

Abstract
Background
Cyclin‐dependent kinase 13 plays a critical role in the regulation of gene transcription. Recent evidence suggests that heterozygous variants in CDK13 are associated with a syndromic form of mental deficiency and developmental delay, which is inherited in an autosomal dominant manner.
Methods
A mentally retarded mother (33‐year‐old) and son (10‐year‐old boy) in our hospital with CDK13 variant (c.2149 (exon 4) G>A. p.Gly717Arg) were detected by whole‐exome sequencing (WES). All published CDK13 variant syndrome cases as of November 11, 2021, were searched, and their clinical information was recorded and summarized.
Results
We studied two patients in a Chinese family with a heterozygous constitutional CDK13 variant (c.2149 (exon 4) G>A. p.Gly717Arg), exhibiting the classical characteristics of dysmorphic facial features and intellectual developmental disorder (CHDFIDD, OMIM # 617360), without congenital heart defects. This is the first reported case of an adult patient with a CDK13 variant that gave birth to the next generation with the same variant. Preimplantation genetic testing for monogenic disease (PGT‐M) was performed for the proband and her husband with full informed consent and successfully blocked the inheritance of the disease.
Conclusion
Our study is of great significance for molecular diagnosis and genetic counseling of patients with CDHFIDD and extends the variant spectrum of CDK13.
Keywords: CDK13 gene, CHDFIDD, novel variant, PGT‐M, whole‐exome sequencing
This is the first reported case of an adult patient with a CDK13 variant that gave birth to the next generation with the same variant. Our study is of great significance for molecular diagnosis and genetic counseling of patients with CDHFIDD and extends the variant spectrum of CDK13.
1. INTRODUCTION
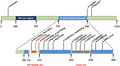
The CDK13 (cyclin‐dependent kinase 13) gene, located on chromosome 7p14.1, contains 16 exons and encodes the CDK13 protein, a member of the cyclin‐dependent serine/threonine protein kinase (STK) family (Hamilton & Suri, 2019). Members of this family are vital master switches in gene expression and cell cycle progression (Bostwick, 2019). Furthermore, this kinase uses adenosine triphosphate (ATP) as a source of phosphate groups and forms, in association with cyclin K, a protein complex that actively regulates transcription by phosphorylation of RNA polymerase (Pol) II (Greenleaf, 2019). Recent work has shown that CDK13 is related to RNA processing and its mouse homolog plays a role in converting the transcribing Pol II from an initiating to an elongating form (Chen et al., 2014; Liang et al., 2015). CDK13 cooperates with CDK12 to control global RNA polymerase II processivity through phosphorylating the C‐terminal domain (Fan et al., 2020; Quereda et al., 2019). The genetic targets of CDK13 activity seem to be mainly involved in processes associated with extracellular and growth signal transduction (Even et al., 2016). Embryonic lethality of mouse model with non‐functional CDK13 has been observed by the embryonic day 16.5, while live embryos were observed on embryonic day 15.5 (Novakova et al., 2019). At this stage, improper development of multiple organs has been documented, partly resembling defects observed in patients with mutated CDK13 (Novakova et al., 2019). To date, pathogenic variants in CDK13 are mainly clustered in the highly conserved STK domain, through molecular modeling, to predict the perturbations of each interaction with cyclin K, ATP, and magnesium binding (Deciphering Developmental Disorders, 2017) (Figure 1).
FIGURE 1.
Schematic of CDK13 domain composition, annotated with the position of reported pathogenic variants. NBR, nucleotide‐binding region; SR‐rich domain, serine‐arginine‐rich domain
The CDK13 protein is predominantly expressed in plasma, monocytes, lymphocytes, T cells, natural killer (NK) cells, blood mononuclear cells, lymph nodes, bone marrow mesenchymal stem cells, the brain, retina, heart, lungs, breast, pancreatic juice, placenta, ovaries, and testes (https://gtexportal.org/home/gene/CDK13). Until the recent analysis of exome sequencing data in the Deciphering Development Disorders study (Firth et al., 2011; Kohoutek & Blazek, 2012), the variants of CDK13 were not reported to be associated with human diseases. Mutations in CDK13 were reported to cause congenital heart defects, dysmorphic facial features, and intellectual developmental disorder (CHDFIDD, OMIM # 617360) (Bostwick et al., 2017). All patients had a history of developmental delay and craniofacial dysmorphism, characterized by a wide nasal bridge and a narrow mouth, hypertelorism, and upslanted palpebral fissures, with additional features including clinodactyly and joint hypermobility, structural brain abnormalities, and skin changes (Ito et al., 2018; Sifrim et al., 2016).
Although CHDFIDD, caused by CDK13 variants, was initially considered to be an extremely rare disease, 30 patients were diagnosed in less than three years after the discovery of the initial disease gene (Bostwick et al., 2017; Hamilton et al., 2018; Hamilton & Suri, 2019; Sifrim et al., 2016; Turner et al., 2019; Yakubov et al., 2019). At the moment, more than 50 patients have been diagnosed up to now including current study in the last few years and the number is increasing every year. Here, we report two patients in a Chinese family with a heterozygous constitutional CDK13 variant (c.2149 (exon 4) G>A. p.Gly717Arg), exhibiting the classical features of CHDFIDD without congenital heart defects.
Preimplantation genetic testing for monogenic (PGT‐M) was employed as a part of the in vitro fertilization (IVF) process to select the unaffected embryo from patients with single genetic diseases, which can effectively prevent the transmission of genetic diseases from patents to the offspring before pregnancy (De Rycke et al., 2017; Treff et al., 2013). To prevent the pathogenic mutation from passing down to the next generation, this couple was counseled and suggested to receive IVF using PGT‐M. After the precise screening, a normal embryo was transferred and a healthy male was born at full term.
1.1. Case presentation
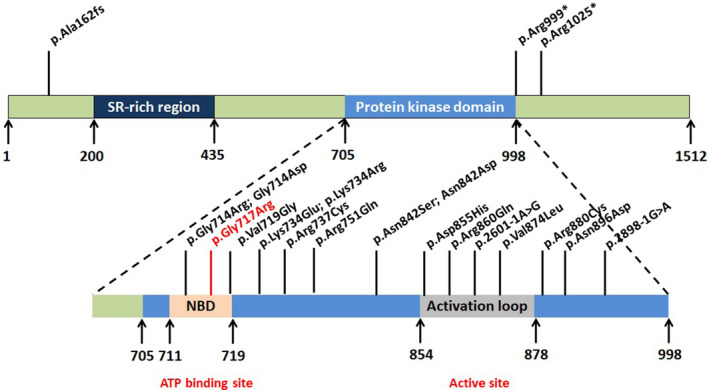
A 33‐year‐old mother and a 10‐year‐old boy were referred to the genetic counseling clinic at the reproductive medicine center. The karyotype of the maternal peripheral blood was 46, XX, and that of the son was 46, XY. However, both the mother and child had intellectual and learning disabilities and autistic features. Interestingly, there have been at least three patients with Cdk13 mutations diagnosed without heart defects in Bostwick’ research (Bostwick et al., 2017) and three more patients in other studies (Uehara et al., 2018). Apart from the fact that she also had a mentally impaired boy, her family history was typical. She was the first child of healthy, non‐consanguineous Chinese parents. Physical examination revealed craniofacial dysmorphism (narrow mouth, thin upper vermilion, epicanthus, telecanthus, low‐set ears, a depressed nasal ridge with a wide nasal bridge, strabismus, ptosis, upwards palpebral fissure oblique, and micrognathia), delayed speech and language development, dysgnosia, and hypotonia; however, the remaining examination was unremarkable (Table 1). No cardiac or musculoskeletal abnormalities (including skull and vertebral column) were noted (Figure 2).
TABLE 1.
Summary of major clinical features of two patients with variants in CDK13
| Patient | Proband | Son |
|---|---|---|
| CDK13 variant | c.2149 G>A (p. Gly717Arg) | c.2149 G>A (p. Gly717Arg) |
| Sex | Female | Male |
| Age at examination | 33 y, 1 m | 8 y |
| Gestational age at birth | 38/40 | 40/40 |
| Developmental delay | + | + |
| Intellectual disability | + | + |
| Autism | + | + |
| Seizures | − | − |
| Facial dysmorphism | + | + |
| Structural heart anomaly | − | − |
| Structural brain abnormality | − | − |
| Digital anomalies | + | + |
FIGURE 2.
Craniofacial and dysmorphology features in individuals with pathogenic CDK13. Both mother (a) and child (b) had intellectual disability, learning disabilities and autistic features
1.2. Genetic testing
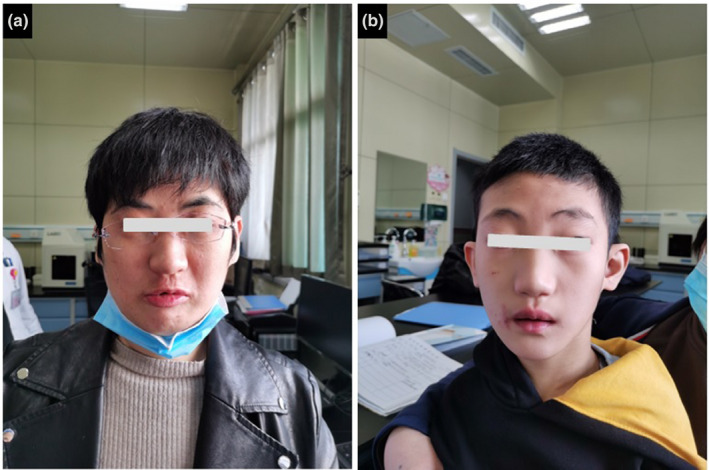
Informed consent was signed by the parents of the patients; 5 ml of peripheral blood was extracted from the patients, parents and husband of the female patient. Genomic DNA was extracted according to manufacturer instructions (Kangweishiji, China). The Chigene Translational Medicine Research Center (Beijing, China, https://www.chigene.cn) conducted sequencing and basic bioinformatic analyses, including read mapping, variant detection, filtering, and annotation. This test is divided into three main steps: variant screening (using high‐throughput sequencing technology), gene data analysis (using bioinformatics and clinical information analysis technology), and suspected pathogenic variant verification (using Sanger sequencing technology). Whole‐exome sequencing (WES) was performed using IDT xGen Exome Research Panel V1.0, and the whole exon cores were captured and sequenced using an Illumina NovaSeq 6000 series sequencer. The target sequencing coverage was not less than 99%. Through molecular biology annotation, biology genetics, and clinical characteristics analysis for the integration of genetic disease screening cloud platform system for precise diagnosis analysis, combined with the pathogenic variant database normal human genome database known for thousand kinds of hereditary disease clinical features, such as database and data analysis algorithms, for hundreds of thousands of genetic variants were classified, grade of variation USES, the three elements of grading system, and the genetic variant grading system of the American College of Medical Genetics and Genomics (ACMG, journal of the American medical society). After PCR, Sanger sequencing was performed using an ABI3730 sequencer to verify the target sequence, and the verification results were analyzed using sequence analysis software. Minor allele frequency (MAF) data were used to distinguish between common and rare variants. The smaller the MAF, the lower the variant rate in the population, indicating a rare variant. A MAF < 1%, against the 1000 Genomes Project, was used to screen candidate variants. According to the 2015 interpretation guidelines recommended by the ACMG, a pathogenic variant of the CDK13 gene was identified as the cause of the condition. The sequencing results showed that a missense variant in c.2149 (exon 4) G>A of the CDK13 gene, resulted in glycine being replaced by arginine (p.Gly717Arg), which was verified by Sanger sequencing. (Figure 3). In addition, the sequencing results revealed that the genotypes of the parents and sister were wild type (WT), indicating that it was a de novo variant. The three‐dimensional (3D) structure of the WT CDK13 protein was generated using the SWISS‐MODEL web and analyzed using the UniProt database (https://www.uniprot.org/). The database demonstrated that amino acids 711–719 are nucleotide‐binding domains. A nucleotide‐binding domain plays a vital role in normal functioning of the CDK13 protein (Akker et al., 2018). According to ACMG guideline (Richards et al., 2015), this missense variant can be classified as “pathogenic” (PS1 + PS2 + PM2 + PP3; Table 2).
FIGURE 3.
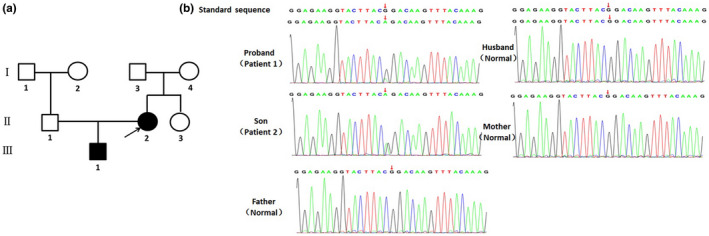
Pedigree of the family (a) and electropherograms (b) of normal, the patient and his parents; c.2149 (exon 4) G>A substitution causes Gly to Arg at position 717 of protein.
TABLE 2.
Criteria for classifying pathogenic variants
| Evidence of pathogenicity | Category |
|---|---|
| Very strong |
PVS1 null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where LOF is a known mechanism of disease Caveats:
|
| Strong |
PS1 Same amino acid change as a previously established pathogenic variant regardless of nucleotide change Example: Val→Leu caused by either G>C or G>T in the same codon Caveat: Beware of changes that impact splicing rather than at the amino acid/protein level PS2 De novo (both maternity and paternity confirmed) in a patient with the disease and no family history Note: Confirmation of paternity only is insufficient. Egg donation, surrogate motherhood, errors in embryo transfer, and so on, can contribute to nonmaternity PS3 Well‐established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product Note: Functional studies that have been validated and shown to be reproducible and robust in a clinical diagnostic laboratory setting are considered the most well established PS4 The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls Note 1: Relative risk or OR, as obtained from case–control studies, is >5.0, and the confidence interval around the estimate of relative risk or OR does not include 1.0. See the article for detailed guidance Note 2: In instances of very rare variants where case–control studies may not reach statistical significance, the prior observation of the variant in multiple unrelated patients with the same phenotype, and its absence in controls, may be used as moderate level of evidence |
| Moderate |
PM1 Located in a mutational hot spot and/or critical and well‐established functional domain (e.g., active site of an enzyme) without benign variation PM2 Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium Caveat: Population data for insertions/deletions may be poorly called by next‐generation sequencing PM3 For recessive disorders, detected in trans with a pathogenic variant Note: This requires testing of parents (or offspring) to determine phase PM4 Protein length changes as a result of in‐frame deletions/insertions in a nonrepeat region or stop‐loss variants PM5 Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before Example: Arg156His is pathogenic; now you observe Arg156Cys Caveat: Beware of changes that impact splicing rather than at the amino acid/protein level PM6 Assumed de novo, but without confirmation of paternity and maternity |
| Supporting |
PP1 Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease Note: May be used as stronger evidence with increasing segregation data PP2 Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease PP3 Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.) Caveat: Because many in silico algorithms use the same or very similar input for their predictions, each algorithm should not be counted as an independent criterion. PP3 can be used only once in any evaluation of a variant PP4 Patient's phenotype or family history is highly specific for a disease with a single genetic etiology PP5 Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation |
To further detect the effects of variant c.2149 (exon 4) G>A on the function of CDK13 protein, I‐TASSER was used to predict the WT and mutated type of the CDK13 protein (Berro et al., 2008; Fan et al., 2020; Roy et al., 2010; Trinh et al., 2019). Visualization and analysis of the 3D protein structure were performed using SWISS‐MODEL (Figure 4). The substitution of Gly717Arg leads to a change in the topological structure of the glycine‐rich loop on the surface of the ligand‐binding pocket, resulting in a change in the hydrogen bonding in this region. It is also possible that since the Gly717Arg substitution occurs in a glycine‐rich region, which reveals a high degree of evolutionary conservation, the effect of this variant on protein folding in vivo is more profound than that in comparative modeling in silico.
FIGURE 4.
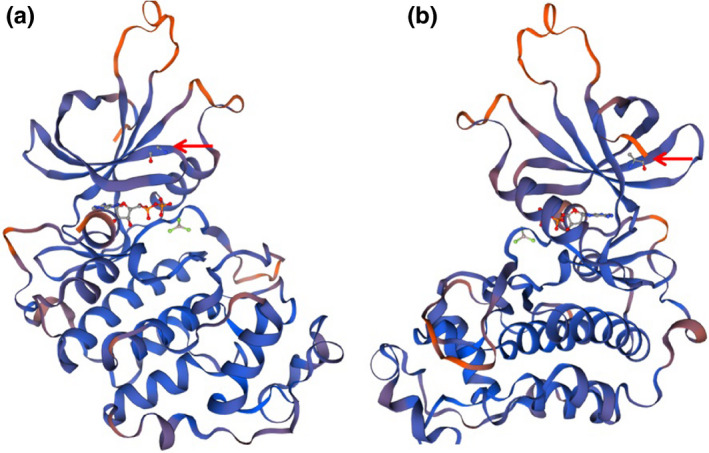
Local structure of CDK13 in wild type (a) and G717P variant type (b). The Gly717Arg substitution results in altered topology of the glycine‐rich loop at the upper surface of the ligand‐binding pocket, leading to altered hydrogen bonding within this region
1.3. Embryo preparation and genetic testing
The IVF and embryo biopsy were processed with the conventional protocol at Center of Reproductive Medicine of Children's Hospital of Shanxi and Women Health Center of Shanxi. Intracytoplasmic sperm injection (ICSI) was performed to detect genetic traits in embryo biopsies, allowing the selection and transfer of embryos without carrying the genetic disease. Embryos were cultured in vitro to the blastocyst stage (Day 5 or Day 6), following by biopsying 5–10 trophectoderm (TE) cells from each blastocyst for analysis. Biopsied TE cells were placed in EP tubes containing cell lysis buffer and then adapted MALBAC (multiple annealing and looping‐based amplification cycles) for whole‐genome amplification (WGA). WGA and sequencing process was done at laboratory of Yikon Genomics. Finally, we selected a euploidy embryo without carrying the mutation (c.2149 G>A) to implant into the uterus. The amniocentesis of this couple of proband was performed at 20 weeks of gestation and the PGT‐M results have been confirmed by the CDK13 gene mutation detection of gDNA in amnion cells (Figure 5).
FIGURE 5.
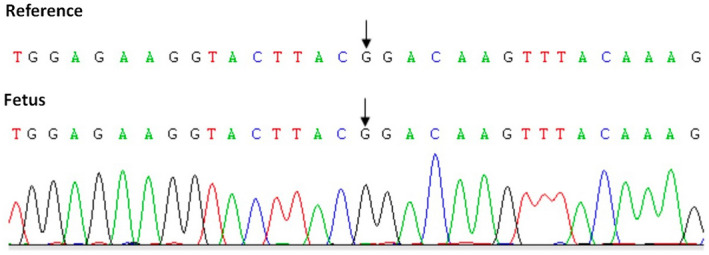
CDK13 gene mutation detection of gDNA in amnion cells.
2. DISCUSSION AND CONCLUSION
CHDFIDD syndrome is an autosomal dominant inheritance demonstrated by the identification of heterozygous variants of CDK13 (Bostwick et al., 2017). To date, 19 variants have been verified to be associated with the CHDFIDD syndrome, with the missense variant of c.2525 A>G (p.Asn842Ser) being the most common one (Hamilton & Suri, 2019; Turner et al., 2019; Yakubov et al., 2019). Most variants occur in the highly conserved protein kinase domain (PKD), and molecular modeling predicted that the variants would damage ATP binding, and binding of the magnesium ion is necessary for interactions with cyclin K and enzyme activity. The catalytic PKD of CDK13 extends from amino acids 705 to 998. The nucleotide‐binding site of the lysine residue extends from positions 711 to 719 in the polypeptide chain, and the activation loop of PKD extends from amino acids 854 to 878 (Bartkowiak et al., 2010; Deciphering Developmental Disorders, 2017; Dong et al., 2018; Even et al., 2016; Greenleaf, 2019; Liu et al., 2020). Homozygous or heterozygous variants of the CDK13 gene can cause CHDFIDD.
All the patients in the previous study were newborns of a single individual, and the vast majority had congenital heart defects. Here, we reported two patients in a Chinese family with a heterozygous constitutional CDK13 variant (c.2149 (exon 4) G>A. p. Gly717Arg), exhibiting the classical features of CHDFIDD without congenital heart defects. We speculated that congenital cardiac defects are not essential phenotypic features of constitutional CDK13 variants. Distinctive facial features, consisting of a depressed nasal ridge with a wide nasal bridge, widely spaced and peg‐shaped teeth, and a short and broad columella may be linked to the diagnosis of this newly discovered syndrome that has not been taken seriously before. In conclusion, our study reports two patients in a Chinese family with a heterozygous constitutional CDK13 variant. To the best of our knowledge, this is the first reported case of an adult patient with a CDK13 variant that gave birth to the next generation without the same variant by PGT‐M to screen a normal embryo.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICAL APPROVAL
Informed consent was obtained from each participant. The entire experiments have already obtained the approval of the Ethics Committee of Shanxi Medical University (Ethical code: 201922021).
Cui, X. , Wu, X. , Wang, H. , Zhang, S. , Wang, W. , & Jing, X. (2022). Genetic of preimplantation diagnosis of dysmorphic facial features and intellectual developmental disorder (CHDFIDD) without congenital heart defects. Molecular Genetics & Genomic Medicine, 10, e1863. 10.1002/mgg3.1863
Funding information
This study was supported by the China Postdoctoral Science Foundation (grant no. 2020M670703), Scientific Research Project of Shanxi Provincial Department of Health (grant no. 201601070), Natural Science Foundation of Shanxi (grant nos. 201901D211519 and 201901D211546), Research Project Supported by Shanxi Scholarship Council of China (grant no. HGKY2019092), Initial Scientific Research Fund of PhD in Shanxi Provincial People's Hospital (grant no. b201635), Natural Science Foundation of China (grant nos. 82000722 and 82000302), Non‐profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019PT310002), and Medical Science and Technology Innovation Team of Shanxi Province (2020TD19).
DATA AVAILABILITY STATEMENT
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding authors.
REFERENCES
- Bartkowiak, B. , Liu, P. , Phatnani, H. P. , Fuda, N. J. , Cooper, J. J. , Price, D. H. , Adelman, K. , Lis, J. T. , & Greenleaf, A. L. (2010). CDK12 is a transcription elongation‐associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes & Development, 24, 2303–2316. 10.1101/gad.1968210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berro, R. , Pedati, C. , Kehn‐Hall, K. , Wu, W. , Klase, Z. , Even, Y. , Genevière, A.‐M. , Ammosova, T. , Nekhai, S. , & Kashanchi, F. (2008). CDK13, a new potential human immunodeficiency virus type 1 inhibitory factor regulating viral mRNA splicing. Journal of Virology, 82, 7155–7166. 10.1128/JVI.02543-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostwick, B. (2019). CDK13‐related disorder. In Adam M. P., Ardinger H. H., & Pagon R. A. et al, (eds). GeneReviews((R)).(pp. 1993–2022). [Google Scholar]
- Bostwick, B. L. , McLean, S. , Posey, J. E. , Streff, H. E. , Gripp, K. W. , Blesson, A. , Powell‐Hamilton, N. , Tusi, J. , Stevenson, D. A. , Farrelly, E. , Hudgins, L. , Yang, Y. , Xia, F. , Wang, X. , Liu, P. , Walkiewicz, M. , McGuire, M. , Grange, D. K. , Andrews, M. V. , … Lalani, S. R. (2017). Phenotypic and molecular characterisation of CDK13‐related congenital heart defects, dysmorphic facial features and intellectual developmental disorders. Genome Medicine, 9, 73. 10.1186/s13073-017-0463-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. R. , Lin, G. T. , Huang, C. K. , & Fann, M. J. (2014). Cdk12 and Cdk13 regulate axonal elongation through a common signaling pathway that modulates Cdk5 expression. Experimental Neurology, 261, 10–21. 10.1016/j.expneurol.2014.06.024 [DOI] [PubMed] [Google Scholar]
- De Rycke, M. , Goossens, V. , Kokkali, G. , Meijer‐Hoogeveen, M. , Coonen, E. , & Moutou, C. (2017). ESHRE PGD Consortium data collection XIV‐XV: cycles from January 2011 to December 2012 with pregnancy follow‐up to October 2013. Human Reproduction, 32, 1974–1994. 10.1093/humrep/dex265 [DOI] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders . (2017). Prevalence and architecture of de novo mutations in developmental disorders. 542, Nature, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, X. , Chen, G. , Cai, Z. , Li, Z. , Qiu, L. , Xu, H. , Yuan, Y. , Liu, X.‐L. , & Liu, J. (2018). CDK13 RNA over‐editing mediated by ADAR1 associates with poor prognosis of hepatocellular carcinoma patients. Cellular Physiology and Biochemistry, 47(6), 2602–2612. 10.1159/000491656 [DOI] [PubMed] [Google Scholar]
- Even, Y. , Escande, M. L. , Fayet, C. , & Geneviere, A. M. (2016). CDK13, a kinase involved in pre‐mRNA splicing, is a component of the perinucleolar compartment. PLoS One, 11, e0149184. 10.1371/journal.pone.0149184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, Z. , Devlin, J. R. , Hogg, S. J. , Doyle, M. A. , Harrison, P. F. , Todorovski, I. , Cluse, L. A. , Knight, D. A. , Sandow, J. J. , Gregory, G. , Fox, A. , Beilharz, T. H. , Kwiatkowski, N. , Scott, N. E. , Vidakovic, A. T. , Kelly, G. P. , Svejstrup, J. Q. , Geyer, M. , Gray, N. S. , … Johnstone, R. W. (2020). CDK13 cooperates with CDK12 to control global RNA polymerase II processivity. Science Advances, 6, 5041. 10.1126/sciadv.aaz5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth, H. V. , & Wright, C. F. (2011). The Deciphering Developmental Disorders (DDD) study. Developmental Medicine & Child Neurology, 53(8), 702–703. 10.1111/j.1469-8749.2011.04032.x [DOI] [PubMed] [Google Scholar]
- Greenleaf, A. L. (2019). Human CDK12 and CDK13, multi‐tasking CTD kinases for the new millenium. Transcription, 10, 91–110. 10.1080/21541264.2018.1535211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, M. J. , Caswell, R. C. , Canham, N. et al (2018). Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. Journal of Medical Genetics, 55, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, M. J. , & Suri, M. (2019). CDK13‐related disorder. Advances in Genetics, 103, 163–182. [DOI] [PubMed] [Google Scholar]
- Ito, M. , Tanaka, T. , Toita, A. , Uchiyama, N. , Kokubo, H. , Morishita, N. , Klein, M. G. , Zou, H. , Murakami, M. , Kondo, M. , & Sameshima, T. (2018). Discovery of 3‐Benzyl‐1‐(trans‐4‐((5‐cyanopyridin‐2‐yl)amino)cyclohexyl)‐1‐arylurea derivatives as novel and selective cyclin‐dependent kinase 12 (CDK12) inhibitors. Journal of Medicinal Chemistry, 61, 7710–7728. [DOI] [PubMed] [Google Scholar]
- Kohoutek, J. , & Blazek, D. (2012). Cyclin K goes with Cdk12 and Cdk13. Cell Division, 7, 12. 10.1186/1747-1028-7-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, K. , Gao, X. , Gilmore, J. M. , Florens, L. , Washburn, M. P. , Smith, E. , & Shilatifard, A. (2015). Characterization of human cyclin‐dependent kinase 12 (CDK12) and CDK13 complexes in C‐terminal domain phosphorylation, gene transcription, and RNA processing. Molecular and Cellular Biology, 35, 928–938. 10.1128/MCB.01426-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Hao, M. , Leggett, A. L. , Gao, Y. , Ficarro, S. B. , Che, J. , He, Z. , Olson, C. M. , Marto, J. A. , Kwiatkowski, N. P. , Zhang, T. , & Gray, N. S. (2020). Discovery of MFH290: A potent and highly selective covalent inhibitor for cyclin‐dependent kinase 12/13. Journal of Medicinal Chemistry, 63, 6708–6726. 10.1021/acs.jmedchem.9b01929 [DOI] [PubMed] [Google Scholar]
- Nováková, M. , Hampl, M. , Vrábel, D. , Procházka, J. , Petrezselyová, S. , Procházková, M. , Sedláček, R. , Kavková, M. , Zikmund, T. , Kaiser, J. , Juan, H.‐C. , Fann, M.‐J. , Buchtová, M. , & Kohoutek, J. (2019). Mouse model of congenital heart defects, dysmorphic facial features and intellectual developmental disorders as a result of non‐functional CDK13. Frontiers in Cell and Developmental Biology, 7, 155. 10.3389/fcell.2019.00155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quereda, V. , Bayle, S. , Vena, F. , Frydman, S. M. , Monastyrskyi, A. , Roush, W. R. , & Duckett, D. R. (2019). Therapeutic targeting of CDK12/CDK13 in triple‐negative breast cancer. Cancer Cell, 36(5), 545–558.e7. 10.1016/j.ccell.2019.09.004 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, A. , Kucukural, A. , & Zhang, Y. (2010). I‐TASSER: a unified platform for automated protein structure and function prediction. Nature Protocols, 5, 725–738. 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifrim, A. , Hitz, M.‐P. , Wilsdon, A. , Breckpot, J. , Turki, S. H. A. , Thienpont, B. , McRae, J. , Fitzgerald, T. W. , Singh, T. , Swaminathan, G. J. , Prigmore, E. , Rajan, D. , Abdul‐Khaliq, H. , Banka, S. , Bauer, U. M. M. , Bentham, J. , Berger, F. , Bhattacharya, S. , Bu'Lock, F. , … Hurles, M. E. (2016). Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nature Genetics, 48(9), 1060–1065. 10.1038/ng.3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treff, N. R. , Fedick, A. , Tao, X. , Devkota, B. , Taylor, D. , & Scott, R. T. Jr (2013). Evaluation of targeted next‐generation sequencing‐based preimplantation genetic diagnosis of monogenic disease. Fertility and Sterility, 99(1377–1384), e1376. [DOI] [PubMed] [Google Scholar]
- Trinh, J. , Kandaswamy, K. K. , Werber, M. , Weiss, M. E. R. , Oprea, G. , Kishore, S. , Lohmann, K. , & Rolfs, A. (2019). Novel pathogenic variants and multiple molecular diagnoses in neurodevelopmental disorders. Journal of Neurodevelopmental Disorders, 11(1), 10.1186/s11689-019-9270-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, T. N. , Wilfert, A. B. , Bakken, T. E. , Bernier, R. A. , Pepper, M. R. , Zhang, Z. , Torene, R. I. , Retterer, K. , & Eichler, E. E. (2019). Sex‐based analysis of de novo variants in neurodevelopmental disorders. The American Journal of Human Genetics, 105(6), 1274–1285. 10.1016/j.ajhg.2019.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara, T. , Takenouchi, T. , Kosaki, R. , Kurosawa, K. , Mizuno, S. , & Kosaki, K. (2018). Redefining the phenotypic spectrum of de novo heterozygous CDK13 variants: Three patients without cardiac defects. European Journal of Medical Genetics, 61, 243–247. 10.1016/j.ejmg.2017.12.004 [DOI] [PubMed] [Google Scholar]
- van den Akker, W. M. R. , Brummelman, I. , Martis, L. M. , Timmermans, R. N. , Pfundt, R. , Kleefstra, T. , Willemsen, M. H. , Gerkes, E. H. , Herkert, J. C. , van Essen, A. J. , & Rump, P. (2018). De novo variants in CDK13 associated with syndromic ID/DD: Molecular and clinical delineation of 15 individuals and a further review. Clinical Genetics, 93, 1000–1007. [DOI] [PubMed] [Google Scholar]
- Yakubov, R. , Ayman, A. , Kremer, A. K. , & van den Akker, M. (2019). One‐month‐old girl presenting with pseudohypoaldosteronism leading to the diagnosis of CDK13‐related disorder: a case report and review of the literature. Journal of Medical Case Reports, 13, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding authors.