

Abstract
Glioblastoma (GBM) is the most lethal primary brain cancer characterized by therapeutic resistance, which is promoted by GBM stem cells (GSCs). Here, we interrogated gene expression and whole genome CRISPR/Cas9 screening in a large panel of patient-derived GSCs, differentiated glioblastoma cells (DGCs), and neural stem cells (NSCs) to identify master regulators of GSC stemness, revealing an essential transcription state with increased RNA polymerase II-mediated transcription. The YY1 and transcriptional CDK9 complex was essential for GSC survival and maintenance in vitro and in vivo. YY1 interacted with CDK9 to regulate transcription elongation in GSCs. Genetic or pharmacological targeting of YY1-CDK9 complex elicited RNA m6A modification-dependent interferon responses, reduced regulatory T cell infiltration, and augmented efficacy of immune checkpoint therapy in glioblastoma. Collectively, these results suggest that YY1-CDK9 transcription elongation complex defines a targetable cell state with active transcription, suppressed interferon responses, and immunotherapy resistance in glioblastoma.
Keywords: Glioblastoma, cancer, YY1, transcription elongation, CDK9, METTL3, m6A, immunotherapy
INTRODUCTION
Glioblastoma (GBM, World Health Organization grade IV glioma) ranks among the most aggressive and lethal types of human cancers. Treatment options for GBM patients remain ineffective and only palliative (1). GBMs display extensive intratumoral and microenvironmental heterogeneity, in which GBM stem cells (GSCs) play an important role in the tumor hierarchy (2–4). GSCs, driven by both genetic and epigenetic alterations, contribute to sustained tumor growth, invasion into normal brain, evasion of immune surveillance, and therapeutic resistance; thus, representing a crucial target for GBM treatment (4–7).
Recent advances in profiling GBM at the bulk and single cell levels have deciphered different cell states associated with GSCs, which can be regulated by the genetic, epigenetic or microenvironmental factors (8). Although genetic driver alterations, such as EGFR amplification, has failed to provide effective therapies in GBM (9), examinations of epigenetic and transcriptional regulations have identified master regulatory circuitry that defines cell state plasticity, as well as immune response in malignant cells (10–12). Dysregulated transcriptional programs in cancer cells open avenues to novel cancer therapies, illustrated by targeting MYC with BRD4 inhibitors or enhancer-associated gene expression with inhibitors of transcriptional cyclin-dependent kinases (CDKs) (13–15). Cell-cycle-related CDK4/6 has been well studied in cancers; however, the transcriptional CDKs, including CDK7, CDK9, and CDK12, which are involved in RNA polymerase (Pol) II-mediated transcriptional regulation, are not fully explored as therapeutic targets (16).
Among the epigenetic regulations, the role of chromatin interactions in cancers remains elusive. The CCCTC-binding factor (CTCF) insulator establishes topologically-associated domains (TADs) (17). In gliomas, IDH1 mutations induce hypermethylation to disrupt CTCF binding sites and chromosomal neighborhoods, leading to the interaction between a constitutive enhancer with platelet-derived growth factor receptor A (PDGFRA) and the subsequent upregulation of PDGFRA expression (18). Yin Yang 1 (YY1) is another structural regulator of chromatin interaction loops that controls gene expression (19). Disrupted chromosomal neighborhoods and loops in cancers represent potential targets, but the therapeutic opportunities underlying how chromatin structural proteins regulates cancer stemness states are poorly understood (20).
Although immune checkpoint inhibitors have transformed care for several solid tumors, including those with brain metastases, their clinical efficacy in GBM has been disappointing. Effective strategies of immune modulation to enhance response to immunotherapy are not available in GBM (21,22). Considering the lack of effective treatment and inconsistent response to immunotherapy in GBM, we hypothesized that a systematic dependency analysis of chromatin regulators in GSCs could identify targetable cell states and associated master epigenetic regulators linked to therapeutic response for GBM.
RESULTS
Chromatin regulator landscapes identify YY1 transcriptional dependency in glioblastoma stem cells
To investigate core chromatin regulators in glioblastoma, we interrogated the landscape of chromatin regulators across GSCs and matched differentiated GBM cells (DGCs), as well as their normal counterparts, neural stem cells (NSCs), by considering both transcription activities and cellular dependencies (Fig. 1A) (23). By integrating differentially expressed genes (DEGs) between 38 GSCs and 5 NSCs (24) with chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) profiling of 160 chromatin regulators, 39 chromatin regulators showed active programs inferred by their target gene expression in GSCs, including transcriptional programs driven by MYC, Polycomb Repressive Complex 2 (PRC2, including EZH2 and SUZ12), and YY1, supporting potential roles in gliomagenesis (Fig. 1B and C; Supplementary Fig. S1A; Supplementary Table S1). To identify stemness-associated regulators, we compared expression data from GSCs and matched DGCs (25), revealing 12 regulators of which programs were enriched in GSCs, including MYC, EZH2, and YY1 (Fig. 1D and E; Supplementary Table S1). Translating master regulators as targets requires an optimal therapeutic index; i.e., selective dependency of a target in neoplastic cells (in this case GSCs) relative to normal counterparts (e.g., NSCs). We then interrogated CRISPR/Cas9 drop-out screens from 8 GSCs and 2 NSCs to identify GSC-specific dependencies (26). Among 160 chromatin regulators, 10 were selectively essential in GSCs compared to NSCs, including JUN, NRF1, and YY1 (Fig. 1F and G; Supplementary Fig. S1B). Integrating these analyses indicated that MYC and PRC2 complex were important for shaping gene expression profiles, but failed to show differential dependencies between GSCs and NSCs (Fig. 1B-G). Surprisingly, several components related to RNA polymerase (Pol)-mediated transcription, including YY1, GTF2B, GTF3C2, and POLR3A, showed differential dependencies in GSCs (Fig. 1F; Supplementary Fig. S1C). Among these regulators, YY1, a master regulator important for shaping the regulatory circuitries and conferring selective dependencies in GSCs (Fig. 1H), was selected for further studies.
Figure 1. Chromatin regulator landscapes identify transcriptional dependencies in glioblastoma stem cells.
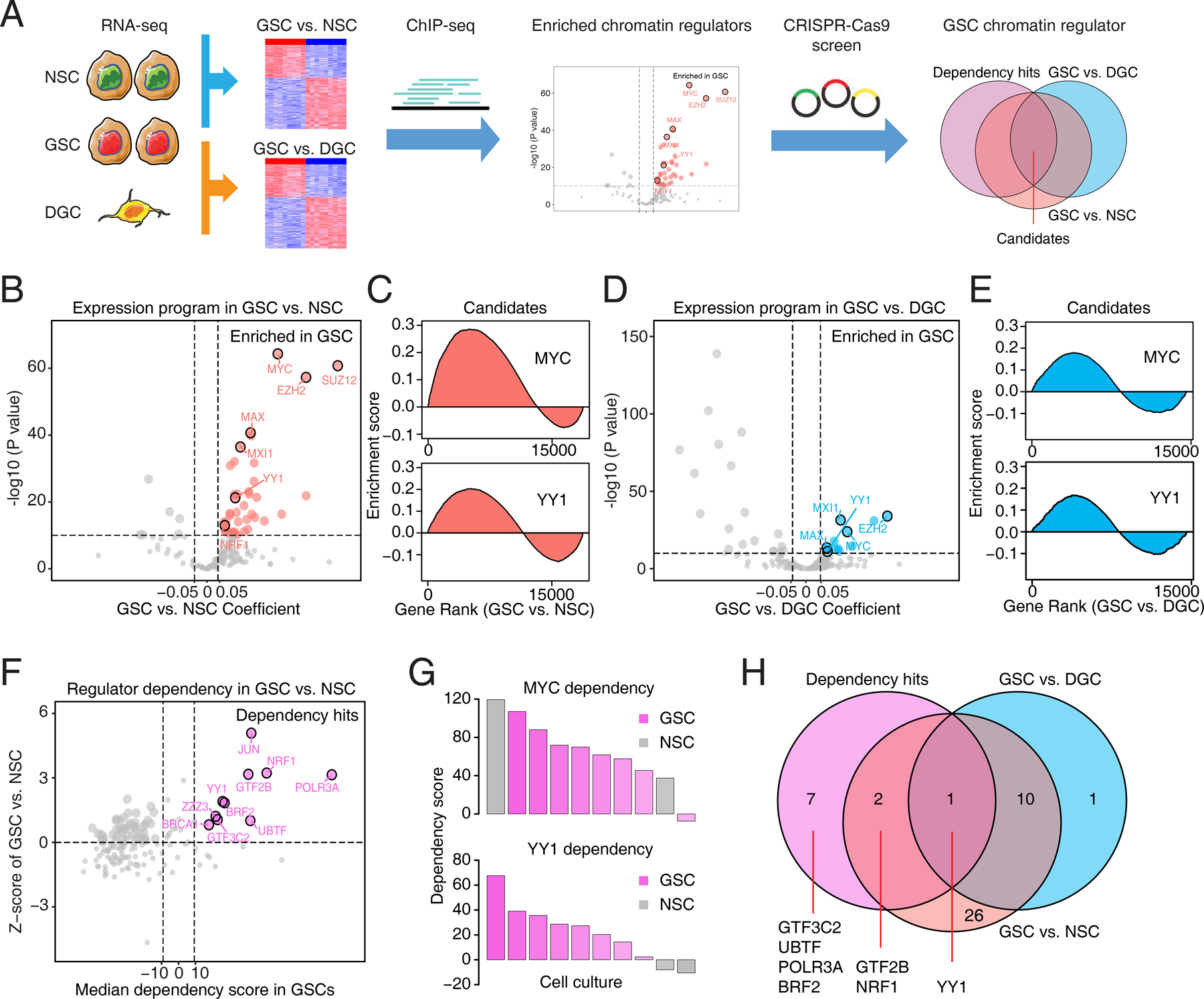
A, Diagram depicting the screening strategy to identify the enriched chromatin regulators with selective dependency in glioblastoma stem cells (GSCs). NSC, neural stem cell. DGC, differentiated glioblastoma cell. B, Volcano plot showing enrichment of 160 transcription regulator programs in 38 GSCs and 5 NSCs (GSE119834). Genes with its target program highly expressed in GSCs were labeled as red. C, Enrichment map visualization of candidate regulator programs (MYC and YY1) in GSCs compared to NSCs. D, Volcano plot showing enrichment of 160 transcription regulator programs in three pairs of GSCs and DGCs (GSE54791). Genes with its target program highly expressed in GSCs were labeled as blue. E, Enrichment map visualization of candidate regulator programs (MYC and YY1) in GSCs compared to DGCs. F, Scatter plot showing relative dependencies of 160 transcription regulators in 8 GSCs and 2 NSCs from whole genome CRISPR/Cas9 loss-of-function screening. Dependency genes specifically for GSCs were labeled as pink. G, Bar plots showing dependency scores of MYC and YY1 from whole genome CRISPR/Cas9 loss-of-function screening in a panel of GSCs (pink) and NSCs (gray). H, Venn diagram plot showing overlapping of transcription program and dependency hits for 160 regulators in GSCs.
GSC master regulator YY1 is essential for the maintenance of glioblastoma stem cells
YY1 is a zinc-finger transcription factor and interacts with other chromatin regulators, including MYC and EZH2, suggesting YY1 may function as a regulatory hub with selective dependency in GSCs (27–29). The dependency of GSCs on YY1 was further tested. Knockdown of YY1 using three non-overlapping shRNAs strongly decreased cell proliferation in a panel of five patient-derived GSCs, compared to non-neoplastic NSCs (Fig. 2A-F; Supplementary Fig. S1D and S1E). YY1 has been implicated in chromatin interactions, especially those connecting active regulatory elements, such as enhancer-enhancer, enhancer-promoter, and promoter-promoter interactions (19). CTCF is a structural protein that generally insulates neighborhoods and global structures, as well as long-range interactions (17). In contrast to effects of YY1 targeting, knockdown of CTCF only modestly inhibited cell proliferation in GSCs (Fig. 2A-E; Supplementary Fig. S1D and S1E). CRISPR/Cas9-based knockout validated selective dependency on YY1 over CTCF in GSCs (Supplementary Fig. S1F and S1G). Stem cell frequency and self-renewal of GSCs were diminished upon YY1 depletion, as revealed by extreme limiting dilution assays and tumor sphere formation (Fig. 2G-L). To assess the in vivo tumor initiation dependency of GSCs on YY1, two patient-derived GSCs were transduced with a control shRNA sequence (shCONT) or one of the two non-overlapping shRNAs targeting YY1, then implanted intracranially into immunodeficient mice. YY1 knockdown prolonged survival of intracranial tumor-bearing mice relative to the shCONT (p < 0.01 by log-rank test; Fig. 2M and N), suggesting that YY1 is a master regulator controlling proliferation and self-renewal of GSCs in vivo.
Figure 2. YY1 is essential for glioblastoma stem cells both in vitro and in vivo.
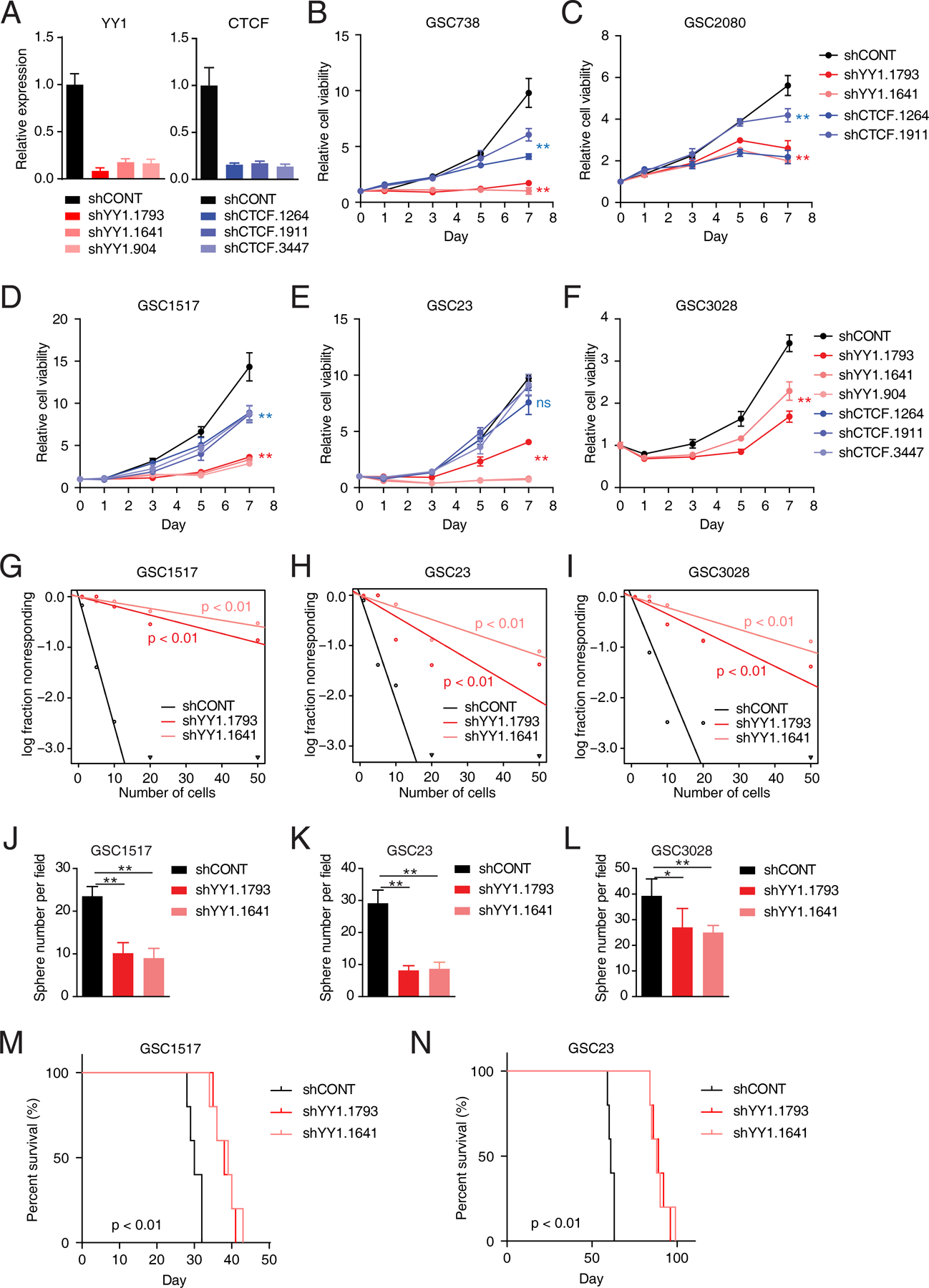
A, Bar plots showing YY1 and CTCF expression levels in GSC1517 with shRNA-mediated knockdown of YY1 or CTCF, respectively, as determined by qRT-PCR. Data are presented as mean + SD. B-F, Relative cell viabilities of a panel of GSCs with shRNA-mediated knockdown of YY1 or CTCF at different time points. Data are presented as mean ± SD. G-I, Tumor sphere formation efficiency and self-renewal capacity were measured by extreme in vitro limiting dilution assays (ELDA) in three GSCs. P-values were determined by likelihood ratio test. J-L, Bar plots showing the number of spheres from different GSCs after YY1 knockdown at day 7. Data are presented as mean + SD. M and N, Kaplan-Meier curves showing survival of immunocompromised mice bearing intracranial tumors from GSC1517 (M) and GSC23 (N) following transduction with indicated shRNAs. P-values were determined by log-rank test. *p < 0.05, **p < 0.01 by one-way ANOVA with Dunnett multiple comparison test; ns, not significant.
YY1 regulates a cell state with active transcription and RNA N6-methyladenosine programs
As YY1 is a structural regulator in chromatin loop formation (19), we integrated RNA-seq, ChIP-seq, and Bridge-Linker-Hi-C (BL-Hi-C) analysis to dissect the YY1-regulated program (30). Gene set enrichment analysis (GSEA) of RNA-seq data identified several pathways downregulated after YY1 knockdown, including Pol II transcription, RNA processing, MYC activity, and cell cycle, which were enriched in YY1 targets from ChIP-seq data and GSC specific gene expression signatures (Fig. 3A; Supplementary Fig. S2A-S2C; Supplementary Table S2). The expression levels of genes involved in Pol II-mediated transcription (PAPOLA and MYC), RNA splicing (SRSF1–3 and HNRNPU), and RNA N6-methyladenosine (m6A) modification (METTL3 and YTHDF2) were downregulated by shRNA-mediated YY1 knockdown and CRISPR/Cas9-mediated knockout (Fig. 3B; Supplementary Fig. S2D). Whole genome CRISPR/Cas9 drop-out screens also supported that YY1 target genes were enriched as GSC-specific dependencies (Supplementary Fig. S2E).
Figure 3. YY1 regulates transcription and RNA m6A methylation programs and suppresses interferon signaling.
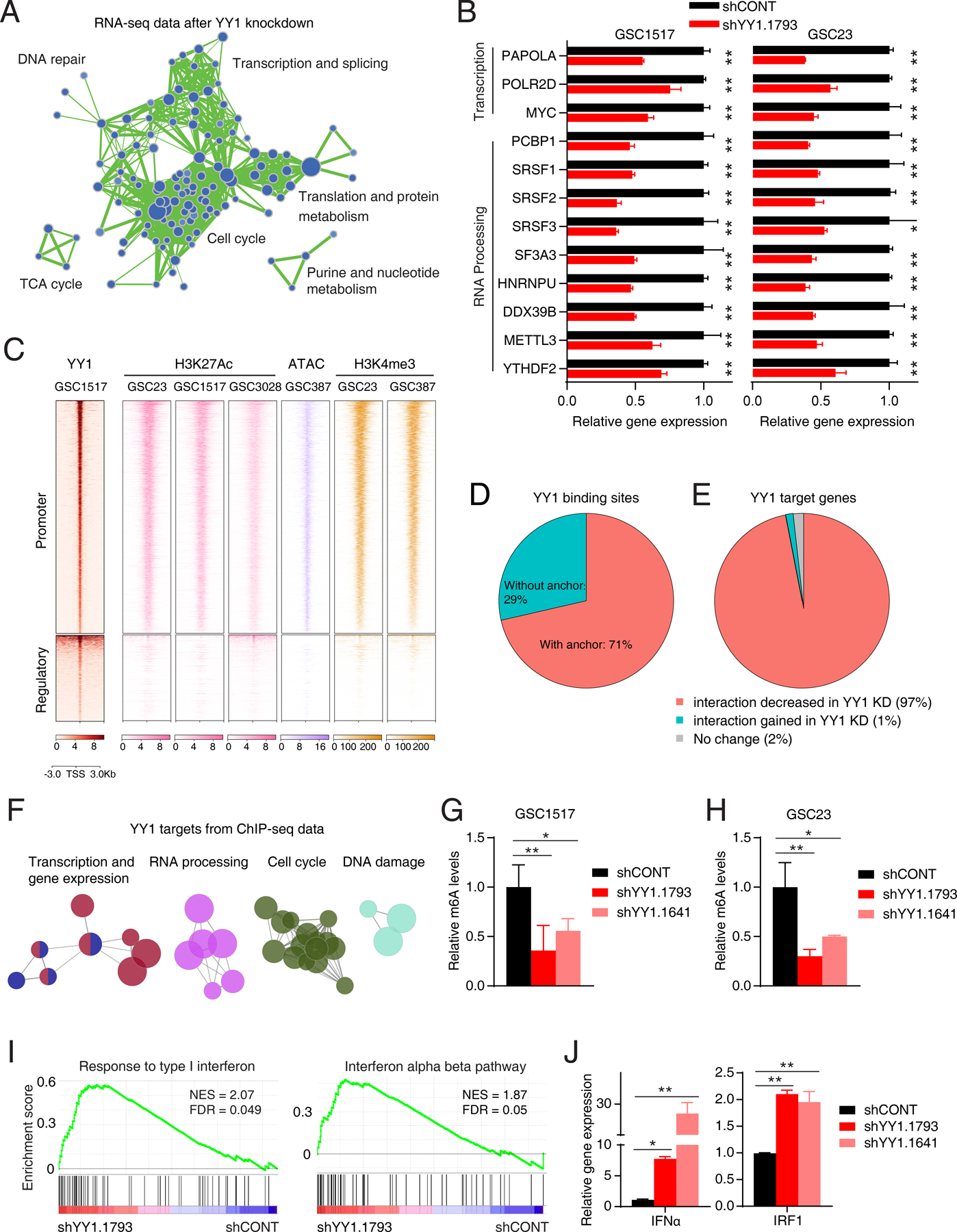
A, Enrichment map visualization of downregulated gene sets in GSEA after YY1 knockdown. B, Bar plots showing selected down-regulated genes involved in RNA Pol II transcription and RNA processing after YY1 knockdown. Data are presented as mean + SD. *p < 0.05, **p < 0.01 by unpaired Student’s t-test. C, Heatmaps showing ChIP-seq signals of YY1, H3K27Ac, H3K4me3 and ATAC-Seq signals in GSCs. The signals are displayed within a region spanning ±3 kb around center of YY1 binding peaks. D, Pie plot showing the overlapping between YY1 binding sites and chromatin interaction anchors inferred by BL-Hi-C sequencing in GSC cells. E, Pie plots showing the percentage of YY1 target genes with decreased chromatin interactions after YY1 knockdown. F, ClueGO plot of canonical pathways enriched in the YY1 putative target genes inferred from ChIP-seq profiling in GSC1517. G and H, Bar plots showing relative m6A levels after YY1 knockdown in GSC1517 (G) and GSC23 (H). Data are presented as mean + SD. *p < 0.05, **p < 0.01 by one-way ANOVA with Dunnett multiple comparison test. I, GSEA plots of representative gene sets involved in immune response after YY1 knockdown. The normalized enrichment score (NES) and false discovery rate (FDR) were indicated. J, Bar plots showing expression levels of IFNα and IRF1 in GSC1517 after YY1 knockdown determined by qRT-PCR. Data are presented as mean + SD. *p < 0.05, **p < 0.01 by one-way ANOVA with Dunnett multiple comparison test.
At the regulatory level, interrogation of active transcription marks, including histone 3 lysine 27 acetylation (H3K27Ac), histone 3 lysine 4 trimethylation (H3K4me3), and open chromatin profile by Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq), showed that YY1 largely bound to the active transcriptional regions in GSCs (Fig. 3C). About 70% of YY1 binding peaks overlapped with loop anchors (Fig. 3D), and almost all the YY1 target genes (97%) showed decreased number of chromatin interactions (Fig. 3E; Supplementary Table S3), indicating a regulatory role of 3D genome for YY1 targets. Pathway analysis using putative YY1 target genes from ChIP-seq profiling showed the similar enrichment of YY1-regulated biological processes in RNA-seq, including RNA Pol II transcription, RNA processing and splicing, cell cycle and DNA damage (Fig. 3F). Taken together, these results suggest that YY1 acts as a checkpoint to maintain a GSC transcription state through controlling RNA Pol II transcription and RNA processing (31).
Genetic targeting of the YY1 program triggers m6A-mediated interferon signaling and antigen presentation
We next focused on the transcriptional regulation of RNA processing programs, especially the m6A program, and its effect on GSCs. Using BL-HiC technology, the chromatin interaction frequencies decreased after YY1 knockdown, as shown at the METTL3 locus (Supplementary Fig. S2F). Two YY1 target genes, METTL3 and YTHDF2, showed decreased promoter-anchored chromatin interactions and decreased expression levels after YY1 knockdown (Fig. 3B; Supplementary Fig. S2G and S2H), supporting the essential role of YY1-mediated chromatin interactions for transcription of RNA m6A regulators. In line with the downregulation of METTL3, the total m6A levels were decreased after YY1 knockdown in GSCs (Fig. 3G and H).
Inhibiting METTL3 or YTHDF2 stabilizes interferon-related genes (e.g., IFNB1, STAT1, and IRF1) and activates interferon signaling in other cell types (32). m6A-seq profiling of GSCs showed m6A modifications on a panel of interferon related genes (Supplementary Fig. S2I). Accordingly, GSEA analysis of RNA-seq with YY1 knockdown revealed enrichment of interferon signaling after YY1 knockdown (Fig. 3I). The qPCR analysis confirmed that YY1 knockdown increased expression levels of IFNα and IRF1 (Fig. 3J). Supporting the role of m6A in interferon response, GSEA analysis of RNA-seq after YTHDF2 knockout in GSC revealed a strong enrichment of immune response, including interferon signaling and antigen presentation, upon YTHDF2 knockout (Supplementary Fig. S2J) (33). In contrast, YY1 ChIP-seq profiles showed that the interferon genes were not bound by YY1, further confirming an indirect link between YY1-mediated transcription and interferon signaling (Supplementary Fig. S2K). Taken together, these results suggest that YY1 regulated transcription process suppressed interferon signaling through regulators of m6A modification.
YY1-associated transcriptional CDK9 inhibitors show efficacy to suppress glioblastoma stem cells
Despite of the strong dependency of GSCs on YY1, targeting YY1, along with other transcription factors, remains challenging, as YY1 is not a readily druggable target. To better understand the YY1 associated mechanism and therapeutic opportunity, we interrogated drug screening data from the Cancer Therapeutics Response Portal (CTRP) to identify YY1 expression-associated drug sensitivity (34,35). We filtered drugs whose area-under-curve (AUC) values correlated with YY1 expression in brain cancer cell lines, then clustered these drugs based on their AUCs (Fig. 4A; Supplementary Table S4). The top candidates which correlated with YY1 expression levels included ferroptosis inducers (ML162, erastin, and 1S,3R−RSL−3), transcriptional CDK inhibitors (alvocidib and dinaciclib), NAMPT inhibitors (CAY10618 and STF-31), and an Aurora kinase inhibitor (alisertib). YY1 expression negatively correlated with AUCs of these drugs (Fig. 4B), suggesting that YY1-driven transcription state was sensitive to these compounds. In contrast, there was a moderate association between high YY1 expression levels and resistance to EGFR or BRAF/MEK inhibitors (Supplementary Fig. S3A and S3B).
Figure 4. A pharmacogenomic analysis identifies YY1-associated therapeutic response in brain cancers.
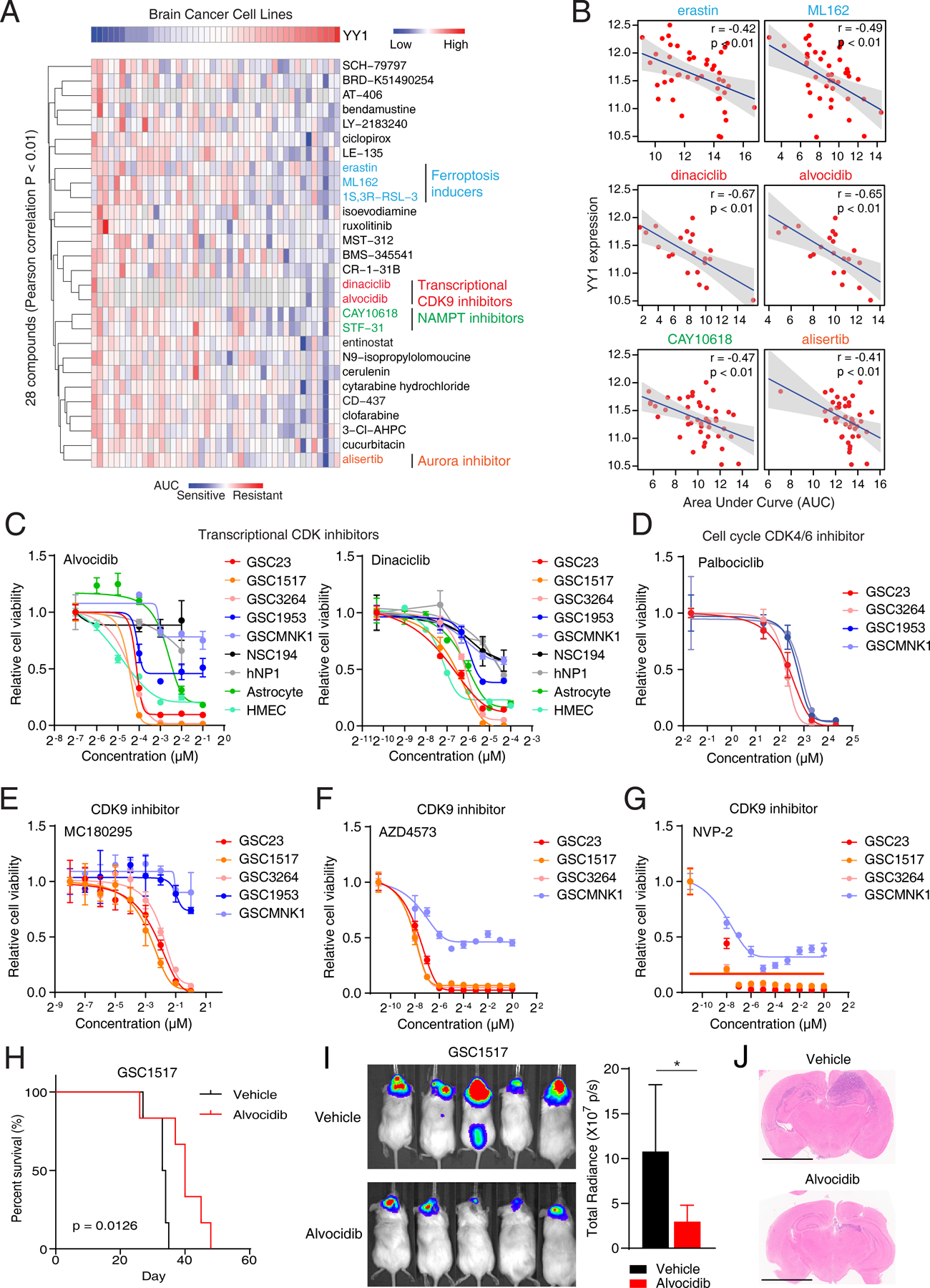
A, Hierarchical clustering of drug AUC values from brain cancer cell lines. YY1 expression levels were shown on the top of the heatmap. Only drugs with Pearson correlation p-value < 0.01 were included. A low AUC value (blue) indicates sensitivity to drug treatment. B, Scatter plots showing correlation between YY1 expression levels and drug AUC values in brain cancer cell lines. Pearson correlation coefficients and p values were shown. C, Dose-response curves of transcriptional CDK inhibitors, alvocidib and dinaciclib, in YY1-dependent GSCs (GSC23, GSC1517 and GSC3264), YY1-independent GSCs (GSC1953 and MNK1) and non-malignant cells (astrocyte and HMEC). HMEC, Human Mammary Epithelial Cells. Data are presented as mean ± SD. D, Dose-response curves of cell cycle-related CDK4/6 inhibitor in 4 GSCs. Data are presented as mean ± SD. E-G, Dose-response curves of selective CDK9 inhibitors in GSCs. Data are presented as mean ± SD. H, Kaplan-Meier curves showing survival of immunocompromised mice bearing intracranial tumors from GSC1517 with vehicle or alvocidib (10 mg/kg) treatment. P-values were determined by log-rank test. I and J, In vivo bioluminescent imaging (I) and H&E-stained coronal sections (J) of immunocompromised mice bearing intracranial tumors with vehicle or alvocidib treatment. Scale bars, 2 mm. Data are presented as mean + SD. *p < 0.05 by unpaired Student’s t-test.
To confirm the therapeutic opportunities targeting YY1, 6 compounds associated with YY1 expression, including 2 transcriptional CDK inhibitors, 2 NAMPT inhibitors, 1 ferroptosis inducer, and 1 Aurora kinase inhibitor underwent validation studies against 5 GSCs using dose response curves (Fig. 4A). GSCs dependent on YY1 were exclusively sensitive to 5 of these drugs, especially transcriptional CDK9 inhibitors (alvocidib and dinaciclib) (Fig. 4C; Supplementary Fig. S3C-S3E). Differential sensitivity was not observed for the cell cycle-related Aurora kinase inhibitor (Supplementary Fig. S3E). Moreover, sensitivity to inhibitors targeting other types of CDKs was limited, including the cell cycle-related CDK4/6 inhibitor palbociclib (Fig. 4D; Supplementary Fig. S3F). In vitro extreme limiting dilution assay (ELDA) showed decreased sphere frequencies in GSCs upon alvocidib or dinaciclib treatment (Supplementary Fig. S3G). Consistently, alvocidib treatment decreased the expression levels of stemness markers SOX2 and OLIG2 (Supplementary Fig. S3H). Considering alvocidib and dinaciclib also inhibit other CDKs, we evaluated three potent selective CDK9 inhibitors, MC180295, AZD4573, and NVP-2, against GSCs (36–38). GSCs dependent on YY1 displayed preferential sensitivity to selective CDK9 inhibitors (Fig. 4E-G). Moreover, knockdown of CDK9 using two non-overlapping shRNAs decreased cell proliferation (Supplementary Fig. S3I). These data suggest a correlation between YY1 dependency and transcription regulation through CDK9 in GSCs.
We next tested the in vivo anti-tumor efficacy of one transcriptional CDK inhibitor, alvocidib. Mice bearing intracranial patient-derived GSC tumors were treated with vehicle or alvocidib. Alvocidib treatment extended the survival of mice compared to vehicle control (Fig. 4H). Both in vivo imaging and histological analysis of tumor-bearing brains indicated the impaired tumor growth after alvocidib treatment (Fig. 4I and J). These findings suggest a pharmacologic vulnerability of targeting YY1-driven GSC state using transcriptional CDK9 inhibitors in a cell-autonomous manner both in vitro and in vivo.
YY1 interacts with CDK9 and transcription elongation complex to regulate m6A program expression
Considering the regulation of RNA Pol II-mediated transcription program by YY1 and the correlation between YY1 expression and response to transcriptional CDK inhibitors, we examined the potential link between YY1 and one of YY1-associated transcriptional CDK inhibitors, alvocidib, using RNA-seq profiling. Similar to findings upon YY1 inhibition, alvocidib downregulated genes were enriched in RNA Pol II transcription, RNA processing, and cell cycle (Fig. 5A). Cross-examination of alvocidib and YY1 knockdown induced gene expression changes showed that genes downregulated after alvocidib treatment were enriched in YY1 knockdown downregulated genes (Supplementary Fig. S4A). A panel of YY1 target genes, including METTL3 and YTHDF2, were downregulated by alvocidib (Fig. 5B). Alvocidib treatment of GSCs decreased total m6A levels, consistent with the downregulation of METTL3 (Fig. 5C). Similarly, shRNA-mediated knockdown of CDK9 and selective CDK9 inhibitors decreased METTL3 and YTHDF2 expression levels in GSCs, suggesting a role of CDK9 in regulating YY1 targets (Fig. 5D-F; Supplementary Fig. S4B).
Figure 5. YY1 is involved in transcription elongation through interacting with CDK9 and other elongation factors.
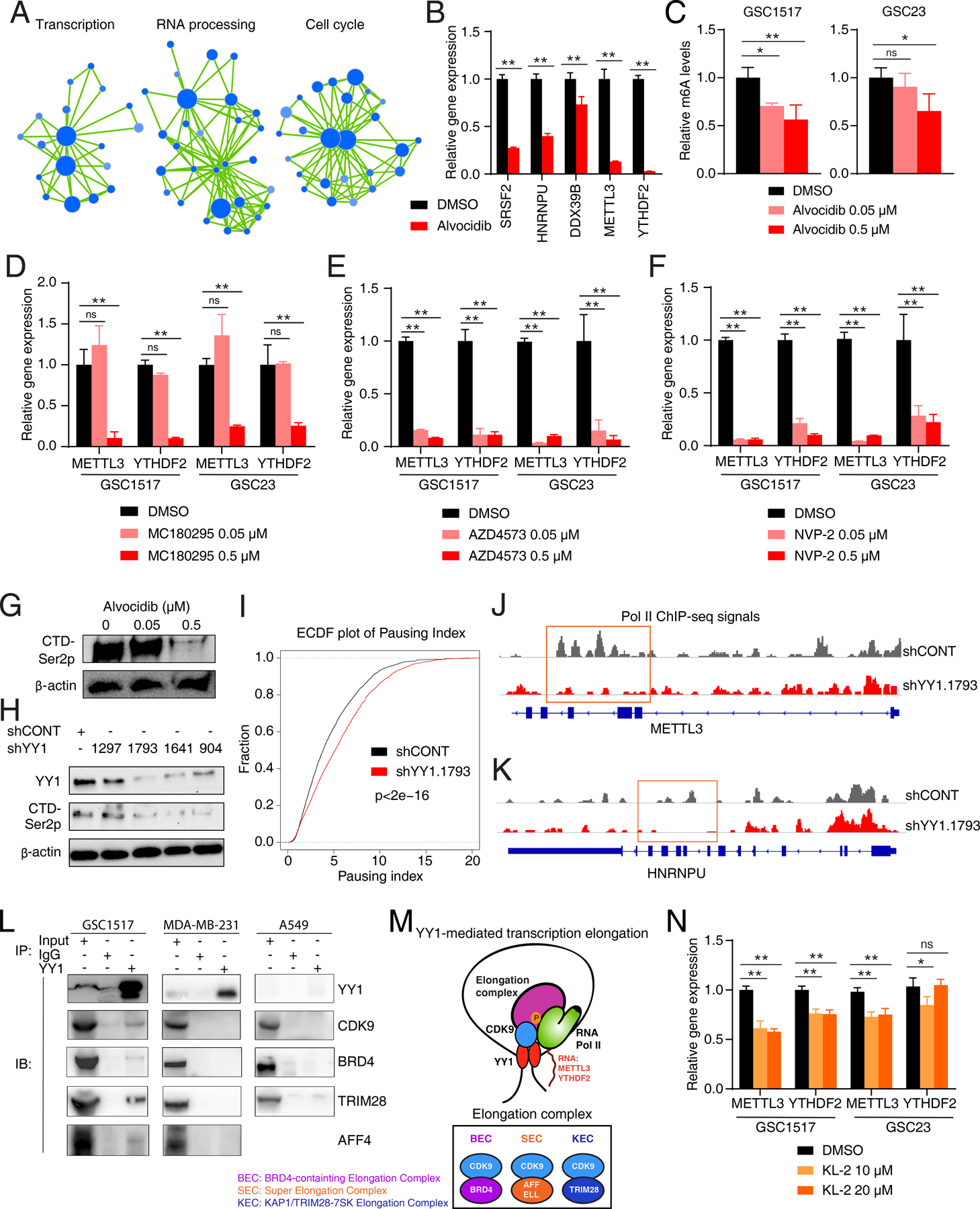
A, Enrichment map visualization of downregulated genes in GSEA after alvocidib treatment in GSC1517 cells. B, Bar plots showing relative expression levels of RNA processing and m6A genes after alvocidib treatment in GSC1517 determined by qRT-PCR. C, Bar plots showing relative m6A levels after alvocidib treatment in GSC1517 and GSC23. D-F, Bar plots showing relative expression levels of METTL3 and YTHDF2 after treatment of selective CDK9 inhibitors in GSC1517 determined by qRT-PCR. G, Immunoblot showing Ser2 phosphorylation (Ser2p) of RNA Pol II CTD (C-terminal domain) in GSC after alvocidib treatment. Data are representative results of three independent experiments. H, Immunoblot showing YY1 and Ser2 phosphorylation (Ser2p) of RNA Pol II CTD in GSCs after YY1 knockdown. I, Empirical cumulative density function (ECDF) plots of Pol II pausing index after YY1 knockdown in GSCs. J and K, Visualization of Pol II occupancy at representative genomic loci, METTL3 (J) and HNRNPU (K). L, Co-immunoprecipitation (IP) assay followed by immunoblot (IB) showing the interactions between YY1 and CDK9 or other transcription elongation complexes (BRD4, TRIM28, and AFF4). M, A working model for YY1-mediated transcription elongation complex. N, Bar plots showing relative expression levels of METTL3 and YTHDF2 after treatment of SEC inhibitor KL-2 determined by qRT-PCR. Data are presented as mean + SD. *p < 0.05, **p < 0.01 by one-way ANOVA with Dunnett multiple comparison test; ns, not significant.
Transcriptional CDKs mediate serine (Ser) phosphorylation of RNA Pol II carboxy-terminal domain (CTD) (16). Immunoblotting showed that both alvocidib treatment and YY1 loss-of-function dramatically downregulated Ser2 phosphorylation of Pol II CTD, a key regulatory step in transcription elongation regulated by transcriptional CDK9 (Fig. 5G and H). To evaluate transcription elongation after YY1 knockdown, we performed ChIP-seq profiling using RNA Pol II antibody to study Pol II distribution across transcripts to study the elongation process. A pausing index was calculated based on the ratio of the Pol II signal in gene promoters to the gene bodies to identify Pol II pausing (39–41). YY1 knockdown increased promoter-proximal pausing of Pol II, indicating a defect in transcription elongation, as shown by the empirical cumulative distribution function (ECDF) plot and the decreased Pol II signal in gene bodies of METTL3 and HNRNPU (Fig. 5I-K; Supplementary Fig. S4C).
To directly detect the interaction between YY1 and CDK9, endogenous co-IP and immunoblotting were performed in GSCs, a breast cancer cell line (MDA-MB-231), and a lung cancer cell line (A549). YY1-CDK9 interaction was detected in GSCs, but not in MDA-MB-231 or A549 cell lines (Fig. 5L), indicating a potential GSC-specific YY1-CDK9 interaction. CDK9 is involved in different transcription elongation complexes, including BRD4-containing Elongation Complex (BEC), Super Elongation Complex (SEC), and KAP1–7SK Elongation Complex (KEC) (Fig. 5M) (40,42,43). To determine which CDK9-containing complexes contain YY1, we performed an in silico analysis of TCGA GBM datasets, revealing YY1 expression correlated with BRD4 (BEC), AFF4 (SEC), MLLT1 (SEC), and TRIM28 (KEC) (Supplementary Fig. S4D). In GSCs, endogenous co-IP experiments showed that YY1 interacted with multiple complexes, including those containing BRD4, AFF4, and TRIM28, which was not detected in MDA-MB-231 or A549 (Fig. 5L). Depletion of transcription elongation complexes showed that GSCs were most dependent on these complexes, compared to MDA-MB-231 (intermediate dependent) and A549 (least dependent) (Supplementary Fig. S4E-S4K). Consistent with YY1 and CDK9 inhibition, depletion of transcriptional elongation complexes decreased expression levels of METTL3 and YTHDF2 (Supplementary Fig. 4L), which was also observed in treatment of BEC inhibitor, JQ1, and SEC inhibitor, KL-2 (Fig.5N; Supplementary Fig. 4M) (41,44). These data suggest a context-specific transcription elongation complex with YY1 and CDK9 functioning in GSCs (Fig. 5M).
Targeting transcription elongation complexes induces interferon response through m6A program
Similar to YY1 knockdown (Fig. 3I and J), several immune response-related gene sets, including interferon response and antigen presentation, were enriched after treatment with the transcriptional CDK inhibitor alvocidib, as revealed by GSEA (Fig. 6A). Genes involved in the major histocompatibility complex (MHC), including HLA-A, HLA-C, HLA-H, were upregulated (Fig. 6A and B). Concordantly, treatment with alvocidib and dinaciclib upregulated IFNα and IFNβ, as well as other interferon-stimulated genes (Fig. 6C-E; Supplementary Fig. S5A). While knockdown of individual transcription elongation complex components induced a weak upregulation of IFNα and IFNβ, potent inhibition of transcription elongation by selective CDK9 inhibitors and SEC inhibitor consistently induced a robust activation of IFNα and IFNβ (Fig. 6F-J; Supplementary Fig. S5B). To explore the potential mechanism of interferon response activation, m6A levels on IFNB1 after CDK inhibitor treatment were determined using m6A RNA immunoprecipitation (Me-RIP) followed by qPCR, showing the decreased m6A level on IFNB1 after alvocidib treatment (Supplementary Fig. S5C), which resulted in the increased stability of IFNB1 mRNA (Supplementary Fig. S5D) (32). Rescue experiments showed that METTL3 or YTHDF2 overexpression partially rescued the activation of interferon signaling by alvocidib treatment (Supplementary Fig. S5E), supporting the role of m6A in mediating the activation of interferon response.
Figure 6. Targeting transcription elongation complexes elicits interferon response in glioblastoma stem cells.
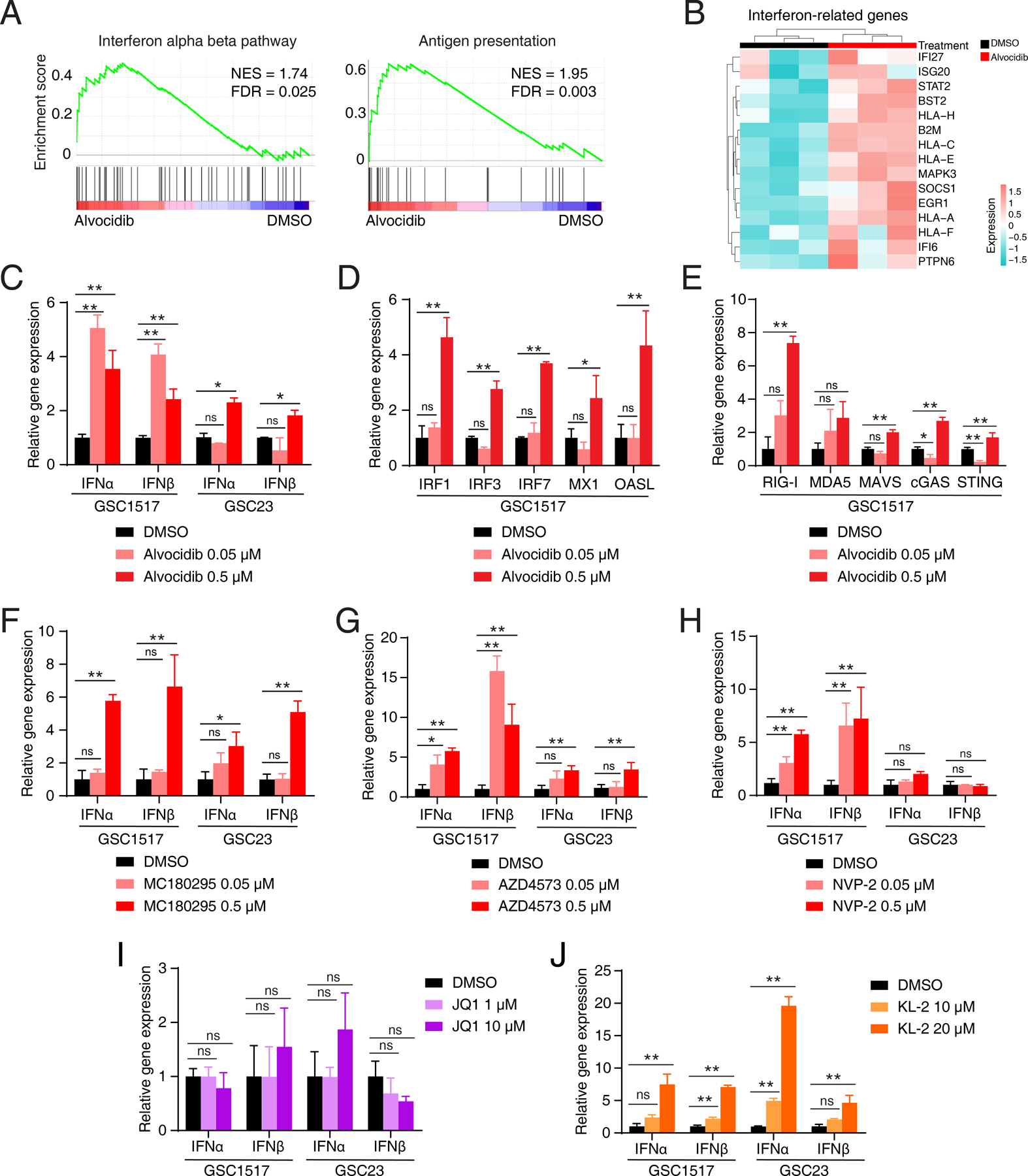
A, GSEA plots of representative gene sets involved in immune response after alvocidib treatment. The normalized enrichment score (NES) and false discovery rate (FDR) were indicated. B, Heatmap showing immune response related differentially expressed genes after alvocidib treatment in GSC1517 cells. C, Bar plots showing expression levels of IFNα and IFNβ in two GSC models (GSC1517 and GSC23) with alvocidib treatment determined by qRT-PCR. D and E, Bar plots showing expression levels of interferon-stimulated genes with alvocidib treatment in GSC1517 determined by qRT-PCR. F-H, Bar plots showing expression levels of IFNα and IFNβ in two GSC models (GSC1517 and GSC23) with selective CDK9 inhibitor treatment determined by qRT-PCR. I and J, Bar plots showing expression levels of IFNα and IFNβ in two GSCs (GSC1517 and GSC23) with BEC inhibitor JQ1 (I) and SEC inhibitor KL-2 (J) treatment determined by qRT-PCR. Data are presented as mean + SD. *p < 0.05, **p < 0.01 by one-way ANOVA with Dunnett multiple comparison test; ns, not significant.
Targeting the YY1-CDK9 transcription elongation complex potentiates anti-PD-1 response through rewiring immunosuppressive microenvironment in GBM
Although immune checkpoint inhibitors have transformed care for several solid tumors, including for patients with brain metastases, clinical efficacy in GBM has been disappointing (21,22). Effective strategies to rewire the immunosuppressive microenvironment and enhance immunotherapy response are still lacking in GBM (21,22). Considering the activation of interferon response after YY1 inhibition and transcriptional CDK inhibitor treatment, we tested whether targeting this mechanism in vivo could reprogram the tumor microenvironment. To extensively characterize the impact of transcriptional CDK inhibitor on tumor microenvironment, we applied cytometry by time-of-flight (CyTOF) with 34 metal-conjugated antibodies to detect major immune cell populations in a syngeneic mouse glioma model with intracranial implantation of GL261 cells (Fig. 7A) (45,46). Cell clusters were computationally defined using FlowSOM clustering and ConsensusClusterPlus, and visualized using t-stochastic neighbor embedding (t-SNE) plots. Major immune cell populations were identified, including CD4+ T, CD8+ T, natural killer (NK), macrophage, and myeloid-derived suppressor cells (MDSCs) (Fig. 7B; Supplementary Fig. S6A and S6B). Most CD4+ T cells were positive for FR4 staining, a marker for regulatory T (Treg) cells (Supplementary Fig. S6B). Compared to the vehicle treated tumors, alvocidib/anti-PD-1 antibody treated tumors showed decreased immunosuppressive Treg cells and MDSCs, and increased anti-tumor cytotoxic CD8+ T cells (Fig. 7B and C; Supplementary Fig. S6C). Immunohistochemistry validated that alvocidib treatment reduced the infiltration of Treg cells and increased the infiltration of CD8+ T cells (Supplementary Fig. S6D). YY1 expression levels were positively correlated with CD4+ T cell levels, but negatively correlated with CD8+ T cell levels in TCGA GBM dataset (Supplementary Fig. S6E). These data suggested that transcriptional CDK inhibitor treatment rewired the tumor microenvironment in GBM models.
Figure 7. Pharmacological Targeting of the YY1-CDK9 complex reprograms microenvironment and enhances anti-PD-1 response in gliomas.
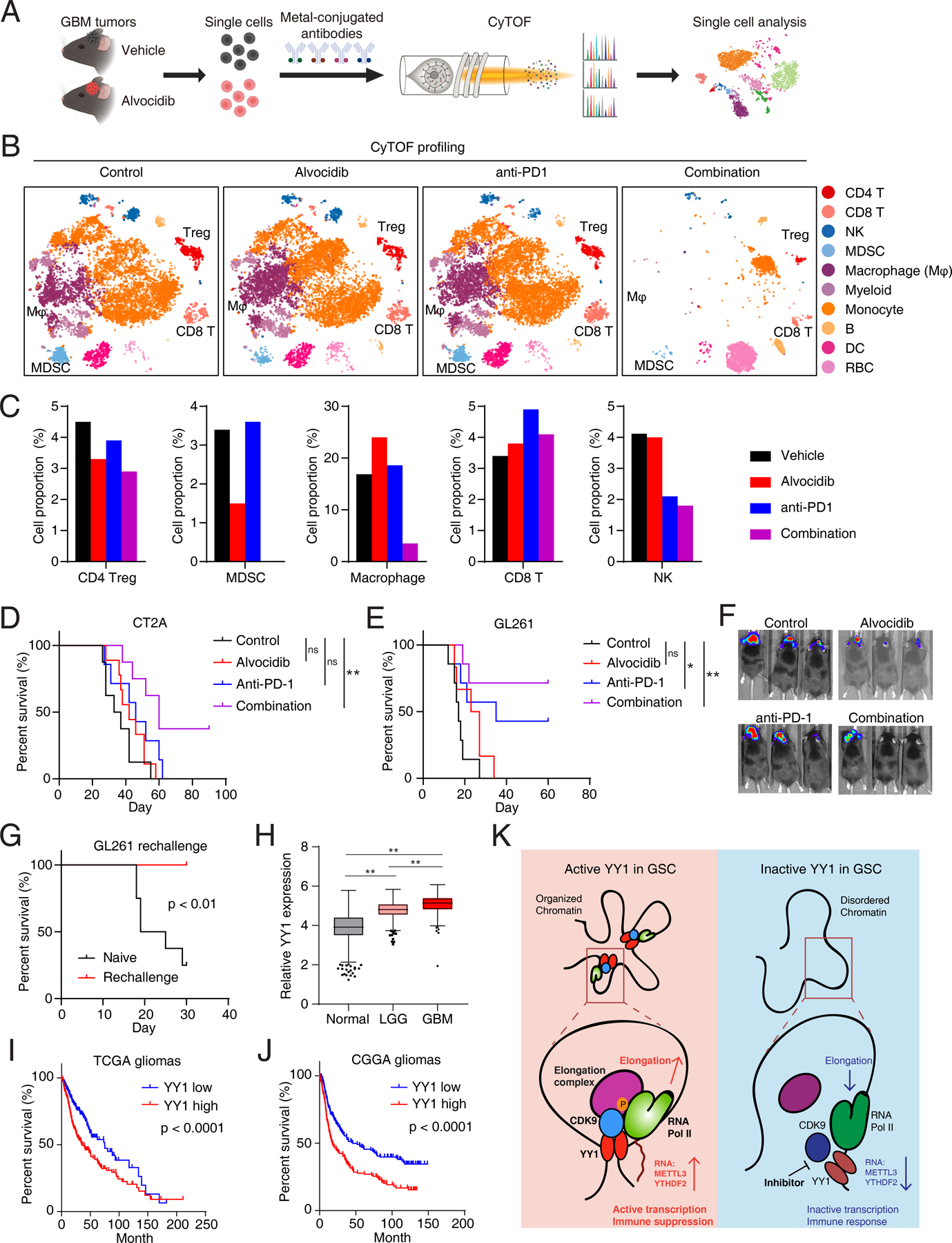
A, The workflow of the cytometry by time of flight (CyTOF) experiment to analyze the microenvironment in GBM models. B, The t-SNE plots of immune cell clusters from GL261 tumors treated with vehicle, alvocidib, anti-PD1 and combination, colored by cell clusters, from CyTOF analysis. C, Proportion of cells from GL261 tumors for the indicated cell types. D and E, Kaplan-Meier curves showing survival of immunocompetent mice bearing intracranial tumors from syngeneic CT2A (D) and GL261 (E) cells with alvocidib and anti-PD-1 treatment. *p < 0.05, **p < 0.01 by log-rank test; ns, not significant. F, In vivo bioluminescent imaging of immunocompetent mice bearing intracranial syngeneic tumors from mouse glioma CT2A cells with alvocidib and anti-PD-1 treatment. G, Kaplan-Meier curves showing survival of immunocompetent mice from Fig. 7D re-challenged with GL261 cells. P value was determined by log-rank test. H, Box-and-whisker plot showing expression pattern of YY1 in normal brains, low grade gliomas and glioblastomas from TCGA and GTEx databases. **p < 0.01 by one-way ANOVA with Dunnett multiple comparison test. I and J, Survival analysis of glioblastoma and low-grade gliomas in TCGA (I) and CGGA (J) databases divided by median value of YY1 expression levels. P value was determined by log-rank test. K, A working model showing the role of YY1-CDK9 complex in transcription elongation regulation and interferon response in GSCs.
Considering the rewiring of the immunosuppressive microenvironment by alvocidib, we hypothesized that alvocidib could enhance response to immunotherapy in vivo. Syngeneic mouse glioma models (CT2A and GL261) were treated with control, alvocidib, anti-PD-1 antibody, or the combination. While treatments using single agent modestly prolonged the overall survival of mice bearing intracranial tumors, the combination of alvocidib and anti-PD-1 antibody showed the longest survival compared to control (p < 0.01 by log-rank test; Fig. 7D and E). Correspondingly, single agents alone reduced tumor growth measured by in vivo bioluminescent imaging, but the combination of alvocidib and anti-PD-1 achieved a greater suppression of tumorigenesis without evidence of increased toxicity at the indicated doses (Fig. 7F). To assess memory response, mice with long-term survival after drug treatment were re-challenged with GL261 cells (47). In contrast to the short survival in naive mice, the re-challenged group showed extended survival, indicating the development of memory response for the antitumor effect (Fig. 7G). Taken together, these data suggest that the transcriptional CDK inhibitors enhance anti-PD-1 therapy by rewiring the immunosuppressive microenvironment in GBM.
YY1 informs poor prognosis and is associated with transcription regulation and interferon response in glioblastoma patients
To establish the clinical relevance of YY1 program in patient samples, we interrogated YY1 expression in glioma patient databases. Compared to non-tumor brain tissues, GBM and low-grade glioma (LGG) tissues showed upregulated expression levels of YY1 (Fig. 7H). Expression levels of YY1 were highly correlated with tumor grade (Fig. 7H). In both TCGA glioma and Chinese Glioma Genome Atlas (CGGA) datasets, high YY1 expression levels were associated with poor prognosis (Fig. 7I and J). In a pan-cancer analysis, GBM is one of the cancer types with the most prominent YY1 upregulation (Supplementary Fig. S7A). To further dissect the YY1-regulated program in glioblastoma, we constructed the co-expression network of YY1 followed by GSEA. In line with the role of YY1 in RNA Pol II-mediated transcription and RNA processing, YY1 expression positively correlated with gene sets involved in Pol II transcription and RNA processing, but negatively correlated with immune response gene sets (Supplementary Fig. S7B and S7C), which was also observed in CGGA glioblastoma dataset (Supplementary Fig. S7D). YY1 expression was negatively correlated with IFNB1, IFNG, and CCL27, which are involved in interferon signaling and immune regulation (Supplementary Fig. S7E).
Considering the observed dependency in GSCs on transcription components, YY1 and CDK9, we proposed a transcription state in GSCs. GSCs showed heterogeneity in transcription score, which was defined by the RNA Pol II related transcription regulation signature (Supplementary Fig. S7F). This transcription state was negatively correlated with the interferon response score in GSCs (Supplementary Fig. S7F). The presence of this transcription state with the suppressed interferon response was confirmed in vivo using an independent panel of patient-derived xenografts from glioblastoma patients (Supplementary Fig. S7G), as well as TCGA GBM samples and scRNA-seq datasets from five primary glioblastomas (48,49) (Supplementary Fig. S7H and S7I). Taken together, our findings suggest that transcription and immune axes underline the GBM trajectory and define a transcription state with suppressed interferon response, where YY1-CDK9 complex plays a role as a gatekeeper by regulating RNA Pol II-mediated transcription elongation and RNA m6A modification (Fig. 7K).
DISCUSSION
Through integrative analysis of gene expression programs and dependencies underlying GSCs, DGCs, and NSCs, we surveyed the chromatin regulator landscape in GBM and identified YY1 as a selective transcriptional dependency in GSCs. YY1 regulates RNA Pol II transcription machinery through interaction with transcriptional CDK9 and mediating chromatin loops, thus acting as a gatekeeper for this transcription cell state (modeled in Fig. 7K). RNA processing programs, including those involved in RNA splicing (SRSF1–3) and m6A modification (METTL3 and YTHDF2), were the main downstream targets regulated by YY1-mediated transcription and chromatin interactions. Our study uncovers a role of YY1 in transcription elongation by interacting with multiple transcription elongation complexes. Targeting the YY1-CDK9 complex or other transcription elongation complexes decreased expression levels of METTL3 and YTHDF2, which induced interferon responses. Treatment with YY1-associated transcriptional CDK inhibitors or other transcription elongation inhibitor augmented interferon response, providing a combination strategy with immunotherapy to treat GBM.
Profiles of chromatin interactions in tumor specimens have been interrogated to identify rational therapeutic targets, including CD276 as a target in GBM (20). As structural proteins, including CTCF, cohesin, and YY1, are challenging drug targets, methods to pharmacologically inhibit the global oncogenic chromatin interactions have not been established. Pharmacogenomic modelling using the large datasets of drug response in hundreds of genetically validated cancer cell lines provides an opportunity to successfully identify potential gene expression correlated drug sensitivity or synthetic lethality (34). Drug response correlated genes might be the direct target of the drug in gene-drug pairs or represent a cell state with functional relationship to the queried drug response. Using this approach, we propose compounds targeting a specific class of transcriptional CDKs as inhibitors of the YY1 program. An apparent overlap of transcriptional CDKs and YY1 in transcription regulation may represent a targetable cell state (14,50).
Transcriptional CDKs -- CDK7, CDK9, and CDK12, among others -- promote RNA Pol II-mediated transcription initiation and elongation by phosphorylating serine residues of the carboxy-terminal domain (CTD) of RNA Pol II (16,51). The value and mechanism of transcriptional CDKs in treating cancers remain unresolved (38,52–54). THZ1, a covalent CDK7 and CDK12/13 inhibitor, inhibits phosphorylation of RNA Pol II and abolishes super-enhancer-driven oncogenic transcription (54). However, specific targeting of CDK7 shows no effect on RNA Pol II phosphorylation, but inhibits CDK1 and CDK2 phosphorylation (53). Although CDK9 contributes to RNA Pol II phosphorylation and transcription elongation, CDK9 inhibition dephosphorylates BRG1 and leads to reactivation of tumor suppressor genes (38). In our study, targeting transcriptional CDKs by alvocidib decreased RNA Pol II phosphorylation and showed a strong inhibitory effect on RNA m6A machinery, in addition to previously reported inhibitory effects on cell cycle, DNA damage, and DNA synthesis (53). These findings highlight the different expression programs regulated by crosstalk among transcriptional CDKs (16). The dysregulated RNA processing machineries (METTL3, YTHDF2, or HNRNPC) might regulate m6A modification of interferon genes or yield improperly processed RNA products, which could trigger interferon response and partially explain the interferon response in our study (32,55). Moreover, the toxicity of transcriptional CDK inhibitors, especially multi-CDK inhibitors, remains an obstacle in applying these inhibitors into clinic, which could be overcome by developing more selective small molecule inhibitors (56). Indeed, several recently developed CDK9 inhibitors are being tested in clinical trials of different cancer types (56).
The immunosuppressive microenvironment in GBM tumors may contribute to the unfavorable outcome from immunotherapy (22,57). There is strong enrichment of immune-suppressive Treg cells and tumor-associated macrophages, which release inhibitory cytokines and inhibit T-cell mediated anti-tumor immune responses (21,22). However, certain therapeutic strategies, such as radiation, may be able to convert immunologically “cold” microenvironments into “hot” for GBM (58). Our data indicate a crosstalk between the YY1-CDK9 driven cell state in GSC and the immunosuppressive microenvironment in GBM. Perturbation of the transcription cell state with transcriptional CDK inhibitors may rewire tumor microenvironment to enhance response to immunotherapy in GBM. Transcriptional CDK inhibitors could have direct effects on the microenvironment cells to simultaneously target tumor cells and microenvironment in vivo. Future studies to test a panel of transcriptional CDK inhibitors with different target profiles may elicit robust response to immunotherapies, including anti-PD-1, anti-CTLA-4, or chimeric antigen receptor (CAR) T-cell therapies.
In summary, our study demonstrates that YY1, YY1-associated transcriptional CDK9 and transcription elongation complexes are essential in GSC maintenance and presents a targetable transcription cell state with suppressed interferon response in GSCs. Both genetic and pharmacogenomic targeting of this transcription cell state suppress self-renewal and proliferation of GSCs and could be combined with immunotherapy to treat GBM patients synergistically.
METHODS
GBM stem cell derivation and maintenance
GBM tissues were obtained from excess surgical resection samples from patients at the Case Western Reserve University after review by neuropathology with written informed consent from the patients and in accordance with an IRB-approved protocol (090401). All specimens were reviewed by a neuropathologist. All patient studies were conducted in accordance with the Declaration of Helsinki. To minimize in vitro cell culture-based artifacts, patient-derived xenografts were propagated as a renewable source of GSCs. STR analyses were performed on the tumor model for authentication. GSC lines GSC387 and GSC3565 were derived by our laboratory and transferred via a material transfer agreement from Duke University. GSC23 was derived from a recurrent glioblastoma biopsy from a 63-year-old male patient and was provided as a generous gift by Dr. Erik Sulman. GSC3028 was derived from a recurrent glioblastoma from a 65-year-old female patient. 1919 GSCs were derived from a glioblastoma from a 53-year-old male patient. All GSC lines were cultured in Neurobasal media (Invitrogen) supplemented with B27 without vitamin A (Invitrogen), EGF, and bFGF (20 ng/ml each; R&D Systems), sodium pyruvate (Life Technologies Cat # 11360070), and Glutamax (Life Technologies Cat# 35050061) at 37°C with 20% oxygen and 5% carbon dioxide.
Xenograft generation
All mouse experiments were performed under an animal protocol approved by Institutional Animal Care and Use Committee, University of California, San Diego. Investigators were not blinded for randomization or treatment. Power analysis has been done according to publications and prior experience. Studies were done in female animals. Intracranial xenografts were created by implanting 10,000 human-derived GSCs into the right cerebral cortex of NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, IMSR Cat# JAX:005557, RRID:IMSR_JAX:005557, The Jackson Laboratory, Bar Harbor, ME, USA) mice at a depth of 3.5 mm. Healthy, wild-type female mice of NSG background, 4–6 weeks old, were randomly selected for intracranial injection. Housing conditions and animal status were supervised by a veterinarian. Animals were monitored until neurological signs were observed, at which point they were sacrificed. Neurological signs included hunched posture, gait changes, lethargy and weigh loss. Mouse survival was analyzed in GraphPad Prism (RRID:SCR_002798) and the statistical significance was tested by log-rank test.
Tumor dissociation and GSC culture
Dissociation of xenografted tumors was performed using a papain dissociation system according to the manufacturer’s instructions. Cells were then cultured in Neurobasal medium supplemented with 2% B27, 1% L-glutamine, 1% sodium pyruvate, 1% penicillin/streptomycin, 10 ng/ml basic fibroblast growth factor (bFGF), and 10 ng/ml epidermal growth factor (EGF) for at least 6 hours to recover expression of surface antigens. GSCs were isolated immediately following dissociation or following transient xenograft passage in immunocompromised NSG mice (The Jackson Laboratory, Bar Harbor, ME, USA) using prospective sorting followed by assays to confirm stem cell marker expression, sphere formation, and secondary tumor initiation. When experiments were performed, we isolated AC133-positive populations using CD133/1 antibody-conjugated magnetic beads. Neurobasal media was used to wash specimens, which were then acutely dissociated to remove non-tumor tissue and subjected to enzymatic dissociation using the Papain dissociation system (Worthington Biomedical Corp, Cat# LK003150). The isolated tumor cells were briefly placed in neurobasal media with B27 supplement (Life Technologies Cat# 12587010) to permit recovery following enzymatic dissociation. Cells were labeled with CD133/2(293C)-Allophycocyanin (APC) antibody kit (Miltenyi Biotec Cat# 130–098-826, RRID:AB_2660882), and the CD133+ cells were sorted and analyzed by flow cytometry. The sorted CD133+ cells were cultured in NBM-B27 medium containing 20 ng/mL of both epidermal growth factor (EGF) (R&D system Cat# 236-EG-01M) and recombinant human FGF basic (FGF) (R&D system Cat#4114-TC-01M) for a short period before treatment and analysis (7,59) . During routine culture, all GSC models were cultured in Neurobasal media (Invitrogen) supplemented with B27 without vitamin A (Invitrogen), EGF, and bFGF (20 ng/ml each; R&D Systems), sodium pyruvate (Life Technologies Cat# 11360070), and Glutamax (Life Technologies Cat# 35050061). Expression of stem cell markers (SOX2 and OLIG2) and functional assays of self-renewal (serial neurosphere passage), and tumor propagation using in vivo limiting dilution assays were used to validate GSC phenotypes.
GSC dataset interrogation
RNA-sequencing data for 38 GSC and 5 NSC cell lines were downloaded from GSE119834 (24). Sequencing reads were trimmed using Trim Galore (RRID:SCR_011847, https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and cutadapt (http://cutadapt.readthedocs.io/en/stable/guide.html). Trimmed reads were quantified using Salmon (RRID:SCR_017036) in the quasi mapping-based mode using – seqBias, --gcBias, and --validateMappings parameters (60). The R package “tximport” (RRID:SCR_016752) was used to import Salmon quant files into R package “DESeq2” (RRID:SCR_015687) for differential expression analysis to obtain the fold change ranking metric for GSC vs. NSC comparison.
RNA-sequencing data for three pairs of GSCs and DGCs were downloaded from GSE54791 (25). RNA-sequencing derived counts-per-million (CPM) matrix were obtained and differential expression analysis was performed using R package “edgeR” to obtain the fold change ranking metric for GSC vs. DGC comparison.
Whole-genome CRISPR loss-of-function screening data in 8 GSCs and 2 NSCs were obtained from a published study (26). The BAGEL algorithm was used to derive a Bayes factor (BF) for each tested gene. A greater BF value indicates increased confidence that the knockout of the gene results in a decrease in fitness. Z-scores of BF value difference between GSCs and NSCs were used to indicate the specificity of gene dependency in GSCs.
Public glioma patient databases
The public databases, including The Cancer Genome Atlas (TCGA), Chinese Glioma Genome Atlas (CGGA) and The Genotype-Tissue Expression (GTEx) project, were used to interrogate YY1 expression levels in glioma patients and normal tissues. UCSC Xena project recomputed TCGA and GTEx RNA-Seq data was obtained from GEPIA (http://gepia.cancer-pku.cn/). YY1 expression and survival of TCGA glioblastoma and low-grade glioma patients were obtained from GlioVis (http://gliovis.bioinfo.cnio.es/) (2). Chinese glioma datasets with 325 glioma patients were obtained from CGGA (http://www.cgga.org.cn/) web server. Expression levels were categorized into two groups with the median value as a cutoff. The survival analysis was analyzed using Kaplan-Meier curve and log-rank test. The R package “GSVA” was used to calculate gene signature score with ssgsea method (61).
Transcription regulator analysis
Enrichment of transcription regulator target program was performed using RePhine algorithm (https://github.com/coexps/Rephine) (23). RePhine analyzed enrichment score of 160 transcription regulator programs in a pre-ranked gene list. First, ChIP-Seq data for 160 transcription regulators were downloaded from ENCODE database (62). To quantitatively determine the targets of a transcription regulator, we applied regulatory potential (RP) score by considering binding site’s distance to TSS (transcription start site) of a gene and the signal strength of ChIP-Seq peaks (63). Therefore, RP score indicates the regulatory strength of a transcription regulator on the specific gene. The higher the score is, the stronger the regulatory strength is. Next, a gene list ranked by their differential expression (log2 fold change) between 44 GSC and 5 NSC was obtained. Third, we determined the concordant enrichment of a transcription regulator target genes in the gene list aforementioned using elastic-net regression. The coefficient from elastic net model was calculated to indicate a positive or negative enrichment in the gene list. The p value was calculated by the likelihood ratio test to indicate the statistical significance. Finally, to visualize the enrichment of a transcription regulator program, we adopted canonical GSEA for the continuous variable (RP score) in the calculation and plot of enrichment score (64). The activity of one regulator in one sample is calculated by Regression Analysis with Background InTegration (RABIT) algorithms using normalized gene expression dataset (65).
Proliferation and neurosphere formation assay
Cell proliferation experiments were performed by plating cells at a density of 1000 cells/well in a 96-well plate with three to six replicate wells. CellTiter-Glo (Promega, Madison, WI, USA) was used to measure cell viability at days 0, 1, 3, 5, and 7. All data were normalized to day 0. In vitro limiting dilution assays were used to assess neurosphere formation capacity. Cells with decreasing numbers per well (50, 20, 10, 5, and 1) were plated into each well of a 96-well plate. The presence and number of neurospheres in each well were recorded seven days after plating. Extreme limiting dilution analysis was performed using software available at http://bioinf.wehi.edu.au/software/elda to calculate stem cell frequency and statistical significance.
Plasmids and lentiviral transduction
Lentiviral clones expressing non-overlapping shRNAs against human YY1 (TRCN0000019895, TRCN0000019894, TRCN0000019896 and TRCN0000019898), human CTCF (TRCN0000230190, TRCN0000230191 and TRCN0000218498), human CDK9 (TRCN0000199892, TRCN0000000494), human BRD4 (TRCN0000021427 and TRCN0000318771), human AFF4 (TRCN0000015825 and TRCN0000426769), human MLLT1 (TRCN0000019290 and TRCN0000019291), human TRIM28 (TRCN0000018001 and TRCN0000017998) and a non-targeting control shRNA (shCONT, Sigma-Aldrich, Cat# SCH002) were obtained from SigmaAldrich. Non-overlapping shRNAs were selected based on knockdown efficiency and were used for the experiments. Human YY1-expressing plasmid was obtained from VectorBuilder. The pcDNA3.1 HA-YY1 was a gift from Richard Young (Addgene plasmid #104395, RRID:Addgene_104395). 293FT cells (ATCC Cat# PTA-5077, RRID:CVCL_6911) were used to generate lentiviral particles through co-transfection of the packaging vectors pCMV-dR8.2 (Addgene plasmid #8455, RRID:Addgene_8455) and pCI-VSV-G (Addgene plasmid #1733, RRID:Addgene_1733) using the transfection method based on polyethylenimine (PEI, Polysciences Cat# 23966–1) or LipoD293 In Vitro DNA Transfection Reagent (SignaGen Laboratories Cat# SL100668). Media were changed after 16 hours. Virus was harvested 48 hours after media change. Viral supernatants were filtered through 0.45 µm filter and used immediately or stored at −80 for future use. GSCs were infected with shRNA lentivirus and the media were replaced after 24 hr. Knockdown efficiency was measured by quantitative RT-PCR (qRT-PCR) after 48 hr.
RNA isolation and quantitative RT-PCR
Cellular total RNA was isolated using Trizol (Life Technology Cat# 15596026) and reversetranscription to cDNA was performed using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific Cat# 4368814). Briefly, 1 µg total RNA was used for cDNA synthesisusing random primers according to the manufacturer’s instructions. Quantitative real-time PCR was performed using SYBR Green Master mix (Thermo Fisher Scientific Cat# 4309155) on an Applied Biosystems 7900HT Real-Time PCR System and normalized to 18S ribosomal RNA or GAPDH. The primers in qRT-PCR were designed using Primer3 (RRID:SCR_003139, http://bioinfo.ut.ee/primer3-0.4.0/).
RNA m6A quantification
GSC cells after shRNA-mediated knockdown or drug treatment were subject for total RNA isolation using RNeasy Plus Mini Kit (Qiagen Cat# 74136). The total RNA was used to determine the quantification of m6A using EpiQuik m6A RNA Methylation Quantification Kit (EpiGentek Cat# P-9005).
Co-Immunoprecipitation analysis (CoIP)
CoIP was performed following the previously reported protocol (66). Briefly, cells were lysed in IP buffer (50 mM Tris-HCl [pH 7.4], 125 mM NaCl, 1 mM EDTA, 0.1% Triton X-100) with inhibitors (50 mM NaF, 1 mM PMSF, 1 mM Na3VO4, 1 µg/mL Aprotinin, 1 µg/mL Leupeptin, 1 µg/mL Pepstatin) followed by sonication and centrifugation to clear insoluble debris. Lysates were then incubated with protein G agarose beads and IgG antibody of the same species as the IP antibody for 2 hr at 4°C to reduce non-specific binding. The cleared lysates were then incubated with IP antibody overnight at 4°C. IP antibody used was YY1 (Active Motif Cat# 61779, RRID:AB_2793763). Beads were washed 3x with IP buffer, boiled in 1x SDS gel loading buffer, and subjected to western blot analysis.
Western blot
Cells were collected and lysed with RIPA buffer (50 mM Tris PH8.0, 1 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1% SDS. 0.1% sodium doxycholate) supplemented with EDTA-free proteinase inhibitor cocktail (Roche Cat# 4693159001) 48 hr after lentivirus infection to extract protein. Lysate was incubated on ice for 15 minutes and quantified using BCA reaction (Thermo Fisher Scientific Cat# 23227) after centrifugation for 10 min at 4°C. 25 ug lysate was resolved on SDS-PAGE and transferred to Immobilon-P PVDF membrane (Millipore Cat# IPVH00010). After blocking with 3% BSA (bovine serum albumin) in TBST buffer (20 mM Tris pH 8.0, 150 mM NaCl, 0.1% Tween 20), membranes were incubated with indicated antibodies at 4°C overnight. The antibodies used were YY1 (Active Motif Cat# 61779, RRID:AB_2793763), CDK9 (Santa Cruz Biotechnology Cat# sc-484, RRID:AB_2275986; Santa Cruz Biotechnology Cat# sc-13130, RRID:AB_627245), phospho-RNA Polymerase II Ser2 (BETHYL Cat# A300–654A, RRID:AB_519341), β-Actin (Sigma Cat# A2228, RRID:AB_476697), BRD4 (Proteintech Cat# 67374–1-Ig, RRID:AB_2882622), AFF4 (Proteintech Cat# 14662–1-AP, RRID:AB_2242609), TRIM28 (Proteintech Cat# 15202–1-AP, RRID:AB_2209890). Afterward, membranes were incubated with HRP-linked secondary (Cell Signaling Technology, anti-mouse IgG, Cat# 7076, RRID:AB_330924; Cat# 7074 is anti-rabbit IgG, RRID:AB_2099233) for 1 hr at room temperature. Then chemical signal was detected by chemiluminescent ECL buffer (Thermo Fisher Scientific Cat# PI34080).
YY1-associated drug response
YY1 associated drug response was identified by interrogation of drug screening datasets in 835 cancer cell lines. The cancer cell line information was retrieved from Cancer Cell Line Encyclopedia (CCLE) project (35). The expression level of YY1 was downloaded from CCLE web portal (https://portals.broadinstitute.org/ccle). The drug screening data (481 drugs and 835 cancer cell lines) were obtained from Cancer Therapeutics Response Portal (CTRP) project (https://portals.broadinstitute.org/ctrp.v2.1/) (34). Area-under-curve (AUC) was used to represent drug response. A small AUC value indicates sensitivity, while a large AUC value indicates resistance. To identify lineage specific gene-drug interactions, only 44 brain cancer associated cell lines were considered in downstream analysis. Pearson correlation between gene expression levels and drug response AUC values was calculated to rank gene-drug correlation. A total of 28 drugs with P < 0.01 were filtered for hierarchical analysis and heatmap visualization using “pheatmap” package.
Drug sensitivity test
Cells were seeded in 96-well plates at a density of 2,000 cells/well. The cells were then treated by a 2-fold serial diluted series of drug with at least three replicate wells per dose. The plates were transferred to an incubator (37°C, 5% CO2) for 72 hr. At the end point of drug treatment, 50 µL CellTiter-Glo reagent (Promega) was added to each well. After 10 min incubation at room temperature, the luminescent signals were measured by an EnVision Multilabel Reader (PerkinElmer) to determine the cell viabilities. The cell viabilities were normalized to the average viability of DMSO control wells. The fitted drug response curve was plotted using GraphPad Prism software (RRID:SCR_002798).
ChIP-seq
The chromatin immunoprecipitation (ChIP) assay was performed using Magna ChIP™ A/G Chromatin Immunoprecipitation Kit (MilliporeSigma Cat# 17–10085) according to the manufacturer’s instructions. Formaldehyde-fixed cells were lysed and sheared. The sheared chromatin was cleared and incubated overnight at 4°C with YY1 antibody (Active Motif Cat# 61779, RRID:AB_2793763) or RNA polymerase II antibody (Millipore Cat# 05–623, RRID:AB_309852). Antibody-chromatin complexes were immunoprecipitated with protein G magnetic Dyna beads (Life Technologies), washed, eluted, reverse crosslinked, and treated with RNAse A followed by proteinase K. ChIP DNA was purified using Ampure XP beads (Beckmann Coulter) and then used to prepare sequencing libraries by NEB NEBNext® Ultra™ DNA Library Prep Kit for Illumina for sequencing.
Raw FASTQ files were trimmed using TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and aligned to the hg19 human genome using Bowtie2 (67). SamTools (RRID:SCR_002105) was used to sort and index the BAM files. Peak calling was performed using MACS2 with the narrow peak mode (68). For subsequent visualization of ChIP-seq signal, bigwig files were generated using the DeepTools (RRID:SCR_016366) “bamCoverage” script using default settings (http://deeptools.readthedocs.io/). DeepTools “computeMatrix”, “plotHeatmap”, and “plotProfile” functions were used to generate heatmaps and profile plots. Signal tracks were visualized using R package “Gviz” and Integrative Genomics Viewer (IGV). Annotation of ChIP-seq peaks was performed using R package “ChIPseeker” (69). GO enrichment of YY1 target genes was analyzed and visualized by ClueGO (RRID:SCR_005748) (70). For pausing indexes, RPKM values were generated for hg19 transcription start site (TSS) loci (from 50 bp upstream to 200 bp downstream of TSS) and for the gene body of these transcripts (from 500 bp downstream of the TSS to the transcription termination site. The ratio of the average coverage of the promoter over the average coverage of the gene body was then taken to be the pausing index (41,71).
RNA-seq
Total RNA was extracted using the Zymo Direct-zol RNA MiniPrep Kit (Cat# R2052) and TRI reagent (Cat# R2050) according to the manufacturer’s instructions. Total RNA was used for library construction using Illumina TruSeq Stranded Total RNA Library Prep Kit. The library was sequenced as paired-end 150 bp read. Raw FASTQ reads were trimmed using Trim Galore and transcript quantification was performed using Salmon in the quasi-mapping mode (60). Salmon “quant” files were converted using Tximport functionand differentially expressed gene (DEG) analysis was performed using DESeq2 (72). Gene set enrichment analysis (GSEA, RRID:SCR_003199) was performed on a pre-ranked gene list using the gene expression fold change as the ranking metric from differentially expressed gene analysis by GSEA desktop application (64). Visualization of GSEA results was performed by Enrichment Map application in Cytoscape (RRID:SCR_003032, http://www.cytoscape.org).
BL-Hi-C Library Construction and Data Analysis
BL-Hi-C library construction and data analysis were performed as described previously (30). Approximately, 1 × 106 cells were chemically crosslinked by addition of 1/36 volume of fresh 37% formaldehyde solution to the medium and incubation for 10 min at room temperature with gentle shaking. Crosslinking was stopped by adding 2.5 M glycine to a final concentration of 0.2 M and incubating for 10 min at room temperature. After rinsing twice with PBS, cells were harvested in a 1.5 mL tube by scraping and centrifugation, and stored at −80°C until use. Cell pellets were resuspended in 1 mL BL-Hi-C Lysis Buffer 1 (50 mM HEPES-KOH pH7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 1X protease inhibitor cocktail) and incubated for 15 min on ice. After centrifugation at 1500 g for 5 min at 4°C, supernatant was removed, and after washing once with 1 mL BL-Hi-C Lysis Buffer 1, cell pellet was resuspended in 1 mL BL-Hi-C Lysis Buffer 2 (50 mM HEPES-KOH pH7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 1% SDS, 1X protease inhibitor cocktail) and rotated for 15 min at 4°C. Supernatant was removed after centrifugation. Cell pellet was washed once with 1 mL BL-Hi-C Lysis Buffer 1, re-suspended in 50 μL of 0.5% SDS and incubated for 5 min at 62°C. At the end of incubation, SDS was quenched by adding 145 μL of ddH2O and 25 μL of 10% Triton X-100 and incubation for 10 min at 37°C. Chromatin was cleaved by adding 25 μL of 10x NEBuffer 2 and 100 U HaeIII (NEB) for 2 h at 37°C followed by additional cleavage with another 30 U HaeIII for 3 hr. Cleaved chromatin was A-tailed by adding 2.5 μL of 10 mM dATP solution (Thermo Fisher Scientific) and 2.5 μL Klenow Fragment (3′→5′ exo-) (NEB) with rotation at 900 rpm for 40 min at 37°C in ThermoMixer C. Proximity ligation was performed by adding 750 μL ddH2O, 120 μL of 10X T4 DNA ligase buffer, 100 μL of 10% Triton X-100, 5 μL T4 DNA ligase (NEB) and 4 μL of 200 ng/μl biotinylated Bridge Linker S2 (annealed by 5Phos-CGCGATATC/iBIOdT/TATCTGACT and 5Phos-GTCAGATAAGATATCGCGT) with rotation for 4 hr at 16°C. After centrifugation at 3500 g for 5 min at 4°C and removal of supernatant, the pellet was resuspended in 309 μL ddH2O, 35 μL of Lambda Exonuclease buffer, 3 μL Lambda Exonuclease, 3 mL Exonuclease I and rocked at 900 rpm for 1 hr at 37°C in the ThermoMixer C. After the addition of 45 μL of 10% SDS and 55 μL of 20 mg/mL Proteinase K, sample was incubated overnight at 55°C, and added in the next day with 65 μL of 5 M NaCl and incubated for 2 hr at 68°C. DNA was recovered by extraction with 500 μL of phenol:chloroform:isoamyl alcohol (25:24:1) and ethanol precipitation with 1 μL glycoblue. After centrifugation and washing with 75% ethanol, DNA pellet was resolved in 130 μL Buffer EB (10 mM Tris-HCl pH8.0). DNA was sheared with Covaris S220 with the setting for DNA size of 300 bp. After washing twice with 2X B&W buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 2 M NaCl), 30 μL Dynabeads M-280 Streptavidin (Thermo Fisher Scientific) were blocked with 100 μL of 1X I-Block buffer (2% I-Block Protein-Based Blocking Reagent (Thermo Fisher Scientific), 0.5% SDS) for 45 min at room temperature in a rotating wheel. Beads were washed twice with 100 μL of 1X B&W buffer (5 mM Tris-HCl pH7.5, 0.5 mM EDTA, 1 M NaCl) followed by resuspension in 1X B&W buffer containing 1 μg of pre-heated Salmon Sperm DNA solution and rotation for 30 min at room temperature. After washing twice with 100 μL of 1X B&W buffer, beads were resuspended with 130 μL of 2X B&W buffer, combined with sonicated DNA and rotated for another 45 min at room temperature. Beads were washed five times with 500 μL of 2X SSC containing 0.5% SDS, twice with 500 μL of 1X B&W buffer and once with 100 μL Buffer EB (QIAGEN). DNA on beads was end-repaired in the reaction containing 75 μL ddH2O, 10 μL of 10X T4 DNA ligase buffer, 5 μL of 10 mM dNTP, 5 μL of T4 Polynucleotide Kinase, 4 μL of T4 DNA Polymerase and 1 μL of Large (Klenow) Fragment with shaking at 900 rpm for 30 min at 37°C in Thermomixer C. After washing twice with 500 μL of 1X TWB (5 mM Tris-HCl pH7.5, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween-20) for 2 min at 55°C, DNA on beads was A-tailed in the reaction containing 80 μL of ddH2O, 10 μL of NEBuffer 2, 5 μL of 10 mM dATP and 5 μL of Klenow Fragment (3′→5′ exo-) with shaking at 900 rpm for 30 min at 37°C in ThermoMixer C. Beads were washed twice with 500 μL 1X TWB for 2 min at 55°C and once with 50 μL of 1X Quick Ligase buffer. DNA on beads was ligated with adaptor in the reaction containing 6.6 μL ddH2O, 10 μL of 2X Quick Ligase buffer, 2 μL of Quick ligase (NEB) and 0.4 μL of 20 μM Y-Adaptor (annealed by /5Phos/GATCGGAAGAGCACACGTCTGAACTCCAGTCAC and TACACTCTTTCCCTACACGACGCTCTTCCGATCT) for 45 min at room temperature. Beads were washed twice with 500 μL 1X TWB for 2 min at 55°C and once with 100 μL of Buffer EB. After resus-pending in 60 μL of Buffer EB, bead suspension was aliquoted in 3X 20 μL for storage at −20°C. One aliquot of bead suspension was used as a template for PCR amplification with Q5 Hot Star DNA Polymerase, universal primer (AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGAC) and index primer (CAAGCAGAAGACGGCATACGAGAT/index/GTGACTGGAGTTCAGACGTGT) for 10 cycles. PCR products between 300–700 bp were purified as BL-Hi-C library using Ampure XP beads and subjected to Illumina HiSeq X10 for paired end 150 bp sequencing.
The ChIA-PET2 v0.9.3 software (73) was used for quality control, mapping to hg19 genome and identification of chromatin interactions with the following parameter setting: -A ACGCGATATCTTATC -B AGTCAGATAAGATAT -s 1 -m 1 -t 24 -k 2 -e 1 -l 15 -S 500 -M “-q 0.05.” HiC-Pro (RRID:SCR_017643) hicpro2juicebox.sh utility was used to generate hic file for further visualization of interaction matrix (74). A/B compartment analysis at 50 kb resolution was performed using Juicer Tools eigenvector function and visualized in The Integrative Genomics Viewer (IGV). The interaction matrix and chromatin interactions were visualized in WashU Epigenome Browser (https://epigenomegateway.wustl.edu/) or Juicebox v1.11.08. The overlapping between YY1 binding peaks and chromatin interaction anchors were analyzed using bedtools (RRID:SCR_006646) pairtobed function with default parameters and further analyzed in R.
In vivo drug treatment and bioluminescence
For alvocidib treatment experiment, intracranial xenografts were created by implanting 6,000 human-derived GSC1517 cells into female immunodeficient mice. After 7 days, immunodeficient mice were randomly selected into two groups and administrated with saline or alvocidib (10 mg/kg, MedChemExpress Cat# HY-10006) three times a week until neurological signs were observed.
For alvocidib and anti-PD-1 treatment experiment, intracranial xenografts were created by implanting 20,000 mouse glioma cells (CT2A) into the right cerebral cortex of 4–6-week-old C57BL/6 female mice (Jackson Laboratory) at a depth of 3.5 mm. After 5 days, mice were randomly selected into four groups (control, alvocidib only, anti-PD-1 only, alvocidib+anti-PD-1). During each of the following 5 weeks, mouse were treated with alvocidib (10 mg/kg) or saline for two consecutive days, followed by anti-PD-1 (200 µg, Bio X Cell Cat# BP0273) or IgG2a isotype control (200 µg, Bio X Cell Cat# BP0089) on the third day. Animals were monitored until neurological signs were observed, at which point they were sacrificed.
Mouse brains implanted with GSCs or mouse glioma cells which were labeled with firefly luciferase were monitored by the bioluminescent imaging. Animals were treated with D-Luciferin (120 mg/kg, Biosynth Carbosynth Cat# L-8220) intraperitoneally and anesthetized with isoflurane for the imaging analysis. The bioluminescent images were captured by an IVIS imaging system (Spectrum CT, PerkinElmer).
CyTOF (cytometry by time of flight)
Mouse glioma tissues were harvested according to the following protocol. Briefly, half of the mouse brain with tumors was chopped with a scalpel in RPMI-1640 media with 50 μg/mL Liberase TL (Roche) and 0.25 mg/mL DNase I (Roche) and incubated for 35 min, 190 rpm at 37 °C. The digestion was stopped with cold complete RPMI-1640 media. Tumor infiltrating immune cells were separated by a 30%−37%−70% Percoll gradient centrifugation. The red blood cells were removed by ACK Lysing Buffer (Thermo Fisher Scientific). The remaining single cell suspension was analyzed by a routine CyTOF protocol in Flow Cytometry Core Facility in La Jolla Institute for Immunology. Briefly, after staining with cisplatin viability staining reagent, cells are stained with antibodies conjugated to metal isotopes. Cells are also stained with DNA intercalators for detection of viable cells. The single cell signal was recorded by a Helios CyTOF Mass Cytometer (Fluidigm). The normalized FCS files were analyzed by R package “cytofWorkflow”. Cell population clusters were identified using the FlowSOM (RRID:SCR_016899) and ConsensusClusterPlus (RRID:SCR_016954) packages and visualized by t-distributed stochastic neighbor embedding (t-SNE) algorithm. The antibody panel used in this work consisted of 34 antibodies, which were designed by Flow Cytometry Core Facility in La Jolla Institute for Immunology to identify major immune cell populations, including antibodies to CD3 (BioLegend Cat# 100302, RRID:AB_312667), CD4 (Fluidigm Cat# 3172003B, RRID:AB_2811242), CD8a (Fluidigm Cat# 3153012B), CD11b (BioLegend Cat# 101202, RRID:AB_312785), CD11c (Fluidigm Cat# 3142003B, RRID:AB_2814737), CD64 (BioLegend Cat# 139302, RRID:AB_10613107), CD25 (Fluidigm Cat# 3151007B, RRID:AB_2827880), FR4 (BioLegend Cat# 125102, RRID:AB_1027724), MERTK (BioLegend Cat# 151502, RRID:AB_2566624), NK1.1 (BioLegend Cat# 108702, RRID:AB_313389), IgM (BioLegend Cat# 406502, RRID:AB_315052), B220 (Fluidigm Cat# 3160012B), Ly6c (BioLegend Cat# 128002, RRID:AB_1134214) and Ly6g (BioLegend Cat# 127602, RRID:AB_1089180).
Statistical analysis
Statistical tests, including Student’s t-test, one-way ANOVA, Pearson or Spearman correlation test, and Kolmogorov-Smirnov normality test were performed using R or GraphPad Prism v8 software (RRID:SCR_002798). Data in the barplot or curve are presented as mean±SD or mean+SD as denoted in each analysis. For the box-and-whisker plot in the Tukey style: the middle line of box, median value; the top and bottom lines of box, the upper and lower quartiles (Q1 and Q3), respectively. All the statistical tests were two-tailed. P < 0.05 was taken to indicate statistical significance unless otherwise stated.
Data availability
All raw sequencing data and selected processed data are available on GEO through accession number GSE155839. Additional data will be provided upon request.
Supplementary Material
SIGNIFICANCE:
Effective strategies to rewire immunosuppressive microenvironment and enhance immunotherapy response are still lacking in glioblastoma. YY1-driven transcriptional elongation machinery represents a druggable target to activate interferon response and enhance anti-PD-1 response through regulating the m6A modification program, linking epigenetic regulation to immunomodulatory function in glioblastoma.
Acknowledgments
We thank our funding sources: NIH: J.N.R., CA238662, CA197718, NS103434; B.C.P., CA217066; R.C.G, CA217065; K.Y., T32CA094186; DoD BCRP: C.S., W81XWH-15–1-0383; ASCO Conquer Cancer Foundation: K.Y., Young Investigator Award in Glioblastoma. We thank Tissue Technology Shared Resource in UCSD and Histology Core Facility in Sanford Burnham Prebys Medical Discovery Institute for histology analysis, Flow Cytometry Core Facility in La Jolla Institute for Immunology for CyTOF analysis, Erik Sulman for providing the GSC23 glioblastoma cell model and Mary E. Donohoe for critical reading of the manuscript. Tissue Technology Shared Resource is supported by a National Cancer Institute Cancer Center Support Grant (CCSG Grant P30CA23100). The CyTOF Helios was acquired through the Shared Instrumentation Grant (SIG) Program CyTOF Mass Cytometer S10 OD018499. Several panels were prepared in part using images from Servier Medical Art by Servier (https://smart.servier.com/), which is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Additional information:
Financial support: NIH: J.N.R., CA238662, CA197718, NS103434; B.C.P., CA217066; R.C.G, CA217065; K.Y., T32CA094186; DoD BCRP: C.S., W81XWH-15–1-0383; ASCO Conquer Cancer Foundation: K.Y., Young Investigator Award in Glioblastoma.
Footnotes
Conflict of interests: The authors declare no financial or competing interests.
REFERENCES
- 1.Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-oncology 2015;17 Suppl 4:iv1–iv62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016;164(3):550–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer cell 2018;33(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004;432(7015):396–401 [DOI] [PubMed] [Google Scholar]
- 5.Wakimoto H, Kesari S, Farrell CJ, Curry WT Jr., Zaupa C, Aghi M, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer research 2009;69(8):3472–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012;488(7412):522–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444(7120):756–60 [DOI] [PubMed] [Google Scholar]
- 8.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019;178(4):835–49 e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Touat M, Idbaih A, Sanson M, Ligon KL. Glioblastoma targeted therapy: updated approaches from recent biological insights. Annals of oncology : official journal of the European Society for Medical Oncology 2017;28(7):1457–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2017;169(2):361. [DOI] [PubMed] [Google Scholar]
- 11.Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018;174(3):549–63 e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wainwright EN, Scaffidi P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends in cancer 2017;3(5):372–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradner JE, Hnisz D, Young RA. Transcriptional Addiction in Cancer. Cell 2017;168(4):629–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sengupta S, George RE. Super-Enhancer-Driven Transcriptional Dependencies in Cancer. Trends in cancer 2017;3(4):269–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zanconato F, Battilana G, Forcato M, Filippi L, Azzolin L, Manfrin A, et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nature medicine 2018;24(10):1599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chou J, Quigley DA, Robinson TM, Feng FY, Ashworth A. Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer Therapy. Cancer discovery 2020;10(3):351–70 [DOI] [PubMed] [Google Scholar]
- 17.Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nature reviews Genetics 2014;15(4):234–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016;529(7584):110–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017;171(7):1573–88 e28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnston MJ, Nikolic A, Ninkovic N, Guilhamon P, Cavalli FMG, Seaman S, et al. High-resolution structural genomics reveals new therapeutic vulnerabilities in glioblastoma. Genome research 2019;29(8):1211–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGranahan T, Therkelsen KE, Ahmad S, Nagpal S. Current State of Immunotherapy for Treatment of Glioblastoma. Current treatment options in oncology 2019;20(3):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Medikonda R, Dunn G, Rahman M, Fecci P, Lim M. A review of glioblastoma immunotherapy. Journal of neuro-oncology 2021;151(1):41–53 [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Zhang Z, Qin W, Liu S, Liu C, Genchev GZ, et al. RePhine: An Integrative Method for Identification of Drug Response-related Transcriptional Regulators. Genomics, proteomics & bioinformatics 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mack SC, Singh I, Wang X, Hirsch R, Wu Q, Villagomez R, et al. Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. The Journal of experimental medicine 2019;216(5):1071–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014;157(3):580–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacLeod G, Bozek DA, Rajakulendran N, Monteiro V, Ahmadi M, Steinhart Z, et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell reports 2019;27(3):971–86 e9 [DOI] [PubMed] [Google Scholar]
- 27.Gordon S, Akopyan G, Garban H, Bonavida B. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene 2006;25(8):1125–42 [DOI] [PubMed] [Google Scholar]
- 28.Sarvagalla S, Kolapalli SP, Vallabhapurapu S. The Two Sides of YY1 in Cancer: A Friend and a Foe. Frontiers in oncology 2019;9:1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varum S, Baggiolini A, Zurkirchen L, Atak ZK, Cantu C, Marzorati E, et al. Yin Yang 1 Orchestrates a Metabolic Program Required for Both Neural Crest Development and Melanoma Formation. Cell stem cell 2019;24(4):637–53 e9 [DOI] [PubMed] [Google Scholar]
- 30.Liang Z, Li G, Wang Z, Djekidel MN, Li Y, Qian MP, et al. BL-Hi-C is an efficient and sensitive approach for capturing structural and regulatory chromatin interactions. Nature communications 2017;8(1):1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obeng EA, Stewart C, Abdel-Wahab O. Altered RNA Processing in Cancer Pathogenesis and Therapy. Cancer discovery 2019;9(11):1493–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winkler R, Gillis E, Lasman L, Safra M, Geula S, Soyris C, et al. m(6)A modification controls the innate immune response to infection by targeting type I interferons. Nature immunology 2019;20(2):173–82 [DOI] [PubMed] [Google Scholar]
- 33.Dixit D, Prager BC, Gimple RC, Poh HX, Wang Y, Wu Q, et al. The RNA m6A Reader YTHDF2 Maintains Oncogene Expression and Is a Targetable Dependency in Glioblastoma Stem Cells. Cancer discovery 2021;11(2):480–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nature chemical biology 2016;12(2):109–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483(7391):603–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nature chemical biology 2018;14(2):163–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M, et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clinical cancer research : an official journal of the American Association for Cancer Research 2020;26(4):922–34 [DOI] [PubMed] [Google Scholar]
- 38.Zhang H, Pandey S, Travers M, Sun H, Morton G, Madzo J, et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018;175(5):1244–58 e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katagi H, Takata N, Aoi Y, Zhang Y, Rendleman EJ, Blyth GT, et al. Therapeutic targeting of transcriptional elongation in diffuse intrinsic pontine glioma. Neuro-oncology 2021;23(8):1348–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bacon CW, Challa A, Hyder U, Shukla A, Borkar AN, Bayo J, et al. KAP1 Is a Chromatin Reader that Couples Steps of RNA Polymerase II Transcription to Sustain Oncogenic Programs. Molecular cell 2020;78(6):1133–51 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang K, Smith ER, Aoi Y, Stoltz KL, Katagi H, Woodfin AR, et al. Targeting Processive Transcription Elongation via SEC Disruption for MYC-Induced Cancer Therapy. Cell 2018;175(3):766–79 e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bacon CW, D’Orso I. CDK9: a signaling hub for transcriptional control. Transcription 2019;10(2):57–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen FX, Smith ER, Shilatifard A. Born to run: control of transcription elongation by RNA polymerase II. Nature reviews Molecular cell biology 2018;19(7):464–78 [DOI] [PubMed] [Google Scholar]
- 44.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature 2010;468(7327):1067–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khalsa JK, Cheng N, Keegan J, Chaudry A, Driver J, Bi WL, et al. Immune phenotyping of diverse syngeneic murine brain tumors identifies immunologically distinct types. Nature communications 2020;11(1):3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spitzer MH, Nolan GP. Mass Cytometry: Single Cells, Many Features. Cell 2016;165(4):780–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mathios D, Kim JE, Mangraviti A, Phallen J, Park CK, Jackson CM, et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Science translational medicine 2016;8(370):370ra180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008;455(7216):1061–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344(6190):1396–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Usheva A, Shenk T. TATA-binding protein-independent initiation: YY1, TFIIB, and RNA polymerase II direct basal transcription on supercoiled template DNA. Cell 1994;76(6):1115–21 [DOI] [PubMed] [Google Scholar]
- 51.Sharifnia T, Wawer MJ, Chen T, Huang QY, Weir BA, Sizemore A, et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nature medicine 2019;25(2):292–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hossain DMS, Javaid S, Cai M, Zhang C, Sawant A, Hinton M, et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. The Journal of clinical investigation 2018;128(2):644–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang H, Christensen CL, Dries R, Oser MG, Deng J, Diskin B, et al. CDK7 Inhibition Potentiates Genome Instability Triggering Anti-tumor Immunity in Small Cell Lung Cancer. Cancer cell 2020;37(1):37–54 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014;511(7511):616–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu Y, Zhao W, Liu Y, Tan X, Li X, Zou Q, et al. Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. The EMBO journal 2018;37(23) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu T, Qin Z, Tian Y, Wang J, Xu C, Li Z, et al. Recent Developments in the Biology and Medicinal Chemistry of CDK9 Inhibitors: An Update. Journal of medicinal chemistry 2020;63(22):13228–57 [DOI] [PubMed] [Google Scholar]
- 57.Ricklefs FL, Alayo Q, Krenzlin H, Mahmoud AB, Speranza MC, Nakashima H, et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Science advances 2018;4(3):eaar2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sevenich L. Turning “Cold” Into “Hot” Tumors-Opportunities and Challenges for Radio-Immunotherapy Against Primary and Metastatic Brain Cancers. Frontiers in oncology 2019;9:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nature neuroscience 2013;16(10):1373–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nature methods 2017;14(4):417–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489(7414):57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang Q, Chen Y, Meyer C, Geistlinger T, Lupien M, Wang Q, et al. A comprehensive view of nuclear receptor cancer cistromes. Cancer research 2011;71(22):6940–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 2005;102(43):15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang P, Freedman ML, Liu JS, Liu XS. Inference of transcriptional regulation in cancers. Proceedings of the National Academy of Sciences of the United States of America 2015;112(25):7731–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen JZ, Qiu Z, Wu Q, Finlay D, Garcia G, Sun D, et al. FBXO44 promotes DNA replication-coupled repetitive element silencing in cancer cells. Cell 2021;184(2):352–69 e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods 2012;9(4):357–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome biology 2008;9(9):R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015;31(14):2382–3 [DOI] [PubMed] [Google Scholar]
- 70.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009;25(8):1091–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miller TE, Liau BB, Wallace LC, Morton AR, Xie Q, Dixit D, et al. Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 2017;547(7663):355–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li G, Chen Y, Snyder MP, Zhang MQ. ChIA-PET2: a versatile and flexible pipeline for ChIA-PET data analysis. Nucleic acids research 2017;45(1):e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome biology 2015;16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequencing data and selected processed data are available on GEO through accession number GSE155839. Additional data will be provided upon request.