

Abstract
Spondylometaepiphyseal dysplasia short limb-abnormal calcification type (SMED-SL/AC) is a rare autosomal recessive disorder. It is a severe dwarfism syndrome with a characteristic feature of progressive calcification of epiphyseal and other cartilaginous tissues. It is caused by pathogenic variants in the DDR2 gene encoding the discoidin domain receptor tyrosine kinase 2. Thus far, 37 cases and 8 pathogenic variants have been reported. Most of the reported cases are of Middle Eastern and Puerto Rican origins. Only one Turkish case has been reported previously with a novel truncating variant p.(R489*). Here, we report 2 new cases, 1 with a novel variant p.(S311G) and 1 with a splice site variant c.2283+1G>A. In addition, we reviewed a previously reported case, and sequencing of stored DNA revealed the recently reported nonsense variant p.(R489*) as the underlying cause. Therefore, our data increase the number of SMED-SL/AC Turkish patients with molecular results to 4. Furthermore, we compared the features of Turkish patients with other reported cases and expanded the characteristics of the disorder with new features such as triventricular hydrocephalus, intracranial hemorrhage, hypopigmentation of hair, dry and scaly skin, arthralgia, and hypocalcemia. We also compared the pathogenic variants of Turkish patients with other variants, aiming to explain the mechanism leading to a more severe and early fatal course in Turkish patients.
Keywords: Spondylometaepiphyseal dysplasia, Short limb-abnormal calcification type, DDR2, Progressive calcification, Skeletal dysplasia
Introduction
Spondylometaepiphyseal dysplasia short limb-abnormal calcification type (SMED-SL/AC, OMIM #271665) is an early onset, severe, and rare form of skeletal dysplasia. It is an autosomal recessive disorder characterized by prenatal onset of short stature and short limbs with short hands and feet, narrow thoracic cage, and short ribs. Progressive calcification and destruction of distal ends of tubular bones, rib cartilages and several other cartilaginous tissues are characteristic features of the disorder and usually the onset occurs around the age of 1 year [Borochowitz et al., 1993; Langer et al., 1993].
SMED-SL/AC has been observed mainly in Middle Eastern and Puerto Rican communities. So far 37 cases have been reported [Borochowitz et al., 1993; Langer et al., 1993; Al-Gazali et al., 1996; Fano et al., 2001; Bargal et al., 2009; Dias et al., 2009; Smithson et al., 2009; Tüysüz et al., 2009; Ali et al., 2010; Rozovsky et al., 2011; Al-Kindi et al., 2014; Mansouri et al., 2016; Ürel-Demir et al., 2018]. It is usually a severe condition which may be fatal due to atlantoaxial instability and spinal cord compression or recurrent bronchopulmonary infections [Langer et al., 1993; Al-Gazali et al., 1996]. It has been found that homozygous and biallelic loss-of-function variants in the discoidin domain receptor 2 (DDR2, MIM # 191311) gene result in this severe dwarfism syndrome Bargal et al. [2009]. Eight pathogenic variants have been reported so far with only one found in a Turkish child with an early fatal phenotype Ürel-Demir et al. [2018].
In this study, we present 2 new cases with one of them harbouring a novel variant. In addition, we reviewed a previously reported case by Tüysüz et al. [2009] who was found to harbour the same identified truncating pathogenic variant of the other recently reported Turkish child reported by Ürel-Demir et al. [2018]. We defined the course and features of this disorder in Turkish patients. The findings further highligt the importance of delineating the molecular mechanism leading to this phenotype.
Materials and Methods
All patients were evaluated by a medical geneticist and their X-ray images and magnetic resonance imagings (MRI) were obtained and evaluated.
Genomic DNA was isolated from the peripheral blood leukocytes. Exon-specific primers of DDR2 were designed using Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/) and used to amplify the coding exons 3–18 and their flanking regions (listed in online suppl. Table 1; for online suppl. material, see www.karger.com/doi/10.1159/000517848). Amplification was done with 1 U of Taq polymerase (Qiagen, GmbH), 0.2 m of each of the dNTPs, 1X PCR reaction buffer, 1.5 mM MgCl2, 5 pmol of each forward and reverse primers, and 50 ng of genomic DNA template in 25 µL reactions. The PCR was performed by an initial 5 min denaturation at 95°C followed by 40 cycles of 95°C for 30 s, 58–62°C for 35 s and 72°C for 45 s with a final elongation at 72°C for 10 min.
Sanger sequencing was performed using dideoxy method by fluorescent automated sequencing on the ABI 3130xl analyzer (Applied Biosystems, Foster City, CA, USA). Variants are designated on the coding DDR2 sequence with reference to Genbank accession number NM_001014796.1 with the A of the initiation codon ATG as number +1. Sanger sequencing of case 3 was done from his stored DNA after his death.
The missense variant, p.(S311G), has been introduced using DDR2-S311G-F: CTACTTCCGCTCTGAAGCCGGTGAGTGGGAACCTAATG and DDR2-S311G-R: CATTAGGTTCCCACTCACCGGCTTCAGAGCGGAAGTAG as mutagenesis primers to the HA-tagged DDR2 cDNA in mammalian expression vector as described previously [Ali et al., 2010; Al-Kindi et al., 2014]. The correct introduction of the variant has been confirmed by Sanger DNA Sequencing.
For immunocytochemistry, HeLa cells were grown on sterile coverslips in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS (Invitrogen) and 100 U per mL penicillin/streptomycin at 37°C with 5% CO2. 1 ug of HA-tagged wild-type and mutant plasmids were cotransfected with the plasma membrane marker GFP-H-Ras plasmid using FuGENE HD transfection reagent. 24 h after transfection, cells were fixed with methanol at −20°C for 5 min. The fixed cells were washed with PBS and blocked in 1% BSA for 30 min. After blocking, the cover slips were washed with PBS and incubated with mouse monoclonal anti-HA antibody (1 in 200 dilution in 1% BSA, Cell Signaling Technologies, Cat. No. 2368) and rabbit polyclonal anti-calnexin antibody (1 in 50 dilution in 1% BSA, Cell Signaling Technologies, Cat. No. 2679) for 1 h at room temperature. The cells were co-stained with anti-calnexin antibodies to visualize the ER network. The cells were washed with PBS and incubated with Alexa 555 conjugated rabbit anti -mouse IgG (Cell Signaling Technologies, Cat. No. 4408S, 1 in 200 dilution in 1% BSA), Alexa Fluor 647 conjugated goat anti-rabbit IgG (1: 200; Invitrogen) or Alexa 488 conjugated goat anti-rabbit IgG (1 in 200 dilution in 1% BSA, Cell Signaling Technologies, Cat. No. 4413S) for 30 min at room temperature. Following antibody incubation, the cover slips were washed several times in PBS and mounted using DACO fluorescent mounting medium. Images were captured with a ×100 oil immersion objective on a Nikon Eclipse system (Nikon Instruments Inc., Tokyo, Japan) equipped with FITC and TRITC filters. The represented images are single sections in the z-plane. The images were pseudocoloured, colour enhanced, and merged using IMAGEJ software [Ali et al., 2010; Al-Kindi et al., 2014].
Clinical Presentation and Results
Case 1
Our first case was a newborn girl born at 33–34 gestational weeks following cesarian section. The birth weight was 1,510 g (10th percentile), length was 42 cm (10th percentile), and head circumference was 29 cm (10th percentile). On her first examination, she was 5 days old and presented with predominantly rhisomelic short extremities, narrow thoracic cage, narrow biparietal diameter of her forehead, wide anterior fontanelle, frontal bossing, depressed nasal root, hypertelorism, propitosis, and a broad nasal tip with anteverted wide nostrils. She was the second living child from the fourth pregnancy of a consanguineous couple from Northern Anatolia. The couple's first child was a stillborn in the seventh month of pregnancy but was not investigated, and their second child was born at term, died 2 hours after birth, and was reported to have short extremities but also was not investigated. Antenatal ultrasonography of the current affected girl revealed short extremities, narrow thoracic cage and polyhydramniosis on 22nd week of pregnancy. Chromosome analysis and FGFR3 gene sequencing from amniotic fluid were unremarkable. After birth, she was further investigated and no cranial, abdominal, or cardiac abnormalities were detected. On her first X-ray, short ribs, narrow thoracic cage, short and dumbbell-shaped femora and humeri, and metaphyseal flaring of other long bones were detected. On her second visit when she was 1 month old, her hands and feet had become shorter and broader, her fingers and toes got relatively shorter and tapered, and she had macrocephaly. During 1 year follow-up, her gross motor development was delayed, no sitting was achieved. Passive movement of the big joints like knees were painful during examination. Her bone survey revealed diffuse microcalcifications and diffuse moth-eaten destructive appearance of all epiphyses of long bones and costal cartilages. Short thickened long bones and short ribs, metaphyseal flaring, and platyspondyly were prominent. Metabolic testing for rhizomelic chondrodysplasia punctata type 1 (RCDP1; OMIM # 215100) was normal. Because of platyspondyly, typical calficified severe epiphyseal and metaphyseal involvement, and small trident hands and small short feet, the clinical diagnosis of SMED-SL/AC was made. Triventricular hydrocephalus and spinal cord compression due to foramen magnum stenosis developed during her follow-up, she was operated on when she was 18 months old. After decompression and ventriculoperitoneal shunt operation, cervical MRI detected an anteriorly displaced odontoid process and syrinx image which resulted in an atlantoaxial subluxation and compressive myelopathy. Molecular analysis including Sanger DNA sequencing of all the coding regions and splice sites of the DDR2 gene revealed a homozygous splice site variant affecting the donor splice site in intron 17 (c.2283+1G>A). During her visits, her skin was dry and scaly. As she grew older, her hair became hypopigmented, dry and brittle. She had a hoarse high-pitched voice, and her audiometric tests revealed bilateral sensoryneural hearing loss and serous otitis media requiring tympanocentesis and hearing aids. Assisted sitting was achieved at 2.5 years of age. She was prone to recurrent lower airway infections and was hospitalized several times. She suddenly passed away due to intracranial hemorrhage when she was 3.5 years old. She had no history of bleeding diathesis (shown in Fig. 1 and Fig. 2).
Fig. 1.
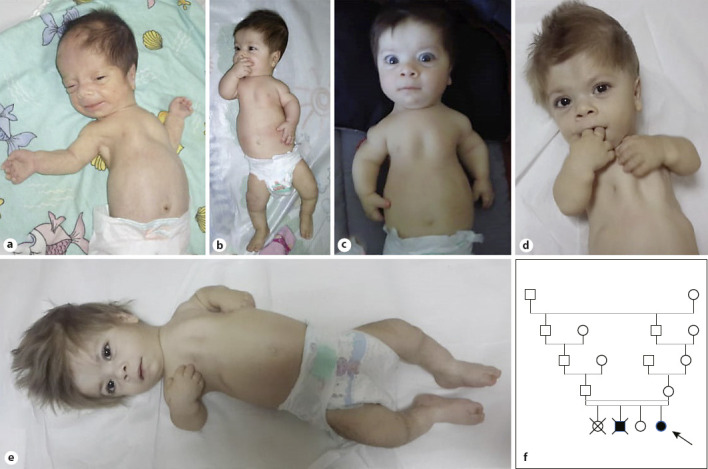
Photographs of case 1. a 1.5 months, b 8 months, c 9 months, d 2 years and 3 months, and e 2 years and 5 months of age. f Pedigree of case 1.
Fig. 2.
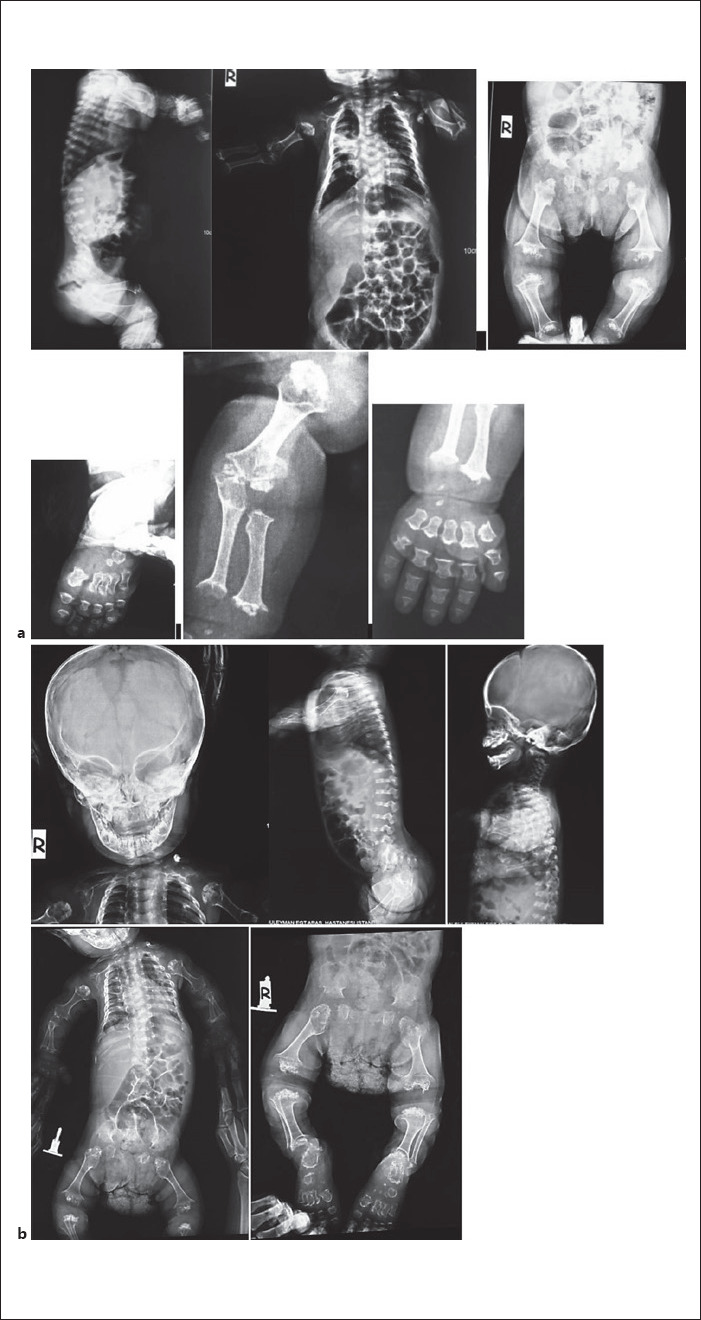
X-rays of case 1. a 1 year and b 1 year and 8 months of age.
Case 2
This female patient was first examined when she was nine and a half months old, due to short extremities, hypotonia, and motor delays. She was the third child of consanguineous parents from Southern Anatolia. No other affected relatives were reported. She was born at the 41st gestational week, her birth weight was 2,850 g (10th percentile), birth height was 47 cm (3rd percentile), and birth head circumference was 35 cm (75th percentile). She was hospitalized for 14 days due to hypocalcemia. During her newborn period, one calcificied subcutanoues nodule was detected on ultrasonography of her left forearm. Renal and transfontanelle ultrasonography were unremarkable. Hip ultrasonography at 1 month of age revealed irregular borders of acetabuli and diffuse increased hyperechogenicity of the femoral head and trochanteric processes mimicking an accumulative osteopathy. She had predominantly rhizomelic short extremities, narrow thoracic cage, micromelia, trident hands, short fusiform fingers, bowed legs, macrocephaly, frontal bossing, flat face with depressed nasal root and bridge, anteverted wide nostrils, and hypertelorism with long palpebral fissures. FGFR3 gene analysis was normal. Her bone survey revealed metaphyseal and epiphyseal irregularities in long bones, short and thick short bones, square shaped iliac bones, with a short pelvic rim and flattened acetabular roofs and platyspondyly. During follow-up, her hypotonia was significant; she achieved head control at 1 year of age, and when she was 14 months old, diffuse calcifications of epiphyses and costal cartilages were detected on X-ray radiographs. Upon this finding she was diagnosed as SMED-SL/AC. Sanger sequencing revealed a novel homozygous single nucleotide substitution (NM_001014796.1:c.931A>G) in the DDR2 gene leading to a missense variant, p.(S311G). However, this residue is not conserved and was predicted as tolerated and likely benign in databases. The variant was heterozygous in both parents. On her brain and spinal MRI scanning, mild hypomyelination, narrow posterior cranial fossa with decreased anterior and posterior subarachnoid space, severe narrowing of the foramen magnum, and myelopathic changes in the spinal cord were detected. C1 laminectomy operation for decompression was done when she was 18 months old. After this intervention she was able to sit with assistance at the age of 19 months. Her medical condition progressively deteriorated, but she did not receive enough medical care. She was hospitalized and intubated because of respiratory insufficiency and pneumonia when she was 5 years old. Her thoracic CT scan was consistent with diffuse infection and bronchial wall thickening, mucus plaques due to aspiration pneumonia, also calcifications of tracheal and main bronchi were significant. Nasogastric tube feeding was started. Follow-up brain and spinal MRI revealed low cerebral volume, delayed myelination of deep white matter, encephalomalacia and gliotic changes of posterior parts of the cerebral hemispheres, refusion of C1 vertebra and restenosis of foramen magnum, increased anterior angulation of odontoid process causing craniocervical stenosis and spinal cord compression, and myelomalacia in the central part of spinal cord. A second laminectomy operation was performed for decompression of the spinal cord and craniocervical stenosis. Following the operation, she stayed in an intensive care unit for 9 months. She died at 5 years and 10 months after respiratory failure secondary to pneumonia (Fig. 3 and Fig. 4).
Fig. 3.
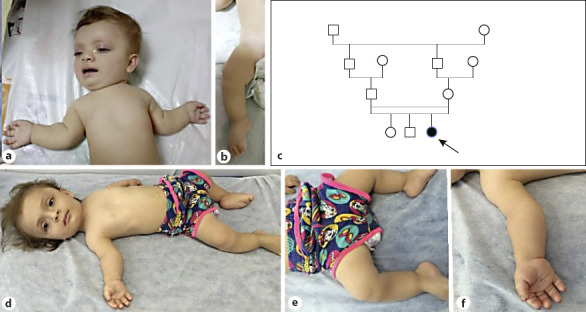
Photographs of case 2. a, b 9.5 months and d, f 4.5 years of age. c Pedigree of the case.
Fig. 4.
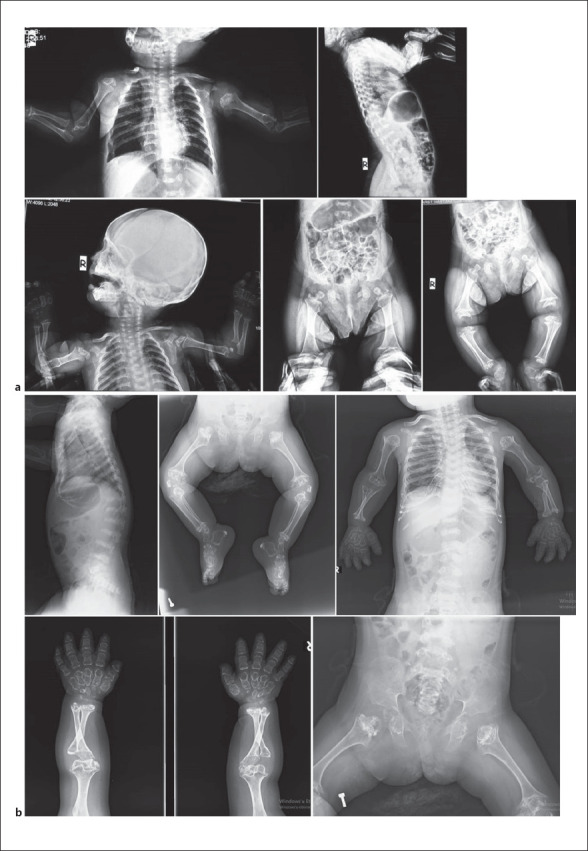
X-rays of case 2. a 9.5 months and b 4.5 years of age.
Case 3
Our third case was reported previously with his clinical findings by Tüysüz et al. [2009]. The boy was from Northern Anatolia. Since the publication, we were able to establish the molecular diagnosis, and we compared the clinical findings with other cases. A homozygous nonsense variant in DDR2 NM_001014796.1:c.1465C>T (p.R489*) was detected (Fig. 5).
Fig. 5.
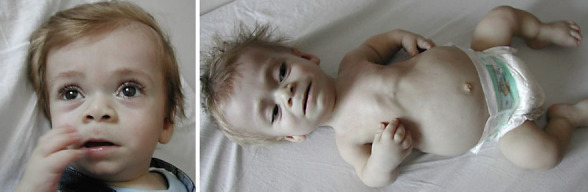
Photographs of case 3.
The Pathogenic Variants Found in SMED-SL/C Turkish Patients Involve Different DDR2 Domains
As shown in Figure 6, the identified pathogenic variants involve different domains in DDR2. The novel missense variant, p.(S311G), involves an amino acid change on discoidin-like domain of extracellular domain of the protein, while p.(R489*) nonsense variant and IVS17+1G>A splice site variant involve the intracellular domains of the protein.
Fig. 6.
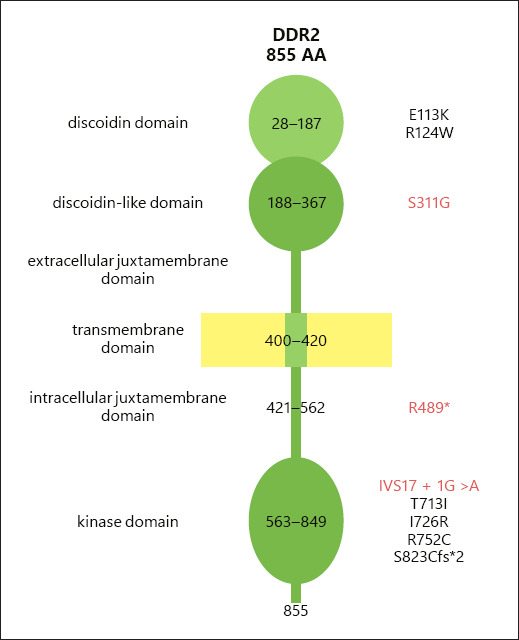
Schematic structure of DDR2 protein domains and amino acid residues written on each domain. The positions of disease-causing variant are written on the left. The variants of Turkish patients are written in red.
The Missense Variant p.(S311G) Seems to Traffic to the Plasma Membrane
We evaluated the effects of the missense variant p.(S311G) on the subcellular localization of DDR2 in HeLa cells using confocal immunoflorescence microscopy as described previously [Ali et al., 2010; Al-Kindi et al., 2014]. After inspecting many transfected cells, it was possible to conclude that the missense variant did not change the subcellular localization of DDR2 (i.e., the mutant protein localized to the plasma membrane similar to wild type as shown in the representative confocal images) (Fig. 7). Since this variant is affecting the extracellular discoidin domain (Fig. 6), it is possible that it might interfere with collagen binding in similar fashion to p.(E113K) reported previously [Ali et al., 2010].
Fig. 7.
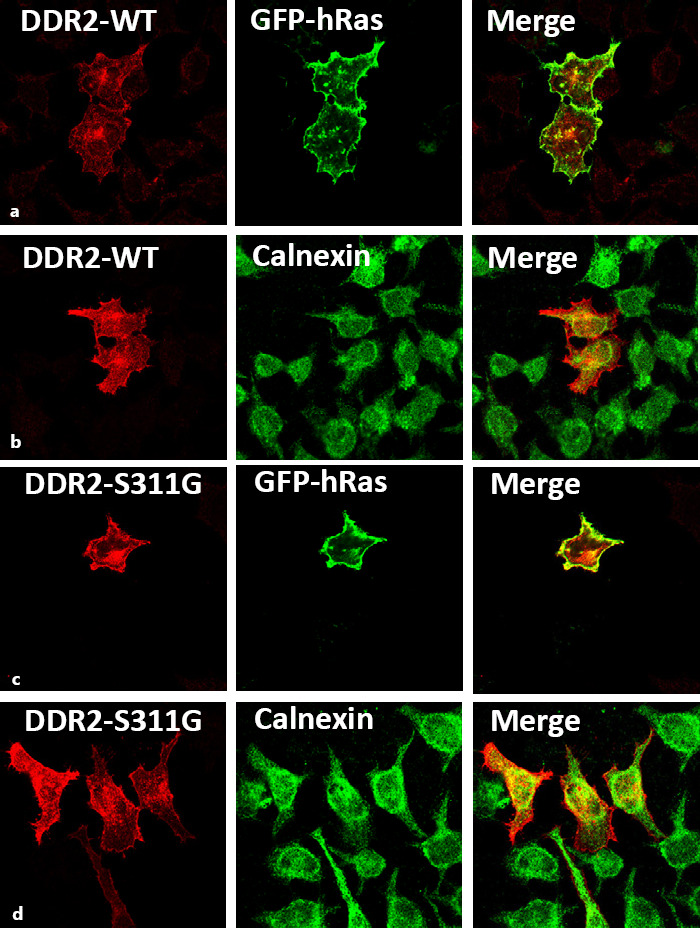
Confocal microscopic images showing localization of DDR2-wild type and the S311G mutant in HeLa cells. Representative panel of WT-DDR2 (a, b) and DDR2-S311G (c, d) localizaton in transfected HeLa cells. DDR2-WT and S311G exhibit similar localization patterns as evident by its colocalization with the plasma membrane-specific GFP-tagged hRas and ER-specific marker, Calnexin.
Discussion and Conclusions
Although it shows great variability, the SMED-SL/AC phenotype first appears antenatally with fetal short limbs on ultrasonography. Disproportionate short stature with short limbs, narrow chest, short fingers, macrocephaly with a broad forehead, and wide, flat nasal base and bridge with wide anteverted nostrils are the typical features in the newborn period and first months of life; thus, it is difficult to differentiate from other short stature dwarfism syndromes. Although platyspondyly, which could be recognized as early as the intrauterine period, gives a clue about a spondyloepiphyseal or spondylometaepiphyseal dysplasia, typical diagnostic features as precoccious chondral calcifications especially of ribs and epiphyses with widespread epiphyseal stippling, metaphyseal flaring and short and bowed long bones usually come to attention after the first year of life [Bargal et al., 2009; Rozovsky et al., 2011]. While phenotype and X-ray findings may resemble RCDP1, severe platyspondyly with typical face and short hands differentiate the disorder from RCDP1 (Table 1).
Table 1.
Features of Turkish patients compared to other reported cases
| Case 1 | Case 2 | Case3 |
Case of |
Reported cases (n = 35) | |
|---|---|---|---|---|---|
| Tüysüz et al. [2009] | Ürel-Demir et al. [2018] | ||||
| Antenatal findings | + | − | + | − | 4 |
| Shortening of extremities | + | + | + | + | 35 |
| Short broad hands | + | + | + | + | 35 |
| Short puffy broad fingers | + | + | + | + | 25 |
| Bowed legs | + | + | + | − | 19 |
| Narrow thoracic cage | + | + | + | + | 26 |
| Pectus excavatum | + | + | + | − | 12 |
| Respiratory problems | + | + | − | + | 20 |
| Macrocephaly | + | + | + | + | 15 |
| Large anterior fontanelle | + | + | + | − | 19 |
| Large broad face | + | + | + | + | 15 |
| Hypertelorism | + | + | + | + | 27 |
| Wide anteverted nostrils | + | + | + | + | 21 |
| Long and wide philtrum | + | − | + | + | 21 |
| Midfacial hypoplasia | + | + | + | + | 26 |
| Short flat nose | + | + | + | + | 28 |
| Abnormal external ear | − | − | + | − | 0 |
| Short neck | + | + | + | + | 12 |
| Neck hyperextension | + | − | − | − | 9 |
| Peculiar hoarse voice | + | − | − | + | 6 |
| Delayed motor development | + | + | + | + | 18 |
| Hypotonia | + | + | + | + | 13 |
| Painful joints | + | − | − | − | 0 |
| Short and broad long bones | + | + | + | + | 35 |
| Short and broad round bones | + | + | + | + | 35 |
| Rib abnormalities | + | + | + | + | 35 |
| Costochondral calcifications | + | + | + | + | 21 |
| Epiphyseal and metaphyseal abnormalities | + | + | + | + | 35 |
| Platyspondyly | + | + | + | + | 35 |
| C1–C2 instability | + | − | + | − | 19 |
| Scoliosis | + | + | − | + | 12 |
| Falx cerebri calcifications | − | − | − | + | 4 |
| Foramen magnum stenosis | + | + | + | + | 11 |
| Triventricular hydrocephalus | + | − | − | − | 0 |
| Spinal cord compression | + | + | + | + | 11 |
| Otitis media | + | − | − | − | 3 |
| Optic atrophy-anisocoria | − | − | − | − | 4 |
| Hearing loss | + | − | − | − | 2 |
| Facial nerve palsy | − | − | − | + | 1 |
| Hypocalcemia during newborn period | − | + | − | − | 0 |
| Calcification of thyroid, hyoid, trachea, main bronchi | − | + | − | − | 10 |
| Dry scaly skin | + | − | − | − | 0 |
| Hypopigmented hair | + | − | + | − | 0 |
Until now, only 4 cases were reported with antenatal findings. Borochowitz et al. [1993] reported a patient with antenatal ultrasonographic findings, polyhydramniosis and short extremities detected on 25th gestational week. Rozovsky et al. [2011] identified platyspondyly by X-ray film on 30th week of gestation, and Ali et al. [2010] reported 2 siblings with antenatal ultrasonographic findings of short extremities. Two of our patients demonstrated antenatal ultrasonographic findings, both had short extremities and one had polyhydramniosis and narrow thoracic cage additionaly detected on a very early week (22nd gestational week). Therefore, detailed fetal ultrasonography could identify the first signs of the disorder as early as the 22nd week of gestation as in our patient. Polyhydramniosis may be the result of inadequate swallowing and breathing activity of the fetus due to narrow thoracic cavity.
With our new cases, the number of patients reported increases to 39 (Table 2). Most of the patients reported are from Puerto Rico or from the Jerusalem region with additional cases from Egypt, Pakistan, Oman, United Arabic Emirates, Morocco, and Algeria with similar pathogenic variants, depicting a founder effect in the Middle Eastern and Northern African-Mediterranean region [Borochowitz et al., 1993; Langer et al., 1993; Al-Gazali et al., 1996; Bargal et al., 2009; Smithson et al., 2009; Ali et al., 2010; Rozovsky et al., 2011; Al-Kindi et al., 2014]. We have previously reported one Turkish case [Tüysüz et al., 2009] with only one additional patient reported by Ürel-Demir et al. [2018], and therefore, the new cases reported here bring the total Turkish cases to 4 patients from 4 different families (Table 2).
Table 2.
Comparison of disease severity, progress and mutations of the cases
| Author | Origin | Age of first calcification | Age of cord compression | Operations, age | Age of death | Cause of death | Mutation | |
|---|---|---|---|---|---|---|---|---|
| 1 | Borochowitz et al., 1993 | Sephardic Jewish, Egypt | 3.5 yrs | − | − | − | − | IVS17+1G>A¥ |
|
| ||||||||
| 2 | Sephardic Jewish, Iraq | 4 yrs | − | Thoracic widening, 4 yrs | 9 yrs | Cardiorespiratory failure | IVS17+1G>A¥ | |
|
| ||||||||
| 3 | Langer et al., 1993 | Puerto Rico | Newborn | − | − | 27 mo | Cardiorespiratory failure | − |
|
| ||||||||
| 4 | Puerto Rico | 2 yrs | − | − | 6 yrs | Sudden death during sleep | − | |
|
| ||||||||
| 5 | Puerto Rico | 15 mo | 37 mo | − | 39 mo | Sudden death/cord compression | − | |
|
| ||||||||
| 6 | Puerto Rico | 3 mo | − | − | − | − | − | |
|
| ||||||||
| 7 | ¥¥ | Puerto Rico | 27 mo | 27 mo | − | 29 mo | Pneumonia and cord compression | − |
|
| ||||||||
| 8 | Puerto Rico | Between 4 and 5 yrs | − | − | − | − | − | |
|
| ||||||||
| 9 | Puerto Rico | − | − | − | − | − | − | |
|
| ||||||||
| 10 | Na | 10 mo | − | − | − | − | − | |
|
| ||||||||
| 11 | Al Gazali et al., 2010¥¥¥ | Egypt | 2 yrs | − | − | 13 yrs | Respiratory failure | p.R752C |
|
| ||||||||
| 12 | Egypt | 1 year | 4 yrs | − | 8 yrs | Cord compression | p.R752C | |
|
| ||||||||
| 13 | Fano et al., 2001 | Hispanic | 10 mo | 15 mo | − | 21 mo | Sudden death/cord compression | − |
|
| ||||||||
| 14 | Smithson et al., 2009 | Pakistan | 5 yrs | − | − | − | − | p.T713I¥ |
|
| ||||||||
| 15 | Dias et al., 2009 | Northwestern Europe | − | 14 mo | − | 16 mo | Pneumonia and cord compression | − |
|
| ||||||||
| 16 | Northwestern Europe | − | 8 mo | − | 8 mo | Sudden death/cord compression | − | |
|
| ||||||||
| 17 | Bargal et al., 2009 | Jerusalem | After 8 yrs | After 3 yrs | Decompression, 10 yrs | − | − | p.R752C |
|
| ||||||||
| 18 | Jerusalem | After 8 yrs | After 3 yrs | Decompression, 11 yrs | − | − | p.R752C | |
|
| ||||||||
| 19 | Jerusalem | After 8 yrs | After 3 yrs | Decompression, 16 yrs | − | − | p.R752C | |
|
| ||||||||
| 20 | Jerusalem | After 8 yrs | After 3 yrs | − | − | − | p.R752C | |
|
| ||||||||
| 21 | Jerusalem | After 8 yrs | − | − | − | − | p.R752C | |
|
| ||||||||
| 22 | Jerusalem | After 8 yrs | − | − | − | − | p.R752C | |
|
| ||||||||
| 23 | Ali et al., 2010¥¥¥ | Pakistan | 6 yrs | − | − | − | − | p.E113K |
|
| ||||||||
| 24 | Pakistan | 8 yrs | − | − | − | − | p.E113K | |
|
| ||||||||
| 25 | Rozovsky et al., 2011 | Jerusalem | 16 yrs | 17 yrs | Decompression, 17 yrs | − | − | − |
|
| ||||||||
| 26 | Jerusalem | 1.5 yrs | 10 yrs | Decompression, 10 yrs | − | − | − | |
|
| ||||||||
| 27 | Jerusalem | − | − | − | − | − | − | |
|
| ||||||||
| 28 | Jerusalem | 6 yrs | 16 yrs | Decompression, 16 yrs | − | − | − | |
|
| ||||||||
| 29 | Jerusalem | − | 3.5 yrs | Decompression, 3.5 yrs | − | − | − | |
|
| ||||||||
| 30 | Jerusalem | − | − | − | − | − | − | |
|
| ||||||||
| 31 | Jerusalem | 6 yrs | 10 yrs | Decompression, 10 yrs | − | − | − | |
|
| ||||||||
| 32 | Algeria | 3 yrs | − | − | − | − | p.I726R¥ | |
|
| ||||||||
| 33 Al Kindi et al., 2014 | Oman | 10 yrs | − | − | − | − | p.S823Cfs*2 | |
|
| ||||||||
| 34 | Oman | 7 yrs | − | − | − | − | p.S823Cfs*2 | |
|
| ||||||||
| 35 Mansouri et al., 2016 | Morocco | − | − | − | − | − | p.R124W | |
|
| ||||||||
| 36 Ürel-Demir et al., 2018 | Turkey | 4 mo | 4 yrs | − | 5 yrs | Cardiorespiratory failure | p.R489* | |
|
| ||||||||
| 37 Tüysüz et al., 2009 (case 3) | Turkey | 1 y | 2 yrs | − | 2.5 yrs | Spinal cord injury | p.R489* | |
|
| ||||||||
| 38 Case 1 | Turkey | 1 y | 1 yr and 18 mo | Decompression, 18 mo | 3.5 yrs | Intracranial hemorrhage | IVS17+1G>A | |
|
| ||||||||
| 39 Case 2 | Turkey | Newborn | 1 yr and 18 mo | Decompression, 18 mo and 5 yrs | 5 yrs and 10 mo | Cardiorespiratory failure, pneumonia | p.S311G | |
yrs, years; mo, months.
Bargal et al. [2009] defined the mutations in one Algerian, one Pakistani and parents of reported Jewish patients. Probably these are the mentioned cases.
Third case of Borochowitz et al. [1993] and fifth case of Langer et al. [1993] are the same patient.
Ali et al. [2010] studied and reported the variants of the Al Gazali et al. [1996] cases and gave information about the disease prognosis of these siblings in their article.
Although it is a progressive disorder, disease severity and phenotype show great variability between patients even in the same family [Langer et al., 1993; Al-Gazali et al., 1996; Rozovsky et al., 2011]. The onset of calcifications vary greatly, Rozovsky et al. [2011] had not seen the calcifications before age of 1.5 years in their 8-patient series, while Langer et al. [1993] demonstrated costochondral calcification in a 2-week-old baby (Table 2). While the disorder follows an early fatal course in some cases, it may display a fairly mild phenotype as in the Smithson et al., [2009] reported case having only calcification around the hallux at 5 years of age. One of our patient (case 2) had a calcified subcutaneous nodule on her left arm during the newborn period; she also had hypocalcemia during her stay in the newborn intensive care unit. During her first months of life, signs of accumulative osteopathy were noticed on hip ultrasonography. Although calcifications may be detected in the newborn period, accompanying hypocalcemia was not reported before. The other Turkish patient reported also had a severe course just as our patients. Our second case had very early signs of calcifications detected at 4 months of age [Ürel-Demir et al., 2018].
The clinical course of the disorder is more severe in Turkish patients. Widespread calcifications and severe epiphyseal and metaphyseal involvement were prominent in all of our cases around 1 year of age. In one of our cases (case 1) passive movement of the big joints were painful as in RCDP1, probably due to arthropathy as a result of calcifications and epiphyseal destruction. This feature was not mentioned before. In the same patient, a hoarse high-pitched voice was also present, probably due to the accumulative nature of the disorder on vocal cords. This feature was also present in the reported Turkish patient by Ürel-Demir et al. [2018] (Table 1).
Spinal cord compression especially at the level of C1–C2 due to dysplastic and hypoplastic dens with persisting synchondrosis and anterior dislocation of C1 and atlantoaxial subluxation is a grave and mortal complication of the disease course [Rozovsky et al., 2011]. Foramen magnum stenosis and spinal cord compression were present in all of our patients after the age of 1 year. Two were operated for spinal cord decompression when they were 18 months old. The stenosis was significant in case 1 causing triventricular hydrocephalus requiring a ventriculoperitoneal shunt. Severe foramen magnum stenosis causing obtructive hydrocephalus was not reported before in this disorder (Table 1). Although there have been some cases with early and severe atlantoaxial pathology reported before [Dias et al., 2009], usual age of onset of atlantoaxial instability and foramen magnum stenosis and spinal cord compression is after 3 years of age [Langer et al., 1993; Bargal et al., 2009; Rozovsky et al., 2011]. In the series of Rozovsky et al. [2011], spinal cord compression and foramen magnum stenosis were observed in 5 of 8 cases after 3.5 years of age and most of the patients were able to walk. Atlantoaxial instability was also observed in 4 of 6 patients [Bargal et al., 2009] and decompression operations were performed in 3 of them at rather older ages (10, 11, and 16 years). The other Turkish patient also had early onset of atlantoaxial pathology with spinal cord compression as seen in our cases [Ürel-Demir et al., 2018].
Combined conductive and sensorineural hearing loss and recurrent otitis media were present in our first case. Al Kindi et al. [2014] reported a moderate conductive type hearing loss in their 7 and 10-year-old siblings as well as additional optic atrophy in one of them. Also Borochowitz et al. [1993] and Al Gazali et al. [1996] reported optic atrophy in their patients. Ürel-Demir et al. [2018] reported facial nevre palsy in their 1-year-old patient. Thus, cranial nevre palsies and hearing loss could be detected during disease course probably due to calcifications on the pathways of the nerves (Table 1). Recurrent upper airway infections and otitis media, which were mentioned in additional patients, also play a role in the etiology of hearing loss [Langer et al., 1993; Al-Gazali et al., 1996].
In case 1 and 3, as the patients grew older, their hair got hypopigmented; case 1 also had dry and mild scaly skin. This unreported feature was perhaps due to defective collagen-binding activity of the DDR2 protein.
The disease was mortal in early ages in all of our cases and the reported Turkish patient by Ürel-Demir et al. [2018]. None of them were able to walk. All experienced recurrent lower airway infections and were hospitalized several times. In the reported cases, calcification of tracheal cartilage, dorsal respiratory muscles, and diaphragma were reported [Langer et al., 1993; Al-Gazali et al., 1996; Bargal et al., 2009; Rozovsky et al., 2011]. Tracheal or diaphragmatic calcifications usually occur after 8–10 years of age [Bargal et al., 2009; Rozovsky et al., 2011]. In our case (case 2), diffuse calcifications of trachea and main bronchi were detected at 5 years of age causing respiratory distress.
The DDR2 protein is an extracellular matrix receptor and belongs to a subfamily of receptor tyrosine kinases DDRs (discoidin domain receptors), possesing a catalytic kinase domain and a distinct extracellular discoidin homology domain [Rammal et al., 2016] (Fig. 6). It is activated upon binding to fibrillar collagen [Labrador et al., 2001]. DDRs control cell and tissue homeostasis via transducing signals regulating cell polarity, tissue morphogenesis, and cell differentiation [Valiathan et al., 2012]. Studies showed that lack or defects of DDR proteins play a detrimental role in skeletal development, reproduction, inflammation, in the cardiovascular system, and cancer progression [Valiathan et al., 2012; Rammal et al., 2016]. Labrador et al. [2001] showed that DDR2 was expressed along chondrocyte columns in the proliferative region of the growth plate of newborn mice, and they also showed that DDR2-deficient mice exhibit dwarfism and short bones. Pathogenic variants of DDR2 are known to cause 2 distinct disorders in humans. While gain-of-function pathogenic variants [e.g., p.(L610P), p.(Y740C)] cause an autosomal dominant disorder (Warburg-Cinotti sydrome, mainly affecting skin, teeth, and joints), loss-of-function pathogenic variants cause autosomal recessive SMED-SL/AC syndrome [Xu et al., 2018]. Bargal et al. [2009] identified the first pathogenic variants causing SMED_SL/AC (IVS17+1G>A; c.2254C>T (p.R752C); c. 2177T>G (p.I726R); c.2138C>T (p.T713I). They defined 3 missense variants and 1 splice site variant on exon 17 coding for the active site of the DDR2 tyrosine kinase domain which is a highly conserved region. Ali et al. [2010] identified an additional variant (c.337G>A; p.E113K), and they also defined the exact mechanism of all the missense pathogenic variants defined before. They demonstrated that missense variants defined by Bargal et al. [2009] cause a trafficking defect with the affected proteins mislocalizing to the endoplasmic reticulum while the variant they had found causes a protein defective in transmembrane signaling due to a ligand-binding defect interfering with collagen binding on the discoidin domain. Our first patient had the reported splice site variant on exon 17 [Bargal et al., 2009]. It was defined in parents of first reported Jewish patients. The disease was fatal in one of the children, but not as early as our patient [Borochowitz et al., 1993; Bargal et al., 2009]. Ali et al. [2010] had not studied this variant, but as it resides in the regulatory region of the same exon, we supposed its pathogenic effect would mimic the missense variants or even would cause a much more severe phenotype as it controls the trancription of the whole exon 17. This scenario may explain the severe phenotypes both in our and in the Bargal et al., [2009] cases.
Ürel-Demir et al. [2018] demonstrated a nonsense variant first in a Turkish patient, c.1465C>T (p.R489*). In our third patient, we found the same variant on exon 13. This may be confined to the Turkish population as a result of a founder effect. As it was a truncating variant, it causes a defective and ineffective protein lacking part of the intracellular juxtamembrane domain and all of the intracellular kinase domain creating a severe outcome.
In case 2, we defined a novel missense variant on exon 10, c.931A>G; p.S311G (Fig. 8). Our novel variant was not on a highly conserved residue, but it was not defined as a benign variant in another database and was heterozygous in both parents (Fig. 8, 9). As long as most of the missense variants of DDR2 were defined as pathogenic and the patient demonstrated the typical clinicial picture, we concluded this variant as the causative pathogenic variant. Additional tests were done whether this variant causes a trafficking defect as the ones on exon 17, but no abnormality in the subcellular localization of the protein was observed (Fig. 7). The DDR2 protein has 6 main domains: extracellular discoidin domain, extracellular discoidin-like domain, extracellular juxtamembrane domain, transmembrane domain, intracellular juxtamembrane domain, and intracellular kinase domain [Rammal et al., 2016] (Fig. 6). Reported pathogenic variants affecting extracellular residues were all in the discoidin domain affecting collagen binding directly [Ali et al., 2010; Mansouri et al., 2016] (Fig. 9). Our patient's pathogenic variant was coding for an amino acid change on the discoidin-like domain perhaps through a different mechanism still causing defective collagen binding or affecting the conformation of the protein. Another possible explanation is that the variant may result in a less stable non-functional protein. As it is not on a highly conserved residue, additional data still are need to confirm the pathogenicity of this variant, and further functional and expression studies should be done.
Fig. 8.
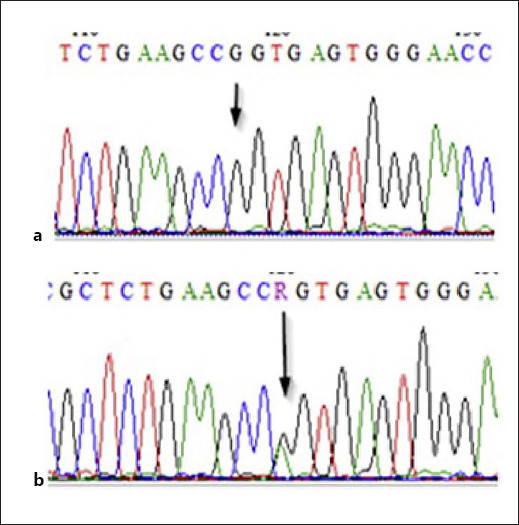
Novel variant in case 2. a c.931A>G, c.1369A>G, g.129914A>G, p.S311G. b Carrier state in the parent.
Fig. 9.

Novel S311G variant. Protein multiple sequence alignments (PMSA) of the corresponding residues for serine in 311th position in different species. Although it is conserved in primates and mouse, amino acid serine is replaced with asparagine in other species.
When evaluating the pathogenic variants and the phenotype of the Turkish children with SMED-AC/SL, we conclude that the pathogenic variants in our population cause more severe and fatal phenotypes with severe neurological signs such as hydrocephalus, intracranial hemorrhage, and cranial nerve palsies, and a rapidly progressive phenotype. This may be related to the pathogenic variants observed in our population;one of them is a nonsense variant which is reasonable to expect a more severe phenotype. It may be the result of additional genetic/epigenetic factors playing role in the disease mechanism; perhaps other coding variants of the same gene may have interacted with the disease-making mechanism of the variants. Deep intronic regulatory variants may have worked together with the coding variants. Coexistence of mild collagenopathies or vitamin and nutrient deficiencies affecting collagen, bone and cartilage structure may have aggravated the phenotype. However, it should be kept in mind that the disease itself is a highly lethal skeletal dysplasia and it shows variety in disease severity between cases with the same mutation even in the same family. The number of defined cases overall is low due to the small number of Turkish patients. With the widespread use of whole-exome and whole-genome analysis in the near future, milder cases with different phenotypes may be defined molecularly and change our opinion.
In conclusion, in severe cases, symptomatic foramen magnum stenosis, spinal cord compression, and neurological signs start around the age of 1 year. In cases carrying the pathogenic variants of Turkish patients, urgent neurological and systemic evaluation of the patients are vital as soon as the diagnosis is confirmed.
Statement of Ethics
The research was conducted ethically in accordance with the World Medical Association Declaration of Helsinki. This study protocol was reviewed and approved by ethics committee of Istanbul University Cerrahpaşa, Cerrahpaşa School of Medicine, approval number [09/08/2018-41,511]. Written informed consent was obtained from the parent/legal guardian of the patient for publication of the details of their medical case and any accompanying images.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Funding Sources
The protein trafficking project at BRA lab is currently funded by Abu Dhabi Department of Education and Knowledge (ADEK) through the Abu Dhabi Award for Research Excellence (AARE-2019-086).
Author Contributions
Elif Yilmaz Gulec: developed the protocol, abstracted and analyzed data, wrote the manuscript. Bassam R Ali: contributed to the development of the protocol, methodology, review and editing. Anne John: contributed to the editing and methodology. Beyhan Tuysuz: developed the original idea, contributed to the development of the protocol, review and editing.
Data Availability Statement
All data generated or analyzed during this study are included in this article and/or its supplementary material files. Further enquiries can be directed to the corresponding author.
Supplementary Material
Supplementary data
References
- Al-Gazali LI, Bakalinova D, Sztriha L. Spondylo-meta-epiphyseal dysplasia, short limb, abnormal calcification type. Clin Dysmorphol. 1996;5((3)):197–206. [PubMed] [Google Scholar]
- Al-Kindi A, Kizhakkedath P, Xu H, John A, Sayegh AA, Ganesh A, et al. A novel mutation in DDR2 causing spondylo-meta-epiphyseal dysplasia with short limbs and abnormal calcifications (SMED-SL) results in defective intra-cellular trafficking. BMC Med Genet. 2014;15:42. doi: 10.1186/1471-2350-15-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali BR, Xu H, Akawi NA, John A, Karuvantevida NS, Langer R, et al. Trafficking defects and loss of ligand binding are the underlying causes of all reported DDR2 missense mutations found in SMED-SL patients. Hum Mol Genet. 2010;19((11)):2239–50. doi: 10.1093/hmg/ddq103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargal R, Cormier-Daire V, Ben-Neriah Z, Le Merrer M, Sosna J, Melki J, et al. Mutations in DDR2 Gene Cause SMED with Short Limbs and Abnormal Calcifications. Am J Hum Genet. 2009;84((1)):80–4. doi: 10.1016/j.ajhg.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borochowitz ZL, Langer LO, Gruber HE, Lachman R, Katznelson MB, Rimoin DL. Spondylo-Meta-Epiphyseal Dysplasia (SMED), Short Limb-Hand Type: A Congenital Familial Skeletal Dysplasia With Distinctive Features and Histopathology. Am J Med Genet. 1993;45((3)):320–6. doi: 10.1002/ajmg.1320450308. [DOI] [PubMed] [Google Scholar]
- Dias C, Cairns R, Patel MS. Sudden death in spondylo-meta-epiphyseal dysplasia, short limb-abnormal calcification type. Clin Dysmorphol. 2009;18((1)):25–9. doi: 10.1097/MCD.0b013e3283186907. [DOI] [PubMed] [Google Scholar]
- Fano V, Lejarraga H, Barreiro C. Spondylo-meta-epiphyseal dysplasia, short limbs, abnormal calcification type: a new case with severe neurological involvement. Pediatr Radiol. 2001;31((1)):19–22. doi: 10.1007/s002470000359. [DOI] [PubMed] [Google Scholar]
- Labrador JP, Azcoitia V, Tuckermann J, Lin C, Olaso E, Mañes S, et al. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep. 2001;2((5)):446–52. doi: 10.1093/embo-reports/kve094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer LO, Wolfson BJ, Scott CI, Reid CS, Schidlow DV, Millar EA, et al. Further Delineation of Spondylo-Meta-Epiphyseal Dysplasia, Short Limb-Abnormal Calcification Type, With Emphasis on Diagnostic Features. Am J Med Genet. 1993;45((4)):488–500. doi: 10.1002/ajmg.1320450419. [DOI] [PubMed] [Google Scholar]
- Mansouri M, Kayserili H, Elalaoui SC, Nishimura G, Iida A, Lyahyai J, et al. Novel DDR2 mutation identified by whole exome sequencing in a Moroccan patient with spondylo-meta-epiphyseal dysplasia, short limb-abnormal calcification type. Am J Med Genet. 2016;170((2)):460–5. doi: 10.1002/ajmg.a.37426. [DOI] [PubMed] [Google Scholar]
- Rammal H, Saby C, Magnien K, Van-Gulick L, Garnotel R, Buache E, et al. Discoidin domain receptors: Potential actors and targets in cancer. Front Pharmacol. 2016;7:55. doi: 10.3389/fphar.2016.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozovsky K, Sosna J, Le Merrer M, Simanovsky N, Koplewitz BZ, Bar-Ziv J, et al. Spondyloepimetaphyseal dysplasia, short limb-abnormal calcifications type: Progressive radiological findings from fetal age to adolescence. Pediatr Radiol. 2011;41((10)):1298–307. doi: 10.1007/s00247-011-2123-2. [DOI] [PubMed] [Google Scholar]
- Smithson SF, Grier D, Hall CM. Spondylo-meta-epiphyseal dysplasia, short limb-abnormal calcification type. Clin Dysmorphol. 2009;18((1)):31–5. doi: 10.1097/MCD.0b013e3283189762. [DOI] [PubMed] [Google Scholar]
- Tüysüz B, Gazioğlu N, Üngür S, Aji DY, Türkmen S. The time of onset of abnormal calcification in spondylometaepiphyseal dysplasia, short limb-abnormal calcification type. Pediatr Radiol. 2009;39((1)):84–9. doi: 10.1007/s00247-008-1036-1. [DOI] [PubMed] [Google Scholar]
- Ürel-Demir G, Simsek-Kiper PO, Akgün-Doğan Ö, Göçmen R, Wang Z, Matsumoto N, et al. Further expansion of the mutational spectrum of spondylo-meta-epiphyseal dysplasia with abnormal calcification. J Hum Genet. 2018;63((9)):1003–7. doi: 10.1038/s10038-018-0473-4. [DOI] [PubMed] [Google Scholar]
- Valiathan RR, Marco M, Leitinger B, Kleer CG, Fridman R. Discoidin domain receptor tyrosine kinases: New players in cancer progression. Cancer Metastasis Rev. 2012;31((1-2)):295–321. doi: 10.1007/s10555-012-9346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Jensen H, Johnston JJ, Di Maria E, Kloth K, Cristea I, et al. Recurrent, Activating Variants in the Receptor Tyrosine Kinase DDR2 Cause Warburg-Cinotti Syndrome. Am J Hum Genet. 2018;103((6)):976–83. doi: 10.1016/j.ajhg.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Data Availability Statement
All data generated or analyzed during this study are included in this article and/or its supplementary material files. Further enquiries can be directed to the corresponding author.