

Abstract
Aims
Assessment of endothelial function in humans by measuring flow-mediated dilation (FMD) risk-stratifies individuals with established cardiovascular disease, whereas its predictive value is limited in primary prevention. We therefore aimed to establish and evaluate novel markers of FMD at the population level.
Methods and results
In order to identify novel targets that were negatively correlated with FMD and investigate their contribution to vascular function, we performed a genome-wide association study (GWAS) of 4175 participants of the population based Gutenberg Health Study. Subsequently, conditional knockout mouse models deleting the gene of interest were generated and characterized. GWAS analysis revealed that single-nucleotide polymorphisms (SNPs) in the tubulin-folding cofactor E (TBCE) gene were negatively correlated with endothelial function and TBCE expression. Vascular smooth muscle cell (VSMC)-targeted TBCE deficiency was associated with endothelial dysfunction, aortic wall hypertrophy, and endoplasmic reticulum (ER) stress-mediated VSMC hyperproliferation in mice, paralleled by calnexin up-regulation and exacerbated by the blood pressure hormone angiotensin II. Treating SMMHC-ERT2-Cre+/−TBCEfl/fl mice with the ER stress modulator tauroursodeoxycholic acid amplified Raptor/Beclin-1-dependent autophagy and reversed vascular dysfunction.
Conclusion
TBCE and tubulin homeostasis seem to be novel predictors of vascular function and offer a new drug target to ameliorate ER stress-dependent vascular dysfunction.
Keywords: Genome-wide association study, Single-nucleotide polymorphism, Tubulin-folding cofactor E, Flow-mediated dilation, Endothelial function, ER stress, Vascular inflammation
Graphical Abstract
See the editorial comment for this article ‘Identification of a novel therapeutic target in vascular dysfunction: a showcase of reverse and forward translational research linking bench to bedside’, by Hiroshi Iwata and Tohru Minamino, https://doi.org/10.1093/eurheartj/ehab263.
Introduction
Vascular dysfunction is a hallmark of cardiovascular disease (CVD). The specific properties of endothelial, smooth muscle and immune cells converge on the vasculature and determine vascular tone and homeostasis. Endothelial and/or smooth muscle cell dysfunction as well as activated immune cells contribute to progression of CVD,1 , 2 the leading cause of mortality worldwide.3
Translational perspective
With this translational research project, we report the hitherto unknown relation between tubulin-folding cofactor E (TBCE) and vascular function discovered in a genome-wide association study of 4175 individuals and further investigated the putative mechanisms in three cell-specific conditional knockout animals. This is the first bedside-to-bench study correlating TBCE with vascular dysfunction, establishing endoplasmic reticulum (ER) stress as a key-mediator of the phenotype. Importantly, administration of the clinically relevant ER stress modulator tauroursodeoxycholic acid reversed vascular dysfunction in vivo. Our work establishes the grounds for further evaluating the role of TBCE and tubulin homeostasis in vascular dysfunction in both the pre-clinical and clinical setting.
Disruption of endothelial homeostasis leads to disturbed nitric oxide-dependent regulation of the vascular tone.4 Myeloid cells are important contributors to the initiation of vascular dysfunction, inflammation, and remodelling,5 in part by their ability to secrete chemokines and cytokines.5 Vascular smooth muscle cells (VSMCs) are mural cells continually exposed to biochemical and mechanical stress generated in the circulation. They adapt to hemodynamic stimuli by switching to a hypertrophic, proliferative phenotype, and by secreting extracellular matrix proteins.6 Thereby, they participate in the regulation of vascular tone and blood pressure.7
Numerous non-invasive techniques including flow-mediated dilation (FMD), venous occlusion plethysmography, or peripheral arterial tone/pulse waveform analysis have been developed for assessment of endothelial function.8 FMD as the most frequently used technique, is useful for risk stratification in cohorts afflicted by CVD,9 , 10 whereas data from various population-based studies have yielded equivocal results on the prognostic value of FMD concerning future cardiovascular events.9 , 11 , 12 The Gutenberg Health Study (GHS) is a population-based prospective cohort study and has previously shown that mid-regional pro-adrenomedullin and mid-regional pro-atrial natriuretic peptide were associated with FMD.13
In the GHS, FMD was strongly associated with age, sex, body mass index, and biomarkers of arterial hypertension or heart failure,13 but classical risk factors accounted for only 15.4% of the variability of the reflective hyperaemic response in FMD.14 The scope of our current study was to discover novel single-nucleotide polymorphisms (SNPs) that correlate with FMD through genome-wide association study (GWAS) at the population level. Furthermore, we aimed to identify putative mechanisms by the generation and vascular phenotyping of cell-specific conditional knockout mouse models.
Materials and methods
Full methods are available in the Supplementary material online.
Genome-wide association study
SNP information was gained from the genome data available from the GHS as previously described,15 employing Affymetrix Genome-Wide Human SNP 6.0 arrays (Affymetrix, Santa Clara, CA, USA); after quality control, genetic data of 4,175 individuals were available for analysis. Correlation analysis by linear regression was performed in order to identify SNPs that were associated with FMD.
Animals
Three different strains of cell-specific, conditional knockout mouse models of tubulin-folding cofactor E (TBCE) were generated by serial crossings. TBCEtm1c mice were first generated according to EUCOMM® instructions.16 , 17 LysMCre+/−TBCEfl/fl mice were generated for myeloid cell-specific deficiency of TBCE,18 Tie2-ERT2Cre+/−TBCEfl/fl mice as a conditional knockdown model in endothelial cells (ECs),19 and SMMHC-ERT2-Cre+/−TBCEfl/fl as a conditional knockdown model in smooth muscle cells.20 For all the above-mentioned strains, age-matched Cre+/−TBCEwt/wt littermates were used as controls in all experiments. A total of 60 LysMCre+/−TBCEfl/fl, 80 Tie2-ERT2-Cre+/−TBCEfl/fl, and 140 SMMHC-ERT2-Cre+/−TBCEfl/fl mice were used in the experiments. All animals were inbred and received proper care in compliance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1985) and were approved by the Ethical Committee of the Prefecture of Rheinland-Pfalz (G17-1-031). All animal experiments were performed in line with the ARRIVE guidelines.21 The breeding strategy, genotyping, and workflow are described in Supplementary material online, Figure S2. Animals that did not meet the wellbeing criteria regarding the 3Rs were excluded from the experiments, while no outliers were removed from the experiments. The TBCEtm2c (e.g. TBCEfl/fl) strain has been accepted for archiving in the INFRAFRONTIER/EMMA repository (EMMA ID: EM13235) and is available for research purposes (https://www.infrafrontier.eu/search).
Vascular relaxation and constriction studies
To assess vasodilator potential of isolated aortic segments (3 mm), they were mounted to force transducers in organ chambers to test their response to acetylcholine (ACh) and nitroglycerine (GTN). The aortic rings were pre-constricted with prostaglandin F2α (PGF2α, 3 nM), a VSMC-dependent constrictor22 to reach 50–80% of the tone induced by KCl. Concentration–relaxation curves were recorded in response to the endothelium-dependent vasodilator ACh (1 nM to 3 mM) as well as GTN (1 nM to 30 mM).23 Aortic tissue was snap frozen for further molecular and histological analyses.
Statistical analysis
Data are presented as means ± standard deviation. Continuous variables were compared among groups using one-way analysis of variance (ANOVA) and post hoc comparisons were made using Tukey’s test, while comparisons concerning the ACh and GTN relaxation and tension data were analysed by two-way ANOVA and Tukey’s test. No assumption of equal variability of differences was performed and data were corrected with Greenhouse-Geisser correction. A P-value of <0.05 was considered statistically significant. All statistical analyses and graph preparation were performed using GraphPad Prism 8.5 analysis software (GraphPad Software, Inc., La Jolla, CA, USA). No outliers due to biological diversity were excluded. Samples that did not meet our technical criteria were not included into the analyses a priori. The absence of outlying values was confirmed by GraphPad Prism analysis software, using ROUT method and Q = 1%. Complete statistical analysis is included in the Supplementary material online.
Results
Single-nucleotide polymorphisms rs6675944 and rs12405889 are negatively correlated with TBCE expression and flow-mediated arterial dilation
GHS revealed that SNPs rs6675944 (T→C) and rs1205889 (T→G) in chromosome 1 were negatively correlated with FMD of the brachial artery (rs6675944 β = −0.4841, P = 5.9e−27, R 2 tot = 0.006 and β = −0.4841, P = 2.3e−12, R 2 tot = 0.006; rs12405889 β = −0.5028, P = 8.8e−14, R 2 tot = 0.006 and β = −0.5028, P = 1.0e−25, R 2 tot = 0.006) (Table 1). Moreover, rs6675944 was associated with decreased reflection index pre occlusion and post occlusion and increased intima-media thickness (IMT; mm), showing an increased arterial stiffness phenotype in the SNP-bearing individuals24 (Table 1).
Table 1.
Linear regression model representing the correlation of the single-nucleotide polymorphisms (SNPs) rs6675944 and rs12405889 in the tubulin-folding cofactor E (TBCE) gene with measures of vascular function and morphology from 4175 individuals in the Gutenberg Health-Study
| SNP | Gene | bp | Phenotype | R 2 | Beta | P-value |
|---|---|---|---|---|---|---|
| rs6675944 | TBCE (intron) Chr1 | 233639292 | FMD | 0.006 | −0.4841 | 5.92E−27 |
| Augmentation index | 0.255 | −0.135 | 0.61 | |||
| Stiffness index pre occlusion (m/s) | 0.242 | −0.0055 | 0.84 | |||
| Stiffness index post occlusion cc (m/s) | 0.3 | −0.0241 | 0.38 | |||
| Reflection index pre occlusion | 0.145 | −0.359 | 0.066 | |||
| Reflection index post occlusion | 0.206 | −0.46 | 0.028 | |||
| IMT (mm) | 0.34 | 0.00262 | 0.053 | |||
| rs12405889 | TBCE (intron) Chr1 | 233663875 | FMD | 0.006 | −0.5028 | 8.78E−25 |
| Augmentation index | 0.257 | 0.0541 | 0.84 | |||
| Stiffness index pre occlusion (m/s) | 0.242 | 0.00133 | 0.96 | |||
| Stiffness index post occlusion (m/s) | 0.3 | 0.0176 | 0.53 | |||
| Reflection index pre occlusion | 0.146 | 0.308 | 0.12 | |||
| Reflection index post occlusion | 0.206 | 0.34 | 0.11 | |||
| IMT (mm) | 0.34 | −0.0022 | 0.12 |
bp, base pair; Chr1, chromosome 1; FMD, flow-mediated dilation; IMT, intima-media thickness. Bold values indicate significant values with P < 0.05.
Both SNPs were found in the TBCE (tubulin folding cofactor E, or tubulin-specific chaperone E) gene region. mRNA expression analysis of peripheral blood mononuclear cells obtained from individuals that were enrolled in GHS revealed that SNPs were negatively associated with TBCE expression (β estimate: −0.4841, R 2 tot = 0.010) (Figure 1A and B). GTEx Portal25 and NIH U.S. National Library of Medicine26 data analysis showed that both SNPs have been identified in various human population studies, with a high occurrence in the general population (Supplementary material online, Figure S1). In a pooled human tissue expression analysis—as performed from TBCE gene expression data in various analysed tissues and attained from the GTEx Portal—both homozygous and heterozygous genotypes were associated with down-regulation of TBCE expression, being more pronounced in the homozygous genotype (Figure 1E and F; GTEx Portal). Concerning the individual tissue analysis, skeletal muscle TBCE expression appeared to be mostly affected by the presence of SNPs (logP = −12.6 and −15.2, respectively), while SNPs were also found to negatively affect TBCE expression in aortic and tibial arterial tissue (Figure 1C and D; GTEx Portal). Conclusively, our GWAS as well as GTEx human data show that the presence of TBCE SNPs is correlated with decreased transcripts of the gene in various tissues. This decreased gene expression is negatively associated with FMD, leading to the assumption that TBCE down-regulation might be causally involved in vascular dysfunction.
Figure 1.

Association of the SNPs rs6675944 and rs12405889 in population studies and TBCE expression data in pooled tissue analyses. (A). Expression level of TBCE gene in the presence of rs6675944 SNP and (B). rs12405889 in peripheral blood mononuclear cells of the enrolled individuals (mean±SD). (C). Normalized Effect Size (NES) heatmap of rs6675944 and rs12405889 SNPs in various human tissues. (D). LogP value heatmap of rs6675944 and rs12405889 SNPs in various human tissues. (E and F) Normalized expression of TBCE in the presence of SNP rs6675944 and SNP rs12405889, stratified by genotype. Pooled data from tested human tissues. (One-Way ANOVA, Tukey’s comparison Test, Kruskal-Wallis, Dunn’s multiple comparison test).
Conditional knockout of TBCE in smooth muscle cells leads to endothelial dysfunction, vascular stiffness, and increased vessel wall thickness
As the GWAS analysis was performed in the peripheral blood mononuclear cells of the enrolled individuals, we initially generated the LysMCre+/−TBCEfl/fl mice (Supplementary material online, Figure S2) to check the phenotype of mice with myeloid cell-specific TBCE deficiency. LysMCre+/−TBCEfl/fl mice did not show any signs of altered body weight and serum postprandial blood glucose, triglycerides and total cholesterol compared to control mice (Supplementary material online, Figure S3). Vascular relaxation and tension in response to ACh (endothelium-dependent) and GTN (endothelium-independent) were not significantly different between LysMCre+/−TBCEfl/fl mice and their controls. Moreover, angiotensin II (AngII)-derived increase in systolic blood pressure, media thickness, and collagen/media thickness ratio were not significantly different between LysMCre+/−TBCEwt/wt and LysMCre+/−TBCEfl/fl mice (Supplementary material online, Figure S4). These data suggest a minor role of TBCE in lysozyme M+ cells in vascular (dys)function.
To further elucidate the mechanisms of TBCE-associated endothelial dysfunction, we generated Tie2-ERT2-Cre+/−TBCEfl/fl mice (Supplementary material online, Figure S2) in which endothelial-specific TBCE deletion was confirmed both in microcirculation and in the aortas (Supplementary material online, Figures S5 and S6). Tie2-ERT2-Cre+/−TBCEfl/fl mice presented a decreased body weight compared to Tie2-ERT2-Cre+/−TBCEwt/wt, both being lower than C57BL/6J controls (Supplementary material online, Figure S3). These mice had a decreased vascular tone both at baseline and in response to infusion with AngII. Vascular relaxation in response to ACh and GTN were comparable between Tie2-ERT2-Cre+/−TBCEfl/fl mice and controls (Supplementary material online, Figure S7A–G). Although the vascular resistance and pulsatility indexes as measures of vascular stiffness did not differ between groups (Supplementary material online, Figure S7H–J), a significant increase in wall thickness was detected only in Tie2-ERT2-Cre+/−TBCEfl/fl+AngII mice, independently of changes in (relative) collagen content (Supplementary material online, Figure S7K–M).
CC-chemokine ligand 2 (Ccl2), interleukin-6 (Il6), and tumour necrosis factor alpha (Tnfα) mRNA expression was significantly up-regulated in Tie2-ERT2-Cre+/−TBCEfl/fl mice compared to controls and in part amplified by AngII (Supplementary material online, Figure S8A). Staining of CD45−/CD31+ pulmonary ECs isolated from the Tie2-ERT2-Cre+/−TBCEfl/fl animals revealed an up-regulation of NACHT, LRR and PYD domains-containing protein 3 (NLRP3, Supplementary material online, Figure S8B). NLRP3 inflammasome activation in Tie2-ERT2-Cre+/−TBCEfl/fl was confirmed by immunoblotting for NACHT, LRR, FIIND, CARD domain, and PYD domains-containing protein 1 (NALP1), NLRP3 and their downstream target cleaved caspase 1 which was increased by AngII (Supplementary material online, Figure S8C). Endoplasmic reticulum (ER) stress markers calnexin, CHOP and protein kinase RNA-like endoplasmic reticulum kinase (PERK) as well as cleaved caspase-1 were increased in aortas and in ECs isolated from Tie2-ERT2-Cre+/−TBCEfl/fl mice, in part augmented by AngII (Supplementary material online, Figure S8D and E). The slightly increased susceptibility of Tie2-ERT2-Cre+/−TBCEfl/fl to AngII-induced hypertension (Supplementary material online, Figure S9) may be related to this pro-inflammatory vascular phenotype of Tie2-ERT2Cre+/−TBCEfl/fl mice, without a significant accumulation of inflammatory cells at baseline (Supplementary material online, Figure S10). These findings suggest that endothelial-specific deletion of TBCE is associated with endothelial inflammasome activation/ER stress, which may explain the increased wall thickness via a possible cross-talk between ECs and VSMCs.
rs6675944 polymorphism was associated with increased carotid IMT in our GWAS (Table 1), suggesting a putative role of TBCE in VSMCs. We therefore sought to elucidate the role of TBCE in VSMCs in more detail by generating the SMMHC-ERT2-Cre+/−TBCEtm2c mice (Supplementary material online, Figure S2). The VSMC-specific TBCE deficiency was confirmed in microcirculation and in the aortas (Supplementary material online, Figure S11). In contrast to myeloid and EC-specific knockouts, the SMMHC-ERT2-Cre+/−TBCEfl/fl mice presented a decreased survival rate compared to their SMMHC-ERT2-Cre+/−TBCEwt/wt controls (Supplementary material online, Figure S3C).
Compared to controls, SMMHC-ERT2-Cre+/−TBCEfl/fl aortas showed reduced response to PGF2α induced constriction with an even lesser response in SMMHC-ERT2-Cre+/−TBCEfl/fl+AngII group (Figure 2A). SMMHC-ERT2-Cre+/−TBCEfl/fl mice were characterized by increased aortic stiffness, further exacerbated by AngII in vivo (Figure 2B and C). SMMHC-ERT2-Cre+/−TBCEfl/fl aortas had endothelial dysfunction at baseline compared to their controls (Figure 2D–G). Vascular dysfunction was accompanied by increased left common carotid artery (LCCA) resistance and pulsatility indexes (Figure 2H–J). Histological evaluation of the aortas showed that wall thickness was increased in SMMHC-ERT2-Cre+/−TBCEfl/fl mice, independent of AngII treatment. AngII infusion induced collagen deposition both in SMMHC-ERT2-Cre+/−TBCEwt/wt and SMMHC-ERT2-Cre+/−TBCEfl/fl mice, but more prominently in the SMMHC-ERT2-Cre+/−TBCEfl/fl + AngII group (Figure 2K–M). Taken together, VSMC-targeted TBCE ablation leads to vascular dysfunction and stiffness, which is exacerbated by AngII.
Figure 2.

Smooth muscle-specific TBCE deficiency leads to endothelial dysfunction, vascular stiffness and increased vessel wall thickness. (A–G) Concentration/relaxation curves of isolated aortic rings. (A) Maximal contraction (g) in response to PGF2α. (B and C) Vascular tension (g) curves in response to ACh and GTN (Two-way ANOVA, Tukey’s multiple comparison test). Vascular concentration-relaxation curves in response to ACh and GTN of sham-operated (D and F) and AngII infused (E and G; 1 mg/kg/day) mice (n = 6–8 per group) (two-way Student’s t-test). (H) Representative ultrasound images of B-mode, M-mode and pulsed-wave Doppler of the murine aortas and graphs of left common carotid artery (LCCA). (I) Resistance index and (J) Pulsatility index (n = 5–6 per group). (K) Sirius red (collagen) and haematoxylin–eosin staining of murine aortas and graphs of L. Media thickness (µm) and (M) Collagen/media thickness (n = 4–5 per group); bars represent 20μm and images were gained at a 40 × magnification (n = 3–5 per group) (one-way ANOVA, Tukey’s multiple comparison test).
AngII infusion led to a significant increase in LCCA wall thickness in the SMMHC-ERT2-Cre+/−TBCEfl/fl mice reflective of increased vascular stiffness. It increased transverse aortic diameter in both SMMHC-ERT2-Cre+/−TBCEwt/wt and SMMHC-ERT2-Cre+/−TBCEfl/fl mice by trend (Supplementary material online, Figure S12). Moreover, AngII led to a significant increase in systolic and diastolic blood pressure in both SMMHC-ERT2-Cre+/−TBCEwt/wt and SMMHC-ERT2-Cre+/−TBCEfl/fl mice, while only the SMMHC-ERT2-Cre+/−TBCEfl/fl+AngII animals had a significant increase in heart rate, left ventricular ejection fraction and left ventricular posterior wall thickness (Supplementary material online, Figure S13).
VSMC-specific deficiency of TBCE modulates vascular inflammatory gene expression, promotes augmentation of endoplasmic reticulum stress and causes a proliferative phenotype paralleled by calnexin up-regulation
Expression analysis of the aortas confirmed down-regulation of the Tbce mRNA in SMMHC-ERT2-Cre+/−TBCEfl/fl mice. AngII led to an up-regulation of vascular inflammation markers, such as Ccl2, Il6, Il1β, Tnfα and Vcam1 in controls and was significantly attenuated in the SMMHC-ERT2-Cre+/−TBCEfl/fl mice (Figure 3A). This phenotype was not accompanied by a significant immune cell infiltration in the SMMHC-ERT2-Cre+/−TBCEfl/fl aortas (Supplementary material online, Figure S14). Interestingly, AngII treatment of the floxed mice led to a 28-fold increase of Lcn2, encoding for lipocalin-2 or neutrophil gelatinase-associated lipocalin, a growth factor and iron sequester involved in innate immune responses.27 Immunofluorescence staining showed an increased abundance of calnexin in the aortic vasculature of SMMHC-ERT2-Cre+/−TBCEfl/fl mice, indicating increased ER stress (Figure 3B). This was confirmed in VSMCs isolated from the murine aortas, showing a significant increase in calnexin, protein-disulfide isomerase (PDI) and CHOP in response to AngII (100 nM in vitro) and more pronounced in the SMMHC-ERT2-Cre+/−TBCEfl/fl group, showing signs of ER stress already at baseline. Inositol-requiring enzyme 1α (IRE-1α) is an intracellular sensor protein of misfolded protein response and was decreased in both AngII-treated and sham-treated VSMCs from SMMHC-ERT2-Cre+/−TBCEfl/fl mice, compatible with ER stress (Figure 3C). In aortic lysates, immunoblotting revealed an AngII-mediated increase in calnexin, CHOP and PDI expression, which was more pronounced in SMMHC-ERT2-Cre+/−TBCEfl/fl mice (Figure 3D).
Figure 3.

Pivotal role of ER stress for the vascular phenotype of SMMHC-ERT2-Cre+/−TBCEfl/fl mice. (A) RT-PCR data of mRNA expression of the differentially regulated genes from the murine aortas (n = 5–6 per group). (B) Immunofluorescent images of SMMHC-ERT2-Cre+/−TBCEfl/fl aortas stained against DAPI (blue) and Calnexin (cyan) (64 × magnification, white bar represents 20 µm). Protein expression analysis of ER stress markers in vascular smooth muscle cells (C, n = 3 per group) and murine aortas (D, n = 4–5 per group). Top: Representative Western blot images; Bottom: Relative densitometry analysis of proteins normalized to α-actinin. Tukey’s multiple comparison test; One-way ANOVA.
To further pinpoint putative mechanisms of TBCE-related vascular dysfunction, primary VSMCs were isolated from aortas and analysed by immunofluorescence confocal microscopy. Decreased TBCE expression along with double-nucleated cells positive for α-tubulin staining indicating a proliferative phenotype were observed in SMMHC-ERT2-Cre+/−TBCEfl/fl mice (Figure 4A, C and D). Furthermore, immunofluorescence staining revealed up-regulated calnexin in VSMCs from SMMHC-ERT2-Cre+/−TBCEfl/fl mice, independently of AngII treatment (Figure 4B and E) and in line with augmented ER stress. We found significantly higher proliferation of VSMCs from SMMHC-ERT2-Cre+/−TBCEfl/fl mice compared to controls at baseline which was increased by AngII (100 nM) in VSMCs from both groups (Figure 4F). Using RNA silencing, we confirmed in human aortic smooth muscle cells (HAoSMCs), that decreased TBCE expression is associated with increased α-tubulin expression (Figure 4G–J). AngII treatment caused an increase in calnexin expression in control-transfected HAoSMCs, while the TBCE-silenced HAoSMCs had an increased calnexin expression at baseline, which was further up-regulated by AngII (Figure 4H and K). AngII increased proliferation of HAoSMCs in both the control and TBCE-siRNA group, while siRNA-transfected VSMCs exhibited a proliferative phenotype even in the absence of AngII (Figure 4L). Taken together, these data support a calnexin-associated increase in ER stress in VSMC-specific TBCE deficiency, which is linked to vascular inflammation and proliferation.
Figure 4.

TBCE knockdown led to a proliferating phenotype in both primary murine VSMCs and in TBCE-silenced HAoSMCs. Confirmation of ER stress induction in vitro. Representative immunofluorescence images of (A) primary VSMCs isolated from murine aortas, stained for DAPI (blue), TBCE (green) and α-tubulin (magenta) (upper panel) or phalloidin (green, F-actin; n = 3 representative images per group), α-tubulin (magenta) and DAPI (blue) (lower panel) as cellular morphology markers; and (B) primary VSMCs isolated from murine aortas (±AngII treatment) were stained for DAPI (blue), α-tubulin (magenta) and calnexin (cyan). Graphs of integrated fluorescence intensity of (C) TBCE/DAPI (n = 3–4 per group) (D) α-tubulin/DAPI (n = 7–8 per group) (E) Calnexin/DAPI (n = 5–9 per group) (F) MTT proliferation assay of primary VSMCs (±AngII treatment) (n = 3–7 per group). Representative immunofluorescence images of (G) human aortic VSMCs (HAoSMCs) transfected with negative control siRNA (NgCTL) or with TBCE siRNA stained for DAPI (blue), TBCE (green), and α-tubulin (magenta) (upper panel) or phalloidin (green, F-actin; n = 3 representative images per group), α-tubulin (magenta) and DAPI (blue) (lower panel) as cellular morphology markers; and of (H) transfected HAoSMCs (± AngII treatment) stained for DAPI (blue), α-tubulin (magenta), and calnexin (cyan). Graphs of integrated fluorescence intensity of (I) TBCE/DAPI (n = 4 per group) (J) α-tubulin/DAPI (n = 6 per group) (K) Calnexin/DAPI (n = 4–3 per group) (L) MTT proliferation assay of primary VSMCs (±AngII treatment) (n = 4 per group) (Tukey’s multiple comparison test; one-way ANOVA; different numbers in the in vitro experiments refer to individual experiments).
TUDCA reverses vascular dysfunction in mice with VSMC-specific deficiency of TBCE and decreases VSMC proliferation by amplification of Raptor/Beclin-1-mediated autophagy
To test whether the phenotype of TBCE-deficient mice could be modulated pharmacologically, we administered the chemical chaperone and ER stress modulator tauroursodeoxycholic acid (TUDCA, 300 mg/kg/day) to SMMHC-ERT2Cre+/−TBCEfl/fl mice. TUDCA reversed vascular dysfunction, as presented by restored ACh-mediated relaxation (Figure 5A and B). TUDCA administration decreased PGF2a induced maximal constriction both in SMMHC-ERT2-Cre+/−TBCEfl/fl and SMMHC-ERT2-Cre+/−TBCEwt/wt mice (Figure 5C). Western blot analysis revealed that TUDCA decreased the ER stress markers Bip and CHOP in the SMMHC-ERT2-Cre+/−TBCEfl/fl mice, while it led to an enhancement of the Raptor/Beclin-1 pathway (which plays an important role in autophagy28) independently of microtubule-associated proteins 1A/1B light chain 3B (LC3B, Figure 5D and E). In vitro, TUDCA treatment reversed the hyper-proliferative phenotype of SMMHC-ERT2-Cre+/−TBCEfl/fl VSMCs, decreasing their proliferation and cellular confluency (Figure 5F and G). Protein expression analysis revealed that TUDCA decreased CHOP expression and caused an up-regulation of Raptor/Beclin-1, independently of LC3B in vitro (Figure 5H and I). Therefore, TBCE deficiency in VSMCs drives vascular dysfunction, which can be attenuated by TUDCA-induced ER stress modulation and amplification of Raptor/Beclin-1-mediated autophagy.
Figure 5.

The chemical chaperone TUDCA reverses vascular dysfunction and VSMCs proliferation in mice with SMC-specific TBCE deficiency. Concentration/relaxation curves of isolated aortic rings, in response to (A) ACh and (B) GTN (Two-Way ANOVA, Tukey’s multiple comparison test) (C) Maximal contraction (g) in response to PGF2α (D) Representative western blot images and (E) Graph of relative densitometry analysis of ER stress and autophagy markers of the murine aortas. (F) MTT absorbance (570 nm) graph of the SMMHC-ERT2-Cre+/−TBCEwt/wt and SMMHC-ERT2-Cre+/−TBCEfl/fl VSMCs with/without TUDCA treatment (1 mM) (G) Confluency images of the primary VSMCs in culture with/without TUDCA treatment (1 mM). (H) Representative Western blot images and I. graph of relative densitometry analysis of ER stress and autophagy markers of the primary VSMCs normalized to α-actinin. (Tukey’s multiple comparison test; 1 way ANOVA; Different n numbers in the in vitro experiments refer to individual experiments; Dotted line on western blot images represent a readjustment of the representative image).
Discussion
In the current study, we present novel evidence that SNPs in the TBCE gene and TBCE down-regulation are important determinants of vascular dysfunction both in humans and mice. While endothelial-targeted TBCE deficiency induces a moderate inflammatory phenotype in the aorta, VSMCs-targeted TBCE deficiency leads to exacerbated vascular stiffness and inflammation, smooth muscle cell proliferation, and endothelial dysfunction associated with ER stress.
GWAS analyses have provided an invaluable tool to identify novel targets and mediators of CVD, including acute and chronic coronary syndrome.29 Among those studies, novel non-coding SNPs,30 as well as genetic mutations31 have been associated with vascular dysfunction and hypertension in the general population.32 GWAS on the FMD axis are scarce, however, some state-of-the-art GWAS analyses have identified an association of cardiac and vascular functional parameters with distinct genomic profiles.33 It is noteworthy that throughout these genomic associations molecular targets that are already known to effect vascular homeostasis, such as endothelial nitric oxide synthetase or vascular endothelial growth factor have emerged as leading targets of interest.29 The GWAS analysis derived from the GHS presented here is the first that reveals an association of the SNPs rs6675944 and rs12405889 in the TBCE gene with vascular function. rs6675944 polymorphism was additionally associated with increased carotid wall thickness showing an increased susceptibility of the SNP-bearing individuals to arterial stiffness.24
Both rs6675944 and rs12405889 SNPs are found in intron domains of the TBCE gene and lead to the generation of TBCE intron variants. Mutations at a specific site at which intron splicing occurs can cause genetic insertion, deletion or frame shift. Mutations in these sequences may lead to retention of large segments of intronic DNA during mRNA maturation, or to entire exon removal. These changes can result in the generation of a non-functional protein and result in the manifestation of diseases.34 Alternative Splice Site Predictor (ASSP) software35 revealed that the rs6675944 site is a highly probable alternative isoform/cryptic 5' splice site (activation score: 0.879; confidence: 0.908) while rs12405889 is a highly probable alternative isoform/cryptic 3' splice site (activation score: 0.674; confidence: 0.533). Therefore, SNPs are highly probable to mediate alternative or cryptic splicing of the TBCE gene. Cryptic splice sites are usually not spliced during transcription leading to the accumulation of intronic DNA and therefore might lead to the generation of a non-functional protein.36 Finally, the Ensembl genome database37 revealed that rs6675944 SNP can result in 37 transcripts that undergo nonsense-mediated decay and 6 non-coding transcript exon variants out of the total of 61 transcripts, indicating that rs6675944 has a 70% probability of leading to a non-functional or truncated protein. rs12405889 can result in 39 transcripts that undergo nonsense-mediated decay and 8 non-coding transcript exon variants out of the total of 61 transcripts, indicating that rs12405889 has a 77% probability of leading to a non-functional or truncated protein.
TBCE specifically modulates α-tubulin incorporation in microtubule networks, which are responsible for cellular integrity, protein transportation and signal transduction.38 Deletion and truncation of TBCE in humans are known to lead to the Sanjad-Sakati or Kenny-Caffey syndrome, which is characterized by congenital hypoparathyroidism, mental retardation, facial dysmorphism and extreme growth deficits,39 while it is also associated with the prevalence of amyotrophic lateral sclerosis.40 So far, research on TBCE has focused on the central nervous system,41 where it has been shown that down-regulation of TBCE leads to Golgi microtubule deficits, which in turn leads to accumulation of ER-Golgi SNAREs.40 , 42 In addition, it was shown in yeast that TBCE can secure the connection between microtubules and proteasome, protecting cells against misfolded protein stress in a cAMP-independent regulatory protein (PAC2)-dependent manner.43
Conclusively, in silico data show that both rs6675944 and rs12405889 SNPs—despite the fact that they are intronic SNPs—are characterized by an epigenetic regulatory trait, while having high probability to result in the generation of a truncated or dysfunctional protein. These findings prompted us to investigate TBCE-associated vascular effects in a translational approach in vivo.
With the present studies, we identified that endothelial-specific knockdown of TBCE leads to increased vessel wall thickness. Mechanistically, endothelial-specific TBCE knockdown was found to be associated with an inflammatory phenotype of the aortas and induction of ER stress. ER stress, a physiological response to unfolded protein accumulation, was associated with NLRP3 activation in the endothelium44 and could act as a mediator of CVDs and hypertension.45 In our model, activation of ER stress may be responsible for activation of NLRP3 inflammasome, which leads to production and release of cytokines, such as interleukin-6. The aforementioned inflammatory stimuli may therefore orchestrate the crosstalk between ECs and VSMCs46 and induce VSMC proliferation and hypertrophy causing the thickened vessel wall47 as seen in the Tie2-ERT2Cre+/−TBCEfl/fl transgenic mice.
The VSMC-targeted transgenic mice showed vascular dysfunction and stiffness already at baseline, which was aggravated by AngII treatment. Lipocalin-2 (LCN2) was up-regulated in the SMMHC-ERT2-Cre+/−TBCEfl/fl aortas, especially after AngII treatment (Figure 3A). LCN2 is associated with increased VSMC proliferation and migration48 or pro-fibrotic effects49 and is correlated with symptomatic carotid atherosclerosis in humans.50 LCN2 is a key inducer of ER stress in human pulmonary arterial smooth muscle cells and augments the progression of pulmonary hypertension.51
By investigating aortas and VSMCs isolated from the transgenic mice, we found that vascular stiffness and dysregulation were related to ER stress. Both in vivo and in vitro, ER chaperones and indicators of ER stress such as calnexin and PDI52 were up-regulated when TBCE was diminished in VSMCs, leading to increased VSMC proliferation.
To confirm that ER stress is involved in mediating the vascular phenotype of SMMHC-ERT2-Cre+/−TBCEfl/fl mice, we used the chemical chaperone TUDCA, shown to mitigate ER stress in multiple cell types53 including VSMCs.54–56 Oral administration of TUDCA reversed vascular dysfunction at the selected dose of 300 mg/kg. Furthermore, it decreased PGF2α-induced contractility, paralleled by the induction of Raptor/Beclin-1 related autophagy.57 Evidence on TUDCA-induced autophagy are manifold; it may both inhibit and induce ER stress-mediated autophagy in nerve cells.58 However, it is also established that TUDCA is capable of modulating the autophagy machinery and induce/amplify autophagy signalling in familial amyloidotic polyneuropathy.59 It may also lead to Beclin-1-dependent autophagic flux,58 supporting our in vivo and in vitro data.
The role of TUDCA in the treatment of vascular dysfunction is currently addressed by ongoing clinical studies [NCT03331432: Effects of Dietary Supplement Tauroursodeoxycholic Acid on Vascular Function (completed); NCT04001647: Targeting ER Stress in Vascular Dysfunction (recruiting); data from https://ClinicalTrials.gov]. TUDCA has shown to be beneficial in patients with amyotrophic lateral sclerosis in terms of cumulative survival and Amyotrophic Lateral Sclerosis Functional Rating Scale Revised score60 and in obese-diabetic patients in terms of insulin reactivity,61 apparently with little side effects. It therefore seems to be a safe dietary supplement and may be a promising agent against arterial hypertension. This is supported by the notion that in translational murine models, TUDCA has anti-hypertensive effects. In ob/ob mice, TUDCA mitigated arterial hypertension and cardiac hypertrophy.62 In spontaneously hypertensive rats and in type 2 diabetic mice, TUDCA exhibited anti-hypertensive potential and improved coronary arterial function, at least in part by ameliorating ER stress.63 , 64
Conclusively, TBCE down-regulation leads to a multi-faceted vascular dysfunction in mice and humans. Most significantly, down-regulation of the chaperone TBCE in VSMCs leads to severe vascular dysfunction and stiffness characterized by an aberrant ER response and VSMC hyperplasia. Modulation of ER stress by the chemical chaperone TUDCA, attenuated the phenotype through enhancement of Beclin-1-induced autophagy. The effectiveness of TUDCA (approved in the USA for the treatment of primary biliary cirrhosis53) suggests putative translational value of our findings.
This is the first study investigating the role of TBCE from bedside to bench, highlighting the role of microtubule dynamics and ER stress in the vascular homeostasis (see Graphical Abstract). It represents a translational approach to identify novel targets of vascular dysfunction through a GWAS analysis of the general population, leading to the discovery of up-to-now unknown contributors to vascular dysfunction.
Graphical Abstract.
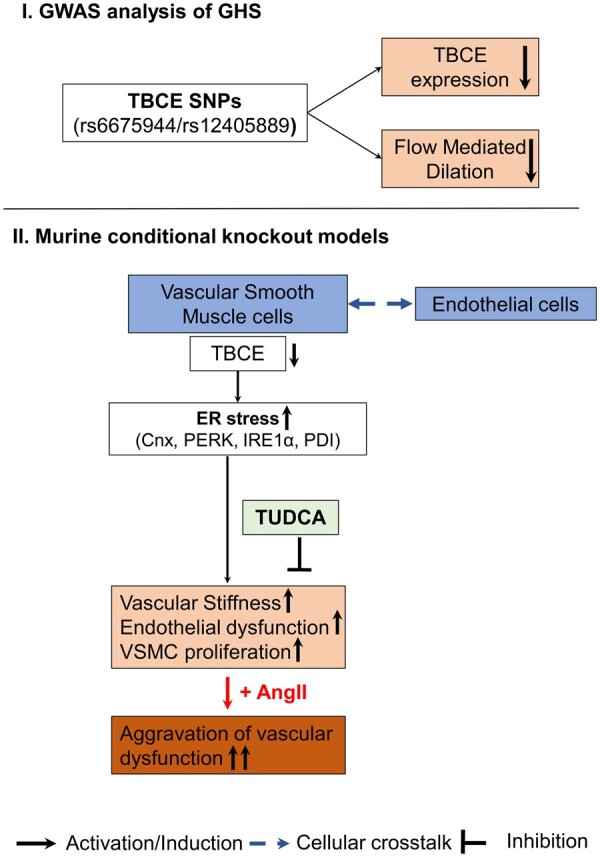
Schematic representation of the main findings regarding (i) the genome-wide association study (GWAS) analysis of the Gutenberg Health Study (GHS), and (ii) the translational approach in the transgenic vascular smooth muscle cell-specific tubulin-folding cofactor E (TBCE) knockout murine model. Arrows represent activation/induction pathways, T lines present inhibition pathways, while dotted arrows present cellular crosstalk between endothelial and smooth muscle cells as observed in vivo. Cnx, calnexin; ER, endoplasmic reticulum; IRE1α, inositol-requiring enzyme 1α; PDI, protein disulfide-isomerase; PERK, protein kinase RNA-like endoplasmic reticulum kinase; SNP, single-nucleotide polymorphism; TBCE, tubulin-folding cofactor E; TUDCA, tauroursodeoxycholic acid.
Limitations of the study
Despite the fact that our study extensively investigated the role of TBCE in vascular function in a translational manner, there are some study limitations. It should be noted that the data presented here may be limited by small sample sizes; a non-significant difference cannot be interpreted as a lack of association. Blood pressure in vivo was assessed by a non-invasive methodology, namely tail-cuff measurements and not by the invasive telemetry catheter implantation regarded as gold standard of blood pressure monitoring. Moreover, ER stress is a general term, describing more complex pathophysiological processes than we assessed in this work. More functional experiments will be needed to define the role of TBCE in ER stress in more detail.65 Even though TUDCA has a well-established effect on ER stress, it has a multifaceted mechanism and we cannot rule out that its protective effects against vascular dysfunction are mediated—at least in part—by other modes of action that are not related to TBCE deficiency. To strengthen the findings from our GWAS study, it should be considered to validate our associations in an independent cohort in order to increase the translational value of TBCE SNPs as mediators of vascular dysfunction.
Supplementary material
Supplementary material is available at European Heart Journal online.
Data availability
The data underlying this article are available in the article and in its online supplementary material.
Supplementary Material
Acknowledgements
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal and dbGaP accession number phs000424.vN.pN on June 2019.
The expert technical assistance of Katharina Perius and Anne-Kristin Conze is gratefully acknowledged.
Funding
This work was supported by grants of the German Federal Ministry for Education and Research (BMBF01EO1003 and BMBF01EO1503), the DFG Major Research Instrumentation Programme (DFG INST 371/47-1 FUGG), as well as the Boehringer Ingelheim Foundation. M.M. is supported by a grant of the Else-Kröner-Fresenius-Stiftung (2020_EKEA.144). P.W. received funds from the German Research Foundation in support of his work (DFG WE4361-4-1 and WE 4361/7-1). K.S., P. Wild, T.M. and P. Wenzel are Principal Investigators of the DZHK.
Conflict of interest: none declared.
Contributor Information
Panagiotis Efentakis, Department of Cardiology, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Michael Molitor, Department of Cardiology, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; German Center for Cardiovascular Research (DZHK)—Partner site Rhine-Main, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Sabine Kossmann, Department of Cardiology, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Magdalena L Bochenek, Department of Cardiology, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; German Center for Cardiovascular Research (DZHK)—Partner site Rhine-Main, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Johannes Wild, Department of Cardiology, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Jeremy Lagrange, Department of Cardiology, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Stefanie Finger, Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Rebecca Jung, Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Susanne Karbach, Department of Cardiology, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; German Center for Cardiovascular Research (DZHK)—Partner site Rhine-Main, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Katrin Schäfer, Department of Cardiology, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; German Center for Cardiovascular Research (DZHK)—Partner site Rhine-Main, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Andreas Schulz, Department of Cardiology—Preventive Cardiology and Medical Prevention, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Philipp Wild, Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; German Center for Cardiovascular Research (DZHK)—Partner site Rhine-Main, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; Department of Cardiology—Preventive Cardiology and Medical Prevention, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Thomas Münzel, Department of Cardiology, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; German Center for Cardiovascular Research (DZHK)—Partner site Rhine-Main, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
Philip Wenzel, Department of Cardiology, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; Center for Thrombosis and Hemostasis, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany; German Center for Cardiovascular Research (DZHK)—Partner site Rhine-Main, University Medical Center Mainz, Langenbeckstraße 1, 55131 Mainz, Germany.
References
- 1. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000;87:840–844. [DOI] [PubMed] [Google Scholar]
- 2. Corban MT, Lerman LO, Lerman A. Endothelial dysfunction. Arterioscler Thromb Vasc Biol 2019;39:1272–1274. [DOI] [PubMed] [Google Scholar]
- 3. Savira F, Magaye R, Liew D, Reid C, Kelly D, Kompa AR, Sangaralingham SJ, Burnett JC Jr, Kaye D, Wang BH. Cardiorenal syndrome: multi-organ dysfunction involving the heart, kidney and vasculature. Br J Pharmacol 2020;177:2906–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation 2007;115:1285–1295. [DOI] [PubMed] [Google Scholar]
- 5. Efentakis P, Varela A, Chavdoula E, Sigala F, Sanoudou D, Tenta R, Gioti K, Kostomitsopoulos N, Papapetropoulos A, Tasouli A, Farmakis D, Davos CH, Klinakis A, Suter T, Cokkinos DV, Iliodromitis EK, Wenzel P, Andreadou I. Levosimendan prevents doxorubicin-induced cardiotoxicity in time- and dose dependent manner: implications for inotropy. Cardiovasc Res 2020;116:576–591. [DOI] [PubMed] [Google Scholar]
- 6. Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res 2012;95:194–204. [DOI] [PubMed] [Google Scholar]
- 7. Bezie Y, Lacolley P, Laurent S, Gabella G. Connection of smooth muscle cells to elastic lamellae in aorta of spontaneously hypertensive rats. Hypertension 1998;32:166–169. [DOI] [PubMed] [Google Scholar]
- 8. Flammer AJ, Anderson T, Celermajer DS, Creager MA, Deanfield J, Ganz P, Hamburg NM, Luscher TF, Shechter M, Taddei S, Vita JA, Lerman A. The assessment of endothelial function: from research into clinical practice. Circulation 2012;126:753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Witte DR, Westerink J, de Koning EJ, van der Graaf Y, Grobbee DE, Bots ML. Is the association between flow-mediated dilation and cardiovascular risk limited to low-risk populations? J Am Coll Cardiol 2005;45:1987–1993. [DOI] [PubMed] [Google Scholar]
- 10. Yeboah J, Folsom AR, Burke GL, Johnson C, Polak JF, Post W, Lima JA, Crouse JR, Herrington DM. Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: the multi-ethnic study of atherosclerosis. Circulation 2009;120:502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matsuzawa Y, Kwon TG, Lennon RJ, Lerman LO, Lerman A. Prognostic value of flow-mediated vasodilation in brachial artery and fingertip artery for cardiovascular events: a systematic review and meta-analysis. J Am Heart Assoc 2015;4:e002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ras RT, Streppel MT, Draijer R, Zock PL. Flow-mediated dilation and cardiovascular risk prediction: a systematic review with meta-analysis. Int J Cardiol 2013;168:344–351. [DOI] [PubMed] [Google Scholar]
- 13. Schnabel RB, Wild PS, Schulz A, Zeller T, Sinning CR, Wilde S, Kunde J, Lubos E, Lackner KJ, Warnholtz A, Gori T, Blankenberg S, Munzel T. Gutenberg Health Study Investigators. Multiple endothelial biomarkers and noninvasive vascular function in the general population: the Gutenberg Health Study. Hypertension 2012;60:288–295. [DOI] [PubMed] [Google Scholar]
- 14. Schnabel RB, Schulz A, Wild PS, Sinning CR, Wilde S, Eleftheriadis M, Herkenhoff S, Zeller T, Lubos E, Lackner KJ, Warnholtz A, Gori T, Blankenberg S, Munzel T. Noninvasive vascular function measurement in the community: cross-sectional relations and comparison of methods. Circ Cardiovasc Imaging 2011;4:371–380. [DOI] [PubMed] [Google Scholar]
- 15. Panova-Noeva M, Schulz A, Hermanns MI, Grossmann V, Pefani E, Spronk HMH, Laubert-Reh D, Binder H, Beutel M, Pfeiffer N, Blankenberg S, Zeller T, Münzel T, Lackner KJ, ten Cate H, Wild PS. Sex-specific differences in genetic and nongenetic determinants of mean platelet volume: results from the Gutenberg Health Study. Blood 2016;127:251–259. [DOI] [PubMed] [Google Scholar]
- 16. Coleman JL, Brennan K, Ngo T, Balaji P, Graham RM, Smith NJ. Rapid knockout and reporter mouse line generation and breeding colony establishment using EUCOMM conditional-ready embryonic stem cells: a case study. Front Endocrinol (Lausanne) 2015;6:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011;474:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 1999;8:265–277. [DOI] [PubMed] [Google Scholar]
- 19. Forde A, Constien R, Grone HJ, Hammerling G, Arnold B. Temporal Cre-mediated recombination exclusively in endothelial cells using Tie2 regulatory elements. Genesis 2002;33:191–197. [DOI] [PubMed] [Google Scholar]
- 20. Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 2008;14:64–68. [DOI] [PubMed] [Google Scholar]
- 21. Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG; National Centre for the Replacement, Refinement and Reduction of Amimals in Research. Animal research: reporting in vivo experiments—the ARRIVE guidelines. J Cereb Blood Flow Metab 2011;31:991–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lockette WE, Webb RC, Bohr DF. Prostaglandins and potassium relaxation in vascular smooth muscle of the rat. The role of Na-K ATPase. Circ Res 1980;46:714–720. [DOI] [PubMed] [Google Scholar]
- 23. Kossmann S, Lagrange J, Jackel S, Jurk K, Ehlken M, Schonfelder T, Weihert Y, Knorr M, Brandt M, Xia N, Li H, Daiber A, Oelze M, Reinhardt C, Lackner K, Gruber A, Monia B, Karbach SH, Walter U, Ruggeri ZM, Renne T, Ruf W, Munzel T, Wenzel P. Platelet-localized FXI promotes a vascular coagulation-inflammatory circuit in arterial hypertension. Sci Transl Med 2017;9:eaah4923. [DOI] [PubMed] [Google Scholar]
- 24. Koivistoinen T, Virtanen M, Hutri-Kähönen N, Lehtimäki T, Jula A, Juonala M, Moilanen L, Aatola H, Hyttinen J, Viikari JSA, Raitakari OT, Kähönen M. Arterial pulse wave velocity in relation to carotid intima-media thickness, brachial flow-mediated dilation and carotid artery distensibility: the Cardiovascular Risk in Young Finns Study and the Health 2000 Survey. Atherosclerosis 2012;220:387–393. [DOI] [PubMed] [Google Scholar]
- 25. https://gtexportal.org.
- 26. https://www.ncbi.nlm.nih.gov/snp/.
- 27. Schmidt-Ott KM, Mori K, Li JY, Kalandadze A, Cohen DJ, Devarajan P, Barasch J. Dual action of neutrophil gelatinase-associated lipocalin. J Am Soc Nephrol 2007;18:407–413. [DOI] [PubMed] [Google Scholar]
- 28. Mondaca-Ruff D, Riquelme JA, Quiroga C, Norambuena-Soto I, Sanhueza-Olivares F, Villar-Fincheira P, Hernandez-Diaz T, Cancino-Arenas N, San Martin A, Garcia L, Lavandero S, Chiong M. Angiotensin II-regulated autophagy is required for vascular smooth muscle cell hypertrophy. Front Pharmacol 2018;9:1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang X. Use of functional genomics to identify candidate genes underlying human genetic association studies of vascular diseases. Arterioscler Thromb Vasc Biol 2012;32:216–222. [DOI] [PubMed] [Google Scholar]
- 30. Madelaine R, Notwell JH, Skariah G, Halluin C, Chen CC, Bejerano G, Mourrain P. A screen for deeply conserved non-coding GWAS SNPs uncovers a MIR-9-2 functional mutation associated to retinal vasculature defects in human. Nucleic Acids Res 2018;46:3517–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rudemiller NP, Lund H, Priestley JR, Endres BT, Prokop JW, Jacob HJ, Geurts AM, Cohen EP, Mattson DL. Mutation of SH2B3 (LNK), a genome-wide association study candidate for hypertension, attenuates Dahl salt-sensitive hypertension via inflammatory modulation. Hypertension 2015;65:1111–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Han Y, Li L, Zhang Y, Yuan H, Ye L, Zhao J, Duan DD. Phenomics of vascular disease: the systematic approach to the combination therapy. Curr Vasc Pharmacol 2015;13:433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vasan RS, Larson MG, Aragam J, Wang TJ, Mitchell GF, Kathiresan S, Newton-Cheh C, Vita JA, Keyes MJ, O'Donnell CJ, Levy D, Benjamin EJ. Genome-wide association of echocardiographic dimensions, brachial artery endothelial function and treadmill exercise responses in the Framingham Heart Study. BMC Med Genet 2007;8: S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nat Biotechnol 2004;22:535–546. [DOI] [PubMed] [Google Scholar]
- 35. Wang M, Marin A. Characterization and prediction of alternative splice sites. Gene 2006;366:219–227. [DOI] [PubMed] [Google Scholar]
- 36. Roca X, Krainer AR, Eperon IC. Pick one, but be quick: 5' splice sites and the problems of too many choices. Genes Dev 2013;27:129–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, Bhai J, Billis K, Boddu S, Marugán JC, Cummins C, Davidson C, Dodiya K, Fatima R, Gall A, Giron CG, Gil L, Grego T, Haggerty L, Haskell E, Hourlier T, Izuogu OG, Janacek SH, Juettemann T, Kay M, Lavidas I, Le T, Lemos D, Martinez JG, Maurel T, McDowall M, McMahon A, Mohanan S, Moore B, Nuhn M, Oheh DN, Parker A, Parton A, Patricio M, Sakthivel MP, Abdul Salam AI, Schmitt BM, Schuilenburg H, Sheppard D, Sycheva M, Szuba M, Taylor K, Thormann A, Threadgold G, Vullo A, Walts B, Winterbottom A, Zadissa A, Chakiachvili M, Flint B, Frankish A, Hunt SE, IIsley G, Kostadima M, Langridge N, Loveland JE, Martin FJ, Morales J, Mudge JM, Muffato M, Perry E, Ruffier M, Trevanion SJ, Cunningham F, Howe KL, Zerbino DR, Flicek P. Ensembl 2020. Nucleic Acids Res 2020;48:D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Serna M, Carranza G, Martin-Benito J, Janowski R, Canals A, Coll M, Zabala JC, Valpuesta JM. The structure of the complex between alpha-tubulin, TBCE and TBCB reveals a tubulin dimer dissociation mechanism. J Cell Sci 2015;128:1824–1834. [DOI] [PubMed] [Google Scholar]
- 39. Parvari R, Hershkovitz E, Grossman N, Gorodischer R, Loeys B, Zecic A, Mortier G, Gregory S, Sharony R, Kambouris M, Sakati N, Meyer BF, Al AA, Al Humaidan AK, Al Zanhrani F, Al Swaid A, Al Othman J, Diaz GA, Weiner R, Khan KT, Gordon R, Gelb BD; HRD/Autosomal Recessive Kenny-Caffey Syndrome Consortium. Consortium HRARK-CS. Mutation of TBCE causes hypoparathyroidism-retardation-dysmorphism and autosomal recessive Kenny-Caffey syndrome. Nat Genet 2002;32:448–452. [DOI] [PubMed] [Google Scholar]
- 40. Haase G, Rabouille C. Golgi fragmentation in ALS motor neurons. New mechanisms targeting microtubules, tethers, and transport vesicles. Front Neurosci 2015;9:448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schafer MK, Bellouze S, Jacquier A, Schaller S, Richard L, Mathis S, Vallat JM, Haase G. Sensory neuropathy in progressive motor neuronopathy (pmn) mice is associated with defects in microtubule polymerization and axonal transport. Brain Pathol 2017;27:459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bellouze S, Schafer MK, Buttigieg D, Baillat G, Rabouille C, Haase G. Golgi fragmentation in pmn mice is due to a defective ARF1/TBCE cross-talk that coordinates COPI vesicle formation and tubulin polymerization. Hum Mol Genet 2014;23:5961–5975. [DOI] [PubMed] [Google Scholar]
- 43. Voloshin O, Gocheva Y, Gutnick M, Movshovich N, Bakhrat A, Baranes-Bachar K, Bar-Zvi D, Parvari R, Gheber L, Raveh D. Tubulin chaperone E binds microtubules and proteasomes and protects against misfolded protein stress. Cell Mol Life Sci 2010;67:2025–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lenna S, Han R, Trojanowska M. Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life 2014;66:530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Scott TE, Kemp-Harper BK, Hobbs AJ. Inflammasomes: a novel therapeutic target in pulmonary hypertension? Br J Pharmacol 2019;176:1880–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jia LX, Zhang WM, Li TT, Liu Y, Piao CM, Ma YC, Lu Y, Wang Y, Liu TT, Qi YF, Du J. ER stress dependent microparticles derived from smooth muscle cells promote endothelial dysfunction during thoracic aortic aneurysm and dissection. Clin Sci (Lond) 2017;131:1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lacolley P, Regnault V, Segers P, Laurent S. Vascular smooth muscle cells and arterial stiffening: relevance in development, aging, and disease. Physiol Rev 2017;97:1555–1617. [DOI] [PubMed] [Google Scholar]
- 48. Wang G, Ma N, Meng L, Wei Y, Gui J. Activation of the phosphatidylinositol 3-kinase/Akt pathway is involved in lipocalin-2-promoted human pulmonary artery smooth muscle cell proliferation. Mol Cell Biochem 2015;410:207–213. [DOI] [PubMed] [Google Scholar]
- 49. Tarjus A, Martínez-Martínez E, Amador C, Latouche C, El Moghrabi S, Berger T, Mak TW, Fay R, Farman N, Rossignol P, Zannad F, López-Andrés N, Jaisser F. Neutrophil gelatinase-associated lipocalin, a novel mineralocorticoid biotarget, mediates vascular profibrotic effects of mineralocorticoids. Hypertension 2015;66:158–166. [DOI] [PubMed] [Google Scholar]
- 50. Eilenberg W, Stojkovic S, Piechota-Polanczyk A, Kaun C, Rauscher S, Groger M, Klinger M, Wojta J, Neumayer C, Huk I, Demyanets S. Neutrophil gelatinase-associated lipocalin (NGAL) is associated with symptomatic carotid atherosclerosis and drives pro-inflammatory state in vitro. Eur J Vasc Endovasc Surg 2016;51:623–631. [DOI] [PubMed] [Google Scholar]
- 51. Wang G, Liu S, Wang L, Meng L, Cui C, Zhang H, Hu S, Ma N, Wei Y. Lipocalin-2 promotes endoplasmic reticulum stress and proliferation by augmenting intracellular iron in human pulmonary arterial smooth muscle cells. Int J Biol Sci 2017;13:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Minamino T, Kitakaze M. ER stress in cardiovascular disease. J Mol Cell Cardiol 2010;48:1105–1110. [DOI] [PubMed] [Google Scholar]
- 53. Uppala JK, Gani AR, Ramaiah KVA. Chemical chaperone, TUDCA unlike PBA, mitigates protein aggregation efficiently and resists ER and non-ER stress induced HepG2 cell death. Sci Rep 2017;7:3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim SY, Kwon YW, Jung IL, Sung JH, Park SG. Tauroursodeoxycholate (TUDCA) inhibits neointimal hyperplasia by suppression of ERK via PKCalpha-mediated MKP-1 induction. Cardiovasc Res 2011;92:307–316. [DOI] [PubMed] [Google Scholar]
- 55. Luo H, Zhou C, Chi J, Pan S, Lin H, Gao F, Ni T, Meng L, Zhang J, Jiang C, Ji Z, Lv H, Guo H. The role of tauroursodeoxycholic acid on dedifferentiation of vascular smooth muscle cells by modulation of endoplasmic reticulum stress and as an oral drug inhibiting in-stent restenosis. Cardiovasc Drugs Ther 2019;33:25–33. [DOI] [PubMed] [Google Scholar]
- 56. Liang B, Wang S, Wang Q, Zhang W, Viollet B, Zhu Y, Zou MH. Aberrant endoplasmic reticulum stress in vascular smooth muscle increases vascular contractility and blood pressure in mice deficient of AMP-activated protein kinase-alpha2 in vivo. Arterioscler Thromb Vasc Biol 2013;33:595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rani S, Sreenivasaiah PK, Kim JO, Lee MY, Kang WS, Kim YS, Ahn Y, Park WJ, Cho C, Kim DH. Tauroursodeoxycholic acid (TUDCA) attenuates pressure overload-induced cardiac remodeling by reducing endoplasmic reticulum stress. PLoS One 2017;12:e0176071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Miao L, Dong Y, Zhou FB, Chang YL, Suo ZG, Ding HQ. Protective effect of tauroursodeoxycholic acid on the autophagy of nerve cells in rats with acute spinal cord injury. Eur Rev Med Pharmacol Sci 2018;22:1133–1141. [DOI] [PubMed] [Google Scholar]
- 59. Teixeira C, Saraiva MJ. TUDCA as an autophagic modulator of ATTR V30M Amyloidosis. Orphanet J Rare Dis 2015;10:P10. [Google Scholar]
- 60. Elia AE, Lalli S, Monsurro MR, Sagnelli A, Taiello AC, Reggiori B, La Bella V, Tedeschi G, Albanese A. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur J Neurol 2016;23:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, Patterson BW, Horton JD, Mittendorfer B, Hotamisligil GS, Klein S. Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 2010;59:1899–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ceylan-Isik AF, Sreejayan N, Ren J. Endoplasmic reticulum chaperon tauroursodeoxycholic acid alleviates obesity-induced myocardial contractile dysfunction. J Mol Cell Cardiol 2011;50:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63. Choi SK, Lim M, Byeon SH, Lee YH. Inhibition of endoplasmic reticulum stress improves coronary artery function in the spontaneously hypertensive rats. Sci Rep 2016;6:31925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Choi SK, Lim M, Yeon SI, Lee YH. Inhibition of endoplasmic reticulum stress improves coronary artery function in type 2 diabetic mice. Exp Physiol 2016;101:768–777. [DOI] [PubMed] [Google Scholar]
- 65. Fu S, Yalcin A, Lee GY, Li P, Fan J, Arruda AP, Pers BM, Yilmaz M, Eguchi K, Hotamisligil GS. Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Sci Transl Med 2015;7:292ra98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.