

Abstract
Chronic obstructive pulmonary disease (COPD) is a complex, heterogeneous, smoking-related disease of significant global impact. The complex biology of COPD is ultimately driven by a few interrelated processes, including proteolytic tissue remodeling, innate immune inflammation, derangements of the host-pathogen response, aberrant cellular phenotype switching, and cellular senescence, among others. Each of these processes are engendered and perpetuated by cells modulating their environment or each other. Extracellular vesicles (EVs) are powerful effectors that allow cells to perform a diverse array of functions on both adjacent and distant tissues, and their pleiotropic nature is only beginning to be appreciated. As such, EVs are candidates to play major roles in these fundamental mechanisms of disease behind COPD. Furthermore, some such roles for EVs are already established, and EVs are implicated in significant aspects of COPD pathogenesis. Here, we discuss known and potential ways that EVs modulate the environment of their originating cells to contribute to the processes that underlie COPD.
Keywords: COPD, exosomes, microvesicles, ectosomes, extracellular vesicles
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is a syndrome of progressive airflow obstruction due to long-term inhalation of noxious substances, most commonly tobacco smoke. COPD afflicts millions of people worldwide, results in an enormous amount of both morbidity and death, and in fact is one of the top causes of mortality in both the United States and worldwide (1). Despite the high impact of this illness, medical management of COPD is largely limited to interventions that temporarily reduce symptom burden as opposed to the disease-modifying and personalized therapeutic strategies now available for other diseases of comparable impact. Despite the general lack of nuanced, targeted therapies for COPD, decades of scientific inquiry into the biology of COPD have uncovered important pathways that drive this disease. Indeed, recent advances in translational science have allowed the field to progress immensely over the past few years, and advances in the study of extracellular vesicles (EVs) have arguably provided some of the most important recent insights into these pathways.
Clinically, COPD is characterized by shortness of breath, wheezing, and cough and culminates in respiratory failure. Biologically, the disease is heterogeneous with multiple overlapping processes largely occurring downstream of the chronic inhalation of tobacco smoke (or sometimes other stimuli) (2). This contributes to an array of pathways that permanently damage the elastic pulmonary extracellular matrix (ECM), culminating in two major classical mechanisms of disease at the histologic level: loss of alveolar tissue/surface area (emphysema) and sustained airway inflammation (chronic bronchitis). In a given patient, the preponderance of emphysema versus chronic bronchitis is highly variable. Additionally, it is now recognized that there are other histological derangements in COPD, which, although interrelated with them, are distinct from emphysema and chronic bronchitis. These include abnormal deposition of fibrotic tissue in the small airways [now thought to be a key, very early feature of COPD (3, 4)], pulmonary vascular remodeling [now appreciated as an important independent contributor to COPD morbidity (5)], and aberrations of cellular morphology (6) (Figure 1). However, despite this complexity and variability at the pathological level, on the mechanistic level, there are a few major derangements that converge and generally drive these disease manifestations. Of course, these are themselves very complex. Such mediators of COPD pathology include proteolytic ECM remodeling, innate immune activation/inflammation, deranged host-pathogen interactions, aberrant cellular phenotype switching, and cellular senescence. Each contributes to the pathological findings of COPD in multiple ways. These processes are not truly discrete, but rather are highly interconnected and, again, there is wide variability in the respective contributions of these mechanisms to a given patient’s disease status. Nonetheless, focusing on these critical common derangements is a useful way to categorize the extent to which various processes contribute to COPD, and this approach is taken here.
Figure 1:
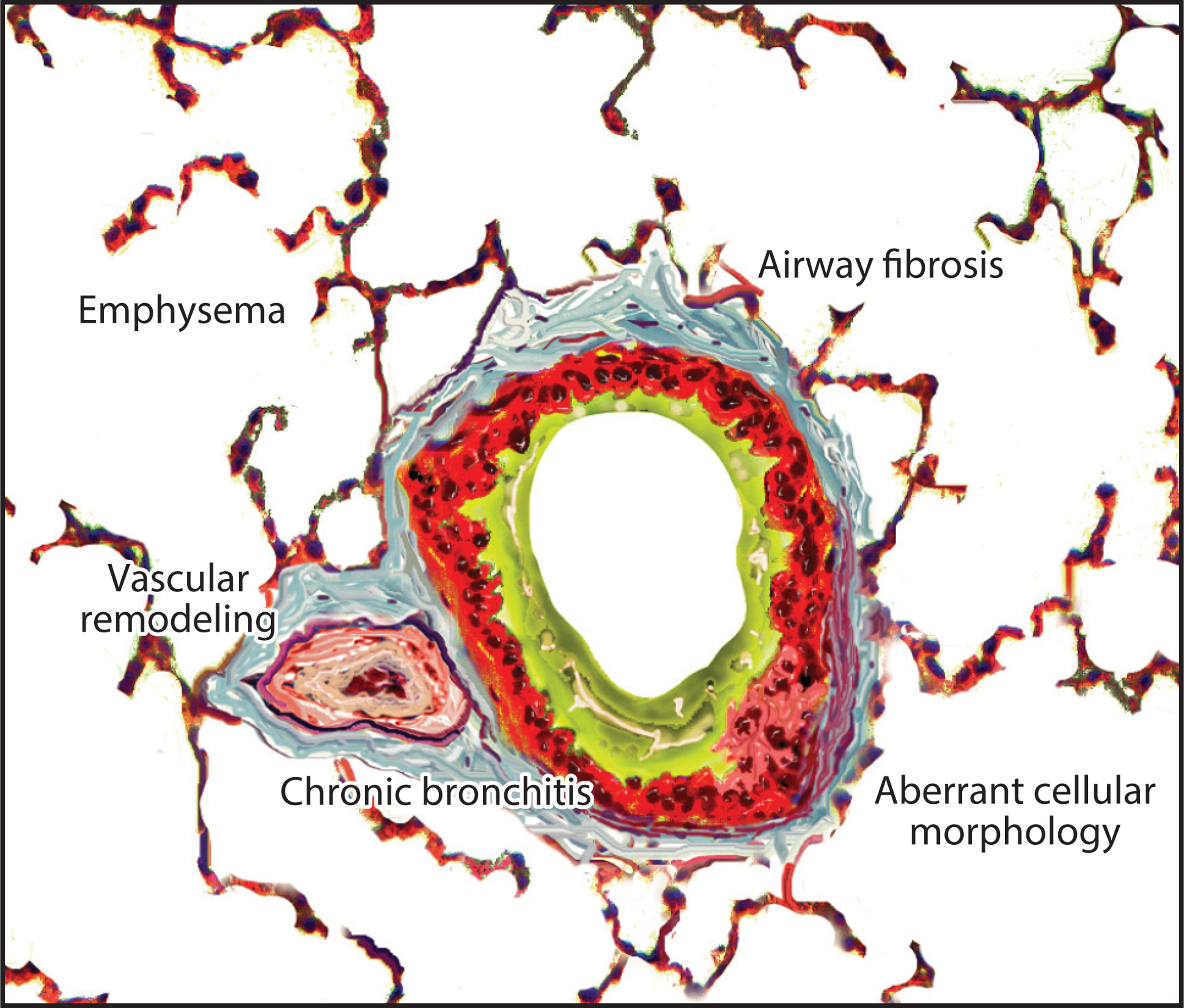
Major histological manifestations of chronic obstructive pulmonary disease (COPD). In the lungs of COPD patients, classical findings include emphysema (e.g., alveolar wall loss, alveolar enlargement) and consequences of chronic bronchitis (e.g., bronchial wall thickening, airway inflammation, and excess mucus). However, other biological derangements are increasingly recognized to be not only characteristic but also significant in the clinical symptoms of this disease. These derangements include fibrosis of the airways (especially small bronchioles), findings of pulmonary hypertension in the vascular bed, and aberrant cellular morphology (exemplified by epithelial-squamous metaplasia, epithelial-mesenchymal transition, and myofibroblast proliferation). All of these are now established pulmonary findings that contribute to COPD. Although most, if not all, of these COPD manifestations are generally present in a given patient, there is wide variability in terms of the predominance of one manifestation or another.
This complexity and heterogeneity of COPD, at both the clinical and pathological levels, have heretofore hamstrung efforts to develop interventions that meaningfully interrupt the disease course as disease-modifying agents. It is hoped that an improved understanding of the underlying biology of COPD will break this stalemate. In part owing to technological advances that allow for more nuanced study of EVs, it is now recognized that cells in the COPD lung interact with each other and with their environment via EVs (Figure 2). In fact, EVs mediate or modulate at least some aspects of each of the aforementioned pathophysiological processes that drive COPD. Knowledge of the roles that EVs play in COPD has greatly increased in recent years, but this can still be expanded considerably by interpolating our understanding of these fundamental mechanisms of disease with study of the contribution of EVs to similar mechanisms in other conditions that share some of these mechanistic pathways. Using these paradigms, this article reviews the known or potential roles of EVs with respect to the critical biological mechanisms that drive COPD.
Figure 2:

Tissues and cells in the lung modulate each other to contribute to the processes that drive chronic obstructive pulmonary disease (COPD). On the biological axis, the histological abnormalities of COPD (see Figure 1) are driven by a complex bidirectional interplay between many cell/tissue types, including most prominently the (alveolar and bronchial) epithelium, the (alveolar and peribronchial) interstitium along with its resident cells, and immune effector cells. Because extracellular vesicles (EVs) mediate intercellular cross talk and can modulate the environments of cells locally and remotely, they serve as important effectors; thus, EVs sit at the center of this interplay. The background shows a colorized electron microscopy image of EVs from polymorphonuclear leukocytes (yellow) bound to type I collagen fibrils (green).
EXTRACELLULAR VESICLES
A brief description of the biology of EVs (Figure 3) and associated terminology is merited. The biogenesis, characteristics, and importance of EVs (also called microparticles and nanovesicles) and EV subclasses (discussed briefly below) have been thoroughly summarized elsewhere (7–9). EVs (as used here) are small particles (<100 nm–1 μm in diameter) bound by a lipid bilayer. The emission of such particles is now thought to occur in virtually all eukaryotic cell types and, indeed, similar vesicles are well described from prokaryotes as well. Although poorly understood for decades, EVs are currently known to play a wide range of roles in the human body and are important for cellular waste removal, intercellular communication/signaling, and as extracellular effectors for originating cells (9). Given their small size and pleiotropic biological roles, it is perhaps little wonder that they remain largely uncategorized on the whole and poorly understood.
Figure 3:
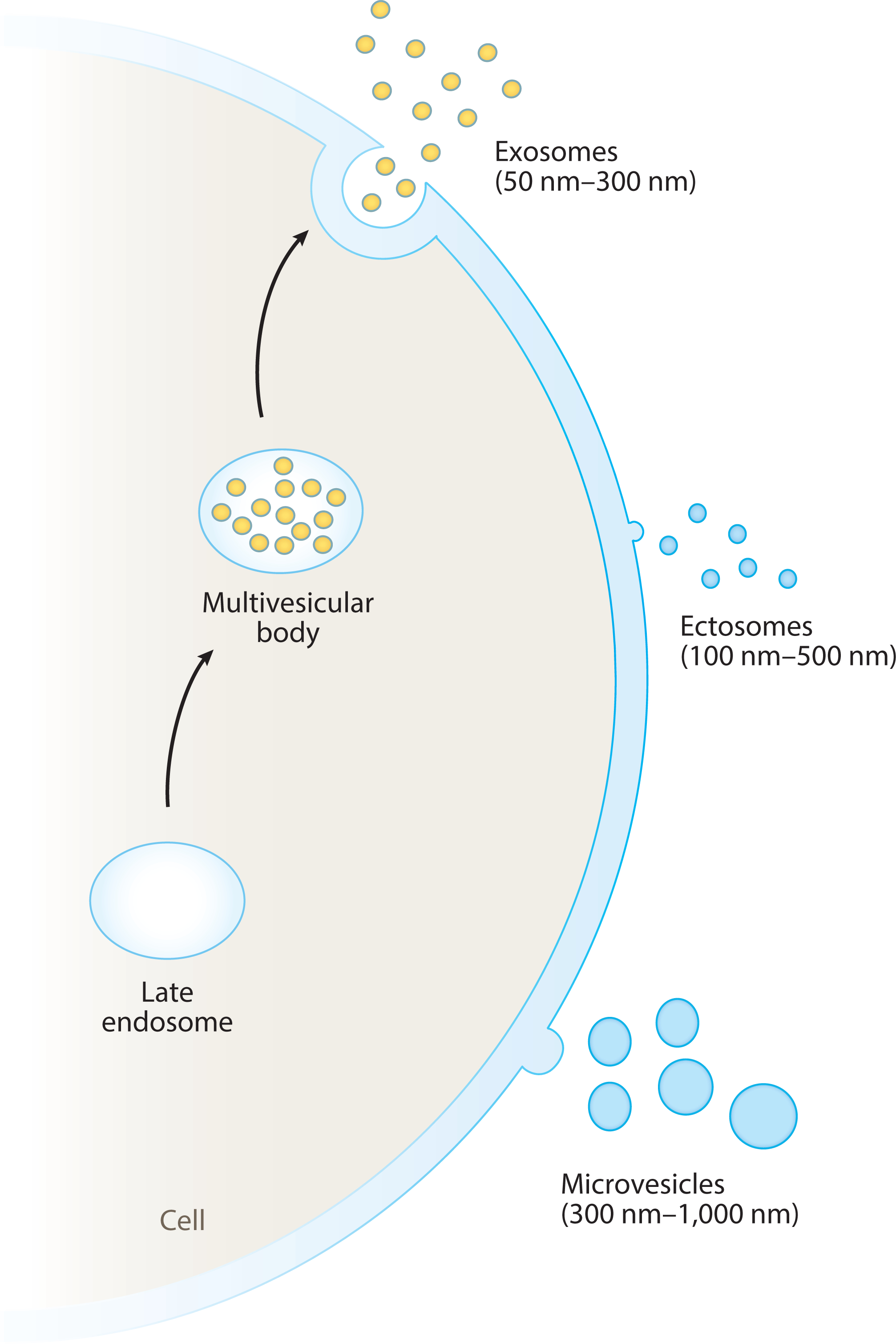
Types of extracellular vesicles (EVs). EVs can be divided into microvesicles, exosomes, and ectosomes, which vary by biogenetic mechanism, size, and other characteristics.
The nomenclature for EVs has evolved considerably as our understanding of their complex nature increased and, indeed, the classification systems and means of characterization of EVs, while increasingly standardized, are still not without controversy. For this review, the generic term EVs is used for all particles wrapped in a lipid bilayer that meet the size constraints mentioned above (<100 nm–1 μm in diameter). There is considerable overlap between the physical and biochemical characteristics of EV categories to the extent that widely available means of separating EV subclasses also are generally subject to some degree of cross-contamination between EV types. For this reason and for the sake of simplicity, this review generally uses the term EVs, unless otherwise specified. Nonetheless, these categories do remain useful as broad descriptors of EV biology, so a word of discussion on the terms used in the literature is merited. In recent years, especially upon the release of guidelines from the International Society of Extracellular Vesicles (10), the nomenclature for and description of EVs have become increasingly standardized in the literature.
Exosomes
Exosome is a term for a small (~50–300 nm in diameter) class of EVs that are released from intracellular vesicles, themselves housed (prior to release) within organelles called microvesicular bodies (MVBs). Upon MVB membrane fusion with the exosomal cell of origin, a population of exosomes is released en masse from the cell into the surrounding environment. This can be done either constitutively or in response to stimuli (11). The complex systems governing the packaging of MVBs and release of exosomes are still incompletely understood, but they involve endosomal sorting complexes required for transport (ESCRT)-dependent and ESCRT-independent pathways (12); as such, certain membrane-bound markers (most notably the tetraspanins CD63, CD9, and CD81) are expressed nearly universally by exosomes. Interestingly, MVBs derive, at least in part, from the endo-lysosomal organelles of the cell. Thus, the contents of exosomes often include previously ubiquitinated proteins and other cellular trash and, indeed, cellular waste disposal seems to be a part of their teleology. Exosomes are also loaded (passively and actively) with a wide variety of bioactive components, including catalytically active enzymes, signaling molecules, and coding and noncoding nucleic acid material, with a preponderance of microRNA (miR) (9, 13–16). Congruent with this, exosomes are capable of communicating a wide variety of signals from cell to cell. Furthermore, exosomes seem to be heavily involved in the intercellular waste disposal and recycling of subcellular components [even large organellar fragments such as from damaged mitochondria (17)]. Exosomes can be taken up by recipient cells via a wide range of mechanisms (pinocytosis, phagocytosis, endocytosis, merging with cellular membrane, etc.), and the mechanism by which they are taken (and thus the ultimate destination in recipient cells) varies by the cell of origin, the recipient cell, and the contents or surface expression pattern of the exosome (9, 11, 18–21).
Ectosomes
Arguably less well described than exosomes are ectosomes, which are similar in size range to the former (~100–350 nm, in some reports up to 500 nm), but which are released directly from the cellular membrane surface by budding. Confoundingly, these seem to play a similar biological role as exosomes in many cases, as they can express some of the same markers and can be released in response to similar cues (such as intracellular Ca++ release) (18). Owing to their biogenesis, these EVs can be released more rapidly by a cell than exosomes, but their release is also governed in part by ESCRT-dependent pathways. Despite these similarities with exosomes, there are significant, measurable biochemical differences between the latter and ectosomes [e.g., the composition and orientation of the vesicle membrane lipids (14, 18)]. Nonetheless, delineation between these two EV types of similar density and size in samples containing both can be very difficult. This contributes to the need for the wider adoption of precise and standardized language describing a population of EVs studied in a given experiment, as well as for more precise analytical techniques for study of individual EVs rather than a population of them (18).
Microvesicles
Similar in terms of biogenesis to the ectosomes (and at times grouped with them), the larger (~300 nm–1 μm) microvesicles, sometimes also called microparticles (the latter being also confusingly used as a synonym for EV), are released from the plasma membrane of the cell (8). Consequently, microvesicles tend to generally reflect the surface composition of their cell of origin and often express cell markers of the latter (8, 22). In recipient cells, microvesicles are more prone to undergo membrane fusion with the cell membrane of the recipient cell than are exosomes, which are generally more apt to go inside the recipient cell (22). Again, however, this also seems to vary somewhat by cell of origin, target, and probably environment (16, 22, 23).
COPD PATHOGENESIS
As mentioned above, the pathogenesis of COPD is complex and heterogeneous. Ultimately, COPD is driven by chronic pathophysiological changes induced or initiated by long-term exposure to noxious inhaled substances such as tobacco smoke. A particular set of pathophysiological and clinical derangements may predominate in a given patient, but these mechanisms generally coexist and often converge. Tobacco smoke contains a complex mixture of carbonaceous compounds of various degrees of oxidation, ranging from microscopic carbon black particles and aromatic hydrocarbons, through an array of partially oxidized substances (aldehydes, alcohols, carbon monoxide) to fully oxidized carbon (carbon dioxide) (24, 25). Each of these are thought to impact the biology of the lungs of tobacco smokers. Similarly, heavy metals, trace bacterial compounds, complex organic substances, nitrosamines, alkaloids such as nicotine, and reactive radical species produced by combustion also likely play a role in the effects of tobacco smoke on the lung (24–26). These substances cumulatively tend to induce a combination of oxidative stress and innate immune cell recruitment and activation into the lung, while also activating a host of deleterious effects on pulmonary tissues (27). This dynamic, over years, can be conceived as disrupting the homeostasis of the lung such that tissue remodeling results in bronchitis and/or emphysema, creating the syndrome known as COPD. However, the reality is somewhat more complex, as many smokers do not develop COPD, and the biological disruptions that induce this remodeling vary from patient to patient. This heterogeneity may be due to the combination of complex and poorly understood environmental and genetic factors that favor COPD pathogenesis. Furthermore, these processes can overlap (e.g., oxidative stress inducing cell death/senescence and vice versa) and, in the right circumstances, these processes can become self-perpetuating in the absence of ongoing smoke exposure. For example, innate immune recruitment/activation causes the breakdown of collagen fibrils into Pro-Gly-Pro peptide fragments that themselves induce innate immune cell recruitment/activation (28) [a process that can continue unabated in former smokers with established COPD (29)].
As pleiotropic and omnipresent mediators of an array of biological processes, EVs might contribute to all of these mechanisms of disease. For the sake of simplicity, we focus this review on the role of EVs in selected processes downstream of tobacco smoke exposure in COPD patients.
PROTEOLYTIC TISSUE REMODELING AND ITS IMPORTANCE IN COPD
Proteolysis resulting in ECM remodeling (classically dominated by tissue degradation) is arguably the biological sine qua non of COPD pathogenesis (30, 31) and inarguably a cardinal feature of this disease. An abundance of proteolysis relative to the protective barrier imposed by antiproteases (i.e., protease-antiprotease imbalance) has been recognized for decades as a major contributor to COPD pathogenesis (30, 32). Many COPD features, including emphysema, chronic inflammation, and obstructive respiratory failure, can be reproduced by instillation of proteases into the lungs of animals when enough is given to overwhelm the native antiprotease barrier (33–35). Although many collagenases, elastases, and other proteases have been associated with COPD progression, severity, or activity, the protease-antiprotease system between neutrophil elastase (NE) and its dominant inhibitor in the pulmonary interstitium, α−1 antitrypsin, remains the best described. In fact, a genetic deficiency of α−1 antitrypsin has been long recognized as an accelerant of tobacco smoke–mediated emphysema (32). So strong is the association of this protease-antiprotease system that α−1 antitrypsin deficiency is even capable of causing COPD at times without exposure to tobacco smoke at all (36). As such, the pathogenetic mechanism of protease-antiprotease imbalance is considered both requisite and (at least facultatively) sufficient to cause COPD. Though a host of other processes contribute to COPD, many of them do so via direct or indirect amplification of ECM degradation. This being the case, there is considerable overlap and cross talk between proteolytic ECM remodeling and the other pathways discussed in this review.
THE ROLE OF EXTRACELLULAR VESICLES
Direct Effects of Extracellular Vesicles
EVs are now recognized as powerful modulators and mediators of tissue remodeling by both direct and indirect means. Due to the combination of their diminutive size, resistance to degradation imparted by the vesicle membrane, and the presence of (as yet incompletely understood) compounds on the EV surface that facilitate homing to a target cell or tissue, EVs are an ideal way for a cell to affect tissues far beyond its own physical reach. Expression of integrins with tropism to a particular tissue bed (11, 21), fibronectin (19), and likely other surface components give EVs from a given cell the ability to target their bioactive cargo to certain tissues remotely to the cell of origin. In fact, in rheumatoid arthritis, EVs released by polymorphonuclear leukocytes (PMNs) have been shown to traffic into arguably the least penetrable tissue of the body, the dense articular cartilage stroma, where they modulate tissue remodeling (23). This capacity, when paired with the fact that EVs can express proteases on the vesicle surface, can give the EV cell of origin significant abilities to directly damage or remodel ECM components even when the cell itself is not in direct contact with this substrate. Enzymes expressed on the surface of an EV are locally concentrated on the EV surface such that, even at a distance from the cell of origin at which simple diffusion of the enzyme would preclude any ability to substantively affect a substrate, sufficient enzyme is present to create a significant quantum of impact on the substrate that the EV encounters. Moreover, as discussed below, EVs can sometimes modulate the properties of their associated enzymes in a variety of ways.
Perhaps unsurprisingly, nonmalignant EVs with a strong capacity for direct proteolysis are to date best demonstrated (albeit not exclusively) for EVs derived from innate immune cells such as PMNs and macrophages. It is well known that, under certain conditions, these cells release (via many mechanisms including degranulation) a wide variety of proteases. What is only more recently appreciated is that such proteases can be released in association with EVs, which can confer important biological effects to the protease. This association can take the form of membrane-bound expression, intravesicular packaging and release, EV membrane extracellular surface expression from solution (e.g., degranulated enzymes binding to an EV membrane outside of a cell upon concurrent release), and other forms. Importantly, proteases bound to or housed within EVs are often able to maintain their proteolytic capacity and, in some cases, become much more potent than the dissolved version of the same protease. In fact, the very role of EV-associated proteases often seems to be distinct from that of the solution-phase enzyme by mere virtue of their association with EVs. This can be due to several factors, including (a) the biological functions of the EV itself (such as the ability of an EV to hold its bound protease in direct apposition to a substrate), (b) the nature of the binding of the protease with the EV (such as the steric hindrance of a binding site for an inhibitor), (c) the presence of other EV constituents (such as the presence of proteases that degrade an inhibitor or bind to a substrate), or (d) a combination of some or all of these factors (11). As such, association with an EV can markedly affect the impact of a protease. For example, matrix metalloproteinase 9 (MMP-9) maintains access to the substrate desmoglein-2 on the surface of EVs released by PMNs. EV-associated MMP-9 has an apparent several-fold increase in potency over the solution-phase version of MMP-9 with respect to its ability to cleave intercellular tight junction, facilitating the transepithelial migration of activated PMNs into inflamed epithelium in the gut (16). Another collagenolytic MMP, MMP-14, is released from macrophages within large EVs in response to tobacco smoke and may directly contribute to ECM damage in the lungs of smokers (37). Airway epithelial cells stimulated with lipopolysaccharide (LPS; a constituent of cigarette smoke) release EVs containing the enzyme prolyl endopeptidase. This EV-associated oligopeptidase, downstream of MMP degradation of collagen fibrils, generates the tripeptide Pro-Gly-Pro, which acts as a PMN chemoattractant (38, 39). This amplifies PMN influx to a site of collagen breakdown, which can lead to a feed-forward cycle of sustained PMN recruitment, activation, degranulation, and tissue damage that contributes to the tissue remodeling seen in COPD (29, 40).
The most definitive evidence that we have on the role of EVs in the pathogenesis of COPD may be our work on PMN-derived EVs involving NE. This protease, as mentioned above, is itself very strongly implicated in COPD pathogenesis (11). Small EVs released from PMNs after activation with degranulating stimuli (but not quiescent PMNs) express catalytically active NE on the EV surface capable of degrading NE substrates present in the pulmonary interstitium, such as elastin and type I collagen. When given intratracheally to mice, we found that these PMN-derived, NE+ EVs recapitulate COPD disease features (emphysema, right ventricular enlargement, and ventilation impairment) in one of the most rapid COPD models published to date. An analogous population of PMN-derived (CD66b+) NE+ EVs can be purified in the lung fluids of patients with COPD. Interestingly, these EVs can transfer COPD from human to mouse when given intratracheally to recipient animals in an NE-dependent manner. Here again, the biological potency of NE was greatly enhanced (several log-fold) by its EV association (relative to solution-phase NE in the same model). This appears at least partially due to the ability of EV-membrane-bound NE to evade α−1 antitrypsin inhibition (likely due to steric hindrance and perhaps also the coexpression of the α−1 antitrypsin–degrading protease MMP-9 on the EV membrane) and to the presence of Mac-1 on the EV surface, an integrin capable of targeting and binding to collagen and elastin fibrils in the alveolar interstitium (shown in Figure 2) (41). Solution-phase NE levels in most COPD patients, although elevated, are not fully sufficient to overcome the α−1 antitrypsin barrier on a molar basis. Other hypothesized explanations for this discrepancy have also fallen short over the years, including a once-theorized acquired form of α−1 antitrypsin deficiency (36, 42). Thus, the finding of a higher-potency, α−1 antitrypsin–resistant form of NE associated with PMN-derived EVs, which may explain how the quantity of NE found in the lung tissue is enough to cause emphysema, represents a breakthrough in our understanding of this long-enigmatic aspect of COPD pathogenesis. We further found that these activated PMN-derived EVs did not differentially affect the RNA expression of recipient pulmonary epithelial cells relative to those derived from quiescent PMNs (11). Thus, the evidence suggests that activated PMN-derived EVs cause emphysema by directly degrading the alveolar interstitial ECM, leading to the indirect loss of the remaining cellular component of the alveolar unit (i.e., the alveolar epithelial layer) by inducing alveolar epithelial cell apoptosis due to loss of underlying ECM cellular anchoring, a phenomenon known as anoikis (43). Furthermore, NE was not the only PMN-associated protease that was carried by the EVs derived from activated PMNs, and some of the others (e.g., cathepsin G, MMP-9) (11) have been previously associated with COPD activity in their own right (44–46). However, the roles that these EV-associated proteases play have not yet been fully elucidated.
One more aspect of these NE+ EVs also bears discussion. Pulmonary hypertension and right ventricular hypertrophy are key features of COPD, belonging to the World Health Organization (WHO) Group 3 pulmonary hypertension category (47). As NE+ EVs caused not only emphysema but also right ventricular hypertrophy in our intratracheal mouse model (11), their effects on the right ventricle and pulmonary vascular bed may also be important in this aspect of COPD. In that vein, it is noteworthy that circulating PMN-derived EVs of patients with primary pulmonary hypertension (WHO Group 1) are also capable of transferring disease to mice, in many ways recapitulating our COPD research in this distantly related disease (48). This confirms our observation of the powerful, disease-inducing effects of EVs in vivo. Moreover, taken together with our EV COPD model, this strongly suggests that EVs themselves play an important role in the pathogenesis of COPD-associated pulmonary hypertension (i.e., WHO Group 3 disease).
The macrophage-derived MMP-12 vies with NE as one of the proteases for which a causative role in COPD is most firmly established (49), but to our knowledge, little published evidence exists on the question of whether MMP-12 may also have an EV-associated form, as appears to be the case for so many other MMPs. This underscores the paucity of previous research published in this field. Nonetheless, taken cumulatively, the aforementioned studies do highlight the importance of this area and the need for further research on the roles of EV-associated proteases in diseases, such as COPD, which are driven by tissue destruction.
Indirect Effects of Extracellular Vesicles
EVs can also indirectly impact tissue remodeling via effects on the recipient cell after being uptaken. This is in part due to the many bioactive components contained within the vesicle cytosol, which become internalized into recipient cells and then join the machinery of that cell either within an organelle or in the cytosol itself. For example, MMP-3 contained within EVs derived from a malignant cell line is transported to the nucleus, where it acts upon a transcription factor to upregulate a tumorigenic/prometastatic compound (connective tissue growth factor) in recipient cells throughout the body in an allogenic mouse transfer model (50). This illustrates that EVs can at times allow their cargo to be trafficked to not only a certain target tissue but also to a specific intracellular compartment. However, EVs are able to indirectly contribute to tissue remodeling by other means as well. One example with clear relevance to COPD is that, in response to tobacco smoke exposure, EVs from pulmonary epithelial cells can autologously induce MMP-1 production via cysteine-rich protein 61, while also inducing interleukin (IL)-8 release, which would be expected to effectively recruit PMNs (and their attendant proteases) in vivo (51–53). Though incompletely understood, miRs and other small, noncoding RNAs are another important bioactive cargo of many EVs (especially small EVs) that can be uptaken into recipient cells and indirectly impact protein expression. Thousands of miRs are now known to exist, and although it is appreciated that they modulate gene transcription, our understanding of them remains rudimentary (54). However, it is known that miRs conveyed by EVs can and do affect proteolysis and other aspects of ECM remodeling via modulation of recipient cell RNA transcription. For example, miR-451a, which is predicted (on the basis of its base pair sequence) to influence expression of MMP-2 and MMP-9, was found to be differentially expressed across EVs derived from alveolar macrophage populations obtained from different zones of the lung (55). Owing to a dearth of comprehensive study, the influence of EV-associated miRs is not yet well established as a mediator of the pathophysiology of COPD itself. Nonetheless, the importance of EV-associated miRs on tissue remodeling is undisputed, as these pathways are often hijacked by malignant cells, a phenomenon that has been more rigorously studied. For example, prostate cancer stem cells can release EVs with miR-21–5p, miR-100–5p, and miR-139–5p, which collectively induce MMP-2, MMP-9, and MMP-13 expression in recipient fibroblasts, shaping the peritumoral niche and facilitating tumoral tissue invasion and metastatic spread (15). Therefore, future work characterizing the complex signal modulations induced by EV-borne miRs that affect pathologic tissue remodeling in COPD may prove pivotal in our understanding of proteolytic ECM remodeling in this disease.
CELLULAR PHENOTYPE SWITCHING
Importance in COPD
In COPD, the cells comprising the bronchial tree epithelial-mesenchymal unit undergo localized phenotypic alterations, including proliferative squamous metaplasia of bronchial epithelial cells (56), epithelial-mesenchymal transition of alveolar epithelial cells (57), fibroblast-myofibroblast transformation (58), and smooth muscle proliferation in the small airways (59, 60). These cellular transformations involve heavy paracrine cross talk between the native cell types of the bronchial tree, in which the epithelium and mesenchyme can each mutually stimulate transformation (61, 62). These processes may contribute to COPD in a few ways. The first involves airflow obstruction via direct mechanical occlusion (63, 64). Second, metaplastic transformation of pulmonary epithelial cells can have significant deleterious consequences on the immunological and inflammatory milieu of the COPD lungs (65). Finally, being a hyperproliferative state associated with oncogene expression, metaplasia and transformation of the pulmonary epithelium are also associated with development of adenocarcinoma of the lung (66). This is important, as COPD patients have a strong predisposition, independently of smoking and other risk factors, to develop lung cancer (67, 68), which in turn causes the death of many COPD sufferers (69).
Role of Extracellular Vesicles
Unfortunately, relatively little is known about EV-mediated paracrine signaling with respect to cellular transformations in COPD itself. However, EVs are nonetheless likely to play prominent roles given their known importance in intercellular communication and their ability to influence protein expression in recipient cells, as mentioned previously. Indeed, PMN-derived EVs are taken up by airway smooth muscle cells to induce a proliferative phenotype, a process postulated to contribute to asthma but potentially relevant to COPD as well (70). In COPD, tobacco smoke exposure encourages this complex process of cellular transformation via inflammatory and profibrotic signal induction, and EVs have been shown to contribute to this. Bronchial epithelial cells exposed to cigarette smoke extract release EVs enriched in miR-210, leading to myofibroblast differentiation in fibroblasts (58). Similarly, EVs containing miR-21, which are elevated in the lungs and serum of COPD patients, can be stimulated by exposure of bronchial epithelial cells to cigarette smoke extract to foster myofibroblast differentiation in vitro (71). Epithelial metaplasia and epithelial-mesenchymal transition exist in continua with pulmonary carcinoma (57). As such, the myriad, important roles that EVs are known to play in shaping the peritumoral niche of non-small-cell lung cancer cells (72) further support the importance of EVs in epithelial transformation in COPD.
CELL SENESCENCE
Cellular senescence is a permanent arrest in the cell cycle due to genetic (extensive telomere shortening) and molecular [reactive oxygen species (ROS), DNA damage, oncogene activation] stressors (73–76). While cell senescence is most often associated with the aging process, patients with COPD exhibit symptoms of premature aging in the lung and the presence of early senescence in lung tissue (77, 78). Tobacco smoke exposure and the associated exposure to ROS in the lung are thought to underlie much of this susceptibility (79). Senescent cells maintain their local environment through a phenomenon known as the senescence-associated secretory phenotype (SASP) (80). SASP consists of various growth factors, proteases, and proinflammatory proteins and cytokines (81) and has been shown to link increased and premature cellular senescence of lung cells to COPD (82). Cultured pulmonary epithelial cells from COPD patients released higher amounts of proinflammatory cytokines [interleukin (IL)-6, IL-8, monocyte chemoattractant protein-1 (MCP-1)] and shorter telomeres than control patients’ cells (83).This indicates that there are both a combination of effects from having a proinflammatory condition such as COPD and an increased senescent cell population, which together drive a progression of increased inflammation in the lung microenvironment.
Recent studies have shown that senescent cells release EVs and that some SASP components may be associated with these EVs, providing the cells with a mechanism to deliver SASP to neighboring cells (84–86). EV-miR-21 is upregulated in senescent cells in vitro and may contribute to the induction of senescence in surrounding cells (87). MiR-21 was also found in EVs of COPD patients (58) and was associated with increased ROS production due to autophagy dysregulation, thereby propagating further inflammatory cell damage (88). MiR-210 in EVs has also been implicated in autophagy arrest, senescence signaling and, as mentioned above, in COPD pathogenesis (58, 89). Additionally, senescence-induced release of miR-210-containing EVs can facilitate a feedback loop of more DNA damage and oxidative stress in recipient cells (90).
A component of SASP, ICAM-1, an adhesion molecule important in trafficking of PMNs and known to be elevated in smokers (91), was found to be released via EVs (92), thus increasing lung inflammatory signals.IL-6 receptor (IL-6R), another SASP component, has been identified within EVs (93). This soluble IL-6R may interact with IL-6 in the alternate IL-6 signaling pathway, known as IL-6 trans-signaling (94), thereby increasing inflammation.
Overall, available data suggest that the release of EVs by senescent cells contributes to increased inflammation and enhanced senescent cell development. The development of COPD in a patient favors the development of senescence in the lung, whereas the accumulation of premature senescent cells in the lung promotes the development of COPD, forming a positive feedback loop that furthers COPD progression. Thus, EVs ultimately contribute to the severity of COPD via both senescence-induced and senescence-inducing mechanisms.
IMMUNE ACTIVATION/INFLAMMATION
Polymorphonuclear Leukocytes
PMNs have long been associated with COPD severity. However, both increased numbers of PMNs and impairments of PMN function (such as impaired chemotaxis and phagocytosis) are well recognized in COPD. PMNs are initially recruited to the site of cellular damage/inflammation via primary chemoattractants (e.g., IL-8 released by tobacco smoke–exposed bronchiolar epithelial cells). En route to such a stimulus, PMNs exhibit a swarming, or clustering, phenotype as they traffic toward the site where the chemoattractant originates. This swarming signal is mediated by paracrine leukotriene B4 (LTB4) signaling by PMNs up the chemotactic gradient, and this behavior seems to be a means of directional fine-tuning by PMNs collectively en route to the initial stimulus (95). Interestingly, this swarming signal is actually released by a trail of EVs left by mobile, activated PMNs. These EVs contain leukotriene-synthesizing enzymes that generate LTB4 in situ (96). Such LTB4-producing EV release is seen uniquely in stimulated PMNs, and not quiescent, unstimulated PMNs. Although several functions of PMN EVs with respect to proteolysis were discussed earlier, it bears mentioning that this sensitivity of the PMN EV cargo to PMN-activating stimuli in COPD is a general theme, perhaps not surprisingly, given the many such stimuli associated with tobacco smoke and COPD itself (53). This includes the aforementioned example that stimulated (but not quiescent) PMNs release EVs containing proteolytically active NE on their surface (11), which, when instilled into the murine bronchi, bind to the lung ECM components and degrade them, generating a COPD-like phenotype (11). Furthermore, PMNs activated with an opsonization stimulus zymosan A, then coated with complement from normal human serum, release medium-sized EVs that amplify inflammation (97). These activated EVs increase the ROS of neighboring PMNs that were similarly activated with phorbol 12-myristate 13-acetate, whereas, again, EVs of quiescent PMNs did not enhance the response of other PMNs to this stimulus. Similarly, IL-8 production in PMNs was increased when in the presence of EVs from activated, but not quiescent PMNs (97). EVs from PMNs can also induce inflammatory signal production in epithelial cells, increasing both IL-8 and IL-6 release by these cells (98). However, this general proinflammatory tendency of EVs from PMNs is not absolute, as EVs from PMNs have been observed to induce anti-inflammatory pathways and prevent inflammatory pathways in macrophages (99, 100).
On the whole, PMN-derived EVs are pleiotropic and powerful modulators of the tissue microenvironment. Synthesizing these findings, the data suggest that the function of PMN-derived EVs depends on the environment to which the PMN is exposed; in the highly activating conditions of the COPD lung (teeming with stimuli such as tobacco smoke, bacteria, cytokines, and inflammatory ECM breakdown products), PMN EVs seem to generally amplify inflammation and cause direct inflammatory tissue destruction, thus linking an initial insult to both end-organ damage and innate immune dysregulation.
Monocytes/Macrophages
Cells of the monocytic line, and especially macrophages, are thought to be pivotal to the lung inflammation seen in COPD. Similar to PMNs, macrophages are abundant in nearly all tissue compartments of the COPD lung and localize to the sites of alveolar septal degradation, and the degree of abundance correlates with COPD activity (101). Despite this, monocyte/macrophage phenotype and function are also abnormal and even impaired in the COPD lung, which likely contributes to the disease (101–103). Despite the apparently pivotal role that this cell type plays in COPD pathophysiology, there are fewer data describing macrophage-derived EVs in COPD than now exist for PMN-derived EVs. Nonetheless, there is some evidence that, similar to PMNs, the contents and biological effects of macrophages also depend upon the signaling milieu. However, the extent to which EVs may mediate some of the effects of macrophages on COPD pathophysiology is not truly known. Cigarette smoke extract (CSE) has been shown to increase Ca++ influx into monocytes, stimulating rapid EV secretion by monocytes. When stimulated with the EVs released by these CSE-stimulated monocytes, cultured human bronchial epithelial cells showed an increase in IL-8 and MCP-1 (104). This increase in IL-8 and MCP-1 release by epithelial cells may be due to an increase in nuclear factor-kappa B (NF-κB) activation induced by the presence of these Ca++-dependent monocyte EV release (105). Cigarette smoke also can cause macrophages to release EVs containing MMP-14, which can degrade collagen and contribute to emphysema (37). The proinflammatory cytokine IL-1β has been identified on macrophage-derived exosomes (106), and the proinflammatory cytokines tumor necrosis factor-alpha (TNF-α) and IL-6 were found in macrophage-derived EVs from mice upon tracheal instillation of LPS (107). Thus, there is ample evidence that monocytes and macrophages utilize EVs to respond to stimuli relevant to COPD pathogenesis and that these EVs can express both inflammatory and proteolytic factors. Further research is clearly merited to investigate the extent to which such EVs might cause or modulate the activity and progression of COPD itself.
HOST-PATHOGEN INTERACTIONS
Importance in COPD
Pulmonary bacterial overgrowth contributes to inflammation in the lungs of COPD patients in many ways. The impact of host-pathogen response and microbial burden on COPD is illustrated well by the recent finding that transcytotic delivery of the antibacterial secretory immunoglobulin A (IgA) by pulmonary epithelial cells into the airway lumen is disrupted uniquely at areas of aberrant epithelial morphology in the COPD lung, leading to bacterial overgrowth, which itself perpetuates inflammation (65, 108). This ultimately culminates in loss of bronchial tethering and the development of emphysema. In fact, mice with a genetic defect in transcytosis of secretory IgA spontaneously develop emphysema and airflow obstruction, an effect ameliorated with either murine germ-free upbringing or administration of antibiotics to sterilize the airways (65). This illustrates an important interplay between bacterial overgrowth and derangements of several key aspects of COPD pathogenesis that have been discussed previously, including epithelial morphology, innate immune function, chronic inflammation, and tissue remodeling. Furthermore, bacteria are known clinically to be important drivers of acute episodes of worsening COPD symptoms [episodes known as acute exacerbations of COPD (AECOPD) (109, 110)], and AECOPD, in turn, are important drivers of both short-term COPD mortality and long-term COPD progression (111, 112). Given this centrality of both bacteria and the host response, the EVs induced by bacteria on human tissues and those expressed by bacteria themselves likely contribute to the pathophysiology of COPD.
Role of Extracellular Vesicles
It has been shown that the presence of bacterial LPS in the lung can signal the release of exosomes and other EVs by pulmonary epithelium and various resident immune cells (38, 113). The LPS released is not just in the form of free LPS but has been identified on EVs from the bacteria themselves. All gram-negative, and some gram-positive, bacteria appear to be capable of releasing EVs (114). The vesicle released by gram-negative bacteria, like Haemophilus influenzae, are termed outer membrane vesicles (OMVs) and possess various virulence factors, as well as LPS, on their surfaces (115, 116). Bacteria present on inhaled dust, such as Escherichia coli and Staphylococcus aureus, can release EVs in the lung and increase PMN inflammation and ECM degradation (117). S. aureus EVs increase IL-17 cytokine and interferon gamma (IFN-γ) production in pulmonary tissue in a Toll-like receptor 2 (TLR-2)-dependent manner (118).
Patients with COPD possess higher amounts of adenosine triphosphate (ATP) in the airways (119, 120), and this increased ATP promotes chronic inflammation in the airways (121) by enhancing the IL-1β and IL-18 production of pulmonary-derived EVs (106, 122). This increased amount of ATP, and subsequent enhancement of inflammatory cytokines by the elevated ATP levels, can be further enhanced by the presence of pulmonary bacterial infection. The LPS present on the surface of OMVs released by H. influenzae in the lung promotes the release of proinflammatory EVs by lung epithelium, and the inflammatory signals are further magnified by the elevated ATP levels in the COPD lung (123).
At least 50% of AECOPD may be caused by bacteria (109, 110). The three most common bacteria found in these exacerbations are H. influenzae, Streptococcus pneumonia, and Moraxella catarrhalis (124). These exacerbations are likely due to the increased inflammatory responses triggered by the bacteria and their released EVs. However, the EVs released by the bacteria not only cause the host to increase its own inflammatory signals, but their EVs possess means to evade and subvert the hosts own immune response to them. The OMVs produced by M. catarrhalis possess ubiquitous surface proteins A1 and A2 (UspA1/A2) on their surface, which binds and inactivates complement protein C3 (125, 126), thus lowering the amount of C3 available to bind to the bacteria itself and signal its destruction by the host immune system. M. catarrhalis OMVs have also been identified and are able to deliver active β-lactamase to neighboring bacteria (127), including H. influenzae, S. pneumoniae, and antibiotic-susceptible M. catarrhalis to confer penicillin and ampicillin resistance to otherwise susceptible infectious strains. EVs released by S. pneumoniae have been identified and possess multiple factors that enable them to minimize the effects of various host immune responses. Pneumococcal surface protein A has been identified on their EV surfaces (128) and shown to bind and sequester the antimicrobial protein lactoferrin, preventing it from destroying the S. pneumoniae (129). In addition, a novel endodeoxyribonuclease has been discovered on the surface of S. pneumoniae EVs that degrades the DNA in neutrophil extracellular traps released by PMNs in response to infection, aiding in the evasion of the bacteria (130). Finally, S. pneumoniae EVs possess pneumococcal surface protein C (PspC) and pneumolysin (131). PspC binds factor H and promotes complement deposition and membrane attack complex formation on the EVs, decreasing the amount of active complement in the surrounding milieu available to act upon the bacteria themselves. Pneumolysin is a cholesterol-binding, pore-forming toxin that can be delivered by the EVs and cause cell death in immune cells, preventing bacterial phagocytosis. Thus, EVs seem to play a significant role in both immune evasion and pathogenicity of bacteria in the lung and do so systemically. This is likely to promote bacterial overgrowth and AECOPD, thereby amplifying and perpetuating inflammation and tissue remodeling downstream.
OTHER HOST-PATHOGEN INTERACTIONS
Mucus Hypersecretion
Mucus hypersecretion is a hallmark of progressive COPD, especially the chronic bronchitis subphenotype (132). PMNs contribute to mucus secretion (133). NE, which as mentioned above has been shown to be present and active in PMN exosomes (11), can induce MUC5AC production in lung epithelial cells (134), contributing to increased mucus production in the lung. Additionally, S100 calcium-binding protein A9 (S100A) is present in PMN exosomes (135), and cigarette smoke has been shown to increase levels of S100A9 in the bronchoalveolar lavage (136). S100A9, among other S100 proteins, increases MUC5A secretion in bronchial epithelial cells via an NF-κB pathway (137). S100A9 in exosomes from leukemia cells also promotes NF-κB activity (138). All of this taken together, it is feasible that chronic inflammation contributes to mucus hypersecretion via EV-directed stimulation of lung epithelial cell mucin production, to which PMN-derived EVs likely contribute.
Systemic Inflammation
Despite the classical conception as a disease limited to the lung, COPD is actually a multisystem syndrome with notable extrapulmonary features, including cachexia (139) and anxiety/depression (140). Given that tobacco smoke is a risk factor for other conditions such as cardiovascular disease, stroke, and extrapulmonary cancer, the morbidity and mortality of COPD patients are also strongly affected by these other ailments (1, 69). These conditions all can contribute to COPD outcomes through a variety of mechanisms, sometimes in surprising ways [e.g., depression and anxiety being strong predictors of mortality in survivors of AECOPD (140)]. Thus, while these are often considered to be comorbid conditions, one could also view them as integral aspects of COPD itself. Indeed, COPD causes systemic inflammation (which itself, of course, contributes to each of these comorbidities), and circulating inflammatory biomarkers correlate with prognosis in COPD patients (141). There is some evidence, albeit preliminary, that EVs are involved in this. The number of CD9+ EVs in circulating blood is higher in COPD patients than in non-COPD controls, tracks with disease activity (increasing during an AECOPD), and correlates with biomarkers of systemic inflammation (C-reactive protein, IL-6, soluble tumor necrosis factor receptor-1) (142). The investigators did not determine the cellular origin or teleologic role of these EVs, but this suggests that EVs are involved in either the development of or the response to systemic inflammation in COPD (if not both). Though few direct data exist on the contribution of EVs to the inflammatory state of COPD patients, it is likely that they do contribute. As mentioned above, EVs have a penchant for widely conducting signals; thus, it may follow that they contribute to this spillover of lung inflammation that occurs in COPD. For example (and perhaps germane to the association between COPD, systemic inflammation, and depression), intravenous injection of EVs derived from LPS-exposed mice into LPS-naive mice led to neurological inflammation and microglial activation in recipient animals (143). Also, as already illustrated in previous examples, EVs can directly contribute to the generation of proinflammatory stimuli in recipient cells, can themselves carry proinflammatory cargoes, or can also induce pathways that favor downstream inflammation (144). Nonetheless, it is noteworthy that EVs also mediate many immunosuppressive and anti-inflammatory effects (144). Although outside the scope of this review, EVs can also contribute to many, if not all, of the COPD comorbidities that occur downstream of systemic inflammation, such as atherosclerosis (145) and cachexia (146). Ultimately, the involvement of EVs in the systemic inflammation of COPD is likely to prove prominent but complex, as it has in other aspects of the disease.
Asthma-COPD Overlap Syndrome
Asthma is in many ways similar to COPD, and sufferers of these two diseases have considerable symptomatology in common. A principle diagnostic distinction between the two diseases is the reversibility of airflow obstruction (in COPD, airflow obstruction is permanent or fixed, but in asthma, airflow obstruction is temporally variable and generally reversible with bronchodilator drugs). However, a subset of COPD patients have reversibility of some portion of their airflow obstruction, whereas a subset of asthma patients go on to develop fixed obstruction (2). However, there are also general differences in the pathophysiology driving these diseases. Relative to COPD, asthma tends to be associated with eosinophilic/T helper 2 (Th2) cell inflammation and atopy. However, some COPD patients also have eosinophilia or atopy, and asthma itself can at times be driven by PMNs or Th1 inflammation reminiscent of that of COPD (2, 147). In fact, there is so much overlap between the pathophysiology of these two conditions that, of the most commonly used drug classes originally developed for asthma, many are used routinely for COPD and vice versa (148).This speaks to the existence of the so-called asthma-COPD overlap syndrome (ACOS), which is a general term for patients with features of both diseases (147). A full review of the biology of EVs in asthma has been undertaken elsewhere (149, 150). However, in light of the existence of ACOS, a brief word on this subject is justified, being relevant to COPD. In general, there is evidence of EV involvement in asthma on many levels, including epithelial cell signaling (151) and proliferation (152, 153), cytokine signaling (13, 20), monocytic cell chemotaxis (151), eosinophilic inflammation (154, 155), mast cell–lymphocyte cross talk (20), and airway smooth muscle cell proliferation (153), among others. Because airway remodeling and neutrophilic inflammation are such characteristic features of COPD, it is especially interesting that EVs from LPS-exposed PMNs induce airway smooth muscle cell proliferation in vitro (70), a finding the reporting investigators proposed to be relevant to neutrophilic asthma. It is not known whether this mechanism contributes to the airway hyper-responsiveness or other ACOS features seen in many COPD patients. One thing does appear certain: Further investigation of EV biology should help shed light on the biology of ACOS.
FUTURE DIRECTIONS
As highlighted in this review, EVs play many known roles in the processes that drive COPD, and a multitude of other roles are probable as this science advances. This is important by sheer virtue of the insights gained into the biology of this complex disease. However, the unusual qualities of EVs, paired with their apparent potency on numerous biological axes, raise the question of whether they might ultimately prove useful for bedside clinical care in the management of this disease. Although our understanding of some aspects of EV biology in COPD is too rudimentary to take this step as of today, there is now sufficient knowledge of other aspects to offer some hope that this could be possible. Measurement of PMN-derived, NE+ EVs in the lung fluids appears to act as a biomarker of ongoing innate immune activation and ECM degradation in COPD irrespective of smoking status (11). The number of such EVs in the lungs may thus be a useful means of prognosticating, or it may serve as a useful index of COPD activity to anticipate patients who would benefit from certain therapeutics. Because it is thought that the mechanisms of disease presage the development of COPD proper (156), perhaps the measurement of these EVs in the lungs of non-COPD smokers could even be used to predict which patients are on their way to developing COPD. This could be a helpful way for physicians to target smoking cessation strategies or to convince patients that smoking is harming their lungs before irreversible damage has set in. Furthermore, therapies designed specifically to abrogate the harmful aspects of EV activity in COPD might prove to be helpful in slowing down the progression of this disease or preventing complications. Although such treatments remain developmental and speculative, the broad effects of EVs demonstrate the promise of future therapeutic avenues for this inexorable disease.
CONCLUSION
COPD is a complex, heterogeneous disease, but the underlying pathophysiology is generally driven by a few major processes that involve cellular cross talk and tissue remodeling. As pleiotropic entities by which cells can mediate a host of intercellular and environmental effects, EVs are candidates to contribute to virtually all of these processes. The study of EV biology in COPD and related conditions is growing, and important pathophysiological insights into these disease mechanisms have been recently gained that were not possible until the role of EVs was better appreciated. In fact, the study of EVs is elucidating the answers to enigmatic questions, such as the now-strong evidence that NE+ EVs derived from activated PMNs contribute to protease-antiprotease imbalance that causes alveolar remodeling. However, the study of EVs in COPD is still nascent, and the picture remains incomplete. A sophisticated understanding of the full spectrum of EV biology in COPD will take rigorous research, but fortunately, advances in EV science are accelerating. Perhaps, in time, medicine will succeed in improving the management of this currently unstoppable respiratory disease through further insights gained from the study of EVs.
ACKNOWLEDGMENTS
D.W.R. was supported by NIH/NHLBI grant 1 K08 HL148514, and K.R.G. and J.E.B. were supported by NIH grant 1 R35HL135710.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Vogelmeier CF, Criner GJ, Martinez FJ, Anzueto A, Barnes PJ, et al. 2017. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report: GOLD Executive Summary. Am. J. Respir. Crit. Care Med. 195:557–82 [DOI] [PubMed] [Google Scholar]
- 2.Russell DW, Wells JM, Blalock JE. 2016. Disease phenotyping in chronic obstructive pulmonary disease: the neutrophilic endotype. Curr. Opin. Pulm. Med. 22:91–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koo HK, Vasilescu DM, Booth S, Hsieh A, Katsamenis OL, et al. 2018. Small airways disease in mild and moderate chronic obstructive pulmonary disease: a cross-sectional study. Lancet Respir. Med. 6:591–602 [DOI] [PubMed] [Google Scholar]
- 4.Russell DW, Wells JM. 2018. COPD ground zero: small airways rather than alveoli as the initial site of injury. Lancet Respir. Med. 6:568–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wells JM, Washko GR, Han MK, Abbas N, Nath H, et al. 2012. Pulmonary arterial enlargement and acute exacerbations of COPD. N. Engl. J. Med. 367:913–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osei ET, Hackett TL. 2020. Epithelial-mesenchymal crosstalk in COPD: an update from in vitro model studies. Int. J. Biochem. Cell Biol. 125:105775. [DOI] [PubMed] [Google Scholar]
- 7.Tkach M, Thery C. 2016. Communication by extracellular vesicles: where we are and where we need to go. Cell 164:1226–32 [DOI] [PubMed] [Google Scholar]
- 8.van Niel G, D’Angelo G, Raposo G. 2018. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19:213–28 [DOI] [PubMed] [Google Scholar]
- 9.Kalluri R, LeBleu VS. 2020. The biology, function, and biomedical applications of exosomes. Science 367:eaau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, et al. 2018. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genschmer KR, Russell DW, Lal C, Szul T, Bratcher PE, et al. 2019. Activated PMN exosomes: pathogenic entities causing matrix destruction and disease in the lung. Cell 176:113–26.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babst M 2011. MVB vesicle formation: ESCRT-dependent, ESCRT-independent and everything in between. Curr. Opin. Cell Biol. 23:452–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gon Y, Maruoka S, Inoue T, Kuroda K, Yamagishi K, et al. 2017. Selective release of miRNAs via extracellular vesicles is associated with house-dust mite allergen-induced airway inflammation. Clin. Exp. Allergy 47:1586–98 [DOI] [PubMed] [Google Scholar]
- 14.Keerthikumar S, Gangoda L, Liem M, Fonseka P, AtukoralaI, et al. 2015. Proteogenomic analysis reveals exosomes are more oncogenic than ectosomes. Oncotarget 6:15375–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez CA, Andahur EI, Valenzuela R, Castellon EA, Fulla JA, et al. 2016. Exosomes from bulk and stem cells from human prostate cancer have a differential microRNA content that contributes cooperatively over local and pre-metastatic niche. Oncotarget 7:3993–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butin-Israeli V, Houser MC, Feng M, Thorp EB, Nusrat A, et al. 2016. Deposition of microparticles by neutrophils onto inflamed epithelium: a new mechanism to disrupt epithelial intercellular adhesions and promote transepithelial migration. FASEB J. 30:4007–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hough KP, Trevor JL, Strenkowski JG, Wang Y, Chacko BK, et al. 2018. Exosomal transfer of mitochondria from airway myeloid-derived regulatory cells to T cells. Redox Biol. 18:54–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cocucci E, Meldolesi J. 2015. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. 25:364–72 [DOI] [PubMed] [Google Scholar]
- 19.Purushothaman A, Bandari SK, Liu J, Mobley JA, Brown EE, Sanderson RD. 2016. Fibronectin on the surface of myeloma cell-derived exosomes mediates exosome-cell interactions. J. Biol. Chem. 291:1652–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie G, Yang H, Peng X, Lin L, Wang J, et al. 2018. Mast cell exosomes can suppress allergic reactions by binding to IgE. J. Allergy Clin. Immunol. 141:788–91 [DOI] [PubMed] [Google Scholar]
- 21.Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, et al. 2015. Tumour exosome integrins determine organotropic metastasis. Nature 527:329–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vituret C, Gallay K, Confort MP, Ftaich N, Matei CI, et al. 2016. Transfer of the cystic fibrosis transmembrane conductance regulator to human cystic fibrosis cells mediated by extracellular vesicles. Hum. Gene Ther. 27:166–83 [DOI] [PubMed] [Google Scholar]
- 23.Headland SE, Jones HR, Norling LV, Kim A, Souza PR, et al. 2015. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci. Transl. Med. 7:315ra190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swartz H 1971. Tobacco smoke: a noxious air pollutant. A review (and comment). II of II. Tobacco smoke: general characteristics and composition. Rev. Allergy 25:490–505 [PubMed] [Google Scholar]
- 25.Stedman RL. 1968. The chemical composition of tobacco and tobacco smoke. Chem. Rev. 68:153–207 [DOI] [PubMed] [Google Scholar]
- 26.Pappas RS, Polzin GM, Zhang L, Watson CH, Paschal DC, Ashley DL. 2006. Cadmium, lead, and thallium in mainstream tobacco smoke particulate. Food Chem. Toxicol. 44:714–23 [DOI] [PubMed] [Google Scholar]
- 27.Iskandar AR, Zanetti F, Kondylis A, Martin F, Leroy P, et al. 2019. A lower impact of an acute exposure to electronic cigarette aerosols than to cigarette smoke in human organotypic buccal and small airway cultures was demonstrated using systems toxicology assessment. Intern. Emerg. Med. 14:863–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, et al. 2006. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat. Med. 12:317–23 [DOI] [PubMed] [Google Scholar]
- 29.Wells JM, O’Reilly PJ, Szul T, Sullivan DI, Handley G, et al. 2014. An aberrant leukotriene A4 hydrolaseproline-glycine-proline pathway in the pathogenesis of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 190:51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turino GM. 2002. The origins of a concept: the protease-antiprotease imbalance hypothesis. Chest 122:1058–60 [DOI] [PubMed] [Google Scholar]
- 31.Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. 2003. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am. J. Pathol. 163:2329–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laurell CB, Eriksson S. 1963. The electrophoretic α;1-globulin pattern of serum in α;1-antitrypsin deficiency. Scand. J. Clin. Lab. Investig. 15:132–40 [Google Scholar]
- 33.Senior RM, Tegner H, Kuhn C, Ohlsson K, Starcher BC, Pierce JA. 1977. The induction of pulmonary emphysema with human leukocyte elastase. Am. Rev. Respir. Dis. 116:469–75 [DOI] [PubMed] [Google Scholar]
- 34.Janoff A, Sloan B, Weinbaum G, Damiano V, Sandhaus RA, et al. 1977. Experimental emphysema induced with purified human neutrophil elastase: tissue localization of the instilled protease. Am. Rev. Respir. Dis. 115:461–78 [DOI] [PubMed] [Google Scholar]
- 35.Gross P, Pfitzer EA, Tolker E, Babyak MA, Kaschak M. 1965. Experimental emphysema: its production with papain in normal and silicotic rats. Arch. Environ. Health 11:50–58 [DOI] [PubMed] [Google Scholar]
- 36.Stockley RA. 2014. Alpha1-antitrypsin review. Clin. Chest Med. 35:39–50 [DOI] [PubMed] [Google Scholar]
- 37.Li CJ, Liu Y, Chen Y, Yu D, Williams KJ, Liu ML. 2013. Novel proteolytic microvesicles released from human macrophages after exposure to tobacco smoke. Am. J. Pathol. 182:1552–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szul T, Bratcher PE, Fraser KB, Kong M, Tirouvanziam R, et al. 2016. Toll-like receptor 4 engagement mediates prolyl endopeptidase release from airway epithelia via exosomes. Am. J. Respir. Cell Mol. Biol. 54:359–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaggar A, Jackson PL, Noerager BD, O’Reilly PJ, McQuaid DB, et al. 2008. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J. Immunol. 180:5662–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wells JM, Gaggar A, Blalock JE. 2015. MMP generated matrikines. Matrix Biol. 44–46:122–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Podolnikova NP, Podolnikov AV, Haas TA, Lishko VK, Ugarova TP. 2015. Ligand recognition specificity of leukocyte integrin αMβ2 (Mac-1, CD11b/CD18) and its functional consequences. Biochemistry 54:1408–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campbell EJ, Campbell MA, Boukedes SS, Owen CA. 2000. Quantum proteolysis by neutrophils: implications for pulmonary emphysema in α1-antitrypsin deficiency. Chest 117:303S. [PubMed] [Google Scholar]
- 43.Mouded M, Egea EE, Brown MJ, Hanlon SM, Houghton AM, et al. 2009. Epithelial cell apoptosis causes acute lung injury masquerading as emphysema. Am. J. Respir. Cell Mol. Biol. 41:407–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao WJ, Li MH, Li JX, Xu X, Ren SX, et al. 2016. High expression of cathepsin E is associated with the severity of airflow limitation in patients with COPD. COPD 13:160–66 [DOI] [PubMed] [Google Scholar]
- 45.Andrault PM, Schamberger AC, Chazeirat T, Sizaret D, Renault J, et al. 2019. Cigarette smoke induces overexpression of active human cathepsin S in lungs from current smokers with or without COPD. Am. J. Physiol. Lung. Cell. Mol. Physiol. 317:L625–38 [DOI] [PubMed] [Google Scholar]
- 46.Wells JM, Parker MM, Oster RA, Bowler RP, Dransfield MT, et al. 2018. Elevated circulating MMP-9 is linked to increased COPD exacerbation risk in SPIROMICS and COPDGene. JCI Insight 3:e123614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, et al. 2009. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J. Am. Coll. Cardiol. 53:1573–619 [DOI] [PubMed] [Google Scholar]
- 48.Taylor S, Contrepois K, Benayoun BA, Jiang L, Isobe S, et al. 2021. Endogenous retroviral elements generate pathologic neutrophils and elastase rich exosomes in pulmonary arterial hypertension. bioRxiv 426001. 10.1101/2021.01.08.426001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. 1997. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 277:2002–4 [DOI] [PubMed] [Google Scholar]
- 50.Okusha Y, Eguchi T, Tran MT, Sogawa C, Yoshida K, et al. 2020. Extracellular vesicles enriched with moonlighting metalloproteinase are highly transmissive, pro-tumorigenic, and trans-activates cellular communication network factor (CCN2/CTGF): CRISPR against cancer. Cancers 12:881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moon HG, Kim SH, Gao J, Quan T, Qin Z, et al. 2014. CCN1 secretion and cleavage regulate the lung epithelial cell functions after cigarette smoke. Am. J. Physiol. Lung. Cell. Mol. Physiol. 307:L326–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aaron SD, Angel JB, Lunau M, Wright K, Fex C, et al. 2001. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 163:349–55 [DOI] [PubMed] [Google Scholar]
- 53.Beeh KM, Kornmann O, Buhl R, Culpitt SV, Giembycz MA, Barnes PJ. 2003. Neutrophil chemotactic activity of sputum from patients with COPD: role of interleukin 8 and leukotriene B4. Chest 123:1240–47 [DOI] [PubMed] [Google Scholar]
- 54.Sato T, Baskoro H, Rennard SI, Seyama K, Takahashi K. 2015. MicroRNAs as therapeutic targets in lung disease: prospects and challenges. Chronic Obstr. Pulm. Dis. 3:382–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Armstrong DA, Nymon AB, Ringelberg CS, Lesseur C, Hazlett HF, et al. 2017. Pulmonary microRNA profiling: implications in upper lobe predominant lung disease. Clin. Epigenet. 9:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rigden HM, Alias A, Havelock T, O’Donnell R, Djukanovic R, et al. 2016. Squamous metaplasia is increased in the bronchial epithelium of smokers with chronic obstructive pulmonary disease. PLOS ONE 11:e0156009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eapen MS, Sharma P, Gaikwad AV, Lu W, Myers S, et al. 2019. Epithelial-mesenchymal transition is driven by transcriptional and post transcriptional modulations in COPD: implications for disease progression and new therapeutics. Int. J. Chron. Obstruct. Pulmon. Dis. 14:1603–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujita Y, Araya J, Ito S, Kobayashi K, Kosaka N, et al. 2015. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. J. Extracell. Vesicles 4:28388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bosken CH, Wiggs BR, Pare PD, Hogg JC. 1990. Small airway dimensions in smokers with obstruction to airflow. Am. Rev. Respir. Dis. 142:563–70 [DOI] [PubMed] [Google Scholar]
- 60.Cosio MG, Hale KA, Niewoehner DE. 1980. Morphologic and morphometric effects of prolonged cigarette smoking on the small airways. Am. Rev. Respir. Dis. 122:265–21 [DOI] [PubMed] [Google Scholar]
- 61.Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, et al. 2007. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J. Clin. Investig. 117:3551–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishioka M, Venkatesan N, Dessalle K, Mogas A, Kyoh S, et al. 2015. Fibroblast-epithelial cell interactions drive epithelial-mesenchymal transition differently in cells from normal and COPD patients. Respir. Res. 16:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sohal SS, Walters EH. 2013. Epithelial mesenchymal transition (EMT) in small airways of COPD patients. Thorax 68:783–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nowrin K, Sohal SS, Peterson G, Patel R, Walters EH. 2014. Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways: fibrosis, remodeling and cancer. Expert Rev. Respir. Med. 8:547–59 [DOI] [PubMed] [Google Scholar]
- 65.Richmond BW, Brucker RM, Han W, Du RH, Zhang Y, et al. 2016. Airway bacteria drive a progressive COPD-like phenotype in mice with polymeric immunoglobulin receptor deficiency. Nat. Commun. 7:11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vu T,Jin L,Datta PK.2016.Effect of cigarette smoking on epithelial to mesenchymal transition (EMT) in lung cancer. J. Clin. Med. 5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park HY, Kang D, Shin SH, Yoo KH, Rhee CK, et al. 2020. Chronic obstructive pulmonary disease and lung cancer incidence in never smokers: a cohort study. Thorax 75:506–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Young RP, Hopkins RJ, Christmas T, Black PN, Metcalf P, Gamble GD. 2009. COPD prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur. Respir. J. 34:380–86 [DOI] [PubMed] [Google Scholar]
- 69.Berry CE, Wise RA. 2010. Mortality in COPD: causes, risk factors, and prevention. COPD 7:375–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vargas A, Roux-Dalvai F, Droit A, Lavoie JP. 2016. Neutrophil-derived exosomes: a new mechanism contributing to airway smooth muscle remodeling. Am. J. Respir. Cell Mol. Biol. 55:450–61 [DOI] [PubMed] [Google Scholar]
- 71.Xu H, Ling M, Xue J, Dai X, Sun Q, et al. 2018. Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking. Theranostics 8:5419–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zheng H, Zhan Y, Liu S, Lu J, Luo J, et al. 2018. The roles of tumor-derived exosomes in non-small cell lung cancer and their clinical implications. J. Exp. Clin. Cancer Res. 37:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hayflick L, Moorhead PS. 1961. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25:585–621 [DOI] [PubMed] [Google Scholar]
- 74.Hayflick L 1965. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 37:614–36 [DOI] [PubMed] [Google Scholar]
- 75.Harley CB, Futcher AB, Greider CW. 1990. Telomeres shorten during ageing of human fibroblasts. Nature 345:458–60 [DOI] [PubMed] [Google Scholar]
- 76.Campisi J, d’Adda di Fagagna F. 2007. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8:729–40 [DOI] [PubMed] [Google Scholar]
- 77.Karrasch S, Holz O, Jorres RA. 2008. Aging and induced senescence as factors in the pathogenesis of lung emphysema. Respir. Med. 102:1215–30 [DOI] [PubMed] [Google Scholar]
- 78.Vogelmeier C, Bals R. 2007. Chronic obstructive pulmonary disease and premature aging. Am. J. Respir. Crit. Care Med. 175:1217–18 [DOI] [PubMed] [Google Scholar]
- 79.Tsuji T, Aoshiba K, Nagai A. 2004. Cigarette smoke induces senescence in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 31:643–49 [DOI] [PubMed] [Google Scholar]
- 80.Munoz-Espin D, Serrano M. 2014. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15:482–96 [DOI] [PubMed] [Google Scholar]
- 81.Campisi J, Andersen JK, Kapahi P, Melov S. 2011. Cellular senescence: A link between cancer and age-related degenerative disease? Semin. Cancer Biol. 21:354–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van Deursen JM. 2014. The role of senescent cells in ageing. Nature 509:439–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amsellem V, Gary-Bobo G, Marcos E, Maitre B, Chaar V, et al. 2011. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 184:1358–66 [DOI] [PubMed] [Google Scholar]
- 84.Coppe JP, Desprez PY, Krtolica A, Campisi J. 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5:99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Takasugi M 2018. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 17:e12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lehmann BD, Paine MS, Brooks AM, McCubrey JA, Renegar RH, et al. 2008. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 68:7864–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dellago H, Preschitz-Kammerhofer B, Terlecki-Zaniewicz L, Schreiner C, Fortschegger K, et al. 2013. High levels of oncomiR-21 contribute to the senescence-induced growth arrest in normal human cells and its knock-down increases the replicative lifespan. Aging Cell 12:446–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tan WSD, Shen HM, Wong WSF. 2019. Dysregulated autophagy in COPD: a pathogenic process to be deciphered. Pharmacol. Res. 144:1–7 [DOI] [PubMed] [Google Scholar]
- 89.Fujii S, Hara H, Araya J, Takasaka N, Kojima J, et al. 2012. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology 1:630–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Faraonio R, Salerno P, Passaro F, Sedia C, Iaccio A, et al. 2012. A set of miRNAs participates in the cellular senescence program in human diploid fibroblasts. Cell Death Differ. 19:713–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hubbard AK, Rothlein R. 2000. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic. Biol. Med. 28:1379–86 [DOI] [PubMed] [Google Scholar]
- 92.Effenberger T, von der Heyde J, Bartsch K, Garbers C, Schulze-Osthoff K, et al. 2014. Senescence-associated release of transmembrane proteins involves proteolytic processing by ADAM17 and microvesicle shedding. FASEB J. 28:4847–56 [DOI] [PubMed] [Google Scholar]
- 93.Schumacher N, Meyer D, Mauermann A, von der Heyde J, Wolf J, et al. 2015. Shedding of endogenous interleukin-6 receptor (IL-6R) is governed by a disintegrin and metalloproteinase (ADAM) proteases while a full-length IL-6R isoform localizes to circulating microvesicles. J. Biol. Chem. 290:26059–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rose-John S, Heinrich PC. 1994. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem. J. 300(Part 2):281–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, et al. 2013. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 498:371–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Majumdar R, Tavakoli Tameh A, Parent CA. 2016. Exosomes mediate LTB4 release during neutrophil chemotaxis. PLOS Biol. 14:e1002336. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 97.Kolonics F, Kajdacsi E, Farkas VJ, Veres DS, Khamari D, et al. 2020. Neutrophils produce proinflammatory or anti-inflammatory extracellular vesicles depending on the environmental conditions. J. Leukoc. Biol. 109:793–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mesri M, Altieri DC. 1999. Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a JNK1 signaling pathway. J. Biol. Chem. 274:23111–18 [DOI] [PubMed] [Google Scholar]
- 99.Eken C, Martin PJ, Sadallah S, Treves S, Schaller M, Schifferli JA. 2010. Ectosomes released by polymorphonuclear neutrophils induce a MerTK-dependent anti-inflammatory pathway in macrophages. J. Biol. Chem. 285:39914–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cumpelik A, Ankli B, Zecher D, Schifferli JA. 2016. Neutrophil microvesicles resolve gout by inhibiting C5a-mediated priming of the inflammasome. Ann. Rheum. Dis. 75:1236–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barnes PJ. 2004. Alveolar macrophages as orchestrators of COPD. COPD 1:59–70 [DOI] [PubMed] [Google Scholar]
- 102.De Cunto G, Cavarra E, Bartalesi B, Lungarella G, Lucattelli M. 2020. Alveolar macrophage phenotype and compartmentalization drive different pulmonary changes in mouse strains exposed to cigarette smoke. COPD 17:429–43 [DOI] [PubMed] [Google Scholar]
- 103.Taylor AE, Finney-Hayward TK, Quint JK, Thomas CM, Tudhope SJ, et al. 2010. Defective macrophage phagocytosis of bacteria in COPD. Eur. Respir. J. 35:1039–47 [DOI] [PubMed] [Google Scholar]
- 104.Cordazzo C, Petrini S, Neri T, Lombardi S, Carmazzi Y, et al. 2014. Rapid shedding of proinflammatory microparticles by human mononuclear cells exposed to cigarette smoke is dependent on Ca2+ mobilization. Inflamm. Res. 63:539–47 [DOI] [PubMed] [Google Scholar]
- 105.Neri T, Armani C, Pegoli A, Cordazzo C, Carmazzi Y, et al. 2011. Role of NF-κB and PPAR-γ in lung inflammation induced by monocyte-derived microparticles. Eur. Respir. J. 37:1494–502 [DOI] [PubMed] [Google Scholar]
- 106.Qu Y, Franchi L, Nunez G, Dubyak GR. 2007. Nonclassical IL-1 β secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J. Immunol. 179:1913–25 [DOI] [PubMed] [Google Scholar]
- 107.Ye C, Li H, Bao M, Zhuo R, Jiang G, Wang W. 2020. Alveolar macrophage-derived exosomes modulate severity and outcome of acute lung injury. Aging 12:6120–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Richmond BW, Mansouri S, Serezani A, Novitskiy S, Blackburn JB, et al. 2020. Monocyte-derived dendritic cells link localized secretory IgA deficiency to adaptive immune activation in COPD. Mucosal Immunol. 14:431–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sethi S, Murphy TF. 2008. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N. Engl. J. Med. 359:2355–65 [DOI] [PubMed] [Google Scholar]
- 110.Shimizu K, Yoshii Y, Morozumi M, Chiba N, Ubukata K, et al. 2015. Pathogens in COPD exacerbations identified by comprehensive real-time PCR plus older methods. Int. J. Chron. Obstruct. Pulmon. Dis. 10:2009–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Spencer S, Calverley PM, Burge PS, Jones PW. 2004. Impact of preventing exacerbations on deterioration of health status in COPD. Eur. Respir. J. 23:698–702 [DOI] [PubMed] [Google Scholar]
- 112.Kanner RE, Anthonisen NR, Connett JE, Lung Health Study Res. Group. 2001. Lower respiratory illnesses promote FEV1 decline in current smokers but not ex-smokers with mild chronic obstructive pulmonary disease: results from the lung health study. Am. J. Respir. Crit. Care Med. 164:358–64 [DOI] [PubMed] [Google Scholar]
- 113.Sarkar A, Mitra S, Mehta S, Raices R, Wewers MD. 2009. Monocyte derived microvesicles deliver a cell death message via encapsulated caspase-1. PLOS ONE 4:e7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim JH, Lee J, Park J, Gho YS. 2015. Gram-negative and Gram-positive bacterial extracellular vesicles. Semin. Cell Dev. Biol. 40:97–104 [DOI] [PubMed] [Google Scholar]
- 115.Brown L, Wolf JM, Prados-Rosales R, Casadevall A. 2015. Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 13:620–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schwechheimer C, Kuehn MJ.2015.Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat. Rev. Microbiol. 13:605–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Park KS, Choi KH, Kim YS, Hong BS, Kim OY, et al. 2010. Outer membrane vesicles derived from Escherichia coli induce systemic inflammatory response syndrome. PLOS ONE 5:e11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim MR, Hong SW, Choi EB, Lee WH, Kim YS, et al. 2012. Staphylococcus aureus-derived extracellular vesicles induce neutrophilic pulmonary inflammation via both Th1 and Th17 cell responses. Allergy 67:1271–81 [DOI] [PubMed] [Google Scholar]
- 119.Lommatzsch M, Cicko S, Muller T, Lucattelli M, Bratke K, et al. 2010. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 181:928–34 [DOI] [PubMed] [Google Scholar]
- 120.Mohsenin A, Blackburn MR. 2006. Adenosine signaling in asthma and chronic obstructive pulmonary disease. Curr. Opin. Pulm. Med. 12:54–59 [DOI] [PubMed] [Google Scholar]
- 121.Barnes PJ. 2008. Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 8:183–92 [DOI] [PubMed] [Google Scholar]
- 122.Gulinelli S, Salaro E, Vuerich M, Bozzato D, Pizzirani C, et al. 2012. IL-18 associates to microvesicles shed from human macrophages by a LPS/TLR-4 independent mechanism in response to P2X receptor stimulation. Eur. J. Immunol. 42:3334–45 [DOI] [PubMed] [Google Scholar]
- 123.Eltom S, Dale N, Raemdonck KR, Stevenson CS, Snelgrove RJ, et al. 2014. Respiratory infections cause the release of extracellular vesicles: implications in exacerbation of asthma/COPD. PLOS ONE 9:e101087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Beasley V, Joshi PV, Singanayagam A, Molyneaux PL, Johnston SL, Mallia P. 2012. Lung microbiology and exacerbations in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 7:555–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.MacDonald IA,Kuehn MJ.2012.Offense and defense: microbial membrane vesicles play both ways. Res. Microbiol. 163:607–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tan TT, Morgelin M, Forsgren A, Riesbeck K. 2007. Haemophilus influenzae survival during complement-mediated attacks is promoted by Moraxella catarrhalis outer membrane vesicles. J. Infect. Dis. 195:1661–70 [DOI] [PubMed] [Google Scholar]
- 127.Schaar V, Nordstrom T, Morgelin M, Riesbeck K. 2011. Moraxella catarrhalis outer membrane vesicles carry β-lactamase and promote survival of Streptococcus pneumoniae and Haemophilus influenzae by inactivating amoxicillin. Antimicrob. Agents Chemother. 55:3845–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Olaya-Abril A, Prados-Rosales R, McConnell MJ, Martin-Pena R, Gonzalez-Reyes JA, et al. 2014. Characterization of protective extracellular membrane-derived vesicles produced by Streptococcus pneumoniae. J. Proteom. 106:46–60 [DOI] [PubMed] [Google Scholar]
- 129.Shaper M, Hollingshead SK, Benjamin WH Jr., Briles DE. 2004. PspA protects Streptococcus pneumoniae from killing by apolactoferrin, and antibody to PspA enhances killing of pneumococci by apolactoferrin. Infect. Immun. 72:5031–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jhelum H, Sori H, Sehgal D. 2018. A novel extracellular vesicle-associated endodeoxyribonuclease helps Streptococcus pneumoniae evade neutrophil extracellular traps and is required for full virulence. Sci. Rep. 8:7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Codemo M, Muschiol S, Iovino F, Nannapaneni P, Plant L, et al. 2018. Immunomodulatory effects of pneumococcal extracellular vesicles on cellular and humoral host defenses. mBio 9:e00559–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vestbo J, Prescott E, Lange P. 1996. Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease morbidity. Copenhagen City Heart Study Group. Am. J. Respir. Crit. Care Med. 153:1530–35 [DOI] [PubMed] [Google Scholar]
- 133.Hoenderdos K, Condliffe A. 2013. The neutrophil in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 48:531–39 [DOI] [PubMed] [Google Scholar]
- 134.Shao MX, Nadel JA. 2005. Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells via a cascade involving protein kinase C, reactive oxygen species, and TNF-α-converting enzyme. J. Immunol. 175:4009–16 [DOI] [PubMed] [Google Scholar]
- 135.Shao S, Fang H, Zhang J, Jiang M, Xue K, et al. 2019. Neutrophil exosomes enhance the skin autoinflammation in generalized pustular psoriasis via activating keratinocytes. FASEB J. 33:6813–28 [DOI] [PubMed] [Google Scholar]
- 136.Railwah C, Lora A, Zahid K, Goldenberg H, Campos M, et al. 2020. Cigarette smoke induction of S100A9 contributes to chronic obstructive pulmonary disease. Am. J. Physiol. Lung. Cell. Mol. Physiol. 319:L1021–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kang JH, Hwang SM, Chung IY. 2015. S100A8, S100A9 and S100A12 activate airway epithelial cells to produce MUC5AC via extracellular signal-regulated kinase and nuclear factor-κB pathways. Immunology 144:79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Prieto D, Sotelo N, Seija N, Sernbo S, Abreu C, et al. 2017. S100-A9 protein in exosomes from chronic lymphocytic leukemia cells promotes NF-κB activity during disease progression. Blood 130:777–88 [DOI] [PubMed] [Google Scholar]
- 139.McDonald MN, Wouters EFM, Rutten E, Casaburi R, Rennard SI, et al. 2019. It’s more than low BMI: prevalence of cachexia and associated mortality in COPD. Respir. Res. 20:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Iyer AS, Holm KE, Bhatt SP, Kim V, Kinney GL, et al. 2019. Symptoms of anxiety and depression and use of anxiolytic-hypnotics and antidepressants in current and former smokers with and without COPD—a cross sectional analysis of the COPDGene cohort. J. Psychosom. Res. 118:18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Agusti A, Edwards LD, Rennard SI, MacNee W, Tal-Singer R, et al. 2012. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLOS ONE 7:e37483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tan DBA, Armitage J, Teo TH, Ong NE, Shin H, Moodley YP. 2017. Elevated levels of circulating exosome in COPD patients are associated with systemic inflammation. Respir. Med. 132:261–64 [DOI] [PubMed] [Google Scholar]
- 143.Li JJ, Wang B, Kodali MC, Chen C, Kim E, et al. 2018. In vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J. Neuroinflamm. 15:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chan BD, Wong WY, Lee MM, Cho WC, Yee BK, et al. 2019. Exosomes in inflammation and inflammatory disease. Proteomics 19:e1800149. [DOI] [PubMed] [Google Scholar]
- 145.Deng W, Tang T, Hou Y, Zeng Q, Wang Y, et al. 2019. Extracellular vesicles in atherosclerosis. Clin. Chim. Acta 495:109–17 [DOI] [PubMed] [Google Scholar]
- 146.Chitti SV, Fonseka P, Mathivanan S. 2018. Emerging role of extracellular vesicles in mediating cancer cachexia. Biochem. Soc. Trans. 46:1129–36 [DOI] [PubMed] [Google Scholar]
- 147.Bujarski S, Parulekar AD, Sharafkhaneh A, Hanania NA. 2015. The asthma COPD overlap syndrome (ACOS). Curr. Allergy Asthma Rep. 15:7. [DOI] [PubMed] [Google Scholar]
- 148.Page C, Cazzola M. 2014. Bifunctional drugs for the treatment of asthma and chronic obstructive pulmonary disease. Eur. Respir. J. 44:475–82 [DOI] [PubMed] [Google Scholar]
- 149.Hough KP, Deshane JS. 2019. Exosomes in allergic airway diseases. Curr. Allergy Asthma Rep. 19:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Huang F, Jia H, Zou Y, Yao Y, Deng Z. 2020. Exosomes: an important messenger in the asthma inflammatory microenvironment. J. Int. Med. Res. 48:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kulshreshtha A, Ahmad T, Agrawal A, Ghosh B. 2013. Proinflammatory role of epithelial cell–derived exosomes in allergic airway inflammation. J. Allergy Clin. Immunol. 131:1194–1203.e14 [DOI] [PubMed] [Google Scholar]
- 152.Haj-Salem I, Plante S, Gounni AS, Rouabhia M, Chakir J. 2018. Fibroblast-derived exosomes promote epithelial cell proliferation through TGF-β2 signalling pathway in severe asthma. Allergy 73:178–86 [DOI] [PubMed] [Google Scholar]
- 153.Canas JA, Sastre B, Rodrigo-Munoz JM, Fernandez-Nieto M, Barranco P, et al. 2018. Eosinophil-derived exosomes contribute to asthma remodelling by activating structural lung cells. Clin. Exp. Allergy 48:1173–85 [DOI] [PubMed] [Google Scholar]
- 154.Mazzeo C, Canas JA, Zafra MP, Rojas Marco A, Fernandez-Nieto M, et al. 2015. Exosome secretion by eosinophils: a possible role in asthma pathogenesis. J. Allergy Clin. Immunol. 135:1603–13 [DOI] [PubMed] [Google Scholar]
- 155.Canas JA, Sastre B, Mazzeo C, Fernandez-Nieto M, Rodrigo-Munoz JM, et al. 2017. Exosomes from eosinophils autoregulate and promote eosinophil functions. J. Leukoc. Biol. 101:1191–99 [DOI] [PubMed] [Google Scholar]
- 156.Woodruff PG, Barr RG, Bleecker E, Christenson SA, Couper D, et al. 2016. Clinical significance of symptoms in smokers with preserved pulmonary function. N. Engl. J. Med. 374:1811–21 [DOI] [PMC free article] [PubMed] [Google Scholar]