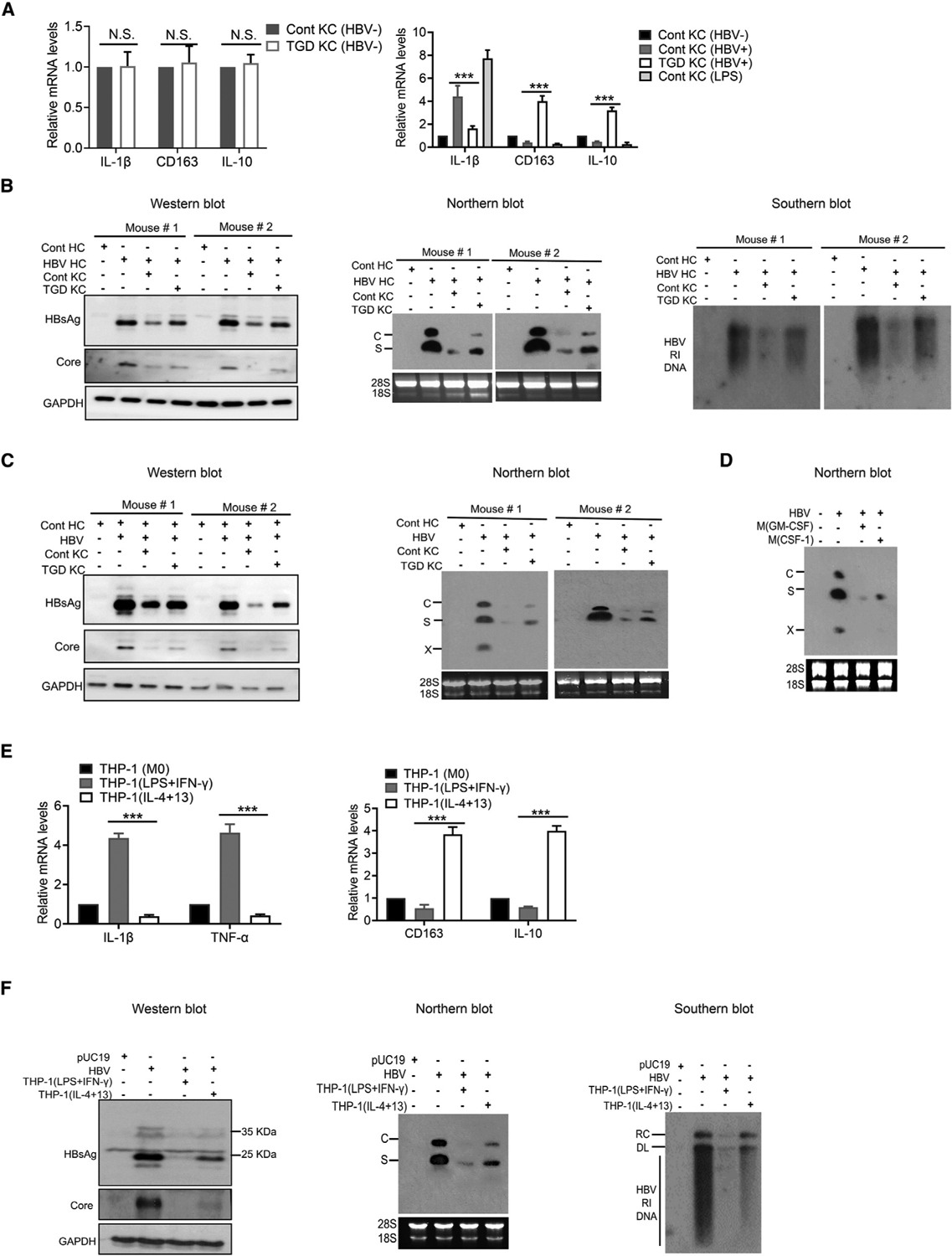
(A) Relative levels of IL-1β, CD163, and IL-10 mRNAs in KCs isolated from control (Cont) or TGD mice as quantified by qRT-PCR. Left panel, KCs without co-culturing with hepatocytes; right panel, KCs co-cultured with HBV-positive hepatocytes. In the right panel, KCs from control mice without co-culturing and those treated with LPSs were also used as the controls. N.S., statistically not significant; ***p < 0.001.
(B) KCs isolated from control and TGD mice were analyzed for their effects on HBV replication in hepatocytes isolated from HBV transgenic mice (HBV HCs) after 3 days of co-culturing. Left panel, western blot of HBV proteins; middle panel, northern blot for HBV RNAs; and right panel, Southern blot of HBV RI DNA. Hepatocytes isolated from control mice (Cont HCs) were used as the negative control. Note that the precore protein was not always visible in the western blot when the core protein was analyzed. In the middle panel, C and S are HBV C and S gene transcripts, respectively, and 28S and 18S rRNAs were used as the loading control.
(C) KCs isolated from control and TGD mice were analyzed for their effects on HBV replication in hepatocytes isolated from mice 1 day after hydrodynamic injection of the 1.3-mer HBV genomic DNA. Hepatocytes were co-cultured with KCs for 3 days and lysed for analysis of HBV proteins by western blot (left panel) or RNAs by northern blot (right panel). The X gene transcript was also detected in the northern blot.
(D) M(GM-CSF) or M(CSF-1) were treated with LPSs for 3 h and then co-cultured with HBV-infected HHs for 14 days. HHs were then lysed for northern blot analysis of HBV RNAs.
(E) THP-1(M0), THP-1(LPS+IFN-γ), and THP-1(IL-4+13) were lysed for analysis of IL-1β, TNF-α, IL-163, and IL-10 RNAs by qRT-PCR.
(F) Huh7 cells were transfected with the control vector pUC19 or the 1.3-mer HBV genomic DNA and, 6 h after DNA transfection, co-cultured with THP-1(LPS+IFN-γ) or THP-1(IL-4+13) for 2 days. Huh7 cells were then lysed for analysis of HBV proteins (left panel), HBV RNA (middle panel), and HBV DNA (right panel). RC, relaxed circular DNA of HBV; DL, double-stranded linear form of HBV DNA.
See also Figure S1.
To determine whether human monocyte-derived macrophages (MDMs) could also suppress HBV replication, we isolated CD14+CD16– classical monocytes from the blood of healthy donors and treated them with granulocyte-macrophage colony-stimulating factor (GM-CSF) to induce their M1-like polarization (i.e., M(GM-CSF)) or with CSF-1 to induce their M2-like polarization (i.e., M(CSF-1)) as previously described (Faure-Dupuy et al., 2019). The purity of CD14+CD16– classical monocytes was verified by flow cytometry (Figure S1D), and their M1- and M2-like phenotypes were confirmed by qRT-PCR using IL-1β and tumor necrosis factor alpha (TNF-α) as M1 markers and CD163 and IL-10 as M2 markers (Figure S1E). M(GM-CSF) and M(CSF-1) cells were then co-cultured with human hepatocytes (HHs) that had been infected by HBV. The possible effects of M(GM-CSF) and M(CSF-1) on HBV were then analyzed. As shown in Figure 1D, both M(GM-CSF) and M(CSF-1) reduced HBV RNA levels, again with the former exhibiting a stronger effect. The analysis of HBsAg, HBeAg, and HBV DNA in the incubation media as well as the analysis of HBV covalently closed circular DNA (cccDNA) in infected HHs similarly demonstrated that both M1-like M(GM-CSF) and M2-like M(CSF-1) macrophages could suppress HBV replication, with the former displaying a stronger suppressive effect than the latter (Figure S1F).
We also determined whether macrophages derived from established human monocytic cell lines such as THP-1 cells could suppress HBV replication. THP-1 cells were treated with phorbol 12-myristate 13-acetate (PMA) for their differentiation into CD14+CD11b+ macrophages (M0) followed by treatment with either LPS and interferon-γ (IFN-γ) for the induction of M1-like polarization (THP-1(LPS+IFN-γ)) or with IL-4 and IL-13 for the induction of M2-like polarization (THP-1(IL-4+13)) as previously described (Genin et al., 2015; Hashimoto et al., 2011; Hong et al., 2018; Mantovani et al., 2004). The M1-like phenotype of THP-1(LPS+IFN-γ)) and the M2-like phenotype of THP-1(IL-4+13) were confirmed by the analysis of M1 markers IL-1β and TNF-α and M2 markers CD163 and IL-10 (Figure 1E). THP-1(LPS+IFN-γ) and THP-1(IL-4+13) were then separately co-cultured with Huh7 cells, a human hepatoma cell line, that had been transfected with pHBV1.3mer. As shown in Figure 1F, both THP-1(LPS+IFN-γ) and THP-1(IL-4+13) could suppress HBV replication by reducing the levels of HBV proteins, RNAs, and RI DNA, with the former exhibiting a stronger suppressive effect than the latter. The same results were obtained when HBsAg, HBeAg, and HBV DNA in the incubation media were analyzed (Figure S1G).
The results shown in Figure 1 indicated that both M1- and M2-like macrophages, whether they were derived from mouse KCs, human monocytes, or THP-1 cells, could suppress HBV replication in hepatocytes, with the former exhibiting a stronger suppressive effect than the latter.
Alteration of gene expression and metabolic profiles of THP-1 macrophages by HBV
As THP-1-derived macrophages could suppress HBV replication, we focused our attention on THP-1 cells for subsequent studies. We first tested whether THP-1(M0) macrophages, without the prior induction of M1- or M2-like polarization, could also suppress HBV replication. As shown in Figure S2A, THP-1(M0) macrophages significantly reduced the levels of HBsAg and HBV core proteins, RNAs, and RI DNA in Huh7 cells that were transfected with pHBV1.3mer. A similar result was observed if THP-1(M0) was co-cultured with HepAD38 cells, which were stable HepG2 cells, a human hepatoblastoma cell line, that contained the HBV genomic DNA under the expression control of tetracycline. In the absence of tetracycline, HBV gene expression was activated, leading to productive HBV replication (i.e., the “Tet-off” mechanism) (Ladner et al., 1997). As shown in Figure S2B, THP-1(M0) similarly reduced HBV proteins, RNAs, and RI DNA levels in HepAD38 cells. The analysis of HBsAg, HBeAg, and HBV DNA in the incubation media generated similar results (Figures S2C and S2D). As the HBV genomic DNA used in our transfection studies belonged to genotype A, and HepAD38 cells contained the genotype D HBV DNA, these results also indicated that the suppressive effect of THP-1 macrophages on HBV was HBV-genotype-independent.
To understand how HBV might affect THP-1 macrophages, we conducted an RNA sequencing (RNA-seq) analysis to determine the gene expression profiles in THP-1 macrophages without co-culturing with HepAD38 cells (THP-1(M0)), THP-1 macrophages co-cultured with HepAD38 cells in the presence of tetracycline (THP-1(HBV−)), and THP-1 macrophages co-cultured with HepAD38 cells in the absence of tetracycline (THP-1(HBV+)). The analysis of RNA-seq results revealed 3,946 differentially expressed genes (DEGs) between THP-1(HBV+) and THP-1(M0) cells, 3,333 DEGs between THP-1(HBV−) and THP-1(M0) cells, and 783 DEGs between THP-1(HBV+) and THP-1(HBV−) cells (Figure 2A). A heatmap analysis revealed that THP-1(HBV+) cells expressed significantly higher levels of M1 markers and lower levels of M2 markers than in THP-1(HBV−) cells (Figure 2B). The induction of IL-1β in THP-1(HBV+) cells was highly prominent when the cytokine expression profiles were analyzed by a volcano plot (Figure 2C). This induction of IL-1β was confirmed by qRT-PCR analysis (Figure S2E). These results indicated that THP-1 macrophages co-cultured with cells containing replicating HBV would undergo M1-like polarization. Interestingly, a heatmap analysis of the top 20 DEGs, based on low false discovery rate and high log2 fold changes, between THP-1(HBV+) and THP-1(HBV−) cells revealed that seven of them were mitochondria-encoded genes that regulate the OXPHOS (Figure 2D). A pathway analysis indicated that the OXPHOS pathway was indeed upregulated in THP-1(HBV+) cells, although some pathways in these cells were also downregulated (e.g., Sirtuin signaling) or not affected (e.g., eIF4 and p70S6K signaling) (Figure 2E).
Figure 2. Effect of HBV on gene expression and mitochondrial metabolism in THP-1 macrophages.
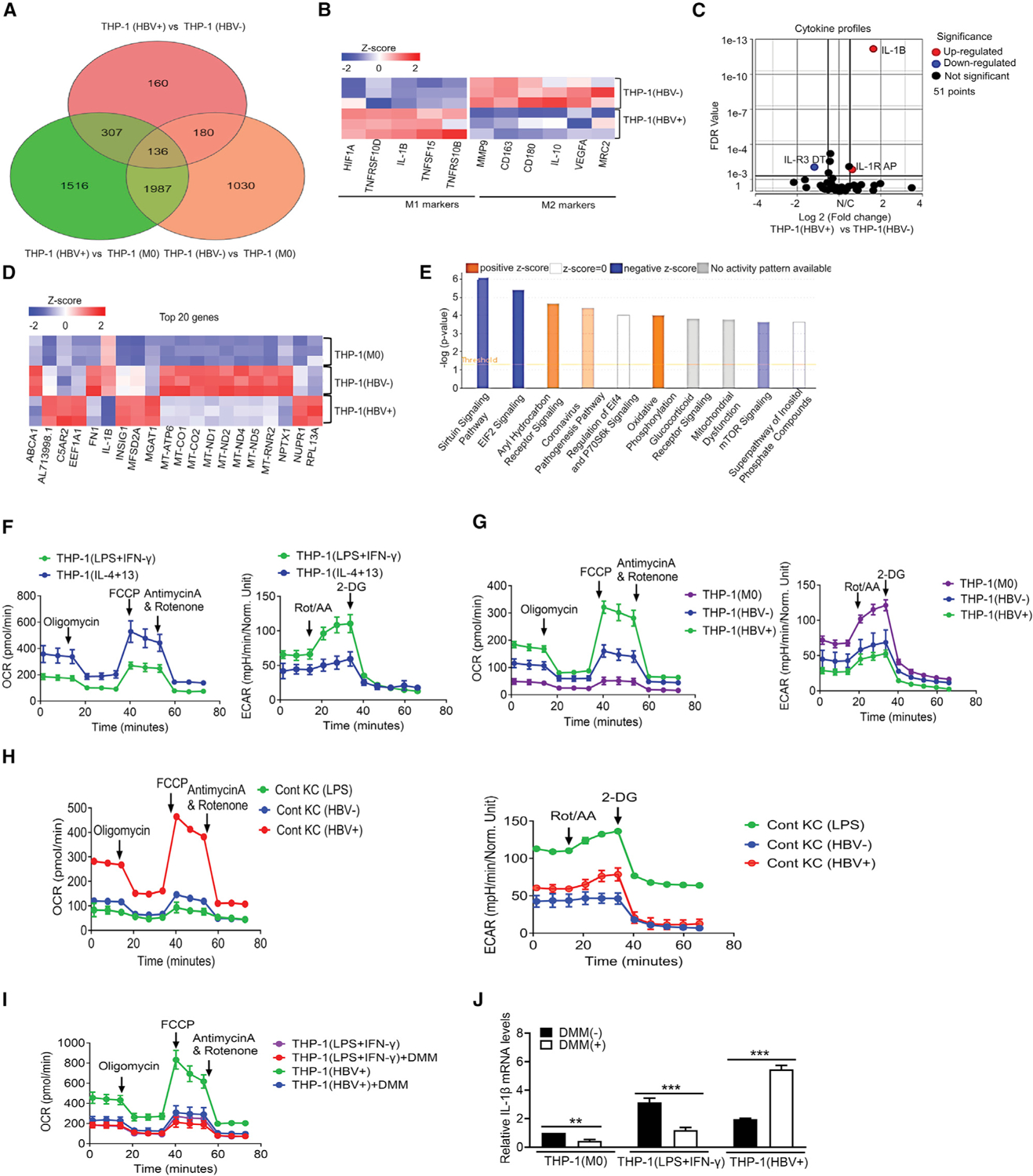
(A) Venn diagram of DEGs between THP-1(HBV+) and THP-1(HBV−), THP-1(HBV+) and THP-1(M0), and THP-1(HBV−) and THP-1(M0).
(B) Heatmap analysis of the relative expression levels of M1 and M2 markers in THP-1(HBV+) and THP-1(HBV−) cells.
(C) Volcano plot of relative cytokine expression levels between THP-1(HBV+) and THP-1(HBV−) cells.
(D) Heatmap and cluster analysis of the top 20 DEGs between THP-1(M0), THP-1(HBV−), and THP-1(HBV+) cells.
(E) Ingenuity pathway analysis (IPA) revealed a strong induction of the oxidative phosphorylation pathway in THP-1(HBV+) cells.
(F) OCRs (left panel) and ECARs (right panel) of M1-like THP-1(LPS+IFN-γ) and M2-like THP-1(IL-4+13) macrophages were determined by Seahorse analysis.
(G) OCRs and ECARs of THP-1(M0), THP-1(HBV−) and THP-1(HBV+) macrophages were compared.
(H) KCs isolated from control mice and treated with LPSs (KC(LPS)) or from control mice that were hydrodynamically injected with pUC19 (KC(HBV−)) or pHBV1.3mer (KC(HBV+)) for 2 days were analyzed for OCRs and ECARs.
(I) DMM slightly inhibited the OCR of THP-1(LPS+IFN-γ) macrophages and strongly inhibited it in THP-1(HBV+) macrophages.
(J) The inhibition of OXPHOS with DMM reduced the expression of IL-1β in THP-1(M0) and THP-1(LPS+IFN-γ) macrophages and enhanced the expression of IL-1β in THP-1(HBV+) macrophages. The results represent the mean ± SEM of three independent experiments. **p < 0.01; ***p < 0.001.
See also Figure S2.
To test whether the OXPHOS was indeed activated in THP-1(HBV+) cells, we measured mitochondrial respiration based on the oxygen consumption rate (OCR) and cellular glycolysis based on the extracellular acidification rate (ECAR) (Zhang and Zhang, 2019). As the control, we first analyzed M1-like THP-1(LPS+IFN-γ) and M2-like THP-1(IL-4+13) macrophages (see Figure 1E). As shown in Figure 2F, THP-1(LPS+IFN-γ) had a lower OCR and a higher ECAR than did THP-1(IL-4+13), which is consistent with the previous report that M1-like macrophages had low OXPHOS and high glycolysis rates and M2-like macrophages had the opposite (Liu et al., 2021). In contrast, THP-1 macrophages activated by HBV, which displayed the M1-like phenotype (Figure 2B), had a high OCR and a low ECAR (Figure 2G). The analysis of mouse KCs with and without in vivo exposure to HBV also revealed that KCs exposed to HBV had a high OCR and a low ECAR, in sharp contrast to those stimulated with LPSs (Figure 2H). These results confirmed that M1-like macrophages activated by HBV indeed had a metabolic profile that was distinct from that of conventional M1-like macrophages. To further determine the biological significance of this HBV-activated OXPHOS, we inhibited OXPHOS with dimethyl malonate (DMM), which inhibits succinate oxidation to block OXPHOS (Mills et al., 2016). As shown in Figure 2I, although DMM only slightly reduced the OCR in THP-1(LPS+IFN-γ) cells, likely due to the low OXPHOS activity in these cells, it significantly reduced the OCR in THP-1(HBV+) cells. This DMM treatment reduced the IL-1β RNA level in THP-1(M0) and THP-1(LPS+IFN-γ) cells, but, surprisingly, it increased the level of IL-1β RNA approximately 3-fold in THP-1(HBV+) cells (Figure 2J). As THP-1(HBV+) produced a lower level of IL-1β than did THP-1(LPS+IFN-γ) (Figure 2J), OXPHOS induced by HBV might play an important role to minimize the production of IL-1β and hence reduce the host inflammatory response against HBV.
Critical role of IL-1β in the suppression of HBV replication both in vitro and in vivo
As macrophages cultured in Transwell could suppress HBV replication in hepatocytes, the effect of macrophages on HBV was likely mediated by a soluble factor. Our cytokine gene profiling study, shown in Figure 2C, revealed a significant induction of IL-1β in THP-1 macrophages by HBV. To test the possible role of IL-1β in the suppression of HBV replication, we co-cultured THP-1 macrophages with Huh7 cells that had been transfected with pHBV1.3mer and added the anti-IL-1β antibody into the incubation media to remove IL-1β. As shown in Figure 3A, THP-1 macrophages reduced HBV protein and RNA levels in Huh7 cells and the amount of viral DNA (i.e., progeny virus) released into the incubation media. These HBV-suppressive effects were not affected by the control antibody, but they were abolished by the anti-IL-1β antibody. This loss of the suppressive effect of THP-1 macrophages on HBV was also observed if the anti-IL-1β antibody was replaced by anakinra, an analog of the IL-1 receptor antagonist (Afonina et al., 2015) (Figure 3A). To confirm that IL-1β could indeed suppress HBV replication in Huh7 cells, we added IL-1β to the incubation media of Huh7 cells that had been transfected with pHBV1.3mer. As shown in Figure 3B, IL-1β reduced the levels of HBV proteins in a dose-dependent manner, which was similarly blocked by the anti-IL-1β antibody or anakinra (Figure 3C). To determine whether IL-1β could also suppress HBV replication in vivo, we injected HBV transgenic mice with IL-1β. As shown in Figure 3D, IL-1β also reduced the level of HBV RNAs in the hepatocytes of HBV transgenic mice, and this reduction was similarly blocked by the anti-IL-1β antibody or anakinra (Figure 3D). The analysis of HBV proteins in mouse hepatocytes generated the same results (see Figure 6C). The anti-IL1β antibody also abolished the suppressive effect of TGD KCs on HBV (Figure 3E), indicating that IL-1β, which was only slightly upregulated in these cells, also mediated the suppressive effect of these KCs on HBV. In contrast to the anti-IL-1β antibody, the anti-TNF-α antibody could not abolish the suppressive effect of control KCs on HBV replication in hepatocytes (Figure 3F), indicating that TNF-α did not play any significant role in the suppression of HBV replication, likely due to its low level produced by macrophages (Figure 3G). Our results thus indicated that the IL-1β produced by macrophages was essential and sufficient to suppress HBV replication.
Figure 3. Suppression of HBV replication by IL-1β.
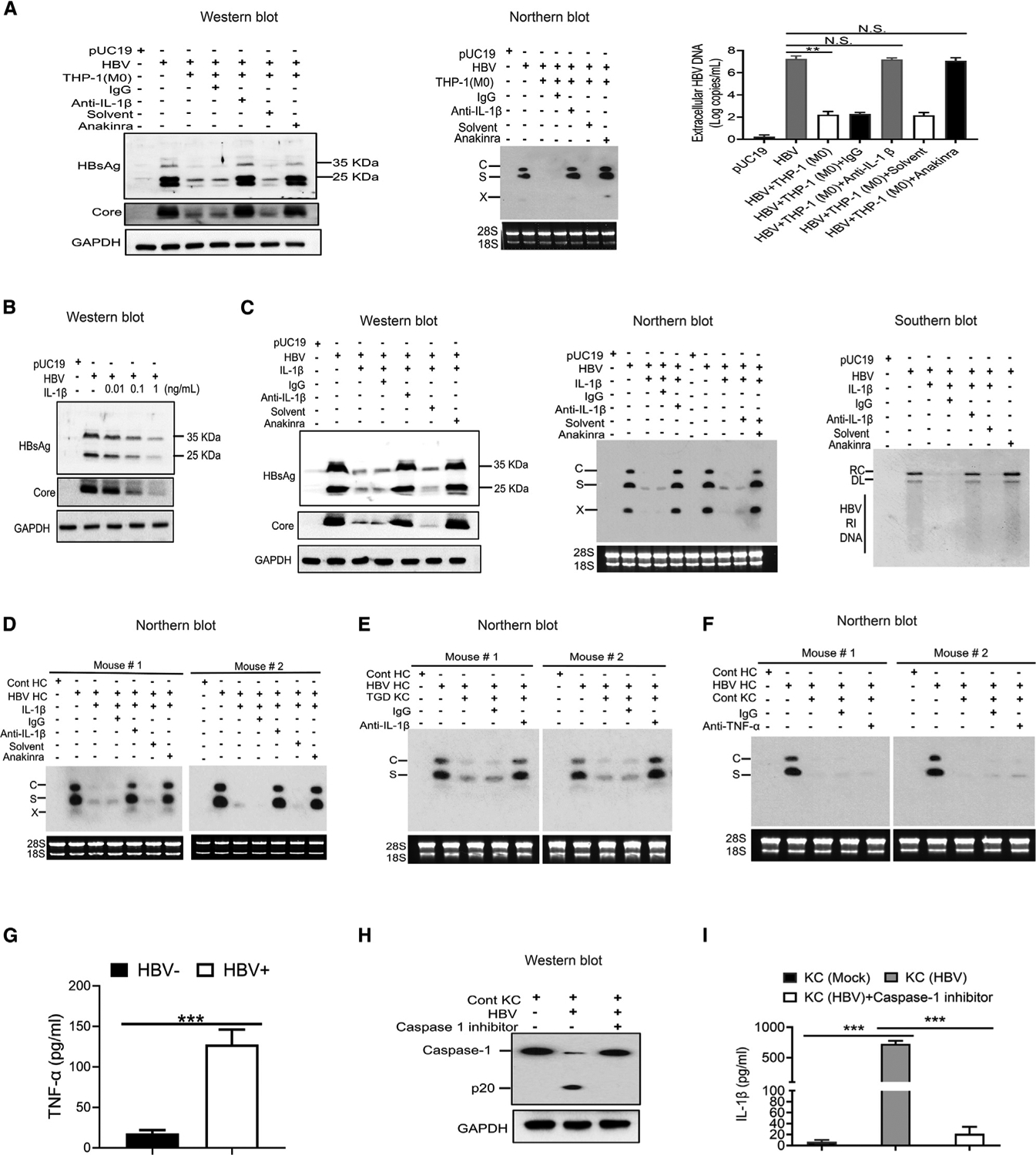
(A) Huh7 cells that had been transfected with pUC19 or pHBV1.3mer were co-cultured with THP-1 macrophages, either in the presence of a control immunoglobulin G (IgG) or the anti-IL-1β antibody (R&D) or in the presence of anakinra or its solvent (i.e., water). Huh7 cells were lysed 2 days after co-culturing for analysis of HBV proteins or HBV RNAs (middle panel). The virion-associated HBV DNA in the incubation media was also quantified by qPCR (right panel). N.S., statistically not significant; **p < 0.01.
(B) Huh7 cells, 2 days after transfection with pHBV1.3mer, were treated with 0.01, 0.1, or 1 ng/mL of IL-1β for 1 day and then lysed for western blot analysis of HBV proteins. Huh7 cells transfected with pUC19 were used as the control.
(C) Huh7 cells transfected with pHBV1.3mer were treated with IL-1β (1 ng/mL) as mentioned above in the presence of control IgG, anti-IL-1β antibody, or anakinra. Cells were then lysed for analysis of HBV proteins (left panel), RNAs (middle panel), and DNA (right panel).
(D) Tg05 HBV transgenic mice were peritoneally injected with IL-1β (200 ng/mouse) with or without the co-injection of control IgG, anti-IL-1β antibody, or anakinra. Hepatocytes were isolated from mice 72 h after injection for the analysis of HBV RNAs.
(E) KCs isolated from TGD mice were co-cultured with hepatocytes isolated from HBV transgenic mice in the presence of a control IgG or the anti-IL-1β antibody. Hepatocytes were lysed 72 h later for analysis of HBV RNAs.
(F) The experiment was conducted the same way as in (E), except that the anti-IL1β antibody was replaced by the anti-TNF-α antibody.
(G) The level of TNF-α in the incubation media of KCs in (F) with (HBV+) or without HBV (HBV−) stimulation was measured by ELISA.
(H) KCs isolated from control mice and co-cultured with HBV-positive hepatocytes in the absence or presence of the caspase 1 inhibitor ex vivo for 72 h were lysed for western blot analysis of caspase-1.
(I) The experiment was conducted as in (H), except that the incubation media were harvested for quantification of IL-1β by ELISA.
See also Figure S3.
Figure 6. Analysis of the effects of PPARα and FOXO3 on HBV gene expression.
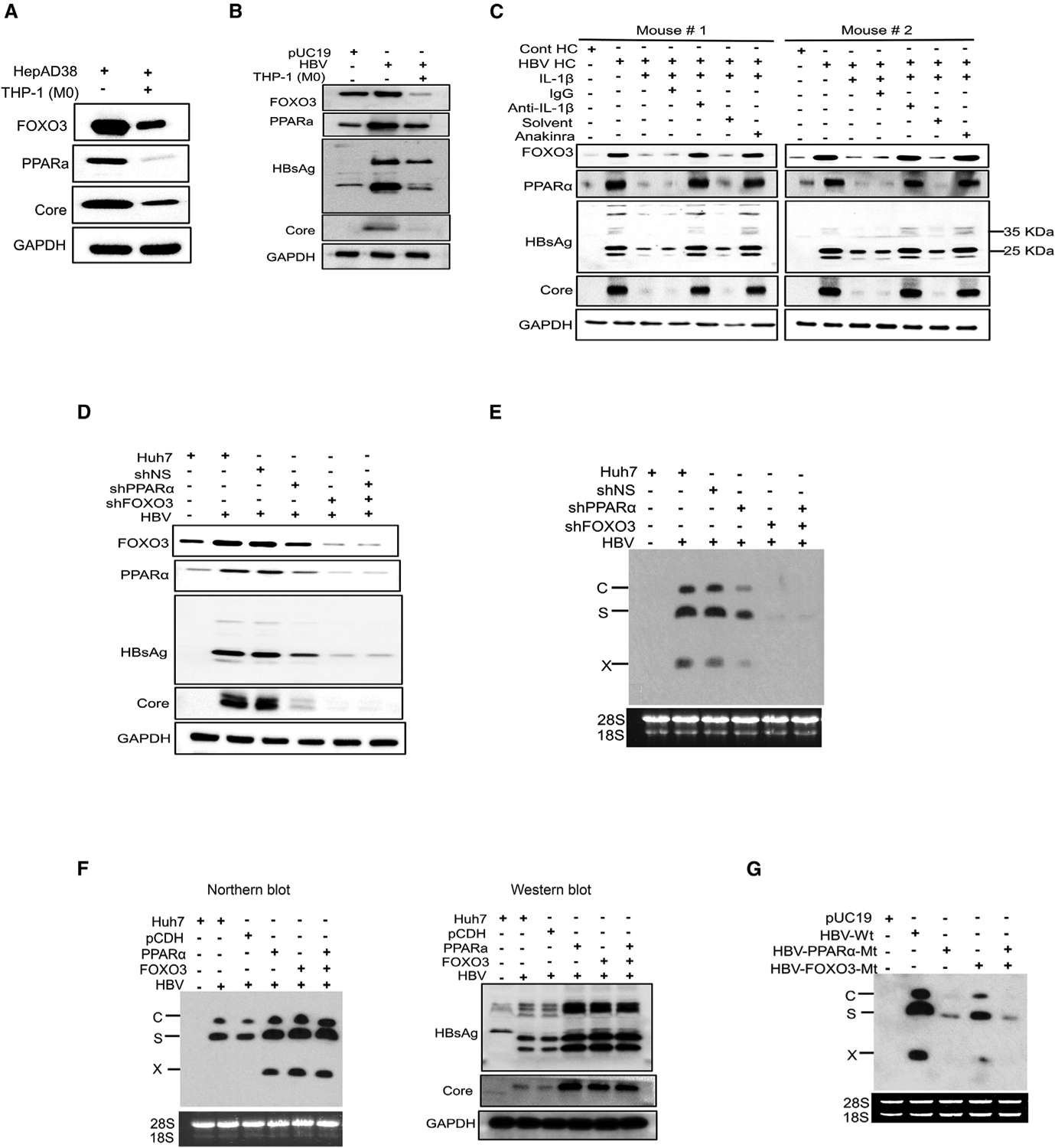
(A) HepAD38 cells with replicating HBV were co-cultured with THP-1 macrophages for 2 days and lysed for western blot analysis of FOXO3, PPARα, and HBV core protein. GAPDH was used as the loading control.
(B) Huh7 cells transfected with the pHBV1.3mer with or without co-culturing with THP-1 macrophages were lysed for western blot analysis of FOXO3, PPARα, HBsAg, and the HBV core protein. Huh7 cells transfected with pUC19 was used as the control.
(C) Tg05 HBV transgenic mice were peritoneally injected with IL-1β (200 ng/mouse) with or without the co-injection of control IgG, anti-IL-1β antibody, or anakinra. Hepatocytes were isolated from mice 24 h later and lysed for western blot analysis.
(D) Huh7 cells transduced with the lentiviral vector that expressed a scrambled shRNA (shNS), PPARα shRNA (shPPARα), or FOXO3 shRNA (shFOXO3) were transfected with the pHBV1.3mer. Cells were lysed 48 h later for western blot analysis.
(E) Experiments were conducted the same way as in (D), followed by northern blot analysis of HBV RNAs.
(F) Huh7 cells were transduced with the lentiviral vector that expressed either PPARα, FOXO3, or both and then transfected with pHBV1.3mer. Cells were lysed 48 h after transfection for analysis of HBV RNAs and proteins. The control lentiviral vector was also used to serve as the control.
(G) Huh7 cells were transfected with pHBV1.3mer or its mutants that contained mutations in the binding site of either PPARα, FOXO3, or both and lysed 48 h later for northern blot analysis of HBV RNAs.
See also Figure S5.
The assembly of the inflammasome complex leads to the cleavage and activation of caspase-1, which then cleaves and activates IL-1β. To test whether caspase-1 was required for the production of IL-1β in KCs, we treated KCs with the caspase-1 inhibitor ex vivo. This caspase-1 inhibitor prevented the cleavage of caspase-1 (Figure 3H) and almost entirely abolished the release of IL-1β from KCs (Figure 3I). To determine whether the injection of HBV DNA into control mice affected KCs and MDMs in the mouse liver, we conducted single-cell RNA-seq (scRNA-seq) of nonparenchymal cells. As shown in Figure S3A, in agreement with the ex vivo results (Figure 1A), KCs of mice injected with the HBV DNA had an increased expression of M1 markers IL-1β and TNF-α and a reduced expression of M2 markers CD206 and CD163. MDMs in mice injected with the pUC19 control vector and those injected with pHBV1.3mer were also compared and are shown in Figure S3B.
Suppression of HBV enhancer I and II activities by macrophages
The observation that THP-1 macrophages could reduce HBV protein and RNA levels indicated that THP-1 macrophages could suppress HBV gene expression, most likely at the transcription step. The HBV gene expression is regulated by two enhancer elements, named enhancer I (ENI) and ENII, in its DNA genome (Marc Flajolet et al., 1998). To test whether the activities of these two enhancer elements could be affected by THP-1 macrophages, ENI and its overlapping X gene promoter (Xp) or ENII and its juxtaposed C gene promoter (Cp) were linked to the firefly luciferase reporter, and these reporter DNA constructs were co-transfected with either pUC19 or pHBV1.3mer into Huh7 cells, which were then co-cultured with THP-1 macrophages. As shown in Figure 4A, THP-1 macrophages could reduce both ENI/Xp and ENII/Cp activities when these reporter plasmids were co-transfected with pHBV1.3mer, but not with pUC19, into Huh7 cells, indicating that THP-1 macrophages could indeed suppress HBV ENI/Xp and ENII/Cp in the presence, but not the absence, of HBV. To further identify the possible macrophage-responsive elements, we conducted deletion-mapping experiments. To bypass the need for including HBV genomic DNA in the co-transfection studies, we transfected the reporter constructs into HepAD38 cells, which were then co-cultured with THP-1 macrophages. As shown in Figure 4B, the deletion of ENI sequence from nucleotide (nt) 1,115 to 1,136 reduced, but did not abolish, the suppressive activity of THP-1 macrophages. However, further deletion of the sequence from nt 1,136 to 1,168 abolished the effect of THP-1 macrophages on the reporter. These results indicated that the sequence between nt 1,136 and 1,168 was essential for THP-1 macrophages to suppress the ENI/Xp activity. The deletion of the ENII/Cp sequence from nt 1,403 to 1,455 abolished the effect of THP-1 macrophages on ENII/Cp, indicating that the sequence between nt 1,403 and 1,455 was essential for macrophages to suppress the ENII/Cp activity (Figure 4C). To test whether nt 1,136–1,168 or 1,403–1,455 would be sufficient to mediate the suppressive effect of macrophages, we inserted these two sequences separately into the plasmid pGL3 promoter in front of the simian virus 40 (SV40) promoter, which drove the expression of the firefly luciferase reporter (Mahmoudi et al., 2009). The resulting plasmids were transfected into HepAD38 cells, which were then co-cultured with THP-1 macrophages. As shown in Figure 4D, although THP-1 macrophages had no effect on the expression of the luciferase reporter from the parental vector, it suppressed its expression when the SV40 early promoter was linked to either nt 1,136–1,168 or 1,403–1,455 of the HBV sequence. These results confirmed that nt 1,136–1,168 and 1,403–1,455 were sufficient to mediate the suppressive effect of macrophages on ENI/Xp and ENII/Cp, respectively.
Figure 4. Suppression of HBV ENI and ENII enhancers by macrophages.
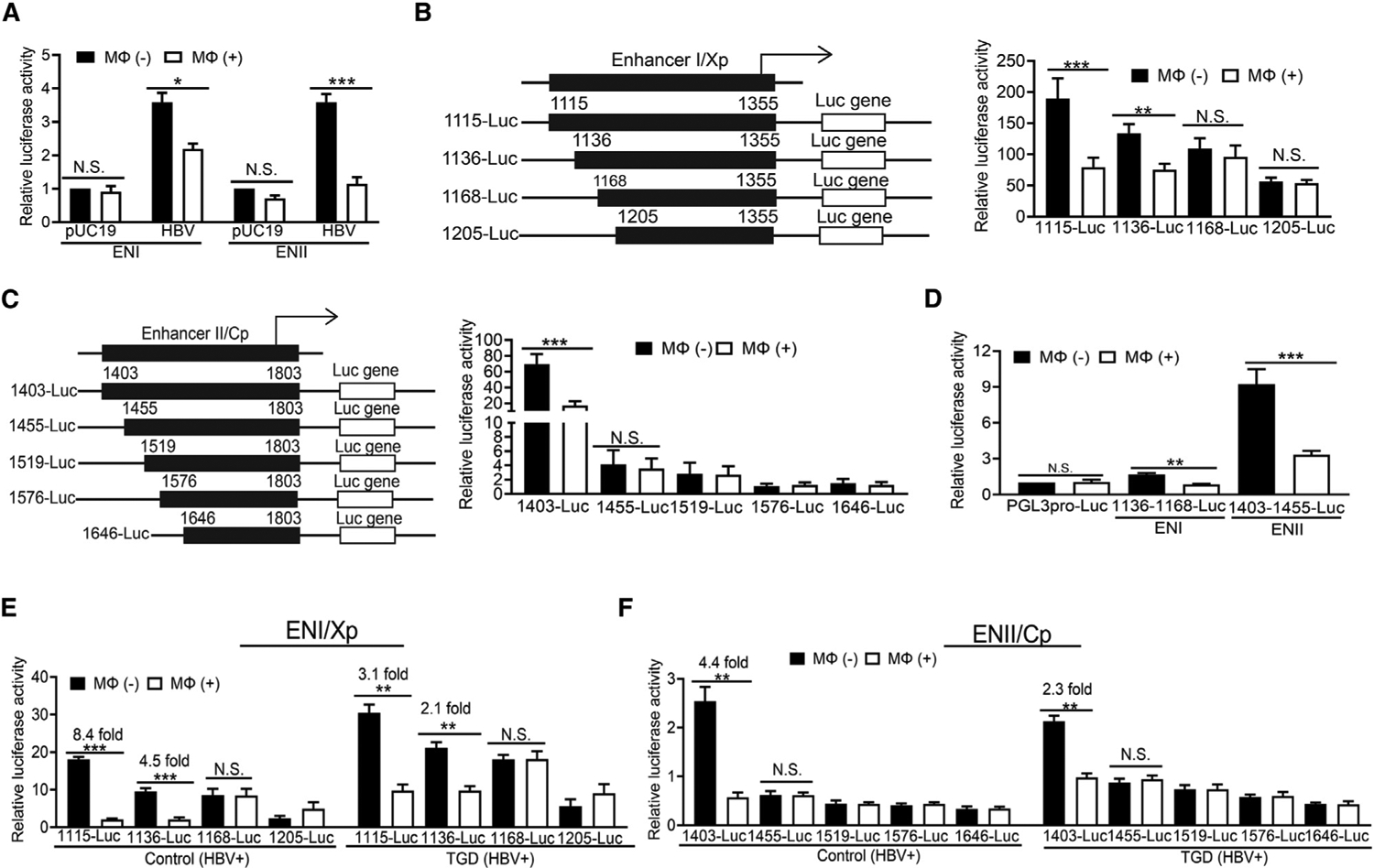
(A) HBV ENI or ENII enhancer reporter construct was co-transfected with pUC19 or pHBV1.3mer into Huh7 cells. The renilla luciferase reporter pRL-TK was also used in the co-transfection to monitor the transfection efficiency. Cells with (Mφ+) or without (Mφ–) co-culturing with THP-1 macrophages for 2 days were then lysed for the measurement of luciferase activities.
(B) The ENI reporter constructs with different deletions are illustrated to the left. These reporter constructs were transfected into HepAD38 cells, and the relative luciferase activities of the reporter constructs in the absence (–) or presence (+) of macrophages are shown in the chart to the right.
(C) The ENII reporter constructs with different deletions are illustrated to the left. The studies were conducted the same way as described in (B).
(D) THP-1 macrophages reduced the activities of 1,136–1,168-luciferase (Luc) and 1,403–1,455-Luc reporter constructs in HepAD38 cells.
(E) Hepatocytes isolated from control or TGD mice that had been injected with 20 μg pHBV1.3mer were transfected with the ENI reporter constructs ex vivo. These hepatocytes, with (+) or without (−) the subsequent co-culturing with their paired KCs isolated from the same mice, were then lysed for analysis of the reporter activities.
(F) The study was conducted the same way as in (E), with the exception that ENII reporter constructs were analyzed. The results represent the mean ± SEM of three independent experiments. N.S., not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figure S4.
The deletion-mapping experiments were also repeated using KCs isolated from control or TGD mice. Hepatocytes were isolated from mice that had been hydrodynamically injected with 20 μg pHBV1.3mer or pUC19. These hepatocytes were transfected with the reporter constructs and co-cultured with KCs isolated from control or TGD mice. In agreement with the THP-1 macrophage results, KCs from control and TGD mice were able to suppress ENI/Xp and ENII/Cp activities in the presence of HBV (Figures 4E and 4F) but not in the absence of it (Figures S4A and S4B). Similarly, nt 1,136–1,168 and 1,403–1,455 were found to be essential for KCs to suppress the activities of ENI/Xp and ENII/Cp, respectively (Figures 4E and 4F). Since control and TGD KCs would undergo M1- and M2-like polarizations, respectively, after exposure to HBV (see Figure 1A), M1- and M2-like KCs likely suppressed ENI/Xp and ENII/Cp via the same mechanism. The results shown in Figure S4 also confirmed that the presence of HBV was essential to activate KCs for the induction of the anti-HBV response. Note that in agreement with the results shown in Figure 2B, M1-like KCs had a higher suppressive effect than did M2-like KCs on ENI/Xp (i.e., 8.4- fold versus 3.1-fold) and ENII/Xp (i.e., 4.4-fold versus 2.3-fold).
Inhibition of PPARα and FOXO3 by macrophages and IL-1β
The sequence of nt 1,136–1,168 contains a nuclear receptor binding site that is recognized by hepatocyte nuclear factor 4 (HNF4), retinoid X receptor alpha (RXRα), PPARα, and COUP-TF, whereas the sequence of nt 1,403–1,455 was not previously known to be recognized by any transcription factors (Oropeza et al., 2020). Computational analysis of the sequence of nt 1,403–1,455 using the JASPAR database led to the identification of a putative FOXO3-binding sequence, GTAAACAA, at nt 1,424–1,431 (Fornes et al., 2020). To test whether THP-1 macrophages would have any effect on the binding of nuclear receptors to the ENI enhancer, we performed a chromatin immunoprecipitation (ChIP) assay. As shown in Figure 5A (top panel), the co-culturing with THP-1 macrophages reduced the binding of PPARα to the ENI enhancer without affecting the binding of HNF4α, RXRα, and COUP-TF1. Similarly, as shown in Figure 5A (bottom panel), the ChIP assay confirmed that FOXO3 could bind to the ENII enhancer and that this binding was abolished by THP-1 macrophages. The same results were obtained if the ChIP assay was conducted using hepatocytes isolated from HBV transgenic mice that had been injected with IL-1β. As shown in Figure 5B (top panel), IL-1β specifically reduced the binding of PPARα to the ENI enhancer without affecting the binding of other nuclear receptors. It also reduced the binding of FOXO3 to ENII enhancer (Figure 5B, bottom panel).
Figure 5. Macrophages and IL-1β suppress the binding of PPARα to ENI and FOXO3 to ENII.
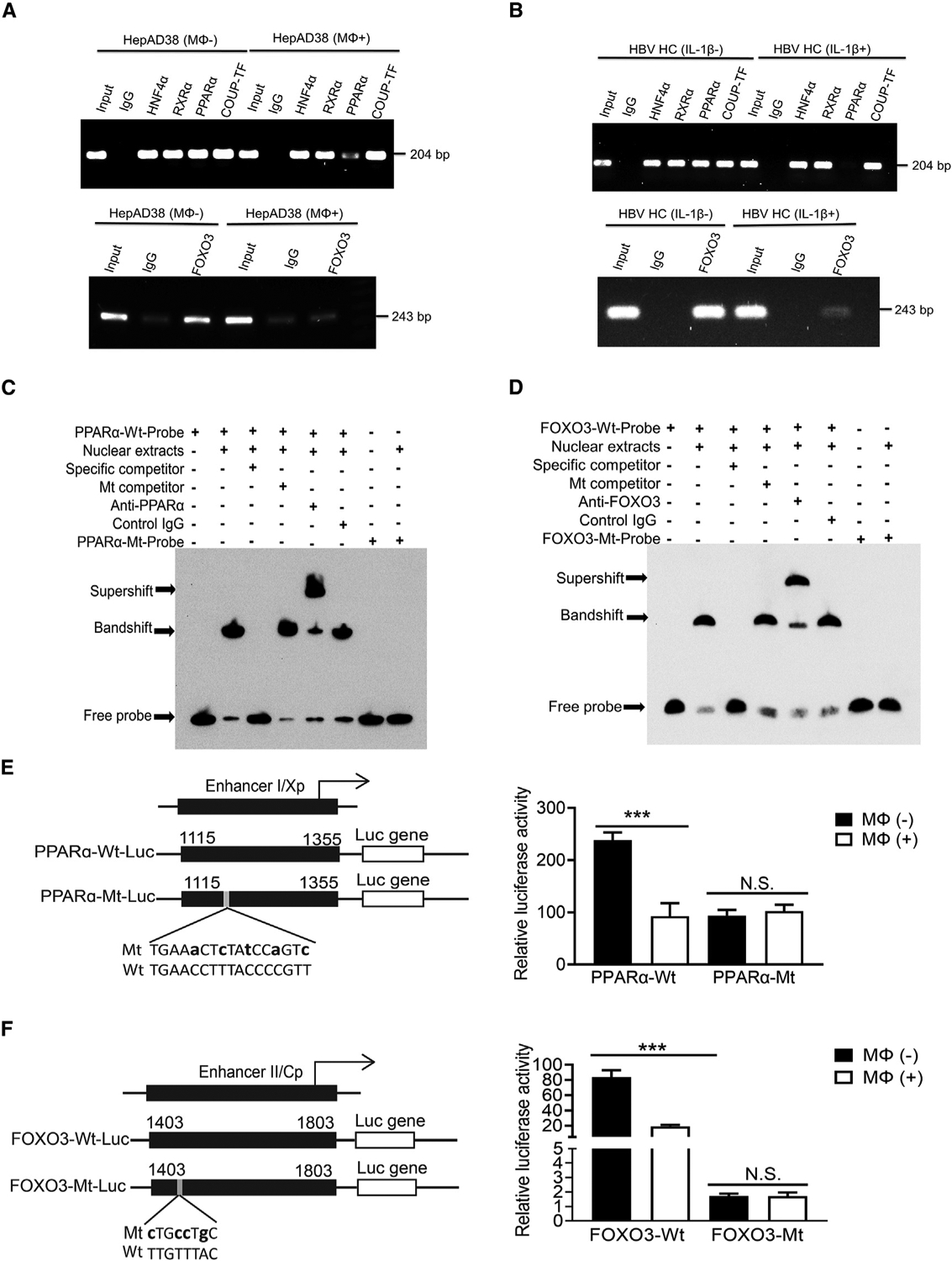
(A) The binding of HNF4α, RXRα, PPARα, and COUP-TF1 to the ENI enhancer (top panel) or FOXO3 to the ENII enhancer (bottom panel) was analyzed by the ChIP assay using HepAD38 cells with (Mφ+) or without (Mφ−) co-culturing with THP-1 macrophages. The control IgG served as the negative control. “Input”, total cell lysates without the step of immunoprecipitation to serve as the positive control.
(B) The same ChIP assay as shown in (A) was conducted using HCs isolated from HBV transgenic mice with (IL-1β+) or without (IL-1β–) the injection of IL-1β.
(C) The binding of PPARα to nt 1,136–1168 of ENI1 was analyzed by the EMSA. The double-stranded oligonucleotide containing the sequence of nt 1,136–1,168 was used as the probe for incubation with the HepG2 nuclear extracts. The nucleotide mutations of the mutant probe are shown in (E).
(D) The binding of FOXO3 to nt 1,403–1,455 of ENII was analyzed by the EMSA. The nucleotide mutations of the mutant probe are shown in (F).
(E) Wild-type (WT) and mutant (Mt) ENI/Xp Luc reporter constructs are shown to the left. The lowercase letters indicate the nucleotide mutations introduced. These reporter constructs were transfected into HepAD38 cells with or without co-culturing with THP-1 macrophages. The Luc reporter results are shown to the right.
(F) WT and Mt ENII/Cp Luc reporter constructs are shown to the left. The study was conducted as shown in (E). N.S., not significant; ***p < 0.001.
The binding of PPARα to nt 1,136–1,168 was further confirmed by the electrophoretic mobility shift assay (EMSA). As shown in Figure 5C, the incubation of an oligonucleotide probe containing this sequence with HepG2 nuclear extracts resulted in a bandshift, which could be removed by a specific competitor that contained the nt 1,136–1,168 sequence but not by a competitor that contained mutations in the PPARα binding site (see below). In addition, this bandshift could be supershifted by the anti-PPARα antibody but not by a control antibody, and the oligonucleotide probe that contained mutations in the PPARα binding site could not generate a bandshift. These results confirmed the binding of PPARα to nt 1,136–1,168. The repeat of the EMSA using a probe containing the sequence of nt 1,403–1,455 confirmed the binding of FOXO3 to this sequence (Figure 5D). Finally, as shown in Figure 5E, the mutations introduced into the ENI enhancer to abolish the binding of PPARα led to the reduction of the ENI/Xp activity that could not be further suppressed by THP-1 macrophages. Similarly, as shown in Figure 5F, the mutations that abolished the binding of FOXO3 to the ENII enhancer also led to the reduction of the ENII/Cp activity that also could no longer be suppressed by THP-1 macrophages. These results together demonstrated that THP-1 macrophages or IL-1β suppressed HBV ENI and ENII enhancers via the suppression of the binding of PPARα and FOXO3 to their respective binding sites in ENI and ENII enhancers.
Critical roles of PPARα and FOXO3 in HBV gene expression
To understand how THP-1 macrophages regulated the activities of PPARα and FOXO3 in HepAD38 cells, we conducted a western blot analysis on these two proteins. As shown in Figure 6A, the co-culturing of HepAD38 cells with THP-1 macrophages reduced the protein levels of PPARα and FOXO3 as well as the HBV core protein. This effect of THP-1 macrophages was apparently post-transcriptional, as the RNA levels of PPARα and FOXO3 were not affected (Figure S5A). A similar result was observed when THP-1 cells were co-cultured with Huh7 cells that were transfected with pHBV1.3mer. As shown in Figure 6B, HBV increased both PPARα and FOXO3 protein levels, although the effect on PPARα was more prominent. The co-culturing with THP-1 macrophages reduced both PPARα and FOXO3 protein levels. As the effect of macrophages on HBV is mediated by IL-1β, we also analyzed the effect of IL-1β on PPARα and FOXO3 in vivo by injecting HBV transgenic mice with IL-1β. As shown in Figure 6C, as compared with control mice, HBV also induced the expression of PPARα and FOXO3 in hepatocytes, and this induction was abolished by IL-1β, which also reduced HBsAg and HBV core protein levels, in agreement with the results shown in Figure 3D. The effect of IL-1β on PPARα and FOXO3 was abolished by the anti-IL-1β antibody or anakinra, which also restored HBsAg and HBV core protein levels. To further examine the effects of PPARα and FOXO3 on HBV gene expression, we silenced the expression of PPARα and/or FOXO3 in Huh7 cells with their respective shRNAs. As shown in Figure 6D, in agreement with the results shown in Figures 6B and 6C, HBV increased PPARα and FOXO3 protein levels. The silencing of PPARα reduced HBV protein levels and also reduced the FOXO3 protein level, which was likely an outcome of reduced HBV replication. Similarly, the silencing of FOXO3 also reduced HBV protein levels and PPARα.The simultaneous silencing of both PPARα and FOXO3 did not further reduce the HBV protein levels. The silencing of PPARα, FOXO3, or both also reduced HBV RNA levels correspondingly (Figure 6E). In addition to gene silencing, we also used the lentiviral expression vector to overexpress PPARα and FOXO3 in Huh7 cells (Figures S5B and 6F). The expression of either PPARα or FOXO3 in Huh7 cells transfected with pHBV1.3mer led to the increase of HBV RNA and protein levels, which were not further increased if PPARα and FOXO3 were co-expressed (Figure 6F).
Finally, to further examine the roles of PPARα and FOXO3 in HBV RNA transcription, mutations shown in Figures 5E and 5F were introduced into the HBV genome to abolish the binding sites of these two protein factors in ENI and ENII enhancers. The 1.3-mer HBV genomic DNA was then transfected into Huh7 cells. As expected, the analysis of HBV RNAs indicated that mutations in either the PPARα binding site, the FOXO3 binding site, or both significantly reduced the HBV RNA levels (Figure 6G).
Activation of macrophages by multiple HBV factors
Our results indicated that HBV could activate macrophages to suppress HBV gene expression. To identify the HBV gene product(s) that might be involved, we produced HBV mutants that were incapable of expressing only HBsAg, HBeAg, the core protein, or HBx. We also produced a mutant that was incapable of expressing both HBsAg and HBeAg. The genomic DNA of these HBV mutants was then transfected into Huh7 cells, with or without the subsequent co-culturing with THP-1 macrophages. As shown in Figure 7A, abolishing the expression of HBsAg did not affect the ability of THP-1 macrophages to suppress the expression of the core protein, nor did abolishing the expression of HBeAg prevent THP-1 macrophages from suppressing the expression of HBsAg and the core protein. Similarly, abolishing the expression of both HBsAg and HBeAg did not prevent macrophages from suppressing the expression of the core protein, nor did abolishing the expression of the core protein affect the ability of the macrophages to suppress the expression of HBsAg. Abolishing the expression of HBx also had no effect on the suppressive activity of macrophages on HBsAg and the core protein. Similarly, as shown in Figures 7B and 7C, THP-1 macrophages were able to reduce the HBV RNA and RI DNA levels of all HBV mutants tested. Note that no HBV RI DNA was detected in cells transfected with the core protein mutant, as the core protein is required for viral DNA replication. These results together indicated that macrophages could be stimulated by multiple HBV factors to suppress HBV replication. It is unlikely that the suppressive effect of macrophages on HBV mutants was caused by DNA transfection, as cells transfected by pUC19 failed to activate macrophages to suppress ENI and ENII enhancers (see Figure S4). To further examine the HBV factors that might activate macrophages, we treated THP-1 macrophages with recombinant human serum albumin (HSA), HBsAg, and the HBV core protein, which were acquired commercially. We also expressed HBeAg in E. coli and subsequently purified the protein. The purity of the protein and its reactivity to HBeAg are shown in Figure S6. As shown in Figure 7D, while HSA had no effect on THP-1 macrophages, HBsAg, the core protein, and HBeAg were all able to induce the expression of IL-1β, confirming that THP-1 macrophages could be activated by multiple HBV proteins. The effect of HBeAg on THP-1 macrophages was specific, as it could be blocked by the anti-HBeAg but not by the control antibody.
Figure 7. Analysis of the response of THP-1 macrophages to various HBV proteins.
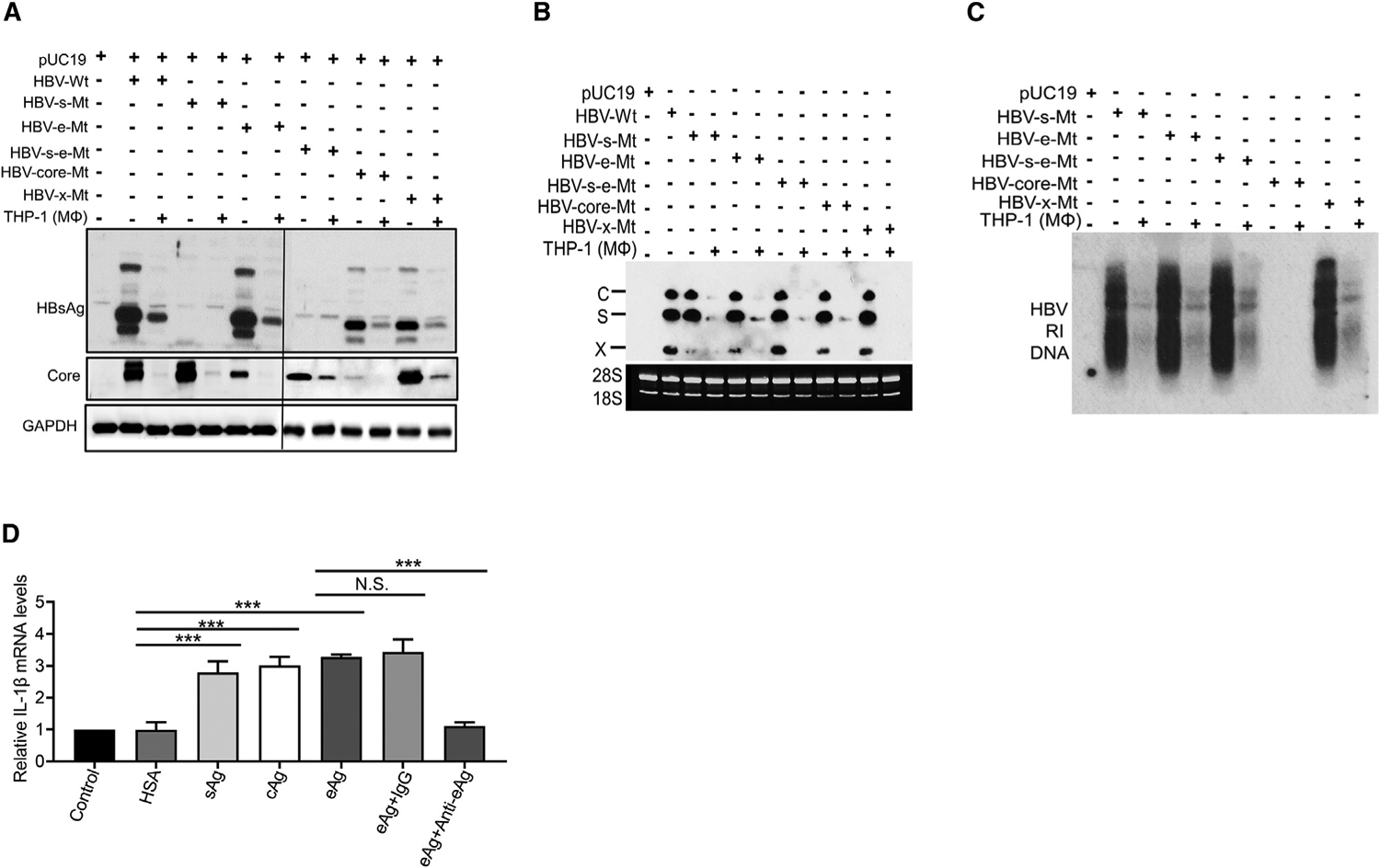
(A–C) Huh7 cells transfected with pUC19 or the HBV genomic DNA with various mutations, and with or without subsequent co-culturing with THP-1 macrophages, were lysed and analyzed by western blot (A), northern blot (B), or Southern blot (C).
(D) THP-1 macrophages not treated (NT) or treated with HSA, HBsAg, HBV core protein, or HBeAg were lysed for qRT-PCR analysis of the IL-1β RNA. The IL-1β RNA level of cells not treated with recombinant proteins was arbitrarily defined as 1. In the HBeAg studies, the addition of the anti-HBeAg antibody, but not the control antibody, abolished the induction of IL-1β by HBeAg in THP-1 macrophages. The results represented the mean ± SEM of three independent experiments.
N.S., not significant; ***p < 0.001.
See also Figure S6.
DISCUSSION
In this article, we demonstrated that both M1- and M2-like KCs stimulated by HBV could directly suppress HBV replication in hepatocytes, with the former exhibiting a stronger suppressive effect on HBV (Figures 1B and 1C). This difference in HBV-suppressive effect was also observed with human MDMs and THP-1 monocytic cells that were induced to undergo M1- and M2-like polarizations (Figure 1D). The observation that M2-like macrophages had reduced HBV-suppressive activity provides an explanation as to why M2-like, and not M1-like, macrophages preferentially accumulate in the liver of chronic HBV patients (Bility et al., 2014).
Our RNA-seq analysis revealed that IL-1β was the most prominent cytokine that was induced by HBV in THP-1 macrophages (Figure 2C). Indeed, IL-1β plays a critical role in the suppression of HBV replication, as the depletion of IL-1β with its specific antibody or the inhibition of its receptor with anakinra abolished the suppressive effect of THP-1 macrophages on HBV replication in hepatocytes (Figure 3A). As IL-1β by itself could also suppress HBV replication both in vitro and in vivo (Figures 3B–3D), our results indicated that IL-1β was essential and sufficient for macrophages to suppress HBV replication. The lower HBV-suppressive effect of M2-like macrophages that we observed was due to its lower expression level of IL-1β (Figure 1A), as this suppressive effect could also be blocked by the anti-IL-1β antibody (Figure 3E). In contrast, TNF-α did not play a significant role in the suppression of HBV replication (Figure 3F). This was likely due to its low level of production by macrophages (Figure 3G), as a much higher level of TNF-α (i.e., 100 ng) was shown to reduce HBV DNA replication by only 30% (Kim et al., 2018). We further investigated the mechanism by which HBV induced the expression of IL-1β in macrophages. Interestingly, we found that HBsAg, HBeAg, or HBcAg could all induce the expression of IL-1β in THP-1 macrophages (Figures 7A–7D). HBsAg has been shown to bind to CD14, the co-factor of toll-like receptor 4 (TLR4) (van Montfoort et al., 2016), and both HBeAg (Lang et al., 2011) and HBcAg (Yi et al., 2020) have been shown to bind to TLR2. Thus, it is possible that HBV antigens activated multiple cellular factors to induce the expression of IL-1β and perhaps also activate inflammasomes, which are important for the activation of IL-1β (Garlanda et al., 2013). We did not analyze HBV DNA and RNA, which may also activate macrophages. However, a recent study suggested that it was unlikely that HBV DNA played any major role in the stimulation of macrophages (Cheng et al., 2017).
An interesting discovery revealed by our gene profiling studies was the activation of the OXPHOS by HBV in macrophages (Figures 2E and 2G). This finding is rather surprising, as previous studies have indicated that M1-like macrophages enhanced glycolysis and an impaired tricarboxylic acid (TCA) cycle and OXPHOS (Liu et al., 2021; Mills et al., 2016; Tannahill et al., 2013), which were also confirmed by our own studies (Figure 2F). The observation that HBV could induce OXPHOS indicates that the metabolic reprogramming in macrophages is complicated, and different external stimuli may lead to different metabolic responses, even in M1-like macrophages. Our further studies indicated that the inhibition of HBV-induced OXPHOS would enhance the production of IL-1β, suggesting that HBV induced OXPHOS in M1-like macrophages to minimize the production of IL-1β for its replication and survival.
We also studied how macrophages suppressed HBV replication in hepatocytes. As the suppressive effect of THP-1 macrophages on HBV was inhibited by anakinra, which is an IL-1RA analog, the effect of IL-1β on HBV was clearly mediated by its receptor (Figures 3A and 3C). We also demonstrated that this IL-1β signaling suppressed ENI and ENII enhancer activities by inhibiting the expression of PPARα and FOXO3 (Figure 4). As THP-1 macrophages did not affect PPARα and FOXO3 RNA levels, the IL-1β signaling likely reduced the protein levels of PPARα and FOXO3 by reducing their stability. In this regard, it is interesting to note that the IL-1β signaling pathway has been shown to activate kinases that phosphorylate and destabilize proteins (Weber et al., 2010). Note that FOXO3 also appeared to be an upstream regulator of PPARα, as its silencing decreased thePPARα protein level (Figure 6D) and its overexpression increased PPARα (Figure S5B).
In conclusion, our studies revealed an interesting interplay between macrophages and HBV. We demonstrated that HBsAg, HBcAg, and HBeAg, which are released from HBV-infected hepatocytes, could all activate macrophages to induce the expression of IL-1β and delineated the molecular pathway used by IL-1β to suppress HBV replication. We also discovered that HBV could metabolically reprogram M1-like macrophages to induce OXPHOS to attenuate this anti-HBV response. How HBV metabolically reprograms macrophages is unclear and may involve the activation or suppression of certain cellular pathways by specific viral factor(s). Further studies in this area will likely generate many more exciting findings.
Limitations of the study
In this study, we demonstrated that HBV could stimulate mouse KCs, human MDMs, and THP-1 macrophages to produce IL-1β, which suppressed HBV gene expression. We also showed that M1-like proinflammatory macrophages had a greater suppressive effect on HBV than did M2-like macrophages. Although our results revealed an interesting interplay between HBV and macrophages both in mice and ex vivo, they will ultimately need to be confirmed in patients with acute and chronic HBV infections. This will require the isolation of KCs from these patients for analysis. This human study is difficult to conduct due to ethical concerns. This is a limitation of our study.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, J.-H. James Ou (jamesou@usc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA sequencing data are available via NCBI Gene Expression Omnibus (GEO) database (GEO: GSE179618).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Tg05 HBV transgenic mouse line, which carries the 1.3mer overlength HBV genomic DNA and produces a serum HBV titer of nearly 109 genome copies per ml, was used for the production of TGD mice as previously described (Tian et al., 2016). Briefly, female hemi-zyous Tg05 mice were crossed to naïve male mice to generate the HBV-negative mouse pups. These HBV-negative mouse pups were termed the transgenic-derived mice or TGD mice in brief. Tg05 mice were also used for testing the effect of IL-1β on HBV replication in vivo. Both Tg05 mice and control mice used in our studies had the C57BL/6 genetic background. Male mice 8–10 weeks of age were used for the studies. Mice were housed on a 12-hour light/dark cycle at 23°C, and standard laboratory mouse diet and water were provided ad libitum. All animal work was conducted under the approval of the University of Southern California Institutional Animal Care and Use Committee in accordance with federal, state and local guidelines.
Isolation of hepatocytes and Kupffer cells from mice
For the isolation of mouse hepatocytes, the liver of 8–10 week-old mice was perfused with 30 mL of the albumin solution (i.e., PBS+0.028 mg/mL EGTA+2% bovine serum albumin (BSA)) via the hepatic portal vein at a flow rate of 7 mL/min at 37°C. After 4 minutes of perfusion, the liquid influx tube of the pump was transferred to a tube containing the pre-warmed collagenase solution (PBS+5.88 mg/mL CaCl2·2H2O+0.4mg/mL collagenase Type IV) for further perfusion until the liver was swollen and became shiny. The liver was then removed quickly from mice, placed in a petri dish with PBS and gently tapped with the help of a tweezer for the release of cells until all liver robes were broken. Cells were filtered with a 40-mm nylon cell strainer (Falcon,#352340), pelleted by centrifugation at 70×g for 2 minutes, and then resuspended with Percoll buffer and centrifuged again in Eppendorf 5804R at 2500 rpm for 3 minutes at room temperature. The top layer of the supernatant, which contained dead cells, was removed, and the rest of the supernatant contained Kupffer cells. The cell pellet contained the purified hepatocytes. For the purification of Kupffer cells, the supernatant was centrifuged at 583×g for 8 minutes and the cell pellet was resuspended and further ultracentrifuged in Beckman SW40Ti rotor at 21,400×g for 20 minutes in a step gradient consisting of OptiPrep Density Gradient Medium (Sigma, #D1556) with densities of 1.08 (2 ml), 1.054 (1.5 ml), 1.043 (1.5 ml) and 1.034 (1.5 ml). It was then overlaid with 5.5 ml culture medium. Kupffer cells at the interface of densities 1.043 and 1.054 were isolated and pelleted at 583×g for 8 minutes. The purified Kupffer cells were then resuspended in 5 ml culture medium for further studies.
Isolation of human monocytes
Human peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors using the Ficoll gradient (Histopaque-1077, Sigma) as previously described (Li et al., 2018). Briefly, the heparin-treated blood was mixed with an equal volume of phosphate-buffered saline (PBS) and then overlaid on 15 mL Ficoll followed by centrifugation at 400×g at 22°C for 45 minutes with the brake off. After the removal of the plasma on the top, the buffy coat, which contained the mononuclear cells and was located at the interface between the plasma (upper layer) and the Ficoll (bottom), were carefully transferred to a 50 mL conical tube and mixed with 30 mL PBS, followed by centrifugation at 400×g for 10 minutes. The cell pellet, which were PBMCs, was resuspended in 10 mL PBS. Classical monocytes were then purified from freshly isolated PBMCs by magnetic-activated cell sorting (MACS®) and negative selection using the Monocyte Isolation Kit II (Miltenyi Biotec) according to the manufacturer’s instructions.
METHOD DETAILS
Human monocytes activation and differentiation
The polarization of human monocytes was conducted as described previously (Du et al., 2019; Faure-Dupuy et al., 2019). Briefly, human monocytes were treated with 50 ng/ml of GM-CSF (R&D) or 50 ng/ml of M-CSF (Peprotech) for seven days to generate M(GM-CSF) and M(CSF-1), respectively, which were then activated with 10 ng/ml of LPS (Invitrogen) for 3 hours, washed with PBS and cultured in fresh media for 24 hours before the experiments.
Human Hepatocyte Culture and HBV Infection
Human hepatocytes (HHs) were isolated from the liver of chimeric uPA/SCID mice that had been grafted with human hepatocytes (PhoenixBio Co.) and plated on type I collagen-coated 6-well plates (2.1×105 cells/cm2) as previously described (Tateno and Kojima, 2020). One day after plating, Chimeric mice-derived HHs were incubated with 2 mL of 2% dimethyl sulfoxide (DMSO)-supplemented hepatocyte clonal growth medium and infected with HBV prepared from the serum of Tg05 HBV transgenic mice (MOI=50) in the presence or absence of 4% polyethylene glycol 8000 (PEG). At day 1 and day 2 after infection, HHs were rinsed with fresh DMSO–supplemented hepatocyte clonal growth medium and further incubated with 2 mL of the same medium. The culture medium was collected and replaced every 5 days.
Differentiation and activation of THP-1 monocytes
THP-1 monocytes maintained in the RPMI medium containing 10% fetal bovine serum (FBS) were differentiated into macrophages with the treatment of 100 nM phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich) for 48 hours, followed by incubation in the PMA-free medium for 24 hours. For the M1 polarization, THP-1 monocytes were treated with 100 nM PMA for 48 hours and then further treated with 20 ng/ml of recombinant human interferon-γ (IFN-γ) and 100 ng/ml of lipopolysaccharides (LPS) (Invitrogen) for another 48 hours to generate THP-1(LPS+IFN-γ) or with 20 ng/ml recombinant human interleukin 13 (IL-13) (Peprotech) and 20 ng/ml of recombinant human IL-4 (Peprotech) for another 48 hours to generate THP-1(IL-4+13).
Flow cytometry
The flow cytometry was carried out using the BD FACSCanto™ II Cell Analyzer (BD Bioscience). The analysis of THP-1 macrophages, human classical monocytes, and Kupffer cells was performed as described previously with some modifications (Tian et al., 2016). Briefly, cells were collected and incubated with primary antibodies IgG2a-PE, IgG1-APC, CD14-PE, APC anti-human CD11b antibody (Biolegend, #301310), APC anti-human CD16 antibody (Biolegend, #360705), F4/80-PE (eBioscience, #12-4801-82), or IgG2a-PE (eBioscience, #12-4321-80) at 4°C for 30 minutes. Cells were rinsed with PBS three times before the flow cytometry analysis. Data were analyzed by FlowJo 10 software (BD Bioscience)
Cell co-culturing
Huh7 cells were seeded at a density of ~0.5×106 cells per well of a 6-well plate (Corning, #353046) and incubated for 18 hours to reach a confluency of 80–90%. Cells were rinsed once with PBS and incubated in Dulbecco’s modified essential medium (DMEM) without FBS and antibiotics. For DNA transfection, cells were transfected with 2 μg 1.3mer HBV genomic DNA or pUC19 per well using Lipofectamine 3000 (Invitrogen, L3000015) following the manufacturer’s instructions. Cells were rinsed once with DMEM four hours after transfection and further incubated in DMEM containing FBS and antibiotics. THP-1 macrophages were seeded at a density of 2×106 cells in the Transwell insert (Corning, #353090). THP-1 macrophages and Huh7 cells were co-cultured in a medium containing 1:1 ratio of DMEM and RPMI for one or two days. Similarly, HepAD38 cells were seeded at a density of ~0.5×106 cells/well in a 6-well plate, incubated in 2 mL DMEM/well for 18 hours, rinsed once with PBS and then incubated in the fresh medium. 2×106 THP-1 macrophages were seeded in each Transwell insert and co-cultured with HepAD38 cells in a 1:1 mixture of RPMI and DMEM for 1–4 days, depending on the experiments. The co-culturing of Kupffer cells and mouse hepatocytes as well as the co-culturing of human monocyte-derived macrophages and HHs were conducted the same way.
Western-blot analysis
For western blotting, cells were chilled on ice, rinsed with ice-cold PBS twice and lysed with RIPA buffer (Sigma-Aldrich, R0278) plus the protease inhibitor cocktail. Protein concentrations were determined using the BCA Protein Assay Kit (BioRad). About 25 μg protein per sample was loaded on a 10% SDS-PAGE gel and transferred to the PVDF membrane, which was washed with PBS Tween 20 buffer (PBST) (Thermo Fisher Scientific, #28360) for 5 minutes three times, blocked with 5% milk in PBST, and incubated with the anti-HBsAg (Novus, #NB100-62652) or the antibody directed against denatured core protein in the cold room overnight. The anti-core antibody was prepared in our lab using the recombinant core protein (Ou et al., 1989). After washing with PBST 3 times for 10 minutes each, the membranes were incubated with the horseradish peroxidase-conjugated secondary antibody (Abcam) at room temperature for one hour. The membranes were washed with PBST three more times with 10 minutes per wash and subjected to chemiluminescent western blot analysis. All experiments were repeated at least three times.
Northern-blot analysis
Total RNA was isolated from Huh7 or HepAD38 cells using Trizol (ThermoFisher Scientific, #15596026).10 μg RNA was mixed with the RNA loading dye (NEB, #B0363S), denatured at 65°C for 5 minutes and then loaded on a 1% agarose gel for gel electrophoresis using 1x MOPS buffer (Fisher Scientific, #BP2900500) as the running buffer. After the staining of 28S and 18S rRNAs with SYBR® safe DNA gel stain, the RNA in the gel was transferred to the nitrocellulose membrane. HBV RNAs were detected using the 32P-labeled (Perki-nelmer, #BLU013H500UC) HBV DNA as the probe and analyzed by autoradiography.
HBV DNA isolation and Southern-blot analysis
The encapsidated HBV DNA was isolated from Huh7 or HepAD38 as previously described (Tian et al., 2011, 2016). Briefly, cells in the 6-well plate were lysed with 700 μL lysis buffer (100 mM Tris-HCl, pH7.5, 1mM EDTA, 50 mM NaCl and 0.2% Nonidet P-40) and, after the addition of 2 μL 2 M MgCl2, and 4 μL 2 M CaCl2, treated with 100 μg DNase I and micrococcal nuclease (5 units) at 37°C for 1 hour. After a brief centrifugation at 3,000 rpm in a microcentrifuge for 5 minutes, 600 μL of the supernatant was transferred to another microfuge tube, and the nuclease digestion reaction was stopped by the addition of 40 μL 0.5 M EDTA and 300 μg proteinase K and further incubated at 65°C for 16 hours. The sample was then extracted with phenol and chloroform and the aqueous phase was transferred to a new microfuge tube. After the addition of 0.1 volume of 3M sodium acetate, 10 μg tRNA carrier and 2 volumes of 100% ethanol, the sample was mixed well and incubated at −80°C for 15 minutes followed by centrifugation at 13,000 rpm in a microcentrifuge for 2 minutes. The pellet was dissolved in 50 μL TE buffer (10 mM Tris-HCl, pH8.0, 1 mM EDTA). 10 μl DNA was used for gel electrophoresis in a 1% agarose gel and transferred to a nitrocellulose membrane. After the hybridization to the 32P-labeled HBV DNA probe, the membrane was exposed to the X-ray film (Bioland, #A03-01) for autoradiography. All of the experiments were repeated at least three times.
Enzyme-linked immunosorbent assay (ELISA)
HBsAg and HBeAg were analyzed using the ELISA kit (International Immuno-Diagnostics) according to the manufacturer’s protocol. The culture medium was diluted 4-fold, and 100 μL of diluted samples were used for ELISA. All experiments were repeated at least three times.
Real-time PCR analysis of HBV DNA in the incubation media
HBV DNA was extracted from the culture media using the QIAamp® MinElute® Virus Spin (Qiagen) following the manufacturer’s protocol. For HBV DNA qPCR analysis, the forward primer 5’-CTGCTGCTATGCCTCATCTT-3’; the reverse primer 5’-GTCCC GTACTGGTTGTTGTT-3’; and the TaqMan probe, 5’-AGGTATGTTGCCCGTTTGTCCTCT-3’ (dyes: FAM at 5’ end, TAMRA at 3’ end) were used. The thermal cycling was conducted using the step one plus system (Applied Biosystems) with the following parameters: 2 minutes at 50°C, 10 minutes at 95°C, and 40 cycles of 15 seconds at 95°C and 60 seconds at 60°C. All of the experiments were repeated at least three times.
Isolation and quantification of HBV cccDNA
HBV DNA was extracted from HHs using the DNeasy Blood & Tissue Kit (Qiagen) following the manufacturer’s protocol. The DNA was then digested with T5 exonuclease (NEB, M0363) for 30 minutes at 37°C for the removal of HBV relaxed circular DNA (rcDNA). The exonuclease was heat inactivated at 99°C for 5 minutes (Faure-Dupuy et al., 2019). The quantification of HBV cccDNA was performed by qPCR as previously described (Takkenberg et al., 2009), using the following primers: forward primer, 5’-CTCCCCGTCTGTGCCTTCT-3’; reverse primer, 5’-GCCCCAAAGCCACCCAAG-3’; TaqMan probe, 5’- CGTCGCATGGARACCACCGTGAACG CC-3’ (dyes: FAM at 5’ end and TAMRA at 3’ end). The thermal cycling was conducted using the step one plus system (Applied Bio-systems) with the following parameters: 2 minutes at 50°C, 10 minutes at 95°C, and 40 cycles of 15 seconds at 95°C and 60 seconds at 60°C. All of the experiments were repeated at least three times.
Dual-luciferase reporter assay
For the construction of pGL3-Basic or pGL3-Promoter firefly luciferase reporter plasmids, wild-type and mutant HBV DNA fragments were isolated by PCR and cloned into the KpnI and XhoI sites of the pGL3-Basic vector (Addgene) or the pGL3-Promoter vector (Promega). All of the reporter constructs were transformed into E. coli for amplification and purified using PureLink HiPure Plasmid Filter Max-iprep Kit (Invitrogen, K210017). HepAD38, HepG2, Huh7 and human hepatocytes were seeded in triplicate in 24-well plates to reach 70%−80% confluency on the next day. 100 ng firefly luciferase reporter constructs and 20 ng pRL-TK, which contained the renilla luciferase reporter under the expression control of the herpesvirus thymidine kinase (TK) promoter and was used as the internal control to monitor the transfection efficiency, were co-transfected with or without the 1.3mer HBV genomic DNA, depending on the experiments, using Lipofectamine 3000 (Invitrogen). After 6 hours of transfection, cells were rinsed once with PBS and incubated with the fresh medium, with or without the co-culturing with macrophages. After 24 hours or 48 hours, cells were lysed and the luciferase activities were determined using the dual-luciferase assay kit (Promega, #E1910). All of the experiments were repeated at least three times.
RNA-seq and related data analyses
Total RNA was isolated from three groups of THP-1 macrophages, designated as THP-1(M0) (no co-culturing), THP-1(HBV-) (co-cultured with HepAD38 in the presence of 0.5 μg/mL tetracycline), and THP-1(HBV+) (co-cultured with HepAD38 without tetracycline) using All-Prep DNA/RNA Mini Kit (QIAGEN, #80204), and the cDNA libraries were prepared using Next Ultra™ II RNA Library Prep Kit for Illumina (New England Biolab). RNA-seq was conducted using NovaSeq 6000. Differentially expressed genes (DEGs) were determined by Partek Flow gene differential filter analysis based on false discovery rate (FDR)<0.05 and Log2 fold change (FC)>1.2. Cellular pathways were analyzed by Ingenuity Pathway Analysis (IPA) software. The RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) repository with the accession number GSE179618. The RNA-seq for each macrophage group was conducted in triplicate.
Single-cell RNA-seq library construction and sequencing
The liver was isolated from mice 4 days after the hydrodynamic injection of 20 μg pUC19 or pHBV1.3. A single-cell suspension of the mouse liver was prepared by the two-step enzyme digestion procedure as described above. Most of parenchymal cells were removed. Cell samples were prepared by following the 10× Genomics Single Cell 3′ v3 Reagent Kit user guide. Briefly, cell samples were washed twice with PBS plus 0.04% BSA, resuspended in the same wash solution and then filtered with a 40-μm cell strainer. The cell viability was assessed using the Trypan Blue exclusion assay (Thermo Fisher) and counted using a hemocytometer. Following cell counting, the appropriate volume for each cell sample was calculated for a target capture of 5000 cells with a cell concentration of 1000 cells/μl. Cells were loaded onto the 10x Genomics single-cell-G chip, and the Chromium Single Cell 3′Reagent Kit v3 was used for the preparation of the cDNA according to the manufacturer’s protocol. Following the library preparation and quantitation, the libraries were sequenced on the Illumina NextSeq 6000 platform.
Single-cell RNA-seq data processing
The raw sequencing data were trimmed to remove tags and aligned to the mouse transcriptome (GRCm38-mm10) using Partek Flow. Cells with low-quality cell barcodes or with high mitochondrial genome were excluded. Qualified cells with detected gene number between 200 and 5,000 were selected for analysis. The unique molecular identifier (UMI) number below 50,000 and the percentage of mitochondrial genes below 10% were kept. After quality control, cells were clustered by graph-based clustering and visualized by t-SNE.
RNA extraction, cDNA synthesis and qPCR
Total RNA from hepatocytes or macrophages were isolated using miRNeasy Mini Kit (QIAGEN, #217004) or TRIzol reagent (Thermo-Fisher Scientific, #15596026) following the manufacturers’ protocols. 200 ng total RNA was used for cDNA synthesis in a 10 μl reaction volume using QuantiTect Reverse Transcription Kit (QIAGEN, #205314) (Su et al., 2018). The quantitative PCR (qPCR) was performed using Power SYBR™ Green PCR Master Mix (ThermoFisher, #4367659) in Step One Plus Real-Time PCR system. The housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. Each qPCR reaction was conducted in triplicate. The primers used in all of the qPCR assays are listed in Key Resources Table. All of the experiments were repeated at least three times.
Seahorse assays
Three groups of THP-1 macrophages, designated as THP-1(M0) (co-culturing only with medium), THP-1(HBV-) (co-cultured with HepAD38 in the presence of 0.5 μg/mL tetracycline), and THP-1(HBV+) (co-cultured with HepAD38 without tetracycline), were subjected to Seahorse assays using the XFe96 Extracellular Flux Analyzer (Agilent). Approximately 105 macrophage cells were seeded on an XF96 plate, and the oxygen consumption rate (OCR) was measured using the mitochondrial stress test kit. The OCR was measured in Seahorse XF Base Medium (i.e., DMEM containing 10 mM glucose, 2 mM L-glutamine, and 1 mM sodium pyruvate) under basal conditions and in response to 2 μM oligomycin, 0.75 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP) and 0.5 μM rote-none/antimycin A (Rot/AA). The extracellular acidification rate (ECAR) was measured using the glycolysis stress test kit. The ECAR was measured in Seahorse XF Base Medium in response to 5 μM Rot/AA and 500 mM 2-deoxyglucose (2-DG). Oligomycin suppresses electron flow through the electron transport chain (ETC), resulting in the reduction of mitochondrial respiration (i.e., OCR). FCCC disrupts mitochondrial membrane potential to allow electron flow through the ETC, optimizing oxygen consumption. Rot and AA are ETC complex I and complex III inhibitors, respectively. Their combined use shut down mitochondrial respiration and enabled the calculation of non-mitochondrial respiration. 2-DG is a glucose analog that inhibits glucose hexokinase and the glycolytic pathway.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using ChIP Kit (Abcam, ab500) following the manufacturer’s protocol with some modifications (Liu et al., 2017). Briefly, HepAD38 cells (about 3×106) with or without co-culturing with THP-1 macrophages were treated with the fresh fixing buffer (1.1% formaldehyde in PBS) at room temperature for 10 minutes for the crosslinking of DNA and proteins. The reaction was stopped with the glycine buffer provided in the kit, and cells were pelleted by centrifugation at 500×g for 5 minutes at 4°C. Cells were then lysed and the chromatin was sheared by sonication to about 200–300 base pairs in length. The DNA fragments were then immunoprecipitated with antibodies directed against HNF-4α (Abcam, #ab181604), PPARα, RXRα (Invitrogen, #433900), COUP-TF1 (Abcam, #ab96846 or Abcam, # ab181137), FOXO3 (Abcam, #ab12162 or Abcam, #ab70315), or the isotype IgG control (Invitrogen, #02_6102). The immunoprecipitated DNA fragments were amplified by PCR using specific primers targeting the HBV DNA sequence from nt. 1136–1168 of the ENI enhancer or nt. 1403–1468 of the ENII enhancer. PCR products (20 μL) were analyzed on a 1.5% agarose gel. 1% of starting chromatin DNA was used for PCR to serve as the input control. The primers used for amplifying the HBV ENI enhancer were: forward primer, 5’-CCTTAATGCCTTTGTATGCATGT-3’, and reverse primer 5’-CCACAAAGGTTC CACGCA-3’. The primers used for amplifying the HBV ENII enhancer were: forward primer, 5’-GCAGGTCTGGAGCAAACATTA-3’, and reverse primer 5’-AGTCCG CGTAAAGAGAGGT-3’. All of the experiments were repeated at least three times. ChIP assays using hepatocytes (3×106) isolated from Tg05 HBV transgenic mice with or without the injection of 200 ng IL-1β (Peprotech, #211-11B) were conducted the same way. Mouse hepatocytes were isolated by liver perfusion 24 hours after the IL-1β injection as mentioned above.
Electrophoretic mobility shift assay (EMSA)
EMSA was conducted as previously described with some modifications (Liu et al., 2017). Briefly, nuclear extracts from HepG2 cells were isolated by the Nuclear Extraction Kit (Abcam, #ab113474). All of the double-stranded DNA probes were synthesized by Integrated DNA Technologies (IDT), and biotin-labeled using the Biotin 3’-end DNA Labeling Kit (ThermoFisher Scientific, #89818). The nucleotide sequence of the PPARα-WT probe was 5’-ACAGTACATGAACCTTTACCCCGTTGCTCGGCAACG-3,’ and the sequence of the PPARα-MT probe was 5’-ACAGTACATGAAACTCTATCCAGTCGCTCGGCAACG-3’. The FOXO3-WT oligonucleotide probe was 5’-GGGACGTCCTTTGTTTACGTCCCGTCGGC-3’, and FOXO3-MT oligonucleotide was 5’GGGACGTCCTCTGCCTGCGTCC CGTCGGC-3’. The EMSA was performed as previously described. Briefly, for the supershift assay, anti-PPARα or anti-FOXO3 (Abcam, #ab12162) was added to the nuclear extracts and incubated on ice for 10 minutes. The oligonucleotide probe was then added and the sample was further incubated at room temperature for 10 minutes. The DNA–protein complexes were separated in a 6% nondenaturing polyacrylamide gels using 0.5X TBE as the running buffer and electrophoresed at 4°C for 60 minutes. The Light Shift Chemiluminescent EMSA Kit (ThermoFisher, #20148) was used to analyze the bandshifts following the manufacturer’s protocol. All of the experiments were repeated at least three times.
Expression of shRNA and proteins using the lentiviral vector
The procedures for the preparation of lentiviral vectors that expressed different shRNAs or proteins were performed as described previously with some modifications (Huang et al., 2018; Su et al., 2018). Lentivirus particles were prepared using the plasmids pLKO.1-shRNA scrambled (a gift of Dr. Lei Gao), pLKO.1-shPPARα (Sigma-Aldrich, #TRCN0000001665), pLKO.1-shFOXO3 (Sigma-Aldrich, #TRCN0000010335), pCDH-puro-cMyc (Addgene), pCDH-PPARα, pCDH-FOXO3, and pCDH-PPARα+FOXO3. pCDH-PPARα and pCDH-FOXO3 were generated by inserting PPARα and FOXO3 cDNA fragments, which were isolated from the PPARA cDNA clone (Sino biological, #HG12080-CH) and the FOXO3a cDNA clone (Addgene, #1787), respectively, into the XbaI and BamHI restriction enzyme sites of the pCDH-puro-cMyc vector. Lentiviral particles were produced by co-transfecting the plasmids mentioned above with the packaging vectors pMD2.G (0.5 μg), pMDLg/pRRE (0.3 μg), and pRSV-Rev (0.7 μg) (gifts of Dr. Yangchan Li) into HEK293T cells in a 60-mm petri dish using X-tremeGENE HP DNA Transfection Reagent (Sigma-Aldrich, #6366236001). Lentiviral particles were harvested and concentrated with PEG-it virus precipitation solution (System Biosciences, # LV810A-1) for 48 or 72 hours, and used to infect target cells in the presence of 4 μg/ml polybrene (Sigma-Aldrich, #H9268). Cells were transduced with lentiviral particles twice and then selected with1 μg/mL puromycin (Sigma-Aldrich, #P8833) for at least two passages. For cells that were transduced by lentiviral particles, they were further selected by flow cytometry based on the expression levels of green fluorescence protein (GFP). Only the top 10% of cells that expressed high levels of GFP were selected.
Construction of HBV 1.3mer mutants
The 1.3mer HBV genomic DNA fragment, which starts from the EcoRV site at nt. 1043 and ends at the BglII site at nt. 1987, contains all of the genomic information of the HBV genome plus a terminal redundancy of nearly 1 Kb. This DNA fragment was isolated and inserted into the HindIII/KpnI site of the pUC19 vector by In-Fusion® Snap Assembly Starter Bundle (TaKaRa, #638945) to generate pHBV1.3mer. For the construction of HBV-s-Mt, a T-to-A mutation at nt. 223, which converted the TTG codon to a TAG translation stop codon, was created to abolish the expression of HBsAg proteins. The HBV-e-Mt had been described before (Tian et al., 2016). This mutant contained a G-to-A mutation at nt. 1896, which converted a TGG codon to the TAG termination codon in the precore sequence and abolished the expression of HBeAg. The HBV-s-e-Mt double mutant contained the nonsense mutations at both nt. 223 and nt. 1896. For the construction of HBV-core-Mt, an AT-to-GC mutation was created at nt. 1904–1905 to remove the translation initiation codon of the core protein. The HBV-X-Mt mutant had been described before (Tian et al., 2016). This mutant contained an A to C missense mutation at nt. 1377, which removed the translation initiation codon of HBx, and a C-to-T mutation at nt. 1398 that created a premature termination codon in the HBx coding sequence. All of the mutations were created using the Q5® Site-Directed Mutagenesis Kit (NEB, # E0554S) and confirmed by nucleotide sequencing.
Expression and purification of HBeAg
The plasmid pET-26b-HBeAg-His6 contained the HBeAg coding sequence from nt. 1873 to nt. 2350 with a 6xHis tag coding sequence at the 3’-end. It did not contain the signal peptide sequence of the precore protein or its C-terminal arginine-rich domain. The HBeAg-His6 sequence was inserted at the NdeI and XhoI sites of the pET-26b+ vector. The expression and purification of the HBeAg recombinant protein were performed as described previously with some modifications (Li et al., 2018; Zhao et al., 2020). The pET-26b-HBeAg-His6 plasmid was transformed into BL21(DE3) cells. Single bacterial colonies were isolated and inoculated into 5 mL Luria broth (LB) and grown in the presence of kanamycin at 37°C overnight. 2 mL of the overnight culture were inoculated into 100 mL LB in a flask. The bacterial culture was grown at 37°C until the OD600 reached 0.8–1.0. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was then added to a final concentration of 0.1 mM and the bacterial culture was then further incubated at 16°C overnight for the induction of HBeAg expression. Cells were then pelleted, resuspended in 10 mL lysis buffer (25 mM Tris-HCl, pH7.5, 300 mM NaCl, 15 mM imidazole) for every gram of bacterial cells, and sonicated. Cell lysates were then centrifuged at 12,000×g at 4°C for 30 minutes. In the meantime, 2 mL Ni-NTA agarose were first washed with 10-gel volumes of Mini-Q H2O and then with 10-gel volumes of lysis buffer for equilibration of the gel. The Ni-NTA agarose beads were then mixed with 50 ml bacterial cell lysates and rocked at 4°C for 2 hours. The supernatant was carefully removed, and the beads were transferred to a column and then successively washed with 10 mL washing buffer 1 (25 mM Tris-HCl, pH7.5; 1M NaCl; 15 mM imidazole), 10 mL washing buffer 2 (25 mM Tris-HCl, pH7.5; 1 M NaCl; 25 mM imidazole), and 10mL washing buffer 3 (25 mM Tris-HCl, pH7.5; 1M NaCl; 40 mM imidazole). The recombinant HBeAg was eluted with 3 mL elution buffer that contained 25 mM Tris-HCl, pH7.5, and 300 mM NaCl and increasing concentrations of imidazole at 100 mM (Eluate 1), 200 mM (Eluate 2), 300 mM (Eluate 3), and 400 mM (Eluate 4). The eluates were collected separately and dialyzed against PBS at 4°C for 2 hours. The dialysis was repeated one more time. The purity of recombinant HBeAg was analyzed on a 12% SDS-PAGE gel and stained by Coomassie brilliant blue. Eluates 3 and 4 produced HBeAg with a high degree of purity and were used for subsequent studies.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed using GraphPad Prism 8 software and presented as mean ± SEM. Two-tailed Student’s t test was used to compare means between groups and p<0.05 was considered statistically significant. For western-blot, northern-blot and Southern-blot studies, the experiments were repeated at least three times and representative results were shown. Densitometry analysis of the signals of these studies were performed using the ImageJ analyzer and normalized against the loading controls. The statistical information of each experiment, including the p values and sample numbers (n) were described above in Western-blot, Southern-blot, ELISA, qPCR, Dual-luciferase Reporter Assay and EMSA sections as well as in figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| F4/80 Monoclonal Antibody (BM8), PE | Invitrogen | Cat# 12-4801-82; RRID:AB_465923 |
| Rat IgG2a kappa Isotype Control (eBR2a), PE | Invitrogen | Cat# 12-4321-80; RRID:AB_1834380 |
| Donkey Anti-Goat IgG H&L (HRP) | Abcam | Cat#ab97110; RRID:AB_10679463 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | Cat# A-11058; RRID: AB_2534105 |
| Mouse IgG2a kappa Isotype Control (eBM2a) | Invitrogen | Cat# 14-4724-82; RRID: AB_470114 |
| Rabbit IgG Isotype Control | Invitrogen | Cat# 02_6102; RRID: AB_2532938 |
| Anti-HNF-4-alpha antibody | Abcam | Cat# ab181604;RRID:AB_2890918 |
| Anti-COUP TF1 | Abcam | Cat# ab96846;RRID:AB_10679404 |
| RXRA Monoclonal Antibody (K8508) | Invitrogen | Cat# 433900; RRID: AB_2532208 |
| Anti-PPAR alpha antibody | Abcam | Cat# ab227074;RRID:AB_227074 |
| APC anti-human CD16 Antibody | Biolegend | Cat# 360705; RRID: AB_2562750 |
| Rabbit Anti-Human IgG H&L (HRP) | Abcam | Cat# ab6759;RRID:AB_955434 |
| Anti-FOXO3A antibody | Abcam | Cat# ab12162;RRID:AB_298893 |
| FITC Mouse Anti-Human CD68 | BD | Cat# 562117;RRID:AB_10896283 |
| FoxO3a (75D8) Rabbit mAb | Cell Signaling Technology | Cat# 2497S; RRID:AB_836876 |
| Human IL-1 beta /IL-1F2 Antibody | R&D systems | Cat# MAB201-500;RRID:AB_358006 |
| Mouse IgG1 Isotype Control | R&D systems | Cat# MAB002; RRID:AB_357344 |
| Anti-FOXO3A antibody | Abcam | Cat# ab70315;RRID:AB_1268913 |
| Anti-COUP TF1 antibody | Abcam | Cat# ab181137;RRID:AB_2890250 |
| Caspase-1 Antibody | Cell Signaling Technology | Cat# 2225T; RRID:AB_2243894 |
| PE anti-human CD14 Antibody | Biolegend | Cat# 301805;RRID:AB_314187 |
| Mouse TNF-alpha Antibody | R&D systems | Cat# MAB4101-SP;RRID:AB_2240643 |
| HBcAg Antibody | This paper | N/A |
| HBsAg Antibody | Novus | Cat# NB100-62652;RRID:AB_963836 |
| Biological samples | ||
| Bovine Serum Albumin solution | Sigma | Cat# A7034 |
| Rabbit Serum | Sigma | Cat# R4505 |
| Recombinant Human Serum Albumin Protein | Sino biological | Cat# 10968-HNAY |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase from Clostridium histolyticum | Sigma | Cat# C5138 |
| Lipofectamine™ 3000 Transfection Reagent | Invitrogen | Cat# L3000015 |
| Pronase | Sigma | Cat# 10165921001 |
| PageRuler™ Prestained Protein Ladder, 10 to 180 kDa | Thermo fisher | Cat# 26616 |
| OptiPrep™ Density Gradient Medium | Sigma | Cat# D1556 |
| Deoxyribonuclease I from bovine pancreas | Sigma | Cat# DN25 |
| Dual luciferase reporter assay system | Promega | Cat# E1910 |
| Taq 2X Master Mix | NEB | Cat# M0270L |
| TaqMan™ Fast Advanced Master Mix | Thermo fisher | Cat# 4444556 |
| VECTASHIELD® Antifade Mounting Medium with DAPI | Vector | Cat# H-1200-10 |
| Quick CIP | NEB | Cat# M0525S |
| MACS BSA Stock Solution | Miltenyi Biotec | Cat# 130-091-376 |
| AutoMACS Rinsing Solution | Miltenyi Biotec | Cat# 130-091-222 |
| Tetracycline | Sigma | Cat# 87128-25G |
| Histopaque®-1077 | Sigma | Cat# 10771 |
| T5 Exonuclease | NEB | Cat# M0663S |
| Power SYBR™ Green PCR Master Mix | Thermo Fisher | Cat# 4367659 |
| TRIzol™ Reagent | Thermo fisher scientific | Cat# 15596026 |
| Hybridization Solution | Sigma | Cat# H7140 |
| RPMI-1640 Medium | ATCC | Cat# ATCC® 30-2001™ |
| PEG-it Virus Precipitation Solution | System Biosciences | Cat# LV825A-1 |
| X-tremeGENE™ HP DNA Transfection Reagent | Sigma | Cat# 6366236001 |
| Recombinant Human GM-CSF Protein | R&D systems | Cat# 215-GM-010 |
| Recombinant Human M-CSF | Peprotech | Cat# 300-25 |
| Lipopolysaccharide (LPS) Solution (500X) | Fisher scientific | Cat# 50-112-2025 |
| Recombinant Human IL-4 | Peprotech | Cat# 200-04 |
| Recombinant Human IFN-γ | Peprotech | Cat# 300-02 |
| Recombinant Human IL-13 | Peprotech | Cat# 200-13 |
| Recombinant Human IL-1β | Peprotech | Cat# 200-01B |
| Anakinra | MCE medchemexpress | Cat# HY-108841 |
| Human IL-1 beta /IL-1F2 Antibody | R&D systems | Cat# MAB201-500 |
| Recombinant Hepatitis B Surface Antigen | Abcam | Cat# ab91276 |
| Recombinant Hepatitis B Virus Core Antigen protein | Abcam | Cat# ab49013 |
| Recombinant Murine IL-1β | Peprotech | Cat# 211-11B |
| Mouse IL-1 beta /IL-1F2 Antibody | R&D systems | Cat# AF-401-SP |
| Caspase 1 inhibitor (Ac-YVAD-cmk) | Sigma | Cat# SML0429-1MG |
| Critical commercial assays | ||
| Q5® Site-Directed Mutagenesis Kit | New England Biolabs | Cat# E0554S |
| Nuclear Extraction Kit | Abcam | Cat# ab113474 |
| Biotin 3’ End DNA Labeling Kit | Thermo fisher Scientific | Cat# 89818 |
| QuantiTect Rev. Transcription Kit | QIAGEN | Cat# 205311 |
| LightShift™ Chemiluminescent EMSA Kit | Thermo Scientific | Cat# 20148 |
| Seahorse XF Glycolytic Rate Assay Kit | Agilent | Cat# 103344-100 |
| Seahorse XF Cell Mito Stress Test Starter Pack | Agilent | Cat# 103708-100 |
| Seahorse FluxPaks | Agilent | Cat# 102601-100 |
| Seahorse XF96 V3 PS Cell Culture Microplates | Agilent | Cat# 101085-004 |
| Seahorse XF Media & Calibrant | Agilent | Cat# 103575-100 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat# 103015-100 |
| TNF alpha Mouse ELISA Kit | Invitrogen | Cat# BMS607-3 |
| IL-1 beta Mouse ELISA Kit | Invitrogen | Cat# BMS6002 |
| Chromium Next GEM Chip G Single Cell Kit (16 rxns) | 10× genomics | Cat# 1000127 |
| Chromium Next GEM Single Cell 3’ Kit v3.1 (4 rxns) | 10× genomics | Cat# 1000269 |
| Dual index kit TT set A (For Gene Expression Libraries) | 10× genomics | Cat# 1000215 |
| Deposited data | ||
| RNA-seq (Raw and analyzed data) | This study | GEO: GSE179618 |
| Experimental models: Cell lines | ||
| Huh7 | This paper | N/A |
| HepG2 | ATCC | Cat# HB-8065 |
| HepAD38 | This paper | N/A |
| THP-1 | ATCC | Cat# TIB-202 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson laboratory | Stock No: 000664 |
| TG05 HBV transgenic mice | This paper | N/A |
| Oligonucleotides | ||
| human GAPDH (Forward Primer) GATTCCACCCATGGCAAATTC | This paper | N/A |
| human GAPDH (Reverse Primer) CTGGAAGATGGTGATGGGATT | This paper | N/A |
| human IL-1β (Forward Primer) ATGACCTGAGCACCTTCTTTC | This paper | N/A |
| human IL-1β (Reverse Primer) TGCACATAAGCCTCGTTATCC | This paper | N/A |
| human TNF-a (Forward Primer) GCGTGGAGCTGAGAGATAAC | This paper | N/A |
| human TNF-a (Reverse Primer) TGAAGAGGACCTGGGAGTAG | This paper | N/A |
| human CD163 (Forward Primer) GGGATGTCCAACTGCTATCAA | This paper | N/A |
| human CD163 (Reverse Primer) GACTCATTCCCACGACAAGAA | This paper | N/A |
| human IL-10 (Forward Primer) GCTGGAGGACTTTAAGGGTTAC | This paper | N/A |
| human IL-10 (Reverse Primer) GATGTCTGGGTCTTGGTTCTC | This paper | N/A |
| mouse GAPDH (Forward Primer) AACAGCAACTCCCACTCTTC | This paper | N/A |
| mouse GAPDH (Reverse Primer) CCTGTTGCTGTAGCCGTATT | This paper | N/A |
| mouse IL-1β (Forward Primer) GAGGACATGAGCACCTTCTTT | This paper | N/A |
| mouse IL-1β (Reverse Primer) GCCTGTAGTGCAGTTGTCTAA | This paper | N/A |
| mouse TNF-a (Forward Primer) CCTCTTCTCATTCCTGCTTGT | This paper | N/A |
| mouse TNF-a (Reverse Primer) TGGGAACTTCTCATCCCTTTG | This paper | N/A |
| mouse CD163 (Forward Primer) CAGACTGGTTGGAGGAGAAATC | This paper | N/A |
| mouse CD163 (Reverse Primer) CAGCTTCCAGAGACAAGTCAA | This paper | N/A |
| mouse IL-10 (Forward Primer) CTATGCTGCCTGCTCTTACTG | This paper | N/A |
| mouse IL-10 (Reverse Primer) GGGAAGTGGGTGCAGTTATT | This paper | N/A |
| Recombinant DNA | ||
| pGL3-promoter | Promega | Cat#E1761 |
| pRL-TK | Promega | Cat# E2241 |
| HA-FOXO3a WT | Addgene | Cat# 1787 |
| Human PPARA cDNA ORF Clone | Sinobiological | Cat# HG12080-CH |
| PPARA MISSION shRNA Bacterial Glycerol Stock | Sigma | Cat# TRCN0000001665 |
| FOXO3 MISSION shRNA Bacterial Glycerol Stock | Sigma | Cat# TRCN0000010335 |
| Scramble shRNA | Addgene | Cat# 1864 |
| pCDH-CMV-MCS-EF1α-GreenPuro | SBI | Cat# CD513-B1 |
| pGL3-Basic | Promega | Cat# E1751 |
| pHBV1.3mer | This paper | N/A |
| Software and algorithms | ||
| FlowJov.10.5.3 | TreeStar | https://www.flowjo.com; RRID: SCR_008520 |
| GraphPad Prism 8.4.3 | GraphPad | https://www.graphpad.com; RRID: SCR_002798 |
| Fiji | ImageJ | https://imagej.net/Welcome |
| JASPAR | N/A | http://jaspar.genereg.net/tools/ |
| Partek Flow | Partek | https://www.partek.com/ |
| Ingenuity Pathway Analysis (IPA) | QIAGEN | https://digitalinsights.qiagen.com/product-login/ |
| Other | ||
| Cell culture insert, 6-well Plate with 0.4 μm Transparent PET Membrane | Corning | Cat# 353090 |
| Sterile Cell Strainers,70 μm | Corning | Cat# 07-201-431 |
| Sterile Cell Strainers,40 μm | Corning | Cat# 07-201-430 |
| Sterilization Pouches, Tubing and Covers | Cardinal health | Cat# 92168 |
| Amersham™ ECL™ Prime Western Blotting Detection | Fisher scientific | Cat# 45-010-090 |
| Microamp Fast optical 96 well reaction plate with Barcode (0.1 ml) | Applied biosystems | Cat# 4346906 |
| 10 × magnetic separator | 10× genomics | Cat# 120250 |
Highlights.
M1- and M2-like macrophages have different inhibitory effects on HBV replication
HBV metabolically reprograms M1 macrophages to enhance oxidative phosphorylation
Macrophages are stimulated by multiple HBV factors to express IL-1β
IL-1β downregulates the expression of PPARα and FOXO3 to inhibit HBV replication
ACKNOWLEDGMENTS
We wish to thank members of J.-h.J.O.’s lab for their help throughout the work described in this article, Dr. Keigo Machida for advice on the Seahorse assays, and members of Dr. Omid Akbari’s lab for their help with the flow cytometry. We also wish to thank Dr. Yibu Chen and USC Norris Library Bioinformatics Services for help with the data analysis. The bioinformatics software and computing resources used in the study are funded by the USC Office of Research and the USC Libraries. This work was supported by NIH grants AI129540, AI145813, and AI148304.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110284.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Afonina IS, Muller C, Martin SJ, and Beyaert R (2015). Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity 42, 991–1004. [DOI] [PubMed] [Google Scholar]
- Bility MT, Cheng L, Zhang Z, Luan Y, Li F, Chi L, Zhang L, Tu Z, Gao Y, Fu Y, et al. (2014). Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. Plos Pathog. 10, e1004032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Ur-dahl KB, Cosma CL, and Ramakrishnan L (2014). Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 505, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Xia Y, Serti E, Block PD, Chung M, Chayama K, Rehermann B, and Liang TJ (2017). Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 66, 1779–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Lin L, Li Q, Liu K, Huang Y, Wang X, Cao K, Chen X, Cao W, Li F, et al. (2019). IGF-2 preprograms maturing macrophages to acquire oxidative phosphorylation-dependent anti-inflammatory properties. Cell Metab 29, 1363–1375.e1368. [DOI] [PubMed] [Google Scholar]
- Faure-Dupuy S, Delphin M, Aillot L, Dimier L, Lebosse F, Fresquet J, Parent R, Matter MS, Rivoire M, Bendriss-Vermare N, et al. (2019). Hepatitis B virus-induced modulation of liver macrophage function promotes he-patocyte infection. J. Hepatol 71, 1086–1098. [DOI] [PubMed] [Google Scholar]
- Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D, et al. (2020). Jaspar 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48, D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlanda C, Dinarello CA, and Mantovani A (2013). The interleukin-1 family: back to the future. Immunity 39, 1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genin M, Clement F, Fattaccioli A, Raes M, and Michiels C (2015). M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 15, 577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto D, Miller J, and Merad M (2011). Dendritic cell and macrophage heterogeneity in vivo. Immunity 35, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D, Ding J, Li O, He Q, Ke M, Zhu M, Liu L, Ou WB, He Y, and Wu Y (2018). Human-induced pluripotent stem cell-derived macrophages and their immunological function in response to tuberculosis infection. Stem Cell Res Ther 9, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al. (2018). Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol 20, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Park ES, Lee AR, Park S, Park YK, Ahn SH, Kang HS, Won JH, Ha YN, Jae B, et al. (2018). Intracellular interleukin-32gamma mediates antiviral activity of cytokines against hepatitis B virus. Nat. Commun 9, 3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenkel O, and Tacke F (2017). Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol 17, 306–321. [DOI] [PubMed] [Google Scholar]
- Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, and King RW (1997). Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemo-ther 41, 1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Lo C, Skinner N, Locarnini S, Visvanathan K, and Mansell A (2011). The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J. Hepatol 55, 762–769. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhou C, Li J, Liu J, Lin L, Li L, Cao D, Li Q, and Wang Z (2018). Single domain based bispecific antibody, Muc1-Bi-1, and its humanized form, Muc1-Bi-2, induce potent cancer cell killing in muc1 positive tumor cells. PLoS One 13, e0191024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST, Cho C, Machida K, Chen D, and Ou JJ (2017). Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol. Cell 68, 281–292.e285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Xu R, Gu H, Zhang E, Qu J, Cao W, Huang X, Yan H, He J, and Cai Z (2021). Metabolic reprogramming in macrophage responses. Biomark Res. 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi S, Henriksson S, Corcoran M, Mendez-Vidal C, Wiman KG, and Farnebo M (2009). Wrap53, a natural p53 antisense transcript required for p53 induction upon DNA damage. Mol. Cell 33, 462–471. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, and Locati M (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686. [DOI] [PubMed] [Google Scholar]
- Marc Flajolet PT, Marie-Annick B, and Geneviève F (1998). Woodchuck hepatitis virus enhancer i and enhancer II are both involved in N-myc2 activation in woodchuck liver tumors. J. Virol 72, 6175–6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tour-lomousis P, Dabritz JHM, Gottlieb E, Latorre I, et al. (2016). Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oropeza CE, Tarnow G, Sridhar A, Taha TY, Shalaby RE, and McLachlan A (2020). The regulation of HBV transcription and replication. Adv. Exp. Med. Biol 1179, 39–69. [DOI] [PubMed] [Google Scholar]
- Ou JH, Yeh CT, and Yen TS (1989). Transport of hepatitis B virus precore protein into the nucleus after cleavage of its signal peptide. J. Virol 63, 5238–5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al. (2018). R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 172, 90–105.e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takkenberg RB, Zaaijer HL, Molenkamp R, Menting S, Terpstra V, Weegink CJ, Dijkgraaf MG, Jansen PL, Reesink HW, and Beld MG (2009). Validation of a sensitive and specific real-time PCR for detection and quantitation of hepatitis B virus covalently closed circular DNA in plasma of chronic hepatitis B patients. J. Med. Virol 81, 988–995. [DOI] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGet-trick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. (2013). Succinate is an inflammatory signal that induces IL-1βeta through HIF-1alpha. Nature 496, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno C, and Kojima Y (2020). Characterization and applications of chimeric mice with humanized livers for preclinical drug development. Lab Anim. Res 36, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Chen WL, and Ou JH (2011). Effects of interferon-alpha/beta on HBV replication determined by viral load. Plos Pathog. 7, e1002159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Kuo CF, Akbari O, and Ou JH (2016). Maternal-derived hepatitis B virus e antigen alters macrophage function in offspring to drive viral persistence after vertical transmission. Immunity 44, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KN, Kuo CF, and Ou JJ (2018). Mechanisms of hepatitis B virus persistence. Trends Microbiol. 26, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Montfoort N, van der Aa E, van den Bosch A, Brouwers H, Vanwolle-ghem T, Janssen HLA, Javanbakht H, Buschow SI, and Woltman AM (2016). Hepatitis B Virus surface antigen activates myeloid dendritic cells via a soluble CD14-dependent mechanism. J. Virol 90, 6187–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Wasiliew P, and Kracht M (2010). Interleukin-1 (IL-1) pathway. Sci. Signal 3, cm1. [DOI] [PubMed] [Google Scholar]
- Xu L, Yin W, Sun R, Wei H, and Tian Z (2014). Kupffer cell-derived IL-10 plays a key role in maintaining humoral immune tolerance in hepatitis B virus-persistent mice. Hepatology 59, 443–452. [DOI] [PubMed] [Google Scholar]
- Yang F, Yu X, Zhou C, Mao R, Zhu M, Zhu H, Ma Z, Mitra B, Zhao G, Huang Y, et al. (2019). Hepatitis B e antigen induces the expansion of mono-cytic myeloid-derived suppressor cells to dampen T-cell function in chronic hepatitis B virus infection. Plos Pathog. 15, e1007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H, Zhang Y, Yang X, Li M, Hu H, Xiong J, Wang N, Jin J, Zhang Y, Song Y, et al. (2020). Hepatitis B core antigen impairs the polarization while promoting the production of inflammatory cytokines of M2 macrophages via the TLR2 pathway. Front Immunol. 11, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Zhang Q (2019). Using seahorse machine to measure OCR and ECAR in cancer cells. Methods Mol. Biol 1928, 353–363. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Li Y, Wu X, Li L, Liu J, Wang Y, Liu Y, Li Q, and Wang Z (2020). Identification of anti-CD16a single domain antibodies and their application in bispecific antibodies. Cancer Biol. Ther 21, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data are available via NCBI Gene Expression Omnibus (GEO) database (GEO: GSE179618).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.