

Abstract
Alzheimer’s disease (ad) adversely affects the health, quality of life and independence of patients. There is a critical need to identify novel blood gene biomarkers for ad risk assessment. We performed a transcriptome-wide association study to identify biomarker candidates for ad risk. We leveraged two sets of gene expression prediction models of blood developed using different reference panels and modeling strategies. By applying the prediction models to a meta-GWAS including 71 880 (proxy) cases and 383 378 (proxy) controls, we identified significant associations of genetically determined expression of 108 genes in blood with ad risk. Of these, 15 genes were differentially expressed between ad patients and controls with concordant directions in measured expression data. With evidence from the analyses based on both genetic instruments and directly measured expression levels, this study identifies 15 genes with strong support as biomarkers in blood for ad risk, which may enhance ad risk assessment and mechanism-focused studies.
Introduction
Alzheimer’s disease (ad) is the most common cause of dementia in older adults (1), which starts with mild memory loss and culminates in severe impairment of individual executive and cognitive functions (2). It is estimated that 5.8 million people have ad in the United States and ad has become the fifth leading cause of mortality among Americans aged 65 or older (3). ad is a slowly progressing neurodegenerative disorder that is estimated to start 20–30 years before the appearance of the first clinical symptoms (4). Effective strategies for risk assessment of ad are critical to decrease the public health burden of this common disease. Multiple approaches have been developed for ad risk assessment. For example, amyloid-β (Aβ) in plasma was reported as a potential biomarker for ad risk, in particular, Aβ40 and Aβ42 (5–7). However, other studies did not support an association between baseline Aβ40 and Aβ42 levels in plasma and the subsequent development of ad (8,9). Aβ and tau levels in cerebrospinal fluid (CSF) have also been reported as potential markers for ad (10,11). However, lumbar puncture results in discomfort or pain for patients, and it is difficult to repeatedly and routinely check patients’ CSF (12). Genetics-informed biomarkers, such as APOE and TOMM40, have also been reported to be associated with risk of ad, but their clinical utility requires further evaluation (13). Newer proposed approaches, such as screening plasma or urine lipid peroxidation biomarkers for ad using the liquid chromatography or ultra-performance liquid chromatography-tandem coupled to mass spectrometry (LC–MS/MS) (14–16), are usually time consuming and cost prohibitive (17). In addition to these approaches, the potential utility of specific genes in blood as candidate biomarkers have been explored (18). Multiple candidate genes have been reported to show differential expression in the blood of ad patients versus healthy controls (19–30). However, findings have been inconsistent across studies. Most studies have assessed only a small list of candidate genes, and those that have taken an agnostic approach have included only a relatively small number of subjects due to cost. Importantly, several limitations are commonly encountered in conventional epidemiologic studies, such as selection bias, potential confounding and reverse causation. These limitations may partially explain some of the inconsistent findings.
One strategy to address these limitations is to use genetic instruments to assess the relationship between gene expression and ad risk. Transcriptome-wide association study (TWAS), a design that integrates gene expression prediction models and genomic datasets of disease cases and controls, has been applied to identify multiple susceptibility genes for human diseases (31–34). Several TWAS have been performed for ad. In an earlier TWAS by Hao et al., which included 17 008 ad cases and 37 154 controls and leveraged gene expression prediction models in brain (dorsolateral prefrontal cortex (DLPFC)), adipose and blood tissues, 25 genes were identified (35). Raj et al., utilizing gene expression prediction models for DLPFC tissue, identified eight associated genes at novel loci by analyzing 25 580 cases and 48 466 controls (36). Leveraging a newly developed TWAS framework, namely, UTMOST, Hu et al. identified significant associations for 69 genes by analyzing 17 008 cases and 37 154 controls (37). In the latest TWAS by Gerring et al., 126 tissue-specific associations involving 50 unique genes were identified (38). Despite these promising findings, these studies primarily aimed at identifying ad susceptibility genes and thus primarily relied on gene expression prediction models developed in tissues beyond blood. Meanwhile, the applications on earlier ad genome-wide association studies (GWAS) with relatively small sample sizes may limit the findings.
Herein, to identify candidate gene biomarkers in blood for ad risk, we performed the most comprehensive TWAS of ad to date to assess the associations between genetically predicted gene expression in blood and ad risk. We leveraged gene expression prediction models using larger reference transcriptome datasets in blood (up to 3344) with state-of-the-art and novel modeling strategies, namely, a modified version of the UTMOST (37) and LASSO (39) methods. To have high statistical power, we analyzed data from the largest ad GWAS available to date involving 71 880 (proxy) cases and 383 378 (proxy) controls of European ancestry from three consortia (Alzheimer’s disease working group of the Psychiatric Genomics Consortium (PGC-ALZ), the International Genomics of Alzheimer’s Project (IGAP) and the Alzheimer’s disease sequencing project (adSP)) and the UK Biobank data (40,41). For identified genes, we further compared their measured expression levels (in blood) between AD cases and controls from an independent dataset. Focusing on significantly associated genes identified in this work, we then conducted pathway gene-set analysis to elucidate enriched pathways.
Results
Blood tissue gene expression genetic prediction models
The overall study flow is presented in Figure 1. Two sets of gene expression prediction models for blood tissue built using different modeling strategies namely, the modified UTMOST and LASSO were leveraged in this TWAS. The number of imputable genes with a performance (R2) of at least 0.01 (≥10% correlation between predicted expression and actual expression) ranged from 7799 to 10 838 (Supplementary Material, Table S1).
Figure 1.
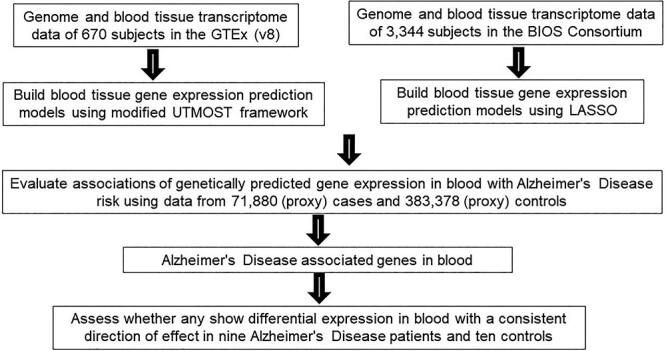
Study design flow chart.
Associations of predicted expression in blood tissue with ad risk
Considering that a different modeling strategy may be more appropriate for the specific genetic regulatory architecture of different genes, we used two sets of prediction models for the association analyses with ad risk. As co-expression is widely prevalent especially among local genes, the stringent Bonferroni correction might result in overadjustment for multiple comparisons. Thus, we used the false discovery rate (FDR) correction in this study (42). Of the 18 603 models tested, we detected 143 significant associations at FDR < 0.05 for 113 genes (Fig. 2). Six genes, namely HLA-DRB1, HLA-DQA2, HLA-DOB, TAP2, DUP160 and CCNT2-AS1, were excluded due to the fact that these genes are located in linkage disequilibrium (LD)-extensive regions. The remaining 137 significant associations, involving 108 unique genes, were listed in Table 1 and Supplementary Material, Table S2. Positive associations between predicted expression level and ad risk were identified for 53 genes, and negative associations were identified for 55 genes. These genes are located in 55 genomic loci. Of the 108 associated genes, 29 genes were identified by both the UTMOST and LASSO prediction models, 37 by only the UTMOST models and 42 by only the LASSO models. Of the 37 genes identified using the UTMOST models, 20 also showed reasonable prediction quality (R2 ≥ 0.01) using the LASSO models. Reassuringly, most of the 20 genes (19/20) showed concordant directions of associations with ad risk in comparison with the LASSO models and the UTMOST models (Supplementary Material, Table S3). Of the 42 genes identified using the LASSO models, for 17, their prediction models with R2 ≥ 0.01 were also established using the UTMOST method. Of these, all except for one showed concordant directions of associations with ad risk in comparison of the UTMOST models and the LASSO models (Supplementary Material, Table S4).
Figure 3.
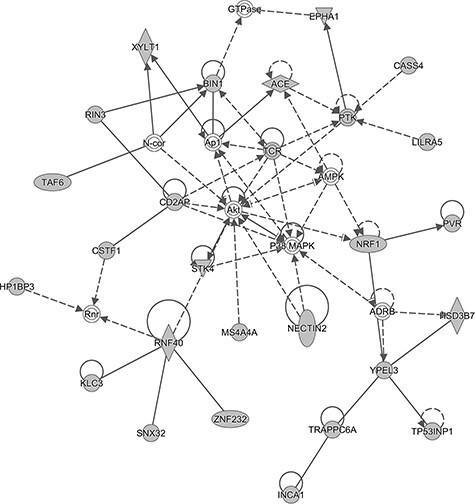
The top molecular networks identified by Ingenuity Pathway Analysis (IPA). Molecule fill color: the white fill color shows molecules from the Knowledge Base—not part of our TWAS identified gene for ad risk; the gray fill color shows molecules of our TWAS identified genes for ad risk. Shape:  Other,
Other,  Complex/Group/Other,
Complex/Group/Other,  Enzyme,
Enzyme,  Kinase,
Kinase,  Peptidase,
Peptidase,  Transcription regulator. Relationship line:
Transcription regulator. Relationship line:  direct interaction,
direct interaction,  indirect interaction. More information of PIA legend can be found in: https://qiagen.secure.force.com/KnowledgeBase/?id=kA41i000000L5rTCAS.
indirect interaction. More information of PIA legend can be found in: https://qiagen.secure.force.com/KnowledgeBase/?id=kA41i000000L5rTCAS.
Table 1.
Forty-two gene expression-trait associations for genes at genomic loci at least 500 kb away from any GWAS-identified Alzheimer’s disease risk variants
| Region | Gene | Typea | Model | R 2 b | Number of SNPs in model | OR (95% CI) | P-valuec | P-value after FDRc | Closest risk SNPd | Distance to the risk SNP (kb) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1p36.12 | HP1BP3 | protein | UTMOST | 0.18 | 5 | 1.29 (1.13–1.47) | 1.31 × 10−4 | 2.28 × 10−2 | rs4575098 | 140 042 |
| 2p22.2 | CEBPZOS | protein | LASSO | 0.09 | 31 | 0.97 (0.96–0.99) | 3.68 × 10−4 | 4.78 × 10−2 | rs4663105 | 90 448 |
| 2q14.2 | TMEM177 | protein | LASSO | 0.03 | 39 | 0.94 (0.92–0.96) | 8.10 × 10 −7 | 3.68 × 10−4 | rs4663105 | 7447 |
| 2q33.2 | CARF | protein | UTMOST | 0.01 | 35 | 1.10 (1.06–1.15) | 5.63 × 10−6 | 1.69 × 10−3 | rs10933431 | 30 129 |
| 3q25.31 | PLCH1 | protein | LASSO | 0.02 | 12 | 0.92 (0.89–0.96) | 1.44 × 10−5 | 3.43 × 10−3 | rs184384746 | 97 867 |
| 4q25 | FAM241A | protein | UTMOST | 0.06 | 23 | 0.96 (0.95–0.98) | 2.59 × 10 −6 | 8.96 × 10−4 | rs6448451 | 102 042 |
| LASSO | 0.10 | 8 | 0.96 (0.94–0.98) | 1.07 × 10−5 | 2.65 × 10−3 | rs6448451 | 102 042 | |||
| 6p25.1 | LY86 | protein | LASSO | 0.19 | 14 | 1.02 (1.01–1.03) | 3.09 × 10−4 | 4.10 × 10−2 | rs9271192 | 25 923 |
| UST | protein | LASSO | 0.08 | 32 | 0.96 (0.95–0.98) | 1.66 × 10−4 | 2.76 × 10−2 | rs10948363 | 101 580 | |
| 6p24.2 | TMEM170B | protein | UTMOST | 0.15 | 28 | 1.02 (1.01–1.04) | 2.27 × 10−4 | 3.37 × 10−2 | rs9271192 | 20 995 |
| 7p22.1 | SLC29A4 | protein | LASSO | 0.03 | 34 | 1.05 (1.02–1.07) | 1.78 × 10−4 | 2.93 × 10−2 | rs2718058 | 32 495 |
| 7p21.3 | TMEM106B | protein | UTMOST | 0.15 | 24 | 0.95 (0.93–0.97) | 6.73 × 10−5 | 1.32 × 10−2 | rs2718058 | 25 559 |
| 7q32.2 | NRF1 | protein | UTMOST | 0.02 | 13 | 1.13 (1.08–1.19) | 1.19 × 10 −6 | 5.03 × 10−4 | rs7810606 | 13 711 |
| 7q32.3 | AC058791.1 | lncRNA | UTMOST | 0.02 | 1 | 0.80 (0.72–0.90) | 1.83 × 10−4 | 2.95 × 10−2 | rs7810606 | 12 484 |
| 8q22.1 | TP53INP1 | protein | UTMOST | 0.08 | 8 | 0.94 (0.92–0.97) | 2.08 × 10−4 | 3.23 × 10−2 | rs9331896 | 68 471 |
| AC087752.3 | lncRNA | UTMOST | 0.02 | 11 | 0.83 (0.76–0.91) | 5.24 × 10−5 | 1.05 × 10−2 | rs9331896 | 68 495 | |
| 9q21.11 | LINC01506 | lncRNA | UTMOST | 0.15 | 32 | 1.11 (1.05–1.18) | 2.58 × 10−4 | 3.63 × 10−2 | – | – |
| 10q21.2 | CCDC6 | protein | LASSO | 0.01 | 8 | 0.87 (0.82–0.92) | 2.80 × 10−6 | 9.30 × 10−4 | rs11257238 | 49 831 |
| 11q13.1 | SNX32 | protein | UTMOST | 0.16 | 15 | 0.98 (0.97–0.99) | 8.45 × 10−6 | 2.28 × 10−3 | rs983392 | 5678 |
| LASSO | 0.30 | 13 | 0.98 (0.98–0.99) | 2.19 × 10−4 | 3.31 × 10−2 | rs983392 | 5678 | |||
| CTSW | protein | LASSO | 0.27 | 26 | 1.02 (1.01–1.03) | 7.82 × 10−6 | 2.17 × 10−3 | rs983392 | 5724 | |
| UTMOST | 0.32 | 25 | 1.02 (1.01–1.02) | 1.07 × 10−5 | 2.65 × 10−3 | rs983392 | 5724 | |||
| 11q24.1 | GRAMD1B | protein | LASSO | 0.08 | 43 | 0.94 (0.91–0.97) | 7.60 × 10−5 | 1.43 × 10−2 | rs11218343 | 1794 |
| 14q11.2 | TRAV35 | other | LASSO | 0.13 | 44 | 1.02 (1.01–1.04) | 2.23 × 10−4 | 3.35 × 10−2 | rs17125944 | 30 710 |
| PRMT5 | protein | LASSO | 0.20 | 36 | 0.98 (0.97–0.99) | 3.59 × 10−4 | 4.71 × 10−2 | rs17125944 | 30 002 | |
| 14q24.3 | RP3-414A15.2 | lncRNA | LASSO | 0.17 | 40 | 0.97 (0.96–0.99) | 3.25 × 10−5 | 6.95 × 10−3 | rs10498633 | 18 930 |
| 15q21.2 | RP11-120 K9.2 | lncRNA | LASSO | 0.03 | 16 | 0.95 (0.92–0.97) | 2.01 × 10−4 | 3.17 × 10−2 | rs442495 | 8170 |
| 15q24.1 | CYP11A1 | protein | LASSO | 0.02 | 14 | 0.94 (0.91–0.97) | 3.00 × 10−4 | 4.08 × 10−2 | rs117618017 | 11 060 |
| 15q25.1 | CTSH | protein | UTMOST | 0.39 | 47 | 1.02 (1.01–1.02) | 5.79 × 10−6 | 1.71 × 10−3 | rs117618017 | 15 643 |
| 16p12.3 | XYLT1 | protein | UTMOST | 0.05 | 3 | 0.69 (0.56–0.84) | 3.16 × 10−4 | 4.17 × 10−2 | rs59735493 | 13 568 |
| 16p11.2 | INO80E | protein | UTMOST | 0.08 | 29 | 1.02 (1.01–1.03) | 3.94 × 10−5 | 8.05 × 10−3 | rs59735493 | 1116 |
| LASSO | 0.08 | 12 | 1.04 (1.02–1.06) | 7.87 × 10−5 | 1.45 × 10−2 | rs59735493 | 1116 | |||
| YPEL3 | protein | UTMOST | 0.11 | 14 | 0.98 (0.97–0.99) | 7.48 × 10−5 | 1.43 × 10−2 | rs59735493 | 1025 | |
| LASSO | 0.10 | 3 | 0.97 (0.95–0.98) | 2.10 × 10−4 | 3.23 × 10−2 | rs59735493 | 1025 | |||
| AC012645.1 | lncRNA | LASSO | 0.05 | 8 | 0.95 (0.92–0.97) | 8.88 × 10−5 | 1.60 × 10−2 | rs59735493 | 1018 | |
| UTMOST | 0.08 | 10 | 0.94 (0.91–0.97) | 1.41 × 10−4 | 2.41 × 10−2 | rs59735493 | 1018 | |||
| AC099524.1 | lncRNA | UTMOST | 0.15 | 1 | 1.06 (1.03–1.09) | 1.33 × 10−4 | 2.30 × 10−2 | rs59735493 | 50 639 | |
| 17p13.3 | C17orf97 | protein | LASSO | 0.33 | 44 | 1.01 (1.01–1.02) | 3.03 × 10−4 | 4.08 × 10−2 | rs9916042 | 4704 |
| 17q21.31 | LSM12 | protein | UTMOST | 0.02 | 18 | 1.22 (1.10–1.36) | 3.03 × 10−4 | 4.08 × 10−2 | rs28394864 | 5306 |
| 17q23.3 | ACE | protein | LASSO | 0.02 | 11 | 0.95 (0.93–0.98) | 2.37 × 10−4 | 3.48 × 10−2 | rs28394864 | 14 104 |
| 18q12.3-q21.1 | EPG5 | protein | UTMOST | 0.07 | 35 | 1.08 (1.04–1.12) | 3.71 × 10−5 | 7.84 × 10−3 | rs76726049 | 12 642 |
| 19q13.31 | KCNN4 | protein | UTMOST | 0.31 | 18 | 0.98 (0.97–0.99) | 1.03 × 10−4 | 1.84 × 10−2 | rs75627662 | 1128 |
| LASSO | 0.24 | 73 | 0.98 (0.97–0.99) | 3.05 × 10−4 | 4.08 × 10−2 | rs75627662 | 1128 | |||
| 19q13.42 | LILRA6 | protein | LASSO | 0.09 | 38 | 0.99 (0.98–0.99) | 2.54 × 10−4 | 3.63 × 10−2 | rs3865444 | 2993 |
| AC008984.7 | other | LASSO | 0.08 | 7 | 0.95 (0.93–0.97) | 2.01 × 10 −6 | 7.79 × 10−4 | rs3865444 | 3085 | |
| LILRA5 | protein | LASSO | 0.14 | 17 | 0.97 (0.95–0.98) | 7.56 × 10−6 | 2.13 × 10−3 | rs3865444 | 3090 | |
| UTMOST | 0.13 | 6 | 0.86 (0.81–0.92) | 9.32 × 10−6 | 2.44 × 10−3 | rs3865444 | 3090 | |||
| AC008984.2 | other | LASSO | 0.07 | 23 | 0.96 (0.94–0.98) | 2.63 × 10−5 | 5.97 × 10−3 | rs3865444 | 3092 | |
| 20q13.12 | TOMM34 | protein | LASSO | 0.25 | 51 | 0.98 (0.97–0.99) | 2.84 × 10−4 | 3.91 × 10−2 | rs6014724 | 11 410 |
| STK4 | protein | LASSO | 0.02 | 4 | 0.93 (0.89–0.96) | 1.99 × 10−4 | 3.17 × 10−2 | rs6014724 | 11 290 |
Protein: protein coding genes; lncRNA: long noncoding RNAs; other: processed transcripts, immunoglobulin genes, or T cell receptor genes.
R 2: prediction performance.
Associations with FDR-corrected P-value ≤0.05 considered significant.
A full list of all risk SNPs and their distances to the genes are presented in Supplementary Material, Table S5.
Figure 2.
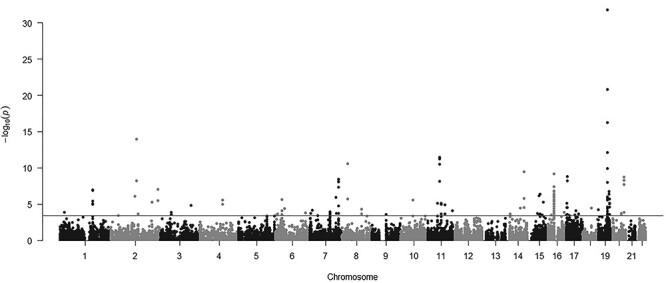
Manhattan plot of association results from the Alzheimer’s disease transcriptome-wide association study using blood tissue gene expression prediction models. The x-axis represents the genomic position of each tested gene, and the y-axis represents −log10-tansformed P-value of the associations. Each dot represents the genetically predicted expression of one specific gene. The red line at P = 3.68 × 10−4 represents the false discovery rate significance threshold.
GWAS-identified ad risk SNPs and their distances to the identified associated genes are shown in Supplementary Material, Table S5. Of these associated genes, 42 are located at least 500 kb away from any GWAS-identified ad risk variants (Table 1). The genetically determined expression of 15 genes, namely, HP1BP3 (1p36.12), CARF (2q33.2), LY86 (6p25.1), TMEM170B (6p24.2), SLC29A4 (7p22.1), NRF1 (7q32.2), LINC01506 (9q21.11), CTSW (11q13.1), TRAV35 (14q11.2), CTSH (15q25.1), INO80E (16p11.2), AC099524.1 (16p11.2), C17orf97 (17p13.3), LSM12 (17q21.31) and EPG5 (18q12.3-q21.1), were found to be positively associated with ad risk. On the other hand, a lower predicted expression was found to be associated with increased ad risk for the remaining 27 genes, namely, CEBPZOS (2p22.2), TMEM177 (2q14.2), PLCH1 (3q25.31), FAM241A (4q25), UST (6q25.1), TMEM106B (7p21.3), AC058791.1 (7q32.3), AC087752.3 (8q22.1), TP53INP1 (8q22.1), CCDC6 (10q21.2), SNX32 (11q13.1), GRAMD1B (11q24.1), PRMT5 (14q11.2), RP3-414A15.2 (14q24.3), RP11-120 K9.2 (15q21.2), CYP11A1 (15q24.1), AC012645.1 (16p11.2), YPEL3 (16p11.2), XYLT1 (16p12.3), ACE (17q23.3), KCNN4 (19q13.31), LILRA5 (19q13.42), AC008984.7 (19q13.42), AC008984.2 (19q13.42), LILRA6 (19q13.42), STK4 (20q13.12) and TOMM34 (20q13.12). One of them, KCNN4 (19q13.31), was identified as an ad susceptibility gene in a previous TWAS (38) (Supplementary Material, Table S4).
Of the remaining 66 genes, 25 had been identified as ad susceptibility genes in previous TWAS (Supplementary Material, Table S6). The associations for 25 identified in the current analyses were consistent with those in previous TWAS, which were largely based on non-blood tissues (Supplementary Material, Table S6). Eight of the genes, namely, BIN1, TAS2R60, PTK2B, MS4A4A, MS4A6A, PICALM, CEACAM19 and PVR, had been identified in previous TWAS using blood prediction models.
Directly measured expression levels of TWAS identified genes in the blood of ad cases versus controls
We compared the directly measured expression of the TWAS identified genes in nine advanced ad cases and ten controls, leveraging an NCBI Gene Expression Omnibus (NCBI-GEO, accession GSE97760) dataset. For 15 of the TWAS identified genes, we observed differential expression in the blood of ad patients and healthy controls (FDR < 0.05) that was consistent with the direction of effect identified in the TWAS analyses (Table 2). For eight of these, namely, HP1BP3 (1p36.12), CD2AP (6p12.3), TMEM170B (6p24.2), NRF1 (7q32.2), PICALM (11q14.2), VKORC1 (16p11.2), EPG5 (18q12.3-q21.1) and BLOC1S3 (19q13.32), higher expression levels were observed in ad cases compared with controls. In contrast, for the remaining seven genes, namely, CCDC6 (10q21.2), CYP11A1 (15q24.1), KAT8 (16p11.2), RNF40 (16p11.2), YPEL3 (16p11.2), ACE (17q23.3) and KLC3 (19q13.32), the expression levels were lower in ad cases.
Table 2.
Fifteen genes with differential expression in ad cases and controls for directly measured levels consistent with association direction in TWAS
| Region | Gene | Type | TWAS | DEGs | |||
|---|---|---|---|---|---|---|---|
| Model | OR (95% CI) | P-value after FDR | log2FC | P-value after FDR | |||
| 1p36.12 | HP1BP3 | Protein | UTMOST | 1.29 (1.13–1.47) | 2.28 × 10−2 | 1.02 | 9.47 × 10−5 |
| 6p12.3 | CD2AP | Protein | UTMOST | 1.13 (1.07–1.20) | 7.86 × 10−3 | 2.12 | 5.82 × 10−5 |
| 6p24.2 | TMEM170B | Protein | UTMOST | 1.02 (1.01–1.04) | 3.37 × 10−2 | 2.83 | 2.14 × 10−4 |
| 7q32.2 | NRF1 | Protein | UTMOST | 1.13 (1.08–1.19) | 5.03 × 10−4 | 0.77 | 1.17 × 10−2 |
| 10q21.2 | CCDC6 | Protein | LASSO | 0.87 (0.82–0.92) | 9.30 × 10−4 | −1.15 | 2.65 × 10−2 |
| 11q14.2 | PICALM | Protein | UTMOST | 1.11 (1.05–1.17) | 3.28 × 10−2 | 1.38 | 9.02 × 10−3 |
| 15q24.1 | CYP11A1 | Protein | LASSO | 0.94 (0.91–0.97) | 4.08 × 10−2 | −0.99 | 9.33 × 10−3 |
| 16p11.2 | KAT8 | Protein | UTMOST | 0.95 (0.94–0.97) | 9.34 × 10−7 | −0.77 | 8.20 × 10−4 |
| LASSO | 0.92 (0.89–0.95) | 3.35 × 10−4 | |||||
| RNF40 | Protein | UTMOST | 0.96 (0.94–0.98) | 4.77 × 10−3 | −0.93 | 2.63 × 10−3 | |
| VKORC1 | Protein | UTMOST | 1.05 (1.03–1.07) | 8.87 × 10−5 | 1.30 | 6.98 × 10−5 | |
| YPEL3 | Protein | UTMOST | 0.98 (0.97–0.99) | 1.43 × 10−2 | −1.00 | 6.87 × 10−3 | |
| LASSO | 0.97 (0.95–0.98) | 3.23 × 10−2 | |||||
| 17q23.3 | ACE | Protein | LASSO | 1.22 (1.10–1.36) | 4.08 × 10−2 | −1.69 | 1.41 × 10−3 |
| 18q12.3-q21.1 | EPG5 | Protein | UTMOST | 1.08 (1.04–1.12) | 7.84 × 10−3 | 0.94 | 8.72 × 10−3 |
| 19q13.32 | BLOC1S3 | Protein | LASSO | 1.06 (1.04–1.08) | 2.59 × 10−9 | 0.87 | 2.04 × 10−2 |
| UTMOST | 1.09 (1.05–1.13) | 5.56 × 10−3 | |||||
| KLC3 | Protein | UTMOST | 0.92 (0.89–0.95) | 6.20 × 10−4 | −3.83 | 4.53 × 10−6 | |
Ingenuity pathway analysis (IPA)
We performed ‘Core Analysis’ function of IPA (Ingenuity System Inc, USA), including ‘Canonical Pathway’, ‘Disease and Functions’, ‘Network’ and ‘Molecules’ analyses. Overall, 17 canonical pathways including 22 of the TWAS identified genes showed significant enrichments at P < 0.05 (Supplementary Material, Table S7). These include the neuroprotective role of THOP1 in Alzheimer’s disease (P = 9.12 × 10−3). One tight junction signaling pathway, four immune-related pathways (Fcγ receptor-mediated phagocytosis in macrophages and monocytes, natural killer cell signaling, Fc epsilon RI signaling and phagosome maturation) and multiple biosynthesis pathways, such as chondroitin sulfate biosynthesis and dermatan sulfate biosynthesis, were also significantly enriched (Supplementary Material, Table S7).
Based on the ‘Disease and Functions’ analysis, 37 neurological disease functional categories were implicated, which involved 52 genes (Supplementary Material, Table S8). Interestingly, the top three neurological disease functional categories were related to ad or dementia. Thirteen associated genes: BIN1 (2q14.3), CD2AP (6p12.3), EPHA1 (7q34-q35), TMEM106B (7p21.3), CHRNA2 (8p21.2), MS4A4A (11q12.2), MS4A6A (11q12.2), MS4A6E (11q12.2), PICALM (11q14.2), CYP11A1 (15q24.1), CHRNE (17p13.2), ACE (17q23.3) and CD33 (19q13.41), contributed to the significant enrichment in neurological disease functional categories related to dementia (14 genes; P = 5.95 × 10−5), ad (13 genes; P = 8.51 × 10−5) and late-onset ad (seven genes; P = 1.21 × 10−11).
A total of nine networks were identified based on the Network Analysis (Supplementary Material, Table S9; Fig. 3 and Supplementary Material, Fig. S1). The top network (Score = 59) containing 25 TWAS identified genes was related to neurological disease (Supplementary Material, Table S9 and Fig. 3). Interestingly, some associated genes that were known risk genes for ad, such as BIN1 (2q14.3), PICALM (11q14.2) and ACE (17q23.3) (43–45), were in nodes of the network, suggesting that these genes could possibly regulate ad development.
Except for AC008984.2 (19q13.42), which is not available in the IPA database, the location, type and biomarker application(s) of the identified associated genes for ad risk were annotated through ‘Molecules’ analysis (Supplementary Material, Table S10). In regards to the cellular location of the products of these genes, 34 were in the cytoplasm, 20 in the plasma membrane, 15 in the nucleus and 1 in extracellular space. Of these molecules, twelve were annotated as enzymes, five transmembrane receptors, six peptidases, four transporters, five transcription regulators, four kinases, two ion channels, one phosphatase and one G-protein coupled receptor. Six genes, VKORC1 (16p11.2), ACE (17q23.3), BLOC1S3 (19q13.32), ERCC2 (19q13.32), VASP (19q13.32) and CD33 (19q13.41), and/or their encoded proteins, have been identified to have potential utility in diagnosis, prognosis, disease progression, efficacy or response to therapy for some diseases (Supplementary Material, Table S10).
Discussion
We have performed the most comprehensive study to date to systematically search for blood gene biomarkers for ad risk using a study design that combines evidence from genetically predicted expression and from directly measured expression. State-of-the-art methods were used to develop blood gene expression prediction models (37,46). Previous work in this area has demonstrated the performance of such methods in establishing prediction models that can well capture the genetically regulated component of gene expression (37,46,47). A total of 108 genes were identified to be associated with ad risk for their genetically predicted expression in blood, including 42 at novel loci. Our study provides substantial novel information to improve our understanding of the genetics and etiology of ad. Notably, 15 of the genes further showed significant differential expression with a consistent direction of effect in the blood of ad patients and controls, suggesting their potential utility for ad risk assessment and warranting further validation. To our knowledge, this study represents the first study of its kind validating disease-associated genetically determined expression changes from GWAS with directly measured expression of genes from a transcriptome study in disease cases and controls to assess the concordance. The evidence from analyses using genetic instruments in TWAS provides additional assurance that such genes could serve as promising markers for improving risk prediction of disease outcome.
Twelve novel genes we identified have been shown to potentially play important roles in ad pathogenesis. CARF (2q33.2) encodes a calcium responsive transcription factor, which regulates the neuronal activity-dependent expression of BDNF (48). CARF was down-expressed in ad brain frontal cortex (49). PLCH1 (3q25.31) encodes phospholipase C eta 1, which is a member of the PLC-eta family of the phosphoinositide-specific phospholipase C (PLC) superfamily of enzymes. The activity of platelet PLC has been reported to be reduced in patients with ad (50). Previous work reported that the expression of LY86 (6p25.1), which encodes lymphocyte antigen 86, was increased in human ad temporal and App NL-G-F/NL-G-F cortex (51). In an ad mouse model, the LY86 protein was reported to be differentially expressed in urinary exosomes (52). TMEM106B (7p21.3) encodes a transmembrane glycoprotein, transmembrane protein 106B, whose risk variant was implicated in the pathologic presentation of ad (53). It was further reported that its mRNA and protein levels were significantly reduced in the frontal cortex and hippocampus tissues of subjects with sporadic ad (54). TP53INP1 (8q22.1) encodes tumor protein p53-inducible nuclear protein 1 and was reported to be associated with ad risk based on the gene-wide analytical approach (55). LINC01506 (9q21.11) is a long non-coding RNA, whose high expression was found to be associated with low risk of ad (56). PRMT5 (14q11.2) encodes protein arginine methyltransferase 5 that belongs to the methyltransferase family. The PRMT5 protein can catalyze the transfer of methyl groups to the amino acid arginine, which is involved in cell growth and apoptosis. In human cells and C. elegans models of ad, PRMT5 had an important role in regulating Aβ-induced toxicity (57). CTSH (15q25.1) encodes cathepsin H, a lysosomal cysteine proteinase. A recent study found that the genetically regulated protein abundance of this gene in brain was associated with ad risk (58). ACE (17q23.3) encodes angiotensin I converting enzyme that is a known risk gene for ad (45). EPG5 (18q12.3-q21.1) encodes ectopic P-granules autophagy protein 5 homolog, a large coiled coil domain-containing protein. Previous work has shown that genetic variants in EPG5 influence the age at onset of ad (59). KCNN4 (19q13.31), also known as KCa3.1, encodes potassium calcium-activated channel subfamily N member 4. The existing evidence supports that KCa3.1 in microglia inhibition can block microglial neurotoxicity and the anti-inflammatory and neuroprotective effects of KCa3.1 blockade could act as a potential therapeutic target for ad (60). TOMM34 (20q13.12) encodes translocase of outer mitochondrial membrane 34, which is involved in granulovacuolar degeneration in ad (61). Besides the above-mentioned known genes (ACE and EPG5), six novel genes (HP1BP3, TMEM170B, NRF1, CCDC6, CYP11A1 and YPEL3) were differentially expressed in the blood of ad cases versus controls for their directly measured levels (Table 2).
The regulatory architecture of gene expression can vary substantially across genes. In the current study, to fully capture the potential diversity of the regulatory architecture, genetic prediction models developed using two different modeling strategies, namely, revised UTMOST and LASSO, were leveraged. By borrowing information across tissues, the UTMOST framework provides a higher prediction accuracy compared with single-tissue approaches (37). As a comparison, we leveraged the LASSO method to establish models when analyzing the BIOS data, which collects transcriptome data in blood. The design that leverages complementary models can maximize the possibility to identify disease-associated genes. It is worth noting, however, that this may also induce false positives. In the current work, of the 108 identified associated genes, 29 were identified by both the UTMOST and LASSO prediction models, 37 only by the UTMOST models and 42 only by the LASSO models. As shown in the Results section, highly concordant association results were found between the two prediction approaches. Additional work is warranted for further validations. For the differential gene expression analysis, we analyzed an independent dataset of nine advanced ad cases and ten matched controls. As the sample size of this dataset is small, future work involving larger datasets is needed to further validate the potential utility of these 15 validated genes for ad risk assessment.
Recent work has reported that plasma P-tau181 is a noninvasive diagnostic and prognostic biomarker of ad (62,63). Plasma P-tau181 was found to be increased in preclinical ad and further increased at the MCI and dementia stages. It also differentiated ad dementia from non-ad neurodegenerative diseases with an accuracy similar to that of Tau PET and CSF P-tau181 (AUC = 0.94–0.98). The purpose of the current study was to identify candidate biomarkers for ad risk assessment. Besides such research, additional work focusing on related traits is also important. For example, it is critical to identify biomarkers that can predict the rate of cognitive decline. This knowledge is critical for planning clinical trials, as a more rapid rate of decline increases the possibility of shorter clinical trials with fewer subjects and thus lower costs. Also, it is important to identify biomarkers for predicting the presence of comorbidities with ad that may interfere with clinical trial success. Further investigation on these questions is needed.
In conclusion, in this large-scale study, we leveraged comprehensive gene expression prediction models in blood to assess the associations between genetically predicted gene expression and ad risk. We identified 108 ad-associated genes, of which the directly measured expression of 15 also showed the same direction of effect. These 15 genes can be further investigated for their potential utility in improving ad risk assessment.
Materials and Methods
Blood tissue gene expression prediction model building
Two different modeling strategies, modified UTMOST and LASSO, were used to build blood tissue gene expression genetic models.
The modified UTMOST models
Transcriptome and genome data from the GTEx v8 were used to develop genetic imputation models for genes expressed in 670 whole blood samples. Details of the GTEx v8 dataset have been described elsewhere (https://gtexportal.org/home/documentationPage). Briefly, genomic information of the subjects was collected using the whole-genome sequencing (WGS), as performed by the Broad Institute’s Genomics Platform. Details of RNA sequencing experiments, quality control (QC) of the gene expression data and genomic data have been described elsewhere (64,65).
The UTMOST framework was used to build gene expression prediction models (37). It has been shown that compared with conventional model building methods such as elastic net or LASSO, such a cross-tissue expression imputation method can substantially improve the accuracy of imputation. Considering the abundance of transcriptome data in GTEx, this framework would provide more accurate prediction by joint-tissue model training. Here the model training approach was modified to obtain a reliable estimate of the prediction performance. SNPs within 1 Mb upstream and downstream of the gene were considered as predictors in the model. It has been shown that there is no significant difference in prediction quality from applying LD pruning (47). Therefore, LD pruning (r2 = 0.9) was performed before model training to reduce the computational burden. In the joint-tissue prediction model, the effect sizes were estimated by minimizing the loss function with a LASSO penalty on the columns (within-tissue effects) and a group-LASSO penalty on the rows (cross-tissue effects). The group penalty term implemented sharing of the information from SNP selection across all the tissues. The optimization problem uses two hyperparameters, λ1 and λ2, for the within-tissue and cross-tissue penalization. Five-fold cross-validation was performed for hyperparameter tuning.
We modified the original model training step by unifying the hyperparameter pairs (λ1 and λ2) to avoid the inflation in the prediction performance. In brief, λ1 and λ2 were initialized using the range of pre-trained lambdas from single-tissue elastic net models. For each gene, 25 lambda pairs were generated. In the modified version, the 25 lambda pairs were consistent across the 5-fold cross-validation, while the original UTMOST assigned different lambdas for each fold. The unified hyperparameter pairs made the different folds directly comparable, thus avoiding the performance overestimation in a retrained model. The optimization of the joint model was initialized by single-tissue weights generated in each fold and the optimization stopped if the training error in each training set or the related tuning error was higher than the previous step. After the 5-fold training, one of the 25 lambda pairs was selected as the best lambda pair according to the average tuning error across the 5-folds. The prediction performance was evaluated by the correlation between the predicted and observed expression in the combined tuning set. Genes with good prediction performance (Pearson’s correlation r > 0.1 and P < 0.05) were included in the subsequent analyses.
LASSO models
Leveraging a reference transcriptome dataset involving 3344 blood samples, we generated prediction models using the LASSO regression method. The detailed information for this set of models has been described elsewhere (39). Briefly, RNA-seq data of these whole-blood samples from the BIOS Consortium were generated using Illumina’s Hiseq2000. The genetic data consisted of 881 977 unambiguous HapMap SNPs with MAF > 5%, minor allele count >10 and imputation info score >0.8. For the 13 870 genes with a significant expression quantitative trait locus (eQTL), a LASSO model was fitted in R with glmnet, to assess the potential predictive value of SNPs within 250 kb of the gene. We derived the unstandardized prediction models leveraging the original standardized models and standard deviation of variants in Europeans of the 1000 Genomes Project data. Notably, blood is the only available tissue/cell type for the BIOS Consortium study. Thus, the cross-tissue imputation method is equivalent to the LASSO method.
Associations between genetically predicted gene expression in blood and ad risk
We investigated the associations of genetically predicted gene expression in blood tissue with ad risk using the summary statistics generated from 71 880 (proxy) cases and 383 378 (proxy) controls of European ancestry. Instead of using the conventional approach of including clinically diagnosed ad alone, this study included both clinically confirmed and parental diagnoses based by-proxy phenotypes, the latter of which has been demonstrated to confer great value in substantially increasing statistical power. This study included 71 880 cases (24 087 clinically diagnosed late-onset ad and 47 793 proxy ad) and 383 378 controls of European ancestry. It has been found that ad-by-proxy, based on parental diagnoses, shows quite strong genetic correlation with ad (rg = 0.81) (41). The data are generated from three consortia (Alzheimer’s disease working group of the PGC-ALZ, the IGAP and the adSP) and the UK Biobank data (40,41). Detailed information on study participants, genotyping and imputation methods have been included in the original paper. The brief information of ad diagnosis methods used in these studies is summarized in the Supplementary Material, File.
Risk estimates for the single variant association analyses were adjusted for sex, batch (if applicable) as well as the top principal components (PCs) for the adSP and PGC-ALZ cohorts. For the PGC-ALZ cohorts, age was also included as a covariate. For the UK Biobank dataset, 12 PCs, age, sex, genotyping array and assessment center were included as covariates. The associations of predicted gene expression with ad risk were further estimated based on gene expression prediction weights, summary statistics of SNP-ad risk associations and an SNP-correlation (LD) matrix, using the S-PrediXcan framework (66). In brief, the formula:
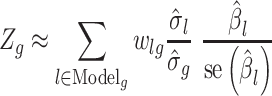 |
was used to estimate the Z-score of the association between predicted gene expression and ad risk. Here 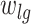 represents the weight of SNP
represents the weight of SNP 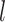 for predicting the expression of gene
for predicting the expression of gene 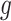 ,
, 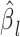 and
and 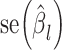 represent the GWAS association regression coefficient and its standard error for SNP
represent the GWAS association regression coefficient and its standard error for SNP 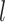 , and
, and 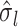 and
and 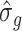 denote the square root of the estimated variances of SNP
denote the square root of the estimated variances of SNP 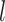 and the square root of the predicted expression of gene
and the square root of the predicted expression of gene 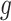 , respectively. The false discovery rate (FDR) corrected P-value threshold of ≤0.05 was used to determine significant associations between genetically predicted gene expression and ad risk.
, respectively. The false discovery rate (FDR) corrected P-value threshold of ≤0.05 was used to determine significant associations between genetically predicted gene expression and ad risk.
Directly measured expression levels of TWAS identified genes in the blood of ad cases versus controls
The detailed information of the dataset we used to assess directly measured expression of the identified genes in ad cases and controls was described elsewhere (67). Briefly, nine advanced female AD cases and ten age-matched female controls (independent of the GWAS samples) were included. Peripheral blood samples were processed to isolate RNA. The transcriptome was assayed using the SurePrint G3 Human Gene Expression 8x60k v2 microarrays (Agilent Technologies, CA). The gene expression data were processed with log transformation followed by quantile normalization using the linear models for microarray data (LIMMA) package in the bioconductor environment in R (68) implemented in the RStudio to identify differentially expressed genes (DEGs). The DEGs were selected based on FDR adjusted P-value < 0.05.
‘Core analysis’ in ingenuity pathway analysis (IPA)
For the genes associated with AD risk, we performed the ‘Core Analysis’ in IPA (69) to determine enriched pathways, diseases and networks. For enriched ‘Canonical Pathway’, the significance of P-value was calculated using the right-tailed Fisher’s Exact Test. ‘Function and Disease’ displayed the annotated biological function and/or linked diseases of the genes. Networks were obtained based on the connectivity of the genes. The network score was based on the hypergeometric distribution and was calculated with the right-tailed Fisher’s Exact Test. We also assessed the ‘Molecule’ function, which encodes knowledge about the molecule from the Ingenuity Knowledge Base.
Funding
University of Hawaii Cancer Center; National Institutes of Health (NIH) Genomic Innovator (R35HG010718, NIH/NHGRI R01HG011138, NIH/NIGMS R01GM140287 to E.G.); U01HG009086 (to N.J.C.); Visiting Research Program for High-Level Talents and Young Excellent Talents (2018-2019) of Fujian Provincial Department of Human Resources and Social Security, P.R. China (to Y.S.) and the Special Fund for Local Science and Technology Development Guided by the Chinese Government (grant 2019L3011).
Supplementary Material
Contributor Information
Yanfa Sun, Department of Animal Science and Veterinary Medicine, College of Life Science, Longyan University, Longyan, Fujian, 364012, P.R. China; Cancer Epidemiology Division, Population Sciences in the Pacific Program, University of Hawaii Cancer Center, University of Hawaii at Manoa, Honolulu, HI 96813, USA; Fujian Provincial Key Laboratory for the Prevention and Control of Animal Infectious Diseases and Biotechnology, Longyan, Fujian 364012, P.R. China; Fujian Province Universities Key Laboratory of Preventive Veterinary Medicine and Biotechnology (Longyan University), Longyan, Fujian, 364012, P.R. China.
Dan Zhou, Vanderbilt Genetics Institute and Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Md Rezanur Rahman, Queensland Brain Institute, The University of Queensland, Brisbane, Qld 4072, Australia.
Jingjing Zhu, Cancer Epidemiology Division, Population Sciences in the Pacific Program, University of Hawaii Cancer Center, University of Hawaii at Manoa, Honolulu, HI 96813, USA.
Dalia Ghoneim, Cancer Epidemiology Division, Population Sciences in the Pacific Program, University of Hawaii Cancer Center, University of Hawaii at Manoa, Honolulu, HI 96813, USA.
Nancy J Cox, Vanderbilt Genetics Institute and Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Thomas G Beach, Banner Sun Health Research Institute, Sun City, AZ 85351, USA.
Chong Wu, Department of Statistics, Florida State University, Tallahassee, FL 32306, USA.
Eric R Gamazon, Vanderbilt Genetics Institute and Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Clare Hall, University of Cambridge, Cambridge CB3 9AL, UK; MRC Epidemiology Unit, School of Clinical Medicine, University of Cambridge, Cambridge CB2 0SL, UK.
Lang Wu, Cancer Epidemiology Division, Population Sciences in the Pacific Program, University of Hawaii Cancer Center, University of Hawaii at Manoa, Honolulu, HI 96813, USA.
References
- 1. Ulrich, J.D., Ulland, T.K., Colonna, M. and Holtzman, D.M. (2017) Elucidating the role of TREM2 in Alzheimer’s disease. Neuron, 94, 237–248. [DOI] [PubMed] [Google Scholar]
- 2. Mathys, H., Davila-Velderrain, J., Peng, Z., Gao, F., Mohammadi, S., Young, J.Z., Menon, M., He, L., Abdurrob, F. and Jiang, X. (2019) Single-cell transcriptomic analysis of Alzheimer’s disease. Nature, 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Association, A.S (2019) 2019 Alzheimer's disease facts and figures. Alzheimers Dement., 15, 321–387. [Google Scholar]
- 4. Goedert, M. and Spillantini, M.G. (2006) A century of Alzheimer's disease. Science, 314, 777–781. [DOI] [PubMed] [Google Scholar]
- 5. Mayeux, R. and Schupf, N. (2011) Blood-based biomarkers for Alzheimer's disease: plasma Aβ40 and Aβ42, and genetic variants. Neurobiol. Aging, 32, S10–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blennow, K., Meyer, G., Hansson, O., Minthon, L., Wallin, A., Zetterberg, H., Lewczuk, P., Vanderstichele, H., Vanmechelen, E., Kornhuber, J. and Wiltfang, J. (2009) Evolution of Aβ42 and Aβ40 levels and Aβ42/Aβ40 ratio in plasma during progression of Alzheimer’s disease: a multicenter assessment. J. Nutr. Health Aging, 13, 205–208. [DOI] [PubMed] [Google Scholar]
- 7. Hansson, O., Zetterberg, H., Vanmechelen, E., Vanderstichele, H., Andreasson, U., Londos, E., Wallin, A., Minthon, L. and Blennow, K. (2010) Evaluation of plasma Aβ40 and Aβ42 as predictors of conversion to Alzheimer's disease in patients with mild cognitive impairment. Neurobiol. Aging, 31, 357–367. [DOI] [PubMed] [Google Scholar]
- 8. Wood, H. (2016) ALZHEIMER DISEASE: biomarkers of AD risk—the end of the road for plasma amyloid-β? Nat. Rev. Neurol., 12, 613. [DOI] [PubMed] [Google Scholar]
- 9. Lövheim, H., Elgh, F., Johansson, A., Zetterberg, H., Blennow, K., Hallmans, G. and Eriksson, S. (2017) Plasma concentrations of free amyloid β cannot predict the development of Alzheimer's disease. Alzheimers Dement., 13, 778–782. [DOI] [PubMed] [Google Scholar]
- 10. Kauwe, J.S., Wang, J., Mayo, K., Morris, J.C., Fagan, A.M., Holtzman, D.M. and Goate, A.M. (2009) Alzheimer’s disease risk variants show association with cerebrospinal fluid amyloid beta. Neurogenetics, 10, 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cruchaga, C., Kauwe, J.S., Harari, O., Jin, S.C., Cai, Y., Karch, C.M., Benitez, B.A., Jeng, A.T., Skorupa, T. and Carrell, D. (2013) GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron, 78, 256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee, J.C., Kim, S.J., Hong, S. and Kim, Y. (2019) Diagnosis of Alzheimer’s disease utilizing amyloid and tau as fluid biomarkers. Exp. Mol. Med., 51, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lutz, M.W., Sundseth, S.S., Burns, D.K., Saunders, A.M., Hayden, K.M., Burke, J.R., Welsh-Bohmer, K.A., Roses, A.D. and Initiative, A.s.D.N (2016) A genetics-based biomarker risk algorithm for predicting risk of Alzheimer's disease. Alzheimer's Dement. Transl. Res. Clin. Interv., 2, 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peña-Bautista, C., Vigor, C., Galano, J.-M., Oger, C., Durand, T., Ferrer, I., Cuevas, A., López-Cuevas, R., Baquero, M. and López-Nogueroles, M. (2019) New screening approach for Alzheimer’s disease risk assessment from urine lipid peroxidation compounds. Sci. Rep., 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peña-Bautista, C., Vigor, C., Galano, J.-M., Oger, C., Durand, T., Ferrer, I., Cuevas, A., López-Cuevas, R., Baquero, M. and López-Nogueroles, M. (2018) Plasma lipid peroxidation biomarkers for early and non-invasive Alzheimer disease detection. Free Radic. Biol. Med., 124, 388–394. [DOI] [PubMed] [Google Scholar]
- 16. García-Blanco, A., Peña-Bautista, C., Oger, C., Vigor, C., Galano, J.-M., Durand, T., Martín-Ibáñez, N., Baquero, M., Vento, M. and Cháfer-Pericás, C. (2018) Reliable determination of new lipid peroxidation compounds as potential early Alzheimer disease biomarkers. Talanta, 184, 193–201. [DOI] [PubMed] [Google Scholar]
- 17. Schoeman, J.C., Fu, J., Harms, A.C., van Wietmarschen, H.A., Vreeken, R.J., Berger, R., Cuppen, B.V., Lafeber, F.P., van der Greef, J. and Hankemeier, T. (2016) Metabolomics profiling of the free and total oxidised lipids in urine by LC-MS/MS: application in patients with rheumatoid arthritis. Virus-host Metabolic Interactions. Anal. Bioanal. Chem., 408, 6307–6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mohr, S. and Liew, C.-C. (2007) The peripheral-blood transcriptome: new insights into disease and risk assessment. Trends Mol. Med., 13, 422–432. [DOI] [PubMed] [Google Scholar]
- 19. Zhu, M. and Wang, L. (2019) Whole blood mRNA expression pattern differentiates AD patients and healthy controls through bioinformatics analysis. J. Biol. Life Sci., 10, 46–57. [Google Scholar]
- 20. Rahman, M.R., Islam, T., Shahjaman, M., Quinn, J.M., Holsinger, R.D. and Moni, M.A. (2019) Identification of common molecular biomarker signatures in blood and brain of Alzheimers disease. BioRxiv, 482828. [Google Scholar]
- 21. Fehlbaum-Beurdeley, P., Jarrige-Le Prado, A.C., Pallares, D., Carrière, J., Guihal, C., Soucaille, C., Rouet, F., Drouin, D., Sol, O. and Jordan, H. (2010) Toward an Alzheimer's disease diagnosis via high-resolution blood gene expression. Alzheimers Dement., 6, 25–38. [DOI] [PubMed] [Google Scholar]
- 22. Booij, B.B., Lindahl, T., Wetterberg, P., Skaane, N.V., Sæbø, S., Feten, G., Rye, P.D., Kristiansen, L.I., Hagen, N. and Jensen, M. (2011) A gene expression pattern in blood for the early detection of Alzheimer's disease. J. Alzheimers Dis., 23, 109–119. [DOI] [PubMed] [Google Scholar]
- 23. Rye, P., Booij, B.B., Grave, G., Lindahl, T., Kristiansen, L., Andersen, H.-M., Horndalsveen, P.O., Nygaard, H.A., Naik, M. and Hoprekstad, D. (2011) A novel blood test for the early detection of Alzheimer's disease. J. Alzheimers Dis., 23, 121–129. [DOI] [PubMed] [Google Scholar]
- 24. Chen, K.-D., Chang, P.-T., Ping, Y.-H., Lee, H.-C., Yeh, C.-W. and Wang, P.-N. (2011) Gene expression profiling of peripheral blood leukocytes identifies and validates ABCB1 as a novel biomarker for Alzheimer's disease. Neurobiol. Dis., 43, 698–705. [DOI] [PubMed] [Google Scholar]
- 25. Lunnon, K., Sattlecker, M., Furney, S.J., Coppola, G., Simmons, A., Proitsi, P., Lupton, M.K., Lourdusamy, A., Johnston, C. and Soininen, H. (2013) A blood gene expression marker of early Alzheimer's disease. J. Alzheimers Dis., 33, 737–753. [DOI] [PubMed] [Google Scholar]
- 26. Xu, H., Stamova, B., Jickling, G., Tian, Y., Zhan, X., Ander, B.P., Liu, D., Turner, R., Rosand, J. and Goldstein, L.B. (2010) Distinctive RNA expression profiles in blood associated with white matter hyperintensities in brain. Stroke, 41, 2744–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Delvaux, E., Mastroeni, D., Nolz, J., Chow, N., Sabbagh, M., Caselli, R.J., Reiman, E.M., Marshall, F.J. and Coleman, P.D. (2017) Multivariate analyses of peripheral blood leukocyte transcripts distinguish Alzheimer's, Parkinson's, control, and those at risk for developing Alzheimer's. Neurobiol. Aging, 58, 225–237. [DOI] [PubMed] [Google Scholar]
- 28. Rahman, M.R., Islam, T., Zaman, T., Shahjaman, M., Karim, M.R., Huq, F., Quinn, J.M., Holsinger, R.D., Gov, E. and Moni, M.A. (2020) Identification of molecular signatures and pathways to identify novel therapeutic targets in Alzheimer's disease: insights from a systems biomedicine perspective. Genomics, 112, 1290–1299. [DOI] [PubMed] [Google Scholar]
- 29. Moradi, E., Marttinen, M., Häkkinen, T., Hiltunen, M. and Nykter, M. (2019) Supervised pathway analysis of blood gene expression profiles in Alzheimer's disease. Neurobiol. Aging, 84, 98–108. [DOI] [PubMed] [Google Scholar]
- 30. Lee, T. and Lee, H. (2020) Prediction of Alzheimer’s disease using blood gene expression data. Sci. Rep., 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mancuso, N., Gayther, S., Gusev, A., Zheng, W., Penney, K.L., Kote-Jarai, Z., Eeles, R., Freedman, M., Haiman, C. and Pasaniuc, B. (2018) Large-scale transcriptome-wide association study identifies new prostate cancer risk regions. Nat. Commun., 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu, L., Wang, J., Cai, Q., Cavazos, T.B., Emami, N.C., Long, J., Shu, X.-O., Lu, Y., Guo, X. and Bauer, J.A. (2019) Identification of novel susceptibility loci and genes for prostate cancer risk: a transcriptome-wide association study in over 140,000 European descendants. Cancer Res., 79, 3192–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu, L., Shi, W., Long, J., Guo, X., Michailidou, K., Beesley, J., Bolla, M.K., Shu, X.-O., Lu, Y. and Cai, Q. (2018) A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nat. Genet., 50, 968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gusev, A., Mancuso, N., Won, H., Kousi, M., Finucane, H.K., Reshef, Y., Song, L., Safi, A., McCarroll, S. and Neale, B.M. (2018) Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet., 50, 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hao, S., Wang, R., Zhang, Y. and Zhan, H. (2019) Prediction of Alzheimer’s disease-associated genes by integration of GWAS summary data and expression data. Front. Genet., 9, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Raj, T., Li, Y.I., Wong, G., Humphrey, J., Wang, M., Ramdhani, S., Wang, Y.-C., Ng, B., Gupta, I. and Haroutunian, V. (2018) Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet., 50, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu, Y., Li, M., Lu, Q., Weng, H., Wang, J., Zekavat, S.M., Yu, Z., Li, B., Gu, J. and Muchnik, S. (2019) A statistical framework for cross-tissue transcriptome-wide association analysis. Nat. Genet., 51, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gerring, Z.F., Lupton, M.K., Edey, D., Gamazon, E.R. and Derks, E.M. (2020) An analysis of genetically regulated gene expression across multiple tissues implicates novel gene candidates in Alzheimer’s disease. Alzheimers Res. Ther., 12, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baselmans, B.M., Jansen, R., Ip, H.F., van Dongen, J., Abdellaoui, A., van de Weijer, M.P., Bao, Y., Smart, M., Kumari, M. and Willemsen, G. (2019) Multivariate genome-wide analyses of the well-being spectrum. Nat. Genet., 51, 445–451. [DOI] [PubMed] [Google Scholar]
- 40. Marioni, R.E., Harris, S.E., Zhang, Q., McRae, A.F., Hagenaars, S.P., Hill, W.D., Davies, G., Ritchie, C.W., Gale, C.R. and Starr, J.M. (2018) GWAS on family history of Alzheimer’s disease. Transl. Psychiatry, 8, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jansen, I.E., Savage, J.E., Watanabe, K., Bryois, J., Williams, D.M., Steinberg, S., Sealock, J., Karlsson, I.K., Hägg, S. and Athanasiu, L. (2019) Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet., 51, 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Storey, J.D. and Tibshirani, R. (2003) Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA., 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M.L., Pahwa, J.S., Moskvina, V., Dowzell, K. and Williams, A. (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet., 41, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Franzmeier, N., Rubinski, A., Neitzel, J. and Ewers, M. (2019) The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat. Commun., 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kunkle, B.W., Grenier-Boley, B., Sims, R., Bis, J.C., Damotte, V., Naj, A.C., Boland, A., Vronskaya, M., Van Der Lee, S.J. and Amlie-Wolf, A. (2019) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet., 51, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou, D., Jiang, Y., Zhong, X., Cox, N.J., Liu, C. and Gamazon, E.R. (2020) A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat. Genet., 52, 1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gamazon, E.R., Wheeler, H.E., Shah, K.P., Mozaffari, S.V., Aquino-Michaels, K., Carroll, R.J., Eyler, A.E., Denny, J.C., Consortium, G.T, Nicolae, D.L. et al. (2015) A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet., 47, 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tao, X., West, A.E., Chen, W.G., Corfas, G. and Greenberg, M.E. (2002) A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron, 33, 383–395. [DOI] [PubMed] [Google Scholar]
- 49. Bennett, J. and Keeney, P. (2017) Micro RNA’s (mirna’s) may help explain expression of multiple genes in Alzheimer’s frontal cortex. J. Syst. Integr. Neurosci., 3, 1–9. [Google Scholar]
- 50. Matsushima, H., Shimohama, S., Fujimoto, S., Takenawa, T. and Kimura, J. (1995) Reduction of platelet phospholipase C activity in patients with Alzheimer disease. Alzheimer Dis. Assoc. Disord., 9, 213–217. [PubMed] [Google Scholar]
- 51. Castillo, E., Leon, J., Mazzei, G., Abolhassani, N., Haruyama, N., Saito, T., Saido, T., Hokama, M., Iwaki, T. and Ohara, T. (2017) Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep., 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Song, Z., Xu, Y., Zhang, L., Zhou, L., Zhang, Y., Han, Y., Li, X., Yu, P., Qu, Y., Zhao, W. and Qin, C. (2020) Comprehensive proteomic profiling of urinary exosomes and identification of potential non-invasive early biomarkers of Alzheimer's disease in 5XFAD mouse model. Front. Genet., 11, 565479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rutherford, N.J., Carrasquillo, M.M., Li, M., Bisceglio, G., Menke, J., Josephs, K.A., Parisi, J.E., Petersen, R.C., Graff-Radford, N.R., Younkin, S.G. et al. (2012) TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology, 79, 717–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Satoh, J., Kino, Y., Kawana, N., Yamamoto, Y., Ishida, T., Saito, Y. and Arima, K. (2014) TMEM106B expression is reduced in Alzheimer's disease brains. Alzheimers Res. Ther., 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Escott-Price, V., Bellenguez, C., Wang, L.S., Choi, S.H., Harold, D., Jones, L., Holmans, P., Gerrish, A., Vedernikov, A., Richards, A. et al. (2014) Gene-wide analysis detects two new susceptibility genes for Alzheimer's disease. PLoS One, 9, e94661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bik-Multanowski, M. and Dobosz, A. (2017) Detection of high expression of complex I mitochondrial genes can indicate low risk of Alzheimer's disease. Int. J. Clin. Exp. Pathol., 10, 1904–1907. [Google Scholar]
- 57. Quan, X., Yue, W., Luo, Y., Cao, J., Wang, H., Wang, Y. and Lu, Z. (2015) The protein arginine methyltransferase PRMT5 regulates Abeta-induced toxicity in human cells and Caenorhabditis elegans models of Alzheimer's disease. J. Neurochem., 134, 969–977. [DOI] [PubMed] [Google Scholar]
- 58. Wingo, A.P., Liu, Y., Gerasimov, E.S., Gockley, J., Logsdon, B.A., Duong, D., Dammer, E.B., Robins, C., Cutler, D.J. and De Jager, P.L. (2020) Integrating human brain proteomes and genome-wide association results implicates new genes in Alzheimer's disease: Functionalizing genetic variants in Alzheimer's disease. Alzheimer's & Dementia, 16, e043865. [Google Scholar]
- 59. Wang, K.S., Liu, X., Xie, C., Liu, Y. and Xu, C. (2016) Non-parametric survival analysis of EPG5 gene with age at onset of Alzheimer's disease. J. Mol. Neurosci., 60, 436–444. [DOI] [PubMed] [Google Scholar]
- 60. Maezawa, I., Jenkins, D.P., Jin, B.E. and Wulff, H. (2012) Microglial KCa3.1 channels as a potential therapeutic target for Alzheimer's disease. Int. J. Alzheimers Dis., 2012, 868972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hondius, D.C., Koopmans, F., Leistner, C., Pita-Illobre, D., Peferoen-Baert, R.M., Marbus, F., Paliukhovich, I., Li, K.W., Rozemuller, A.J.M., Hoozemans, J.J.M. et al. (2021) The proteome of granulovacuolar degeneration and neurofibrillary tangles in Alzheimer's disease. Acta Neuropathol., 141, 341–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Janelidze, S., Mattsson, N., Palmqvist, S., Smith, R., Beach, T.G., Serrano, G.E., Chai, X., Proctor, N.K., Eichenlaub, U. and Zetterberg, H. (2020) Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med., 26, 379–386. [DOI] [PubMed] [Google Scholar]
- 63. Thijssen, E.H., La Joie, R., Wolf, A., Strom, A., Wang, P., Iaccarino, L., Bourakova, V., Cobigo, Y., Heuer, H. and Spina, S. (2020) Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat. Med., 26, 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. The GTEx Consortium, Ardlie, K.G., Deluca, D.S., Segre, A.V., Sullivan, T.J., Young, T.R., Gelfand, E.T., Trowbridge, C.A., Maller, J.B., Tukiainen, T. et al. (2015) The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science, 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Consortium, G (2017) Genetic effects on gene expression across human tissues. Nature, 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gusev, A., Ko, A., Shi, H., Bhatia, G., Chung, W., Penninx, B.W., Jansen, R., De Geus, E.J., Boomsma, D.I. and Wright, F.A. (2016) Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet., 48, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Naughton, B.J., Duncan, F.J., Murrey, D.A., Meadows, A.S., Newsom, D.E., Stoicea, N., White, P., Scharre, D.W., Mccarty, D.M. and Fu, H. (2015) Blood genome-wide transcriptional profiles reflect broad molecular impairments and strong blood-brain links in Alzheimer's disease. J. Alzheimers Dis., 43, 93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Smyth, G.K. and Speed, T. (2003) Normalization of cDNA microarray data. Methods, 31, 265–273. [DOI] [PubMed] [Google Scholar]
- 69. Krämer, A., Green, J., Pollard, J., Jr. and Tugendreich, S. (2014) Causal analysis approaches in ingenuity pathway analysis. Bioinformatics, 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lambert, J.-C., Ibrahim-Verbaas, C.A., Harold, D., Naj, A.C., Sims, R., Bellenguez, C., Jun, G., DeStefano, A.L., Bis, J.C. and Beecham, G.W. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet., 45, 1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.