

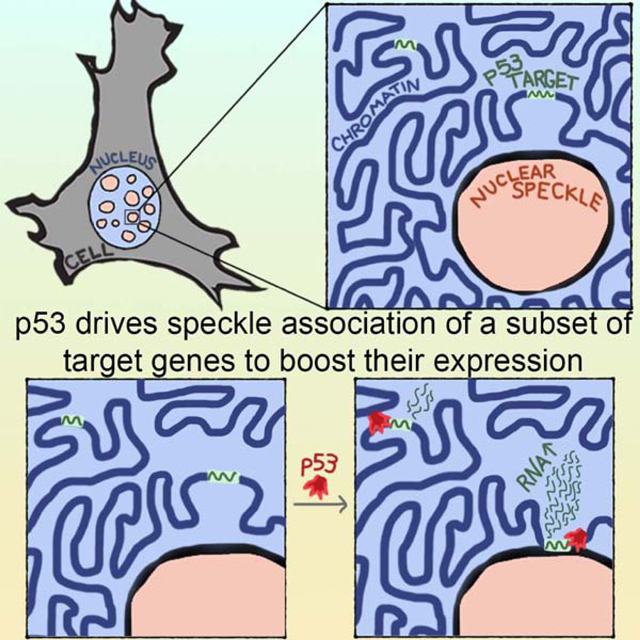
SUMMARY
Nuclear speckles are prominent nuclear bodies that contain proteins and RNA involved in gene expression. While links between nuclear speckles and gene activation are emerging, the mechanisms regulating association of genes with speckles are unclear. We find that speckle association of p53 target genes is driven by the p53 transcription factor. Focusing on p21, a key p53 target, we demonstrate that speckle association boosts expression by elevating nascent RNA amounts. p53-regulated speckle association did not depend on p53 transactivation functions, but required an intact proline-rich domain and direct DNA binding, providing mechanisms within p53 for regulating gene-speckle association. Beyond p21, a substantial subset of p53 targets have p53-regulated speckle association. Strikingly, speckle-associating p53 targets are more robustly activated and occupy a distinct niche of p53 biology compared to non-speckle-associating p53 targets. Together, our findings illuminate regulated speckle association as a mechanism utilized by a transcription factor to boost gene expression.
Keywords: Nuclear speckles, p53, transcription, p21, nuclear positioning, chromosome architecture, phase-separated nuclear bodies, gene activation
eTOC Blurb:
Regulated association between genes and nuclear speckles remains poorly understood. Alexander et al. find that the p53 transcription factor regulates speckle association of certain target genes, demonstrating that speckle association by p53 governs gene expression. Thus, speckle association by transcription factors has the potential to be a major gene-regulatory mechanism.
Graphical Abstract
INTRODUCTION
Nuclear speckles have been identified as a major activating area of the nucleus, with the most highly expressed genes, super-enhancers, and gene-dense regions residing adjacent to nuclear speckles (Chen et al., 2018; Quinodoz et al., 2018). Originally proposed to be storage sites within the nucleus, speckles are nuclear bodies that contain RNA and proteins involved in transcription, splicing, polyadenylation, RNA modification, and RNA export (reviewed in (Galganski et al., 2017)). Although highly expressed genes tend to associate with nuclear speckles (Brown et al., 2008; Chen et al., 2018; Quinodoz et al., 2018; Shopland et al., 2003), ascribing them a functional role beyond storage sites has been elusive due to challenges disrupting nuclear speckles. Speckle perturbations described to date affect cell growth (Saitoh et al., 2012; Sharma et al., 2010), and often affect only a subset of speckle components (Miyagawa et al., 2012; Saitoh et al., 2012). Thus, while correlational data suggest that speckles promote gene expression, functional data supporting this conclusion are limited.
Given that speckles contain a myriad of factors involved in RNA production, positioning of genes next to speckles may be highly consequential for RNA expression. However, the extent to which speckle association of genes can be regulated and the mechanisms of this regulation are unclear. One key case of regulated speckle association is during erythroid differentiation, when both α-globin and β-globin genomic loci increase their localization at nuclear speckles (Brown et al., 2006). Speckle association of α- and β-globin is coincidental with increased transcription, suggesting that the localization of these genes to speckles may be consequential for gene expression. However, the factors that drive speckle association of the globin genes during differentiation are not known.
Another instance of regulated speckle association is of an Hsp70 transgene that becomes speckle-associated upon heat shock (Hu et al., 2009). In contrast, the endogenous Hsp70 gene is pre-positioned at speckles, with 90–100% of Hsp70 loci at the speckle prior to heat shock (Tasan et al., 2018; Zhang et al., 2020). While endogenous regulation of Hsp70 speckle association is limited, genome-wide studies have found that other heat shock response genes increase speckle association upon heat shock (Zhang et al., 2020). Expression studies in live and fixed cells of the Hsp70 transgene, endogenous Hsp70, and the other speckle-associating heat shock response genes support that gene positioning adjacent to speckles boosts gene expression, with higher levels of RNA within speckle-associating transcription sites (Khanna et al., 2014; Kim et al., 2019; Zhang et al., 2020).
The evidence to date is suggestive of speckle association as a gene regulatory mechanism. However, the extent that regulated speckle association is utilized across diverse transcriptional responses, and the mechanisms driving gene speckle association are opaque. Speckle association of the Hsp70 transgene depends on the Hsp70 promoter sequence (Hu et al., 2010), suggesting some specificity to regulated speckle association. However, factors conferring this specificity have yet to be identified.
Thus, outstanding questions are whether and how speckle association is regulated, and in particular, a critical functional matter is whether DNA-binding transcriptional activators have a direct role. Here we investigate whether the p53 tumor suppressor, a key stress-responsive transcription factor, can regulate speckle association of its target genes. Our findings demonstrate that speckle association can be regulated, and via distinct domains within p53, highlighting a novel mechanism by which transcription factors can modulate gene expression. We show that reduced speckle integrity lowers p53-mediated target gene speckle association and RNA expression, and uncover a regulated subset of critical p53 target genes, which are enriched in cell cycle and apoptotic pathways. These findings provide new insight into the functions and mechanisms of nuclear speckles, and identify speckles as key modulators of the p53 transcriptional program.
RESULTS
p53 drives speckle association of its key transcriptional target, p21
p53 is a model for a strong transcriptional activator with direct DNA binding. In response to stress, p53 activates gene pathways that stall the cell cycle, promote DNA repair, and drive apoptosis (reviewed in (Kastenhuber and Lowe, 2017)). To investigate whether p53 regulates speckle association of its target genes, we first focused on p21, a strongly induced target of p53 that regulates cell cycle progression (el-Deiry et al., 1993; Karimian et al., 2016). To induce p53, we utilized two cell culture systems (Figures 1A and 1B). In the first, we used IMR90 primary human fibroblast cells in which endogenous p53 protein is maintained at low levels by Mdm2 (Moll and Petrenko, 2003), an interaction that can be inhibited by the small molecule, Nutlin-3a (Vassilev et al., 2004). Therefore, application of Nutlin-3a results in enhanced levels and genomic binding of p53 as well as transcriptional activation of its target genes (Figures 1 and S1A–S1D)(Catizone et al., 2019). In the second test, we utilized the p53-null Saos2 cell line and introduced p53, as described below (Figure 1B).
Figure 1. p21 becomes speckle associated upon p53 activation.

(A) Model of IMR90 cells treated with Nutlin-3a to activate p53.
(B) Model of Saos2 p53−/− cells treated with doxycycline to activate expression of p53 transgene.
(C) Chromosome locations of p21 and Hmga1 (bottom). RNA-seq FPKM values in IMR90 cells (top left), and qRT-PCR showing expression relative to Gapdh.
(D) ImmunoDNA-FISH of Hmga1 (top; green) and p21 (bottom; green) genes with nuclear speckle immunofluorescence (red) and DAPI DNA staining (blue).
(E) Distribution of Hmga1 loci distance to the nearest speckle in IMR90 cells.
(F) Percentage of Hmga1 loci at the speckle in IMR90 cells.
(G) Distribution of p21 loci distances to the nearest speckle in IMR90 cells.
(H) Percentage of p21 loci at the speckle in IMR90 cells.
(I) ImmunoDNA-FISH in Saos2 p53 null (left) or dox-inducible WT p53 cells (right).
(J) Distribution of Hmga1 loci distance to the nearest speckle in Saos2 cells.
(K) Percentage of Hmga1 loci at the speckle in Saos2 cells.
(L) Distribution of p21 loci distances to the nearest speckle in Saos2 cells.
(M) Percentage of p21 loci at the speckle in Saos2 cells.
*** - p < 0.0001, ** p < 0.01, * - p < 0.05, unlabelled - not significant.
D – DMSO treated.
For additional immunoFISH images, see Figure S1. For number of loci counted, see Table S1.
To determine whether p53 activation alters the localization of the p21 gene relative to nuclear speckles, we performed immunoDNA-FISH using immunofluorescence of nuclear speckles combined with DNA-FISH probes against p21. As a control, we assayed Hmga1, which is not a p53 target, is on the same chromosome as p21, and is expressed at similar levels to p21 after Nutlin-3a treatment (Figure 1C). Treating IMR90 cells with Nutlin-3a over a timecourse of 2, 6, and 9 hours, we assessed the dynamics of speckle association, measuring the distance from the DNA-FISH signal to the edge of the nearest speckle (see Table S1 for number of loci quantified). We evaluated speckle association using two metrics: (1) the distribution of distances of loci to the edge of the nearest speckle, and (2) the percentage of loci that contacted nuclear speckles, defining contact as within 0.26μm, the equivalent of 2 pixels in our imaging data. Hmga1 localization relative to nuclear speckles was unaltered across the Nutlin-3a timecourse (Figures 1D upper, 1E and 1F). In contrast, p21 loci became closer to nuclear speckles (Figures 1D lower, 1G, and S1E–S1H), and the percentage of p21 loci contacting the speckle increased from 29% speckle associated in the DMSO control to 52% after 6 hours of Nutlin-3a (Figure 1H). Thus, p21 underwent a substantial increase in speckle association upon p53 activation.
After 9 hours of Nutlin-3a, speckle association of p21 returned to baseline (Figures 1G and 1H), indicating that p21 speckle association is transiently induced in response to p53 activation. The amount of p53 bound to the p21 enhancer and promoter remained consistent between 6 and 9 hours of Nutlin-3a treatment (Figure S1C). Thus, p21 dissociation from speckles was not due to altered p53 chromatin binding over the timecourse. We also found no substantial difference in speckle size or shape when p21 was associating at 2 and 6 hours, but dramatic alteration in speckle shape at 9 hours, suggesting that p53-induced speckle changes could be involved in p21 dissociation from speckles (Figures S1I–S1L). Hence, p53 drives p21 speckle association independently of major changes in speckle characteristics.
Our data support that p53 may have a direct role in speckle association of p21. To test this in a second assay, we expressed p53 in p53-null Saos2 cells using a doxycycline-inducible system (Figures 1B and 1C). We found that p21 increased speckle association at 3 hours of dox-induction, and, as in IMR90 cells, speckle-association of p21 was transient, decreasing at the 6 hour time point (Figures 1I lower, 1L, 1M, and S1M–S1O). In contrast, Hmga1 did not become speckle-associated (Figures 1I upper, 1J, and 1K). These findings demonstrate that in a p53-null system, induced expression of p53 was sufficient to instigate transient speckle association of the p21 gene.
Increased nascent RNA at speckle-proximal p21 transcription sites
To investigate the consequences of p21 speckle association, we utilized single molecule RNA-FISH, which enables three measurements of RNA expression: (1) the percentage of cells that have active transcription sites, (2) the amount of nascent RNA within each active site, and (3) the total number of RNA molecules in each cell. To examine the relationship between RNA expression and speckle association, we used probes against exons to mark all p21 RNA molecules, against introns to mark the sites of transcription, and against speckle-marker RNA, Malat1, to label nuclear speckles (Figures 2A–2C and S2). As anticipated, Nutlin-3a treatment in IMR90 cells substantially increased the percentage of cells with active p21 transcription sites (Figure 2D). Additionally, the amount of nascent RNA within active transcription sites increased during Nutlin-3a treatment (Figure 2E). Thus, Nutlin-3a increased the percentage of cells with active transcription sites as well as the amount of nascent RNA within each transcription site. This combination likely contributed to the robust increase in the total number of p21 RNA molecules per cell (Figures 2B and 2C green, and 2F).
Figure 2. Relationship between speckle association and p21 expression.

(A) p21 probe locations.
(B and C) Maximum projection images of Malat1 speckle marker (red), p21 introns (white), and p21 exons (green) in IMR90 cells.
(D) Quantification of the number of active transcription sites per cell. Error bars represent standard error.
(E) Amount of nascent RNA. Each dot represents an individual transcription site.
(F) Number of p21 exon spots per cell. Each dot represents a single cell.
(G) Distribution of p21 active transcription site distances to the nearest speckle. Each dot represents an individual transcription site.
(H-K) Nascent RNA amount versus distance to speckle. Each white circle is an individual transcription site. Background color represents the density of data points.
D – DMSO treated.
For additional RNA-FISH images see Figure S2. For number of transcription sites and cells counted, see Table S1.
In the same set of experiments, we measured speckle association by calculating the distance from each active transcription site to the nearest Malat1-labelled speckle. We found that speckle association of active p21 transcription sites occurs at 2 hours of Nutlin-3a treatment (Figure 2G). As observed in our DNA-FISH studies (Figures 1G–H), this increase in p21 speckle association was transient, returning to baseline at 9 hours of Nutlin-3a treatment.
To investigate the relationship between speckle association and RNA expression, we measured the distance to the speckle and the nascent RNA levels of each active transcription site (Figure 2H–K). Under control DMSO conditions, there is no relationship between speckle association and nascent RNA levels (Figure 2H). At 2 hours of Nutlin-3a treatment, most transcription sites are near speckles with low levels of nascent RNA. However, at this time we observe a population of transcription sites that is proximal to the speckle and have higher nascent RNA levels, showing an L-shaped data distribution (Figure 2I; Fisher’s exact p < 0.01). At 6 hours of Nutlin-3a treatment, the transcription sites adjacent to the nuclear speckle increase nascent RNA levels, maintaining the L-shaped data distribution (Figure 2J; Fisher’s exact p < 0.05). By 9 hours, the transcription sites are no longer concentrated next to the speckle and no longer show an L-shaped distribution (Figure 2K). These data demonstrate increased nascent RNA within speckle-adjacent transcription sites during early stages of p53 activation, supporting that speckle association boosts p21 expression.
Knockdown of the speckle-resident proteins compromise p53-mediated induction of p21
To evaluate the relationship between nuclear speckles and p53 induction of p21, we used shRNA knockdown in IMR90 cells to reduce SON (Figure 3A), a speckle-resident protein that has been implicated in nuclear speckle organization (Sharma et al., 2010). We chose a modest level of knockdown due to our observations of cell death resulting from more severe knockdown, findings that are consistent with other studies that perturb SON (Sharma et al., 2010). Similar to previous reports (Sharma et al., 2010), SON knockdown (SON KD) compromised speckle localization of some, but not all speckle markers, with Malat1 diffusely localized in the nucleus and SRRM2 properly localized in a speckle pattern (Figures S3A and S3B). Although SRRM2 was speckle-localized, the total number, area, and perimeter of speckles per cell was reduced upon SON KD compared to the non-targeting control (NTC) (Figures S3C–S3F). Thus, even slight reduction of SON affects the content and abundance of nuclear speckles.
Figure 3. Knockdown of SON compromises p53-mediated induction of p21 expression and speckle association.

(A) qRT-PCR measuring SON RNA levels in IMR90 cells treated with a SON shRNA (SON KD) or non-targeting control (NTC).
(B) Percentage of cells with active transcription sites.
(C) Number of p21 exon spots per cell (p21 molecules per cell) in IMR90 cells.
(D) Nascent RNA amount in IMR90 cells.
(E) Distribution of p21 transcription site distances to the nearest speckle.
(F-K) Nascent RNA amount versus distance to speckle. Each white circle is an individual transcription site. Background color represents the density of data points.
(L) Western blot of p53, p21, and Gapdh. Total protein loading is shown by Ponceau (Pon.) staining.
(M) Quantification of protein levels based on L and Figure S3 of band intensity relative to Gapdh loading control and normalized to NTC 9h levels.
D – DMSO treated; 2h – Nutlin-3h 2 hour treated; 6h – Nutlin-3a 6h treated
* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001
For additional characterization of SON KD and for SRSF1 KD, see Figure S3. For number of transcription sites and cells counted, see Table S1.
To assess the consequences for p53-mediated speckle association and expression of p21, we compared the effects of Nutlin-3a in IMR90 cells with SON KD versus NTC. Both SON KD and NTC-treated IMR90 cells displayed robust increases in the percent of cells with active p21 transcription sites upon Nutlin-3a treatment (Figure 3B). Despite this, the total amount of p21 RNA was reduced in SON KD compared to NTC (Figure 3C). Likewise, the amount of nascent RNA within the site of transcription was significantly lower in SON KD cells after Nutlin-3a treatment (Figure 3D). These results demonstrate that SON KD dramatically reduces the ability of p53 to induce p21 RNA expression, with marked defects in the amount of nascent RNA at active transcription sites.
We next investigated the effects of SON KD on p53-mediated speckle association of p21 (Figure 3E) and found reduced speckle association at baseline as compared to NTC-treated cells. In addition, p21 transcription sites in SON KD cells treated with Nutlin-3a do not increase their speckle association, and are more distal from speckles compared to controls (Figure 3E). Thus, SON is crucial for p53-mediated speckle association of p21 transcription sites.
To explore the relationship between speckle association and nascent RNA amount, we related transcription site speckle distances to levels of nascent RNA upon SON KD. We found that speckle-proximal transcription sites from SON KD fail to accumulate high nascent RNA levels observed in NTC controls, and are defective at producing the L-shaped data distribution of high nascent RNA levels at speckle-proximal transcription sites (Figures 3I–3K compared to Figures 3F–3H). Beyond RNA expression, SON KD results in defective p21 protein levels upon p53 activation (Figures 3L, 3M, and S3G–S3J). Thus, SON is required for p53-mediated speckle association of p21, for accumulation of p21 RNA within transcription sites and within the cell, and for subsequent accumulation of p21 protein. Based on these findings, we propose that speckle integrity is critical for p53-mediated speckle-association and expression of p21.
To gain additional evidence linking speckle integrity to p53 induction of p21, we performed knockdown of SRSF1, which, similar to SON KD, results in perturbation of some, but not all, speckle components (Fei et al., 2017; Tripathi et al., 2012). Consistent with these observations, we found that two SRSF1 shRNAs resulted in disrupted Malat1 speckle localization, showing diffuse and perispeckled (surrounding speckles) localization, while SRRM2 displayed a speckle pattern similar to the NTC (Figures S3K–S3L). Similar to SON KD, SRSF1 KD disrupted speckle association of p21 (Figure S3M), decreased nascent p21 RNA amount (Figure S3N), and decreased total p21 RNA (Figure S3O) upon Nutlin-3a treatment. These results provide additional evidence that intact speckles are important for p53-mediated speckle association and activation of p21.
Regulation of speckle association requires intact DNA-binding by p53
A major area of interest, and lacking in previous analyses of gene association with speckles, is the mechanism(s) underpinning speckle association. In particular, it is not known whether genes can be specifically targeted to nuclear speckles by transcription factors that bind directly to DNA. To investigate the mechanisms governing p53-mediated speckle association, we generated point mutations in p53 targeting specific p53 molecular functions (Figure 4A), and used doxycycline to induce their expression in Saos2 p53-null cells (Figures 1B and S4A–S4G).
Figure 4. p21 speckle association by p53 requires p53 DNA binding and proline rich domains, but not p53 transactivation functions.

(A) p53 protein domains and mutations. TAD – transactivation domain, PRD – proline-rich domain, DBD – DNA-binding domain, TET – tetramerization domain, REG – regulatory domain.
(B) Percentage of cells with active transcription sites as measured by RNA-FISH.
(C) p21 nascent transcript amount as measured by RNA-FISH.
(D) Distribution of the number of p21 molecules per cell as measured by RNA-FISH.
(E) Distribution of p21 loci distances to the nearest nuclear speckle as measured by immunoDNA-FISH.
(F) Percent p21 loci at nuclear speckles measured by immunoDNA-FISH.
(G) ChIP-qPCR of p53 at the p21 enhancer, promoter, and negative region. For primer locations, see Figure S1D.
(H) Distribution of speckle distances of p21 loci measured by immunoDNA-FISH.
(I) Distribution of speckle distances of p21 loci measured by immunoRNA-FISH.
(J) Number of p21 RNA molecules per cell as measured by RNA-FISH.
(K) Percentage of cells with active transcription sites as measured by RNA-FISH.
(L) Nascent RNA amount in p53 wild type and mutant Saos2 cells.
(M-P) Distance to speckle versus transcription site intensity in Saos2 cells induced to express wild type or Δ62–77 p53.
(Q) Model showing the consequences of perturbing specific p53 functions.
*** p < 0.0001, ** p < 0.01, * p < 0.05, unlabeled - not significant.
For additional characterization of p53 mutants, see Figure S4. For number of loci, transcription sites, and cells counted, see Table S1.
We first assessed whether direct DNA binding by p53 was required for speckle association, utilizing the R273H point mutation in the DNA binding domain (Figure 4A; DBD), which disrupts the contact between p53 and its response element, abolishing direct binding of p53 to DNA (Joerger et al., 2006; Kitayner et al., 2006). We also used a L344P point mutation (Figure 4A; TET), which renders p53 unable to dimerize or tetramerize, also disrupting direct DNA binding (Davison et al., 1998; Lomax et al., 1998). Dox-induced expression of p53-R273H or p53-L344P mutants failed to increase p21 expression as assessed by RNA-FISH measurements of the percentage of cells with active p21 transcription sites (Figure 4B), by p21 nascent RNA levels (Figure 4C, blue), and by the number of p21 RNA molecules per cell (Figures 4D and S4H). Importantly, p53-R273H and p53-L344P failed to induce p21 speckle association as measured by DNA-FISH (Figures 4E and 4F). Thus, p53-mediated speckle association of p21 requires the ability of p53 to directly bind DNA. These findings provide a mechanism for the specificity of regulated speckle association, indicating that speckle association of genes can be regulated by a transcription factor based on direct DNA binding.
Transcriptional activation is dispensable for speckle association
One proposed model for speckle association is that transcription and splicing leads to nucleation of speckles through a process of self-assembly (Brown et al., 2008; Guo et al., 2019; Shevtsov and Dundr, 2011). To query the requirement of transcription for speckle association, we utilized point mutations within the two transactivation domains of p53, TAD1 and TAD2 (reviewed in (Raj and Attardi, 2017)) (Figure 4A). We assessed p53 TAD1 (L22Q, W23S; p53-TAD1) and TAD2 (W53Q, F54S, p53-TAD2) mutations separately and in combination (p53-TAD1/2). All three TAD p53 mutants were defective at increasing the number of active transcription sites per cell (Figure 4B), the amount of nascent RNA within active transcription sites (Figure 4C, gold), and the total number of p21 RNA molecules per cell (Figure 4D, gold). Despite these striking transcriptional defects, immunoDNA-FISH showed that each TAD mutant displayed robust increases in p21 speckle association, with comparable speckle association to that of WT p53 (Figure 4E and 4F, gold). These data reveal that speckle association of p21 via p53 does not require p21 transcriptional activation, demonstrating that transactivation and speckle association are distinct molecular functions of p53. Altogether, our findings provide strong evidence that the localization of genes to nuclear speckles is not merely a consequence of transcription, but rather can be a distinct function of a transcription factor.
p53-mediated speckle association of p21 requires the proline-rich domain of p53
Having determined that speckle association of p21 requires p53 DNA binding but not p53 transactivation functions, we sought to identify whether p53 contains a speckle-association domain. To this end, we generated deletions of the two remaining p53 domains: the C-terminal regulatory domain (Figure 4A; REG) and the proline-rich domain (Figure 4A; PRD). We initially deleted the entire p53 PRD (Δ62–91; Figure 4A), and found disruption of speckle association by DNA-FISH (Figure S4I). However, the p53 Δ62–91 PRD deletion also disrupted the ability of p53 to interact with DNA, leading to ~60% reduction of binding at the p21 promoter and upstream enhancer, as measured by p53 ChIP-qPCR (Figure S4J), confounding interpretation. We therefore generated a second PRD deletion, Δ62–77, that retained aa78–91, which is the half of the PRD abutting the p53 DBD (Figure 4A); this p53 Δ62–77 mutant remained competent in DNA binding (Figure 4G). Using DNA-FISH, we assessed speckle association in cells with p53 Δ62–77 or a C-terminal deletion in which a stop codon was introduced into p53 K370 (p53–370*). p53–370* was competent for p21 speckle association (Figure 4H), demonstrating that the p53 C-terminus is dispensable for p53-mediated speckle association. In contrast, p53 Δ62–77 was defective at driving speckle association of p21 (Figure 4H). While the precise mechanisms by which Δ62–77 disrupts the ability of p53 to mediate speckle association remain to be determined, we observed a modest but significant reduction in p53 protein enrichment at speckles in cells expressing Δ62–77 as compared to cells with WT p53 (Figures S4K–S4M). Overall, these findings identify the p53 PRD as an essential domain for speckle association of p21. Additionally, disruption of speckle association without altered DNA binding demonstrates that DNA binding is not sufficient to direct speckle association. Rather, speckle association is a distinct molecular of function of p53 that requires an intact PRD in addition to p53 direct DNA binding.
The speckle-association defective Δ62–77 PRD p53 mutant fails to fully induce p21
Our above findings show that nuclear speckle integrity is crucial for p53-mediated induction of p21 expression (Figures 3 and S3). To independently assess the importance of speckle association for p21 expression, we used RNA-FISH in Saos2 cells expressing the speckle-association-defective p53 Δ62–77 mutant. Consistent with our DNA-FISH results, RNA-FISH showed speckle association of p21 loci at 3 hours of dox-treatment in wild type cells, with p21 transcription sites becoming more distal from speckles at 6 hours of dox-induction, whereas p53 Δ62–77 mutant Saos2 cells were comparatively distal from speckles at both time points (Figure 4I). Looking at p53-mediated induction of p21 expression, we found that both wild type and Δ62–77 increased total p21 RNA expression. However, the amount of p21 was dramatically lower in Δ62–77 compared to wild type (Figure 4J). This decrease was not due to an altered proportion of cells with active transcription sites, which was comparable between wild type and Δ62–77 cells (Figure 4K). Instead, p53 Δ62–77 cells displayed reduced levels of nascent transcripts within active transcription sites (Figure 4L). Assessment of nascent RNA amount versus the distance to the speckle further highlighted this difference, with p21 transcription sites in wild type cells concentrated near the speckle and showing accumulation of nascent transcripts (Figure 4M), and transcription sites in Δ62–77 mutant cells distributed distally from speckles and defective in accumulating high nascent RNA levels (Figure 4N). At 6 hours of dox-induction, nascent transcript levels remained high as transcription sites become distal to speckles in wild type cells (Figure 4O), while transcription sites in Δ62–77 cells retain similar distance distributions and nascent RNA levels (Figures 4I, 4L, and 4P). These data again demonstrate that speckle association by p53 is critical for full induction of p21 expression. In addition, our finding that disruption of speckle association does not affect the proportion of cells with active transcription, but compromises the amount of nascent RNA within active sites, supports that regulated speckle association boosts gene expression by elevating nascent RNA levels.
p53 regulates speckle association of a subset of target genes
p53 has hundreds of direct transcriptional targets across many cell types and conditions (reviewed in (Kastenhuber and Lowe, 2017)). To understand the extent that p53 drives speckle association of different target genes, we performed immunoDNA-FISH in a panel of 11 p53 targets and 10 control genes in IMR90 cells over a Nutlin-3a timecourse. Well-known and variably induced p53 targets were selected, while control genes were selected as those that (1) are greater than 100kb from a p53 ChIP-seq binding peak (Figures 5A and 5B), (2) do not change expression upon Nutlin-3a treatment (Figures 5A and 5C), and (3) have similar expression to the p53 targets in RNA-seq data from Nutlin-3a treated IMR90 cells (Figures 5A and 5D).
Figure 5. Nutlin-3a treatment drives increased speckle association of a subset of p53 targets.

(A) Schematic showing selection criteria for DNA-FISH control genes.
(B) Gene distance to the nearest p53 peak in IMR90 cells treated with Nutlin-3a (data from Sammons eta al., 2015).
(C) RNA fold change in IMR90 cells treated with Nutlin-3a (data from Sammons et al., 2015).
(D) Expression (FPKM) of control genes and p53 targets in IMR90 cells treated with Nutlin-3a.
(E) Heatmap of change in percentage of transcription sites with speckle association in IMR90 cells upon Nutlin-3a treatment relative to control. Plots (right) show examples of the primary data. The grey bar (left) shows the median baseline distance to the speckle, split into quintiles.
(F) Table of p53 target genes with (blue) or without (green) p53-regulated speckle association, and their gene density, HiC subcompartment, and SON TSA-seq decile.
For additional representations of speckle association, see Figure S5. For number of loci counted, see Table S1.
At baseline (DMSO-treated), p53 target genes and the control genes showed varied speckle association (Figure S5A). Upon addition of Nutlin-3a, we found that 7 out of 11 p53 targets increased their speckle association, while 0 of the 10 control genes increased association (Figures 5E, S5B, and S5C). Thus, the ability of p53 to regulate association of its target genes with nuclear speckles is not restricted to p21, and may be a widespread regulatory mechanism. That no control genes increased speckle association underscores the specificity to direct targets of p53.
Our results also show that that not all p53 targets increased speckle association following p53 activation, with 4 p53 targets showing no change in speckle association (Figure 5E). Thus, the ability of p53 to drive gene speckle association is selective to a subset of target genes. We therefore investigated whether existing genomic datasets could distinguish between p53 target genes that do or do not display p53-mediated increases in speckle association. Using three criteria: gene density, chromatin compartment annotations (HiC subcompartments; Rao et al., 2012), and genome-wide speckle association data (SON TSA-seq; Chen et al., 2018), we found that chromatin context is highly co-incident with whether a p53 target became speckle associated upon p53 activation (Figure 5F). Overall, p53 targets that displayed regulated speckle association were more likely to be in gene-dense regions, to reside within the A1 HiC subcompartment, and to have higher speckle association at baseline (10th SON TSA-seq decile). These data suggest that genomic context may be a key variable dictating which p53 target genes have speckle association regulated by p53.
Genome-wide identification of p53-induced changes in speckle association
To further investigate the extent that p53 alters speckle association of target genes, we used TSA-seq, a genome-wide method for mapping chromosomal distances relative to nuclear compartments utilizing an antibody coupled with tyramide free-radical generation to label proximal DNA (Chen et al., 2018; Zhang et al., 2020). To map speckle association changes upon p53 activation, we performed TSA-seq targeting the speckle resident protein, SON, in replicates of IMR90 cells treated with DMSO or Nutlin-3a for 6 hours. We quantified SON TSA-seq signal over 25kb, 50kb, 100kb, and 500kb sized genomic windows, and found strong correlation between replicates for each window size (Figures 6A, 6B, and S6A). Based on diffusion of tyramide free-radicals, TSA-seq displays an exponential decay relationship between SON signal and the mean distance to the speckle as measured by DNA-FISH (Chen et al., 2018). As important controls to evaluate the robustness of our SON TSA-seq dataset and the appropriateness of quantification over different window sizes, we compared SON TSA-seq signal to our previous 42 DNA-FISH measurements of mean speckle distance (11 p53 targets and 10 controls in DMSO and Nutlin-3a conditions)(Figure 6C for 50kb window; Figure S6A for other window sizes). This analysis showed the expected decay of SON TSA-seq signal in relation to mean speckle distances across each window size. Thus, we conclude that our SON TSA-seq data reasonably detects speckle distances across a range of quantified window sizes.
Figure 6. SON TSA-seq mapping of p53-induced changes in speckle association.

(A-B) Correlation of SON TSA-seq replicates quantified over 50kb windows.
(C) SON TSA-seq signal versus mean distance to speckle for 42 DNA-FISH measurements (same measurements as Figure S5).
(D) UCSC genome browser tracks of the p21 locus for p53 ChIP-seq (top) and SON TSA-seq in IMR90 cells treated with DMSO or Nutlin-3a for 6 hours, and no primary antibody control (smoothing of 10). Bars below show merged significant domains with adjusted p-value of 0.01 or 1e-5. Genes are in grey, with IMR90 p53 targets in red.
(E) Proportion DNA-FISHed genes that do (“positive”) or do not (“negative”) increase speckle association upon Nutlin-3a treatment that were correctly called by SON TSA-seq at different adjusted p-value cutoffs.
(F) Number of IMR90 p53 targets that increase SON signal (padj < 0.01; red), do not increase SON signal (padj > 0.1; blue), or have borderline significance (grey).
For additional quality checks on SON TSA-seq data, see Figure S6.
We next assessed the sensitivity and selectivity of SON TSA-seq for detecting p53-induced changes. To this end, we called significant differences in SON signal between DMSO and Nutlin-3a treated IMR90 cells. Visual inspection of genome browser tracks of SON TSA-seq at the p21 gene indicate that called differences reasonably reflect observed changes in SON TSA-seq signal, and we note significant increase of SON TSA-seq signal at the p21 gene upon Nutlin-3a treatment (Figure 6D). By extracting genes within significant regions, we evaluated the performance of SON TSA-seq for detecting changes in speckle association as previously detected by DNA-FISH, assessing calls at different adjusted p-value cutoffs (Figure 6E). Using 7 p53 targets that were detected to increase speckle association by DNA-FISH (“positives”), and 14 genes that did not increase speckle association upon Nutlin-3a treatment (“negatives”; 4 p53 targets and 10 control genes)(Figure 5E), we determined that more stringent cutoffs led to decreased sensitivity of detecting positives (Figure 6E; black), and increased correct SON TSA-seq calling of negatives (Figure 6E; grey). A cutoff of 0.01 resulted in 100% correct negative calls (14/14 correct negatives, and also detected the majority of positives (5/7 correct positives). Relaxing the adjusted p-value to 0.05 improved sensitivity (6/7 correct positives), but compromised selectivity (3/14 incorrect negatives). Based on this, we considered p53 targets with adjusted p-values of less than 0.01 to significantly increase SON signal, those with adjusted p-values of greater than 0.1 to not increase SON signal, and the remainder to be of borderline significance. In total, of the 1694 genes that increase expression upon Nutlin-3a treatment by RNA-seq and are within 200kb of a p53 ChIP-seq binding peak in IMR90 cells (hereafter referred to as “IMR90 p53 target genes”), 303 increased SON signal, 202 were of borderline significance, and 1189 did not increase SON signal (Figure 6F). These results indicate that while the majority of targets do not increase speckle association, a substantial subset of IMR90 p53 target genes increase their speckle association upon p53 activation.
To further validate that SON TSA-seq reflects bona fide p53-induced changes in speckle association, we performed DNA-FISH validation on five p53 targets across a range of p-value cutoffs. We found that two p53 target genes with increased SON signal by TSA-seq (Gdf15 and Traf4) displayed increased speckle association upon 6 hours of Nutlin-3a treatment in IMR90 cells (Figures S6B and S6C). In contrast, Fuca1 (borderline significance) and Polh and Tigar (not increasing SON signal) showed no change in speckle association upon p53 activation (Figures S6D–S6F). This validation provided confidence in our designation of IMR90 p53 target genes that do or do not increase speckle association as measured by SON TSA-seq (Figure 6F), and provide a framework for comparing the characteristics of p53 targets genome-wide.
Genomic context, including speckle association at baseline, predicts which p53 targets display p53-regulated speckle association
Our DNA-FISH findings suggest that genomic context may be deterministic of whether the target gene undergoes p53-mediated speckle association (Figure 5F). With genome-wide measurements from SON TSA-seq (Figure 6F), we comprehensively evaluated genomic predictors of speckle association. To this end, we assessed the ability of HiC subcompartment, gene density, and baseline speckle association to predict whether a p53 target increases speckle association upon Nutlin-3a treatment. We also evaluated the effect of p53 peak density, as measured by the number of p53 ChIP-seq peaks within 200kb of the p53 target gene.Consistent with our previous findings (Figure 5F), we found that IMR90 p53 target genes that increased speckle association upon Nutlin-3a treatment were enriched in the A1 HiC subcompartment compared to other HiC subcompartments (Figure 7A, left), with ~50 times higher odds that a target with p53-regulated speckle association resides within the A1 versus A2 subcompartment (Figure 7A, right; B1 not calculated due to few genes within this subcompartment). Similarly, speckle-associating IMR90 p53 target genes were enriched in gene dense regions (Figure 7B, left), and targets in dense regions were ~35 times more likely to have p53-regulated speckle association (Figure 7B, right). The effect of the number of p53 peaks was more modest, but statistically significant, showing that IMR90 targets with a greater number of nearby p53 peaks are more likely to have p53-regulated increases in SON signal (Figure 7C). Strikingly, IMR90 p53 target genes that increased speckle association upon Nutlin-3a treatment had higher SON TSA-seq signal at baseline compared to p53 targets that did not increase speckle association (Figure 7D, left). Consequently, IMR90 p53 target genes’ SON TSA-seq signal at baseline was highly predictive of whether the target had p53-mediated increase in speckle association (Figure 7D, right). Overall, HiC subcompartment, gene density, and speckle association at baseline are all highly predictive of which p53 target genes displayed p53-regulated increases in speckle association. Because these three attributes are concordant with one another (Chen et al., 2018), we cannot currently distinguish which is the causal variable. However, these results suggest that the ability of p53 to enhance speckle association of its target genes may be restricted to target genes that already have propensity for speckle association.
Figure 7. Characteristics of p53 targets that have p53-regulated increases in speckle association.

(A-C) Stacked barplots (left) and logistic regression odds ratios (right) of IMR90 p53 targets that do (Increased SON) or do not (Not increased) increase speckle association based on HiC subcompartments (A), gene density (B), and number of p53 peaks within 200kb (C).
(D) Baseline (DMSO) SON TSA-seq signal of p53 targets that do or do not increase speckle association (left), and logistic regression predicted probability that a p53 target increases speckle association based on baseline SON TSA-seq signal (right).
(E) Fold change (of Nutlin-3a for 6, 9, or 12 hours relative to DMSO) of p53 targets that increase (red) and do not increase (blue) SON signal.
(F-G) Gene ontology (F) and KEGG pathway (G) comparison of IMR90 p53 targets that do or do not increase SON TSA-seq signal upon Nutlin-3a treatment.
p53 targets that increase speckle association show enhanced expression during later stages of p53 activation
Our separation of IMR90 p53 target genes into those that do or do not increase speckle association allowed us to determine whether these two groups have distinct expression characteristics upon p53 activation. We performed polyA RNA-seq in DMSO- or Nutlin-3a-treated IMR90 cells to compare RNA induction of p53 targets with or without p53-regulated speckle association. From our RNA-FISH studies of p21, a major increase in overall RNA expression occurred after initial p21 speckle association (Figure 2 and 3). For this reason, we performed RNA-seq of a Nutlin-3a timecourse starting at 6 hours of Nultin-3a treatment, the time point at which we measured speckle association by SON TSA-seq, and continued the timecourse to 9 and 12 hours after Nutlin-3a treatment. At 6 hours, IMR90 p53 targets that did or did not increase SON signal (from Figure 6F) each showed increased expression over the DMSO control, but were not significantly different from one another (Figure 7E). At 9 hours of Nutlin-3a treatment, IMR90 p53 targets with p53-regulated speckle association showed enhanced induction compared to p53 targets without speckle association (Figure 7E; 9h blue versus 9h red). This boosted expression of target genes with p53-regulated speckle association was more pronounced at 12 hours (Figure 7E; 12h blue versus 12h red). These findings support that speckle association is not absolutely required for transcriptional activation, but rather provides an expression boost, particularly at time points after speckle association. Because RNA-seq measures total RNA, this expression boost could be due to increased RNA stability and/or increased transcription.
p53 targets that increase speckle association are of distinct functional classes compared to targets that do not increase speckle association
The p53 transcription factor is a critical tumor suppressor that responds to a myriad of cellular stress signals, and is capable of orchestrating diverse cellular responses (reviewed in (Kastenhuber and Lowe, 2017)). Given that p53 targets that have p53-induced increase in speckle association show distinct kinetics and magnitude of induction compared to p53 targets that do not increase speckle association (Figure 7E), we evaluated whether these groups of genes partitioned evenly across p53 functional classes or rather, segregated according to biological function. Using gene ontology (Figure 7F) and KEGG pathway (Figure 7G) analyses, we found that p53 targets that increase speckle association fall into functionally distinct classes from targets that do not increase speckle association. Speckle-associating p53 targets are enriched in apoptosis, cell cycle checkpoint, and unfolded protein response GO pathways, while targets that do not increase speckle association are enriched in autophagy, metabolism, and immunity pathways (Figure 7F). Consistently, KEGG pathways show apoptosis as specific to speckle-associating p53 targets, with autophagy specific to targets that do not increase speckle association (Figure 7G). Thus, strikingly, the target genes that are subject to p53-regulated speckle association occupy a distinct niche of p53 biology, covering the key tumor-suppressor p53 functions of apoptosis and cell cycle checkpoint.
DISCUSSION
In this study we demonstrate that p53, a key stress response transcription factor and tumor suppressor, facilitates speckle association of a subset of its target genes. We provide correlational and functional evidence that gene-speckle association by p53 boosts RNA expression by elevating nascent RNA amounts within transcription sites. Together, these results highlight speckle association as a key step facilitating dynamic activation of specific p53 target genes.
By assessing the ability of p53 mutants to drive speckle association of p21, we dissected which p53 molecular functions contribute to speckle association, with direct DNA binding being essential for speckle association and transactivation being dispensable. Importantly, through identification of a p53 PRD mutant that is competent in DNA binding, but defective in speckle association, we demonstrate that speckle association is a distinct molecular function of p53. These findings reveal key features of regulated speckle association. First, the requirement for DNA binding illustrates how speckle association of specific genes can be regulated through the direct binding of a transcription factor. Second, by demonstrating that p53 regulation of speckle association is distinct from p53 transactivation functions, this study raises a general possibility that transcription factor mediated changes of gene localization to activating nuclear compartments may be a major discrete mechanism of gene regulation.
Extent of regulated speckle association
Fewer than 10 genes have been previously described to display regulated speckle association. In this study, we add over 300 genes, substantially expanding our knowledge of the cadre of genes that can have their speckle association regulated. Our genome-wide approach also enabled us to identify the underlying genomic logic distinguishing between p53 targets that do or do not have p53-regulated speckle association, with baseline speckle association being a key factor (Figure 7D). The extent to which this logic applies to other systems that display regulatable speckle association remains to be determined.
Our investigations of the p53 response substantially adds to understanding the physiological contexts of regulated speckle association—the other two being erythroid differentiation and heat shock. The transcription response to heat shock occurs over the course of minutes, whereas p53 activation occurs on a 2–6 hour time scale, and erythroid differentiation occurs over the course of days. Thus, regulated speckle association is amenable to different time scales of gene activation, indicating utility across a wide variety of biological processes and responses.
Mechanisms of regulated speckle association
Our finding that p53 can drive speckle association of its target genes represents a major mechanistic advance, demonstrating that speckle association of genes can be regulated by a transcription factor. By mapping the protein domains required for speckle association, our results support a model for p53-mediated speckle association where p53 is directed to its target genes via the DNA binding domain, while the PRD provides the biochemical properties required to mediate speckle association (see model; Figure 4Q). Compared to the other domains within p53, the molecular functions of the p53 PRD are not well established. The p53 PRD has been described to be involved in apoptosis (Baptiste et al., 2002), consistent with our functional analysis that speckle-associating p53 targets are enriched in apoptotic pathways (Figures 7F and 7G). A handful of specific PRD protein interacting partners have been identified (Dornan et al., 2003; Zacchi et al., 2002; Zheng et al., 2002; Zilfou et al., 2001). However, a cohesive model for the function of the p53 PRD has not emerged. Mapping the speckle-associating functions of p53 to the PRD thus exposes a new framework through which to understand the molecular functions of the p53 PRD. We anticipate that as additional transcription factors and machinery are identified, these findings will provide a roadmap to guide investigations into the detailed mechanisms of regulated speckle association.
In addition to demonstrating requirements for the p53 DNA binding and PRD domains, we found that p53-mediated speckle association does not required transcriptional activation. This stands in contrast to theories that speckle association is driven by the act of transcription through nucleation of new speckles at the transcription site. The model that speckles form as a consequence of transcription is supported primarily by correlational data (Brown et al., 2008), or relies on global inhibitors of transcription such as 5,6-dichloro-1-β-D-ribobenzimidazole (DRB)(Guo et al., 2019) or α-amanitin (Hu et al., 2010). DRB and α-amanitin also cause mislocalization of the Malat1 speckle marker (Bernard et al., 2010), leaving it unclear whether the loss of speckle association in these studies was a direct or indirect effect of transcription inhibition. Our study of p53 provides a unique approach to query the requirements of transcription for speckle association, using point mutations to assess requirements for transcription. Based on our findings, we conclude that transcription is neither required nor sufficient for p53-regulated speckle association, and that speckle-association by p53 is facilitated by transcription-independent mechanisms.
Consequences of regulated speckle association
By disrupting nuclear speckles, we demonstrate that speckles are critical for the full induction of p21 by p53 (Figures 3 and S3). Mutagenesis of the p53 PRD supports this conclusion, showing that perturbation of speckle association also results in defective p21 induction (Figures 4H–4P). Moreover, genome-wide experiments indicate that this speckle-associated expression boost exists generally for p53 targets (Figure 7E). Using RNA-FISH studies, we identify a likely mechanism for the observed speckle-associated increases in gene expression: through a build-up of nascent RNAs within transcription sites (Figures 2H–2K and 3F–3K). In contrast, we did not observe dramatic changes in the proportion of cells with active transcription sites when speckles or speckle association were disrupted (Figures 3B and 4K), highlighting that the RNA amount within transcription sites is the key contributor to the speckle-dependent boost in p21 expression by p53. The processes underlying speckle-mediated accumulation of RNA within transcription sites are still under investigation, as are other potential consequences of regulated speckle association. Given the contents of nuclear speckles, speckles could potentially contribute to each step of RNA production, ranging from transcription to splicing, polyadenylation and RNA stabilization. By demonstrating the importance of nuclear speckles for the induction of RNA expression, our study exposes them as a major regulator of gene expression, highlighting a need for future studies into the extent, mechanisms, and consequences of regulated speckle association.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Shelley Berger (bergers@pennmedicine.upenn.edu)
Materials Availability
Plasmids generated from this study are available from the corresponding author upon request.
Data and Code Availability
The genomic datasets generated during this study are available through GEO accession numbers GSE139003 (RNA-seq) and GSE154095 (SON TSA-seq).
The imaging data supporting this study have not been deposited in a public repository due to file sizes, but are available from the corresponding author upon request.
The software, instructions, and code generated by this study used for DNA-FISH and RNA-FISH analysis and analysis of speckle characteristics are available at: https://github.com/arjunrajlaboratory/rajlabimagetools/wiki/Blob-Analyzer https://github.com/katealexander/distanceToSpeckle-semiManual https://github.com/katealexander/processProfilesFromFiji
The instructions and code used for analysis of SON TSA-seq data is available at: https://github.com/katealexander/TSAseq-Alexander2020/tree/master/genomicBins_DiffBind
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Nutlin-3a treatment in IMR90 cells
IMR90 cells from PD 25–35 were grown in DMEM media with 10% FBS under 3% oxygen and 5% carbon dioxide. Nutlin-3a (Sigma, SML0580–5MG) was dissolved in DMSO to a 5mM stock solution and added to IMR90 cells to a final concentration of 5uM. Treatment of IMR90 cells with Nutlin-3a resulted in increased p53 protein levels and induction of p21 expression as has been previously described (Vassilev et al., 2004) (Figure S1A). Treatment with Nutlin-3a also led to strong accumulation of p53 in the nucleus, and consistent with previous reports (Choi et al., 2019), this includes locations proximal to nuclear speckles as identified by the speckle-marking antibody, ab11826 (Figure S1B; (Ilık et al., 2020)). Additionally, ChIP-qPCR shows that p53 binds to its consensus sites at the p21 enhancer and promoter throughout 2, 6, and 9 hours of Nutlin-3a treatment (Figures S1C and S1D), demonstrating persistent interaction of p53 with its p21 binding sites throughout the time points examined.
Doxycyclin-induction of p53 in Saos2 cells
Saos2 cells were grown in RPMI media with 10% FBS under atmospheric oxygen and 5% carbon dioxide. To generate Saos2 cells with doxycycline-inducible p53, we packaged the reverse tetracycline controlled transactivator plasmid (Lenti CMV rtTA3 Blast [w756–1]) into viruses, infected Saos2 cells, and performed selection for cells with stably-integrated rtTA using blasticidin. Saos2-rtTA cells were then infected with viruses containing p53 wild type or mutant plasmids under the control of a TRE3G promoter (pVXL-TRE3G p53) and selected for stable integration using puromycin. For virus generation, we transfected HEK-293T cells with pPAX2, pVSV-G, and the lentiviral vector (rtTA or pVXL-TRE3G) using Lipofectamine2000, as described (Cribbs et al., 2013), and infected Saos2 cells with a 1:1 ratio of virus-containing media and Saos2 growth media. To induce p53 expression, doxycyclin (Sigma, D3447–500MG) was dissolved in water to a 30mM stock solution and added to Saos2 cells at a final concentration of 3uM. Upon induction of wild type or mutant p53 with doxycycline for 3 hours, each mutant showed nuclear localization of p53, similar to wild type p53 subcellular localization (Figures S4A–S4F). Each p53 mutant had a similar expression range to wild type p53, and quantification of the immunofluorescence signal showed that none of the mutants had significantly different p53 levels from wild type (Figure S4G).
METHOD DETAILS
Cloning and mutagenesis
Wild type p53 was cloned into pVLX-TRE3G using XhoI and BamHI restriction enzymes, and mutagenesis PCR was performed using the primers show in Table S2. Mutagenesis PCR products were digested with Dpn1 to eliminate original template, and 1uL of the ensuing digestion was used for transformation into Stbl3 chemically competent E. coli. For TAD1/2 mutants, mutation was performed sequentially, first generating TAD1 constructs, then using mutagenesis PCR to generate TAD1/2 mutants.
ChIP-qPCR
Doxycyclin-treated Saos2 cells (3 hours of 3μM doxycyclin) or Nutlin-3a treated IMR90 cells (PD 25–35; 5μM Nutlin-3a or the equivalent volume of DMSO) were crosslinked in formaldehyde (1% final concentration) for 10 minutes. Crosslinked cells were quenched with glycine (125mM final) for 5 minutes, followed by two washes in ice cold PBS. Nuclei were isolated from approximately 20 million cells as previously described (Shah et al., 2013), and chromatin was sheared to an average size of ~250nt using a Covaris S220. Immunoprecipitation was performed using 500μg (for Δ62–91, null, and WT ChIP-qPCR in Figure S4J) or 350μg (for all other ChIP-qPCR experiments) of sheared chromatin lysate and 3μg of antibodies pre-conjugated to protein G beads (Invitrogen): p53 (D01; Santa Cruz SC-126), FLAG (M2; Sigma-Aldrich F1804), IgG (Millipore 06–271). Lysate and antibody-beads were incubated for 16 hours at 4°C with rotation and then washed four times in wash buffer [50 mM Hepes-HCl (pH 8), 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, and 0.5% N-laurylsarcosine], followed by one wash in ChIP final wash buffer (1× tris-EDTA (TE) Buffer and 50 mM NaCl). Immunoprecipitated DNA was eluted from washed beads, reverse cross-linked overnight, purified, and diluted 1:3 (pulldown) or 1:9 (Input) for qPCR using the two primers within the upstream p53 enhancer ChIP-seq peak at p21, two primers within the p53 promoter peak at p21, and two negative-control primers located between the enhancer and promoter p53 binding regions (see Table S2 for primer sequences).
SON and SRSF1 knockdown
Knockdown of SON in IMR90 cells was carried out by first preparing shRNA-expressing virus in HEK-293T cells transfected pPAX2, pVSV-G, and a plasmid containing an shRNA against SON (Sigma TRC ID: TRCN0000083723) or SRSF1 (Sigma TRC IDs: TRCN0000010592 [KD1] and TRCN0000001093 [KD2]) and Lipofectamine2000, as described (Cribbs et al., 2013). IMR90 cells were treated with non-template control or SON knockdown expressing lentivirus for 48 hours, then trypsinized into media-containing plates. At 48 hours after re-plating following trypsinization, cells were treated with DMSO or Nutlin-3a and harvested in Trizol for RNA, attached to coverglass slides for immunoRNA-FISH, and as cell pellets for Western blotting.
qRT-PCR
For qRT-PCR, cells were lysed and RNA was extracted using QIAzol Lysis Reagent (Qiagen) using two chloroform extractions. RNA was purified using RNeasy Mini Kit (Qiagen) with in-column DNAse treatment. cDNA was synthesized using the High Capacity RNA to cDNA kit (Thermo), and qRT-PCR was performed using Power SYBR Green Master Mix (Thermo). For primers used, see Table S2.
Western blotting
Cells were lysed in RIPA buffer containing 1% NP-40, 150 mM NaCl, 50 mM tris-Cl (pH 8.0), and 1% SDS supplemented with cOmplete, EDTA-free protease inhibitor (11873580001, Roche). Protein concentration was determined by bicinchoninic acid assay (BCA) protein assay (#23227, Life Technologies), after which equal amounts of proteins were loaded and separated by polyacrylamide gel electrophoresis. The following antibodies were used: p53 (OP43, Millipore), p21 (ab109520, abcam), and Gapdh (10R-G109a, Fitzgerald).
immunoDNA-FISH
Oligopaint DNA-FISH probes to p53 targets and control genes were designed across a 50kb region centered on the transcription start site as described (Petrovic et al., 2019). ImmunoDNA-FISH was performed sequentially, first with the standard immunofluorescence protocol, followed by a 10 minute fixation in 4% paraformaldehyde to fix the secondary antibody prior to DNA-FISH. Specifically, cells were fixed in 4% paraformaldehyde for 10 minutes, permeabilized using 0.1% Triton-X-100 in PBS for 10 minutes, incubated in primary antibody overnight at 4C, and incubated in secondary antibody for 1 hour at room temperature and fixed with 10% paraformaldehyde for 10 minutes, with three five-minute washes in PBS after each step. For IMR90 cells, only speckle-labelling antibody against SRRM2 was used (abcam ab11826; 1:200). Note that the speckle-marking antibody, ab11826, was originally raised against spliceosomal extract (Fu and Maniatis, 1990), co-localizes with other speckle markers (Ilık et al., 2020), and has recently been determined to predominantly detect the speckle-resident protein SRRM2 between residues 1,360 and 1,884 (Ilık et al., 2020), although it has been marketed as an antibody against SC35 (also called SRSF2), another speckle-resident protein. For Saos2 cells with dox-induced p53 expression, not all cells expressed p53. Thus, to distinguish p53-positive from p53-negative cells, we used immunofluorescence with a polyclonal p53 antibody (Novus, nb200–171; 1:200). After fixation and PBS washes, DNA-FISH was performed as described (Nguyen and Joyce, 2019). Secondary probes for p53 targets were labelled in Cy5, for control genes were labelled in Cy3. Anti-mouse A488 secondary antibody was used for speckle detection, anti-rabbit A594 secondary antibody was used for p53 detection.
RNA-FISH
RNA-FISH probes were designed using Stellaris probe design software, inputting the p21 exons and introns that were present in all isoforms. For Malat1, we used pre-designed probes (Stellaris). For experiments in Saos2 cells, we used immunoRNA-FISH to detect p53 positive cells using immunofluorescence (as in immunoDNA-FISH above) followed by the RNA-FISH protocol as in (Bartman et al., 2016). p21 exon probes were labelled with Quasar 570, p21 intron probes were labelled with Quasar 610, Malat1 probes were labelled with Quasar 670. Anti-Rabbit A488 antibody was used to detect p53.
Image acquisition
Cells were imaged in a series of 30 optical sections, spaced 0.33 microns apart, that spanned the vertical extent of the cell using a Nikon Ti-E Widefield microscope equipped with appropriate filter sets. We used a 100×1.4NA oil-immersion objective, and a deep-depletion CCD camera cooled to between −70 and −80 degrees Celsius (Pixis 1024, Princeton Instruments).
RNA-seq
IMR90 cells at PD28 were treated with DMSO for 9 hours, or Nutlin-3a for 6, 9, or 12 hours. For RNA isolation, cells were lysed in TRIzol (15596018, Thermo Fisher Scientific) and snap frozen. RNA was then isolated with chloroform extraction, followed by QIAGEN RNeasy Mini Kit isolation (#74106), including DNA digestion with DNAse. Poly(A) + RNA was then isolated using double selection with poly-dT beads (E7490, NEB), and RNA-seq libraries were prepared using a NEB-Next Ultra II Directional Library Prep Kit for Illumina (E7760, NEB). Library sizes were determined on a Bioanalyzer, and concentration determined using NEBNext Lib Quant Kit (E7630, NEB). Libraries were sequenced on an Illumina NextSeq 550, using paired-end sequencing of 42 bases per read.
SON TSA-seq in IMR90 cells
TSA-seq was performed on IMR90 cells treated with DMSO or Nutlin-3a for 6 hours using an antibody against SON (ab121759), and in untreated IMR90 cells as a no primary antibody control. SON TSA-seq was carried out using the protocol for SON TSA-seq 2.0 on attached cells, with Condition E as described ((Zhang et al., 2020); labelling with 50% sucrose, 1:300 tyramide-biotin, and 0.0015% hydrogen peroxide for 30 minutes), with the following minor modifications: DNA was fragmented to an average size of 250bp using a Covaris S220 and the resulting DNA fragments were prepared for sequencing using the NEBNext Ultra II DNA Library Prep Kit for Illumina. Library sizes were determined on a Bioanalyzer, and concentration determined using NEBNext Lib Quant Kit (E7630, NEB). Input and pulldown libraries were sequenced for DMSO- and Nutlin-treated SON TSA-seq and for the no primary control using paired-end sequencing with an Illumina NextSeq550 with 42bp per read (total of 84bp) using the NextSeq 500/550 High Output Kit 75-cycle v2.5 kit.
QUANTIFICATION AND STATISTICAL ANALYSIS
qRT-PCR and ChIP-qPCR
CT values for qRT-PCR and qPCR were normalized using standard curves of ten-fold dilutions for each primer, followed by normalization to Gapdh levels (for qRT-PCR) or input-normalized (for ChIP-qPCR). Experiments were performed in triplicate. p-values were calculated using Students unpaired T-test, and error bars represent standard error.
Western blotting
Western blots were quantified in Fiji, relativized to Gapdh protein levels, and normalized to the 9 hour NTC time point (for Figure 3M; two replicates from blots in Figures 3L and S3G) or the 6 hour NTC time point (Figure S3J; four replicates from blots in Figures 3L and S3G–I). Differential protein level p-values were calculated using Student’s T test, and error bars represent standard error.
Distance to speckle and RNA expression RNA- and DNA-FISH image analysis
We used an in-house graphical user interface MATLAB software (https://github.com/arjunrajlaboratory/rajlabimagetools/wiki (Mellis et al., 2017)), which allows a combination of automation with manual review and speckle selection. For DNA-FISH, loci were thresholded and selected using the software, and the location of edge of the nearest speckle was manually selected and stored. Distances to from each locus to the nearest speckle were then calculated. Speckle distance distribution p-values were calculated using Mann-Whitney-Wilcoxon tests. Percent speckle associated p-values were calculated using Students unpaired T-test, and error bars represent standard error. For RNA-FISH, the intron and exon channels were thresholded individually, and transcription sites (overlap of intron spots with exon spots) and the edge of the nearest speckle were manually selected. Based on predominantly co-transcriptional splicing, the overlap between the exon and intron channels from RNA-FISH data identifies the location of the p21 transcription sites (Figures 2B and 2C, white arrowheads in 2C), enabling measurements of the percentage of cells that contain active transcription sites (Figure 2D). The number of overlapping p21 intron/exon spots per cell ranged from zero to four, giving confidence that this method detected bona fide sites of transcription (cells after S-phase DNA replication can have up to four active transcription sites). To measure the amount of nascent RNA within each active transcription site, we calculated the fluorescence intensity of the exon channel at the site of transcription, and divided by the median fluorescence intensity of the single-molecule exon spots from the same cell. The consequent normalized intensity thus represents an approximation of the number of nascent transcripts within the site of transcription (Figure 2E). Fishers exact calculation of variable independence between transcription site intensity and distance to speckle was calculated using speckle distance cutoff of 0.52μm (5 pixels in imaging data, cutoff based on observed variable dependence), and a transcription site intensity cutoff of 5. We did not assess intron transcription site intensities due to the inability to normalized to single-molecule spots (intron signal is largely exclusive to the transcription sites, which represent multiple RNA molecules). Cells without well-formed speckles which were likely in mitosis and early G1 before speckles re-form were excluded from analysis. See https://github.com/katealexander/distanceToSpeckle-semiManual for instructions and code used to measure the distance to speckles from RNA and DNA-FISH data. For the number of cells and loci counted for each replicate in DNA- and RNA-FISH experiments, see Table S1.
Image analysis of speckle number, area, and perimeter
For assessment of immunofluorescence speckle number, area, and perimeter, we developed an additional module for our in-house MATLAB software (rajlabimagetools; https://github.com/arjunrajlaboratory/rajlabimagetools/wiki), called Blob Analyzer (https://github.com/arjunrajlaboratory/rajlabimagetools/wiki/Blob-Analyzer). Blob Analyzer utilizes the built-in MATLAB function, bwselect, to define speckle edges from immunofluorescence data, enabling extraction of the number, area, perimeter, extent (measure of the proportion of the speckle that fits in a bounding box), and eccentricity (measure of speckle elongation) of nuclear speckles. P-values for differences in speckle characteristics were calculated using Mann-Whitney-Wilcoxon tests.
Analysis of p53 fluorescent intensity at transcription sites and speckles
Intensity profiles for SRRM2, introns, exons, and p53 were collected in Fiji (Schindelin et al., 2012) by drawing a 6.5 micron line centered on the transcription site (overlap of intron and exon spot) at a 90 degree angle to the nearest speckle. Profiles were max-min normalized between 0 and 1, and distances between p53 local maxima and the most intense SRRM2 location or the transcription site (center of profile) were calculated in Python (Figure S4L; local maxima analysis). As another form of quantification, the average relative intensity (max-min normalized within each profile) of p53 within speckles (defined as values > 0.5 in max-min normalized SRRM2 profiles) and within transcription sites (defined as the five pixels within the center of the profile) were divided by the average intensity of p53 outside of speckles or transcription sites, respectively (Figure S4M; average intensity analysis). Raw profiles collected, code used to analyze, and analysis instructions can be found at https://github.com/katealexander/processProfilesFromFiji.
Gene density definition
Genomic regions were defined as gene dense or gene sparse based on whether there were greater or fewer than 26 unique genes/Mb (two standard deviations above the genome-wide mean density) as calculated using a 1Mb sliding window across the genome.
RNA-seq data analysis
RNA-seq data was aligned to the reference human genome assembly, GRC37/hg19, using the splice-aware STAR alignment (Dobin et al., 2013). PCR duplicates were removed using Picard, and reads for each gene with RefSeq annotations were counted using htseq-counts (Anders et al., 2015). Normalized counts and significant differences were calculated using Deseq2 (Love et al., 2014). Raw and processed data are available through GEO, accession number: GSE139003.
Defining IMR90 p53 targets
IMR90 p53 targets were functionally defined as genes that significantly increased expression (padj < 0.05) in RNA-seq data from IMR90 cells treated with Nutlin-3a for 6, 9, or 12 hours (included as target if significant at any time point), and were within 200kb of a p53 ChIP-seq peak (from GSM1418970; (Sammons et al., 2015)). See https://github.com/katealexander/TSAseq-Alexander2020/tree/master/IMR90_p53targets for detailed instructions, files, and code used.
SON TSA-seq differential analysis
SON TSA-seq reads were aligned to the human reference genome assembly, GRC37/hg19, using Bowtie2 allowing for a maximum fragment size of 1000 base pairs (Langmead and Salzberg, 2012). PCR duplicates were removed using Picard, and SON TSA-seq signal was quantified over sliding windows of various sizes (25kb, 50kb, and 100kb windows slid by 1/10th of the window size) using DiffBind with input subtraction (Wu et al., 2015). Correlations between replicates and relationship to mean speckle distance measured by DNA-FISH was assessed for each window size using normalized counts from DiffBind analysis. Differential 25kb, 50kb, and 100kb windows identified using DiffBind were merged using BEDTools (Quinlan and Hall, 2010), resulting in differential domains. Genes within differential domains were then extracted using Python. Based on comparison with DNA-FISH results, an adjusted p-value of 0.01 was used as a cutoff to define genes that significantly increased SON signal, while genes falling outside of domains with an adjusted p-value of less than 0.1 were used as a cutoff to define genes that did not change SON signal. Genes within domains that had an adjusted p-value between 0.01 and 0.1 were considered to be of borderline significance, and were excluded from analyses comparing IMR90 p53 targets that increased SON signal versus IMR90 p53 that did not increase SON signal. See https://github.com/katealexander/TSAseq-Alexander2020/tree/master/genomicBins_DiffBind for detailed instruction and code used. Raw and processed data are available through GEO, accession number: GSE154095.
Logistic regression
Logistic regression was used to measure the predictive capacity of the number of nearby p53 peaks (from GSM1418970; (Sammons et al., 2015)), gene density, HiC subcompartments (Xiong and Ma, 2019), and SON TSA-seq signal at baseline on whether p53 targets did or did not increase SON signal. See https://github.com/katealexander/TSAseq-Alexander2020/tree/master/logisticRegression for detailed instructions, files, and code used.
Functional analysis
Using IMR90 p53 targets that did (303 targets within SON TSA-seq domains with padj < 0.01) or did not (1189 targets not within SON TSA-seq domains with padj > 0.1), we performed GO and KEGG analysis using the R package clusterProfiler function compareCluster (Yu et al., 2012). See https://github.com/katealexander/TSAseq-Alexander2020/tree/master/clusterProfiler for detailed instructions and files used.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-p53 (Ab-6) (Pantropic) Mouse mAb (DO1) | Millipore | Cat#OP43; RRID: AB_213402 |
| p21 antibody [EPR362] | Abcam | Cat#ab109520; RRID: AB_10860537 |
| GAPDH antibody | Fitzgerald Industries International | Cat#10R-G109a; RRID: AB_1285808 |
| Mouse Anti-SC35 Monoclonal antibody (against SRRM2) | Abcam; also see Ilık et al., 2020 | Cat#ab11826; RRID: AB_298608 |
| p53 antibody | Novus | Cat#NB200-171; RRID: AB_10003046 |
| SON antibody | Abcam | Cat#ab121759; RRID: AB_11132447 |
| Bacterial and Virus Strains | ||
| Stbl3 Chemically Competent E. coli | Invitrogen | Cat#C7373-03 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nutlin-3a | Sigma | Cat#SML0580-5MG |
| Doxycycline | Sigma | Cat#D3447-500MG |
| Blasticidin | Thermo | Cat#A1113903 |
| Puromycin | Thermo | Cat#A1113803 |
| Critical Commercial Assays | ||
| QIAGEN RNeasy Mini Kit isolation | QIAGEN | Cat#74106 |
| NEBNext® Poly(A) mRNA Magnetic Isolation Module | NEB | Cat#E7490 |
| NEBNext® UltraII Directional RNA Library Prep Kit | NEB | Cat#E7760 |
| NEBNext Ultra II DNA Library Prep Kit | NEB | Cat#E7645 |
| NEBNext Lib Quant Kit | NEB | Cat#E7630 |
| High-Capacity RNA to cDNA kit | Thermo | Cat#4368813 |
| Deposited Data | ||
| SON TSA-seq raw and analyzed data | this paper | GSE154095 |
| RNA-seq raw and analyzed data | this paper | GSE139003 |
| Experimental Models: Cell Lines | ||
| Human: Passage 25–35 IMR90 primary lung fibroblast cell line | ATCC | Cat#CL186 |
| Human: Saos2 cells | ATCC | Cat#HTB-85 |
| Human: 293T cells | ATCC | Cat#CRL-3216 |
| Oligonucleotides | ||
| Primers for ChIP-qPCR, see Table S2 | this paper | N/A |
| Primers for qRT-PCR, see Table S2 | this paper | N/A |
| Recombinant DNA | ||
| Lenti CMV rtTA3 Blast (w756-1) | Eric Campeau Addgene | Cat#26429; RRID: Addgene_26429 |
| pVXL-TRE3G | Takara | Cat#631187 |
| pVXL-TRE3G p53 wild type | this paper | N/A |
| pVXL-TRE3G p53 L22Q, W23S | this paper | N/A |
| pVXL-TRE3G p53 W53Q, F54S | this paper | N/A |
| pVXL-TRE3G p53 L22Q, W23S, W53Q, F54S | this paper | N/A |
| pVXL-TRE3G p53 Δ62–77 | this paper | N/A |
| pVXL-TRE3G p53 Δ62–91 | this paper | N/A |
| pVXL-TRE3G p53 R273H | this paper | N/A |
| pVXL-TRE3G p53 L344P | this paper | N/A |
| pVXL-TRE3G p53 370* | this paper | N/A |
| psPAX2 | Didier Trono Addgene | Cat#12260; RRID: Addgene_12260 |
| pVSV-G | Didier Trono Addgene | Cat#12259; RRID: Addgene_12259 |
| pLKO.1 SON shRNA | Sigma | TRC ID: TRCN0000083723 |
| pLKO.1 SRSF1 shRNA1 | Sigma | TRC ID: TRCN0000010592 |
| pLKO.1 SRSF1 shRNA2 | Sigma | TRC ID: TRCN0000001093 |
| Software and Algorithms | ||
| Rajlabimagetools for analysis of spots from RNA- and DNA-FISH data | Mellis et al., 2017 | https://github.com/arjunrajlaboratory/rajlabimagetools/wiki |
| Blob analysis module of rajlabimagetools for analysis of nuclear speckles | this paper | https://github.com/arjunrajlaboratory/rajlabimagetools/wiki/Blob-Analyzer |
| Use of rajlabimage tools for semi-manual measurement of loci distance to speckle from RNA-FISH and immunoDNA-FISH data | this paper | https://github.com/katealexander/distanceToSpeckle-semiManual |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR/releases |
| Picard | “Picard Toolkit.” 2019. Broad Institute, GitHub Repository. | https://broadinstitute.github.io/picard/ |
| htseq | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/ |
| DESeq2 | Love et al., 2014 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| DiffBind | Wu et al., 2015 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| Use of DiffBind for SON TSA-seq of sliding windows | this paper | https://github.com/katealexander/TSAseq-Alexander2020/tree/master/genomicBins_DiffBind |
| BEDTools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| clusterProfiler | Yu et al., 2012 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| Fiji | Schindelin et al., 2012 | https://imagej.net/Fiji |
Highlights:
The p53 transcription factor drives speckle association of a subset of target genes
p53-mediated speckle association of target genes boosts RNA expression
Regulated speckle association by p53 requires DNA binding, but not transcription
Speckle-associating and non-associating p53 gene targets are functionally distinct
ACKNOWLEGEMENTS
S.L.B. acknowledges support from NIH R01CA078831. K.A.A. acknowledges support from NIH F32CA221010.
A.R. acknowledges support from NIH U01DK127405, NIH 4DN U01 HL129998, and NIH Center for Photogenomics. A.B. acknowledges health grants R01 GM58460 and U54 DK107965. E.F.J. acknowledges support from NIH/NIGMS R35GM128903.
Footnotes
DECLARATION OF INTERESTS
A.R. receives royalties from LGC/Biosearch Technologies related to Stellaris RNA-FISH.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adzhubei AA, Sternberg MJ, and Makarov AA (2013). Polyproline-II helix in proteins: structure and function. J Mol Biol 425, 2100–2132. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W. (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptiste N, Friedlander P, Chen X, and Prives C. (2002). The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene 21, 9–21. [DOI] [PubMed] [Google Scholar]
- Bartman CR, Hsu SC, Hsiung CC, Raj A, and Blobel GA (2016). Enhancer Regulation of Transcriptional Bursting Parameters Revealed by Forced Chromatin Looping. Mol Cell 62, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z, Zhang MQ, Sedel F, Jourdren L, Coulpier F, et al. (2010). A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J 29, 3082–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Green J, das Neves RP, Wallace HA, Smith AJ, Hughes J, Gray N, Taylor S, Wood WG, Higgs DR, et al. (2008). Association between active genes occurs at nuclear speckles and is modulated by chromatin environment. J Cell Biol 182, 1083–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Leach J, Reittie JE, Atzberger A, Lee-Prudhoe J, Wood WG, Higgs DR, Iborra FJ, and Buckle VJ (2006). Coregulated human globin genes are frequently in spatial proximity when active. J Cell Biol 172, 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catizone AN, Good CR, Alexander KA, Berger SL, and Sammons MA (2019). Comparison of genotoxic versus nongenotoxic stabilization of p53 provides insight into parallel stress-responsive transcriptional networks. Cell Cycle 18, 809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang Y, Wang Y, Zhang L, Brinkman EK, Adam SA, Goldman R, van Steensel B, Ma J, and Belmont AS (2018). Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J Cell Biol 217, 4025–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Chen M, Cryns VL, and Anderson RA (2019). A nuclear phosphoinositide kinase complex regulates p53. Nat Cell Biol 21, 462–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, Cremer M, Hübner B, Strickfaden H, Smeets D, Popken J, Sterr M, Markaki Y, Rippe K, and Cremer C. (2015). The 4D nucleome: Evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett 589, 2931–2943. [DOI] [PubMed] [Google Scholar]
- Cribbs AP, Kennedy A, Gregory B, and Brennan FM (2013). Simplified production and concentration of lentiviral vectors to achieve high transduction in primary human T cells. BMC Biotechnol 13, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson S, Macpherson N, and Mitchell JA (2013). Nuclear organization of RNA polymerase II transcription. Biochem Cell Biol 91, 22–30. [DOI] [PubMed] [Google Scholar]
- Davison TS, Yin P, Nie E, Kay C, and Arrowsmith CH (1998). Characterization of the oligomerization defects of two p53 mutants found in families with Li-Fraumeni and Li-Fraumeni-like syndrome. Oncogene 17, 651–656. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornan D, Shimizu H, Burch L, Smith AJ, and Hupp TR (2003). The proline repeat domain of p53 binds directly to the transcriptional coactivator p300 and allosterically controls DNA-dependent acetylation of p53. Mol Cell Biol 23, 8846–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, and Vogelstein B. (1993). WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Fei J, Jadaliha M, Harmon TS, Li ITS, Hua B, Hao Q, Holehouse AS, Reyer M, Sun Q, Freier SM, et al. (2017). Quantitative analysis of multilayer organization of proteins and RNA in nuclear speckles at super resolution. J Cell Sci 130, 4180–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD, and Maniatis T. (1990). Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature 343, 437–441. [DOI] [PubMed] [Google Scholar]
- Galganski L, Urbanek MO, and Krzyzosiak WJ (2017). Nuclear speckles: molecular organization, biological function and role in disease. Nucleic Acids Res 45, 10350–10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall’Agnese A, Hannett NM, Spille JH, Afeyan LK, Zamudio AV, Shrinivas K, et al. (2019). Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Kireev I, Plutz M, Ashourian N, and Belmont AS (2009). Large-scale chromatin structure of inducible genes: transcription on a condensed, linear template. J Cell Biol 185, 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Plutz M, and Belmont AS (2010). Hsp70 gene association with nuclear speckles is Hsp70 promoter specific. J Cell Biol 191, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilık İA, Malszycki M, Lübke AK, Schade C, Meierhofer D, and Aktaş T. (2020). SON and SRRM2 form nuclear speckles in human cells. bioRxiv, 2020.2006.2019.160762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger AC, Ang HC, and Fersht AR (2006). Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc Natl Acad Sci U S A 103, 15056–15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimian A, Ahmadi Y, and Yousefi B. (2016). Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst) 42, 63–71. [DOI] [PubMed] [Google Scholar]
- Kastenhuber ER, and Lowe SW (2017). Putting p53 in Context. Cell 170, 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna N, Hu Y, and Belmont AS (2014). HSP70 transgene directed motion to nuclear speckles facilitates heat shock activation. Curr Biol 24, 1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Venkata NC, Hernandez Gonzalez GA, Khanna N, and Belmont AS (2019). Gene expression amplification by nuclear speckle association. The Journal of Cell Biology 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, and Shakked Z. (2006). Structural basis of DNA recognition by p53 tetramers. Mol Cell 22, 741–753. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomax ME, Barnes DM, Hupp TR, Picksley SM, and Camplejohn RS (1998). Characterization of p53 oligomerization domain mutations isolated from Li-Fraumeni and Li-Fraumeni like family members. Oncogene 17, 643–649. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellis IA, Gupte R, Raj A, and Rouhanifard SH (2017). Visualizing adenosine-to-inosine RNA editing in single mammalian cells. Nat Methods 14, 801–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagawa R, Tano K, Mizuno R, Nakamura Y, Ijiri K, Rakwal R, Shibato J, Masuo Y, Mayeda A, Hirose T, et al. (2012). Identification of cis- and trans-acting factors involved in the localization of MALAT-1 noncoding RNA to nuclear speckles. RNA 18, 738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll UM, and Petrenko O. (2003). The MDM2-p53 interaction. Mol Cancer Res 1, 1001–1008. [PubMed] [Google Scholar]
- Nguyen SC, and Joyce EF (2019). Programmable Chromosome Painting with Oligopaints. Methods Mol Biol 2038, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic J, Zhou Y, Fasolino M, Goldman N, Schwartz GW, Mumbach MR, Nguyen SC, Rome KS, Sela Y, Zapataro Z, et al. (2019). Oncogenic Notch Promotes Long-Range Regulatory Interactions within Hyperconnected 3D Cliques. Mol Cell 73, 1174–1190.e1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, et al. (2018). Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 174, 744–757.e724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj N, and Attardi LD (2017). The Transactivation Domains of the p53 Protein. Cold Spring Harb Perspect Med 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh N, Sakamoto C, Hagiwara M, Agredano-Moreno LT, Jiménez-García LF, and Nakao M. (2012). The distribution of phosphorylated SR proteins and alternative splicing are regulated by RANBP2. Mol Biol Cell 23, 1115–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammons MA, Zhu J, Drake AM, and Berger SL (2015). TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res 25, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, et al. (2013). Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev 27, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Takata H, Shibahara K, Bubulya A, and Bubulya PA (2010). Son is essential for nuclear speckle organization and cell cycle progression. Mol Biol Cell 21, 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevtsov SP, and Dundr M. (2011). Nucleation of nuclear bodies by RNA. Nat Cell Biol 13, 167–173. [DOI] [PubMed] [Google Scholar]
- Shopland LS, Johnson CV, Byron M, McNeil J, and Lawrence JB (2003). Clustering of multiple specific genes and gene-rich R-bands around SC-35 domains: evidence for local euchromatic neighborhoods. J Cell Biol 162, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasan I, Sustackova G, Zhang L, Kim J, Sivaguru M, HamediRad M, Wang Y, Genova J, Ma J, Belmont AS, et al. (2018). CRISPR/Cas9-mediated knock-in of an optimized TetO repeat for live cell imaging of endogenous loci. Nucleic Acids Res 46, e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi V, Song DY, Zong X, Shevtsov SP, Hearn S, Fu XD, Dundr M, and Prasanth KV (2012). SRSF1 regulates the assembly of pre-mRNA processing factors in nuclear speckles. Mol Biol Cell 23, 3694–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- Wells M, Tidow H, Rutherford TJ, Markwick P, Jensen MR, Mylonas E, Svergun DI, Blackledge M, and Fersht AR (2008). Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci U S A 105, 5762–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DY, Bittencourt D, Stallcup MR, and Siegmund KD (2015). Identifying differential transcription factor binding in ChIP-seq. Front Genet 6, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong K, and Ma J. (2019). Revealing Hi-C subcompartments by imputing inter-chromosomal chromatin interactions. Nat Commun 10, 5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai Z, Blandino G, Schneider C, and Del Sal G. (2002). The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 419, 853–857. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang Y, Chen Y, Gholamalamdari O, Wang Y, Ma J, and Belmont AS (2020). TSA-Seq reveals a largely “hardwired” genome organization relative to nuclear speckles with small position changes tightly correlated with gene expression changes. bioRxiv, 824433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP, and Xiao ZX (2002). The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 419, 849–853. [DOI] [PubMed] [Google Scholar]
- Zilfou JT, Hoffman WH, Sank M, George DL, and Murphy M. (2001). The corepressor mSin3a interacts with the proline-rich domain of p53 and protects p53 from proteasome-mediated degradation. Mol Cell Biol 21, 3974–3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genomic datasets generated during this study are available through GEO accession numbers GSE139003 (RNA-seq) and GSE154095 (SON TSA-seq).
The imaging data supporting this study have not been deposited in a public repository due to file sizes, but are available from the corresponding author upon request.
The software, instructions, and code generated by this study used for DNA-FISH and RNA-FISH analysis and analysis of speckle characteristics are available at: https://github.com/arjunrajlaboratory/rajlabimagetools/wiki/Blob-Analyzer https://github.com/katealexander/distanceToSpeckle-semiManual https://github.com/katealexander/processProfilesFromFiji
The instructions and code used for analysis of SON TSA-seq data is available at: https://github.com/katealexander/TSAseq-Alexander2020/tree/master/genomicBins_DiffBind