

Abstract
Autosomal recessive primary microcephaly (MCPH) is a uncommon disorder due to congenital deficiency in the development of the cerebral cortex, characterized by a head circumference below 2 SD. MCPH is a group of diseases with genetic heterogeneity and has been reported by the Online Mendelian Inheritance In Man® (OMIM) database and associated with 25 different genes. It is known that MCPH cases are most frequently associated with abnormal spindle-like, microcephaly-associated (ASPM) gene mutations. The ASPM protein consists of an N-terminal 81 IQ (isoleucine-glutamine) domain, a calponin-homology domain, and a C-terminal domain. It interacts with calmodulin and calmodulin-related proteins via the IQ domain and acts as a part in mitotic spindle function. The basic characteristics of cases with ASPM gene mutations are microcephaly (below −3 SD) present before 1 year of age, intellectual disability, and the absence of other congenital anomalies. Macroscopic organization of the brain is preserved in cases with ASPM mutation, and a decrease in brain volume, particularly gray matter volume loss and a simplified gyral pattern are observed. Cortical migration defects are a very rare finding in patients with ASPM mutations. In the present study, we aimed to discuss the clinical and genetic findings in 2 cases with cortical dysplasia in which truncated variants in the ASPM gene were detected, particularly in terms of genotype-phenotype correlation in comparison with the literature.
Keywords: ASPM, Polymicrogyria, Pachygyria, Whole-exome sequencing, Novel variant
Established Facts
Autosomal recessive primary microcephaly is a rare disorder due to congenital deficiency in the development of the cerebral cortex, characterized by a head circumference below 2 SD.
Neuronal migration defects are very rare findings in patients with ASPM gene mutations.
Until now, polymicrogyria has been found in 4 cases with ASPM gene mutations and pachygyria in 2 cases as reported in the relevant literature.
Novel Insights
Brain MRI of patient 1 showed polymicrogyria located in the right frontotemporal region, and patient 2 had pachygyria.
Patient 1 with polymicrogyria is the fifth case and patient 2 with pachygyria is the third case described in the literature.
Introduction
Microcephaly is defined as a head circumference below 2 SD in the ethnic, age, and sex compatible population. Intellectual disability and seizures may be present in cases of microcephaly, depending on the influence on brain development. It is basically divided into primary and secondary microcephaly. Primary microcephaly, a prenatal developmental neurogenic disease, has an intrauterine onset and is detected at birth. Secondary microcephaly is a type of microcephaly in which the head circumference is normal at birth, and syndromic findings may develope due to progressive neurodegenerative disease [Naveed et al., 2018]. Autosomal recessive primary microcephaly (MCPH) is a rare disease occurring due to congenital deficiency in the development of the cerebral cortex, characterized by a head circumference below 2 SD [Mochida and Walsh, 2001]. Although the frequency of MCPH varies between populations, it occurs as 1/30,000–1/250,000 live births [Van Den Bosch, 1959; Zaqout et al., 2017]. It is more common in Asian and Arab societies where consanguineous marriage is a common practice [Zhou et al., 2013]. MCPH is a genetically heterogeneous group of diseases that has been reported in the OMIM database (http://omim.org/) associated with 25 different genes. The gene responsible for MCPH in the MCPH5 locus was the ASPM gene. It is known that MCPH cases are most frequently associated with ASPM gene mutations. This is frequently followed by mutations in the WDR62 and MCPH1 genes [Woods et al., 2005; Sajid Hussain et al., 2013; Jayaraman et al., 2018; Naveed et al., 2018]. The fact that no pathogenic variation in 25 different OMIM genes known to be associated with the disease was detected in some families with MCPH suggests that there may be genes that are not yet identified related to the phenotype [Mahmood et al., 2011].
ASPM, located in chromosome 1q31.3, is composed of 28 exons and encodes 3,477 amino acids [Saunders et al., 1997; Ponting 2006]. During neurogenesis, the ASPM protein is expressed in different parts of the cerebral cortex and creates transcripts of different sizes that are thought to have different functions according to the size of the IQ (isoleucine-glutamine) domain present in it. The N-terminal region and C-terminal region of the ASPM protein are located in the spindle poles of mitotic cells and the midbody, respectively. Proliferative symmetrical cell divisions have been observed to be decreased in neocortex cells that develop in Aspm knockdown mouse models [Fish et al., 2006]. It has been shown that the ASPM protein forms a complex with ATPase katanin and regulates the mitosis process in the progenitor cells of the nervous system [Jiang et al., 2017]. In addition, the presence of ASPM overexpression in brain tumors has been demonstrated; it has been suggested that it might be associated with malignant progression and could be targeted therapeutically [Bikeye et al., 2010].
In the present study, we aimed to discuss the clinical and genetic findings, particularly in terms of genotype-phenotype correlation, in 2 cases with cortical dysplasia in which truncated variants in the ASPM gene were detected in comparison with the available literature.
Materials and Methods
To investigate the molecular etiology of the diagnosis of primary microcephaly, genomic DNA was isolated from peripheral blood of the patients using the QIAamp DNA Blood Mini QIAcube Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocols. All the coding regions in the human genome of the patient were sequenced using whole-exome sequencing analysis via the Illumina NovaSeq Platform using the Agilent SureSelect V5 kit (Agilent, Santa Clara, CA, USA). Raw data were analyzed using the Sophia DDM® data analysis platform. The following filtering steps were applied to detect the pathogenic variants responsible for the clinical features of the case: (1) all missense, nonsense, frameshift, splice site, indel, in-frame and synonymous variants, and (2) variants with minor allele frequency <1.0% in population studies [1000 Genome (1000G), ESP, ExAC, and Genome Aggregation Database (gnomAD)]. Integrative Genome Viewer was used for sequence data visualization. The novel variant was checked in HGMD® and ClinVar (http://ncbi.nlm.nih.gov/clinvar) databases. The pathogenicity of the novel variants was interpreted using in silico analysis tools [Mutation Taster, CADD (Combined Annotation Dependent Depletion)], probability of being loss-of-function intolerant (pLI) score, and American College of Medical Genetics and Genomics (ACMG) criteria [Richards et al., 2015].
Case Reports and Results
Two patients from 2 different families with a head circumference below 2 SD in the first 6 months after birth were included in the study. Both cases were evaluated with anthropometric and family history evaluation, dysmorphology examination, hearing and visual examination, echocardiography, EEG, and brain MRI.
Family 1
Patient 1, a 9-year-old female, is the first child of healthy parents who are first cousins. The patient, without any anomaly in prenatal history, was born by normal spontaneous vaginal delivery, with a birth weight of 2,500 g. The head circumference was measured as 32 cm at birth. The patient gained head control at 2 months, sat without support at 10 months, and started walking at 18 months of age. The head circumference of the patient was 43 cm (−6.07 SD) at the age of 8, and during her examination at age 9, the height, body weight, and head circumference measurements were 123 cm (−1.57 SD), 25 kg (−0.83 SD), and 43.5 cm (−6.01 SD), respectively. Her dysmorphology examination result revealed a narrow and sloping forehead. She started having secondarily generalized seizures at the age of 7, which stopped at the age of 8 with carbamazepin and valproate therapy. Each EEG revealed focal epileptiform discharges located in the left hemisphere (Fig. 1). She had mild-to-moderate intellectual disability. She could only speak 2 words: “mother” and “father.”
Fig. 1.
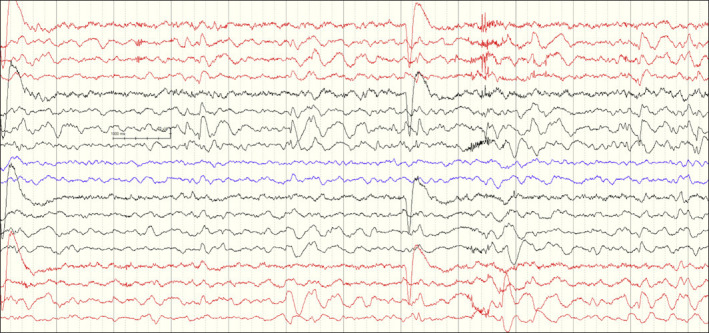
Interictal EEG on patient 1 showing high amplitute slow wave spikes emerging predominantly from centroparietal regions in the left hemisphere and normal background rhythm (Sensitivity: 10 μV/mm; Bandpass 0.5–70 Hz).
Family 2
Patient 2, a 10-year-old male, is the first child of healthy parents who are second cousins. Microcephaly was discovered prenatally at week 32 of gestation. He was born by normal spontaneous vaginal delivery, with a birth weight of 2,900 g. It was reported that the patient, whose head circumference was 31 cm at birth, gained head control at 3 months, sat without support at 11 months, and started walking at 20 months of age. During the examination at age 10, his height, body weight, and head circumference were 131 cm (−1.12 SD), 31 kg (−0.21 SD), and 45 cm (−5.88 SD), respectively. His dysmorphology examination revealed synophrys and a narrow, sloping forehead. He started having focal motor seizures at the age of 5 which stopped at the age of 7 under valproate therapy. His EEG revealed focal epileptiform discharges located in the left hemisphere. He had severe intellectual disability.
The results of both the patients' ophthalmologic and audiological examinations, as well as metabolic screening tests and their karyotype analyses were all normal.
The craniofacial and neurological features of the patients are summarized in Table 1.
Table 1.
Clinical and genetic findings of the previously reported and present cases with polymicrogyria and pachygyria
| Patient 1 | Patient 2 | Passemard et al. [2009] | Ariani et al. [2012] | Hu et al. [2014] | Nakamura et al. [2015] | Letard et al. [2018] | Letard et al. [2018] | |
|---|---|---|---|---|---|---|---|---|
| Sex | F | M | M | M | NA | M | M | M |
|
| ||||||||
| Age, years | 9 | 10 | 20 | 29 | 23 | 8 | 4 | 7 |
|
| ||||||||
| HC at birth, cm | 32 | 31 | NA | NA | NA | 29.5 | NA | NA |
|
| ||||||||
| HC at examination age, SD | −6.01 | −5.88 | −5 | −7.8 | NA | −7.6 | −10.8 | −5.6 |
|
| ||||||||
| Intellecual disability | Moderate | Severe | Mild | Moderate-severe | Moderate | Severe | NA | NA |
|
| ||||||||
| Age of epilepsy onset, years | 7 | 5 | 14 | − | 4 | 4 | − | 6 |
|
| ||||||||
| Type of epilepsy | Secondary generalized tonic-clonic seizures | Focal motor epilepsy | Generalized tonic-clonic seizures | − | Focal and generalized epilepsy | Tonic seizures | − | − |
|
| ||||||||
| Additional neurological findings | − | − | − | − | Fine motor problems and required surgery for strabismus | Increased muscle tone and mild spasticity in the arms and severe spasticity in the legs | Spastic tetraplegia | − |
|
| ||||||||
| Dysmorphic features | Narrow, sloping forehead | Synophrys Narrow, sloping forehead | Narrow bitemporal distance, sloping forehead, oxycephaly | Sloping forehead, long face, thick lips | NA | − | NA | NA |
|
| ||||||||
| MRI findings | Symmetrical ventriculomegaly, thinning of the corpus callosum, simplified gyral pattern located in the left frontotemporal region, and polymicrogyria located in the right frontotemporal region | Symmetrical ventriculomegaly, simplified gyral pattern located in both hemispheres and pachygyria | Extensive unilateral perisylvian polymicrogyria from the frontal pole to the occipital pole; contralateral simplified frontal and occipital gyral pattern | Global reduction in brain size, thin brain stem, normal corpus callosum, temporal pachygyria | Reduced gyration, parietal polymicrogyria and a myelination defect |
Frontal-dominant pachygyria with enlarged lateral ventricles and a slightly thickened corpus callosum | Thick frontal gyri, gyral simplification, thick corpus callosum, extensive bilateral posterior polymicrogyria | Gyral simplification, polymicrogyria in frontoinsular region |
|
| ||||||||
| ASPM mutation | Homozygous c.5219_5225delGAGGATA (p.Arg1740Thrfs*7) | Homozygous c.7792C>T (p.Gln2598*) | Homozygous c.9507delG (p.lle3170Leufs*9) | Compound heterozygous c.3796 G>T (p.Glul266*); c.7815_7816del (p.Glu2605Aspfs*31) | Compound heterozygous c.8227C> Τ (p. Arg2743*); c.7772_7775delAAAA (p.Lys2591Argfs*24) | Compound heterozygous c.3055C>T (p.Argl019*);c.6750delT (p.Phe2250Leufs*10) | Homozygous c.8702delA (p.His2901Leufs*37) | Homozygous c.7744delA (p.lle2582Serfs*34) |
|
| ||||||||
| Mutation location | Exon 18 | Exon 18 | Exon 23 | Exon 16 and 18 | Exon 18 and 18 | Exon 11 and 18 | Exon 18 | Exon 18 |
MRI, magnetic resonance imaging; HC, head circumference; NA;, not available; SD, standard deviation.
The radiological findings of the patients are summarized in Table 1 and illustrated in Figure 2. Patient 1 had symmetrical mild ventriculomegaly, simplified gyral pattern, and cortical dysplasia located in the right temporoparietal lobe on T2 FLAIR coronal image and T2-weighted axial images of brain (Fig. 2a–c). Patient 2 had pachgyria patern of cortical thickness on coronal T2 weighted (Fig. 2d) and axial FLAIR images (Fig. 2e).
Fig. 2.
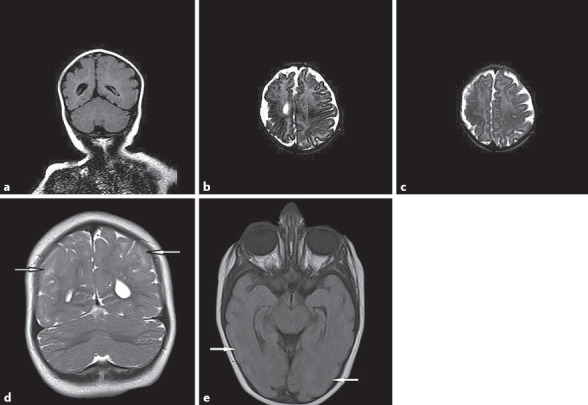
Radiological findings. Patient 1: a Symmetrical mild ventriculomegaly, simplified gyral pattern, and cortical dysplasia located in the right temporaparietal lobe on T2 FLAIR coronal image. b, c T2-weighted axial images of brain. Patient 2: d Pachgyria pattern of cortical thickness on coronal T2-weighted image. e Axial FLAIR image.
In whole-exome sequencing analysis, patient 1 was found to have a homozygous frameshift variant (NM_018136: c.5219_5225delGAGGATA, p.Arg1740Thrfs*7) in exon 18 of ASPM. Patient 2 had a novel homozygous nonsense variant (NM_018136: c.7792C>T, p.Gln2598*) in exon 18 of ASPM (Fig. 3). Due to the restrictions imposed because of the Covid-19 outbreak, segregation analysis could not be performed in the parents and other family members.
Fig. 3.
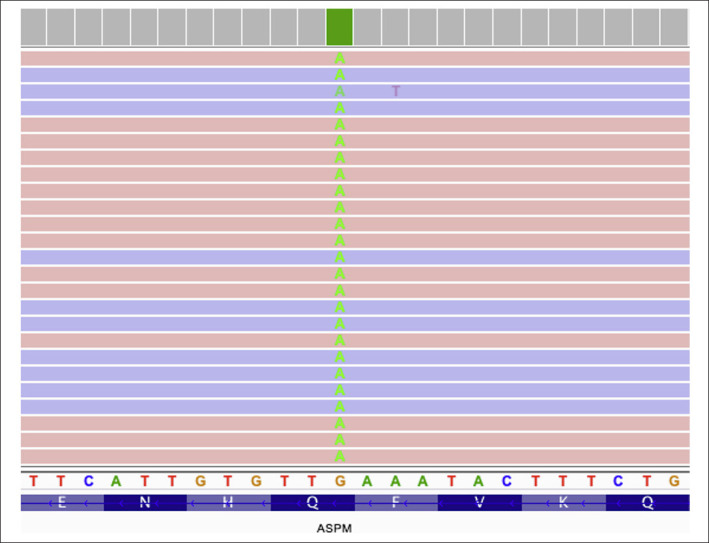
A novel homozygous nonsense variant (NM_018136: c.7792C>T, p.Gln2598*) in exon 18 of the ASPM gene detected in patient 2.
Discussion
The ASPM protein consists of an N-terminal 81 IQ (isoleucine-glutamine) domain, a calponin-homology domain, and a C-terminal domain. It interacts with calmodulin and calmodulin-related proteins through the IQ domain and plays a role in mitotic spindle function. Together, these observations suggest that mutations in the human ASPM gene may cause microcephaly due to the dysregulation of mitotic spindle activity in neuronal progenitor cells [Naveed et al., 2018]. Since the ASPM protein has a role in centriol duplication, it has been stated that primary microcephaly is a “centriolopathy” [Jayaraman et al., 2016]. However, ASPM functions are still not clearly known. Till now, 230 different variants (98 nonsense/missense, 20 splice site, 90 small and 2 gross deletions, 17 small insertions, 2 small indels, and 1 complex rearrangement) have been reported in the ASPM gene registered in Human Gene Mutation Database® (HGMD®) Professional (https://portal.biobase-international.com/hgmd/pro/all.php).
Verloes et al. [2020] described the clinical features of ASPM-associated MCPH phenotype in patients [Verloes et al., 2020]. These criteria are (i) microcephaly (below −3 SD) present at birth and present before the age of 1 year, and (ii) no other congenital anomalies [Nicholas et al., 2009]. Létard et al. [2018] classified the structural brain anomalies seen in the ASPM-associated MCPH phenotype. According to that study, the most common ASPM gene-related MCPH phenotype was characterized by gyral simplification (90%), corpus callosum abnormalities (shape and size anomalies, 52%), and mild-to-moderate cerebellar and/or pontine hypoplasia (10%). Cortical migration defect was found in a few cases [McSherry et al., 2018].
An overview of our patients and patients with cortical dysplasia as reported in the literature suggests that the locations of migration anomalies vary. The simplified gyral pattern specified in other studies is observed in both the patients included in our study. In addition, secondary generalized tonic-clonic seizures were observed in all patients except one patient. In the available literature, the incidence of epilepsy without cortical migration defect in patients with ASPM-associated MCPH phenotype was defined as approximately 3–8%. This suggests the possibility that cortical dysplasia that cannot be detected by MRI images may still be present in these patients. Mochida and Walsh [2001] reported that MCPH is rarely associated with epilepsy. Mutations in ASPM do not seem to affect the later stages of cortical development such as neuronal migration, and this might be responsible for the low epileptogenicity of this malformation [Passemard et al., 2009]. Alternatively, the existence of the “absence of seizure” criterion in the primary microcephaly diagnosis may cause the epilepsy phenotype to be ignored. Although it falls outside the diagnostic criteria in patients with microcephaly and a history of seizures, there is a possibility of mutation in ASPM or other primary microcephaly genes [Shen et al., 2005].
A review of the mutation distribution of the ASPM-associated MCPH cases in the literature suggests that there is no obvious hot-spot region and that the mutations are distributed throughout the gene. Exon 18, which constitutes 45% of the open reading frame of the ASPM gene, is the most frequently mutated exon. Mutations detected in all ASPM-associated MCPH cases cause loss of function. It is predicted that transcription of mutated ASPM may cause nonsense-mediated decay and/or truncated protein formation. A clear genotype-phenotype correlation could not be established between the position of the mutation in the gene and the size of the truncated protein formed, and the head circumference, degrees of mental involvement, and epilepsy phenotypes [Bond et al., 2003; Ariani et al., 2013]. In patient 1, we detected a frameshift variant in exon 18 that was previously reported in literature. In a previously reported case with the same mutation, the patient was a 2-year-old girl with severe microcephaly, narrow frontal area, and large ears phenotype, as well as cortical atrophy, and deep sulci detected on brain computed tomography images [Hu et al., 2014]. Patient 1 included in our study differed from the previous case in findings of polymicrogyria, ventriculomegaly, and a simplified gyral pattern in the right frontotemporal region. In patient-2, the novel homozygous nonsense variant was detected in exon 18. The CADD score of this variation, which was evaluated as likely pathogenic according to the ACMG criteria, was 36, and further, in silico analysis predicted it to be pathogenic. Consistent with the literature, we found mutations in exon 18 in both cases. Bilateral cerebral sulcus shallowness, decreased number of gyri, and pachygyria were detected in brain MRI in patient 2. Until now, polymicrogyria has been found in 4 cases with ASPM mutation and pachygyria in 2 cases as reported in the relevant literature [Passemard et al., 2009; Ariani et al., 2013; Hu et al., 2014; Nakamura et al., 2015; Létard et al., 2018]. Detailed clinical findings of the cases are summarized in Table 1. When all cases with polymicrogyria and pachygyria were evaluated, no correlation was found among the mutation site and truncated protein size and head circumference, severity of intellectual disability, or the presence of epilepsy.
It was emphasized by Abdel-Hamid et al. [2016] that mild-to-severe intellectual disability, speech delay, and hyperactivity disorder may be present in patients with primary microcephaly associated with the ASPM gene. In patients of primary microcephaly, it may be necessary to apply special education and appropriate behavior management strategies.
Primary microcephaly is a group of diseases with genetic heterogeneity, mostly inherited as autosomal recessive [Woods et al., 2005]. Determining the molecular etiology in patients with primary microcephaly provides an opportunity to calculate the risk of recurrence, to analyze the carrier status of the parents, and to present prenatal diagnosis options [Faheem et al., 2015].
In conclusion, neuronal migration defect is a rare finding in patients with ASPM-associated MCPH. Whether this defect detected in the MCPH cases occurs as a result of ASPM gene mutation cannot be clearly stated unless it is proven by in vitro/in vivo experimental studies and animal models. The mechanism of the defect should be investigated with further functional studies. The neuronal migration defect detected in these cases may also be a coincidental finding. The present study contributes to the expansion of the phenotypic spectrum based on the cases with neuronal migration defect in which a truncating mutation in the ASPM gene was detected.
Statement of Ethics
All experimental procedures were performed in terms of the guidelines of the local Ethics Committee and conducted in accordance with the principles of the Declaration of Helsinki, and written informed consent was obtained from patients or their guardians.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Funding Sources
No funding was received for this study.
Author Contributions
A.T. performed the whole-exome sequencing analysis as well as the segregation study and drafted the manuscript. S.G.S. conducted the patient's physical examination and critically reviewed the manuscript.
References
- Abdel-Hamid MS, Ismail MF, Darwish HA, Effat LK, Zaki MS, Abdel-Salam GM. Molecular and phenotypic spectrum of ASPM-related primary microcephaly: Identification of eight novel mutations. Am J Med Genet A. 2016;170((8)):2133–40. doi: 10.1002/ajmg.a.37724. [DOI] [PubMed] [Google Scholar]
- Ariani F, Mari F, Amitrano S, Di Marco C, Artuso R, Scala E, et al. Exome sequencing overrides formal genetics: ASPM mutations in a case study of apparent X-linked microcephalic intellectual deficit. Clin Genet. 2013;83((3)):288–90. doi: 10.1111/j.1399-0004.2012.01901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikeye SN, Colin C, Marie Y, Vampouille R, Ravassard P, Rousseau A, et al. ASPM-associated stem cell proliferation is involved in malignant progression of gliomas and constitutes an attractive therapeutic target. Cancer Cell Int. 2010;10:1. doi: 10.1186/1475-2867-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond J, Scott S, Hampshire DJ, Springell K, Corry P, Abramowicz MJ, et al. Protein-truncating mutations in ASPM cause variable reduction in brain size. Am J Hum Genet. 2003;73((5)):1170–7. doi: 10.1086/379085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faheem M, Naseer MI, Rasool M, Chaudhary AG, Kumosani TA, Ilyas AM, et al. Molecular genetics of human primary microcephaly: an overview. BMC Med Genomics. 2015;(8 Suppl 1):S4. doi: 10.1186/1755-8794-8-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish JL, Kosodo Y, Enard W, Pääbo S, Huttner WB. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci USA. 2006;103((27)):10438–43. doi: 10.1073/pnas.0604066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Suckow V, Musante L, Roggenkamp V, Kraemer N, Ropers HH, et al. Previously reported new type of autosomal recessive primary microcephaly is caused by compound heterozygous ASPM gene mutations. Cell Cycle. 2014;13((10)):1650–1. doi: 10.4161/cc.28706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman D, Kodani A, Gonzalez DM, Mancias JD, Mochida GH, Vagnoni C, et al. Microcephaly Proteins Wdr62 and Aspm Define a Mother Centriole Complex Regulating Centriole Biogenesis, Apical Complex, and Cell Fate. Neuron. 2016;92((4)):813–28. doi: 10.1016/j.neuron.2016.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman D, Bae B-I, Walsh CA. The Genetics of Primary Microcephaly. Annu Rev Genomics Hum Genet. 2018;19:177–200. doi: 10.1146/annurev-genom-083117-021441. [DOI] [PubMed] [Google Scholar]
- Jiang K, Rezabkova L, Hua S, Liu Q, Capitani G, Altelaar AFM, et al. Microtubule minus-end regulation at spindle poles by an ASPM-katanin complex. Nat Cell Biol. 2017;19((5)):480–92. doi: 10.1038/ncb3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Létard P, Drunat S, Vial Y, Duerinckx S, Ernault A, Amram D, et al. Autosomal recessive primary microcephaly due to ASPM mutations: An update. Hum Mutat. 2018;39((3)):319–32. doi: 10.1002/humu.23381. [DOI] [PubMed] [Google Scholar]
- Mahmood S, Ahmad W, Hassan MJ. Autosomal Recessive Primary Microcephaly (MCPH): clinical manifestations, genetic heterogeneity and mutation continuum. Orphanet J Rare Dis. 2011;6((6)):39. doi: 10.1186/1750-1172-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSherry M, Masih KE, Elcioglu NH, Celik P, Balci O, Cengiz FB, et al. Identification of candidate gene FAM183A and novel pathogenic variants in known genes: High genetic heterogeneity for autosomal recessive intellectual disability. PLoS One. 2018;13((11)):e0208324. doi: 10.1371/journal.pone.0208324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida GH, Walsh CA. Molecular genetics of human microcephaly. Curr Opin Neurol. 2001;14((2)):151–6. doi: 10.1097/00019052-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Inui T, Miya F, Kanemura Y, Okamoto N, Saitoh S, et al. Primary microcephaly with anterior predominant pachygyria caused by novel compound heterozygous mutations in ASPM. Pediatr Neurol. 2015;52((5)):e7–8. doi: 10.1016/j.pediatrneurol.2015.01.019. [DOI] [PubMed] [Google Scholar]
- Naveed M, Kazmi SK, Amin M, Asif Z, Islam U, Shahid K, et al. Comprehensive review on the molecular genetics of autosomal recessive primary microcephaly (MCPH) Genet Res (Camb) 2018;100:e7. doi: 10.1017/S0016672318000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas AK, Swanson EA, Cox JJ, Karbani G, Malik S, Springell K, et al. The molecular landscape of ASPM mutations in primary microcephaly. J Med Genet. 2009;46((4)):249–53. doi: 10.1136/jmg.2008.062380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passemard S, Titomanlio L, Elmaleh M, Afenjar A, Alessandri JL, Andria G, et al. Expanding the clinical and neuroradiologic phenotype of primary microcephaly due to ASPM mutations. Neurology. 2009;73((12)):962–9. doi: 10.1212/WNL.0b013e3181b8799a. [DOI] [PubMed] [Google Scholar]
- Ponting CP. A novel domain suggests a ciliary function for ASPM, a brain size determining gene. Bioinformatics. 2006;22((9)):1031–5. doi: 10.1093/bioinformatics/btl022. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17((5)):405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajid Hussain M, Marriam Bakhtiar S, Farooq M, Anjum I, Janzen E, Reza Toliat M, et al. Genetic heterogeneity in Pakistani microcephaly families. Clin Genet. 2013;83((5)):446–51. doi: 10.1111/j.1399-0004.2012.01932.x. [DOI] [PubMed] [Google Scholar]
- Saunders RD, Avides MC, Howard T, Gonzalez C, Glover DM. The Drosophila gene abnormal spindle encodes a novel microtubule-associated protein that associates with the polar regions of the mitotic spindle. J Cell Biol. 1997;137((4)):881–90. doi: 10.1083/jcb.137.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Eyaid W, Mochida GH, Al-Moayyad F, Bodell A, Woods CG, et al. ASPM mutations identified in patients with primary microcephaly and seizures. J Med Genet. 2005;42((9)):725–9. doi: 10.1136/jmg.2004.027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bosch J. Microcephaly in the Netherlands: a clinical and genetical study. Ann Hum Genet. 1959;23((2)):91–116. doi: 10.1111/j.1469-1809.1958.tb01455.x. [DOI] [PubMed] [Google Scholar]
- Verloes A, Drunat S, Passemard S. ASPM Primary Microcephaly. 2020 Apr 2. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 1993–2021. [PubMed] [Google Scholar]
- Woods CG, Bond J, Enard W. Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am J Hum Genet. 2005;76((5)):717–28. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaqout S, Morris-Rosendahl D, Kaindl AM. Autosomal Recessive Primary Microcephaly (MCPH): An Update. Neuropediatrics. 2017;48((3)):135–42. doi: 10.1055/s-0037-1601448. [DOI] [PubMed] [Google Scholar]
- Zhou ZW, Tapias A, Bruhn C, Gruber R, Sukchev M, Wang ZQ. DNA damage response in microcephaly development of MCPH1 mouse model. DNA Repair (Amst) 2013;12((8)):645–55. doi: 10.1016/j.dnarep.2013.04.017. [DOI] [PubMed] [Google Scholar]