

Abstract
Fluorine 18 (18F) fluorthanatrace (18F-FTT) is a PET radiotracer for imaging poly (adenosine diphosphate–ribose) polymerase-1 (PARP-1), an important target for a class of drugs known as PARP inhibitors, or PARPi. This article describes the stepwise development of this radiotracer from its design and preclinical evaluation to the first-in-human imaging studies and the initial validation of 18F-FTT as an imaging-based biomarker for measuring PARP-1 expression levels in patients with breast and ovarian cancer. A detailed discussion on the preparation and submission of an exploratory investigational new drug application to the Food and Drug Administration is also provided. Additionally, this review highlights the need and future plans for identifying a commercialization strategy to overcome the major financial barriers that exist when conducting the multicenter clinical trials needed for approval in the new drug application process. The goal of this article is to provide a road map that scientists and clinicians can follow for the successful clinical translation of a PET radiotracer developed in an academic setting.
Keywords: Molecular Imaging-Cancer, PET, Breast, Genital/Reproductive, Chemistry, Radiotracer Development, PARPi, 18F-FTT, Investigational New Drug
© RSNA, 2022
Keywords: Molecular Imaging-Cancer, PET, Breast, Genital/Reproductive, Chemistry, Radiotracer Development, PARPi, 18F-FTT, Investigational New Drug
Summary
The process to develop and evaluate fluorine 18 fluorthanatrace as a PET radiotracer for cancer imaging requires substantial time and resources, but it may improve the diagnosis and clinical management of cancer.
Essentials
■ This review illustrates the process of poly (adenosine diphosphate–ribose) polymerase inhibitor (PARPi) radiotracer development from ligand identification, to preclinical evaluation, and to early phase clinical trials.
■ Fluorine 18 fluorthanatrace(18F-FTT) showed PARP-1 specific uptake in preclinical models of cancer.
■ In both ovarian and breast cancer trials, the uptake of 18F-FTT varied among study volunteers, and the uptake was blocked by PARPi treatment.
Introduction
PET imaging has several distinct properties that make it a powerful tool in the age of precision medicine (1–3). This noninvasive imaging technique can provide information on disease burden for the whole body. Additionally, imaging can provide results on drug uptake in the targeted and healthy tissues and can enable early assessment of the risk and benefit of the treatment (4). For example, the combination of an imaging agent with a traditional pharmaceutical or radiation therapy (ie, a theranostic pair) allows physicians to tailor the treatment on the basis of an individual’s response (5–7). Furthermore, the development of improved PET imaging capabilities will provide the opportunity to measure the complete pharmacokinetics and pharmacodynamics of targeted cancer drugs by using positron-emitting radionuclide-labeled drugs or analogs.
In the past decade, PET radiotracers have shown promise in preclinical models of cancer, including prostate, breast, and many other types, and some have crossed the threshold to clinical translation to obtain regulatory approval from the Food and Drug Administration (FDA), such as the recent examples of fluorine 18 (18F) fluoroestradiol (Cerianna) for breast cancer and 18F fluciclovine (Axumin) for prostate cancer. However, the steps needed for the identification of a new radiotracer for translational PET imaging studies is an expensive and time-consuming process (8). Hung (9) and Harapanhalli (10) published review articles on the current state and regulatory consideration for PET radiotracer development. For PET radiotracers that are generally considered safe and effective, such as a radioactive version of a known substance or drug, clinical trials may be conducted under an institutional approval via the Radioactive Drug Research Committee (11). Mosessian et al (12) have also presented an example of an investigational new drug (IND) submission and early clinical evaluation of 18F fluoro-arabinofuranosylcytosine.
In recent years, there have been a number of PET radiotracers, including 18F PARP inhibitor (PARPi), 18F fluorthanatrace (18F-FTT), and 18F olaparib, that have been developed for imaging poly (adenosine diphosphate–ribose) polymerase-1 (PARP-1) expression levels in patients with cancer. The development and evaluation of these radiotracers were reviewed by Puentes et al (13). The authors discussed 10 radiotracers targeting PARP-1 presented in the literature in the past 5 years. Of these, only 18F-FTT has been used in translational imaging studies in a variety of tumor types, including hepatocellular carcinoma and breast and ovarian cancer (14–16). In this report, we present a detailed description of the development of 18F-FTT as a PET-based biomarker for imaging PARP-1 expression levels in patients with cancer. To illustrate this process, we have focused on the following steps: (a) identification of PARP-1 as a target for PET imaging studies, (b) identification of lead radiotracers, (c) preclinical evaluation and validation of 18F-FTT as a PET radiotracer for imaging PARP-1, (d) steps in the regulatory process to enable first-in-human studies, and (e) single-site (phase 0 or 1) and (f) multicenter (phase 2) clinical trials. A flowchart demonstrating the steps for PET radiotracer development and evaluation of 18F-FTT is shown in Figure 1.
Figure 1:

Flowchart illustrates the process of radiotracer development from target selection, evaluation, investigational new drug (IND) enabling studies, IND application, and clinical trials. CMC = chemistry, manufacturing, and controls; ICH = International Conference on Harmonization; IRB = institutional review board.
18F-FTT: PET Radiotracer Development Process
Step 1: Identification of PARP-1 as a Target for PET Imaging Studies
PARP-1 is a 116 kDa protein that is widely known for its role in DNA repair mechanisms. Specifically, PARP-1 recognizes and associates with damaged DNA and catalyzes formation of poly-adenosine diphosphate–ribose, which is necessary for recruitment of DNA repair proteins (18). Increased PARP-1 expression has been associated with several pathologic conditions, and an overexpression of PARP-1 has been reported across a diverse set of carcinomas (17–20). For example, overexpression of PARP-1 in germline or somatic mutations of BRCA1 and BRCA2 (breast cancer susceptibility gene) leads to the development of homologous recombination–deficient cancers (21–23). Patients who have these BRCA1 or BRCA2 gene mutations have been shown to have a favorable response to PARPi, and four different PARPi have been approved by the FDA for patients with ovarian and breast cancer. Additionally, PARPi are approved as maintenance therapy in women with advanced epithelial ovarian, fallopian tube, or primary peritoneal carcinoma who are in complete or partial response to first-line platinum chemotherapy independent of BRCA status (24). Ongoing studies of PARPi monotherapy or combination therapy are being tested in multiple other solid tumors, such as pancreatic (25), brain (26), and prostate (17). Despite the success, the mechanism of resistance to PARPi treatment remains unclear, and the timing and duration of treatment still require further investigation (27). This is evidenced by the fact that clinical trials have shown that only approximately 50% of patients with BRCA1/2 mutations respond to treatment with a PARPi (28). This observation, in combination with the high cost associated with PARPi and potential toxicity of the drug (ie, hematologic toxicities), has raised questions regarding the cost-effectiveness of PARPi (17,29). Therefore, a biomarker capable of identifying patients on the basis of tumor expression of PARP-1, the molecular target of PARPi, could guide patient selection and improve function and cost-effectiveness of PARPi in the treatment of patients with breast, ovarian, and other solid tumors. A PET-based biomarker for measuring PARP-1 levels in patients with cancer offers the advantage of measuring drug-PARP-1 binding in vivo across the full burden of disease and measuring potential heterogeneity in PARP-1 expression. This measure would be highly complementary to current selection strategies based largely on genomics rather than tumor expression of the drug target (30).
Step 2: Identification of a Lead Compound for PET Radiotracer Development
A common mechanism used in PET radiotracer development is to select a drug known for interacting with the protein of interest and prepare an analog that can be radiolabeled with 18F, the preferred radionuclide for translational PET imaging studies. For PARP-1, there were four PARPi showing efficacy in clinical trials that were subsequently approved by the FDA for the treatment of cancer: olaparib, rucaparib, niraparib, and talazoparib. The design of 18F-FTT was based on rucaparib (Fig 2), the second PARPi to receive FDA approval for the treatment of patients with ovarian cancer harboring mutations in the BRCA1/2. Iodine 125 (125I) KX-1, another structural analog of rucaparib, is a second radiotracer that was used in the validation of 18F-FTT. This radiotracer has been used in in vitro studies measuring PARP-1 expression levels via homogenate radioligand-binding assays or in vitro autoradiography and is important for cross-validating 18F-FTT PET results against assays of tissue samples from surgery or biopsy (31).
Figure 2:

Structures of rucaparib, 18F fluorthanatrace (18F-FTT) and 125I KX-1. Fluoroethyl and iodo moieties were added to rucaparib scaffold to enable radiofluorination and radioiodination. Both radioactive analogs exhibit high binding affinities toward PARP-1. The PET radiotracer, 18F-FTT, was used for the preclinical and clinical imaging studies, while 125I KX-1 (18F-FTT analog) was used for PET tracer evaluation. FDA = Food and Drug Administration, PARPi = poly (adenosine diphosphate–ribose) polymerase inhibitor.
The radiosynthesis of 18F-FTT involves the simple one-step displacement of the corresponding tosylate precursor with 18F fluoride followed by high-performance liquid chromatography purification (Fig 3). This radiosynthesis method was chosen because it could be readily adapted to commercial PET synthesis modules. Routine radiosynthesis yields are 18%–47% (corrected to start of synthesis), and the radiotracer is produced in high molar activity (0.059–0.367 GBq/µmol or 2.17–13.58 Ci/µmol decay corrected to end of synthesis), which is suitable for both preclinical and clinical imaging studies. Early radiochemistry development included testing the radiolabeling at different automated synthesis platforms. The robust chemistry strategy laid the foundation for later-phase clinical trials and commercialization requiring tracer supplies from multiple sites.
Figure 3:
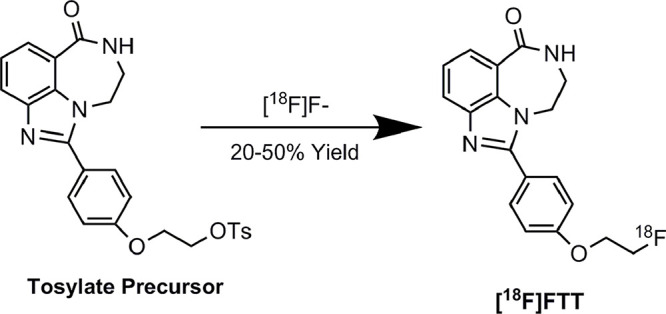
Radiosynthesis of 18F fluorthanatrace (18F-FTT). Radiofluorination was carried out with tosylate precursor. After the drying of 18F F−, the reaction was carried out at 100°C for 10 minutes, followed by semipreparative high-performance liquid chromatography purification. The process was developed on automated synthesis module for clinical studies. The overall reaction time is around 60 minutes with yield between 20% and 50%.
Step 3: In Vitro and Preclinical Imaging Studies
A preclinical imaging study was initially conducted in a murine model of breast cancer; this preliminary study indicated that the high uptake of 18F-FTT could be blocked by olaparib. In a follow-up study, preclinical imaging studies were performed in mice bearing a xenograft of MDA-MB-231 cells, a triple-negative breast cancer cell line (32). Blocking studies were conducted with either olaparib or iniparib, a drug initially thought to be a PARPi but subsequently shown to have no PARP inhibitory potency (15). The imaging studies revealed that olaparib was able to block the uptake of 18F-FTT, whereas iniparib had no effect on radiotracer uptake in the breast tumor xenograft (Fig 4). These studies indicate that 18F-FTT has the potential to measure target engagement of PARPi, a key step in determining the therapeutic efficacy of a drug.
Figure 4:

Initial preclinical PET studies of 18F fluorthanatrace (18F-FTT) demonstrate the ability of the radiotracer to measure occupancy of a poly (adenosine diphosphate–ribose) polymerase inhibitor (PARPi). MDA-MB-231 tumor–bearing female athymic nude mice (n = 25) were injected with 18F-FTT (11.4 MBq ± 0.5), showing baseline tumor uptake (indicated by the circle showing the position of the tumor). The tracer uptake was blocked by the administration of olaparib (n = 14), a known PARPi, but not by iniparib (n = 11), as shown on the images (left panel) and the quantitative measurement as presented as percentage of injected dose per milliliter (right panel). Iniparib has no inhibitory effect on PARP-1 activity.
The specificity of 18F-FTT for PARP-1 versus PARP-2 was confirmed in an in vitro study using fibroblast cells where either PARP-1 or PARP-2 was knocked out using the gene editing technique, CRISPR (Fig 5) (33). A preclinical imaging study was also conducted in three different human breast tumor cell lines: HCC1937 cells (BRCA1 mutation at codon 1755 that forms a truncated BRCA1 protein); MDA-MB-231, a triple-negative breast cancer cell line with wild-type BRCA1/2; and MCF7, a luminal A cell line with a wild-type BRCA1/2 and positive for the estrogen receptor (31). Western blot analysis revealed that the three cell lines have different expression levels of PARP-1 in the rank order of: HCC1937 > MDA-MB-231 > MCF-7 cells. In vivo preclinical studies revealed that the uptake of 18F-FTT paralleled the expression levels of PARP-1 in these cells, which presented the first set of experimental data suggesting that 18F-FTT is capable of measuring PARP-1 expression in solid tumors in vivo. A subsequent study in mice bearing a xenograft of patient-derived ovarian cancer indicated that 18F-FTT uptake was blocked by olaparib, lending further support that 18F-FTT is capable of measuring target engagement of clinically used PARPi (31).
Figure 5:

In vitro and in vivo studies demonstrate the ability of the analog of 18F fluorthanatrace (18F-FTT), 125I-KX1, to provide a measurement of poly (adenosine diphosphate–ribose) polymerase-1 (PARP-1) expression levels in tumor cells or a solid tumor. In vitro cell uptake study was performed using MEF PARP-1 KO−/−, MEF PAR2 KO −/−, and MEF wild-type cell. The cells were incubated with 125I-KX1, and the uptake was abrogated by the knockout of PARP-1 activity, shown in the top left. 125I-KX1 uptake was not decreased in the wild-type and the PARP-2 knockout cell line, showing the specificity of the 18F-FTT uptake to PARP-1 expression. PARP-1 activity was measured in three different cell lines, HCC1937 (BRCA mutation), MD-MBA-231 (triple negative), and MCF (ER positive). In vivo studies using xenografts implanted with three different cell lines revealed that the uptake of 125I-KX1, as shown in tumor to muscle ratio (TMR), paralleled the expression levels of PARP-1 in these cells. AU = arbitrary unit, BRCA = breast cancer gene, ER = estrogen receptor, KO = knockout, MEF = mouse embryonic fibroblast.
Step 4: Preparation of an IND Application
After successful preclinical characterization of 18F-FTT, the next step was to evaluate this radiotracer in patients with cancer, a process that required FDA approval via the filing of an exploratory IND application. There were several steps that had to be completed before filing an exploratory IND application: acute toxicology study to assess the toxicity of the agent using a dose multiple of the injected mass, clinical study protocol development, precursor characterization, radiosynthesis optimization and validation, preclinical dosimetry study, and IND submission and maintenance. These are discussed below.
Acute toxicity studies.—Pharmacology and toxicity information was derived from preclinical safety testing performed in animals followed by tissue histologic studies. The exploratory IND is considered a “microdose proof-of-concept” study that involves limited human exposure to an agent and has no therapeutic or diagnostic intent. A single dose tolerability study used for the exploratory IND application was performed for a cold drug (19F-FTT) in both male and female Sprague-Dawley rats (15). The route of administration for the toxicity studies must be the same as that of the radiotracer. In this toxicology study, animals were injected with the vehicle or 0.865 mg/kg of cold FTT and observed for 15 days after dosing, with an interim necropsy performed on day 2. End points included an evaluation of body weight, clinical pathologic findings, hematologic findings, clinical chemistries, and coagulation. To establish a margin of safety, the study must use at least 100 times the proposed human mass dose (ie, dose multiple = 100) and confirm that this tested dose does not induce adverse effects in the experimental animals. The study demonstrated that cold FTT was well-tolerated in the studied animals. Up to 750 times of 19F-FTT was tested in the toxicity studies, and the results of the toxicity study indicated a high safety margin for this radiotracer. Less or equal to 10 µg of drug mass was used for the clinical trials.
Clinical study protocol development.—The protocol included the following key elements: (a) introduction, background, and rationale for the study of the drug in a particular human population; (b) summary of preclinical data to support the safety and justification for human studies to be conducted; (c) previous human experience on the drug or related compounds, if available; and (d) investigational plan with description of the studied agent, risks and benefits, study design, statistical analyses, safety oversight and monitoring, record keeping, and confidentiality and privacy considerations (34). Once the initial protocol was finalized, additional documents, including informed consent form and case report forms, were created to complete the necessary regulatory submission package. Other information needed for local regulatory submission consisted of documents specific to the study, such as advertising materials and patient questionnaires or instructions. In an academic setting, the institutional regulatory authorities included an Institutional Review Board/Ethics Committee, the Radiation Research Safety Committee, and, in the case of 18F-FTT, Cancer Center Scientific Review Committee.
Precursor characterization and radiosynthesis optimization and validation.—To demonstrate the manufacturing site’s ability to supply a PET drug for the clinical trial and to comply with the Code of Federal Regulations title 21 Part 212 (21CFR Part 212), Current Good Manufacturing Practice for PET drug production in the late phase clinical trials and U.S. Pharmacopeia general chapter 823, Positron Emission Tomography Drugs for Compounding, Investigational, and Research Uses for early phase trials, the FDA requires that sufficient information is submitted to ensure the proper identification, strength, quality, purity, and potency of the investigational radiopharmaceutical (35,36). Therefore, each manufacturing site needs to demonstrate its ability to develop, maintain, and follow written procedures from the beginning of manufacturing process until product release.
For the early phase of the trial, the precursor can be produced in-house using a well-documented procedure. The process for synthesis should demonstrate control and traceability and minimize impurities in the product. After synthesis, the precursor must be fully characterized via spectral data, such as that from nuclear magnetic resonance, mass spectroscopy, and high-performance liquid chromatography. A provisional expiry can be assigned and updated as needed. Written procedures for the radiosynthesis of 18F-FTT were generated to detail the steps for synthesis and purification, as well as the materials used. Sterility-associated critical tasks, such as the final product vial assembly and sterility sampling and testing, must be performed in an International Organization for Standardization class 5 environment with appropriate environmental monitoring.
Quality control release specifications to ensure the product quality were developed to define the attributes of 18F-FTT for human injection. This was done to ensure the identity, strength, quality, purity, and potency of the final product. See the Table for the details on release specification and data from three validation runs. This is also used as the basis for ensuring the quality of radiopharmaceuticals used in the later multicenter trials. Because of the route of administration, the results of sterility testing are of great importance.
18F Fluorthanatrace Quality Control Release Specification and Validation Results

Three consecutive validation batches, defined as successful radiolabeling and quality control test results within predefined ranges, were required to demonstrate the site’s ability to consistently produce high-quality products. The stability testing was also performed over 6 hours to assess product quality during the storage period. This serves as the basis of determining product expiry. Other than following Part 212, for the manufacturing of radiopharmaceuticals for early phase clinical trial, one can also follow the U.S. Pharmacopeia chapter on PET compounding.
Preclinical dosimetry study.—Mice were injected with 18F-FTT and euthanized at 5, 30, 60, 120, or 240 minutes after injection (n = 4 at each time point, except n = 3 for the 120-minute time point). Several samples, such as blood, heart, lung, liver, spleen, fat, kidney, uterus, ovaries, bone, and intestine, plus any produced urine and feces, were collected from each animal and weighed. All organs and excretions were counted in a gamma counter to determine the percentage of injected dose per gram for tracer biodistribution, which is used for human dose estimate.
The mouse organ residence times were calculated by using the standard medical internal radiation dose method. The data were entered into the OLINDA/EXM program (Organ Level INternal Dose Assessment/EXponential Modeling, version 2, Hermes Medical Solutions) to observe the highest radiation dose to the bladder wall and estimate human effective doses of 0.013 and 0.015 mSv/MBq for men and women, respectively. On the basis of this higher estimate, a dose of 10 mCi was selected for the human dosimetry study with an estimated human effective dose of 5.6 mSv (15).
IND submission and maintenance.—The initial submission of an exploratory IND for 18F-FTT was prepared by Washington University in St Louis, followed by subsequent individual submissions by the University of Pennsylvania and MD Anderson Cancer Center. In addition to the sections mentioned above for the exploratory IND, the submissions should also contain form 1572 and credentials of the principal investigator and any co-investigators.
Following submission to the FDA Center for Drug Evaluation and Research, an acknowledgment letter was received, and a 30-day initial FDA review commenced. During this period, the FDA may request clarification or amendment from any sections of the IND. If the requests were not addressed or serious issues were identified, the FDA had the option to place the IND on a clinical hold until the reviewers were satisfied with the additional communication. If no issues were identified or requests were adequately addressed during this period, an IND may-proceed letter was granted.
The responsibilities of the sponsor (or sponsor-investigator) and investigators after the initiation of the trial are defined in the 21 CFR 312.50 and 21 CFR 312.60, respectively. The sponsor must select a monitor qualified by training and experience to monitor the progress of the clinical investigation. Amendments to any portion of the IND, annual reports, and IND safety reports should be submitted to the FDA in a timely manner by the sponsor.
Step 5: Initial Clinical Trials
The first clinical trials of 18F-FTT were all conducted under exploratory INDs, taking advantage of the limited exposure to study volunteers and the diagnostic nature of the agent. These studies mainly took place at Washington University and the University of Pennsylvania. The following sections describe the various first-in-human studies.
First-in-human biodistribution and dosimetry studies.—When designing a phase 0 or 1 clinical trial for new chemical entities intended to be diagnostic radiopharmaceuticals, the main consideration is demonstrating safety of administering trace amounts of the radiopharmaceutical. Exploring initial efficacy is often also incorporated into the study design to maximize the data available for initial evaluation while limiting the number of participants recruited and exposed to radiation. Preclinical dosimetry studies for 18F-FTT indicated that a 374-MBq (10-mCi) injection would yield an estimated effective dose of 5.6 mSv, which was a reasonable starting injected dose for evaluation (15). Healthy volunteers were included as part of the protocol because of the desire to evaluate the biodistribution in participants without any chronic medication exposure that might affect 18F-FTT distribution (15). The study volunteers were scanned with the dynamic PET/CT scan protocol for up to 210 minutes. In this study, participants with cancer (median age, 60 years; five men and three women, all White) were also included to obtain preliminary data regarding the variability of 18F-FTT uptake in various cancer types, including hepatocellular carcinoma, cholangiocarcinoma, and colorectal, pancreatic, and head and neck cancers, that might be treated with PARPi (15). All participants were enrolled into a clinical trial with study imaging time points chosen to estimate the 18F-FTT retention time and total potential radiation exposure from the 18F-FTT injection. Secondary analyses were then conducted to assess tumor uptake in the participants with cancer. The study design also incorporated standard safety assessments, including complete blood counts, complete metabolic profile, urinalysis, and electrocardiograms before and after injection. Symptom assessment was also performed before and after injection. No substantial adverse effects were noted.
Human internal organ estimates were produced by using calculated Medical Internal Radiation Dose organ residence times and measurements of tracer excretion, such as urine counts, using software such as OLINDA/EXM (37). The first step was to assemble individual organ percentage of injected dose. These measurements were indirectly extracted from a series of 18F-FTT PET/CT scans of 16 volunteers. Individual organ residence times are typically calculated for organs exhibiting either high or very low uptake of radiotracer. Individual organ residence times for bone marrow, brain, gallbladder, heart, lungs, kidneys, spleen, liver, stomach, small bowel, large bowel, urinary bladder, pancreas, and muscle were derived from the time-activity curves of individual organs using a variety of area under the curve calculations (15). The resulting percentage injected doses for every measured organ were then used to determine residence times for dosimetry calculations. The difference between the injected activity and activity in the measured organs represents the cumulated activity in the remainder of the body (37). The remainder activity is then used by OLINDA/EXM to estimate absorbed doses in organs not directly measured but commonly reported in human studies based on the selected anthropomorphic model (eg, standard adult male phantom). The human study’s absorbed dose estimates indicated critical organs were spleen and pancreas based on the highest overall estimated absorbed dose in the spleen of 0.054 mSv/MBq in women and the highest estimated absorbed dose in the pancreas of 0.034 mSv/MBq in men. While preclinical dosimetry found the highest radiation dose in the bladder wall, human dosimetry observed the highest absorbed doses in the spleen and pancreas (15). Because interspecies differences in radiotracer biodistribution and metabolic rates are common, this was not surprising and highlights the importance of repeating preclinical dosimetry studies in human volunteers. There was agreement in the human dosimetry estimates of effective dose obtained from both the preclinical (0.013 to 0.015 mSv/MBq) and human (0.014 to 0.019 mSv/MBq) dosimetry studies (15). No adverse effect was observed in this study.
Studies in breast and ovarian cancer.—An early follow-up study to the Chen study was conducted at the University of Pennsylvania and focused on patients with advanced ovarian cancer (31). This study examined the level and heterogeneity of 18F-FTT in a group of patients with ovarian cancer (Fig 6) and showed that the level of radiotracer uptake correlated closely with an in vitro assay of tissue samples for PARP-1 expression by immunofluorescence and radioligand binding using 125I-KX1 autoradiography. Subsequent data analysis at the University of Pennsylvania examined the kinetics of 18F-FTT uptake in 14 patients with ovarian cancer (age range, 21–70 years, all women), leveraging dynamic PET imaging and metabolite measurements to model in vivo tracer binding (38). In this study, the Logan reference tissue distribution volume ratio and total distribution volume from a reversible, two-tissue compartment from injection to 1 hour after injection were used to analyze the uptake of 18F-FTT, and the results correlated with in vitro assay of tumor PARP-1 expression by immunofluorescence (r = 0.83 and 0.76, respectively, P < .05). Tumor maximum and peak standardized uptake value (SUVmax and SUVpeak, respectively) measures at midpoints of 58 minutes, 110 minutes ± 3, and 199 minutes ± 4 were also similarly correlated with PARP-1 expression by immunofluorescence staining (mean ± standard deviation, r ≥ 0.79, P < .05). The study recommended future 18F-FTT uptake quantification using either distribution volume ratio from dynamic PET acquisitions of at least 1 hour or using SUVmax and SUVpeak measures from static PET scans acquired approximately 1 hour or later after injection. This latter finding is important in enabling shorter scan times for more widespread clinical trials, using imaging protocols similar to those used for clinical PET imaging instead of the more sophisticated and precise estimates of tracer kinetics and tumor binding from (less clinically practical) dynamic imaging.
Figure 6:

Clinical PET/CT imaging studies in patients with (A) ovarian and (B) breast cancer. (A) The standardized uptake value (SUV) measurements of 18F fluorthanatrace (18F-FTT) in patients with ovarian cancer (n = 20 women; age range, 22–68 years; median, 60 years) varied greatly, from 2 (background) to 12 (highest). Arrows indicate the site of disease. This is the representation of three images from patients with low, medium, and high uptake. (B) In a follow-up breast cancer trial, four patients (age range, 41–71 years; median age, 52 years) with stage III or IV disease and receiving poly (adenosine diphosphate–ribose) polymerase inhibitor (PARPi) therapy were enrolled. Three of the tumors were triple negative and one was estrogen positive. The initial SUV was moderate, 4.2–6.8, at baseline (left). After 1 week of PARPi treatment, the participants were scanned with 18F-FTT again. The uptake of 18F-FTT was diminished after the PARPi treatment. (Data reproduced from references 16 and 31.) (B reprinted, with permission, from reference 16.)
A second study involved a prospective nonrandomized clinical trial of 30 participants with early and locally advanced breast cancer (39). This trial studied the correlation of 18F-FTT uptake across a range of breast cancer phenotypes with the goal of studying the potential utility of 18F-FTT PET as a functional biomarker for monitoring PARPi response. In this study, patients with estrogen receptor–positive, human epidermal growth factor receptor 2–positive, and triple-negative immunohistochemical phenotypes were enrolled. The SUV from these patients ranged from 2.6 to 11.3 g/mL, which was independent of cancer subtypes. In patients with BRCA1/2 pathogenic variants, the SUV varied greatly, and the range overlapped values from patients without BRCA 1/2. In this preliminary study, pathogenic variant carriers without allele-specific loss of heterozygosity had lower levels of uptake than carriers who retained loss of heterozygosity. These results showed that the level of radioligand binding varied considerably across and within traditional breast cancer subtypes, including germline or tumor mutations in BRCA-related genes. Elevated radiotracer uptake in participants who would not have been considered candidates for PARPi therapy by current selection criteria suggests that some patients with breast cancer outside of current practice might potentially benefit from the PARPi treatment.
In a follow-up study at the University of Pennsylvania in patients with metastatic breast cancer receiving PARPi therapy, as shown in Figure 6, 18F-FTT uptake in known sites of disease was blocked by PARPi treatment (16). Drug PARP-1 occupancy was also measured by in vitro autoradiography radioligand-binding studies of cancer tissue samples using 125I-KX1 with and without pharmacologic levels of olapraib. 125I-KX1 binding was suppressed by greater than 80% by olaparib and matched PARPi blockade measured at pre- and post-PARPi PET. Despite the small sample size (four patients; age range, 41–71 years; median age, 52 years; all women; stage III or IV breast cancer), this study demonstrates the potential of 18F-FTT PET to noninvasively quantify PARP-1 expression and provides early evidence of using this modality to assess PARPi drug-target engagement, indicating its potential as a biomarker for PARPi treatment.
Step 6: Phase 2 Studies
The data presented above strongly suggest that 18F-FTT has the potential to serve as a PET-based biomarker for imaging PARP-1 expression levels in breast and ovarian cancer. 18F-FTT is also capable of measuring drug occupancy of FDA-approved PARPi based on the results in the blocking studies conducted in patients with breast cancer and in the murine models of breast and ovarian cancer (16,31). Therefore, the next step in the clinical evaluation of 18F-FTT is to acquire the body of clinical data required for FDA approval as a new drug application. This will require the standardization of 18F-FTT production for a multicenter clinical study aimed at validating the preliminary findings that demonstrate a positive correlation between tracer uptake, as measured by SUV, and ex vivo measures of PARP-1 expression using immunofluorescence, immunohistochemistry, and autoradiography with 125I-KX1, an iodinated PARP inhibitor tracer (40). In 2006, Nunn estimated that the time required to complete radiotracer development and clinical trials and to obtain the final new drug application approval from the FDA was approximately 10 years (41). The projected cost was $150 million U.S. dollars, an amount that is beyond the resources of academic research laboratories. Therefore, cases where this process has succeeded have required either a partnership with industry, the spinoff of the intellectual property into a small business entity, or a combination of both, to raise the funds needed to support a phase 2 multicenter clinical trial of a new PET radiotracer. With 18F-FTT, Washington University helped spur the creation of Trevarx and gave this small business entity the exclusive rights to the intellectual property of this radiotracer. The initial multicenter clinical trial noted above will be supported through an academic-industrial partnership R01 (R01CA258717) recently awarded to three academic sites and Trevarx. The academic sites involved in this academic-industrial partnership—Washington University in St Louis, University of Pennsylvania, and MD Anderson Cancer Center—all have experience using 18F-FTT and existing exploratory INDs that leverage the original Washington University exploratory IND. The study will be a coordinated partnership between industry and academic experts with the goal of FDA approval of an emerging PET agent. Each partner will play a key role in clinical trial management, regulatory support and oversight, and harmonization and standardization of radiopharmaceutical production, image acquisition, and image analysis.
The first step is to develop coordinated radiopharmaceutical production, in accordance with Current Good Manufacturing Practice regulations, for phase 2 multicenter studies as described above. A Trevarx-sponsored full IND will be obtained as an initial first step with Current Good Manufacturing Practice–compliant synthesis at each of the three sites. A total of 36 patients will be enrolled across the three centers. Patients with primary invasive breast cancers with tumor size measuring at least 1 cm and undergoing upfront surgery will be eligible. Patients will undergo a single static PET/CT scan approximately 60 minutes after injection of 18F-FTT, based on the kinetic studies noted above (38). Approximately six to 10 patients will undergo test-retest 18F-FTT PET/CT imaging with both scans performed prior to any surgery to assess the precision of PET uptake measures. Tumor tissue will be taken at the time of surgery for ex vivo tissue assays of PARP expression.
The trial will use standardized methods of 18F-FTT production that will be developed early in the course of the grant period. Uniform procedures for patient preparation and scanning will be used. PET scans will be performed using a single scanner at each site, which has been prequalified with data collection from a uniform phantom within 3 months of study activation and repeated yearly during the course of the study. Images and tissue will be de-identified and analyzed centrally at the University of Pennsylvania. Images will be reviewed by a nuclear medicine physician expert who will be blinded to any clinical information. This multisite study will provide estimates of 18F-FTT accuracy and study feasibility and will provide the data needed to design a pivotal phase 3 study required for FDA approval. This phase 2 trial will be a real-world test of the methods needed for a future phase 3 registration trial of 18F-FTT as a quantitative diagnostic marker of PARP-1 expression.
Conclusion
Since the first publication of 18F-FTT, the preclinical and clinical evaluation of this PET agent has progressed through early phase clinical trials to the beginning of the phase 2 multicenter trials, which has taken 7 years to complete. Initial studies in patients with breast and ovarian cancer suggest that PET imaging studies with 18F-FTT are able to identify patients who have high PARP-1 expression, a key component for determining a positive response to treatment with a PARPi. Preliminary drug occupancy studies also demonstrate that 18F-FTT is able to measure target engagement through clinically approved PARPi. Although these data are encouraging, there are a number of steps still needed to validate 18F-FTT as an imaging biomarker for measuring PARP-1 levels in tumors. These steps will require substantial effort and resources to complete and will require financial resources beyond what is available through traditional funding mechanisms, such as grants from the National Institutes of Health and other funding agencies. The road map presented above illustrates the steps one must take, as well as the commitment and resources (both human and financial) needed, for the successful clinical translation of a PET radiotracer from preclinical studies in murine models of cancer to clinical research trials in patients with cancer. The key lesson is to involve investigators from different disciplines as early as possible for study planning and execution. In addition, working closely with the regulatory agencies is instrumental, as they provide critical insight into the study design and execution. Although the road is long and contains many obstacles, the recent success stories, such as 18F fluciclovine, 18F-DCFPyL (2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine 3-carbonyl)-amino]-pentyl}-ureido)-pentanedioic acid), 68Ga-PSMA-11 (gallium 68 prostate-specific membrane antigen), and 18F-piflufolastat, demonstrate the need for new molecular imaging agents in improving the characterization and clinical management of cancer.
Authors declared no funding for this work.
Disclosures of conflicts of interest: H.S.L. No relevant relationships. S.W.S. Employed by Trevarx Biomedical for 40% of this author’s time. E.K.S. Consultant for Trevarx. D.L.C. Honorarium from Telix Pharmaceuticals for participation in expert panel discussion on PSMA PET; co-inventor on patent held by Washington University and licensed to Trevarx Biomedical (WO 2015103526 A1: Mach RH, Chu W, Zhou D, Michel LS, Chen DL. Radiolabeled tracers for poly(adp-ribose)polymerase-1(PARP-1)); president-elect for Center for Molecular Imaging, Innovation and Translation, Society of Nuclear Medicine and Molecular Imaging. R.K.D. Grants (paid to institution) from Department of Defense office of the Congressionally Directed Medical Research (W81XWH-20-1-0025: “In Vivo PARP-1 Expression as a Predictive and Pharmacodynamic Tool” and W81XWH-17-1-0092: “Investigation of a Novel PARP Inhibitor PET Tracer in Ovarian Carcinoma”). M.M. No relevant relationships. L.L.L. Support from Marsha Rivkin Foundation and Kaleidoscope of Hope; grants NIH 1R01CA249329-01A1 and NIH 1R01CA258717-01; support from AstraZeneca to travel to attend consortia meeting. E.S.M. Grants from Department of Defense office of the Congressionally Directed Medical Research, National Cancer Institute, Susan G. Komen Foundation, and Metavivor. D.A.M. NIH grant R01 CA258717 (PI); Trevarx has a license to IP for FTT, the topic of this article. This author’s wife is the CEO of Trevarx; Trevarx has a license to IP for FTT from Washington University, the topic of this article. This author’s wife is the CEO of Trevarx; Trevarx has a license to IP for FTT, the topic of this article. This author’s wife is the CEO of Trevarx. Author is owner of Trevarx equity; associate editor for Radiology: Imaging Cancer. R.H.M. Co-founder of Trevarx; patent licensed to Trevarx, issued US patent; stock options in Trevarx; co-founder of Accuronix Therapeutics and Vellum Biosciences (not related to this article).
Abbreviations:
- FDA
- Food and Drug Administration
- 18F-FTT
- fluorine 18 fluorthanatrace
- IND
- investigational new drug
- PARP-1
- poly (adenosine diphosphate–ribose) polymerase-1
- PARPi
- PARP inhibitor(s)
- SUV
- standardized uptake value
References
- 1. Groheux D , Cochet A , Humbert O , Alberini JL , Hindié E , Mankoff D . 18F-FDG PET/CT for Staging and Restaging of Breast Cancer . J Nucl Med 2016. ; 57 ( Suppl 1 ): 17S – 26S . [DOI] [PubMed] [Google Scholar]
- 2. Hampel H , Caraci F , Cuello AC , et al . A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease . Front Immunol 2020. ; 11 456 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ghafoor S , Burger IA , Vargas AH . Multimodality Imaging of Prostate Cancer . J Nucl Med 2019. ; 60 ( 10 ): 1350 – 1358 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barrington SF , Trotman J . The role of PET in the first-line treatment of the most common subtypes of non-Hodgkin lymphoma . Lancet Haematol 2021. ; 8 ( 1 ): e80 – e93 . [DOI] [PubMed] [Google Scholar]
- 5. Cutler CS . Economics of New Molecular Targeted Personalized Radiopharmaceuticals . Semin Nucl Med 2019. ; 49 ( 5 ): 450 – 457 . [DOI] [PubMed] [Google Scholar]
- 6. Altmann A , Haberkorn U , Siveke J . The Latest Developments in Imaging of Fibroblast Activation Protein . J Nucl Med 2021. ; 62 ( 2 ): 160 – 167 . [DOI] [PubMed] [Google Scholar]
- 7. Hofman MS , Lau WF , Hicks RJ . Somatostatin receptor imaging with 68Ga DOTATATE PET/CT: clinical utility, normal patterns, pearls, and pitfalls in interpretation . RadioGraphics 2015. ; 35 ( 2 ): 500 – 516 . [DOI] [PubMed] [Google Scholar]
- 8. Agdeppa ED , Spilker ME . A review of imaging agent development . AAPS J 2009. ; 11 ( 2 ): 286 – 299 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hung JC . Bringing New PET drugs to clinical practice - a regulatory perspective . Theranostics 2013. ; 3 ( 11 ): 885 – 893 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harapanhalli RS . Food and Drug Administration requirements for testing and approval of new radiopharmaceuticals . Semin Nucl Med 2010. ; 40 ( 5 ): 364 – 384 . [DOI] [PubMed] [Google Scholar]
- 11. Jackson IM , Lee SJ , Sowa AR , et al . Use of 55 PET radiotracers under approval of a Radioactive Drug Research Committee (RDRC) . EJNMMI Radiopharm Chem 2020. ; 5 ( 1 ): 24 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mosessian S , Duarte-Vogel SM , Stout DB , et al . INDs for PET molecular imaging probes-approach by an academic institution . Mol Imaging Biol 2014. ; 16 ( 4 ): 441 – 448 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Puentes LN , Makvandi M , Mach RH . Molecular Imaging: PARP-1 and Beyond . J Nucl Med 2021. ; 62 ( 6 ): 765 – 770 . [DOI] [PubMed] [Google Scholar]
- 14. Makvandi M , Pantel A , Schwartz L , et al . A PET imaging agent for evaluating PARP-1 expression in ovarian cancer . J Clin Invest 2018. ; 128 ( 5 ): 2116 – 2126 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Michel LS , Dyroff S , Brooks FJ , et al . PET of Poly (ADP-Ribose) Polymerase Activity in Cancer: Preclinical Assessment and First In-Human Studies . Radiology 2017. ; 282 ( 2 ): 453 – 463 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McDonald ES , Pantel AR , Shah PD , et al . In vivo visualization of PARP inhibitor pharmacodynamics . JCI Insight 2021. ; 6 ( 8 ): e146592 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Bono J , Mateo J , Fizazi K , et al . Olaparib for Metastatic Castration-Resistant Prostate Cancer . N Engl J Med 2020. ; 382 ( 22 ): 2091 – 2102 . [DOI] [PubMed] [Google Scholar]
- 18. He C , Kawaguchi K , Toi M . DNA damage repair functions and targeted treatment in breast cancer . Breast Cancer 2020. ; 27 ( 3 ): 355 – 362 . [DOI] [PubMed] [Google Scholar]
- 19. Ledermann JA . Extending the scope of PARP inhibitors in ovarian cancer . Lancet Oncol 2019. ; 20 ( 4 ): 470 – 472 . [DOI] [PubMed] [Google Scholar]
- 20. Mateo J , Lord CJ , Serra V , et al . A decade of clinical development of PARP inhibitors in perspective . Ann Oncol 2019. ; 30 ( 9 ): 1437 – 1447 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lim D , Ngeow J . Evaluation of the methods to identify patients who may benefit from PARP inhibitor use . Endocr Relat Cancer 2016. ; 23 ( 6 ): R267 – R285 . [DOI] [PubMed] [Google Scholar]
- 22. Zimmer AS , Gillard M , Lipkowitz S , Lee JM . Update on PARP Inhibitors in Breast Cancer . Curr Treat Options Oncol 2018. ; 19 ( 5 ): 21 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Franzese E , Centonze S , Diana A , et al . PARP inhibitors in ovarian cancer . Cancer Treat Rev 2019. ; 73 ( 1 ): 9 . [DOI] [PubMed] [Google Scholar]
- 24. González-Martín A , Pothuri B , Vergote I , et al . Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer . N Engl J Med 2019. ; 381 ( 25 ): 2391 – 2402 . [DOI] [PubMed] [Google Scholar]
- 25. Golan T , Hammel P , Reni M , et al . Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer . N Engl J Med 2019. ; 381 ( 4 ): 317 – 327 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hanna C , Kurian KM , Williams K , et al . Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: results of the phase I OPARATIC trial . Neuro Oncol 2020. ; 22 ( 12 ): 1840 – 1850 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang L , Wang Q , Xu Y , Cui M , Han L . Advances in the Treatment of Ovarian Cancer Using PARP Inhibitors and the Underlying Mechanism of Resistance . Curr Drug Targets 2020. ; 21 ( 2 ): 167 – 178 . [DOI] [PubMed] [Google Scholar]
- 28. Pettitt SJ , Lord CJ . Dissecting PARP inhibitor resistance with functional genomics . Curr Opin Genet Dev 2019. ; 54 ( 55 ): 63 . [DOI] [PubMed] [Google Scholar]
- 29. Sandhu D , Antolin AA , Cox AR , Jones AM . Identification of different side effects between PARP inhibitors and their polypharmacological multi-target rationale . Br J Clin Pharmacol 2021 . 10.1111/bcp.15015. Published online July 30, 2021 . [DOI] [PubMed] [Google Scholar]
- 30. Stover EH , Fuh K , Konstantinopoulos PA , Matulonis UA , Liu JF . Clinical assays for assessment of homologous recombination DNA repair deficiency . Gynecol Oncol 2020. ; 159 ( 3 ): 887 – 898 . [DOI] [PubMed] [Google Scholar]
- 31. Makvandi M , Xu K , Lieberman BP , et al . A Radiotracer Strategy to Quantify PARP-1 Expression In Vivo Provides a Biomarker That Can Enable Patient Selection for PARP Inhibitor Therapy . Cancer Res 2016. ; 76 ( 15 ): 4516 – 4524 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou D , Chu W , Xu J , et al . Synthesis, [18F] radiolabeling, and evaluation of poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors for in vivo imaging of PARP-1 using positron emission tomography . Bioorg Med Chem 2014. ; 22 ( 5 ): 1700 – 1707 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Edmonds CE , Makvandi M , Lieberman BP , et al . [(18)F]FluorThanatrace uptake as a marker of PARP1 expression and activity in breast cancer . Am J Nucl Med Mol Imaging 2016. ; 6 ( 1 ): 94 – 101 . [PMC free article] [PubMed] [Google Scholar]
- 34. Schwarz SW , Oyama R . The role of exploratory investigational new drugs for translating radiopharmaceuticals into first-in-human studies . J Nucl Med 2015. ; 56 ( 4 ): 497 – 500 . [DOI] [PubMed] [Google Scholar]
- 35. Schwarz SW , Dick D , VanBrocklin HF , Hoffman JM . Regulatory Requirements for PET Drug Production . J Nucl Med 2014. ; 55 ( 7 ): 1132 – 1137 . [DOI] [PubMed] [Google Scholar]
- 36. Norenberg JP , Schwarz S , VanBrocklin H . FDA cGMP requirements for PET drugs . J Nucl Med 2011. ; 52 ( 5 ): 16N . [PubMed] [Google Scholar]
- 37. Stabin MG , Sparks RB , Crowe E . OLINDA/EXM: the second-generation personal computer software for internal dose assessment in nuclear medicine . J Nucl Med 2005. ; 46 ( 6 ): 1023 – 1027 . [PubMed] [Google Scholar]
- 38. Young AJ , Pantel AR , Viswanath V , et al . Kinetic and Static Analysis of Poly-(Adenosine Diphosphate-Ribose) Polymerase-1-Targeted 18 F-Fluorthanatrace PET Images of Ovarian Cancer . J Nucl Med 2022. ; 63 ( 1 ): 44 – 50 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McDonald ES , Doot RK , Pantel AR , et al . Positron Emission Tomography Imaging of Poly-(Adenosine Diphosphate-Ribose) Polymerase 1 Expression in Breast Cancer: A Nonrandomized Clinical Trial . JAMA Oncol 2020. ; 6 ( 6 ): 921 – 923 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Anderson RC , Makvandi M , Xu K , et al . Iodinated benzimidazole PARP radiotracer for evaluating PARP1/2 expression in vitro and in vivo . Nucl Med Biol 2016. ; 43 ( 12 ): 752 – 758 . [DOI] [PubMed] [Google Scholar]
- 41. Nunn AD . The cost of developing imaging agents for routine clinical use . Invest Radiol 2006. ; 41 ( 3 ): 206 – 212 . [DOI] [PubMed] [Google Scholar]