

Abstract
Heterozygous activating missense variants of PDGFRB are associated with the phenotype of Kosaki overgrowth syndrome (KOGS). Here, we present a family including a father and 2 siblings with a novel variant, c.2567A>T (p.Asn856Ile), localized in the cytoplasmic tyrosine kinase domain, exhibiting a KOGS phenotype. The coarsening of the facial features, enlargement of the hands/feet, and progressive scoliosis started to appear after an average age of 6. There were no signs of thin/fragile skin, premature aging appearance, myofibroma, white matter findings, and intellectual disability in any of them. Corneal pterygium and evidence of cerebral vasculopathy were only detected in the father. One sibling exhibited café-au-lait spots. Posterior fossa enlargement was revealed only in one sibling. KOGS is an extremely rare overgrowth syndrome. No familial cases of KOGS have been reported so far. Hereby, we demonstrated that the features of KOGS can show mild intrafamilial variability, and the risk of vascular complications may arise with age.
Keywords: Kosaki overgrowth syndrome, PDGFRB, Scoliosis, Tyrosine kinase domain
Introduction
Kosaki overgrowth syndrome (KOGS; OMIM #616592) is a somatic overgrowth syndrome characterized by common findings of coarse facial features; tall stature; accelerated growth with elongated hands and feet; thin, soft and fragile skin; scoliosis; periventricular white matter lesions with or without intellectual disability, and neurological deterioration.
Heterozygous activating missense variants of platelet-derived growth factor receptor beta (PDGFRB), a tyrosine kinase receptor involved in activation of multiple signaling pathways including those related to cell proliferation and survival, are responsible for the phenotype [Andrae et al., 2008]. It was first described by Takenouchi et al. [2015] in 2 unrelated Japanese children. A total of 9 patients with 3 common types of PDGFRB missense variants (c.1751C>G, c.1696T>C, and c.1477A>T) have been reported up to date. However, no familial cases of KOGS have been previously reported. Here, we present 2 siblings, and a father exhibiting the KOGS phenotype in whom a novel missense PDGFRB variant (c.2567A>T) was identified.
Case Presentation
Two siblings, an 8-year-old girl (P1) and a 6-year-old boy (P2), were admitted to the Pediatric Genetic Clinic due to progressive enlargement of hands and feet during the last year. Both were born at term with a weight of 3,000–3,500 g. The parents were consanguineous (first cousins). Both siblings exhibited normal developmental milestones. The 33-year-old father (P3) also had enlargement of the hands and feet, and progressive kyphoscoliosis since the of age 12. He had had an episode of intracerebral hemorrhage one year previously. Growth measurements of the patients were calculated according to growth cards of Turkish children [Neyzi et al., 2015]. On the examination of P1, the weight was 25.2 kg (49.6th centile), height 130 cm (72.5th centile), OFD 50 cm (−2 SD), hand size 17 cm (>97th centile), and foot size was 22 cm (75th–90th centile). The weight of P2 was 21,5 kg (61.8th centile), height 116.5 cm (55.6th centile), OFD 53.5 cm (−2 SD), hand size 15 cm (>97th centile), and foot size was 22 cm (>97th centile). P1 had a narrow frontal hairline, thick eyebrows, synophrys, telecanthus, downslanting palpebral fissures, broad nasal root, large tubular nose, long philtrum, macrodontia, and long-large hands and feet (Fig. 1). Café-au-lait spots were noted on the abdominal skin of P1. Dysmorphic features of thick eyebrows, telecanthus, epicanthus, downslanting palpebral fıssures, large tubular nose, long philtrum, low-set ears, macrodontia, and localized protrusion on the occipital region were noted in P2 (Fig. 1). The father had a prominent supraorbital ridge, malar flattening, linear lines on the forehead skin, and corneal pterygium in the right eye. Dysmorphic features compered with the other patients who have been reported previously are summarized in Table 1. The cardiac examination of the siblings did not reveal any abnormalities. However, the father did not undergo cardiac examination. Both P1 and P2 showed progressive right-convex thoracolumbar scoliosis, evident from 6 years of age in P2 (Fig. 2). On the X-ray of P1 taken 2 years later (at the age of 10), it was observed that scoliosis increased progressively. There were no associated vertebral segmentation anomalies. All patients demonstrated prominent pneumatization of the frontal sinuses with a prominent supraorbital ridge (Fig. 3). These features were more prominent in the father and the older child suggesting progression with age. In addition, there was occipital prominence in all patients, with an occipital ridge visible in the father. Mega cisterna magna was visible only in P2. Brain MRI of the father showed a T2-weighted signal hyperintense area in the left internal capsule. Long bone and pelvic modeling was normal. The father had bony expansion of the distal third proximal phalanges of the 2nd–5th fingers (Fig. 4). This feature was not clearly present in the children.
Fig. 1.

a–d Dysmorphic features of the family members exhibiting narrow frontal hairline, thick eyebrows, synophrys, telecanthus, downslanting palpebral fissures, broad nasal root, and bulbous nose. e, f Large and rough hands of P1 and P2. g Severe kyphoscoliosis present in the father.
Table 1.
The summary of the major dysmorphic features and complications
| Patients | P1 | P2 | P3 |
|---|---|---|---|
| Growth pattern | |||
| Macrosomic birth | − | − | − |
| Tall stature | − | − | − |
| Macrocephaly | − | − | − |
| Overgrowth of hands and feet | + | + | + |
| Craniofacial features | |||
| Occipital prominence | + | + | + |
| Sparse hair | − | − | − |
| Narrow frontal hairline | + | + | + |
| Prominent supraorbital ridge | + | + | + |
| Thick eyebrows | + | + | + |
| Synophrys | + | + | − |
| Downslanting palpebral fissures | + | + | + |
| Malar flattening | + | + | + |
| Wide nasal bridge | + | + | + |
| Large tubular nose | + | + | + |
| Long philtrum | + | + | + |
| Thin upper lip | + | + | + |
| Skeletal features | |||
| Progressive thoracolumbar scoliosis | + | + | + |
| Kyphosis | − | − | + |
| Bony expansion of the proximal phalanges | − | − | + |
| Skin findings | |||
| Hyperelastic skin | − | − | − |
| Thin, soft, fragile skin | − | − | − |
| Lipodystrophy | − | − | − |
| Café au lait spots | + | − | − |
| Brain imaging | |||
| Posterior fossa enlargement | − | + | − |
| Periventricular white matter lesions | − | − | − |
| Sign of the cerebral vasculopathy | − | − | + |
| Other | Corneal pterygium |
Fig. 2.

Anteroposterior spine radiographs in P1 at age 8 (a), P2 at age 6 (b), and the father at age 33 (c). Right-convex thoracolumbar scoliosis is shown, more severe with age, becoming S-shaped in the father.
Fig. 3.

Skull radiographs of P1 at age 8 (a), P2 at age 6 (b), and the father at age 33 (c, d). Prominent pneumatization of the frontal sinuses in the children is shown, which is marked in the father. A prominent supraorbital ridge is visible in the children, which is very marked in the father. Prominence of the occipital bone in the children has developed into an occipital ridge in the father. Cranial CT of P1 exhibiting normal parenchyma and calvarial structures (e). Occipital protrusion, frontal sinus enlargement, and mega cisterna magna on cranial CT of P2 (f, g). Father's cranial MRI cross section, showing periventricular hyperintense area in left internal capsule consistent with prior hemorrhage (h).
Fig. 4.
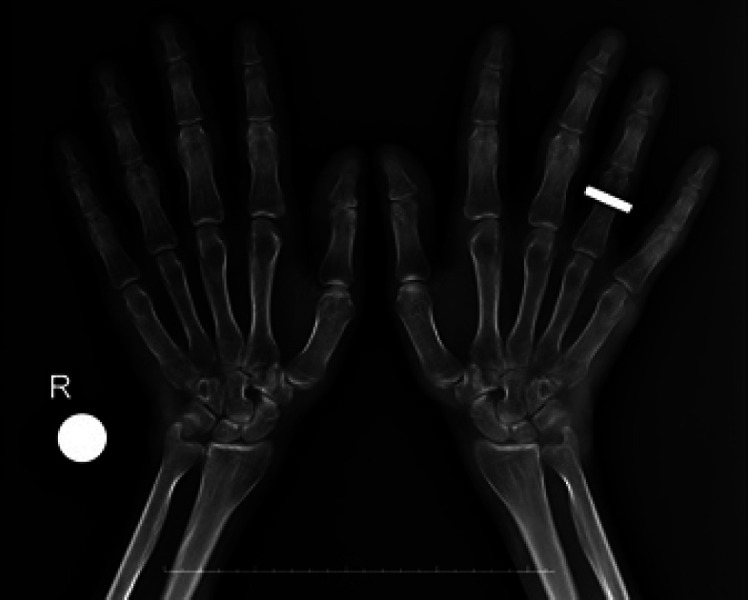
Dorsal-plantar radiograph of both hands of the father at age 33. The hands are generally large. Boneexpansion of the distal thirds of the proximal phalanges of the 2nd–5th fingers in both hands is shown.
Materials and Methods
DNA material was isolated from peripheral blood sample, and PCR products obtained with primer pairs specifically designed for all coding exon regions of the genome were analyzed by DNA sequence analysis technique. Whole-exome sequencing analysis primarily involving analysis of known disease gene sets focused mainly on the patient's clinical presentation was performed for the father. In case of negative results, other coding regions were analyzed that may cause the patient's clinical presentation. The quality score of the analysis was within normal limits. The reading rate of the sequenced genome regions with at least 20x coverage is 98.59%, and the reading rate with at least 4x coverage is 98.97%. A NM_002609.4:c.2567A>T (p.Asn856Ile) heterozygous missense variant was identified in the PDGFRB gene. This variant was not found, neither in gnomAD exomes (good gnomAD exomes coverage = 65.7) nor in gnomAD genomes (good gnomAD genomes coverage = 31.4). Pathogenic computational verdict based on 11 pathogenic predictions from BayesDel_addAF, DANN, DEOGEN2, EIGEN, FATHMM-MKL and 6 more versus 2 benign predictions from MVP and MutationAssessor. PCR products obtained with primer pairs specifically designed for the c.2567A>T variant in the PDGFRB gene were analyzed by DNA sequence analysis technique and the same heterozygous variant was detected in both siblings. No other pathogenic variants were found contributing to the phenotype.
Discussion
The receptor for PDGFB is a member of the type III class of tyrosine-protein kinase receptors, which are characterized by 5 Ig-like domains extracellularly, a transmembrane domain, a juxtamembrane domain (JMD), and a split tyrosine kinase domain (TKD) intracellularly [Heldin and Lennartsson, 2013]. The c.1751C>G (p.Pro584Arg) variant (exon 12), which is located in the JMD of PDGFRB and negatively regulates the catalytic activity of cytoplasmic kinases, was described in 2 cases (P5, P6) with a KOGS phenotype for the first time [Takenouchi et al., 2015]. Later, it was identified in 3 more patients (P7, P8, and P9) [Gawlinski et al., 2018; Foster et al., 2020]. There were some common dysmorphic features and clinical findings in these patients with c.1751C>G; they all had typical coarse facial features of KOGS: downslanting palpebral fissures; prominent supraorbital ridges, wide nasal bridge, large tubuler nose, underdeveloped malae, thin upper lip, long philtrum, and a pointed chin together with skeletal overgrowth. Most of them exhibited scoliosis; hyperelastic, thin and fragile skin, and neurologic deterioration associated with perivetricular white matter lesions. Although some had a premature aging appearance, dystrophic scars and joint contractures, none of them showed acroosteolysis. Only one patient (P5) had infantile myofibroma (IMF). Dandy-Walker malformation/hydrocephalus, and arachnoid cysts were the distinct additional features of P8 and P9, who were recently reported by Foster et al. [2020]. Neither of these 2 patients showed either intellectual disability or neurological deterioration. Dolichoectasia of cerebral arteries was an accompanying finding in P8. P9 had additional findings of visual impairment and carpal tunnel syndrome.
A number of novel findings were described after identification of 3 new patients (P10, P11, P12) with the variant of c.1696T>C (p.Trp566Arg) in exon 12 localized to JMD [Minatogawa et al., 2017; Zarate et al., 2019]. They had a classical phenotype of KOGS. Furthermore, pectus excavatum, premature aging findings with wrinkled skin, dystrophic scars, arachnoid cyst, and IMF were the accompanying clinical findings of P10. Cardiac valvulopathy was described as a novel finding in P11. Dolichoectasia of the cerebral arteries and coronary artery aneurysms were vascular complications which were found in a patient who died at the age of 19 (P12). The c.1477A>T (p.Ser493Cys) variant in exon 10 localized to a transmembrane domain was the latest variant which was identified in P13 [Foster et al., 2020]. This patient died due to vascular complications.
A novel variant, c.2567A>T (p.Asn856Ile), localized in the cytoplasmic TKD (exon 18), was identified in the current family. Unlike the most patients reported in the literature, our patients did not have a history of macrosomic births. Furthermore, height, weight, and OFD of the siblings were between the 50th and 75th centile. The coarsening of the facial features of the patients, enlargement of the hands and feet, and the signs of scoliosis started to appear after an average age of 6. The patients did not show any skin findings such as hyperelastic or thin/soft/fragile skin which have been previously described in KOGS patients. Lipodystrophy, sparse hair, and premature aging appearance which have been also reported in most of the KOGS patients, were not found in any of the family members. There were multiple café-au-lait spots in P1 as a cutaneous finding located on the anterior chest, which was not previously reported in any case of KOGS. The first patient reported by Takenouchi et al. [2015] demonstrated a striking copper beaten appearance of the skull. Radiological images of the skull and cranium were evaluated for this feature in index patients. None of them exhibited copper beaten appearance and calvarial thickness. However, mild scalloping of the inner table could be seen in the CT of P2. On the other hand, there was occipital prominence in all patients, with an occipital ridge visible in the father. Enlargement of the posterior fossa was shown only in P2. The father had a lesion revealed by brain MRI secondary to the history of intracerebral hemorrhage. However, he did not show neurological deterioration.
On the vertebral radiography of P1 taken 2 years later (age 10), it was seen that scoliosis showed significant progression. P1 and P2 had no signs indicating cardiac or valvular pathology. Nevertheless, some vascular complications associated with high mortality and morbidity such as coronary artery aneurysms, and dolichoectasia of cerebral arteries have been reported in adult patients (minimum age 19) [Foster et al., 2020; Zarate et al., 2019]. Therefore, due to the father having a history of intracerebral hemorrhage, close follow-up of the siblings for possible cerebrovascular disease was planned. On ophthalmologic examination of the father, a corneal pterygium on the right eye was detected. Ocular pterygium has been recently described in a 53-year-old male patient (P8); the pterygia first became visible on the right eye at the age of 10 and on the left eye at the age of 32 [Foster et al., 2020]. For this reason, this ocular finding was not considered age-dependent.
Infantile myofibromatosis (OMIM #228550), and premature aging (Penttinen) syndrome (PS) (OMIM #601812) are the other well-defined disorders linked to the activating variants of PDGFRB. The c.1978C>A (p.Pro660Thr) and c.1994T>C (p.Val665Ala) variants localized to the TKD have been previously described in association with the IMF and PS, respectively [Martignetti et al., 2013; Johnston et al., 2015]. PS is characterized by some cutaneous findings overlapping with KOGS such as thin translucent skin, visible vessels, atrophic scars and a premature aging apprearence due to lipoatrophy, acro-osteolysis, and normal linear growth. However, none of the patients with KOGS exhibited brachydactyly with acro-osteolysis, which are the most known features in PS. Activating germ-line mutations in PDGFRB can cause a spectrum of complex phenotypes. There was also a reported patient (P4) with the c.1996A>C (p.Asn666His) variant localized to TKD, who exhibited overlapping phenotypes of PS-KOGS characterized by coarse facial appearance, skeletal overgrowth, acroosteolysis, and posterior fossa enlargement as well as dermal abnormalities such as thin skin, flesh-colored nodules, and palmar/plantar thickening [Pond et al., 2018]. Therewithal, no overlapping signs of PS were found in any of the index family members.
The index patients exhibited coarse facial features with thick eyebrows, synophrys, and thick hair which also resemble Cantu syndrome. In addition, pachydermoperiostosis and acromegaloid facial appearance syndrome were also considered in the differential diagnosis due to having prominent supraorbital ridge and overgrowth of hands/feet. Cantu syndrome and pachydermoperiostosis can be distinguished by their typical radiological findings. Acromegaloid facial appearance syndrome was ruled out after confirmation of the genetic results by whole-exome sequencing.
It has been established that the KOGS-specific pathogenic variants in PDGFRB are mutations that activate the PI3K-AKT pathway and are responsive to imatinib, suggesting that individuals with KOGS might also be candidates for treatment with tyrosine kinase inhibitors [Zarate et al., 2019]. Two individuals with other PDGFRB-related disorders were reported who have been treated with imatinib, of which one had a complex phenotype, c.1996A>C (p.Asn666His), and showed clinical improvement in contractures of the hands and decrease in coarseness of the facial features [Pond et al., 2018]. Another patient with c.1681C>A p.(Arg561Cys) exhibiting generalized IMF showed positive response to a combination of sunitinib and vinblastine [Mudry et al., 2017]. Vertebral deformity and the intractable pain due to progressive scoliosis were the main complaints which the current family members suffered from in addition to the enlargement of the hands-feet. Therefore, further in vivo and in vitro studies are required to find out whether imatinib will be beneficial for these patients in ameliorating the progression of scoliosis.
Statement of Ethics
Written informed consent was obtained from the parent/legal guardian of the patient for publication of the details of their medical case and any accompanying images. The investigations were conducted in line with the principles detailed by the Declaration of Helsinki. This study protocol was reviewed and the need for approval was waived by ethical committee of Gaziantep University.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Funding Sources
The authors did not receive support from any organization for the submitted work.
Author Contributions
H.M.A. contributed to the design and writing of the article, and A.D.C. contributed to the interpretation of the radiological images of the patients.
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.
Acknowledgement
The authors thank the Gaziantep Ersin Arslan Research and Training Hospital, Medical Genetics Laboratory for performing the genetic analysis of the patients.
References
- Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22((10)):1276–312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster A, Chalot B, Antoniadi T, Schaefer E, Keelagher R, Ryan G, et al. Kosaki overgrowth syndrome: A novel pathogenic variant in PDGFRB and expansion of the phenotype including cerebrovascular complications. Clin Genet. 2020;98((1)):19–31. doi: 10.1111/cge.13752. [DOI] [PubMed] [Google Scholar]
- Gawlinski P, Pelc M, Ciara E, Jhangiani S, Jurkiewicz E, Gambin T, et al. Phenotype expansion and development in Kosaki overgrowth syndrome. Clin Genet. 2018;93((4)):919–24. doi: 10.1111/cge.13192. [DOI] [PubMed] [Google Scholar]
- Heldin C-H, Lennartsson J. Structural and functional properties of platelet-derived growth factor and stem cell factor receptors. Cold Spring Harb Perspect Biol. 2013;5((8)):a009100. doi: 10.1101/cshperspect.a009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Sanchez-Contreras MY, Keppler-Noreuil KM, Sapp J, Crenshaw M, Finch NA, et al. A Point Mutation in PDGFRB Causes Autosomal-Dominant Penttinen Syndrome. Am J Hum Genet. 2015;97((3)):465–74. doi: 10.1016/j.ajhg.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martignetti JA, Tian L, Li D, Ramirez MC, Camacho-Vanegas O, Camacho SC, et al. Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. Am J Hum Genet. 2013;92((6)):1001–7. doi: 10.1016/j.ajhg.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minatogawa M, Takenouchi T, Tsuyusaki Y, Iwasaki F, Uehara T, Kurosawa K, et al. Expansion of the phenotype of Kosaki overgrowth syndrome. Am J Med Genet A. 2017;173((9)):2422–7. doi: 10.1002/ajmg.a.38310. [DOI] [PubMed] [Google Scholar]
- Mudry P, Slaby O, Neradil J, Soukalova J, Melicharkova K, Rohleder O, et al. Case report: rapid and durable response to PDGFR targeted therapy in a child with refractory multiple infantile myofibromatosis and a heterozygous germline mutation of the PDGFRB gene. BMC Cancer. 2017;17((1)):119. doi: 10.1186/s12885-017-3115-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyzi O, Bundak R, Gökçay G, Günöz H, Furman A, Darendeliler F, et al. Reference values for weight, height, head circumference, and body mass index in Turkish children. J Clin Res Pediatr Endocrinol. 2015;7((4)):280–93. doi: 10.4274/jcrpe.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond D, Arts FA, Mendelsohn NJ, Demoulin JB, Scharer G, Messinger Y. A patient with germ-line gain-of-function PDGFRB p.N666H mutation and marked clinical response to imatinib. Genet Med. 2018;20((1)):142–50. doi: 10.1038/gim.2017.104. [DOI] [PubMed] [Google Scholar]
- Takenouchi T, Yamaguchi Y, Tanikawa A, Kosaki R, Okano H, Kosaki K. Novel overgrowth syndrome phenotype due to recurrent de novo PDGFRB mutation. J Pediatr. 2015;166((2)):483–6. doi: 10.1016/j.jpeds.2014.10.015. [DOI] [PubMed] [Google Scholar]
- Zarate YA, Boccuto L, Srikanth S, Pauly R, Ocal E, Balmakund T, et al. Constitutive activation of the PI3K-AKT pathway and cardiovascular abnormalities in an individual with Kosaki overgrowth syndrome. Am J Med Genet A. 2019;179((6)):1047–52. doi: 10.1002/ajmg.a.61145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.