

Abstract
Three siblings born to Turkish parents from the same village had normal brain development until acute neurological deterioration between 12 months and 8 years of age. Consequent loss of all acquired motor, social, and language functions following infections was associated with a pontine cyst, calcification, and cerebellar atrophy. Exome sequencing revealed a homozygous c.1297G>A (p.Gly433Ser) alteration in BEND4, which was predicted to be deleterious in in silico analysis tools and segregated in multiple affected individuals in the family. BEND4 has not been associated with any existing disease. Immunofluorescence microscopy analysis of wild-type and mutant BEND4 expressing Vero cells showed nuclear and cytoplasmic localization. Wild-type BEND4 displayed a network-like distribution, whereas mutant BEND4 showed a juxtanuclear distribution pattern. Differential proteome analysis of Vero cells expressing BEND4 revealed that mutant BEND4 expression caused selective increase in reticulocalbin-1 and endoplasmic reticulum resident protein-29. Both proteins are associated with the endoplasmic reticulum and are primarily involved in protein processing and folding pathways. Any defect or stress in protein folding creates stress on cells and may cause chronic damage. This is the first study showing that pathogenic BEND4 variants may lead to an infection-induced acute necrotizing encephalopathy as demonstrated in characteristic neuroimaging findings.
Keywords: BEND4, Infection-induced acute encephalopathy, Pontine cyst, Pontine calcification, Cerebellar atrophy
Introduction
Infection-induced acute encephalopathies (IIAEs) or acute necrotizing encephalopathies are characterized by rapidly progressive neurological deterioration that can occur in healthy children following common viral infections in infancy or early childhood [Neilson et al., 2009]. Neuroimaging plays an important role in the diagnosis of IIAEs with characteristic symmetric lesions, which can turn into necrosis over time, present in the thalami, putamen, internal and external capsules, cerebellar white matter, pons, and brainstem [Neilson et al., 2009]. An isolated intrinsic pontine cyst and pontine calcification and/or cerebellar atrophy without leukodystrophy are not reported as being characteristic radiological findings for diseases with intracranial calcification and cysts [Livingston et al., 2014a].
BEND4, a BEN domain-containing protein, was first characterized in 2003 [Dogan et al., 2016]. The BEN domain is a conserved DNA-binding domain, and BEN domain-containing transcription factors mediate chromatin organization and transcription [Abhiman et al., 2008; Sathyan et al., 2011]. Peptide sequence alignment reveals that BEND4 in human (Q6ZU67) have 5 potential isoforms produced by alternative splicing (534 amino acids [aa], 441 aa, 416 aa, 409 aa, and 316 aa) [UniProt Consortium, 2019]. BEND4 is highly expressed in the granular layer and Purkinje cells of the cerebellum in the brain, parathyroid, and adrenal glands in the endocrine tissues, heart muscle, gallbladder, salivary gland, stomach, duodenum, small intestine, colon, rectum, kidneys, testis, epididymis, cervix, uterus, and fallopian tubes [The Human Protein Atlas; http://www.proteinatlas.org/ENSG00000188848-BEND4/tissue). The protein is mainly located in the nucleus and cytoplasm [Uhlén et al., 2010, 2015]. Some BEN superfamily proteins are known as transcriptional repressors, including SMAR1, NAC1, BEND3, BEND5, and BEND6 [Sathyan et al., 2011]. However, the role of BEND4 in the physiological state, and the endogenous target genes whose expression is modulated by BEND4, are not known.
We present 3 siblings born to Turkish parents from the same settlement with acute-onset neurological deterioration triggered by infections in infancy to late childhood, intrinsic pontine cysts and calcification, and cerebellar atrophy. Studies on the investigation of genetic basis, immunofluorescence microscopy, and proteomics analysis of the candidate gene are presented.
Materials and Methods
Study Family
A 9-year-old boy born to nonconsanguineous parents was referred to the Division of Child Neurology, Department of Pediatrics, Kocaeli University Medical Faculty because he had siblings with a severe neurological disability that began during infancy to late childhood (Fig. 1). His medical records revealed no significant findings on examination, but his 3 siblings had an unspecified phenotype characterized by IIAE associated with pontine cysts and calcification and cerebellar atrophy. An experimental study was designed to investigate the genetic cause of this novel phenotype. The study was conducted in accordance with the Declaration of Helsinki and national guidelines. Research for genetic analysis and proteomic studies was approved by the Ethical Review Board of Kocaeli University. The pedigree of the family was drawn in detail and signed informed consent was obtained to isolate DNA before the collection of blood samples and inclusion of the family members in the study.
Fig. 1.
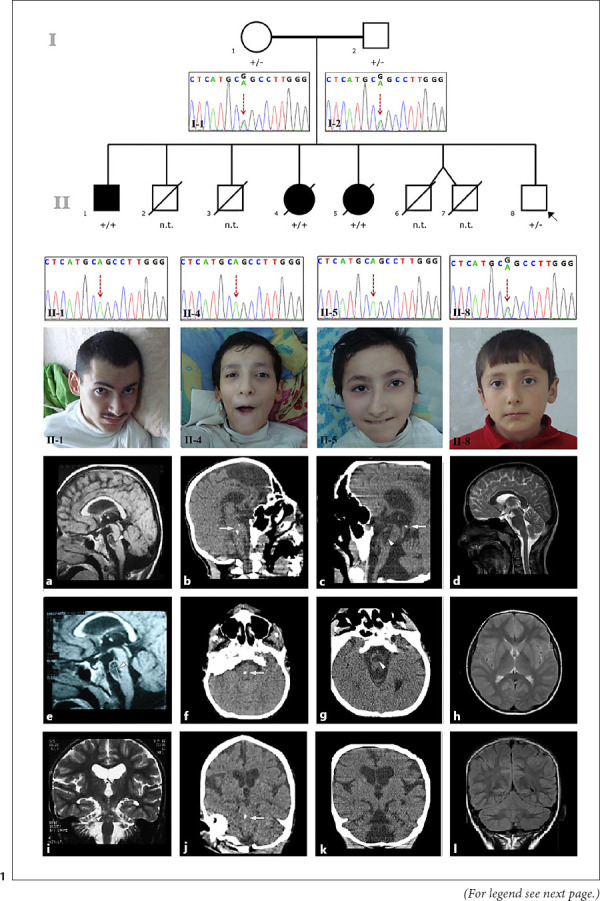
Pedigree and electropherograms of the study family and photographs of the affected siblings (brother, II-1; older sister, II-4; and younger sister, II-5) and the proband, II-8. Brain MRI of the brother showed a 20.4 × 12.7 mm intraparenchymal pontine cyst on sagittal T1-weighted and fluid attenuation inversion recovery images (a, e, arrowhead) and cerebellar atrophy on a coronal T2-weighted image at the age of 22 years (i). Cranial CT image of the older sister showed pontine calcification on sagittal (b), axial (f), and coronal (j) images (arrows) at the age of 22 years. Cranial CT image of the younger sister showed a pontine cyst (arrowhead) and calcification on a sagittal image (c, arrow), pontine cyst on an axial image (g, arrowhead), and cerebellar atrophy on a coronal image (k) at the age of 20 years. Brain MRI of the proband showed normal sagittal T2-weighted (d), axial T2-weighted (h), and coronal fluid attenuation inversion recovery (l) images at the age of 10 years. n.t., not tested; +/+, homozygous for the variant; +/−, heterozygous for the variant.
Genetic Analyses
DNA was extracted from 2 mL of peripheral blood leukocytes from each family member (Fig. 1; I:1-2, II:1,4,5,8) using a commercial kit following the manufacturer's instructions (Mammalian Blood DNA Isolation Kit, Roche). Linkage, microarray, and exome sequencing were all performed successively to find an associated gene.
Linkage analysis was performed by Mapping by Affymetrix GeneChip Human Mapping 250K NspI SNP array (Affymetrix-Ayka, Ankara, Turkey) followed by the conversion of the scanned genotype data of the Merlin parametric analysis into linkage and haplotype information by Alohomora (Affymetrix) [Rüschendorf and Nürnberg, 2005]. Autosomal recessive, autosomal dominant with decreased penetrance, and gonadal mosaicism in parent's models were used for analysis.
Affymetrix 6.0 SNP chip array (Affymetrix) is used for deletion and duplication analysis. Data retrieved in CN5.CNCHP files from Genotype Console (version 4.0) were visualized on Chromosome Analysis Suite (ChAS).
Exome capture of the subjects (I:1-2 and II:1-4-5-8) were performed using Agilent Sure Select Human All Exom v4.0 kit, sequenced on Illumina HiSeq2000 platform using TruSeq v3 chemistry at a mean coverage of 50X, reads provided in Fastq files mapped to the human genome (hg19) using Burrows-Wheeler Aligner (BWA package version 0.6.2), local realignment was performed by Genome Analysis Toolkit (GATK), duplicated reads were marked for exclusion from further analysis, using Picard (version 1.107) tool. Further alignment manipulations were performed by Samtools (version 0.1.18) and base quality (Phred scale) scores were recalibrated using GATK's covariance recalibration for each sample (Oxford Gene Technology, UK). Interpretations of the variants were performed from VarSome Community site, and gnomAD was referred for frequency data [Kopanos et al., 2018; Karczewski et al., 2020].
Mitochondrial genome analysis was performed by mtDNA amplification in 2 overlapping polymerase chain reaction (PCR) fragments (9731 bp and 12,083 bp) using Roche Expand Long Range PCR dNTPack (Roche Applied Science Indianapolis, IN, USA) from the blood of the family members. Next-generation sequencing was performed using the Illumina MiSeq Next Generation Sequencer. The sequence reading was aligned with the revised Cambridge Reference Sequence (rCRS) (NC_012920), using CLCBIO Genomics Workbench v5.1 (CLCBIO) [Andrews et al., 1999]. SNPs and deletion-insertion polymorphisms were determined for each patient. All data were compared to MITOMAP and PubMed [Kogelnik et al., 1996].
Protein Study
Cloning of the Wild-Type and the Mutant BEND4 Genes
The BEND4 full-length gene sequence was retrieved from NCBI (Accession number NM_207406.3). The sequence was modified to create c.1297G>A mutation and both the wild-type (WT) and the mutant gene sequences were synthesized and cloned into pCDNA4/TO by Eurofins (https://www.eurofinsgenomics.eu/). The clones were re-sequenced to confirm in-frame expression.
Expression of the Wild-Type and the Mutant BEND4 Proteins
Vero cells isolated from the kidney of an African green monkey, Cercopithecus aethiops, are of epithelial origin and used in many studies [Ammerman et al., 2008]. The cells were acquired from ATCC (CCL-81) and propagated as recommended. Vero cells were grown to 60% confluence in DMEM supplemented with 10% (vol/vol) fetal bovine serum, 100 U/mL penicillin-streptomycin, and 2 mML-glutamine at 37°C in a humidified 5% CO2 atmosphere. pCDNA4/TO harboring BEND4 gene sequences was transfected using Lipofectamine 3000 (LifeTech, USA). Empty pCDNA4/TO transfected cells were used as the control.
Immunofluorescence Microscopy
The WT and the mutant BEND4 transfected cells were cultured under standard tissue culture conditions in culture plates containing glass coverslips. After 48 h of BEND4 expressions, the cells were fixed with formaldehyde and stained for BEND4 with an anti-BEND4 antibody (BEND4 (T-12): sc-137377, Santa Cruz, Dallas, TX, USA). The nuclei of the cells were stained with DAPI (LifeTech, USA). Coverslips were mounted in Mowiol before the analysis. Imaging was performed with an inverted microscope (Olympus CKX41) using appropriate filter sets.
Western Blot Analysis
Cell-free extracts were prepared as described by Ozgul et al. [2015]. Western blot analysis was performed using an anti-BEND4 (BEND4 (T-12): sc-37377, Santa Cruz, USA) and an anti-beta actin (Santa Cruz; ACTBD, sc-81178) antibodies as described by Kasap et al. [2018]. Untransfected Vero cells were used to determine the level of endogenous BEND4 expression. Proteins were separated by 12% SDS-PAGE and transferred to a nitrocellulose membrane (Roche) using a Semi-Dry Transfer Cell (BioRad, Hercules, CA, USA). The membrane was blocked in TBS-T buffer (Tris.HCl 25 mM pH 7.2, NaCl 150 mM, and 0.1% Tween 20) containing 5% non-fat dry milk for 1 h at room temperature. After blocking, the membranes were incubated overnight at 4°C with appropriately diluted primary antibodies in TBS-T. The membranes were washed 3× with TBS-T and then incubated with HRP-labeled secondary antibodies (BioRad) for 1 h at room temperature. Following 3 washes with TBS-T, the membranes were visualized with the ECL detection system (BioRad) using X-ray films and the VersaDoc imaging system (BioRad). The bands were quantified with Quantity One 1D image analysis software (BioRad). Each experiment was performed at least 3 times.
2D Gel electrophoresis experiments, image analysis, protein identification, and bioinformatics analysis, see online supplementary material (for all online suppl. information, see www.karger.com/doi/10.1159/000517541).
Results
Clinical Findings
A 9-year-old boy (Fig. 1; II:8) was brought to be assessed in terms of the disease of the siblings. He had no complaints. His neurological examination was normal. The parents reported they were not consanguineous but were from the same village. The mother was 35 years old. She had type 1 diabetes mellitus and arterial hypertension for 5 years with a history of surgery for gallbladder stones. Her exercise capacity was limited and echocardiography showed mild mitral valve insufficiency. A brother and a sister of the mother died in infancy, and another sister had a history of an unexpected death at the age of 35 years. The father was 37 years old and had a history of urolithiasis. Two boys born to the parents had died of unknown cause when they were 45 days and 7 months old, respectively. They were born at term and appeared healthy until death. In addition, twin boys were born prematurely at 24 weeks gestation and died 1 h after delivery. The siblings of the boy were evaluated in a house visit. The older brother and the 2 sisters were bedridden, and all had a similar history (Fig. 1). The older brother was 23 years old (II:1), the older sister was 18 years old (II:4), and the younger sister was 15 years old (II:5) at the first visit. All patients were born at term, and their prenatal, natal, and early postnatal periods were uneventful.
The older brother (II:1) showed normal neurological development until he suffered an acute encephalopathy attack at the age of 8 years. Preceding this acute neurological insult, he had a history of upper respiratory infections. Then, he lost all his acquired motor, social, and language development and became bedridden. When he reached the age of 15 years, he started bleeding in the upper gastrointestinal system. Endoscopic examination showed a duodenal ulcer, esophagitis, and hiatus hernia. Physical examination at the age of 23 years revealed that he could not sit or walk, and spoke only 2 words, “mother” and “father”. He could understand conversations, watch television, play computer games, and indicate his toilet needs (online suppl. video). He did not have cardiac or respiratory problems. He had severe generalized muscle atrophy, flexor joint contractions of the elbows, wrists, and knees, and finger deformities. Cranial nerve examinations were normal. He had upper motor neuron signs, such as increased muscle tonus in all extremities, increased deep tendon reflexes, ankle clonus, and muscle weakness. Pathologic reflexes could not be elicited. Cerebellar signs and unintentional movements were not observed. There was no organomegaly. His vision and hearing were normal. Eye fundus examination was normal. He had no dysmorphism. Whole blood count, serum biochemistry, urine analysis, blood gases, plasma ammonia, and lactate levels were normal. Tandem mass spectrometry, urine organic acids, and plasma amino acid chromatography did not reveal any evidence in support of an inborn error of metabolism. An IQ test could not be performed due to a lack of cooperation. Visual examination and hearing screening tests were normal. Brain MRI of the older brother showed an intrinsic pontine cyst and moderate cerebellar atrophy (Fig. 1a, e, i) at the age of 21 years. Cranial CT could not be performed. Currently, the older brother is 30 years old and has had stable neurological findings for the last 6 years of the survey.
The older sister (II:4) showed normal neurological development until the age of 18 months. At that age, she suffered acute encephalopathy following a gastroenteritis infection. The causative agent was unknown. She lost all motor, social, and language development after this acute insult and became bedridden. Physical examination at the age of 18 years showed that she could neither sit, walk nor speak. She had severe failure to thrive. Her mental performance was much worse than her older brother. No meaningful relationship could be established with the patient. She had difficulties swallowing. Her muscle bulk was severely reduced, and there were flexor contractures in the large joints of the upper and lower extremities and finger deformities. She had crossed legs and scoliosis (online suppl. video). Cranial nerve examinations were normal and eye movements were intact. She had spastic tetraparesis with increased deep tendon reflexes, ankle clonus, and extensor plantar reflexes. There were no cerebellar and extrapyramidal signs. Her vision and hearing were intact. Eye fundus examination was normal. She had no organomegaly and other system examinations were normal. Facially she looked like her sister and mother. Routine laboratory tests were normal. At the age of 22 years, cranial CT showed spot calcification in the pons and mild cerebellar atrophy without a pontine cyst (Fig. 1b, f, j). Brain MRI could not be obtained. At follow-up, she was hospitalized many times for upper gastrointestinal system bleeding and anemia necessitating blood transfusion. Her endoscopic examination showed erosive gastritis; coagulation parameters were normal. She died at the age of 24 years after an excessive hemorrhagic vomiting attack at home according to the history taken from the parents.
The younger sister's (II:5) neurological development was defined as normal until the age of 12 months. At that age, she had an acute encephalopathy attack following an upper respiratory tract infection and lost all her achieved neurological milestones. The results of her physical examination and routine laboratory examinations were similar to her sister's at 15 years of age. She had an intrinsic pontine cyst, spot calcification in the pons, and moderate cerebellar atrophy on cranial CT at the age of 20 years (Fig. 1c, g, k). Brain MRI could not be obtained. The younger sister had a stable clinical status until the age of 22 years when she died in a hospital in another city. We learned from the doctor who followed the patient that she died as a result of sudden-onset multiple organ failure, but the underlying cause could not be clarified.
Brain MRI of the younger brother (II:8) was normal (Fig. 1d, h, l). He currently is 16 years old and asymptomatic.
Genetic Analyses
Analysis with 250K NspI SNP array for autosomal recessive (AR) and autosomal dominant (AD) models showed no significant linked regions, with LOD (logarithm of the odds) scores above 3.0 and 2.0, respectively. SNP 6.0 chip array showed no duplications (>200 kb disclosed by a minimum of 20 copy number probes) or deletions (>20 kb disclosed by a minimum of 5 copy number probes) that are shared by the affected individuals in the family.
Exome sequencing analysis of variants sequenced equal to or above 20X in depth is considered for interpretation. For AD inheritance model shared variants between II:1, II:4, and II:5 which are not found in I:1, I:2 or II:8, and AR inheritance form, compound heterozygose, and homozygous variants shared in II:1, II:4, and II:5 which are not have a call in compound heterozygous or homozygous form in II:8 are searched. There were no variants in conformity with AD or compound heterozygosity models. Nevertheless, 86 variants were found in homozygous form in the affected individuals and heterozygous in the unaffected parents and heterozygous or absent in healthy sibling, II:8 (online suppl. Table). Among all those variants, c.1297G>A altering glycine to serine (p.Gly433Ser, NP_997289) in exon 5 of the BEND4 gene annotated with reference number rs779411414, distinguished from all others by having no frequency data and interpreted as a variance of unknown significance according to VarSome since it was not found in gnomAD exome and genome populations and predicted pathogenic based on 9 prediction programs (BayesDel_addAF, DANN, EIGEN, FATHMM-MKL, LIST-S2, M-CAP, MutationTaster, PrimateAI, and SIFT) and benign on 4 (DEOGEN2, MVP, MutationAssessor, and REVEL). The frequency data for all the remaining 85 variants were high in rate, with the lowest with 1.8% in gnomAD-genome (567:31,322 alleles). The parents and a single healthy sibling were heterozygous for the same variant in BEND4. The c.1297G>A variant was not found in DNA samples from 50 independent individuals searched initially and this followed by investigating 100 in-house trio groups from the Advanced Genomics and Bioinformatics Research Group of the National Research Institute of Electronics and Cryptology of Turkey (AGBRG), plus 128 independent samples from our in-house exome sequencing collection data. This variant was also not found in the Greater Middle East (GME) Variome Project database, including 2,497 individuals from 6 different Middle East countries [Scott et al., 2016]. Altogether we confirmed that the c.1297G>A variant of BEND4 is not found in 5,950 alleles from related geographical regions of Turkey and Middle Eastern country populations. This variant was only shown in a single allele in an ExAc Aggregated Population of 120,680 alleles, obtained from dbSNP [Sherry et al., 1999; Lek et al., 2016]. Mitochondrial DNA sequencing of all siblings and parents did not reveal any pathogenic variations.
Proteomics Study
Protein extracts from Vero cells transiently expressing the WT and the mutant BEND4 proteins were subjected to western blot analysis to assess any changes that may occur in the BEND4 profile (Fig. 2). The level of BEND4 expression has significantly increased after transient expression of the WT and the mutant clones (p < 0.05). There was no change in molecular weights of both proteins indicating that the proteins were not post translationally modified. On the other hand, a weak band in protein extracts prepared from non-transfected cells was observed. Vero cells have endogenous BEND4 expression at a detectable level. To test whether the expression level of endogenous BEND4 was constant in time, we performed several western blots. The results showed that the expression of endogenous BEND4 protein was stable with no change in time. The exogenous expression of the WT and the mutant BEND4 proteins was time-dependent and increased in time. We confirmed that 24 h of BEND4 expression produces a moderate level of BEND4 proteins which were suitable to study their effects on cell metabolism.
Fig. 2.
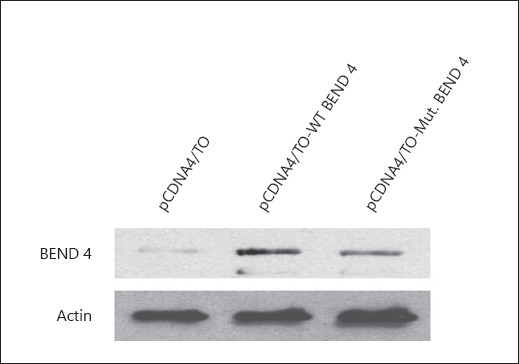
Western blot analysis of protein extracts prepared from Vero cells expressing the wild-type (WT) and the mutant BEND4 proteins. The weak band in vector-only transfected cells (pCDNA4/TO) illustrates the level of endogenous BEND4 expression in Vero cells. Beta-actin was used as the internal control.
In some cases, mutations cause changes in the cellular localization of the proteins. Therefore, we performed an immunofluorescence microscopy study to determine the localization of the WT BEND4 and if there is any effect of the mutation on its localization (Fig. 3). The WT BEND4 displayed an interesting distribution pattern within the cells. A distinct rim around the nucleus with some network-like cytoplasmic distribution was evident. On the other hand, the mutant BEND4 protein had a significant loss in its cytoplasmic distribution pattern. A more pronounced nuclear localization without a distinct rim was evident. More interestingly, in a juxtanuclear position, a thread-like distribution resembling the distribution of Golgi apparatus was observed. The overall evaluation of the immunofluorescence microscopy results implied a change in the distribution pattern of BEND4 upon the introduction of the mutation.
Fig. 3.
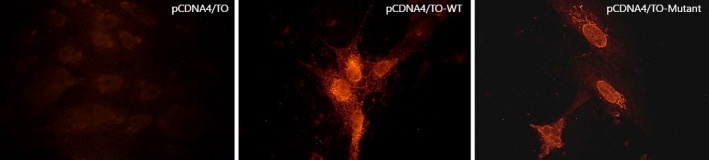
Immunofluorescence microscopy analysis of Vero cells expressing either the wild-type (WT) or the mutant BEND4 proteins. The weak staining in vector-only transfected cells (pCDNA4/TO) illustrates the level of endogenous BEND4 expression in Vero cells. The cellular distribution of the WT-BEND4 differed from the distribution of the mutant BEND4 protein. The WT-BEND4 protein formed a distinct rim around the nucleus with some network-like cytoplasmic distribution pattern while the mutant BEND4 had a significant loss in its cytoplasmic distribution and a more pronounced nuclear localization without a distinct rim.
To further assess the effect of expressing the WT and the mutant BEND4 proteins, we studied the changes at the proteome level using 2DE gels (Fig. 4). 750 protein spots were evaluated. There was a great deal of similarity among the protein distribution patterns. The overall mean coefficient of variation was 31.8%. Based on the 2-fold up-or downregulation criteria, in comparison to non-transfected cells, 4 differentially regulated proteins in BEND4 WT expressing cells and 5 differentially regulated proteins in BEND4 mutant expressing cells were detected (Table 1). However, only 2 proteins displayed changes in their levels upon expression of the mutant BEND4. Those proteins were reticulocalbin-1 and ER-resident protein-29. On the other hand, there was one protein that displayed regulation in its level upon expression of the WT BEND4. That protein was Rab GDP dissociation inhibitor beta (GDIβ).
Fig. 4.

Representative 2D gel images of the cell-free extracts prepared from the wild-type (WT) and the mutant BEND4 expressing Vero cells. The control gel is prepared from the Vero cells transfected with the empty plasmid DNA, pCDNA4/TO. The regulated protein spots were marked with black circles.
Table 1.
The results of MALDI-TOF/TOF analysis for regulated protein spots
| SSP No. | AC No. | Best protein Acc | Best protein score | Expect | Matches | Seq. cov., % | Best protein description | Upregulated in | Downregulated in |
|---|---|---|---|---|---|---|---|---|---|
| 105 | P62258 | 1433E_HUMAN | 294 | 8.1e–026 | 21 | 56 | 14-3-3 protein epsilon | Wild-type and mutant | |
| 303 | Q15293 | RCN1_HUMAN | 146 | 5.1e–011 | 9 | 18 | Reticulocalbin-1 | Mutant | |
| 502 | P27797 | CALR_HUMAN | 743 | 1e–070 | 31 | 47 | Calreticulin | Wild-type and mutant | |
| 3205 | P07195 | LDHB_HUMAN | 133 | 1e–009 | 10 | 15 | l-lactate dehydrogenase Β chain | Wild-type and mutant | |
| 5104 | P30040 | ERP29_HUMAN | 338 | 3,20E–30 | 17 | 32 | Endoplasmic reticulum resident protein 29 | Mutant | |
| 5502 | P50395 | GDIB_HUMAN | 146 | 5,10E–11 | 17 | 28 | Rab GDP dissociation inhibitor beta | Wild-type |
The TOF spectra were recorded in the positive ion reflector mode with a mass range from 400 to 2,000 Da. The Peptide Mass Fingerprints (PMFs) search was performed using MASCOT version 2.5 (Matrix Science). The following search criteria were used: species restricted to human; enzyme of trypsin; at least two independent peptides matched; at most one missed cleavage site; MS tolerance of ±50 ppm and MS/MS tolerance of ±0.4 Da; fixed modification being carbamidomethyl (Cys) and variable modification being oxidation (Met); peptide charge of 1+; and being monoisotopic. Only significant hits, as defined by the MASCOT probability analysis (p < 0.05), were accepted
Discussion
A total of 16,368 genes have been reported in OMIM. Nevertheless, only 5,921 of those have currently been revealed with molecular basis and associated with a phenotype (OMIM; https://www.omim.org/statistics/entry; October 31, 2020). Many genes still cannot be associated with any disease. Here, we have presented a new IIAE phenotype in 3 siblings that may be associated with a variant of the BEND4 gene, which was not reported to cause disease previously. Combined Annotation Dependent Depletion (CADD) score of c.1297G>A within the “mutation significance cutoff (MSC)” value at a 99% confidence interval was 8.429, and the MSC-CADD_score was 3.313, indicating a high-impact prediction for damage for this missense alteration at the gene level by using data from ClinVar and the Human Genome Mutation database [Itan et al., 2016; Requena et al., 2018]. We were curious as to why the 4p13 region was not linked in our first study with 250K NspI SNP array and inferred that the BEND4 region was encompassed with only 3 markers (rs16854011, rs7677472, and rs7684103), which are not sufficient for powerful analysis. In fact, missing heritability in genome-wide association studies were well recognized in previous genetic studies since gaps between markers was the critical factor liable for decreased statistical power for the linkage analysis [López-Cortegano and Caballero, 2019].
There were no known functional human disorders associated with BEND4 according to a detailed survey of the literature. Because our variant was located in the coding region of the BEN domain, which is known to have novel sequence-specific DNA-binding properties in neural transcription, we considered this variant highly responsible for the phenotype in our family [Dai et al., 2013].
Although sharing of the same variant by 3 similarly affected siblings provides multiple verifications and is a strong supportive factor for genetic associations, the establishment of a cause-and-effect relationship is not an easy task for novel or rare variants that have not been associated with any phenotype before. Legitimate verification needs functional studies or identification of a second unrelated case with a similar phenotype with the same variant. Our cases had an unspecified phenotype, and similar cases have not yet been reported. The presence of the BEND4 gene in Gene Matcher (GM - Submissions - Identifier: bend4 (genematcher.org) (https://genematcher.org/) was checked, and no submissions of the clinically relevant variant were found. We studied immunofluorescence microscopy and proteomics analysis to show the distribution pattern and changes in protein expression profiles of WT and mutant BEND4 in Vero cells from kidney epithelium.
BEND4 is a BEN domain-containing protein with a calculated molecular weight of 58.338 Da. The physiological function of BEND4 is not known. Based on the Human Protein Expression Atlas (https://www.ebi.ac.uk/gxa/home/), BEND4 is moderately expressed in brain tissue and in the spinal cord as well as the reproductive organs. BioGrid analysis of BEND4 to elucidate its interaction partners did not yield significant hits, indicating that BEND4 has not been described as an interaction partner in any of the known metabolic events. Further bioinformatic analysis using the STRING database with a text-mining tool predicted connections between BEND4 and Ribonuclease T2, BTG3 associated nuclear protein, and Zinc finger protein 583. A common property of these 3 BEND4-connected proteins is that they are involved in the transcriptional regulation of several genes. Our immunofluorescence microscopy analysis demonstrated nuclear localization of BEND4 and in a way verified the prediction made by the STRING analysis.
Expression of both the WT and the mutant BEND4 proteins caused changes in the abundance of 3 proteins: 14-3-3 protein epsilon, calreticulin, and L-lactate dehydrogenase B chain. We assume that none of these changes were associated with the effect of the mutant BEND4 expression. STRING analysis with these 3 proteins detected no significantly enriched pathway.
The expression of the WT BEND4 distinctively altered the level of the Rab GDIβ. Rab GDIβ is a member of the GDP dissociation inhibitor family, which controls the recycling of Rab GTPases involved in membrane trafficking and is related to tumorigenicity, development, and invasion [Sedlacek et al., 1994; Ming et al., 2014]. This protein regulates the GDP/GTP exchange reaction of most Rab proteins by inhibiting the dissociation of GDP from them, and the subsequent binding of GTP to them. BioGRID analysis revealed 58 interactors and 71 interactions of GDIβ. This finding puts GDIβ at the center of several metabolic pathways and makes it a hub in the regulation of various interactomes.
To predict the effect of Gly433 on Ser mutation, we used 2 different bioinformatics approaches. The first approach involved modeling BEND4 and its mutant using Swiss Modeller. The most appropriate template structure that was found by the server belonged to inositol polyphosphate 5-phosphatase (3qis.1.A). However, the region that showed similarity was short, covering only 97 amino acids (starting from Glu378, ending at Thr475), thus the model did not provide any evidence for distortion in the 3D structure of BEND4. Prediction of the domain structure using the Pfam database located the BEN domain between L415 and E480. Computational analysis suggests that the BEN domain mediates protein-DNA and protein-protein interactions during chromatin organization and transcription, and thus may have a significant effect on protein structure and function. The mutation discovered in this study is located within the BEN domain and thus may have a deleterious effect on BEND4 function. Indeed, PROVEAN predicted a score of −3.92 for Gly-Ser mutation, indicating that the mutation ought to have a deleterious effect on protein structure or function.
The mutant BEND4 expression caused a selective increase in the abundance of reticulocalbin-1 (RCN1) and ER resident protein-29 (ERP29). These 2 proteins do not appear to be functionally connected to each other. RCN1 may regulate calcium-dependent activities in the endoplasmic reticulum (ER) lumen or post-ER compartment. The mutant BEND4 expression regulates calcium-dependent activities in the ER lumen or post-ER compartment. ERP29 plays an important role in the processing of secretory proteins within the ER, possibly by participating in the folding of proteins in the ER [Shnyder and Hubbard, 2002]. However, both proteins are associated with the ER and are primarily involved in protein processing and folding pathways. Any defect or stress in protein folding creates pressure on cells and may cause chronic damage to the cells.
Gene ontology analysis did not reveal any significant pathway or a biological process in which BEND4 might play a role. However, BIOGRID analysis indicated that BEND4 interacts with activator protein-1 (AP-1). The interaction was elucidated with affinity capture-MS analysis and listed as a high-confidence interaction [Li et al., 2015]. AP-1 is a transcription factor and a proto-oncogene. It binds to an enhancer motif to promote the activity of NR5A1 when phosphorylated with HIPK3 [Qing et al., 2000; Ji et al., 2012]. Phosphorylation of AP-1 leads to activation of the cAMP signaling pathway [Lan et al., 2007]. The cAMP is well established as a potent regulator of innate and adaptive immune cell functions. Like BEND4, any factor that affects cAMP generation should have immunoregulatory potential in autoimmune and inflammatory disorders [Raker et al., 2016].
Initial motor and intellectual development of our cases were normal, followed by acute-onset neurological deterioration triggered by infections in infancy to late childhood. Attacks resulted in irreversible loss of motor, verbal, social, and intellectual functions with patients becoming bedridden with a stable neurological status. This course resembles vanishing white matter disease, and IIAE susceptibility syndromes, triggered by some viruses such as influenza A, influenza B, parainfluenza II viruses, and Herpes simplex virus-1 [Bugiani et al., 2010; Gika et al., 2010]. In our cases, no further deterioration was observed, and there were no episodes of lethargy or coma after minor infection or minor head trauma in the following years. Moreover, brain MRI findings were not compatible with vanishing white matter disease. Clinical and neuroimaging findings of our cases were more consistent with IIAEs. The detection of a cystic lesion in the pons in an unpublished case in our clinic with an RANBP2 mutation increased the probability of IIAE. However, there was no significant variation in all known IIAEs genes.
Brain MRI findings of our cases were specific. All siblings had an intrinsic pontine cyst and/or spot calcification in the pons. Isolated intrinsic pontine cysts and pontine calcification are extremely rare radiological findings in pediatric patients with neurological disorders. Cystic tumors, particularly epidermoid cysts, are rarely located in the pons. Their clinical findings are limited to the mass effect of the lesion and/or hydrocephalus, and they generally grow slowly with a good course [Patibandla et al., 2016]. Pontine cysts in our cases had no mass effect, and the clinical findings could not be explained solely by the cystic lesion in the pons. Coats plus disease (CP) due to CTC1 or STN1 mutations and leukoencephalopathy with calcification and cysts (LCC) due to SNORD118 mutation are 2 different disorders with largely identical neuroimaging findings [Livingston et al., 2014b; Jenkinson et al., 2016; Simon et al., 2016]. Intrinsic brainstem cysts and intracranial calcifications may be seen in these disorders. Cysts are generally multiple and may develop in different parts of the brain as well as the brainstem, frequently causing a mass effect. Calcification was spot-like and restricted to the pons in our cases, not rock-like or blush-like, and widely located in the basal ganglia, thalami, dentate nucleus, deep gyri, deep white matter as in CP or LCC. Also, the absence of leukodystrophy in our cases was not compatible with CP or LCC. Congenital cytomegalovirus infection, cystic leukoencephalopathy without megalencephaly (RNASET2 mutation), and COL4A1 mutation-related disease are other disorders presenting with an intraparenchymal brain cyst and calcification and were excluded with the radiological features and genetic analysis [Livingston et al., 2014b]. IIAEs progress with necrotizing changes in the affected brain regions. Since pons involvement can also be seen in IIAEs, theoretically, cysts may develop in the pons. However, isolated pontine cysts and calcification are not defined in the radiological findings of known IIAEs.
In this study, we present 3 siblings with a new IIAE associated with a pontine cyst and calcification, an unspecified phenotype, probably due to a deleterious effect of a BEND4 variation. Proteomics studies support the disease-causing effect of this variation, but new cases need to be identified to confirm the disease-causing effect of the BEND4 mutation.
Statement of Ethics
Written informed consent was obtained from the parents of the patients for publication of the details of their medical case and any accompanying images and videos. This study protocol was reviewed and approved by Ethical Review Board of Kocaeli University (approval number 2015/52).
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Funding Sources
This study received support from Kocaeli University-BAP.
Author Contributions
This study was coordinated by B.K.; clinical investigations were done by B.K., H.M.G. and E.U.Y; linkage analysis, microarray and exome sequencing were performed by O.U. and G.T.; proteomic studies were performed by E.E.İ., M.K., and G.A.; mitochondrial genom analysis was performed by D.Ü.; cranial CT and MRI images were evaluated by Y.A.; the manuscript was written by B.K. and edited by O.U., H.M.G., M.K., G.A., and D.Ü.
Supplementary Material
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Acknowledgments
We would like to thank all the participating patients and their families for their support in this ongoing research. We appreciate AGBRG (IGBAM, Bilgem, TUBITAK) for sharing their parallel sequencing results of 100 private individuals with us.
References
- Abhiman S, Iyer LM, Aravind L. BEN: A novel domain in chromatin factors and DANN viral proteins. Bioinformatics. 2008;24:458–61. doi: 10.1093/bioinformatics/btn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerman NC, Beier-Sexton M, Azad F. Growth and maintenance of Vero cell lines. Curr Protoc Microbiol. 2008 doi: 10.1002/9780471729259.mca04es11. Appendix4: Appendix 4E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23((2)):147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- Bugiani M, Boor I, Powers JM, Scheper GC, van der Knaap MS. Leukoencephalopathy with vanishing white matter: a review. J Neuropathol Exp Neurol. 2010;69:987–96. doi: 10.1097/NEN.0b013e3181f2eafa. [DOI] [PubMed] [Google Scholar]
- Dai Q, Ren A, Westholm JO, Serganov AA, Patel DJ, Lai EC. The BEN domain is a novel sequence-specific DNA-binding domain conserved in neural transcriptional repressors. Genes Dev. 2013;27:602–14. doi: 10.1101/gad.213314.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan T, MacDougall A, Saidi R, Poggioli D, Bateman A, O'Donovan C, et al. UniProt-DAAC: domain architecture alignment and classification, a new method for automatic functional annotation in UniProtKB. Bioinformatics. 2016;32:2264–71. doi: 10.1093/bioinformatics/btw114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gika AD, Rich P, Gupta S, Neilson DE, Clarke A. Recurrent acute necrotizing encephalopathy following influenza A in a genetically predisposed family. Dev Med Child Neurol. 2010;52:99–102. doi: 10.1111/j.1469-8749.2009.03405.x. [DOI] [PubMed] [Google Scholar]
- Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. 2016;13:109–10. doi: 10.1038/nmeth.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson EM, Rodero MP, Kasher PR, Uggenti C, Oojageer A, Goosey LC, et al. Mutations in SNORD118 cause the cerebral microangiopathy leukoencephalopathy with calcifications and cysts. Nat Genet. 2016;48:1185–92. doi: 10.1038/ng.3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Donaldson IJ, Liu J, Hayes A, Zeef LA, Sharrocks AD. The forkhead transcription factor FOXK2 promotes AP-1-mediated transcriptional regulation. Mol Cell Biol. 2012;32((2)):385–98. doi: 10.1128/MCB.05504-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581((7809)):434–43. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasap M, Akpınar G, Karaosmanoğlu KY, Çöl B. Utilization of bioinformatics tools for understanding proteomics data. Turk Mol Biol Biotech. 2018;3:41–53. [Google Scholar]
- Kogelnik AM, Lott MT, Brown MD, Navathe SB, Wallace DC. MITOMAP: a human mitochondrial genome database. Nucleic Acids Res. 1996;24((1)):177–9. doi: 10.1093/nar/24.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2018;35((11)):1978–80. doi: 10.1093/bioinformatics/bty897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan HC, Li HJ, Lin G, Lai PY, Chung BC. Cyclic AMP stimulates SF-1-dependent CYP11A1 expression through homeodomain-interacting protein kinase 3-mediated Jun N-terminal kinase and c-Jun phosphorylation. Mol Cell Biol. 2007;27((6)):2027–36. doi: 10.1128/MCB.02253-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang W, Wang J, Malovannaya A, Xi Y, Li W, et al. Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol Syst Biol. 2015;11((1)):775. doi: 10.15252/msb.20145504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston JH, Mayer J, Jenkinson E, Kasher P, Stivaros S, Berger A, et al. Leukoencephalopathy with calcifications and cysts: a purely neurological disorder distinct from coats plus. Neuropediatrics. 2014b;45:175–82. doi: 10.1055/s-0033-1364180. [DOI] [PubMed] [Google Scholar]
- Livingston JH, Stivaros S, Warren D, Crow YJ. Intracranial calcification in childhood: a review of aetiologies and recognizable phenotypes. Dev Med Child Neurol. 2014a;56((7)):612–26. doi: 10.1111/dmcn.12359. [DOI] [PubMed] [Google Scholar]
- López-Cortegano E, Caballero A. GWEHS: A Genome-Wide Effect Sizes and Heritability Screener. Genes (Basel) 2019;10((8)):558. doi: 10.3390/genes10080558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming Z, Guo C, Jiang M, Li W, Zhang Y, Fan N, et al. Bioinformatics analysis of Rab GDP dissociation inhibitor beta and its expression in non-small cell lung cancer. Diagn Pathol. 2014;9:201. doi: 10.1186/s13000-014-0201-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilson DE, Adams MD, Orr CM, Schelling DK, Eiben RM, Kerr DS, et al. Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am J Hum Genet. 2009;84((1)):44–51. doi: 10.1016/j.ajhg.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgul S, Kasap M, Akpinar G, Kanli A, Güzel N, Karaosmanoglu K, et al. Linking a compound-heterozygous Parkin mutant (Q311R and A371T) to Parkinson's disease by using proteomic and molecular approaches. Neurochem Int. 2015;85–86:1–13. doi: 10.1016/j.neuint.2015.03.007. [DOI] [PubMed] [Google Scholar]
- Patibandla MR, Yerramneni VK, Mudumba VS, Manisha N, Addagada GC. Brainstem epidermoid cyst: An update. Asian J Neurosurg. 2016;11:194–200. doi: 10.4103/1793-5482.145163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing J, Zhang Y, Derynck R. Structural and functional characterization of the transforming growth factor-beta -induced Smad3/c-Jun transcriptional cooperativity. J Biol Chem. 2000;275((49)):38802–12. doi: 10.1074/jbc.M004731200. [DOI] [PubMed] [Google Scholar]
- Raker VK, Becker C, Steinbrink K. The cAMP pathway as therapeutic target in autoimmune and inflammatory diseases. Front Immunol. 2016;7:123. doi: 10.3389/fimmu.2016.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Requena D, Maffucci P, Bigio B, Shang L, Abhyankar A, Boisson B, et al. CDG: an online server for detecting biologically closest disease-causing genes and its application to primary immunodeficiency. Front Immunol. 2018;9:1340. doi: 10.3389/fimmu.2018.01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüschendorf F, Nürnberg P. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21((9)):2123–5. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- Sathyan KM, Shen Z, Tripathi V, Prasanth KV, Prasanth SG. A BEN-domain-containing protein associates with heterochromatin and represses transcription. J Cell Sci. 2011;124((Pt 18)):3149–63. doi: 10.1242/jcs.086603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EM, Halees A, Itan Y, Spencer EG, He Y, Azab MA, et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016;48:1071–6. doi: 10.1038/ng.3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlacek Z, Konecki DS, Korn B, Klauck SM, Poustka A. Evolutionary conservation and genomic organization of XAP-4, an Xq28 located gene coding for a human rab GDP-dissociation inhibitor (GDI) Mamm Genome. 1994;5:633–9. doi: 10.1007/BF00411459. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward M, Sirotkin K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999;9:677–9. [PubMed] [Google Scholar]
- Shnyder SD, Hubbard MJ. ERp29 is a ubiquitous resident of the endoplasmic reticulum with a distinct role in secretory protein production. J Histochem Cytochem. 2002;50:557–66. doi: 10.1177/002215540205000413. [DOI] [PubMed] [Google Scholar]
- Simon AJ, Lev A, Zhang Y, Weiss B, Rylova A, Eyal E, et al. Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. J Exp Med. 2016;213:1429–40. doi: 10.1084/jem.20151618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28:1248–50. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47((D1)):D506–15. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data
Supplementary data
Supplementary data